

ABSTRACT
Lipid accumulation often leads to lipotoxic injuries to hepatocytes, which can cause nonalcoholic steatohepatitis. The association of inflammation with lipid accumulation in liver tissue has been studied for decades; however, key mechanisms have been identified only recently. In particular, it is still unknown how hepatic inflammation regulates lipid metabolism in hepatocytes. Herein, we found that PA treatment or direct stimulation of STING1 promoted, whereas STING1 deficiency impaired, MTORC1 activation, suggesting that STING1 is involved in PA-induced MTORC1 activation. Mechanistic studies revealed that STING1 interacted with several components of the MTORC1 complex and played an important role in the complex formation of MTORC1 under PA treatment. The involvement of STING1 in MTORC1 activation was dependent on SQSTM1, a key regulator of the MTORC1 pathway. In SQSTM1-deficient cells, the interaction of STING1 with the components of MTORC1 was weak. Furthermore, the impaired activity of MTORC1 via rapamycin treatment or STING1 deficiency decreased the numbers of LDs in cells. PA treatment inhibited lipophagy, which was not observed in STING1-deficient cells or rapamycin-treated cells. Restoration of MTORC1 activity via treatment with amino acids blocked lipophagy and LDs degradation. Finally, increased MTORC1 activation concomitant with STING1 activation was observed in liver tissues of nonalcoholic fatty liver disease patients, which provided clinical evidence for the involvement of STING1 in MTORC1 activation. In summary, we identified a novel regulatory loop of STING1-MTORC1 and explain how hepatic inflammation regulates lipid accumulation. Our findings may facilitate the development of new strategies for clinical treatment of hepatic steatosis.
Abbreviations: AA: amino acid; ACTB: actin beta; cGAMP: cyclic GMP-AMP; CGAS: cyclic GMP-AMP synthase; DEPTOR: DEP domain containing MTOR interacting protein; EIF4EBP1: eukaryotic translation initiation factor 4E binding protein 1; FFAs: free fatty acids; GFP: green fluorescent protein; HFD: high-fat diet; HT-DNA: herring testis DNA; IL1B: interleukin 1 beta; LAMP1: lysosomal associated membrane protein 1; LDs: lipid droplets; MAP1LC3: microtubule associated protein 1 light chain 3; MAP1LC3B: microtubule associated protein 1 light chain 3 beta; MEFs: mouse embryonic fibroblasts; MLST8: MTOR associated protein, LST8 homolog; MT-ND1: mitochondrially encoded NADH: ubiquinone oxidoreductase core subunit 1; mtDNA: mitochondrial DNA; MTOR: mechanistic target of rapamycin kinase; MTORC1: MTOR complex 1; NAFL: nonalcoholic fatty liver; NAFLD: nonalcoholic fatty liver disease; NASH: nonalcoholic steatohepatitis; NPCs: non-parenchymal cells; PA: palmitic acid; PLIN2: perilipin 2; RD: regular diet; RELA: RELA proto-oncogene, NF-kB subunit; RPS6: ribosomal protein S6; RPS6KB1: ribosomal protein S6 kinase B1; RPTOR: regulatory associated protein of MTOR complex 1; RRAGA: Ras related GTP binding A; RRAGC: Ras related GTP binding C; SQSTM1: sequestosome 1; STING1: stimulator of interferon response cGAMP interactor 1; TBK1: TANK binding kinase 1; TGs: triglycerides; TREX1: three prime repair exonuclease 1.
KEYWORDS: Lipophagy, MTORC1, NAFLD, STING1, TBK1
Introduction
As a spectrum of liver abnormalities ranging from nonalcoholic fatty liver (NAFL) to nonalcoholic steatohepatitis (NASH), nonalcoholic fatty liver disease (NAFLD; recently re-termed as metabolic associated fatty liver disease, MAFLD), is highly prevalent globally and can lead to cirrhosis and liver cancer [1]. NAFL is characterized by hepatic steatosis (fatty liver) and can progress to NASH when inflammation and hepatocyte damage occur in the liver [2]. Despite its widespread occurrence, there are currently no approved pharmacotherapies for NASH. Thus, more studies to elucidate NASH pathogenesis and identify potential molecular targets for NASH treatment are necessary [3]. Currently, the pathogenesis of NASH is controversial. Although the “two-hit” or “multiple parallel hits” theory is proposed to explain NASH pathogenesis, the specific mechanism of lipid accumulation and inflammation in NASH is still unclear [3].
Hepatic steatosis and other NASH-related conditions begin with the abnormal accumulation of hepatocellular lipids. In the hepatocytes, free fatty acids (FFAs) are taken up and converted to triglycerides (TGs) for storage in lipid droplets (LDs). Normally, LDs are degraded via hydrolysis, catalyzed by the LIPE-PNPLA2-MGLL axis, and lipophagy. However, excessive intake of FFAs results in the abundance of intracellular LDs, which causes lipotoxicity and further promotes ER stress, apoptosis, and inflammation [4]. As the major perpetrators of lipotoxic injuries to hepatocytes, LD clearance is important for the maintenance of liver function. Numerous studies have been conducted to determine the role of MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) in LD degradation. Saturated FFA, such as palmitic acid (PA), can increase the activity of MTORC1, whereas unsaturated FFA has opposite effects on MTORC1 [5]. The inhibition of MTORC1 by rapamycin or RPTOR (regulatory associated protein of MTOR complex 1) ablation promotes lipid accumulation in cells. The activation of MTORC1 by the overexpression of RHEB inhibits lipolysis [6]. RPS6KB1 (ribosomal protein S6 kinase B1) is downstream of MTORC1, and hence, rps6kb1 KO (knockout) mice have elevated rates of lipolysis, and loss of EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1) reduces this effect [7,8]. Altogether, these results suggest that the MTORC1-RPS6KB1 axis negatively regulates lipid clearance and TGs breakdown. Recent studies suggested that MTORC1 promotes lipid accumulation by regulating the activity of SREBF1/SREBP1, which mediates the transcription of lipogenesis-related genes [9]. Herein, we propose that MTORC1 also influences lipophagy through STING1 (stimulator of interferon response cGAMP interactor 1).
Recent studies found that STING1 plays an important role in hepatic steatosis-related inflammation and the progression of NASH. STING1 expression is increased in the liver tissues of patients with NAFLD or mice with high-fat diet (HFD)-induced hepatic steatosis, and loss of STING1 ameliorates HFD-induced inflammatory response and fibrosis [10]. PA treatment activates STING1-TBK1 (TANK binding kinase 1) signaling and promotes inflammatory response in hepatocytes. The STING1-TBK1 axis has been proposed to be associated with increased inflammation in the liver tissues of HFD-treated mice [11]. STING1 may act as a mitochondrial DNA (mtDNA) sensor in the Kupffer cells of the liver under lipid overload and promotes inflammation in NASH [12]. Although it is known that lipid accumulation in hepatocytes causes cell death and inflammation through STING1 [13], it is unclear whether the induction of inflammation by the STING1 pathway is involved in LD metabolism.
Given the important roles of STING1 and MTORC1 in hepatic steatosis, this study determined the relationship between STING1 and MTORC1-related LD degradation. We found that PA stimulated MTORC1 and consequently blocked lipophagy. Loss of STING1 or the inhibition of MTORC1 by rapamycin could rescue lipophagy. STING1 was directly involved in the complex formation of MTORC1 and promotes the SQSTM1 (sequestosome 1)-dependent activity of MTORC1. The present study provides a new insight into the relationship between STING1 and MTORC1. Further, our findings propose the possible mechanism underlying the effect of hepatic inflammation on lipid clearance and provide new strategies for controlling LD clearance in NAFLD treatment.
Results
STING1 mediates PA-induced activation of MTORC1
PA treatment can increase the phosphorylation level of RPS6KB1, which is downstream of MTORC1 [14]. PA-induced activation of STING1 promotes the phosphorylation of SQSTM1, which is necessary for MTORC1 activity [11,15]. Altogether, we proposed that STING1 may be involved in MTORC1 activation under PA treatment. Cyclic GMP-AMP (CGAMP) synthesized by the CGAS (cyclic GMP-AMP synthase) is an upstream regulator of STING1 [16]. Thus, we treated Hep3B cells with cGAMP to activate STING1. In accordance with previous studies, our results showed that STING1 activation by cGAMP enhanced the phosphorylation of RELA (RELA proto-oncogene) and TBK1 in Hep3B cells (Figure 1A). As the downstream signaling of MTORC1, RPS6KB1-RPS6 (ribosomal protein S6) signaling, but not EIF4EBP1 signaling, has been detected when LD accumulation is increased [7,8]. Thus, the phosphorylated level of RPS6KB1 or RPS6 is considered the marker of MTORC1 activity. Interestingly, cGAMP treatment promoted the phosphorylation of RPS6KB1, indicating increased activity of MTORC1 (Figure 1A). As a stimulus of CGAS, upstream of STING1, herring testis DNA (HT-DNA) is used to activate CGAS-STING1 signaling [17]. Similar to cGAMP treatment, HT-DNA and PA also promoted the phosphorylation of RPS6KB1, RPS6, and TBK1 (Figure 1B). These results suggest that direct stimulation by cGAMP or indirect stimulation by HT-DNA and PA can promote MTORC1 activity in Hep3B cells.
Figure 1.
