

ABSTRACT
While the functions of STING1 (stimulator of interferon response cGAMP interactor 1) during DNA virus infection had been well documented, the roles STING1 plays during RNA viruses infection is obscure. Infection with foot-and-mouth disease virus (FMDV), a well-known picornavirus, induces endoplasmic reticulum (ER) stress response and autophagy. Here, we found that the FMDV-induced integrated stress response originates from the cellular pattern recognition receptor DDX58/RIG-I (DExD/H-box helicase 58). DDX58 transmits signals to the ER-anchored adaptor protein STING1, which specifically activates the EIF2AK3/PERK (eukaryotic translation initiation factor 2A)-dependent integrated stress response and finally leads to reticulophagy and degradation of STING1 itself. Knockdown/knockout of STING1 or EIF2AK3 suppresses FMDV genome replication and viral protein expression. Reticulophagy induction by STING1 does not require its translocation to the Golgi or IFN response activation. However, STING1 polymerization is necessary for the FMDV-induced integrated stress response and reticulophagy. Our work illustrated the signaling cascades that mediate the cellular stress response to FMDV infection and indicated that induction of autophagy in response to both DNA and RNA virus infection may be an evolutionarily conserved function of STING1. Abbreviations: ATF6: activating transcription factor 6; CGAS: cyclic GMP-AMP synthase; DDX58/RIG-I: DExD/H-box helicase 58; EIF2A/eIF2α: eukaryotic translation initiation factor 2A; EIF2AK3/PERK: eukaryotic translation initiation factor 2 alpha kinase 2; ER: endoplasmic reticulum; ERN1/IRE1: endoplasmic reticulum to nucleus signaling 1; FMD: foot-and-mouth disease; FMDV: foot-and-mouth disease virus; IFIH1/MDA5: interferon induced with helicase C domain 1; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MAVS: mitochondrial antiviral signaling protein; MOI: multiplicity of infection; RETREG1/FAM134B: reticulophagy regulator 1; STING1: stimulator of interferon response cGAMP interactor 1; TCID50: 50% tissue culture infectious dose; XBP1: X-box binding protein 1.
KEYWORDS: Autophagy, EIF2AK3, integrated stress response, RNA virus, STING1
Introduction
The endoplasmic reticulum (ER)-localized protein STING1 (stimulator of interferon response cGAMP interactor 1) plays pivotal roles in host defenses against DNA viruses and some bacteria [1]. Pathogen-derived DNA can be detected by cytosolic DNA sensor CGAS (cyclic GMP-AMP synthase), and binding to DNA leads to its activation and catalyzes the synthesis of cyclic GMP-AMP (cGAMP) from ATP and GTP [2]. After binding to cGAMP, STING1 undergoes a conformational change and self-activation [3]. Subsequently, activated STING1 traffics from the ER through the Golgi complex and finally to perinuclear compartments to form large punctate structures. During the trafficking process, STING1 recruits TBK1 (TANK binding kinase 1) and NFKB/NF-κB (nuclear factor kappa B), inducing the activation of IFNs (type I interferons) and other inflammatory cytokines, such as IL1B/IL-1β and IL6. In addition to its pivotal roles in anti-DNA virus activity, STING1 deficiency also increases cell or mouse sensitivity to several RNA viruses, including vesicular stomatitis virus (VSV), dengue virus (DENV), and West Nile virus (WNV) [4,5], indicating that STING1 also modulates cellular responses to RNA viruses infection. In contrast to its important signaling adaptor function in the activation of type I IFN responses to infection with DNA viruses and bacteria, STING1 is less involved in regulating RNA virus-induced IFN expression and cytokine production. Recently, STING1 was identified as an inhibitory modulator of translation during VSV infection and upon transfection of synthetic RLR ligands [6], leading to restricted production of viral proteins as well as host proteins. There is no doubt that STING1 plays important roles in modulating cellular responses to RNA virus infection, but the mechanisms by which STING1 modulates cellular responses to infection with RNA viruses remain to be fully elucidated.
FMDV is a member of the picornaviridae family, with a positive-sense RNA genome of approximately 8.5 kb. FMDV infection leads to a cellular stress response and autophagy, but it is not known how FMDV initiates the signaling cascades and whether the stress responses coordinate with or counteract innate immune responses. Previously, we showed that FMDV infection activates the EIF2AK3/PERK (eukaryotic translation initiation factor 2 alpha kinase 3)-EIF2A/eIF2α (eukaryotic translation initiation factor 2A) signaling pathway and ultimately leads to autophagy. Blocking autophagy by ATG5 knockdown suppressed FMDV replication [7]. EIF2AK3 is localized in the ER, which is the site for protein synthesis and folding. In addition to being a “protein factory,” the ER is also a signaling hub that transduces antiviral signals. We wondered whether the ER-localized antiviral protein STING1 is perturbed in response to picornavirus infection.
In this work, using a series of gene knockdown (KD)/knockout (KO) cells, we had revealed that the innate immune sensor STING1 initiated integrated stress response and autophagy during FMDV infection via its polymerization but without its translocation to the Golgi. Our research indicates that induction of autophagy by STING1 may be a conserved mechanism that cell responds to both DNA and RNA viruses.
Results
FMDV infection leads to STING1 degradation through reticulophagy
Our previous study showed that FMDV infection induced autophagy in PK15 cells in a manner dependent on the activation of the EIF2AK3-EIF2A axis, and 3 h post-FMDV infection, cells with FMDV had significantly higher level of autophagy (Fig. S1A-D). Because EIF2AK3 localizes to the ER membrane, we reasoned that FMDV infection would lead to disruption of ER-localized antiviral proteins. At the protein level, STING1 was significantly decreased at 3 h post-FMDV infection (from 4 h to 6 h, STING1 protein level gradually recovered, Fig. S1E), while the mRNA level of STING1 remained unchanged (Figure 1A, 1B and S1F). This result indicated that FMDV-induced STING1 loss was due to perturbation of protein turnover. We then tested different inhibitors of protein degradation pathways to identify the pathway responsible for STING1 loss. As shown in Figure 1C, both the proteasome inhibitor MG132 and caspase inhibitor Z-VAD-FMK failed to block STING1 loss after FMDV infection. However, when the autophagy inhibitor chloroquine (CQ) was used, FMDV infection was unable to induce STING1 loss (Figure 1C,D). Moreover, bafilomycin A1 (Baf-A1), another inhibitor of the autophagy-mediated degradation pathway, also blocked the FMDV-induced STING1 degradation (Figure 1E). These results indicate that FMDV induces STING1 degradation through the induction of autophagy.
Figure 1.
