

ABSTRACT
Until recently, the ubiquitin-proteasome system (UPS) and macroautophagy/autophagy were considered to be two independent systems that target proteins for degradation by proteasomes or via lysosomes, respectively. Here, we report that TRIM44 (tripartite motif containing 44) is a novel link that connects the UPS system with the autophagy degradation pathway. Suppressing the UPS degradation pathway leads to TRIM44 upregulation, which further promotes aggregated protein clearance through the binding of K48 ubiquitin chains on proteins. TRIM44 expression activates autophagy via promoting SQSTM1/p62 oligomerization, which rapidly increases the rate of aggregate protein removal. Overall, our data reveal that TRIM44 is a newly identified link between the UPS system and the autophagy pathway. Delineating the cross-talk between these two degradation pathways may reveal new mechanisms of targeting aggregate-prone diseases, such as cancer and neurodegenerative disease.
Abbreviations: 3-MA: 3-methyladenine; ACTB: actin beta; ATG5: autophagy related 5; BB: B-box domain; BECN1: beclin1; BM: bone marrow; CC: coiled-coil domain; CFTR: cystic fibrosis transmembrane conductance regulator; CON: control; CQ: chloroquine; DOX: doxycycline; DSP: dithiobis(succinimidly propionate); ER: endoplasmic reticulum; FI: fluorescence intensity; FL: full length; HIF1A/HIF-1#x3B1;: hypoxia inducible factor 1 subunit alpha; HSC: hematopoietic stem cells; HTT: huntingtin; KD: knockdown; KD-CON: knockdown construct control; MM: multiple myeloma; MTOR: mechanistic target of rapamycin kinase; NP-40: nonidet P-40; NFE2L2/NRF2: nuclear factor, erythroid 2 like 2; OE: overexpression; OE-CON: overexpression construct control; PARP: poly (ADP-ribose) polymerase; SDS: sodium dodecyl sulfate; SQSTM1/p62: sequestosome 1; Tet-on: tetracycline; TRIM44: tripartite motif containing 44; UPS: ubiquitin-proteasome system; ZF: zinc-finger
KEYWORDS: Aggregates, autophagy, deubiquitinase, misfolded proteins, protein homeostasis, TRIM44, ubiquitin-proteasome system
Introduction
Protein homeostasis is orchestrated by coordinated protein synthesis, folding, transport and degradation [1]. Inappropriate protein assembly or modification promotes protein misfolding, which can lead to not only disruptions to protein homeostasis but also to normal cellular functions [2]. Chaperones are regulators of protein folding processes and include the heat shock proteins. Misfolded proteins that escape these control mechanisms must be targeted for degradation, either through the UPS or by autophagic processes [3].
One crucial mechanism that marks the target protein for degradation in both the UPS and autophagy pathways is ubiquitination [4]. UPS-mediated protein degradation is mediated by the recognition of the protein substrates marked through polyubiquitination. E1-E2-E3 ubiquitin activators, conjugases and ligases work together to conjugate polyubiquitin chains to the substrate targeted for degradation [5]. Polyubiquitinated proteins, including misfolded proteins, are recognized by the ATP-dependent proteasome complex and subsequently degraded [6]. In the autophagy-mediated degradation pathway, the polyubiquitin chain is recognized by proteins such as SQSTM1/p62 and receptor proteins that recruit substrates to autophagosomes for degradation [7].
When misfolded proteins are not efficiently degraded by proteasome complexes, they form toxic aggregates, and their removal becomes critical for cell survival [8]. The accumulation of misfolded proteins and protein aggregates is a central pathological feature of many neurodegenerative diseases, such as Alzheimer, Parkinson, and Huntington [9]. Balanced proteostasis is also important for cancer cell survival. In cancer cells that have uncontrolled protein synthesis, misfolded protein accumulation suppresses growth [10]. Therefore, understanding the regulatory mechanisms of proteostasis control is critical for understanding not only normal cell growth but also malignant cell survival.
We previously discovered a member of tripartite motif (TRIM) family, TRIM44, which is highly upregulated in quiescent multiple myeloma (MM) cells isolated from the osteoblastic niches of the bone marrow. Since TRIM44 was first cloned in 2001 [11], only a few studies have reported on TRIM44 functions. We found that TRIM44 deubiquitinates HIF1A/HIF-1α to stabilize it, which in turn promotes quiescent MM cell survival. Upregulated TRIM44 promotes bone destruction in a similar manner to that observed in symptomatic MM patients [12]. Silencing TRIM44 reverses bone destruction [12].
In this study, we revealed novel TRIM44 functions in proteostasis control. TRIM44 associated with ubiquitinated aggregates via its CC domains and played critical functions in misfolded protein clearance. Upregulated TRIM44 expression in aggregate-prone models showed a remarkably reduced number of protein aggregates with decreased overall aggregate volume. In a cell model that localized misfolded proteins to aggresomes, TRIM44 colocalized with aggresome components, revealing that TRIM44 is part of an aggresome complex. In addition, TRIMM44, a deubiquitination enzyme, activated autophagy via promoting SQSTM1 oligomerization and played critical roles in aggregate removal. Taken together, our data supported a novel function of TRIM44 in aggregate binding and removal and were the first to show that TRIM44 plays an essential function in protein homeostatic control.
Results
TRIM44 protein associates with ubiquitin proteins upon proteotoxic stress
Misfolded proteins are normally degraded by the proteasome system. When the proteasomes are overloaded or the action is inhibited, misfolded proteins accumulate and form toxic aggregates [13]. Bortezomib, the first therapeutic proteasome inhibitor, is used in humans to suppress MM growth. In addition, the up-regulation of the deubiquitinase TRIM44 in quiescent MM cells supports their drug resistance [14]. In order to study the role of TRIM44 in proteostasis, we generated MM cells that in which TRIM44 was overexpressed (TRIM44[OE]) or knocked down (TRIM44[KD]) via a lentivirus (Fig. S1A) to examine the effects of TRIM44 on cell viability under the treatment of bortezomib, since the lethality caused by TRIM44 knockout [12]. Control cells were infected with relevant control vectors corresponding to the infecting virus vector (TRIM44[OE-CON] or TRIM44[KD-CON]) (Fig. S1A). Dose-dependent cell viability was measured 5 days after drugs treatment. Compared with corresponding vector transfected cells (TRIM44[OE-CON]), TRIM44[OE] cells were much more resistant to cell death induced by bortezomib (Fig. S1B). Contrary to these results, knockdown of TRIM44 led to decreased resistance to bortezomib (Fig. S1C), indicating that elevated expression level of TRIM44 increased the resistance of MM cells to bortezomib.
To analyze the role of TRIM44 in proteostasis, we treated MM (U266) cells and A549 adenocarcinoma cells with bortezomib, or MG132, another proteasome inhibitor. The concentration had been predetermined to induce aggregates in more than 40% of the cells in each lineage. In both U266 and A549 cells, TRIM44 colocalized with ubiquitin dots (Figure 1(a), Fig. S1D to F). Protein aggregates can also be concentrated near microtubule-dependent dynein motors in a perinuclear inclusion body called the aggresome [15]. TRIM44 was colocalized with ubiquitin proteins in aggresomes, which were positive for aggresome markers, such as VIM/vimentin, TUBG/γ-Tubulin, and the 20S proteasomes (Figure 1(a), Fig. S1D and E, G and H). TRIM44 was also colocalized with SQSTM1, an autophagy receptor in aggresomes (Figure 1(a), Fig. S1D and F). MG132 is an indirect ER stressor, and ER stress increases misfolded protein formation [16]. Therefore, we treated the cells using the direct ER stressors tunicamycin or thapsigargin. TRIM44 also colocalized with ubiquitin proteins in the MM and A549 cells upon ER stressor treatment (Figure 1(b), Fig. S1I).
Figure 1.
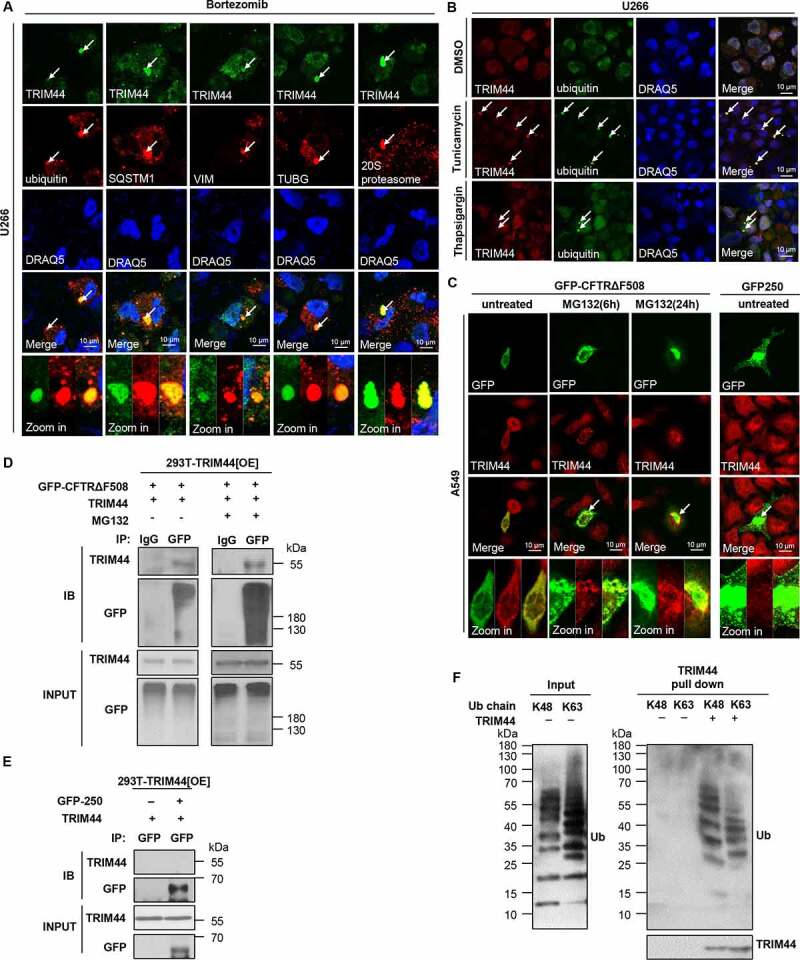
TRIM44 is associated with ubiquitin proteins. (a) U266 cells were treated with bortezomib (5 nM) for 24 h, and visualized with an antibody to ubiquitin, SQSTM1, VIM, TUBG/γ-tubulin, and 20S proteasome. Scale bars: 10 μm. (b) U266 cells were treated with DMSO or tunicamycin (1 µg/mL) for 24 h or thapsigargin (2.5 µM) for 6 h, and immunostained with antibodies against to TRIM44 and ubiquitin. Scale bars: 10 μm. (c) A549 cells were transfected with expression plasmids for GFP-CFTRΔF508 or GFP-250 as indicated. GFP-CFTRΔF508-transfected cells were treated with MG132 (5 μM) for 6 or 24 h, and visualized with an antibody to TRIM44. The colocalization of TRIM44 with GFP-250 aggresomes was assessed at 24 h after GFP-250 transfection. (d) 293 T-TRIM44[OE] cells were transfected with GFP-CFTRΔF508 plasmids, and in the presence or absence of MG132 (5 μM, 6 h) as indicated. TRIM44 was immunoprecipitated with an antibody to GFP (IgG), followed by immunoblotting with a TRIM44 or GFP antibody. (e) 293 T-TRIM44[OE] cells were transfected with GFP-250 plasmids. TRIM44 were immunoprecipitated with an antibody to GFP, followed by immunoblotting with a TRIM44 or GFP antibody. (f) Immunopurified TRIM44 produced in U266-TRIM44[OE] cells were incubated with unanchored ubiquitin chains (K48 or K63-linked Ub 1–7). The input and bound fractions were analyzed by immunoblotting with the indicated antibodies.