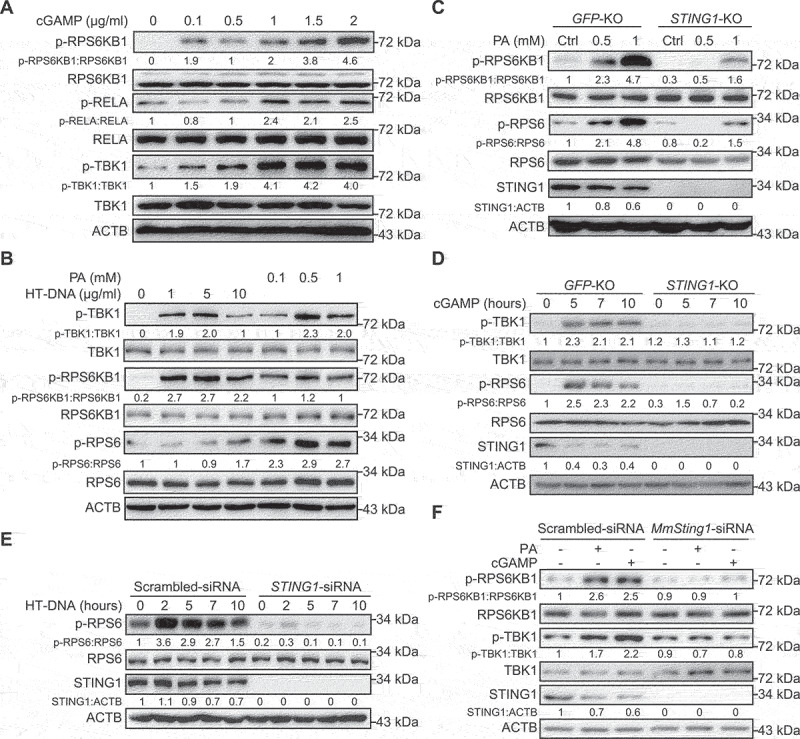
PA treatment stimulates MTORC1 activity through STING1. (A) Hep3B cells were transfected with 2ʹ3’-cGAMP for indicated concentration. After 6 h, lysates were immunoblotted for p-RPS6KB1 (T389), RPS6KB1, p-RELA (S536), RELA, p-TBK1 (S172), TBK1, and ACTB. (B) Lysates of Hep3B cells transfected with the indicated concentration of HT-DNA for 12 h, or treated with the indicated concentration of PA for 24 h, were used for immunoblotting with the p-RPS6KB1 (T389), RPS6KB1, p-TBK1 (S172), TBK1, p-RPS6 (S235/236), RPS6, and ACTB antibodies. (C) Control (GFP-KO) and STING1-KO Hep3B cells were treated with PA (24 h) for indicated concentration, and lysates were immunoblotted for p-RPS6KB1 (T389), RPS6KB1, p-RPS6 (S235/236), RPS6, STING1, and ACTB. (D) Immunoassay of extracts of control (GFP-KO) or STING1-KO Hep3B cells treated with 2ʹ3’-cGAMP (cGAMP, 2 μg/ml) for the indicated time interval. (E) Hep3B cells with or without STING1-siRNA (STING1-siRNA) transfection were treated with HT-DNA (10 μg/ml) for the indicated time interval were used for immunoblotting with the indicated antibodies. Cells treated with scrambled siRNA were used as control. (F) Mouse primary hepatocytes with or without mouse-Sting1-siRNA (MmSting1-siRNA) transfection were treated with PA (0.5 mM, 24 h), or transfected with cGAMP (2 μg/ml, 6 h), and the lysates of cells were performed for immunoblotting. Cells treated with scrambled siRNA were used as control.
Considering PA-induced STING1 activation, we determined the role of STING1 in PA-stimulated MTORC1 activation. Using Cas9 technology, we established STING1-KO cells and treated them with PA or STING1 stimulus. We also established control cells by transferring GFP (green fluorescent protein)-targeted guide RNA into cells. Compared with that in normal cells (GFP-KO), the phosphorylation levels of RPS6KB1 and RPS6 were lower in STING1-KO cells treated with PA, indicating low activity of MTORC1 in STING1-KO cells (Figure 1C). To exclude the influence of PA on other pathways, we used cGAMP to directly stimulate STING1. Based on our results, the phosphorylation of RPS6 was significantly promoted, indicating increased activity of MTORC1. Similarly, the effect of cGAMP on MTORC1 activity was hindered in STING1-KO cells (Figure 1D). To further confirm the effects of STING1 deficiency on MTORC1 activity, we transfected small interfering RNA (siRNA) to knockdown STING1 in Hep3B cells; this impaired MTORC1 activation induced by HT-DNA transfection (Figure 1E). Similar observations were made in isolated mouse hepatocytes. PA or cGAMP treatment enhanced the phosphorylation of RPS6KB1 and TBK1. STING1 deficiency impaired the effects of PA and cGAMP on the phosphorylation of these proteins, indicating that STING1 is critical for PA-induced activation of TBK1 and MTORC1 (Figure 1F). Similar observations were also made in mouse primary hepatocytes derived from wild-type (WT) or sting1-KO mice (data not shown). Some studies report the activation of TBK1 through the CGAS-STING1 pathway [11,18], which is not observed in CGAS or STING1 knockdown cells. These studies also show that the amount of cytosolic mtDNA significantly increases in cells treated with PA compared with that in untreated cells. To confirm these results, we determined the functions of PA in Hep3B cells and found that PA could actually increase the levels of p-TBK1 and p-RPS6KB1 concomitant with an increase in cytosolic mtDNA level, which revealed by the DNA levels of MT-ND1 (mitochondrially encoded NADH: ubiquinone oxidoreductase core subunit 1) (Fig. S1A and S1B). When STING1-siRNA was used to block CGAS signaling, PA lost its functions (Fig. S1B). Altogether, these results suggest that STING1 plays an important role in MTORC1 activation.
Activated STING1 interacts with the components of the MTORC1 complex
Considering the function of STING1 in MTORC1 activation, we also investigated whether STING1 is directly involved in the MTORC1 complex. We transfected HEK293T cells with expressed plasmids of STING1 and the main members of MTORC1 complex. Based on our findings, there was a strong interaction between STING1 and MLST8 (MTOR associated protein, LST8 homolog), DEPTOR (DEP domain containing MTOR interacting protein), or RRAGA (Ras related GTP binding A) and RRAGC (Ras related GTP binding C) (Figure 2A). Direct activation by cGAMP or indirect activation by HT-DNA/PA increased the interaction of STING1 and RPTOR, the main activator of the MTORC1 complex (Figure 2B). STING1 activation impaired the interaction of STING1 and DEPTOR, a critical inhibitor of MTORC1 (Fig. S2A). A previous study demonstrates that PA or amino acid (AA) treatment can significantly promote the lysosome-localization of MTOR [5]. MTORC1 is the core component of the MTOR complex, and hence, MTOR staining may not specifically reveal its localization in cells. Instead of detecting the colocalization of MTOR and STING1, we detected the colocalization of STING1 and RPTOR puncta, which is an indicator of the localization of active MTORC1 on the lysosome [19]. Thus, we utilized confocal microscopy to capture the involvement of STING1 in the MTORC1 complex by detecting the colocalization of STING1 and RPTOR puncta. The results showed that the numbers of RPTOR puncta were rare in the control cells. PA or cGAMP treatment increased the level of RPTOR puncta in control cells but not in STING1-deficient cells, indicating that the promotion of MTORC1 activation by PA and cGAMP was dependent on STING1. Furthermore, the results revealed a rare colocalization of STING1 and RPTOR puncta in control cells and the enhancement of such localization by PA or cGAMP treatment (Figure 2C and D). STING1 could colocalize with RPTOR puncta at the lysosome under PA or cGAMP treatment (Figure 2E). Besides, PA or cGAMP treatment increased the interaction of RPTOR-RRAGA or MLST8-RRAGA, indicating that direct or indirect activation of STING1 promoted the formation of activated MTORC1 complex (Figure 2F, G and S2B). These results demonstrate that activated STING1 is associated with the MTORC1 complex.
Figure 2.