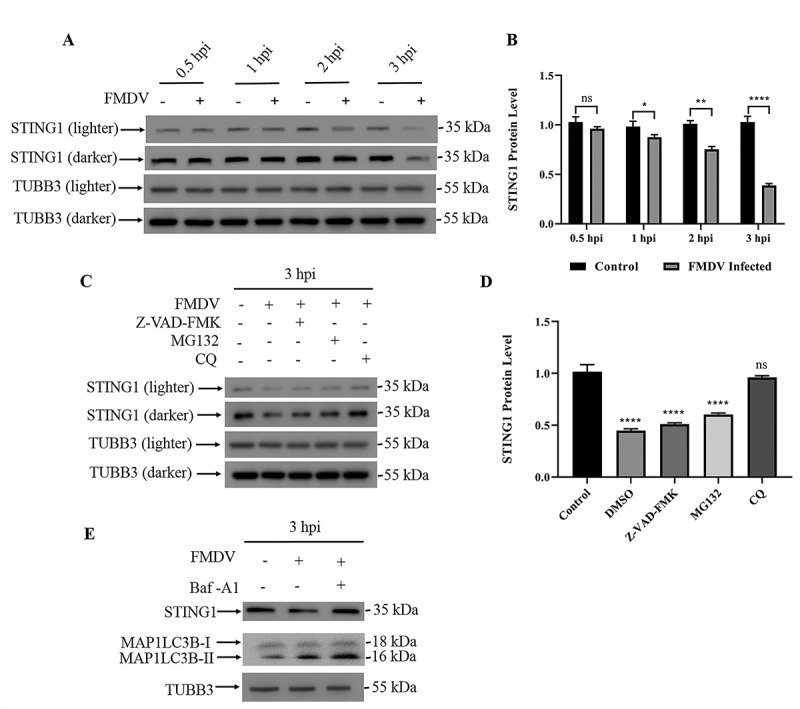
FMDV infection leads to STING1 degradation through autophagy. (A) Western blot detection of STING1 protein in FMDV-infected PK15 cells (MOI = 1) and uninfected controls (0.5 hpi, 1 hpi, 2 hpi and 3 hpi). TUBB3/β-tubulin was used as a loading control. (B) Gray values of western blot bands in (A) were analyzed by ImageJ, and STING1 values were normalized to TUBB3 intensity. The data show the mean ± SD; n = 3; ns, no significance, *P < 0.1, **P < 0.01, ****P < 0.0001. (C) Western blot detection of STING1 in cells with the indicated treatment. TUBB3 was used as a loading control. PK15 cells were either untreated or treated with the pan-caspase inhibitor Z-VAD-FMK, protease inhibitor MG-132 or autophagy inhibitor CQ when infected with FMDV (MOI = 1). (D) Gray values of western blot bands in (C) were analyzed by ImageJ, and STING1 values were normalized to TUBB3 intensity. The data show the mean ± SD; n = 3; ns, no significance, ****P < 0.0001. (E) Western blot detection of STING1 and MAP1LC3B in PK15 cells infected with FMDV (3 hpi). PK15 cells were treated with or without Baf-A1 when infected with FMDV (MOI = 1). TUBB3 was used as a loading control.
To further confirm the finding that the STING1 protein decrease was due to autophagy induction, we knocked down the expression of two autophagy pathway components, ATG7 (Figure 2A) and ATG5 (Fig. S2A). ATG7 works at the initiation of autophagy and acts similar to an E1 enzyme for ubiquitin-like proteins, such as ATG12. Once activated by ATG7, ATG12 forms a complex with ATG5 and ATG16L1, which is responsible for elongation of the phagophore. Knockdown of ATG7 or ATG5 can block the autophagy process [8]. As shown in Figure 2, knockdown of either ATG7 (Figure 2B) or ATG5 (Fig. S2B and S2C) blocked the autophagy process and decrease of STING1 protein induced by FMDV infection. However, phosphorylated EIF2AK3 and EIF2A, which are considered to be induced by FMDV that upstream of autophagy, were not affected by ATG7 knockdown (Figure 2B). We also tested whether ATG7 or ATG5 knockdown affected FMDV viral replication and VP1 protein expression. Both ATG7 (Figure 2C-E) and ATG5 (Fig. S2D-F) knockdown significantly repressed FMDV replication and viral envelope protein VP1 expression. Moreover, inhibition of autophagy with Baf-A1 also suppressed FMDV VP1 protein expression (Fig. S2G). The ER can be fragmented and delivered to the lysosome for degradation via a specific type of autophagy called reticulophagy. We wondered whether FMDV infection triggers pan-autophagy or reticulophagy. RETREG1/FAM134B (reticulophagy regulator 1) is the ER-resident receptor that binds to the autophagy modifier MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) and is required for reticulophagy but not pan-autophagy [9]. We then constructed RETREG1 knockdown PK15 cells (Figure 2F). In RETREG1 knockdown cells, the FMDV-induced autophagy induction and STING1 decrease were abolished (Figure 2G and S2H), and FMDV VP1 expression declined (Figure 2H). Our data showed that FMDV infection induced reticulophagy to degrade STING1.
Figure 2.
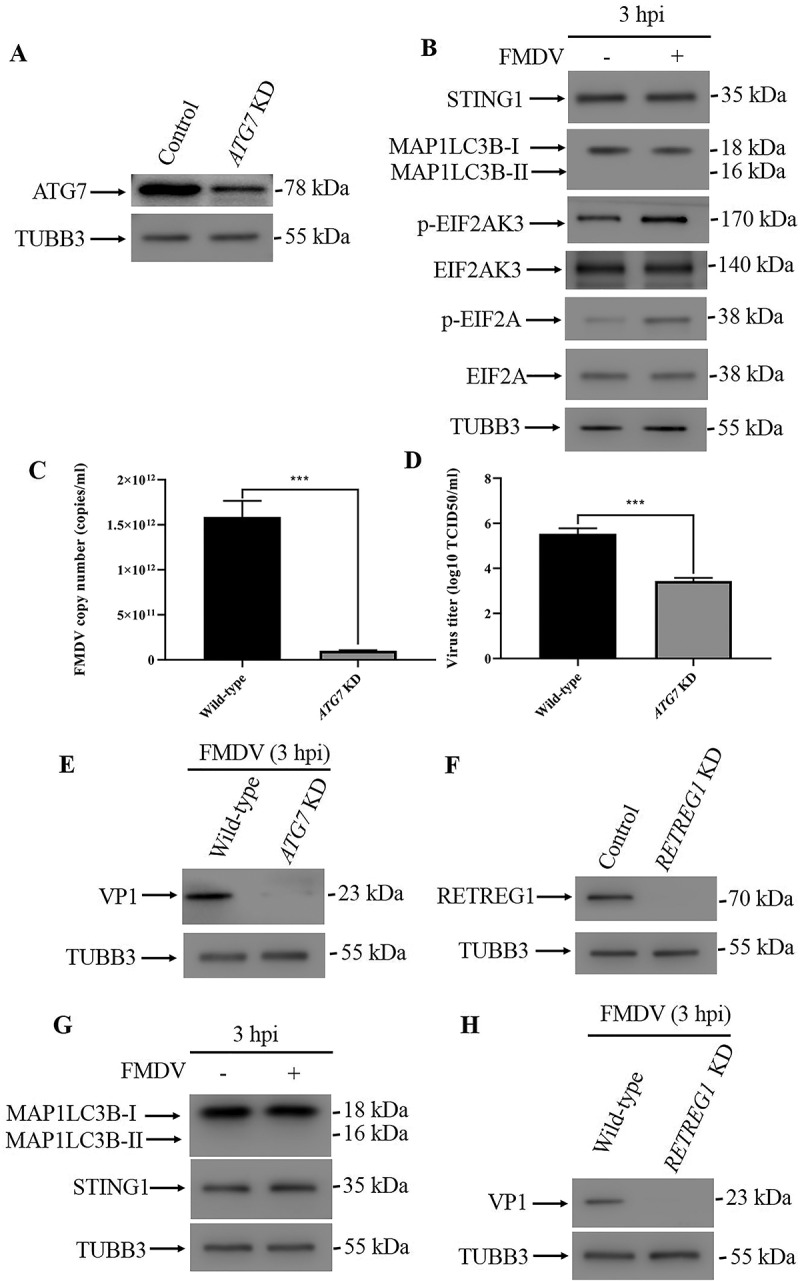
FMDV infection induced reticulophagy. (A) Stable ATG7 knockdown (KD) PK15 cells were verified by western blot. TUBB3 was used as a loading control. (B) Western blot analysis of STING1, MAP1LC3B, phosphorylated EIF2AK3 and EIF2A levels in FMDV-infected (MOI = 1) and uninfected control cells (3 hpi). TUBB3 was used as a loading control. (C) Copy numbers of FMDV in FMDV-infected wild-type and ATG7 KD cells (MOI = 1, 3 hpi) were measured by qPCR. The data show the mean ± SD; n = 3; ***P < 0.001. (D) Wild-type and ATG7 KD cells were infected with FMDV (MOI = 1), and virus titers were measured by TCID50 (3 hpi). The data show the mean ± SD; n = 3; ***P < 0.001. (E) Western blot analysis of FMDV VP1 protein in FMDV-infected wild-type and ATG7 KD cells (MOI = 1, 3 hpi). (F) Stable RETREG1 KD PK15 cells were verified by western blot. TUBB3 was used as a loading control. (G) Western blot analysis of MAP1LC3B and STING1 in FMDV-infected (MOI = 1) and uninfected RETREG1 KD cells (3 hpi). TUBB3 was used as a loading control. (H) Western blot analysis of FMDV VP1 protein in FMDV-infected wild-type and RETREG1 KD cells (MOI = 1, 3 hpi).