Aggresomes are classified as polyubiquitin-positive and polyubiquitin-negative [8]. To investigate whether TRIM44 associates with a specific type of aggresome, we used confocal microscopy to examine the colocalization of TRIM44 with CFTR (cystic fibrosis transmembrane conductance regulator) with phenylalanine 508 deleted (CFTRΔF508)-induced polyubiquitin-enriched aggresomes and with GFP-250-induced polyubiquitin-deficient aggresomes. TRIM44 colocalized with the CFTRΔF508-positive aggresomes (Figure 1(c)). On the other hand, little or no TRIM44 was found in the GFP-250-containing aggresomes, despite the formation of prominent aggresomes upon GFP-250 expression (Figure 1(c)). Results from immunoprecipitation experiments confirmed this finding. In TRIM44-expressing 293 T cells immunoprecipitated with GFP, the ubiquitin proteins were pulled down and subjected to immunoblotting with TRIM44 or GFP antibodies. The results showed that TRIM44 clearly associated with ubiquitin-positive aggresomes (Figure 1(d)) but not with ubiquitin-negative aggresomes (Figure 1(e)). Taken together, these data support the supposition that TRIM44 associates with ubiquitin proteins and autophagy receptors, such as SQSTM1, in response to proteotoxic stress.
Different polyUb linkages regulate a variety of cellular processes. lysine-48 (K48)-linked ubiquitin chains regulate protein levels by acting as signals on proteins that target them for degradation by the proteasome, while K63-linked chains are involved with “proteasome-independent” processes such as endocytic trafficking, inflammation and DNA repair [17]. Due to the colocalization of the TRIM44 and ubiquitin proteins, we analyzed the types of ubiquitin chains targeted by TRIM44. TRIM44 purified from human embryonic kidney (HEK) 293 T cells binds to free ubiquitin chains at K48 and K63 (Figure 1(f)). These data suggest that TRIM44 may regulate protein levels by binding of ubiquitin chains on proteins.
TRIM44 expression increases aggregate clearance in different aggregate-prone models
Previously, we reported that TRIM44 is highly expressed in quiescent MM stem-like cells isolated from the osteoblastic niche [12]. Since TRIM44 colocalized with ubiquitins in aggresomes and its expression led to cell resistance to bortezomib, we analyzed the roles of TRIM44 in proteotoxic stress controls. We induced endogenous aggregates in MM cells using bortezomib or MG132 and calculated the number of cells containing aggregates. Figure S2 showed the examples of MM cells without aggregates (Fig. S2A), and cells with aggregates (Fig. S2B). TRIM44 expression decreased the number of cells containing aggregates (Figure 2(a,b) Fig. S2C and D). Conversely, TRIM44 silencing increased the number of cells containing aggregates compared to the number of control cells (Figure 2(a,b), Fig. S2C and D). As a result, the total volume of aggregates was significantly reduced in the TRIM44[OE] cells but was increased in the TRIM44[KD] MM cells (Figure 2(c), Fig. S2E). We further isolated the NP40-soluble non-aggregates (non-aggregates) and 1% SDS soluble aggregates (aggregates) proteins using the differences in their solubility in NP-40 and SDS buffers and quantified their levels in MM cells. On the one hand, TRIM44 expression reduced the levels of aggregate proteins; on the other hand, the levels of non-aggregate proteins were not changed (Figure 2(d), Fig. S2F and G). Similar results were also observed in the TRIM44[Tet-on] MM cells. Following induction of TRIM44 expression with doxycycline (DOX) for 24 h (Figure 2(e)), upregulated TRIM44 significantly decreased the number of cells containing aggregates induced by bortezomib or MG132 (Figure 2(f,g), Fig. S2H and I), and the total volume of aggregates was significantly reduced (Figure 2(h), Fig. S2J). Moreover, DOX induced TRIM44 expression reduced the levels of aggregate proteins (Fig. S2K). Dose-dependent cell viability was measured 5 days after bortezomib treatment using TRIM44[Tet-on] MM cells after DOX induction. Compared with the cells without DOX induction, the cells pretreated with DOX were much more resistant to cell death induced by bortezomib (Fig. S2L).
Figure 2.
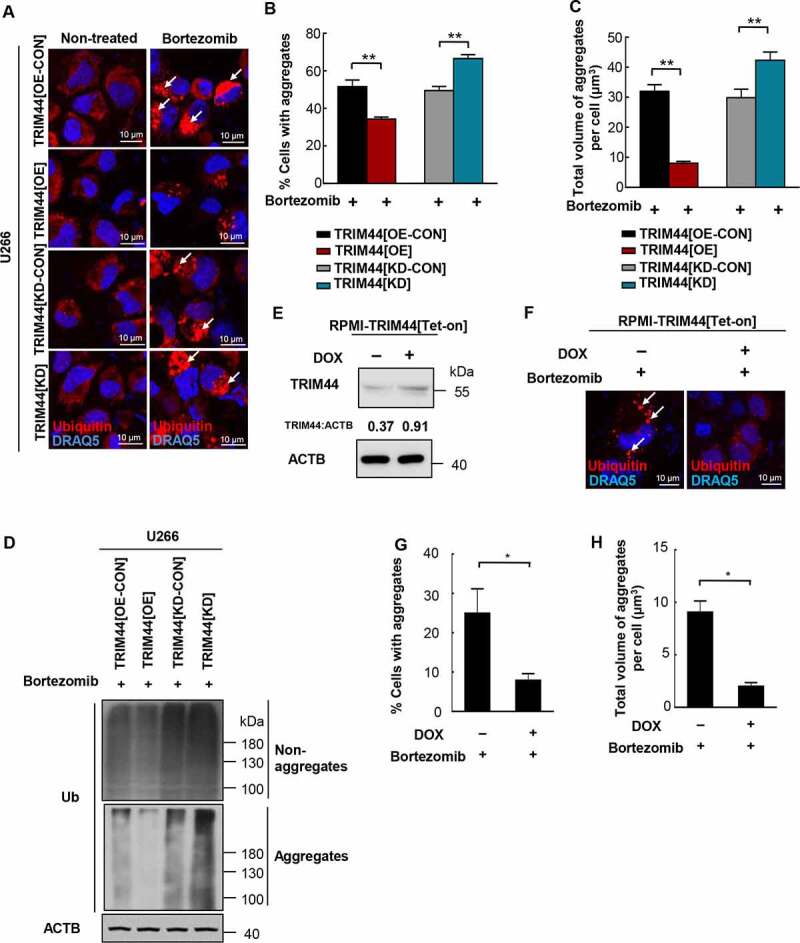
TRIM44 is required for aggregates deaggregation and clearance in MM cells. (a, b) U266 cells (TRIM44[OE-CON], TRIM44[OE], TRIM44[KD-CON] and TRIM44[KD]) were treated withor without bortezomib (5 nM) for 24 h to induce aggregates formation. Aggregates (marked by arrows) were identified by staining with the antibody against ubiquitin (a). Scale bars: 10 μm. The status of aggregates after MG132 treatment was quantified in the histogram (b). *, P < 0.05; **, P < 0.01. (c) Quantification of the total volume of aggregates in U266 cells (TRIM44[OE-CON], TRIM44[OE], TRIM44[KD-CON] and TRIM44[KD]). The volume of aggregates in 100 cells was counted for each data point shown. (d) Western blot analysis of non-aggregates and aggregates fractions of U266 cells (TRIM44[OE-CON], TRIM44[OE], TRIM44[KD-CON] and TRIM44[KD]) treated with bortezomib (5 nM, 24 h) and probe with indicated antibodies. Non-aggregates and aggregates protein were fractionated using NP40 lysis buffer. After centrifugation, the supernatant was used as non-aggregates fraction and the pellet was extracted in 1% SDS to obtain aggregates protein fraction. (e-g) RPMI-TRIM44[Tet-on] cells were treated with or without DOX (1 µg/mL, 24 h) together with bortezomib (10 nM, 24 h) treatment to induce aggregates formation. Aggregates (marked by arrows) were identified by staining with the antibody against ubiquitin. Scale bars: 10 μm. The protein level of TRIM44 were assayed by western blot (e). The status of aggregates after bortezomib treatment was quantified in the histogram (g). *, P < 0.05. (h) Quantification of the total volume of aggregates in RPMI-TRIM44[Tet-on] cells treated with or without DOX (1 µg/mL, 24 h) together with bortezomib (10 nM, 24 h) treatment to induce aggregates formation. The volume of aggregates in 100 cells was counted for each data point shown. *, P < 0.05.
To further analyze the role of TRIM44 in the proteotoxic stress control, aggregate-prone cell models were explored. Protein aggregates were induced in A549 adenocarcinoma, HeLa and N2A neuroblastoma cells using CFTRΔF508, MAPT/TauP301L and HTT (huntingtin) Q94 plasmids, respectively. We followed the protocols published for each cell model to induce aggregates [8,18,19]. The transfection of these plasmids led to protein aggregates in each cell model (Figure 3(a)).
Figure 3.