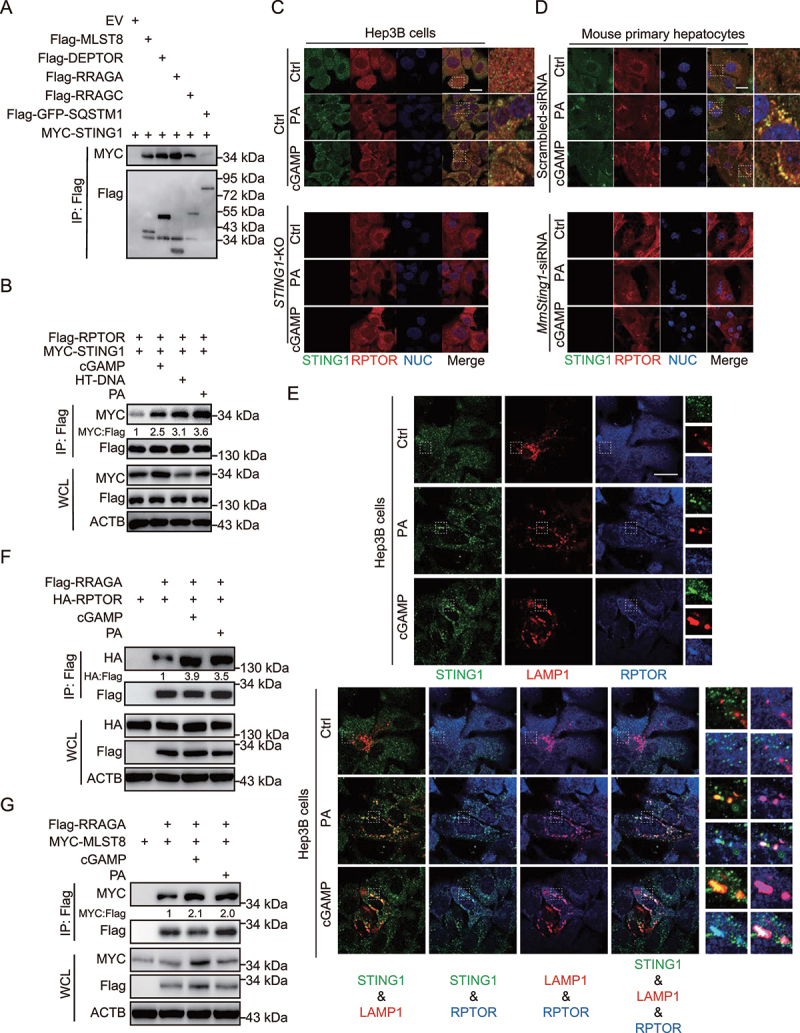
STING1 interacts with the components of the MTORC1 complex. (A) Coimmunoprecipitation and immunoassay of extracts of HEK293T cells transfected with various combinations of expression vector for MYC-STING1 and other indicated plasmids. (B) Coimmunoprecipitation and immunoassay of extracts of Hep3B cells transfected with Flag-RPTOR, MYC-STING1, and transfected with cGAMP (2 μg/ml, 6 h), HT-DNA (10 μg/ml, 6 h), or treated with PA (0.5 mM, 24 h). (C-E) Confocal microscopy of Hep3B cells (C and E), or mouse primary hepatocytes with or without mouse-Sting1-siRNA (MmSting1-siRNA) transfection (D) were treated with PA (0.5 mM, 24 h) or cGAMP (2 μg/ml, 6 h). Cells treated with scrambled siRNA were used as control. The cells were fixed 8 h later and stained with indicated antibodies. Hoechst 33342 was used for nucleus staining. Representative images are shown. Scale bar: 10 μm. For (E), the right panels of enlarged images correspond to rectangular frames in the left single or overlapped results from the same group. For the overlapped results, the enlarged images are listed in the following order: the left-upper image in the enlarged part is associated with the overlapped STING1-LAMP1; the left-lower image is associated with the overlapped STING1-RPTOR; the right-upper image is associated with the overlapped LAMP1-RPTOR; the right-lower image is associated with the overlapped STING-LAMP1-RPTOR. (F) Coimmunoprecipitation and immunoassay of extracts of Hep3B cells transfected with HA-RPTOR, Flag-RRAGA, and transfected with cGAMP (2 μg/ml, 6 h), or treated with PA (0.5 mM, 24 h). (G) Coimmunoprecipitation and immunoassay of extracts of Hep3B cells transfected with MYC-MLST8, Flag-RRAGA, and transfected with cGAMP (2 μg/ml, 6 h), or treated with PA (0.5 mM, 24 h).
MTORC1 activity is highly sensitive to AAs; the addition of AAs to cells markedly promotes MTORC1 activity [20]. Thus, we sought to determine whether STING1 is involved in the mechanism through which AAs regulate MTORC1 activity. We found that essential or non-essential AAs, which cannot activate STING1, did not exert any influence on the interaction of STING with the components of the MTORC1 complex (Fig. S2C-G), suggesting that STING1-regulated MTORC1 activation is independent of AA signaling.
Activated STING1 promotes MTORC1 activity through SQSTM1 and TBK1
The aforementioned results indicated that STING1 can interact with SQSTM1. SQSTM1 upregulates MTORC1 activity, and this effect is dependent on the interaction of SQSTM1 with RPTOR, the main member of the MTORC1 complex [15]. Thus, we determined whether SQSTM1 is necessary for the role of STING1 in MTORC1 activity. PA treatment increased the phosphorylation of RPS6 and the STING1-RPTOR interaction; however, SQSTM1 knockdown impaired this effect (Figure 3A). Additionally, PA or cGAMP could not promote the phosphorylation of RPS6KB1 and could not enhance the interaction between STING1 and the components of the MTORC1 complex in SQSTM1-deficient hepatocytes (Figure 3B, S3A and S3B). Confocal imaging also revealed increased SQSTM1 colocalization with RPTOR-positive puncta under PA or cGAMP treatment. SQSTM1 knockdown by siRNA treatment attenuated PA or cGAMP-induced formation of RPTOR puncta (Figure 3C). These results indicate that SQSTM1 is critical for the role of STING1 in MTORC1 activation.
Figure 3.
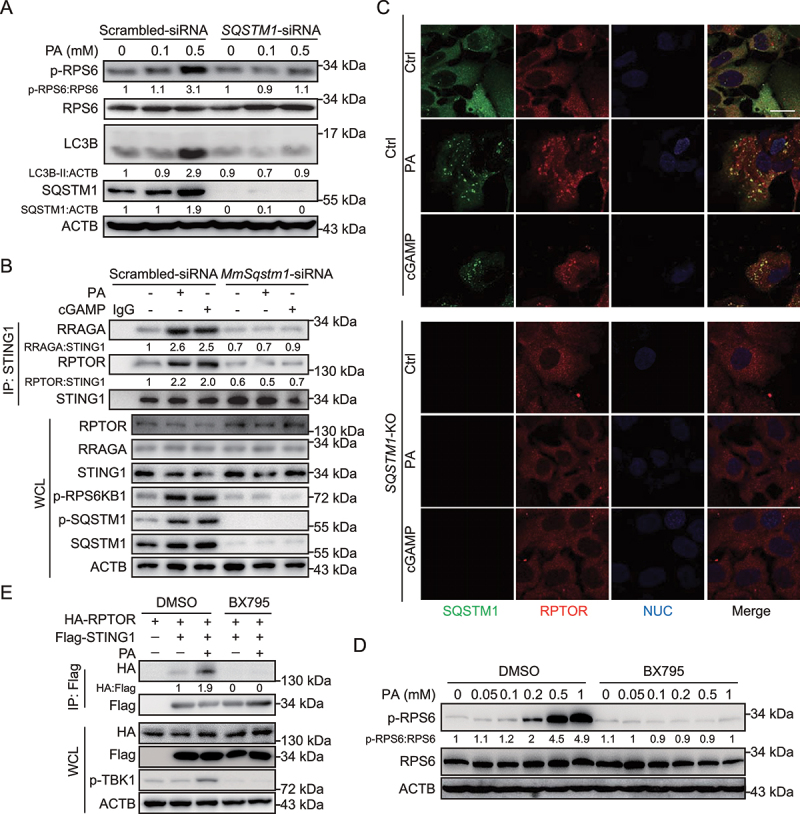
PA-induced STING1 activation stimulates MTORC1 relied on SQSTM1. (A) Hep3B cells with or without SQSTM1-siRNA transfection were treated with the indicated concentration of PA for 24 h, and the lysates of cells were performed for immunoblotting with p-RPS6, RPS6, LC3B, SQSTM1, and ACTB. Cells treated with scrambled siRNA were used as control. (B) Mouse primary hepatocytes were treated with or without PA (0.5 mM, 24 h), or transfected with cGAMP (2 μg/ml, 6 h) in the presence or absence of mouse-Sqstm1-siRNA (MmSqstm1-siRNA) transfection for 48 h. STING1 was immunoprecipitated from whole-cell lysates, and levels of STING1 and RPTOR and RRAGA in the precipitates were evaluated by immunoblotting. Cells treated with scrambled siRNA were used as control. (C) Confocal microscopy of Hep3B cells treated with PA (0.5 mM, 24 h) or cGAMP (2 μg/ml, 6 h). The cells were fixed 8 h later and stained with antibodies against SQSTM1 and RPTOR. Hoechst 33342 was used for nucleus staining. Representative images are shown. Scale bar: 10 μm. (D) Hep3B cells were treated with the indicated concentration of PA (0.5 mM) for 24 h with DMSO or BX795 (10 μM), and lysates were immunoblotted for p-RPS6, RPS6, and ACTB. (E) Coimmunoprecipitation and immunoassay of extracts of Hep3B cells transfected with HA-RPTOR, Flag-STING1 followed with PA (0.5 mM, 24 h) with or without BX795 (10 μM).
Activated STING1 had increased effect on MTORC1 activity (Figs. 1 and 2) and promoted activation of TBK1, which is a kinase of SQSTM1. As a result, we speculated that STING1-mediated MTORC1 activity is dependent on TBK1. We used BX795 to inhibit TBK1 kinase activity and found that the phosphorylation of RPS6 and the interaction of STING1-RPTOR induced by PA were significantly blocked (Figure 3D and E). In summary, our results suggest that activated STING1 promotes MTORC1 activity. Further, this effect is found to be dependent on SQSTM1 and TBK1.