EIF2AK3-mediated integrated stress responses are upstream of autophagy
Previously, we showed that FMDV-induced autophagy is downstream of ER unfolded protein responses. The ER stress response generally activates three branches of signaling pathways: EIF2AK3-EIF2A-ATF4 (activating transcription factor 4), ERN1/IRE1 (endoplasmic reticulum to nucleus signaling 1)-XBP1 (X-box binding protein 1) and ATF6 signaling. Whether the three signaling pathways are all required for autophagy initiation is not known. When the expression of EIF2AK3 was inhibited, during FMDV infection (Figure 3A and S3A), no autophagy occurred, and the STING1 level remained unchanged (Figure 3B, 3C, S3B and S3C). Furthermore, the absence of EIF2AK3 significantly inhibited FMDV replication and viral structural protein VP1 expression (Figure 3D-F and S3D), implying that EIF2AK3 is required for the induction of autophagy and the STING1 protein decrease induced by FMDV. To test whether other branches of ER unfolded protein responses are involved in autophagy induction or not, we constructed ATF6 stable knockdown cells (Figure 3G). As shown in Figure 3, in comparison to wild-type cells, knockdown of ATF6 made no difference to FMDV-mediated autophagy induction and STING1 decrease (Figure 3H); meanwhile, the absence of ATF6 had no effect on viral replication or VP1 protein expression (Fig. S3E-G). To block ERN1-XBP1 signaling, we treated PK15 cells with the ERN1-specific inhibitor 4μ8 c, which could not block autophagy and rescue the decrease in STING1 protein during FMDV infection (Fig. S3H). Our results suggest that FMDV infection does not activate general ER unfolded protein responses but specifically activates the EIF2AK3-mediated integrated stress response.
Figure 3.

EIF2AK3 is required for FMDV-induced autophagy and STING1 degradation. (A) Stable EIF2AK3 KD PK15 cells were verified by western blot. TUBB3 was used as a loading control. (B) FMDV-infected (MOI = 1) and uninfected EIF2AK3 KD cells were analyzed by immunofluorescence using anti-MAP1LC3B antibodies at 3 hpi. The fluorescence signals were visualized by confocal immunofluorescence microscopy. Scale bar: 10 μm. (C) Western blot analysis of STING1 and MAP1LC3B in FMDV-infected (MOI = 1) and uninfected EIF2AK3 KD cells (3 hpi). TUBB3 was used as a loading control. (D) qPCR was used to measure the copy numbers of FMDV in FMDV-infected wild-type and EIF2AK3 KD cells (MOI = 1, 3 hpi). The data show the mean ± SD; n = 3; ***P < 0.001. (E) Wild-type and EIF2AK3 KD cells were infected with FMDV (MOI = 1), and virus titers were measured by TCID50 (3 hpi). The data show the mean ± SD; n = 3; ***P < 0.001. (F) Western blot analysis of FMDV VP1 protein in FMDV-infected wild-type and EIF2AK3 KD cells (MOI = 1, 3 hpi). (G) Stable ATF6 KD PK15 cells were verified by western blot. TUBB3 was used as a loading control. (H) Western blot analysis of STING1 and MAP1LC3B in FMDV-infected (MOI = 1) and uninfected ATF6 KD cells (3 hpi). TUBB3 was used as a loading control.
STING1 itself is required for the activation of the EIF2AK3-dependent integrated stress response during FMDV infection
Since STING1 is degraded during FMDV infection, we speculated that loss of STING1 would promote FMDV replication. To test this hypothesis, we constructed PK15 cell lines with STING1 deficiency (Figure 4A and S4A). Surprisingly, in STING1-deficient cells, both the RNA level of FMDV and the expression of VP1 decreased (Figure 4B-D and S4B). We tested autophagy levels in STING1-deficient cells during FMDV infection and found that autophagy was not induced by FMDV in either the STING1 knockdown or knockout cells (Figure 4E, 4F, S4C and S4D). Meanwhile, phosphorylation of EIF2AK3 and EIF2A was not induced by FMDV in STING1 knockdown or knockout cells (Figure 4F and S4D). These results suggest that STING1 is upstream of EIF2AK3-EIF2A signaling to induce autophagy, finally leading to a negative feedback degradation of STING1 itself. Since both STING1 and EIF2AK3 are localized in the ER, we speculated that STING1 may interact with EIF2AK3 to specifically activate the EIF2AK3-EIF2A signaling cascade. Co-immunoprecipitation results showed that EIF2AK3 interacts with STING1 (Fig. S4E).
Figure 4.