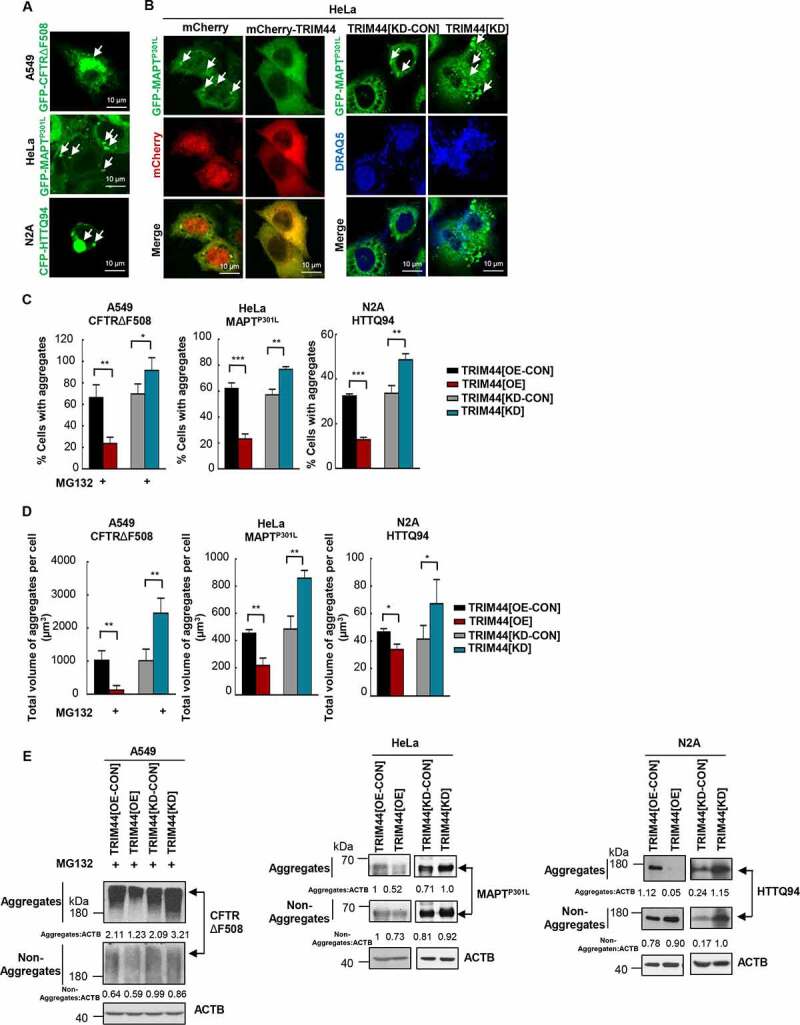
TRIM44 promotes the clearance of aggregates. (a) Aggregates prone models. A549 cells were transfected with GFP-CFTRΔF508; HeLa cells were transfected with GFP-MAPTP301L; and N2A cells were transfected with CFP-HTTQ94. Scale bars: 10 μm. (b) Representative fluorescence images of transfected cells. Left: HeLa cells were transfected with GFP-MAPTP301L, mCherry or mCherry-TRIM44. Right: HeLa cells (TRIM44[KD-CON] and TRIM44[KD]) were transfected with GFP-MAPTP301L. Forty-eight hours later, cells were fixed and then assayed for confocal. Arrowheads indicate aggregates. Scale bars: 10 μm. (c) Relative percentage of cells with aggregates (means + SD, n = 3) (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Left: CFTRΔF508-expressing A549 cells; Middle: MAPTP301L-expressing HeLa cells; Right: HTTQ94-expressing N2A cells. (d) Quantification of the total volume of aggregates per cell. The volume of aggregates in 100 cells was counted for each data point shown. Left: CFTRΔF508-expressing A549 cells; Middle: MAPTP301L-expressing HeLa cells; Right: HTTQ94-expressing N2A cells. (e) Steady-state levels of CFTRΔF508, MAPTP301L and HTTQ94 expressed in TRIM44[OE-CON], TRIM44[OE], TRIM44[KD-CON] and TRIM44[KD] cells, analyzed by western blots. Non-Aggregates: NP-40-soluble; Aggregates: NP40 insoluble, 1% SDS-soluble. Relative ratios of non-Aggregates or aggregates versus ACTB are indicated.
Consistent with the results in MM cells, TRIM44[OE] A549 cells dramatically reduced CFTRΔF508 aggregates, whereas TRIM44 silencing increased the number of cells containing aggregates (Figure 3(c), Fig. S3A). Similar results were also observed in the HeLa (Figure 3(b,c)) and N2A (Figure 3(c) Fig. S3B) cells. As a consequence, TRIM44 expression reduced the total volume of aggregates, whereas silencing TRIM44 increased the total volume of aggregates in all the aggregate cell models (Figure 3(d)). The aggregates likely removed by direct binding. TRIM44 was found to bind to the ubiquitin-positive CFTRΔF508 protein aggregates (Figure 1(d)) as well as MAPTP301L-induced aggregates (Fig. S3C). MAPT protein can be ubiquitinated, and the ubiquitination of MAPT can increase its aggregation [20]. Additionally, ubiquitin can also be recruited to mutant huntingtin aggregates [21]. Ubiquitination of the N-terminus of HTT is found to affect aggregation [22]. Moreover, MAPT and HTT appear to be targets for both the UPS and autophagy, with a key role for ubiquitin to target MAPT and HTT for degradation [23,24].These various results may indicate together that TRIM44-mediated MAPT and HTT aggregates clearance could base on its affinity to polyubiquitinated proteins. Therefore, we further isolated the aggregate and non-aggregate proteins using the differences in their solubility in NP-40 and SDS buffers and quantified their levels in cells. TRIM44 expression reduced the levels of aggregate proteins in these aggregates prone models. Consistently, silencing TRIM44 increased the level of protein aggregates, indicating that TRIM44 plays a role in protein aggregate removal (Figure 3(e)).
Different TRIM44 domains are responsible for aggregate binding and removal
TRIM44 has several known functional domains (Figure 4(a)). We generated mutant protein constructs to identify the TRIM44 domains responsible for colocalization and binding with ubiquitin proteins. The following deletional mutant constructs were generated: (i) a full-length TRIM44 (FL, 1–344); (ii) TRIM44 with only a zinc finger domain (ZF, 13–48); (iii) TRIM44 with only a B-box domain (BB, 174–215); (iv) TRIM44 with only a coiled-coil domain (CC, 290–325); (v) TRIM44 with a deleted CC domain, a construct without the CC domain (dCC, 1–290); (vi) and TRIM44 with a deleted ZF domain, a construct missing the ZF domain (dZF, 48–344) (Figure 4(a)). These mutants were transfected into HeLa cells and examined for TRIM44 and TRIM44 mutant colocalization with ubiquitins. Transfected full-length TRIM44 colocalized with ubiquitins at perinuclear aggregates. However, the TRIM44 ZF and BB mutants failed to colocalize with ubiquitins, suggesting that the ZF and BB domains are not essential domains for ubiquitin binding (Figure 4(a,b)).
Figure 4.
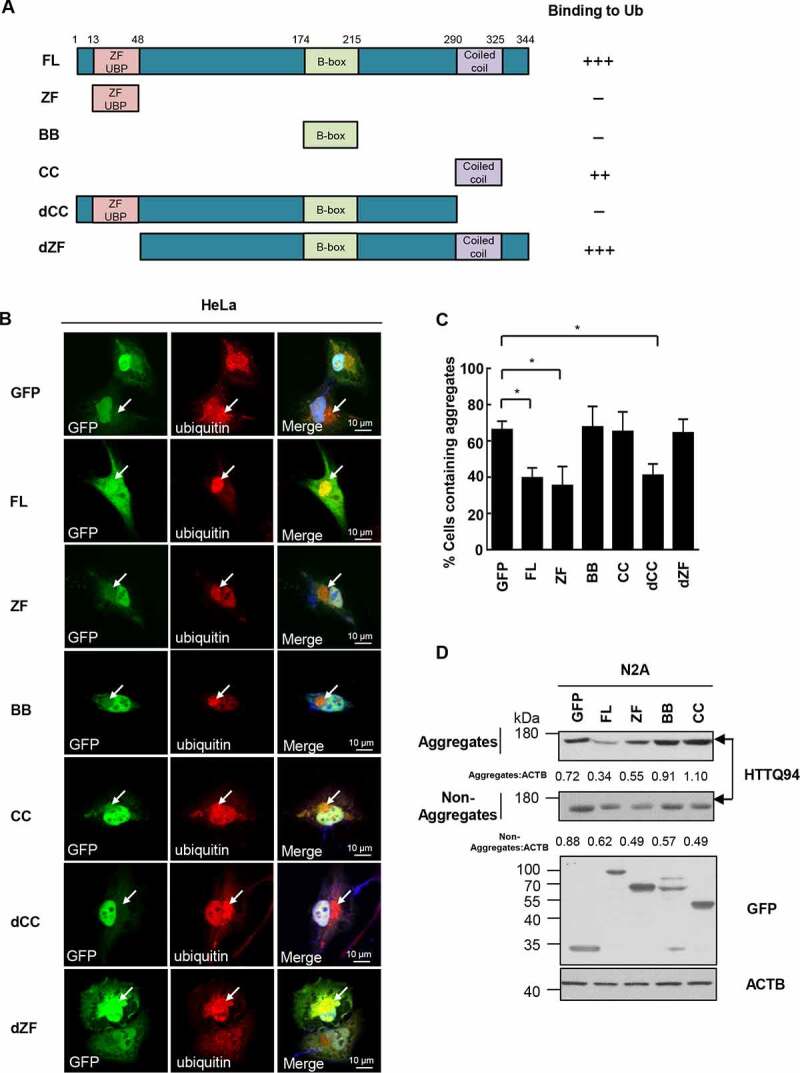
CC domain binds aggregates and ZF domain is required for aggregate clearance. (a) The TRIM44 constructs employed were GFP-tagged wild-type (WT) TRIM44; full-length (FL) TRIM44 (amino acids 1 to 344); ZF domain, a construct amino acids 13 to 48; BB domain, a construct amino acids 174 to 215; CC domain, a construct amino acids 290 to 325; dCC domain, a construct missing the CC domain (amino acids 1 to 290); and dZF domain, a construct missing the ZF domain (amino acids 48 to 344). (b) Shown are laser scanning confocal microscopy images of immunofluorescence staining for exogenous GFP-TRIM44 truncates (green) and Ubiquitin (red) with proteasomal inhibitor MG132 (5 µM) for 24 h. Cells were incubated with merged images with overlapping immunoreactivity are shown in yellow. All experiments were replicated three independent times with similar results. Scale bars: 10 μm. (c) Relative percentage of cells with aggregates (means + SD, n = 3) (*, P < 0.05). (d) Steady-state levels of HTTQ94 expressed in N2A cells transfected with GFP-tagged TRIM44 truncates (full-length, ZF domain, BB domain and CC domain), analyzed by western blots. Non-Aggregates: NP-40-soluble; Aggregates: SDS-soluble, NP40 insoluble. Relative ratios of non-aggregates or aggregates versus ACTB are indicated.