Phosphorylated SQSTM1 activated by TBK1 binds to STING1 following PA treatment
Under various stress conditions, TBK1 phosphorylates SQSTM1 on the Ser403 site, a modification essential for increased affinity between SQSTM1 and poly-ubiquitin chain; this results in efficient interaction between SQSTM1 and poly-ubiquitinated proteins [21]. Meanwhile, PA treatment can significantly stimulate STING1-TBK1 and consequently increase SQSTM1 phosphorylation on Ser403 in hepatocytes [11]. As the promotion of MTORC1 activity by activated STING1 is dependent on both SQSTM1 and TBK1, we hypothesized that with PA treatment, TBK1 acts as SQSTM1 kinase to participate in STING1-regulated activity of MTORC1. These results were consistent with the notion that phosphorylated SQSTM1 on the Ser403 site and p-RPS6KB1 were activated by PA treatment in a concentration-dependent manner but were blocked in TBK1 knockdown cells (Figure 4A). Such findings indicate that TBK1 is essential for SQSTM1 phosphorylation and MTORC1 activity under PA treatment. A previous study indicates that STING1 has a stronger interaction with SQSTM1 phosphorylated on Ser403 [22]. Consistent with this result, we found that TBK1 increased the interaction between SQSTM1 and STING1 by promoting SQSTM1 phosphorylation on Ser403 (Figure 4B). PA also enhanced SQSTM1-STING1 interaction by activating p-SQSTM1; however, BX795 could block this interaction (Figure 4C). Endogenous immunoprecipitation also revealed that BX795 or TBK1 siRNA treatment significantly impaired the interaction of STING1 with RPTOR and SQSTM1, and attenuated PA- or cGAMP-induced phosphorylation of RPS6KB1 (Figure 4D). Confocal imaging also demonstrated the interaction between TBK1 of RPTOR, which has been previously reported [23]. TBK1 knockdown blocked the formation of RPTOR puncta. Moreover, SQSTM1 deficiency inhibited the colocalization of TBK1 and RPTOR under PA or cGAMP treatment, indicating that TBK1 colocalization with RPTOR was dependent on SQSTM1 (Figure 4E). Furthermore, we detected MTORC1 phosphorylation (Ser2159) in PA-treated WT or SQSTM1-KO cells and found that PA could induce the phosphorylation of p-TBK1 and p-MTOR (Ser2159) in WT cells, but not in SQSTM1-KO cells (Fig. S4A). We also found that PA could induce the colocalization of TBK1 and MTOR in WT cells, but not in SQSTM1-KO cells (Fig. S4B). These data suggest that SQSTM1 provides a bridge between TBK1 and MTORC1.
Figure 4.
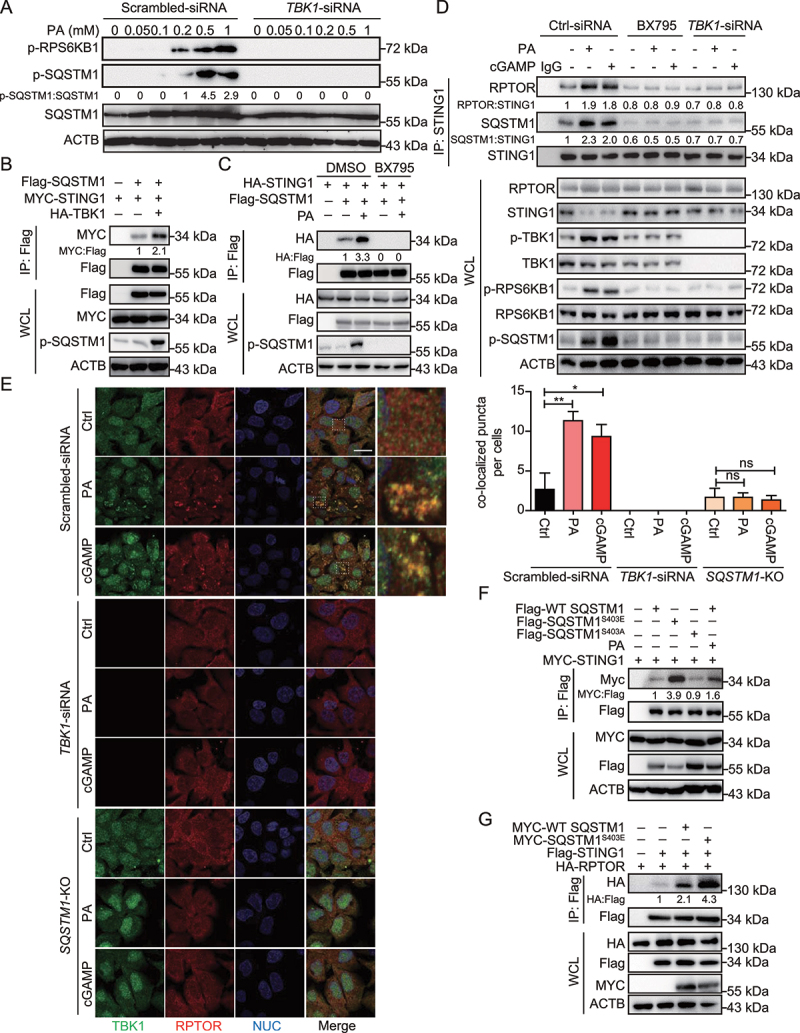
PA treatment stimulates SQSTM1 phosphorylation and the interaction of STING1 and SQSTM1. (A) Hep3B cells with TBK1-siRNA transfection for 48 h were treated with the indicated concentration of PA for 24 h, and lysates were immunoblotted for p-RPS6KB1 (T389), p-SQSTM1 (S403), SQSTM1, and ACTB. Cells treated with scrambled siRNA were used as control. (B) Coimmunoprecipitation and immunoassay of extracts of Hep3B cells transfected with MYC-STING1, Flag-SQSTM1, and HA-TBK1. (C) Coimmunoprecipitation and immunoassay of extracts of Hep3B cells transfected with HA-STING1, Flag-SQSTM1, and followed with PA treatment (0.5 mM, 24 h) with or without BX795 (10 μM). (D) Hep3B were treated with or without PA (0.5 mM, 24 h), or transfected with cGAMP (2 μg/ml, 6 h) in the presence or absence of TBK1-siRNA transfection for 48 h, or in the presence of absence of BX795 (10 μM, 8 h). Cells treated with scrambled siRNA were used as control (Ctrl-siRNA). STING1 was immunoprecipitated from whole-cell lysates, and levels of STING1, RPTOR and SQSTM1 in the precipitates were evaluated by immunoblotting. (E) Confocal microscopy of Hep3B cells treated with PA (0.5 mM, 24 h) or cGAMP (2 μg/ml, 6 h). Cells treated with scrambled siRNA were used as control. The cells were fixed 8 h later and stained with antibodies against TBK1 and RPTOR. Hoechst 33342 was used for nucleus staining. Representative images are shown. Scale bar: 10 μm. Quantitative analysis of colocalized puncta of TBK1 and RPTOR is shown on the right. The numbers of puncta per cell were quantified by blind counting. The data are presented as means of three views of counting. **p < 0.01, *p < 0.05, ns = not significant. (F) Coimmunoprecipitation and immunoassay of extracts of Hep3B cells transfected with various combinations of expression vector for MYC-STING1 and other indicated plasmids, and treated with PA (0.5 mM, 24 h). (G) Coimmunoprecipitation and immunoassay of extracts of Hep3B cells transfected with various combinations of expression vector for Flag-STING1, HA-RPTOR and other indicated plasmids.
To further determine SQSTM1 binding to STING1 through its phosphorylation on Ser403, we constructed phosphorylation-resistant (SQSTM1S403A) and phosphorylation-mimic (SQSTM1S403E) mutants. Based on our findings, STING1 tended to bind to p-SQSTM1 (Figure 4F). Moreover, SQSTM1S403E enhanced STING1-RPTOR interaction (Figure 4G). Next, we constructed cell line with stable expression of WT SQSTM1 or SQSTM1-mutants. Accordingly, p-SQSTM1 was more likely to bind to STING1 and could enhance the interaction of STING1 and RPTOR (Fig. S4C and S4D). These results suggest that activated STING1 promotes the phosphorylation of TBK1 and SQSTM1, and p-SQSTM1 binds to STING1 and influences MTORC1 activity.
STING1 is involved in lipophagy inhibition under PA treatment
Lipophagy is an important metabolic pathway for lipid degradation; however, the associated mechanism is unknown. Based on the aforementioned results, PA activated STING1, which in turn influenced MTORC1 activity. However, the mechanism through which STING1 or MTORC1 influences the metabolism of intracellular LDs is not clear. Both STING1 and MTORC1 regulate lipid metabolism. Thus, we proceeded to determine whether they could regulate lipophagy of intracellular PA. First, we determined the number of LDs using oil red O staining. Increased numbers of LD were detected using PA treatment. Both rapamycin treatment and STING1 KO reduced the number of intracellular LDs. Meanwhile, MTORC1 activation via AA treatment inhibited the effect of STING1 deficiency on amount of LDs (Figure 5A). AAs could not stimulate STING1-TBK1 (Fig. S2G), and hence, we considered using AAs as a STING1-independent activator of MTORC1. We proceeded to detect whether PA and AAs have a synergistic effect on MTORC1 activation in hepatocytes. AA treatment could enhance PA-induced activation of MTORC1, but did not influence PA-induced TBK1 phosphorylation. In STING1-deficient hepatocytes, AAs, not PA, promoted MTORC1 activation (Fig. S5A). By detecting the number of LDs in mouse hepatocytes, we found that higher activation of MTORC1 induced more accumulation of LDs in cells treated with both AAs and PA than in cells treated with PA alone. Although STING1 deficiency decreased the number of LDs, the re-activation of MTORC1 by AA treatment in STING1-deficient hepatocytes also enhanced the number of LDs (Fig. S5B). Such findings demonstrate that MTORC1 activity enhances the accumulation of LDs.
Figure 5.