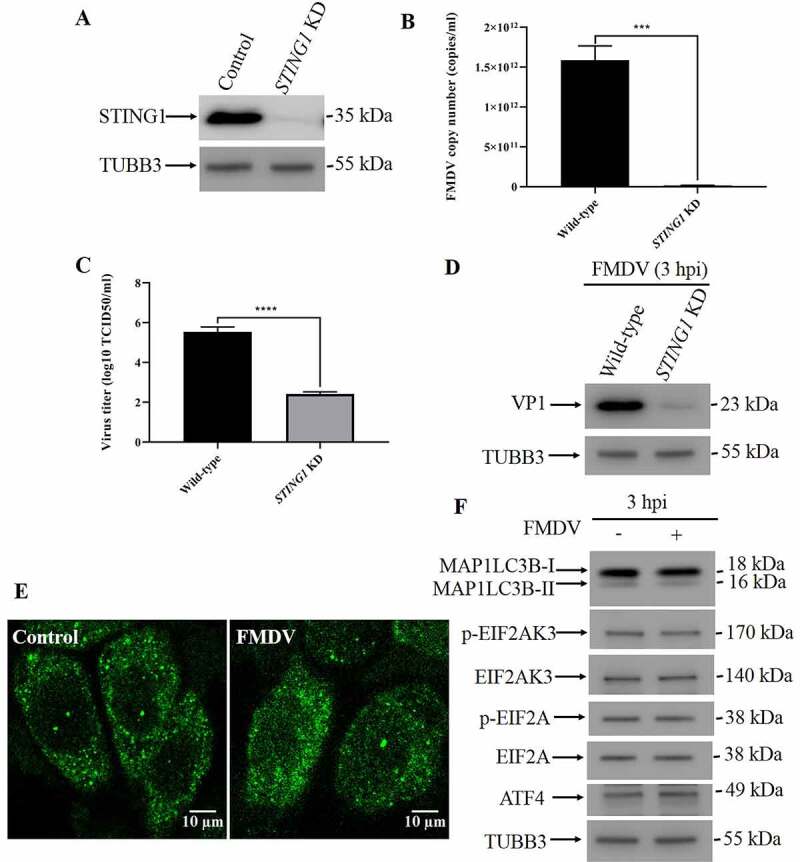
STING1 serves as an upstream molecule of EIF2AK3-EIF2A signaling to induce autophagy. (A) Stable STING1 KD PK15 cells were verified by western blot. TUBB3 was used as a loading control. (B) qPCR-measured copy numbers of FMDV in FMDV-infected wild-type and STING1 KD cells (MOI = 1, 3 hpi). The data show the mean ± SD; n = 3; ***P < 0.001. (C) Wild-type and STING1 KD cells were infected with FMDV (MOI = 1), and virus titers were measured by TCID50 (3 hpi). The data show the mean ± SD; n = 3; ****P < 0.0001. (D) Western blot analysis of FMDV VP1 protein in FMDV-infected wild-type and STING1 KD cells (MOI = 1, 3 hpi). (E) FMDV-infected (MOI = 1) and uninfected STING1 KD cells were analyzed by immunofluorescence using anti-MAP1LC3B antibodies at 3 hpi. The fluorescence signals were visualized by confocal immunofluorescence microscopy. Scale bar: 10 μm. (F) Western blot analysis of MAP1LC3B, phosphorylated EIF2AK3 and EIF2A levels in FMDV-infected (MOI = 1) and uninfected STING1 KD cells (3 hpi). TUBB3 was used as a loading control.
DDX58 but not CGAS is essential for STING1-mediated autophagy
STING1 is best known for its pivotal roles in the foreign DNA-induced IFN response, such as DNA viruses. We wondered whether upstream nucleotide sensors are required for the STING1-induced integrated stress response during FMDV infection. To test this hypothesis, we silenced the expression of CGAS (Figure 5A), the pattern recognition receptor for foreign DNA in mammalian cells. In CGAS knockdown cells, FMDV infection still led to a decrease in STING1 protein, which suggests that CGAS is not necessary for FMDV-induced, STING1-dependent autophagy (Figure 5B,5C). Recently, it has also been suggested that STING1 can transmit signaling downstream of the RNA sensors DDX58/RIG-I (interferon induced with helicase C domain 1) and IFIH1/MDA5 (interferon induced with helicase C domain 1) [10]. We also constructed DDX58 (Figure 5D), IFIH1(Figure 5G) and MAVS (mitochondrial antiviral signaling protein) knockdown cells (Fig. S5C). In DDX58 knockdown cells, FMDV infection did not induce autophagy or STING1 degradation (Figure 5E,5F). In contrast, knockdown of either IFIH1 or MAVS had no effect on FMDV-induced STING1 degradation and autophagy induction (Figure 5H, 5I and S5D). In summary, DDX58 (Fig. S5A and S5B) but not CGAS or MAVS (Fig. S5D) is the upstream molecule of STING1 for FMDV-induced autophagy.
Figure 5.
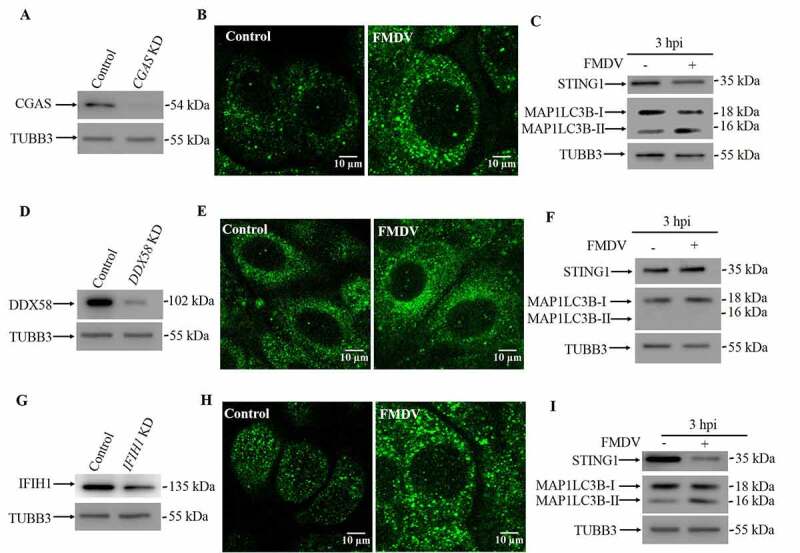
DDX58 initiated the FMDV-induced STING1-dependent autophagy response. (A) Stable CGAS KD PK15 cells were verified by western blot. TUBB3 was used as a loading control. (B) FMDV-infected (MOI = 1) and uninfected CGAS KD cells were analyzed by immunofluorescence using anti-MAP1LC3B antibodies at 3 hpi. The fluorescence signals were visualized by confocal immunofluorescence microscopy. Scale bar: 10 μm. (C) Western blot analysis of STING1 and MAP1LC3B in FMDV-infected (MOI = 1) and uninfected CGAS KD cells (3 hpi). TUBB3 was used as a loading control. (D) Stable DDX58 KD PK15 cells were verified by western blot. TUBB3 was used as a loading control. (E) FMDV-infected (MOI = 1) and uninfected DDX58 KD cells were analyzed by immunofluorescence using anti-MAP1LC3B antibodies at 3 hpi. The fluorescence signals were visualized by confocal immunofluorescence microscopy. Scale bar: 10 μm. (F) Western blot analysis of STING1 and MAP1LC3B in FMDV-infected (MOI = 1) and uninfected DDX58 KD cells (3 hpi). TUBB3 was used as a loading control. (G) Stable IFIH1 KD PK15 cells were verified by western blot. TUBB3 was used as a loading control. (H) FMDV-infected (MOI = 1) and uninfected IFIH1 KD cells were analyzed by immunofluorescence using anti-MAP1LC3B antibodies at 3 hpi. The fluorescence signals were visualized by confocal immunofluorescence microscopy. Scale bar: 10 μm. (I) Western blot analysis of STING1 and MAP1LC3B in FMDV-infected (MOI = 1) and uninfected IFIH1 KD cells (3 hpi). TUBB3 was used as a loading control.