On the other hand, the CC domain of TRIM44 bound to ubiquitins, supporting that the CC domains in TRIM44 are critical for ubiquitin binding. The CC domains and dZF domains also colocalized with ubiquitins (Figure 4(a,b)). We then transfected the mutants with deleted domains into HeLa cells, treated with MG132 to induce endogenous aggregates, to determine the domains responsible for aggregate removal. The number of cells containing aggregates was then quantified. The number of aggregates in the cells transfected with full-length TRIM44 and the TRIM44 ZF or dCC mutants, which consisted of zinc finger domains, was low, indicating that the zinc finger domains are essential for aggregate removal (Figure 4(c)). We further isolated aggregate and non-aggregated HTTQ94 proteins and quantified their levels in N2A cells transfected with TRIM44 truncates. Only the cells expressing full-length TRIM44 and ZF had reduced numbers of aggregates (Figure 4(d)), which confirmed the results found using the HeLa cells. Taken together, these findings indicate that TRIM44 binds to aggregates via the CC domains, but aggregate removal requires the ZF domains.
TRIM44 expression increases autophagy processes
Aggresomes provide a platform for congregating misfolded proteins [25], which are then removed by autophagic processes [26]. Of the 67 human TRIMs, knocking down 21 TRIMs, including TRIM44, suppressed autophagy induction [27]. However, detailed TRIM44 roles in autophagy processes have not been known. TRIM44 expression increased the level of LC3B-II derived from LC3B-I (Figure 5(a,b)), indicating increased basal autophagy levels. Notably, in the presence of chloroquine (CQ), an lysosome inhibitor widely used to block autophagic flux [28], the LC3B-II level in the TRIM44[OE] cells was higher than it was in the TRIM44[OE-CON] cells, suggesting increased autophagic flux in the TRIM44[OE] cells (Figure 5(c,d)). In contrast, the LC3B-II level was markedly decreased in the TRIM44[KD] cells (Figure 5(a,b)), indicating that autophagic flux was suppressed in the TRIM44[KD] cells. To confirm our findings, we analyzed autophagy levels based on the number of LC3B-II puncta, which showed similar results that TRIM44 expression increased autophagy levels (Figure 5(e)), and down-regulation of TRIM44 decreased the number of LC3B-II puncta (Figure 5(f)).
Figure 5.
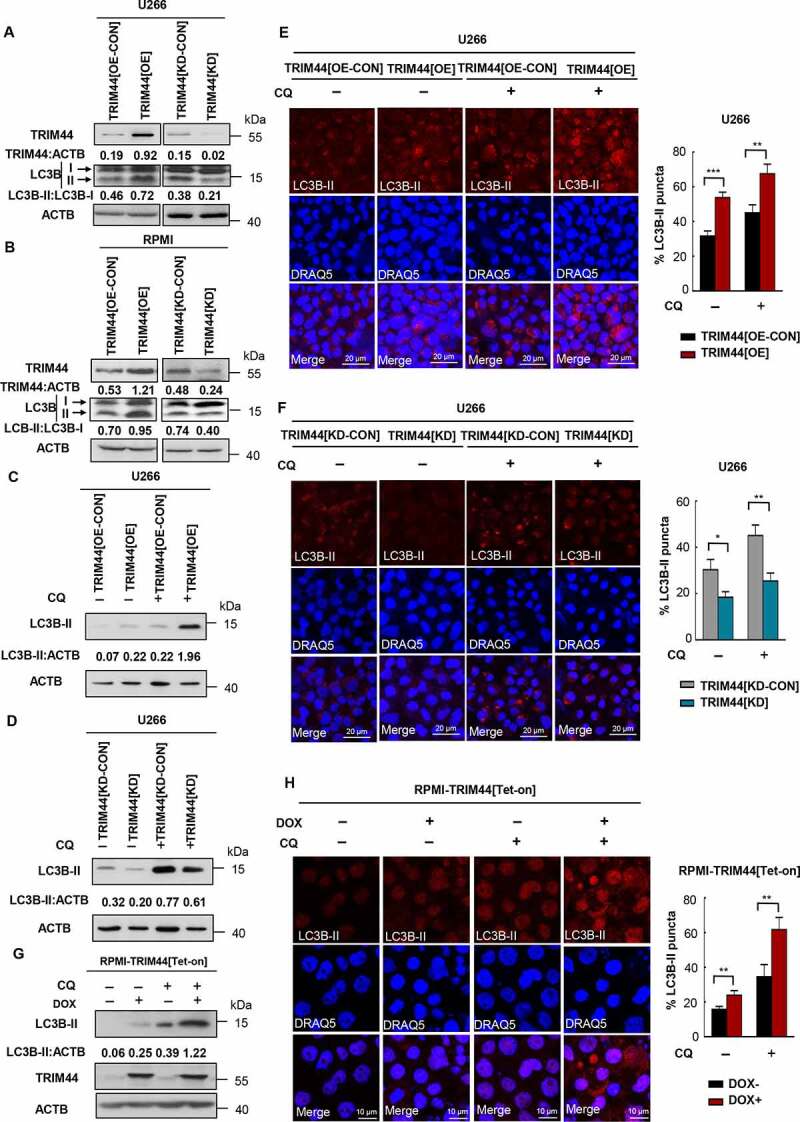
TRIM44 leads to autophagy activities in MM cells. (a, b) The protein levels of LC3B and SQSTM1 of TRIM44[OE-CON], TRIM44[OE], TRIM44[KD-CON] and TRIM44[KD] MM cells (U266 and RPMI) were assayed by western blots. (c, d) U266 cells (TRIM44[OE-CON], TRIM44[OE], TRIM44[KD-CON] and TRIM44[KD]) were treated with or without CQ (10 μM, 24 h). The protein levels of LC3B were assayed by western blots. (e) TRIM44[OE-CON] and TRIM44[OE] MM cells (U266) were treated with or without chloroquine CQ (10 μM, 24 h). And then cells were fixed and assayed for the appearance of autophagosomes by confocal microscopy. LC3B puncta were analyzed by LC3B-II antibody (magnification is 10 × 60 oil). Scale bars: 20 μm. Quantification of autophagosome formation. Cells with eight or more LC3B puncta were considered to have accumulated autophagosomes. Data are presented as mean % LC3B-positive cells ± SEM in three independent experiments. In each treatment at least 100 cells were analyzed (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (f) TRIM44[KD-CON] and TRIM44[KD] MM cells (U266) were treated with or without CQ (10 μM, 24 h). And then cells were fixed and assayed for the appearance of autophagosomes by confocal microscopy. LC3B puncta were analyzed by LC3B-II antibody (magnification is 10 × 60 oil). Scale bars: 20 μm. Quantification of autophagosome formation. Cells with eight or more LC3B puncta were considered to have accumulated autophagosomes. Data are presented as mean % LC3B-positive cells ± SEM in three independent experiments. In each treatment at least 100 cells were analyzed (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (g) RPMI-TRIM44[Tet-on] cells were treated with or without DOX (1 µg/mL) together CQ (10 μM, 24 h). The protein levels of LC3B were assayed by western blots. (h) RPMI-TRIM44[Tet-on] cells were treated with or without DOX (1 µg/mL) together with CQ (10 μM, 24 h). And then cells were fixed and assayed for the appearance of autophagosomes by confocal microscopy. LC3B puncta were analyzed by LC3B-II antibody (magnification is 10 × 60 oil). Scale bars: 20 μm. Quantification of autophagosome formation. Cells with eight or more LC3B puncta were considered to have accumulated autophagosomes. Data are presented as mean % LC3B-positive cells ± SEM in three independent experiments. In each treatment at least 100 cells were analyzed (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Similar results were also observed in the TRIM44[Tet-on] MM cells. Following induction of TRIM44 expression with DOX for 24 h (Figure 2(e)), up-regulated TRIM44 significantly increased the level of LC3B-II with or without CQ treatment (Figure 5(g)). Moreover, LC3B-II puncta number also confirmed that DOX induced TRIM44 expression increased autophagy levels (Figure 5(h)). When we analyzed the levels of autophagy in TRIM44[OE] or TRIM44[KD] HeLa cells in the presence of CQ, the LC3B-II level in the TRIM44[OE] cells was increased compared to that in the TRIM44[OE-CON] cells. In contrast, the LC3B-II level was markedly decreased by CQ in the TRIM44[KD] cells (Fig. S3D). We further analyzed whether the flux of SQSTM1 is affected by over expression or deletion of TRIM44. The SQSTM1 level was downregulated in TRIM44[OE] cells, while SQSTM1 level increased in TRIM44[KD] cells. However, there was not significant different between TRIM44[OE-CON] and TRIM44[OE], or TRIM44[KD-CON] and TRIM44[KD] cells in the presence of bafilomycin A1 (Baf A1) (Fig. S3E). In addition, we assessed a half-life of ubiquitin in TRIM44[OE] or TRIM44[KD] cells. The expression of TRIM44 resulted in shortening of ubiquitin’s half-life (Fig. S3F), while TRIM44 silencing stabilized ubiquitin and extended its half-life (Fig. S3G). Together, these support TRIM44 directly contribute increasing autophagy processes in mammalian cells, and its downregulation decreases autophagy processes.
To further discover the roles of TRIM44 during autophagic vacuole formation and related processes, we transfected HeLa cells with the GFP-RFP-LC3B plasmid. In acidic vesicles (autolysosomes), only it emits red fluoresces, and in neutral structures (autophagosomes), it emits yellow fluoresces (Fig. S3H and I). We counted an average of approximately 17 red dots and 15 yellow dots per TRIM44[OE-CON] cell. On the other hand, the numbers of both yellow and red dots were increased in the TRIM44[OE] cells, especially the numbers of red dots (40 red dots and 22 yellow dots per cell). In TRIM44[KD] cells, an average of approximately 2 red dots and 23 yellow dots per cell was counted (Fig. S3H and I). These data indicated that autolysosome step was significantly affected by TRIM44 silencing.