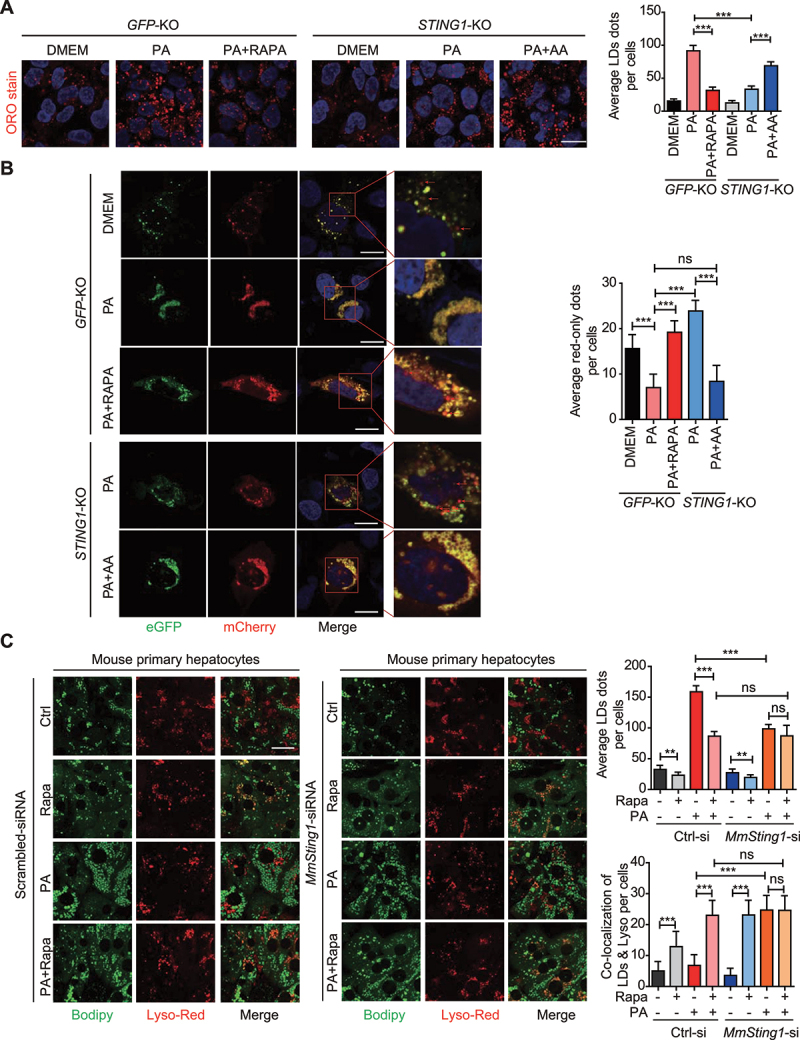
STING1 involves in inhibition of lipophagy under PA treatment. (A) Control (GFP-KO) and STING1-KO Hep3B cells with indicated treatment were fixed and stained with Oil red O (ORO stain), and followed by confocal fluorescence microscopy. The numbers of red-only puncta per cell were quantified by blind counting. Shown on the right is the mean plus SD of ten views of counting. ***p < 0.001. (B) Control (GFP-KO) and STING1-KO Hep3B cells transfected with mCherry-eGFP-PLIN2 plasmid for 48 h were treated with or without PA (0.5 mM, 24 h) followed with rapamycin (200 nM, 6 h) and AA (60 min). Cells were fixed and followed by confocal fluorescence microscopy. Representative images are shown. Right panels are enlarged photographs from the boxed areas. Arrow denotes the red-only puncta. Scale bar: 10 μm. The numbers of red-only puncta per cell were quantified by blind counting. Shown on the right is the mean plus SD of ten views of counting. ***p < 0.001, ns = not significant. (C) Confocal microscopy of mouse primary hepatocytes with or without mouse-Sting1-siRNA (MmSting1-siRNA) transfection followed with treatment of PA (0.5 mM, 24 h) or rapamycin (200 nM, 6 h). Cells treated with scrambled siRNA were used as control. For the combination treatment of PA and rapamycin, PA was treated for 18 h followed with 6 h of rapamycin treatment. The cultured cells were stained with Bodipy-FL and LysoTracker Red. Representative images are shown. Scale bar: 10 μm. The numbers of colocalized puncta per cell were quantified by blind counting. Shown on the right is the mean plus SD of ten views of counting. Ctrl-si referred to scrambled siRNA. MmSting1-si refereed to mouse-Sting1-siRNA. ***p < 0.001, **p < 0.01. ns = not significant.
We proceeded to investigate whether the STING1-regulated activity of MTORC1 influenced lipophagy. GFP loses its structure in the acidic and/or proteolytic conditions of the lysosome lumen, whereas mCherry is more stable and can be detected in the lysosome. Therefore, colocalization of GFP and mCherry fluorescence is considered to indicate a compartment that has not fused with a lysosome. Tandem mRFP/mCherry-GFP fluorescence microscopy is a useful technology for monitoring lysosome-related degradation [24], and PLIN2 (perilipin 2) is a known protein localized on the surface of LDs in hepatic cells [25]. Combining tandem mCherry-GFP microscopy and PLIN2, we constructed a tandem mCherry-GFP-PLIN2 fusion protein for monitoring the lysosomal degradation of LDs. Compared with that in cells in normal condition, PA treatment significantly increased the accumulation of lipid drops, but inhibited lipophagy, which was revealed by decreased number of red puncta (Figure 5B). The lipophagy was rescued with the blockage of MTORC1 signaling using rapamycin, a specific inhibitor of MTORC1, as shown by increase in the number of red puncta (Figure 5B), suggesting that MTORC1 and its downstream signaling are involved in lipophagy inhibition by PA. As shown in aforementioned results, we found that STING1 is involved in the regulation of MTORC1 activity. Furthermore, we determined the function of STING1 in lipophagy. The fusion of LDs and lysosomes was observed in STING1-KO cells upon treatment with PA. The knockout of STING1 significantly rescued lipophagy. However, the activation of MTORC1 by AA treatment blocked lipophagy in STING1-KO cells (Figure 5B). To further validate the role of STING1 and MTORC1 activation in lipophagy, we isolated primary hepatocytes from mouse liver. After PA treatment, the live hepatocytes were labeled for LDs and lysosomes using BODIPY and LysoTracker Red probe, respectively. Thereafter, the labeled live cells were subjected to confocal imaging to determine the colocalization of LDs and lysosome, which is an indicator of lipophagy [26]. Rapamycin treatment could significantly promote the colocalization of LDs and lysosome in control cells under normal conditions. Compared with normal hepatocytes, hepatocytes with STING1 knockdown showed increased lipophagy and lower number of LDs under PA treatment. Rapamycin could enhance lipophagy and decrease the number of LDs in normal hepatocytes under PA treatment, but not in STING1-deficient hepatocytes (Figure 5C). Similar observations were made in mouse primary hepatocytes derived from WT or sting1-KO mice (data not shown). These results indicate that STING1 and MTORC1 can inhibit lipophagy by PA treatment.
STING1 is reported to promote autophagic flux through its direct interaction with MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) [27]. To further assess whether lipophagy inhibition by STING1 was canonical macroautophagy/autophagy specifically, we detected the autophagic flux under STING1 activator or FFA treatment. Consistent with the previous study, stimulating STING1 by cGAMP or HT-DNA enhanced autophagic flux. STING1 KO suppressed basal and cGAMP/HT-DNA-stimulated autophagy (Fig. S5C). However, treatment with PA, but not oleic acid, promoted LC3B-II and SQSTM1 accumulation, indicating that PA induced the blockade of autophagic flux. STING1 KO did not improve SQSTM1 accumulation, suggesting that STING1 was involved in lipophagy independent of canonical autophagy (Fig. S5D). These results demonstrate that the effect of STING1 on lipophagy differs from that previously reported on autophagy flux.
Concomitant enhancement of the phosphorylated levels of RPS6KB1 and TBK1 correlates with inflammatory levels in liver tissues with NAFLD
Based on the correlation between STING1 and MTORC1 activity, we determined the activation of STING1 and MTORC1 in human liver tissues with or without NAFLD. As the main downstream effector of STING1 or MTORC1, we used TBK1 phosphorylation to demonstrate STING1 activation. Moreover, MTORC1 activity was determined by measuring RPS6KB1 phosphorylation. The results of immunohistochemical assay revealed higher levels of RPS6KB1 phosphorylation in liver tissues with NAFLD, indicating increased MTORC1 activity (Figure 6A). The phosphorylated levels of TBK1 were also enhanced in liver tissues with NAFLD (Figure 6B and S6A). The copy numbers of cytosolic mtDNA, which were revealed by the DNA levels of MT-ND1, were increased in liver tissues with NAFLD (Fig. S6B). The copy numbers of cytosolic mtDNA and the activation of STING1 and MTORC1 were also increased in the liver tissue of MCD-treated mice (Fig. S6C and S6D). Furthermore, a strong positive correlation was found between the staining scores of p-RPS6KB1 and p-TBK1 from the surgically removed samples of liver tissues with NAFLD (Figure 6C). In addition, we determined the levels of lipophagy by detecting the colocalization of PLIN2, a typical protein located on the LD surface, and LAMP1 (lysosomal associated membrane protein 1), a lysosomal marker. By using NAFLD samples with high or low phosphorylation of RPS6KB1 and TBK1, confocal imaging revealed that liver tissue with high phosphorylation of RPS6KB1 and TBK1 had reduced colocalization of PLIN2 and LAMP1, indicating that the ability of lysosome-clearance of LDs was impaired in the liver of NAFLD patients with high MTORC1 activity (Figure 6D).
Figure 6.