STING1 polymerization but not translocation is required for autophagy induction
Recently, STING1 has been shown to induce autophagy upon activation by DNA viruses or foreign DNA mimics [11,12]. To test whether STING1 activation is necessary for the induction of autophagy during FMDV infection, we first treated cells with two different STING1 traffic inhibitors, golgicide A (GCA) and brefeldin A (BFA). Both inhibitors failed to block FMDV-induced autophagy (Fig. S6A and S6B). In fact, co-staining of STING1 and ERGIC marker LMAN1/ERGIC53 (lectin, mannose binding 1) showed that during FMDV infection, there is no trafficking of STING1 to ERGIC (Fig. S6C). Because these two inhibitors affect the general trafficking process, they may have side effects that make the results hard to interpret; therefore, we sought to use a STING1-specific inhibitor. Activation of STING1 requires palmitoylation at the Golgi. H-151 exerts its inhibitory action by covalently binding to STING1 at the transmembrane cysteine residue at position 91, which blocks STING1 palmitoylation [13]. However, H-151 works only in human and mouse cells. To make PK15 cells respond to H-151, we reconstituted human STING1 in STING1 knockout PK15 cells (Fig. S6D). We validated that upon FMDV infection, the reconstituted cells underwent autophagy and STING1 degradation the same as wild-type PK15 cells (Fig. S6E). In STING1-reconstituted PK15 cells, H-151 could not block autophagy and STING1 degradation (Figure 6A). Additionally, inhibition of STING1 activation did not affect viral replication or VP1 protein expression (Figure 6B-D). STING1 activation of TBK1 and NFKB depends on its C-terminal domain, and a truncation mutant of STING11−340 could not activate TBK1 and NFKB [14]. In STING11−340-reconstituted cells, FMDV infection still activated autophagy and caused STING1 degradation (Figure 6E,6F). Our results thus point to the hypothesis that STING1 trafficking and activation are not necessary for the induction of autophagy during FMDV infection.
Figure 6.
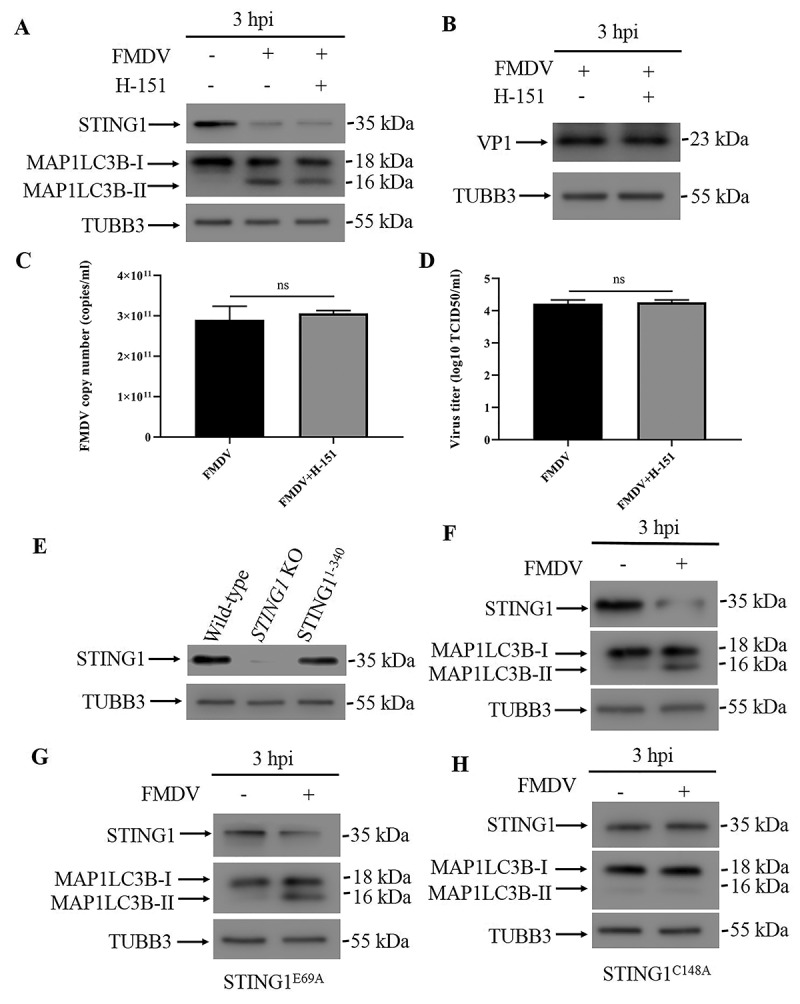
STING1 activation is not required for autophagy induction. (A) Western blot detection of STING1 and MAP1LC3B in human STING1-reconstituted STING1 KO PK15 cells infected with FMDV (3 hpi). PK15 cells were pre-treated with or without H-151 for 24 h before being infected with FMDV (MOI = 1). TUBB3 was used as a loading control. (B) Western blot detection of FMDV VP1 protein in FMDV-infected human STING1-reconstituted PK15 cells with or without H-151 treatment (3 hpi). (C) qPCR-measured copy numbers of FMDV in FMDV-infected human STING1-reconstituted PK15 cells with or without H-151 treatment (MOI = 1, 3 hpi). The data show the mean ± SD; n = 3; ns, no significance. (D) Human STING1-reconstituted PK15 cells with or without H-151 treatment were infected with FMDV (MOI = 1), and virus titers were measured by TCID50 (3 hpi). The data show the mean ± SD; n = 3; ns, no significance. (E) Stable STING1 KO PK15 cells were transfected with STING1 truncations (residues 1–340) and validated by western blot. (F) Western blot analysis of STING1 and MAP1LC3B in FMDV-infected (MOI = 1) and uninfected PK15 cells that expressed the STING1 truncation (residues 1–340) (3 hpi). TUBB3 was used as a loading control. (G) Western blot analysis of STING1 and MAP1LC3B in FMDV-infected (MOI = 1) and uninfected PK15 cells that expressed the STING1 mutant (STING1E69A) (3 hpi). TUBB3 was used as a loading control. (H) Western blot analysis of STING1 and MAP1LC3B in FMDV-infected (MOI = 1) and uninfected PK15 cells that expressed the STING1 mutant (STING1C148A) (3 hpi). TUBB3 was used as a loading control.
In recent years, the crystal structures of STING1 and STING1 binding with its ligands cGAMP and CDN have been determined. Based on the structural information, key residues required for STING1 function have also been revealed. The N-terminal segment of STING1 sticks into a deep pocket in the surface groove of STING1 and forms a charge–charge interaction with conserved the charged residues Glu68, Glu69, Arg76, Glu149 and Arg293. Substitution of Glu69 with alanine disrupts cGAMP-triggered translocation of STING1 from the ER to post-Golgi compartments [15]. Ligand-activated STING1 polymerization is stabilized by a disulfide bridge at C148, and mutation of C148 to alanine disrupts STING1 dimerization and polymerization [16]. To further illustrate how STING1 activated EIF2AK3 and autophagy during FMDV infection, we reconstituted different STING1 mutants in STING1 knockout cells (Fig. S6D). While STING1E69A cells responded to FMDV infection and underwent autophagy and STING1 degradation (Figure 6G), FMDV failed to induce autophagy in STING1C148A-reconstituted cells (Figure 6H). These results suggest that STING1 polymerization but not translocation and activation of the innate immune response is required for activating the integrated stress response and autophagy. Indeed, our native-page result showed that FMDV infection leads to STING1 polymerization (Figure 7G).