TRIM44 facilitates aggregate clearance by upregulating autophagy
A previous study showed that large aggregates are excluded from the proteasome but then cleared from the cytosol by autophagic processes. Autophagy improves cell viability by removing misfolded proteins [29,30]. TRIM44-containing aggregates contained the ubiquitin-binding autophagy receptor SQSTM1 and the autophagosome marker LC3B (Figure 1(a), Figure 6(a)). Treatment with proteasome inhibitor led to aggregate formation, and these aggregates were significantly reduced during the recovery period with fresh medium in TRIM44[OE] cells.
Figure 6.
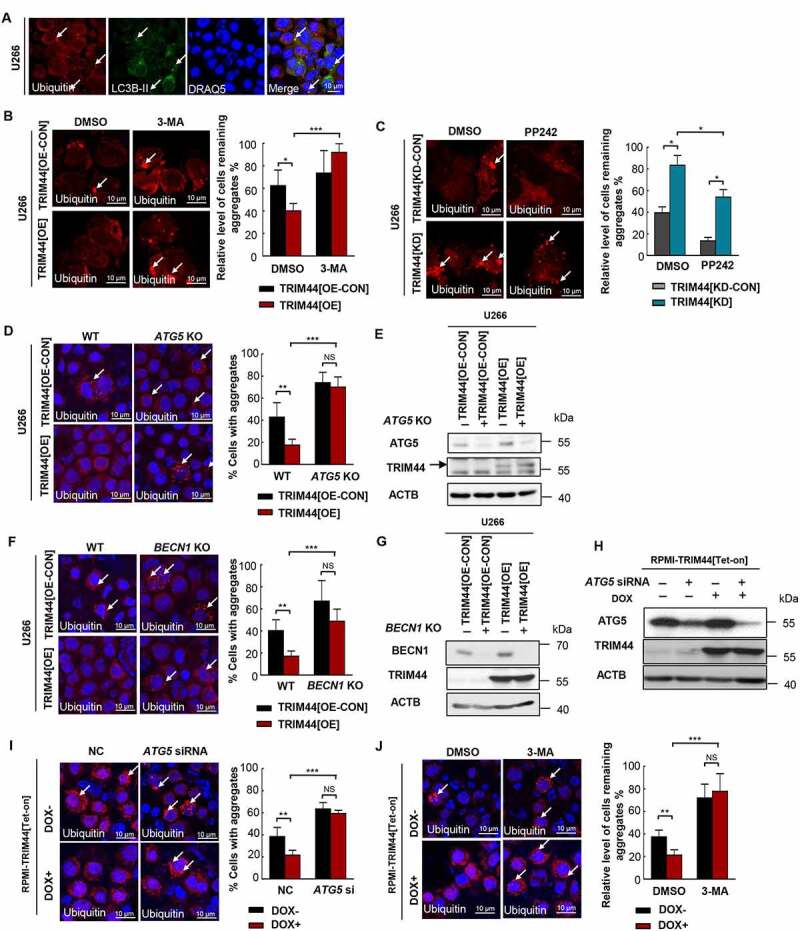
TRIM44 promtoes aggregates deaggregation and clearance via autophagy. (a) Cells were treated with MG132 and immunostained with antibodies to ubiquitin (red) and LC3B-II (green). Arrows indicate ubiquitin-positive aggregates that colocalize with LC3B-positive autophagosomes. Scale bars: 10 μm. (b) Confocal images of TRIM44[OE-CON] and TRIM44[OE] U266 cells after treatment with MG132 (0.5 µM) for 16 h followed by a 24-h chase period in normal culture media with DMSO (vehicle), or 3-MA. Arrows indicate cells with remaining aggregates. The relative level of remaining aggregates is determinded by quantifying the percentage of cells with remaining aggregates after a 24-h chase period in normal culture media with DMSO (vehicle), or 3-MA (10 mM) and normalized to the percentage of cells with aggregates formed by the 16 h MG132 (0.5 µM) treatment in corresponding cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bars: 10 μm. (c) Confocal images of TRIM44[KD-CON] and TRIM44[KD] U266 cells after treatment with MG132 (0.5 µM) for 16 h followed by a 24-h chase period in normal culture media with DMSO (vehicle), or PP242 (10 nM). Arrows indicate cells with remaining aggregates. Scale bars: 10 µm. The relative level of remaining aggregates is determinded by quantifying the percentage of cells with remaining aggregates after a 24-h chase period in normal culture media with DMSO (vehicle), or PP242 (10 nM) and normalized to the percentage of cells with aggregates formed by the 16 h MG132 (0.5 µM) treatment in corresponding cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bars: 10 μm. (d, e) Confocal images of WT or ATG5 KO TRIM44[OE-CON] and TRIM44[OE] U266 cells transfected with NC or ATG5 siRNA after treatment with MG132 (0.5 µM) for 16 h. Aggregates (marked by arrows) were identified by staining with the antibody against ubiquitin (d). Scale bars: 10 µm. The status of aggregates after MG132 treatment was quantified in the histogram. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bars: 10 μm. The protein level of ATG5 and TRIM44 were assayed by western blots (e). (f, g) Confocal images of WT or BECN1 KO TRIM44[OE-CON] and TRIM44[OE] U266 cells were treated with MG132 (0.5 µM) for 16 h. Aggregates (marked by arrows) were identified by staining with the antibody against ubiquitin (f). Scale bars: 10 µm. The status of aggregates after MG132 treatment was quantified in the histogram. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bars: 10 μm. The protein level of BECN1 and TRIM44 were assayed by western blot (g). (h, i) Confocal images of RPMI-TRIM44[Tet-on] cells treated with or without DOX (1 µg/mL) transfected with NC or ATG5 siRNA after treatment with MG132 (0.5 µM) for 16 h. The status of aggregates after MG132 treatment was quantified in the histogram. Scale bars: 10 µm. The status of aggregates after MG132 treatment was quantified in the histogram. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bars: 10 μm. The protein level of ATG5 and TRIM44 were assayed by western blots (h). (j) Confocal images of RPMI-TRIM44[Tet-on] cells treated with or without DOX (1 µg/mL) together with MG132 (5 µM) for 16 h followed by a 24-h chase period in normal culture media with DMSO (vehicle), or 3-MA. Arrows indicate cells with remaining aggregates. The relative level of remaining aggregates is determinded by quantifying the percentage of cells with remaining aggregates after a 24-h chase period in normal culture media with DMSO (vehicle), or 3-MA and normalized to the percentage of cells with aggregates formed by the 16 h MG132 (5 µM) treatment in corresponding cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bars: 10 μm.
3-Methyladenine (3-MA) blocks autophagy by inhibiting Class III PI3-kinases, which are involved in the formation of the phagophore, an early step in autophagy induction [31]. In order to demonstrate whether the autophagy induced by TRIM44 is responsible for the clearance of aggregates, we added 3-MA (10 mM) to the culture medium during the recovery period, the aggregates were retained in approximately 90% of the cells, however, the aggregates in most of the cells without 3-MA treatment were removed (Figure 6(b)). Activator of autophagy, PP242, MTOR kinase inhibitor, PP242 largely increased the removal of aggregates in TRIM44[KD] cells. Moreover, the remaining aggregates in TRIM44[KD] cells showed significantly reduced volume (Figure 6(c)). We also knocked out ATG5, an essential gene in the autophagy process, in the TRIM44[OE] MM cells. In the absence of ATG5, the aggregates were not cleared (Figure 6 (d) and E, Fig. S4A and B). Knockout BECN1/Beclin1, another essential autophagy gene, in the TRIM44[OE] MM cells could also decrease the TRIM44 mediated aggregates clearance (Figure 6 (f) and G, Fig. S4C and D). Similar results were also observed in the TRIM44[Tet-on] MM cells. Both ATG5 silencing (Figure 6(h,i)) and 3-MA treatment (Figure 6(j)) could decrease aggregates clearance in TRIM44-upregulated cells.
We also analyzed the level of MAPTP301L aggregates clearance in the HeLa cells silencing BECN1 or treated with 3-MA. TRIM44-induced degradation of the MAPTP301L aggregates was inhibited (Fig. S4E and F). When we treated the HeLa cells with the autophagy inhibitors, SAR405 and SBI-0206965, and the similar trend was observed (Fig. S4G and H). The effect of 3-MA on the level of HTTQ94 aggregate clearance in the N2A cells was also similar. TRIM44-induced degradation of the HTTQ94 aggregates was inhibited by the autophagy inhibitor 3-MA (Fig. S4I). Moreover, temporally enhancing aggrephagy could make MM cells more resistant to bortezomib. The results of ANXA5/annexin V-PI assay indicated that apoptosis rates increased under the treatment of bortezomib (Fig. S5A). Few small molecules, for example PP242, rapamycin, and torin1, induce aggrephagy by inhibiting the MTOR [32]. Compared with single agents, a combination of PP242 and bortezomib resulted in a significant decrease in apoptosis (Fig. S5A). Cleaved PARP (poly (ADP-ribose) polymerase) levels also confirmed this result (Fig. S5B). Together, these results support that supposition that TRIM44-induced autophagy contributes to aggregate clearance.
TRIM44 leads to autophagy activity via promoting SQSTM1 oligomerization
Not only TRIM44 and SQSTM1 colocalization was observed (Figure 1(a)), we also found that TRIM44 expression increased the polymerization of SQSTM1 (Figure 7(a), Fig. S5C). Furthermore, the volume of polymerized SQSTM1 was significantly reduced in the TRIM44 silencing cells compared to that of control cells (Figure 7(a), Fig. S5C). To further analyze the effects of TRIM44 on SQSTM1 oligomerization, TRIM44[OE], TRIM44[KD], and control MM cells were cross-linked with 0.4 mg/ml DSP at 4°C for 2 h, and the lysates were run under reducing or nonreducing conditions. The data showed that TRIM44 expression increased the amount of oligomerized SQSTM1 (Figure 7(b)). The ratios of oligomerized and monomeric SQSTM1 under nonreducing versus reducing conditions were quantified by immunoblotting. Taken together, these findings indicate that TRIM44 promotes SQSTM1 oligomerization.
Figure 7.