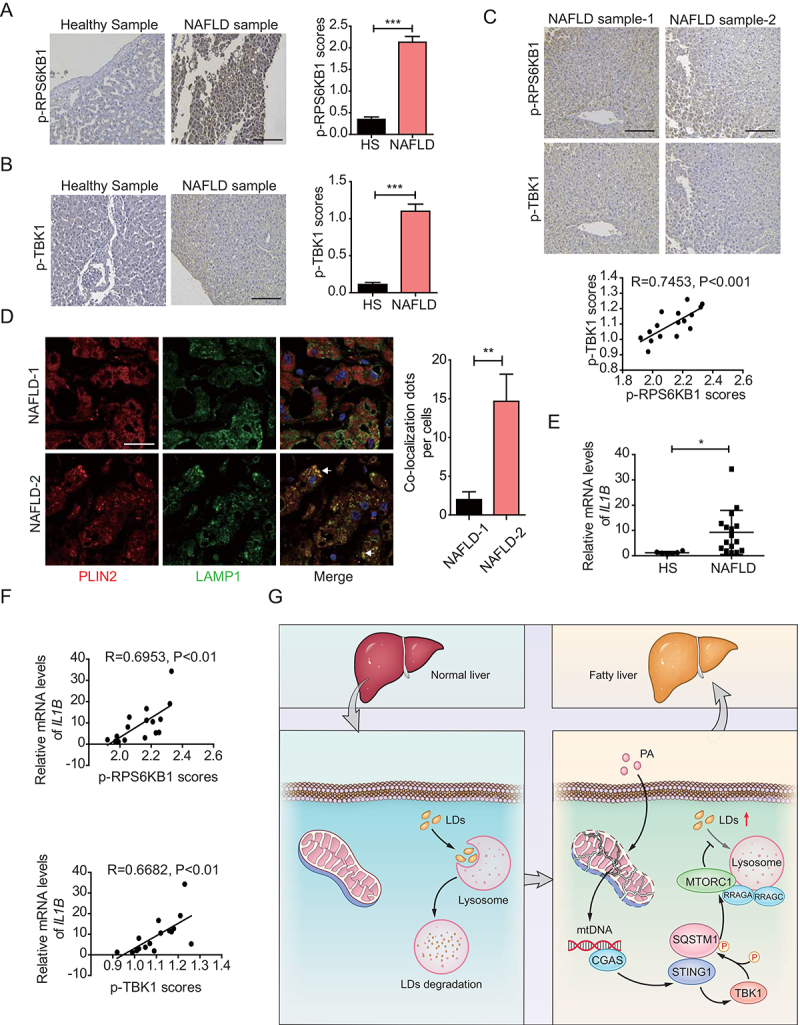
Concomitant enhancement of phosphorylated levels of RPS6KB1 and TBK1 correlates with inflammatory levels in liver tissues with NAFLD. (A) Immunohistochemical staining of 6 cases of normal human liver tissue specimens (Healthy Sample, HS) and 16 cases of NAFLD specimens (NAFLD Sample, NAFLD) with the anti-p-RPS6KB1 antibody. Scale bar: 100 μm. Quantitative analysis of p-RPS6KB1 staining is shown as scores. The data are presented as means ± SD. ***p < 0.001. (B) Immunohistochemical staining of normal human liver tissue specimens (Healthy Sample, HS) and NAFLD specimens (NAFLD Sample, NAFLD) with the anti-p-TBK1 antibody. Scale bar: 100 μm. Quantitative analysis of p-TBK1 staining is shown as scores. The data are presented as means ± SD. ***p < 0.001. (C) Human NAFLD specimens were immunohistochemically stained with anti-p-RPS6KB1 or anti-p-TBK1 antibodies. Representative images are shown. Scale bar: 100 μm. The correlation of p-RPS6KB1 and p-TBK1 was statistically significant among different specimens (n = 16, r = 0.7435, p < 0.001). (D) Confocal microscopy of human NAFLD specimens using in above. NAFLD-1 is related to the NAFLD sample-1 in (C). NAFLD-2 is related to the NAFLD sample-2 in (C). Representative images are shown. Scale bar: 10 μm. The quantitative data are presented as means ± SD. **p < 0.01. (E) The IL1B mRNA expression level in the normal liver (healthy sample, HS) (n = 6) and NAFLD tissue (NAFLD) (n = 16) samples was determined by qPCR assay. The data are presented as means ± SD. *p < 0.05. (F) The correlation of IL1B mRNA expression and p-RPS6KB1 (upper) and p-TBK1 (lower) was statistically significant among different specimens (n = 16). (G) Schematic representation of STING1-regulated MTORC1 activity and lipophagy under PA treatment.
We confirmed that the mRNA levels of IL1B (interleukin 1 beta), an inflammatory factor, were slightly increased in NAFLD samples compared with those in normal liver tissues (Figure 6E). Notably, a significant positive correlation was observed between p-RPS6KB1 levels and IL1B mRNA in the NAFLD tissues. TBK1 phosphorylation also correlated with IL1B mRNA, indicating that STING1 was involved in the initiation of inflammation in the liver tissues with NAFLD (Figure 6F). Collectively, these results provided evidence to support the clinical relevance of STING1 activation and MTORC1 activity in NAFLD progression.
Discussion
Steatosis and inflammation in hepatocytes severely endanger the normal function of the liver, resulting in chronic damage of the liver tissue. Lipid accumulation in hepatocytes causes liver inflammation, and the dysfunction of lipid metabolism has been associated with lipid accumulation [3]. Initiation of liver inflammation is a marker of NASH, which typically occurs after lipid accumulation [28]. Although lipid accumulation contributes to the initiation of liver inflammation, the particular mechanism associated with the role of inflammation in the clearance of lipids, especially LDs, and the reason for the difficulty in reversing NASH, remain unclear. Herein, we demonstrated that STING1 activation by PA treatment prevents the lysosomal degradation of LDs and proposed an explanation for the difficulty associated with reversing liver inflammation of NASH.
Novel roles of STING1 in LD clearance
As a central pivot of sensing exogenous and intracellular DNA, STING1 is a critical receptor that links the upstream signal of DNA to activation of the downstream IRF3 and NFKB pathways. By stimulating the transcription of interferon and inflammatory factors, STING1 signaling has been demonstrated to be essential for protecting cells against a variety of pathogens [29]. STING1 activation by self-DNA, which may be derived from necrotic cells or dysfunctional mitochondria, plays an important role in auto-inflammatory disease and chronic inflammation involving the sensing of self-DNA [30]. PA treatment also stimulates STING1 and enhances the inflammatory response in hepatic cells [11]. We also found that PA can significantly promote STING1 activation. In hepatic steatosis-related disease, the activation of the STING1-TBK1 pathway promotes hepatocyte injury and dysfunction by inducing inflammation and apoptosis and impairing glucose and lipid metabolism [13]. A recent study reported that the elevated expression of STING1 promotes macrophage-mediated hepatic inflammation and fibrosis [10]. STING1 deficiency markedly attenuates hepatic steatosis, fibrosis, and inflammation in both MCD- and HFD-fed mice. Such roles of STING1 mainly rely on the sensing of intracellular mtDNA derived from dysfunctional mitochondria in Kupffer cells [12]. These findings indicate that STING1 activation contributes to the inflammation that occurs in the liver. However, it is unclear whether STING1 activation has other roles in hepatic steatosis.
Some studies concluded that STING1 is not expressed in mouse hepatocytes [10,12], whereas several studies found that STING1 plays a role in mouse hepatocytes [11,18]. Using western blot, qPCR, IF and IHC, we confirmed STING1 expression in hepatocytes and liver non-parenchymal cells (NPCs). Further, STING1 was found to have a relatively weaker expression in hepatocytes than in NPCs (data not shown). Because of the low expression of STING1 in hepatocytes, some reports concluded that STING1 is not expressed in mouse hepatocytes, which may be attributed to the weak immunoblotting signals.
In this study, STING1 activation by PA treatment promoted MTORC1 activation (Figure 1), which has been associated with enhanced accumulation of intracellular lipids [31]. Recently, STING1 knockdown was reported to reduce lipid deposition in hepatocytes [32]. We also demonstrated that STING1 KO could reduce the number of LDs (Figure 5A). We also found that STING1 deficiency promoted the lysosomal-related degradation of LD. The STING1-regulated activity of MTORC1 impairs lipophagy, indicating the roles of STING1 and MTORC1 in the lysosomal-associated clearance of hepatic LD (Figure 5B). Such findings suggest that STING1 activation may not only enhance the inflammation of liver tissue, but also increase lipid accumulation by regulating MTOR activity in the progression of hepatic steatosis.
Synergetic axis of STING1-TBK1-SQSTM1 in MTORC1 activation
As a central pivot of intracellular signaling, MTOR senses upstream stress and change in energy level, and controls cellular behaviors by targeting different types of substrates [31]. There are at least five major activators of MTORC1, namely insulin, stress, glucose, oxygen, and AAs. With regard to lipids, it is known that saturated FFAs, but not unsaturated FFAs, can activate MTORC1 signaling by enhancing MTORC1 translocation onto lysosomal membranes [5]. Furthermore, saturated FFA results in apoptosis through the hyperactivation of MTORC1, but not of MTORC2 [5]. However, the particular mechanism associated with saturated FFA-stimulated activation of MTORC1 is unknown. Herein, we found that PA stimulated the activation of MTORC1 through STING1 (Figure 1). STING1 could interact with the members of the MTORC1 complex. The stimulator of STING1 and PA could reinforce the complex formation of MTORC1 (Figure 2). Mechanistic studies revealed that STING1 was involved in MTORC1 activation mainly through the recruitment of SQSTM1 (Figure 3), a known regulator of MTORC1 [15]. Without SQSTM1, PA could not stimulate MTORC1 activity (Figure 3). The phosphorylation of SQSTM1 by TBK1 was important for both SQSTM1-STING1 interaction and the involvement of STING1 in MTORC1 complex (Figure 4). Previous studies have mainly focused on the degradative role of SQSTM1 in SQSTM1-STING1 interaction [22], and thus, our work may provide new insights into the role of STING1 in lipid homeostasis in relation to the regulation of MTORC1 signaling.