Figure 7.
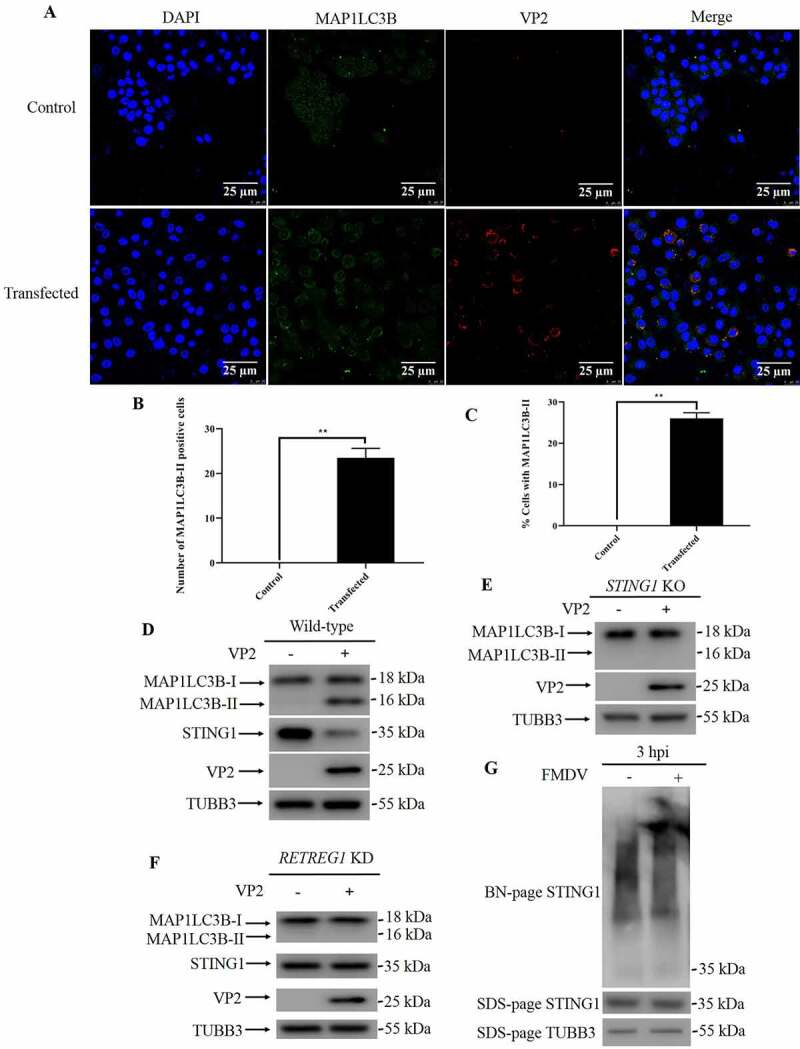
FMDV infection leads to STING1 polymerization. (A) PK15 cells were transfected with VP2 plasmids or empty vector for 24 h, followed by immunofluorescence and observed under confocal microscopy. Scale bar: 25 μm. (B) Cells from A were quantified for the number of cells with MAP1LC3B dot. The data show the mean ± SD; n = 3; **P < 0.01. (C) Cells from A were quantified for % cells with MAP1LC3B dot. The data show the mean ± SD; n = 3; **P < 0.01. (D) Western blot detection of STING1, MAP1LC3B and VP2 in PK15 cells transfected with VP2 or empty vector (24 hpt). TUBB3 was used as a loading control. (E) Western blot detection of MAP1LC3B and VP2 in STING1 KO PK15 cells transfected with VP2 or empty vector (24 hpt). TUBB3 was used as a loading control. (F) Western blot detection of STING1, MAP1LC3B and VP2 in RETREG1 KD PK15 cells transfected with VP2 or empty vector (24 hpt). TUBB3 was used as a loading control. (G) Native-page detection of STING1 in PK15 cells infected with FMDV (3 hpi). STING1 and TUBB3 protein levels were analyzed by SDS-page.
Discussion
FMDV, member of picornavirus, is one of the most notorious animal viruses for livestock and still represents a long-lasting threat in many less developed countries. It has been reported by several groups that FMDV infection leads to autophagy in host cells. We and others have found that FMDV infection leads to activation of EIF2AK3-EIF2A cascades and then autophagy. However, the upstream events of EIF2AK3 activation are not clear. In our previous work, we showed that overexpression of VP2 is able to induce autophagy. In this work, we found that STING1 KO or RETREG1 KD attenuated VP2 transfection-induced autophagy (Figure 7A-F). This result suggests that VP2 also induces reticulophagy. During FMDV infection, viral protein gradually accumulated, but reticulophagy only occurred at 3 hpi, so factors other than VP2 must regulate the reticulophagy process. In the present study, we found that STING1 is responsible for FMDV-induced integrated stress responses and autophagy (Fig. S7).
It has recently been shown that the induction of autophagy is an evolutionarily conserved function of CGAS-STING1 signaling cascades. Gui et al found that during DNA virus infection or HT-DNA transfection, cGAMP binding and STING1 translocation are necessary for autophagy initiation [12]. Liu et al reported that STING1 directly recruits MAP1LC3B to initiate autophagy [11]. These studies suggest that STING1 counteracts DNA viruses and bacteria by inducing autophagy and IFN responses. Many RNA viruses can also induce autophagy during infection, but whether STING1 plays a role in RNA virus-induced autophagy has not been reported. Here, we show that FMDV-induced autophagy also requires STING1.
Unlike DNA virus-induced autophagy, in FMDV-induced autophagy, STING1 translocation is not necessary. However, polymerization of STING1 and activation of the EIF2AK3-dependent integrated stress response are essential prerequisites. In a recent study using the gram-positive bacterium L. innocua as a pathogen, Julien et al found that STING1 engagement by the vita-PAMP c-di-AMP controls the integrated stress response and finally leads to reticulophagy [17]. Our results showed that during RNA virus infection, STING1 engagement by DDX58 also induces the integrated stress response and reticulophagy. It has not been tested whether activation of the integrated stress response precedes STING1-dependent autophagy during DNA virus infection. Thus, STING1 could induce autophagy during both DNA virus infection and RNA virus infection through different mechanisms. We provided evidence that STING1 can interact with EIF2AK3, but whether STING1 polymerization specifically and directly activates EIF2AK3 requires further investigation. In a recent work reported by Dr. Kagan, they found that RNA virus infection leads to a DDX58-mediated and STING1-dependent pathway to translation shutdown, independent of STING1 trafficking and MAVS. While the precise mechanism by which STING1 influences translation is unclear, their result suggests that this process is distinct from that mediated by PRKRA/PKR (protein activator of interferon induced protein kinase EIF2AK2) and EIF2A phosphorylation. Their results support a model where RLRs detect a common upstream ligand (viral RNA) and then engage distinct sets of downstream factors to induce IFN expression or translation inhibition [6]. Our results are in accordance with Dr. Kagan’s work. Upon detection of RNA virus infection by DDX58, it engages two different sets of downstream pathways to induce IFN expression and integrated stress response and reticulophagy. Next, it will be important to uncover the mechanisms by which DDX58 transmits signals to STING1 and downstream translation inhibition/integrated stress response.