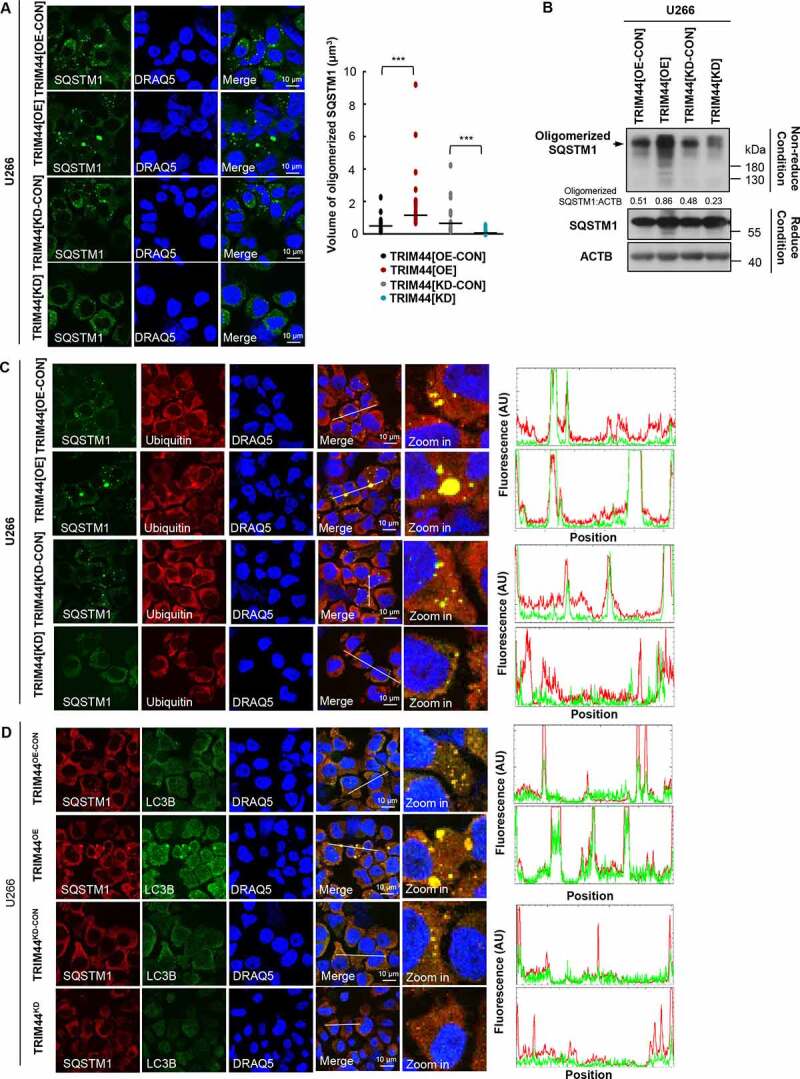
TRIM44 leads to autophagy activity via promoting SQSTM1 oligomerization. (a) TRIM44 promotes SQSTM1 oligomerization. U266 cells (TRIM44[OE-CON], TRIM44[OE], TRIM44[KD-CON] and TRIM44[KD]) and immunofluorescence imaging were analyzed using confocal microscopy (left). Graphic presentation of the average volume of SQSTM1 oligomerization 48 h after transfection. The volume of SQSTM1 oligomerization in 50 cells was counted for each data point shown (each bar) (right). Student’s t test was used for statistical analysis. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Scale bars: 10 μm. (b) MM cells (TRIM44[OE-CON], TRIM44[OE], TRIM44[KD-CON] and TRIM44[KD]) were cross-linked with 0.4 mg/ml DSP at 4°C for 2 h, and the lysates were run under reducing (with βME) or nonreducing conditions (without βME). (c) TRIM44 promotes colocalization between oligomerized SQSTM1 and substrates. HA-SQSTM1 plasmids were transfected into HeLa cells (TRIM44[OE-CON], TRIM44[OE], TRIM44[KD-CON] and TRIM44[KD]), and cells were immunostained with HA (green), ubiquitin (red) and DRAQ5 (blue) 48 h later. Relative fluorescence intensity (FI) along a white line is shown. Scale bars: 10 μm. (d) TRIM44 promotes colocalization between oligomerized SQSTM1 and LC3B. HA-SQSTM1 and mCherry-LC3B plasmids were co-transfected into HeLa cells (TRIM44[OE-CON], TRIM44[OE], TRIM44[KD-CON] and TRIM44[KD]), and cells were immunostained with mCherry (green), HA (red) and DRAQ5 (blue) 48 h later. Relative FI along a white line is shown. Scale bars: 10 μm.
It is reported that SQSTM1 is a multi-domain protein and functions in an oligomeric state in cell, and oligomeric state of SQSTM1 could enhance the binding affinity to substrates [33]. Therefore, we generated HA-tagged wild-type SQSTM1 plasmid and monomeric mutant K7A D69A. The binding between SQSTM1 or SQSTM1K7A,D69A mutant and ubiquitinated substrates was performed in HeLa cells by confocal analysis. We found that wild type SQSTM1 formed a large oligomer in HeLa cells, while the K7A D69A mutant was mostly observed as monomeric forms (Fig. S5D). Furthermore, oligomerized wild type SQSTM1 showed higher binding affinity to substrates than monomeric K7A D69A mutant (Fig. S5D).
Since TRIM44 promoted SQSTM1 oligomerization and oligomerized wild type SQSTM1 showed higher binding affinity to substrates, we examined whether TRIM44 could enhance the binding affinity of SQSTM1 with substrates. When TRIM44 was over-expressed in MM cells or HeLa cells, more SQSTM1 could be found colocalized with ubiquitin substrates compared to that of control cells, and TRIM44 silencing could disrupt this colocalization (Figure 7(c), Fig. S5E). Oligomerization of SQSTM1 was reported to be essential for its localization to the autophagosome [34], we then tested whether TRIM44 could promote SQSTM1 localize to autophagy-related structures using LC3B as a marker. As shown in Figure 7D, more colocalizations between SQSTM1 and LC3B were found in TRIM44 expressing cells, however, this colocalization decreased in TRIM44 silencing cells. These results suggest that TRIM44 promotes SQSTM1 oligomerization and targeting oligomerized SQSTM1 to autophagy-related structures, which could enhance the degradation of SQSTM1 substrates.
Since TRIM44 promotes SQSTM1 oligomerization, we examined whether TRIM44 interacts with oligomerized SQSTM1. We used the reversible crosslinking agent DSP to treat cell lysates and subjected them to co-IP, which showed that TRIM44 interacts with both the SQSTM1 monomer and polymer (Fig. S6A). We then cotransfected HA-SQSTM1 and the TRIM44 mutants with deleted domains into HeLa cells to determine the domains responsible for SQSTM1 oligomerization (Fig. S6B). The percentage of cells containing SQSTM1 aggregates was then quantified. The number of aggregates in the cells transfected with full-length TRIM44 was 60% higher than in the cells transfected with GFP alone, indicating that TRIM44 is essential for SQSTM1 aggregation (Fig. S6C). In contrast, the BB and dZF mutants significantly decreased the level of SQSTM1 aggregation (Fig. S6C). Taken together, these findings indicate that TRIM44 promotes SQSTM1 oligomerization via the ZF domain.
It has been reported that the ubiquitylation of SQSTM1 prevents its dimerization and sequestration [35]. Since TRIM44 functions as a deubiquitinase, we performed in vivo deubiquitination assays with SQSTM1. TRIM44 reduced the levels of ubiquitinated SQSTM1 (Fig. S6D and E). These data suggest that TRIM44 promotes SQSTM1 oligomerization through its deubiquitination.
TRIM16, another member of TRIM family, could stabilize and active NFE2L2 to govern the process of protein aggregates degradation [21]. Hence, we test whether TRIM44 could assess any potential role in the SQSTM1-KEAP1-NFE2L2 complex activating autophagy. We found that TRIM44 interacted with NFE2L2, especially under the treatment of MG132 (Fig. S6F). Furthermore, TRIM44 overexpression led to increased NFE2L2, whereas TRIM44 silencing decreased NFE2L2 (Fig. S6G). Taken together, these data support that TRIM44 play potential roles in SQSTM1-NFE2L2 axis modulation.
Discussion
TRIM family members are involved in many cellular functions, including regulation of the immune system, antiviral responses, autophagy-related receptor regulation, and cancer initiation [36]. Several TRIM family genes are linked to cancer as either oncogenes or tumor suppressors [37]. In gastric tumors, overexpression of TRIM28 [38] and TRIM29 [39] is associated with increased cell proliferation and poor prognosis. TRIM13, TRIM19, TRIM24 and TRIM28 regulate levels of TP53/p53 by controlling the degradation of MDM2 [40] or by inducing stabilization of TP53 by ubiquitination of MDM2 [41]. In blood cancers, TRIM62 expression is associated with reduced overall survival in acute myeloid leukemia (AML) patients [42]. TRIM67 and TRIM58 can suppress cancer initiation and progression in colorectal cancers [43,44]. Conversely, TRIM14 promotes colorectal cancer cell migration and invasion through the STAT3 pathway [45].
Unique among TRIM family proteins, TRIM44 contains a zinc finger ubiquitin protease domain (ZF-UBP), instead of a RING domain, in the N-terminus, functioning as a deubiquitinating enzyme [46]. Since TRIM44 was first cloned in 2001 [11], few studies have reported on it. TRIM44 is upregulated and linked to poor prognosis in many human tumors, such as ovarian cancer [47], esophageal squamous cell carcinoma [48], breast cancer [49], testicular germ cell cancer [50], and hepatocellular carcinoma [51]. However, no mechanisms of action for TRIM44 in tumor initiation and/or progression have been proposed. We found that TRIM44 expression increased quiescent MM cell engraftment in the OS niche and increased the ability of MM cells to compete with HSCs in the BM. As a result, TRIM44-expressing MM cells caused lytic bone lesions and bone destruction in xenograft mice [12]. One TRIM44 deubiquitination substrate is HIF1A [12], which is a key hypoxia-induced transcription factor that regulates MM tumor growth, angiogenesis and bone destruction [52]. Collectively, our data suggest that TRIM44 supports MM cancer cell survival as an oncogene and/or by stabilizing oncoproteins via deubiquitination.
In this report, we reveal novel TRIM44 functions in protein homeostasis control. Cells encounter various disruptions to the efficiency of protein folding in the ER, which leads to the accumulation of misfolded proteins. When misfolded proteins accumulate above a critical threshold, cells employ a control system called the unfolded protein response to restore protein homeostasis. Activation of the unfolded protein response triggers two distinct cellular events to decrease protein misfolding: signaling pathways to reduce protein synthesis and to enhance degradation of the misfolded proteins [53].