Several studies have indicated that TBK1 has controversial roles in MTORC1 activation. Consistent with the results of this study, two recent studies revealed that TBK1 enhances the activation of MTORC1 in response to growth factors and innate immune agonists [23,33]. Moreover, although many studies reported that TBK1 interacts with the main components of the MTORC1 complex, they concluded that TBK1 directly inhibits MTORC1 [34–36]. The activation of MTORC1 signaling is strictly and timely regulated in response to various conditions in cells. It is highly possible that TBK1 influences MTORC1 signaling directly or indirectly in certain physiologic or pathological contexts. One hypothesis is that TBK1 may regulate MTORC1 signaling in specific stress, which re-wires cell signaling and positively impacts the MTORC1 pathway. Upon continuous stimulation, such as PA-induced lipotoxicity or long-term nutrient oversupply, active TBK1 may indirectly promote MTORC1 signaling through SQSTM1. However, specific stress or condition may result in the negative engagement of MTORC1. For example, one study found that KO of the Trex1 (three prime repair exonuclease 1) in mouse embryonic fibroblasts (MEFs) activates the CGAS-STING1-TBK1 pathway. Active TBK1 in trex1−/- MEFs directly inhibits MTORC1 signaling [34]. The continuous accumulation of self-DNA or glycans in trex1−/- MEFs or mice tissue may stimulate not only CGAS-STING1 signaling, but also protective signaling in cells, such as endoplasmic reticulum stress signaling or macroautophagy, which stimulates the reduction of MTORC1 activity. Interestingly, trex1−/- mice have lower MTORC1 activity in liver and lower body fat in normal condition and lower weight under HFD condition. This result partially supports our conclusion that high MTORC1 activity increases the accumulation of LDs. Other studies also detected the relationship between TBK1 and MTORC1 activity under no stimulus and concluded that TBK1 inhibits MTORC1 signaling [35,36]. However, some studies determined that TBK1 supports MTORC1 activity under specific stimuli, such as that with EGF (epidermal growth factor) [23] and AAs [33]. In this study, cells were mainly treated with PA or cGAMP, which can partially preclude MTORC1 engagement in negative feedback. However, the effect of PA and TBK1 on MTORC1 could vary depending on the cell contexts. In this study, the TBK1-dependent activation mechanism overrides the negative impacts on MTORC1 signaling, and this may rely on the unique intracellular context of hepatocytes.
Another hypothesis is that TBK1 may have indirectly regulated MTORC1 signaling through other regulators of MTORC1, such as SQSTM1. Other studies that have reported the controversial effect of TBK1 on MTORC1 activity mainly focus on the direct regulation of the MTORC1 complex by TBK1. We found that SQSTM1 is critical for TBK1-regulated MTORC1 activation under PA or cGAMP stimulus (Figure 4E). SQSTM1 is reported to promote MTORC1 signaling through the regulation of the formation of the active Rag heterodimer and the recruitment of MTOR to lysosomes [15], which may represent the missing link between TBK1 and MTORC1. Without SQSTM1, TBK1 could not colocalize with RPTOR puncta upon PA or cGAMP treatment (Figure 4E). Our data also revealed that TBK1 was involved in the PA-induced activation of MTORC1, which was dependent on the phosphorylation of SQSTM1 (Ser403) (Figure 4). Although the mechanism is not clear, our data indicate that TBK1 indirectly contributes to MTORC1 activation in response to PA or cGAMP-induced STING1 activation in hepatic cells.
STING1-regulated lipophagy is dependent on MTORC1
Although it is clear that MTORC1 signaling regulates lipid homeostasis, the detailed mechanism remains unclear. In 2008, it is first identified that MTORC1 promotes lipogenesis by regulating SREBP activity and the expression of genes involved in lipogenesis [37]. Nonetheless, mice with a liver-specific deletion of Tsc1 shows enhanced MTORC1 activity but defective SREBP1c activation and lipogenesis, indicating that MTORC1 increases the cellular accumulation of lipid through other catabolic processes. Lipolysis is defined as the catabolism of triacylglycerols stored in cellular LD, and more recent studies have proposed the role of MTORC1 in this process. The link between MTORC1 and lipolysis of adipose tissue has been observed in various experimental conditions, such as under rapamycin treatment [6,8]. MTORC1 may control lipolysis by regulating the expression and activity of lipid hydrolases, such as PNPLA2 and LIPE [38]. However, the roles of MTORC1 in lysosome-related LD clearance (lipophagy) are unknown. Herein, we demonstrated that MTORC1 inhibition by rapamycin could rescue lipophagy inhibition under PA treatment (Figure 5). Moreover, we identified that MTORC1 activity was increased in liver tissues with NAFLD compared with that in normal liver tissues. Importantly, MTORC1 activity has a high correlation with STING1 activation and transcription of inflammatory factors (Figure 6), suggesting that STING1 activation contributes to MTORC1 signaling and consequent accumulation of lipid in the hepatocytes.
Although STING1 has been reported to promote autophagic flux, the effect of STING1 on autophagy is not dependent on its downstream signaling. PA was demonstrated to promote CGAS-STING1 activation and TBK1 activation. However, according to previous studies, STING1 promotes autophagy by directly interacting with MAP1LC3, independently of TBK1, the downstream factor of CGAS-STING1 signaling [27,39]. We found that STING1 regulated the activity of MTORC1 through TBK1 activation and TBK1-induced SQSTM1 phosphorylation (Figs. 3 and 4). Therefore, STING1 activation has two different effects on autophagic degradation in the liver. On one hand, STING1 activation promotes autophagic initiation by directly interacting with MAP1LC3. On the other hand, STING1-TBK1 signaling can sustain a high level of MTORC1 activation, which suppresses the function of lysosome and lipophagy. This hypothesis is partially supported by the results of studies conducted on animals subjected to short- or long-term HFD treatment. Autophagic degradation increases during the first few weeks, which was followed by a gradual decrease in autophagic degradation in long-term HFD- and MCD-treated animals [40]. This fluctuation in autophagy may partially be attributed to the activation of STING1-TBK1 signaling and MTORC1 in the liver.
The role of CGAS-STING1 signaling in lipophagy may not be strictly related to the role of STING1 in autophagic flux. Autophagic flux comprises various steps that are differentially regulated by different signaling processes. Some proteins regulate autophagic degradation by influencing the fusion of autophagosome and lysosome, but have little effect on autophagic flux [41]. However, lipophagy is selective and is more dependent on the fusion of LDs and lysosomes. According to previous studies, the oversupply of fatty acid induces the dysfunction of lysosomes. Autophagosomes and lysosomes isolated from the livers of mice maintained on regular diet (RD) or HFD for 16 weeks exhibit significantly different fusion rates [42]. The fusion rate between the fractions isolated from the HFD group are up to 40% lower than that between those isolated from the RD mice [42]. Moreover, the fusion rates of autophagosomes from the RD group and lysosome from the HFD group are 75% lower than those isolated from the RD mice [42]. This may contribute to the increased activity of MTORC1, which suppresses TFEB and reduces the levels of lysosome-associated proteins [43]. Activated MTORC1 may also inhibit lysosomal function through other mechanisms [44]. Our results also showed that PA-induced activation of MTORC1 blocks the colocalization of LDs and lysosome as well as lipophagy in normal hepatocytes, but not in STING1-deficient hepatocytes with lower activation of MTORC1. Stimulating MTORC1 activity with AAs in STING1-deficient hepatocytes caused an increase in the number of LDs and a concomitant inhibition of lipophagy (Figure 5). Although STING1 can directly interact with MAP1LC3 and facilitate the maturation of autophagosome, STING1-mediated activation of MTORC1 can suppress lysosomal function in the course of autophagosome-lysosome fusion and further inhibits lipophagy. Thus, the role of STING1-MTORC1 in lipophagy does not conflict with that of STING1 in autophagic flux.
Based on our findings, we propose herein a working model to illustrate how directional communication between STING1 and MTORC1 suppresses the clearance of LDs (Figure 6F). Saturated fatty acid, such as PA, causes mitochondrial damage and mtDNA release. By sensing mtDNA, CGAS activates STING1 and promotes SQSTM1 phosphorylation on the Ser403 site. STING1, through phosphorylated SQSTM1, facilitates the complex formation of MTORC1 and increases the activation of MTORC1 signaling. Finally, activated MTORC1 suppresses the lysosomal degradation of LDs. Our study unveiled a synergetic suppressing effect of STING1 and MTORC1 on LD clearance. STING1 activation may contribute to MTORC1-inhibited lipid degradation. The interaction of STING1 and MTORC1 signaling may explain the difficulty associated with clinically treating patients with NASH than patients with NAFL. Manipulating STING1 activation in the clinical treatment of NASH may not only be a viable therapeutic option against excessive inflammation, but it may also mitigate lipid accumulation and lipid-related toxicity.
Materials and methods
Cell culture and transfection
HEK293T (GNHu17) and Hep3B (SCSP-5045) cells were purchased from the Cell Bank at the Institute of Biochemistry and Cell Biology, China Academy of Science, and were cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco, 11965092) supplemented with 10% fetal bovine serum (PAN, P30-3302) and 1% GlutaMAX Supplement (Gibco, 35050061), and maintained at 37°C under 5% CO2. Transient transfections of cells were performed with Lipofectamine 3000 reagent (Invitrogen, L3000075) according to the manufacturer’s instructions. For cGAMP transfection, the concentration of cGAMP is the dose diluted in the culture medium.
Isolation of mouse primary hepatocytes
The method of isolating mouse hepatocytes is based on previously reported with slight modifications [45]. Briefly, the two-step collagenase perfusion procedure was used. The mice were perfused with Ca2+ and Mg2+-free Hanks’ buffered salt solution (HBSS; Gibco, 14185052) containing EGTA (Sigma, E4378) (2.5 mM) through portal vein cannulation (Step 1), followed by the enzymatic solution containing collagenase P (Sigma, 11249002001) (Step 2). The cell suspensions were filtered through a 70 μm nylon mesh, centrifuged at 50 g for 1 min at 4°C, and the cell pellets (hepatocytes) were collected.