In conclusion, our research revealed that in addition to mediating autophagy in response to foreign DNA, STING1 also plays an important role in induction of integrated stress responses and autophagy in response to RNA viruses. Thus, STING1 sits at the intersection of cellular stress responses and innate immune responses. Modulation of STING1 represents an attractive approach to counteract both DNA and RNA viruses.
Materials and methods
Cell culture and maintenance
Porcine kidney (PK15) cells were from the ATCC (ATCC CCL-33), and Lenti-X™ 293 T cells were from Clontech (632180). The cells were all grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, C14190500BT) supplemented with 10% FBS (Gibco, 10099–141), 100 U/ml penicillin and 50 μg/ml streptomycin (Gibco, 15140–122). Cells were grown in monolayers in tissue culture flasks or dishes at 37°C in a humidified atmosphere containing 5% CO2 (Binder, CB160).
Antibodies and reagents
Rabbit polyclonal antibody against MAP1LC3B/LC3B (2775s), rabbit monoclonal antibody against STING1 (D1V5L) (50494S), were from Cell Signaling Technology. Mouse monoclonal antibody against TUBB3/β-tubulin (66240-1-Ig), rabbit polyclonal antibody against ATF4 (10835-1-AP), mouse monoclonal antibody against GFP (66002-1-Ig), rabbit polyclonal antibody against EIF2A/EIF2S1 (11170-1-AP), rabbit polyclonal antibody against EIF2AK3/PERK (24390-1-AP), rabbit polyclonal antibody against IF1H1/MDA5 (21775-1-AP), rabbit polyclonal antibody against LMAN1/ERGIC53 (13364-1-AP), rabbit polyclonal antibody against DDX58/RIG-I (20566-1-AP), rabbit polyclonal antibody against MAVS (14341-1-AP) and rabbit polyclonal antibody against RETREG1/FAM134B (21537-1-AP) were from Proteintech. Mouse monoclonal antibody against ATF6 (ab11909), rabbit polyclonal antibody against phospho-EIF2A/EIF2S1 [E90] (Ser51) (ab32157), donkey anti-mouse IgG (Alexa Fluor® 488, ab150105), goat anti-rabbit IgG (Alexa Fluor® 594, ab150080) and rabbit monoclonal antibody against ATG7 [EPR6251] (ab133528) were from Abcam. Rabbit polyclonal antibody against phospho-EIF2AK3/PERK (Thr981) (sc-32577) was obtained from Santa Cruz Biotechnology. Rabbit polyclonal antibody against CGAS (ABF124) was from EMD Millipore Corp. Rabbit polyclonal antibody against ATG5 (NB110-53818) was from Novus Biologicals. Mouse monoclonal antibody against DDDDK/FLAG (M20008) were from Abmart. Goat anti-rabbit IgG (Alexa Fluor® 488, A11070) and goat anti-mouse IgG (Alexa Fluor® 594, A21125) were from Invitrogen. Mounting medium, antifading (with DAPI [S2110]) was from Solarbio. Mouse monoclonal antibody against FMDV serotype O VP1 was prepared by our laboratory. Secondary goat anti-rabbit polyclonal antibody (170–6515) and secondary goat anti-mouse antibody (170–6516) were from Bio-Rad. MG132 (474787) and Z-VAD-FMK (627610) were purchased from Merck & Co. CQ (509272) was from Sigma-Aldrich. Baf-A1 (B-1080) was from LC Laboratories. 4µ8 c (14003–96-4), GCA (1139889–93-2) and BFA (20350–15-6) were from Selleck.cn. H-151 (941987–60-6) was from InvivoGen.
Virus propagation
FMDV O strain was obtained from OIE/National Foot-and-Mouth Disease Reference Laboratory (Lanzhou, Gansu, P.R. China). To prepare viral stock, adsorption of FMDV on PK15 cells at 37°C for 1 h, unbound viruses were removed by washing three times with phosphate-buffered saline (PBS, Hyclone, SH302561.01). Coated cells with DMEM containing 2% FBS and incubated at 37°C for 10 h in an atmosphere of 5% CO2. Viral stock was clarified by centrifugation at 1000 g for 10 min. The titer was determined by Reed-Muench method [18] and expressed as 50% tissue culture infectious does (TCID50).
Pharmaceutical treatment and virus infection
Viral supernatant containing certain concentration of above-mentioned inhibitor was adsorbed into PK15 cells with multiplicity of infection (MOI) of 1 at 37°C for 1 h and the unbound virus was removed via washing with PBS. Overlaid cells with DMEM containing 2% FBS and definite concentration of described inhibitor and cultured under the condition of 5% CO2 at 37°C. 4µ8 c and H-151 was used to pretreat the PK15 cells for 24 h before infected with FMDV as described above. Cells were harvested at indicated time for the preparation of the protein samples of western blot.
Construction of vectors
Human STING1 cDNA (accession no. 3. NM_198282.4) was cloned from HEK293 cDNAs with KOD One PCR Master Mix (Toyobo, KMM-101), and are validated by sequencing. cDNAs was cloned into pLVX-puro lentiviral vector with NEBuilder HiFi DNA Assembly Cloning Kit (Neb, E5520). Mutants of STING1 were obtained using Quick Change Site-Directed Mutagenesis Kit (Stratagene, 200519) according to manufacturer’s specification, primers for PCR and mutagenesis are listed below:
STING1 (Human) Forward: 5′-CTAGCTCGAGATGCCCCACTCCAGCCTGCAT-3′
STING1 (Human) Reverse: 5′-CTAGGGATCCCTTGTACAGCTCGTCCATGCC-3′
STING1E69A: 5′-GCAGCCTGGCTGAGGCGCTGCGCCACATCCA-3′
STING1C148A: 5′-TGAGATCTCTGCAGTGGCTGAAAAAGGGAATTTC-3′
For construction of lentiviral shRNA vectors, shRNA are designed using BLOCK-iT™ RNAi Designer (Invitrogen). Primers are synthesized and cloned into pLKO.1 vector by digestion and ligation, shRNA sequences are shown in the following:
ATF6: 5′-GCTGTCCAATACACAGAAACC-3′
ATG5: 5′-GCACACCACTGAAATGGCATT-3′
ATG7: 5′-GCTGGTTTCCTTACTTAAACA-3′
EIF2AK3: 5′-GCAGATCACTAGTGATTATCA-3′
RETREG1: 5′-GCGAAAGCTGGGAAGTTATCA-3′
STING1: 5′-GCTCGGATCCAAGCTTATAAT-3′
CGAS: 5′-GCGGAGAGTCCGAGTTCAAAG-3′
IFIH1: 5′-GCAGAAGAAGGTCTGGATATT-3′
DDX58: 5′-GCAAAGATATATTGTGCTAGG-3′
MAVS: 5′-GGATGGATAGCCAGCCTTTCT-3′
For construction of lentiviral CRISPR-Cas9 vectors, sgRNA are designed using sgRNA Designer from Feng Zhang’s lab. Primers are synthesized and cloned into Lenti CRISPR v2 vector by ligation, sgRNA sequences are as follows:
STING1: 5′-CCCCCAAAGGGCCACCAAGC-3′
EIF2AK3: 5′-GCGCCGCGCGACCACCGTTC-3′
DDX58: 5′- GCGGAATCTGCACGCTTTCG-3′
Lentivirus packing and infection
For packaging lentiviruses, 4 µg pLKO.1/ plasmid, 2 µg psPAX2 packaging plasmid (Addgene, 12260) and 1 µg pMD2.G envelope plasmid (Addgene, 12259) were co-transfected into 4 × 106 Lenti-X™ 293 T cells with 21 µl Lipofectamine™ 3000 Transfection Reagent (Thermo Fisher Scientific, L3000015). The supernatants containing lentiviruses were collected, filtered and stored at −80°C as aliquots. For infection, PK15 cells were incubated with viral stocks supplemented with 8 mg/mL polybrene (Solarbio, H8761) for 24 h, and then supplied with fresh medium. Cells were selected with hygromycin (Invitrogen, 10687010) or puromycin (Invitrogen, A1113803) at 24 h post-infection.