The UPS and autophagy were considered to be largely independent systems that targeted proteins for degradation in the proteasome and lysosome, respectively. The UPS is predominantly driven by ubiquitin as a degradation tag, which mostly degrades single, unfolded polypeptides able to enter into the narrow channel of the proteasome. Yet autophagy primarily deals with larger, cytosolic structures such as protein complexes, cellular aggregates, organelles, or pathogens [54]. Autophagy, including general and selective autophagy, is critical for cellular homeostasis with intricate links to cell metabolism, growth control, the balance between cell survival and cell death, as well as aging. Selective autophagy requires one or more selective autophagy receptors, which tag the specific cargo for engulfment in an autophagosome and delivery to the lysosome [55]. However, the identification of the crucial roles of molecular players such as ubiquitin and SQSTM1 in both of these pathways, as well as the observation that blocking the UPS affects autophagy flux and vice versa, has generated interest in studying the cross-talk between these pathways. Dysfunction within either of these two proteolytic pathways, the UPS and the autophagic-lysosomal system, which are involved in clearing incompletely folded proteins or aggregates, has been increasingly implicated in neurodegenerative diseases [56–58]. These two degradation pathways are interconnected rather than independently regulated; therefore, it is critical to understand the mechanisms of the cross-talk between these pathways.
In this report, we provide evidence that TRIM44 is a novel link connecting UPS and the autophagy degradation pathway. Suppressing UPS by treating cell models with proteasome inhibitors induced aggregate formation but upregulated TRIM44 remarkably reduced the levels of aggregate proteins and reduced the overall aggregate volumes. TRIM44 colocalized with the autophagy receptor SQSTM1 to aggresomes during proteotoxic stress. This phenomenon was observed in several aggregate-prone disease models, such as the cystic fibrosis CFTR∆F508 model, the Alzheimer disease MAPTP301L model and the huntingtin disease HTTQ94 model. Moreover, TRIM44 promotes SQSTM1 oligomerization and targeting oligomerized SQSTM1 to autophagy-related structures, which could enhance the degradation of SQSTM1 substrates.
Inappropriate protein aggregation and proteostasis imbalance are the central pathological features common to neurodegenerative diseases, including Alzheimer disease, Huntington disease, Parkinson disease, and related disorders [59,60]. Overexpressed TRIM44 leads to autophagy induction, indicating its potential use in protein-aggregation neuropathology. Moreover, TRIM44-induced autophagy can serve as a cytoprotective response by degrading aggregates, a function particularly important in the brains of the elderly and for people with neurodegenerative diseases, thus suggesting that a reduction in TRIM44 function may contribute to the age-associated accumulation of HTT or MAPT-positive aggregates. In line with our results, an Alzheimer data set analysis (http://www.alzdata.org/) revealed a reduction in TRIM44 expression in the temporal cortex of Alzheimer patients (p = 0.022, data not shown).
MM cells are derived from an incurable plasma cell malignancy that is under constitutive ER stress due to its function. Proteasome-associated ubiquitinating/deubiquitinating enzymes play central roles in modulating MM proteotoxic stress. Hence, MM cells are sensitive to compounds targeting protein homeostasis, such as proteasome inhibitors [12]. Bortezomib, the 26S proteasome inhibitor, was the first therapeutic proteasome inhibitor used in humans to suppress cell growth by inducing apoptosis in MM cells [61]. However, most patients treated with proteasome inhibitors develop resistance and eventually relapse, indicating that blocking proteasome-mediated protein degradation causes the cells to use alternative degradation pathways.
We found several ways in which the TRIM44 mechanisms can contribute to misfolded protein removal when the proteasome is not functioning. TRIM44 plays a direct role in removing aggregates by increasing autophagy flux. Silencing TRIM44 prevents autolysosome formation during autophagy processes. Moreover, TRIM44 promotes SQSTM1 oligomerization and targeting oligomerized SQSTM1 to autophagy-related structures. Together, our data suggest that TRIM44 is a novel protein that links the UPS and autophagy processes, and this is the first report of TRIM44 functions in protein homeostasis controls. The roles of TRIM44 in regulating proteotoxic stress in cancers and neurological diseases further support the idea that both of the delineated molecular pathways are involved in these processes.
Materials and methods
Cell lines
A549 (CCL-185), HeLa (CCL-2), HEK293T (CRL-3216), N2A (CCL-131), U266 (TIB-196) and RPMI8226 (CCL-155) cell lines were obtained from ATCC. A549, HeLa and HEK293T cells were cultured in in Dulbecco’s Modified Eagle’s medium (Corning, 10–013-CM) with 10% FBS (Peak serum, PS-FB1). RPMI8226 and U266 cells were cultured in RPMI medium (GE Healthcare Life Sciences, SH30255.01) with 10% FBS. N2A cells were cultured in EMEM (Corning, 10–009-CV) medium with 10% FBS.
Antibodies
Anti-TRIM44 polyclonal antibody (Proteintech Group, 11,511-1-AP); anti-Ub antibody (Biolegend, 646,301); anti-mCherry antibody (ThermoFisher, PA5-34,974); anti-VIM antibody (ThermoFisher, MA5-11,883); anti-NFE2L2/NRF2 antibody (ThermoFisher, PA5-27,882); anti-20S proteasome antibody (MilliporeSigma, ST1049); anti-TUBG/γ-Tubulin antibody (MilliporeSigma, T5326); anti-ATG5 antibody (Novus Biologicals, NBP2-24,389); Anti-GFP antibody (Santa Cruz Biotechnology, sc-9996); anti-ACTB/β-Actin antibody (Santa Cruz Biotechnology, sc-47,778), anti-HA antibody (Santa Cruz Biotechnology, sc-805); anti-BECN1 antibody (Cell Signaling Technology, 4122); anti-HDAC6 antibody (Cell Signaling Technology, 7558); anti-LC3B antibody (Cell Signaling Technology, 3868); anti-SQSTM1/p62 antibody (Cell Signaling Technology, 88,588); anti-cleaved PARP antibody (Cell Signaling Technology, 5625); Alexa Fluor 594 donkey anti rabbit IgG(H + L) (Invitrogen, A21207); Alexa Fluor 546 goat anti mouse IgG(H + L) (Invitrogen, A11003).
Regents
MG132 (Selleck Chemicals, s2619); 3-MA (Selleck Chemicals, s2767); PP242 (Selleck Chemicals, s2218); doxycycline (Selleck Chemicals, S5159); thapsigargin (Selleck Chemicals, S7895); SAR405 (Selleck Chemicals, s7682); Bortezomib (Sigma-Aldrich, 504,314); chloroquine (Sigma-Aldrich, C6628); tunicamycin (Sigma-Aldrich, T7765); SBI-0206965 (APExBIO, A8715).
Plasmids
pEGFP-C1-CFTRΔF508 was provided by Dr. Stanton, Dartmouth. GFP250 was a gift from Dr. Elizabeth S. Sztul, University of Alabama at Birmingham. GFP-MAPTP301L (46,908, Karen Ashe Lab) and CFP-HTTQ94 (23,966, Nico Dantuma Lab) were purchased from Addgene.
Confocal microscopy
The cells were fixed with 4% paraformaldehyde (Thermo Fischer scientific, AAJ61899AK) and permeabilized with 0.2% Triton X-100 (Sigma-Aldrich, 93,443). Following blocking with Animal-Free Blocker (Vector laboratories, SP-5030-250) for 1 h, followed by washing with 1× phosphate-buffered saline (PBS; Corning, 46–013-CM; pH 7.4). The cells were then incubated with indicated antibodies overnight at 4°C, washed 3 times with PBS and incubated with fluorochrome-conjugated secondary antibodies for 1 h at room temperature, washed 3 times with PBS and incubated with DRAQ5 (Cell Signaling Technology, 4084) for 30 min. Slides were analyzed by confocal microscopy (Leica TCS SP5). To quantify indicated protein levels, four or five random images were taken from each slide, and were quantified with ImageJ software (National Institutes of Health).
Co-IP and western blotting
Preparation of cell lysates and immunoblots were performed as described before. For immunoprecipitation, the indicated cells were lysed with cell lysis buffer, and the lysates were incubated with the indicated antibodies or IgG for 1 h, followed by incubation with protein A + G Sepharose beads (Santa Cruz Biotechnology, sc-2003) overnight at 4°C.
CHX chase assay
MM cells were seeded into 6-well plates at a density of 3 × 105 cells/well and incubated overnight at 37°C in a CO2 incubator. Cells were treated with 50 μg/ml of cycloheximide (CHX) dissolved in absolute ethanol, and harvested in ice cold PBS (pH 7.4) at varying chase points by centrifugation at 2500 × g for 10 min at 4°C. Cell pellets were lysed in a lysis buffer. Samples were heated at 95°C for 10 min.
Assessment of aggregates formation
MM cells were incubated in the presence or absence of bortezomib or MG132, and then processed for fluorescence confocal microscopy to assess aggregates formation. The cells containing dispersed ubiquitin were recorded as cells without aggregates. The cells containing ubiquitin spots were recorded as cells with aggregates whose percentage was averaged from at least 100 cells. The data are displayed as mean plus SD of three counts, and statistical significance was calculated and represented as the P value. *, P < 0.05; **, P < 0.01; ***, P < 0.001. And the total volume of aggregates per cell was calculated by ImageJ. At least 100 cells per experimental condition were randomly selected and scored for the presence of an aggregates in a blinded manner, and each experiment was repeated at least three times.
Analysis of aggregates clearance
MM cells were first treated with MG132 to allow formation of aggregates. After washing with the media to remove MG132, one subset of treated cells was immediately processed for fluorescence microscopy to visualize aggregates, and three parallel subsets of identically treated cells were allowed to recover for 24 h in normal media containing 0.1% DMSO (Thermo Fischer scientific, BP231-1), 100 nM PP242, or 10 mM 3-MA. Following 24 h recovery, cells were processed for evaluation of remaining aggregates. Quantification was carried out in a blinded manner from randomly selected 100 cells in each group, and experiments were repeated at least three times.
Cell lysate fraction
Cells were lysed in a NP-40-containing lysis buffer (Boston Bioproducts, BP-119) supplemented with protease inhibitors mixture (Roche Diagnostics, 11,697,498,001) and centrifuged at 15,000 x g into supernatant (Non-aggregates) and pellet (Aggregates) fractions. Both fractions were boiled in buffer containing 1% SDS and analyzed by western blot.
Statistical analysis
Data were analyzed by unpaired two-tailed Student’s t test or one- or two-way ANOVA followed by Tukey’s post hoc analysis, and P < 0.05 was considered statistically significant. Results are expressed as mean±SEM from at least three independent experiments.
Supplementary Material
Acknowledgments
We thank Dr. Bruce A. Stanton (Dartmouth Geisel school of medicine, Hanover, New Hampshire, USA) for kindly providing us with the pEGFP-C1-CFTRΔF508 plasmids. We thank Dr. Elizabeth S. Sztul (University of Alabama at Birmingham, Birmingham, Alabama, USA) for kindly providing us with the GFP-250 plasmid. This work is supported by NIH grant (R01CA181319) and CPRIT (RP200093) given to NM.
Funding Statement
This work was supported by the CPRIT [RP200093]; NIH [R01CA181319].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110(10):1389–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–899. [DOI] [PubMed] [Google Scholar]
- [3].Hipp MS, Park SH, Hartl FU. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014;24(9):506–514. [DOI] [PubMed] [Google Scholar]
- [4].Tai HC, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008;9(11):826–838. [DOI] [PubMed] [Google Scholar]
- [5].Nandi D, Tahiliani P, Kumar A, et al. The ubiquitin-proteasome system. J Biosci. 2006;31(1):137–155. [DOI] [PubMed] [Google Scholar]
- [6].Inobe T, Matouschek A. Protein targeting to ATP-dependent proteases. Curr Opin Struct Biol. 2008;18(1):43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. . [DOI] [PubMed] [Google Scholar]
- [8].Kawaguchi Y, Kovacs JJ, McLaurin A, et al. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115(6):727–738. [DOI] [PubMed] [Google Scholar]
- [9].Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. 2003;4(1):49–60. [DOI] [PubMed] [Google Scholar]
- [10].Clarke R, Cook KL, Hu R, et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012;72:1321–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Boutou E, Matsas R, Mamalaki A. Isolation of a mouse brain cDNA expressed in developing neuroblasts and mature neurons. Brain Res Mol Brain Res. 2001;86(1–2):153–167. [DOI] [PubMed] [Google Scholar]
- [12].Chen Z, Lin TC, Bi X, et al. TRIM44 promotes quiescent multiple myeloma cell occupancy and survival in the osteoblastic niche via HIF-1alpha stabilization. Leukemia. 2019;33(2):469–486. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thibaudeau TA, Anderson RT, Smith DM. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat Commun. 2018;9(1):1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].McCarty N. Battling quiescence for tumor eradication: too good to be true? Oncotarget. 2018;9(99):37276–37277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10(12):524–530. [DOI] [PubMed] [Google Scholar]
- [16].Park HS, Jun Do Y, Cr H, et al. Proteasome inhibitor MG132-induced apoptosis via ER stress-mediated apoptotic pathway and its potentiation by protein tyrosine kinase p56lck in human Jurkat T cells. Biochem Pharmacol. 2011;82(9):1110–1125. [DOI] [PubMed] [Google Scholar]
- [17].Ohtake F, Tsuchiya H, Saeki Y, et al. K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc Natl Acad Sci U S A. 2018;115(7):E1401–E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011;31(21):7604–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yamamoto A, Cremona ML, Rothman JE. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol. 2006;172(5):719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Petrucelli L, Dickson D, Kehoe K, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13(7):703–714. . [DOI] [PubMed] [Google Scholar]
- [21].Jena KK, Kolapalli SP, Mehto S, et al. TRIM16 controls turnover of protein aggregates by modulating NRF2, ubiquitin system, and autophagy: implication for tumorigenesis. Mol Cell Oncol. 2018;5(6):e1532251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Steffan JS, Agrawal N, Pallos J, et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304(5667):100–104. . [DOI] [PubMed] [Google Scholar]
- [23].Sap KA, Reits EA. Strategies to investigate ubiquitination in Huntington’s disease. Front Chem. 2020;8:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015;47(3):e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Taylor JP, Tanaka F, Robitschek J, et al. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12(7):749–757. . [DOI] [PubMed] [Google Scholar]
- [26].Lim J, Lachenmayer ML, Wu S, et al. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015;11(2):e1004987. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mandell MA, Jain A, Arko-Mensah J, et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell. 2014;30(4):394–409. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol. 2009;625(1–3):220–233. [DOI] [PubMed] [Google Scholar]
- [29].Eisenberg-Lerner A, Bialik S, Simon HU, et al. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16(7):966–975. [DOI] [PubMed] [Google Scholar]
- [30].Goldbaum O, Richter-Landsberg C. Proteolytic stress causes heat shock protein induction, tau ubiquitination, and the recruitment of ubiquitin to tau-positive aggregates in oligodendrocytes in culture. J Neurosci. 2004;24(25):5748–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Maiuri MC, Zalckvar E, Kimchi A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–752. [DOI] [PubMed] [Google Scholar]
- [32].Malampati S, Song JX, Chun-Kit Tong B, Nalluri A, Yang CB, Wang Z, Gopalkrishnashetty Sreenivasmurthy S, Zhu Z, Liu J, Su C, Krishnamoorthi S, Iyaswamy A, Cheung KH, Lu JH, Li AM. Targeting Aggrephagy for the Treatment of Alzheimer's Disease. Cells. 2020. Jan 28;9(2):311. doi: 10.3390/cells9020311. PMID: 32012902; PMCID: PMC7072705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kwon DH, Park OH, Kim L, et al. Insights into degradation mechanism of N-end rule substrates by p62/SQSTM1 autophagy adapter. Nat Commun. 2018;9(1):3291. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Itakura E, Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. 2011;192(1):17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pan JA, Sun Y, Jiang YP, et al. TRIM21 Ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate Redox homeostasis. Mol Cell. 2016;62(1):149–151. . [DOI] [PubMed] [Google Scholar]
- [36].Watanabe M, Hatakeyama S. TRIM proteins and diseases. J Biochem. 2017;161:135–144. [DOI] [PubMed] [Google Scholar]
- [37].Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11(11):792–804. [DOI] [PubMed] [Google Scholar]
- [38].Yokoe T, Toiyama Y, Okugawa Y, et al. KAP1 is associated with peritoneal carcinomatosis in gastric cancer. Ann Surg Oncol. 2010;17(3):821–828. . [DOI] [PubMed] [Google Scholar]
- [39].Kosaka Y, Inoue H, Ohmachi T, et al. Tripartite motif-containing 29 (TRIM29) is a novel marker for lymph node metastasis in gastric cancer. Ann Surg Oncol. 2007;14(9):2543–2549. . [DOI] [PubMed] [Google Scholar]
- [40].Wang C, Ivanov A, Chen L, et al. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005;24(18):3279–3290. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Joo HM, Kim JY, Jeong JB, et al. Ret finger protein 2 enhances ionizing radiation-induced apoptosis via degradation of AKT and MDM2. Eur J Cell Biol. 2011;90(5):420–431. . [DOI] [PubMed] [Google Scholar]
- [42].Quintas-Cardama A, Zhang N, Qiu YH, et al. Loss of TRIM62 expression is an independent adverse prognostic factor in acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2015;15(2):115–27 e15. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wang S, Zhang Y, Huang J, et al. TRIM67 activates p53 to suppress colorectal cancer initiation and progression. Cancer Res. 2019;79:4086–4098. [DOI] [PubMed] [Google Scholar]
- [44].Liu M, Zhang X, Cai J, et al. Downregulation of TRIM58 expression is associated with a poor patient outcome and enhances colorectal cancer cell invasion. Oncol Rep. 2018;40:1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jin Z, Li H, Hong X, et al. TRIM14 promotes colorectal cancer cell migration and invasion through the SPHK1/STAT3 pathway. Cancer Cell Int. 2018;18(1):202. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Allen MD, Bycroft M. The solution structure of the ZnF UBP domain of USP33/VDU1. Protein Sci. 2007;16(9):2072–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Liu S, Yin H, Ji H, et al. Overexpression of TRIM44 is an independent marker for predicting poor prognosis in epithelial ovarian cancer. Exp Ther Med. 2018;16:3034–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kawaguchi T, Komatsu S, Ichikawa D, et al. Overexpression of TRIM44 is related to invasive potential and malignant outcomes in esophageal squamous cell carcinoma. Tumour Biol. 2017;39(6):1010428317700409. . [DOI] [PubMed] [Google Scholar]
- [49].Kawabata H, Azuma K, Ikeda K, Sugitani I, Kinowaki K, Fujii T, Osaki A, Saeki T, Horie-Inoue K, Inoue S. TRIM44 Is a Poor Prognostic Factor for Breast Cancer Patients as a Modulator of NF-κB Signaling. Int J Mol Sci. 2017. Sep 8;18(9):1931. doi: 10.3390/ijms18091931. PMID: 28885545; PMCID: PMC5618580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yamada Y, Takayama KI, Fujimura T, et al. A novel prognostic factor TRIM44 promotes cell proliferation and migration, and inhibits apoptosis in testicular germ cell tumor. Cancer Sci. 2017;108(1):32–41. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhu X, Wu Y, Miao X, et al. High expression of TRIM44 is associated with enhanced cell proliferation, migration, invasion, and resistance to doxorubicin in hepatocellular carcinoma. Tumour Biol. 2016;37(11):14615–14628. . [DOI] [PubMed] [Google Scholar]
- [52].Bhaskar A, Tiwary BN. Hypoxia inducible factor-1 alpha and multiple myeloma. International journal of advanced research. 2016;4:706–715. [PMC free article] [PubMed] [Google Scholar]
- [53].Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334(6059):1086–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366(6467):818–822. [DOI] [PubMed] [Google Scholar]
- [55].Farre JC, Subramani S. Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol. 2016;17(9):537–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cuervo AM, Stefanis L, Fredenburg R, et al. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292–1295. [DOI] [PubMed] [Google Scholar]
- [57].Rubinsztein DC, DiFiglia M, Heintz N, et al. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1(1):11–22. . [DOI] [PubMed] [Google Scholar]
- [58].Chu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol. 2006;65(5):423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10(10):1055–1063. [DOI] [PubMed] [Google Scholar]
- [60].Jellinger KA. Recent advances in our understanding of neurodegeneration. J Neural Transm (Vienna). 2009;116:1111–1162. [DOI] [PubMed] [Google Scholar]
- [61].Roy L, Guilhot J, Krahnke T, et al. Survival advantage from imatinib compared with the combination interferon-alpha plus cytarabine in chronic-phase chronic myelogenous leukemia: historical comparison between two phase 3 trials. Blood. 2006;108(5):1478–1484. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.