Western blot
Proteins were separated on a 10% electrophoresis acrylamide gel and then transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, ISEQ00010). Membranes were blocked in in PBS 0.01% Tween 20 (Sigma, P7949-500ML) and (5%) nonfat dry milk (BD, 232100) for 2 h at room temperature (RT) and incubated with primary antibody at 4°C overnight. Washing with PBS-Tween 20 for 10 min (three times) and incubated with peroxidase-conjugated secondary antibody (Bio-Rad, 170–6515 and 170–6516) for 1 h at RT. Membranes were washed and visualized by enhanced chemiluminescence (Bio-Rad, 170–5061). Mouse horseradish peroxidase-coupled monoclonal antibody specific to TUBB3/β-tubulin (Proteintech, 66240-1-Ig) was used as a loading control. Bands were detected using GE healthcare Amersham™ Imager 600 in automatic exposure model to make sure that bands are not saturated. Quantification of western blots was done by normalization to TUBB3 using ImageJ software (NIH). At least 3 independent experiments were performed.
Co-immunoprecipitation
PK15 cells were harvested and lysed in protease and phosphatase inhibitor (Thermo Fisher Scientific, A32959) in RIPA lysis buffer (Beyotime, P0013B) supplemented with 1% 100 mM PMSF (Thermo Fisher Scientific, 36978). The clarified fraction of whole cell extracts was incubated with antibody and protein G-Sepharose beads (GE Healthcare, 17–0618-01) at 4°C overnight. The precipitates were washed with PBS-Tween 20 three times and eluted by boiling in 1× SDS loading buffer. The sample was detected by western blot.
Immunofluorescence
Cell were grown in glass-bottom dishes (NEST, 801001). Cells infected with FMDV at 1 MOI were fixed with 4% paraformaldehyde (Solarbio, P1110) for 15 min and treated with 0.2% Triton X-100 (Solarbio, T8200) for 10 min for permeabilization. Glasses were blocked in 5% bovine serum albumin (MP, 9048–46-8) in PBS for 1 h and then incubated with primary antibody overnight at 4°C. The next day cells were incubated with corresponding fluorochrome-conjugated secondary antibody for 1 h at RT under dark condition. Samples were then imaged using a confocal fluorescence microscope (Leica SP8, Germany).
Quantitative real-time PCR
PK15 cells were harvested at indicated time point (3 hpi) and total RNA was extracted using a RNeasy Plus Universal Mini Kit (QIAGEN, 73404) according to the manufacturer’s protocol. The first strand cDNA was synthesized using a Maxima H Minus cDNA Synthesis Master Mix with dsDNase kit (Thermo Fisher Scientific, M1682). PowerUp SYBR Green Master Mix (Applied Biosystems, 1801040) was used to perform the quantitative real-time PCR and the thermal cycling conditions were set as the manufacturer’s manual indicated. The primers used in qPCR were shown below:
STING1 Forward: 5′-CTCCCAGCAGATAGGACTGC-3′
STING1 Reverse: 5′-CCTTGTCTCGGATGGAGAAG-3′
FMDV 3D Forward: 5′-ACTGGGTTTTAYAAACCTGTGATG-3′
FMDV 3D Reverse: 5′-TCAACTTCTCCTGKATGGTCCCA-3′
GAPDH Forward: 5′-GTCGGTTGTGGATCTGACCT-3′
GAPDH Reverse: 5′-AGCTTGACGAAGTGGTCGTT-3′
STING11−340 Forward: 5′-ATGCCCCACTCCAGCCTGCA-3′
STING11−340 Reverse: 5′-CTCTTCCTTTTCCTCCTGCC-3′
Statistical analysis
Data were expressed as mean ± standard deviation (SD). The significance of the variability between different treatment groups was analyzed by two-way analysis of variance (ANOVA) test via GraphPad Prism software (version 7.0). Differences were considered statistically significant at P < 0.05.
Supplementary Material
Acknowledgments
We thank Dr. Kun Li and Dr. Yuanfang Fu (Lanzhou Veterinary Research Institute) for anti-FMDV antibodies. We thank Dr. Rongzeng Hao (Lanzhou Veterinary Research Institute) for help with FMDV recovery experiments.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Funding Statement
This work was supported by the National Key R & D Program of China (2016YFE0204100), the National Natural Science Foundation of China (31801191), Southwest Minzu University Research Startup Funds (125900/16011211013).
Supplementary material
Supplemental data for this article can be accessed here.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Ishikawa H, Barber GN.. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sun L, Wu J, Du F, et al. Cyclic GMP-AMP synthase is a Cytosolic DNA sensor that activates the Type I interferon pathway. Science. 2013;339(6121):786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhang X, Shi H, Wu J, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. 2013;51(2):226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McGuckin Wuertz K, Treuting PM, Hemann EA, et al. STING is required for host defense against neuropathological West Nile virus infection. PLoS Pathog. 2019;15(8):e1007899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zevini A, Olagnier D, Hiscott J.. Crosstalk between Cytoplasmic RIG-I and STING sensing pathways. Trends Immunol. 2017;38(3):194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Franz KM, Neidermyer WJ, Tan Y-J, et al. STING-dependent translation inhibition restricts RNA virus replication. Proc Nat Acad Sci. 2018;115(9):E2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sun P, Zhang S, Qin X, et al. Foot-and-mouth disease virus capsid protein VP2 activates the cellular EIF2S1-ATF4 pathway and induces autophagy via HSPB1. Autophagy. 2018;14(2):336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169(3):425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Khaminets A, Heinrich T, Mari M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522(7556):354–358. [DOI] [PubMed] [Google Scholar]
- [10].Nazmi A, Mukhopadhyay R, Dutta K, et al. STING mediates neuronal innate immune response following Japanese encephalitis virus infection. Sci Rep. 2012;2(1):347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Liu D, Wu H, Wang C, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 2019;26(9):1735–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gui X, Yang H, Li T, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567(7747):262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Haag SM, Gulen MF, Reymond L, et al. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559(7713):269–273. [DOI] [PubMed] [Google Scholar]
- [14].Zhao B, Du F, Xu P, et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature. 2019;569(7758):718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shang G, Zhang C, Chen ZJ, et al. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature. 2019;567(7748):389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ergun SL, Fernandez D, Weiss TM, et al. STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell. 2019;178(2):290–301.e10. [DOI] [PubMed] [Google Scholar]
- [17].Moretti J, Roy S, Bozec D, et al. STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell. 2017;171(4):809–823.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Epidemiol. 1938;27(3):493–497. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.