Patients and tissue samples
We obtained 16 cases of snap-frozen tissue samples for the extraction of RNA and 16 cases of paraffin-embedded specimens from partial liver donation with biopsy-proven NAFLD at the Third Affiliated Hospital of Sun Yat-Sen University (Table S1). Liver sample obtained from 6 healthy subjects candidate for partial liver donation served as controls (Table S2). The usage of the clinical samples and methods in this study was carried out in accordance with the approved guidelines by the Third Affiliated Hospital of Sun Yat-Sen University, and written informed consent to participate in the study was obtained from all subjects.
Antibodies and reagents
The primary antibodies used were as follows: monoclonal anti-Flag M2-peroxidase (Sigma, A8592), anti-HA-peroxidase (Roche, 12013819001), anti-MYC-horseradish peroxidase (Santa Cruz Biotechnology, sc-40; HRP), monoclonal anti-ACTB/actin beta (Abclonal, AC026), anti-p-MTOR (S2159; Merck, ABS79), anti-p-RPS6KB1 (T389; Cell Signaling Technology [CST], 9234S), anti-RPS6KB1 (CST, 2708S), anti-p-SQSTM1 (S403; CST, 39786S), anti-STING1 (Proteintech, 19851-1-AP; Proteintech, 66680-1-Ig; CST, 13647), anti-RPTOR (CST, 2280S; Santa Cruz Biotechnology-sc-81537), anti-RRAGA (CST, 4357), anti-MTOR (CST, 2983S; Proteintech, 66888-1-Ig), anti-p-TBK1 (S172; CST, 5483S), anti-p-RPS6 (S235/236; CST, 4858S), anti-RPS6 (Immunoway, YT4139), anti-SQSTM1 (Proteintech, 18400-1-AP), anti-MAP1LC3B (Proteintech, 18725-1-AP), anti-PLIN2 (Proteintech, 15294-1-AP), anti-LAMP1 (CST, 15665). The secondary antibodies used in western blotting were as follows: goat anti-rabbit IgG-HRP (CST, 7074S) and goat anti-rabbit IgG-HRP (CST, 7076S). Goat anti-rabbit IgG (H + L; Biotium, CF405M-20373, CF488-20019, CF568-20103), goat anti-mouse IgG (H + L; Biotium, CF488-20018, CF568-20101), Anti-Flag affinity gel (B23102) was purchased from Bimake. Protein A/G agarose (20241) was purchased from Pierce. The chemical reagents used were as follow: 2ʹ3’-cGAMP (Invivogen, tlrl-nacga23), HT-DNA (Sigma, D6898), rapamycin (Selleckchem, S1039), BX795 (Selleckchem, S1274), amino acids solution (Gibco, 11130051), non-essential amino acids (Gibco, 11140076), LysoTracker Red (Beyotime, C1046), BODIPY-493/503 (Invitrogen, D3922). For FFA treatment, de-lipidated low-endotoxin bovine serum albumin (BSA; Shanghai Yeasen Biotechnology, 36104ES25) was loaded with PA (Sigma, P5585) and OA (Sigma, O1008), as described [11].
Plasmid construction and mutagenesis
The human genes STING1, SQSTM1, MLST8, DEPTOR, RRAGA, RRAGC, and RPTOR were generated by PCR amplification from a normal liver cDNA library and cloned into Flag-, HA- and MYC-tagged FG-EH-DEST vectors were derived from Cui Jun lab in Sun Yat-Sen University. Flag-tagged SQSTM1S403E and SQTM1S403A mutants were generated using a site-directed mutagenesis kit (SBS Genetech, SDM-15).
Knockout of STING1 by the CRISPR-Cas9 system
Construction of the lenti-CRISPR-Cas9 vectors targeting STING1 was performed following a standard protocol. Briefly, gRNA was synthesized and annealed and then was ligated into the vector at the BsmBI restriction sites. The gRNA sequences of STING1 were obtained using an online gRNA design tool as follows:
GFP-sgRNA (as control): Forward 5ʹ- CACCGGGGCGAGGAGCTGTTCACCG −3ʹ, reverse 5ʹ- AAACCGGTGAACAGCTCCTCGCCCC −3ʹ;
Human STING1-sgRNA:
Forward 5ʹ- CACCGCGGGCCGACCGCATTTGGGAGGG −3ʹ, reverse 5ʹ- AAACCCCTCCCAAATGCGGTCGGCCCGC −3ʹ;
RNA interference
LipoRNAiMAX (Invitrogen, 13778150) was used for the transfection of siRNAs into cells according to the manufacturer’s instructions. The sequences of siRNAs are listed as follows:
Scramble-siRNA: 5ʹ- UUCUCCGAACGUGUCACGUTT −3ʹ
Human STING1-siRNA: 5ʹ- GCAUCAAGGAUCGGGUUUATT −3ʹ
Human SQSTM1-siRNA: 5ʹ- GAUCUGCGAUGGCUGCAAUTT −3ʹ
Human TBK1-siRNA: 5ʹ- GCAGUUUGUUUCUCUGUAUTT −3ʹ
Mouse Sqstm1-siRNA: 5ʹ- CUCAGCCAAGCAGCUGCUCTT-3ʹ
Mouse Sting1-siRNA-1#: 5ʹ- GCAUCAAGAAUCGGGUUUATT −3ʹ
Mouse Sting1-siRNA-2#: 5ʹ- CAACAUUCGAUUCCGAGAUTT −3ʹ
Mouse Sting1-siRNA-3#: 5ʹ- GAGGUCACCGCUCCAAAUATT −3ʹ
Mouse Sting1-siRNA-4#: 5ʹ- GAUUCUACUAUCGUCUUAUTT −3ʹ
Transfection and generation of stable cell lines
For transient transfection, Hep3B cells were plated in 24-well plates at density of 1.2 × 105 cells per well 24 h before transfection. Lipofectamine 3000 (Invitrogen, L3000015) was used for transfection according to the manufacturer’s instructions. To generate stable cell lines, lentivirus-containing medium was collected from HEK293T cells twice at 24 h intervals beginning 48 h after transfection with the lentiviral overexpression or CRISPR-Cas9-mediated gene knockout vector. The infection of Hep3B cells was performed with serial dilutions of lentivirus in the presence of 5 µg/ml polybrene (Sigma, 107689). Forty-eight hours post-transduction, lentivirus-transduced Hep3B cells were cultured with 2 µg/ml puromycin (Invivogen, ant-pr) for 2 weeks. The puromycin-resistant colonies were then collected and expanded for further analysis under selective conditions.
qPCR
Total RNA was extracted using TRIzol (Invitrogen, 15596026) and was reverse transcribed to cDNA using a PrimeScript RT reagent kit (Vazyme, R233-01). Real-time qPCR was performed using the Lightcyler 480 Real-Time PCR System (Roche) and SYBR Green PCR Master Mix (Genstar, A301). RPL13A was used for normalization. The primers for the qPCR are listed as follow:
RPL13A-Forward: GCCATCGTGGCTAAACAGGTA
RPL13A-Reverse: GTTGGTGTTCATCCGCTTGC
IL1B-Forward: AAATACCTGTGGCCTTGGGC
IL1B-Reverse: TTTGGGATCTACACTCTCCAGCT
Human MT-ND1-Forward: ATACCCATGGCCAACCTCCT
Human MT-ND1- Reverse: GGGCCTTTGCGTAGTTGTAT
Mouse Mt-nd1-Forward: TATCTCAACCCTAGCAGAAA
Mouse Mt-nd1-Reverse: TAACGCGAATGGGCCGGCTG
Immunoprecipitation and immunoblot analysis
After the indicated treatment, cells were rinsed twice with cold PBS (Boster Biological Technology, PYG0021) and were lysed in low salt lysis buffer (150 mM NaCl [Sigma, S5886], 50 mM HEPES [Sigma, H3375], pH 7.4, 1 mM EDTA[Sigma, E6758], 1.5 mM MgCl2 [Sigma, V900020], 1% Triton X-100 [Sigma, X100] containing protease inhibitors [Bimake, B14011] and phosphatase inhibitors [Bimake, B15001]) for 30 min at 4°C. For immunoprecipitation, equal amounts of protein from each group were incubated with anti-Flag antibody-conjugated affinity gel (Bimake, B23102) or primary antibodies with either protein A/G agarose beads (Pierce, 20423) in lysis buffer overnight. The beads were centrifuged at 5000 g for 5 min at 4°C and were extensively washed with lysis buffer. The beads were then re-suspended with loading buffer (62.5 mM Tris-HCl [Solarbio, T8230], pH 6.8, 2.5% SDS [Solarbio, S8010], 0.002% bromophenol blue [Sigma, 114391], 0.7135 M (5%) β-mercaptoethanol [Solarbio, M8210], 10% glycerol [Sigma, G5516]) and subjected to SDS-PAGE. After electrophoresis, the protein bands in the gel were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, IPVH00010) and probed with specific primary antibodies, followed by the appropriate secondary antibodies. The bands were detected using the ChemiDoc XRS system (Bio-Rad Laboratories, Hercules, CA, USA).
Immunohistochemistry
Immunohistochemistry procedures were performed as previously described [46]. Briefly, paraffin-embedded specimens were cut into 4 μm sections and were placed on polylysine-coated slides. After baking at 45°C for 2 h, sections were deparaffinized with xylenes and rehydrated using graded ethanol. Next, the slides were placed in 3% hydrogen peroxide (Aladdin, H112517) to eliminate the activity of endogenous peroxidase and then were processed for antigen retrieval by microwave heating for 15 min in 10 mM citrate buffer (pH 6.0) (Maixin Biological, MVS-0100). The sections were then submerged into 5% goat serum (Boster Biological Technology, AR0009) diluent for 1 h to block nonspecific binding, and were incubated with the indicated antibodies diluted in SignalStain Antibody Diluent (CST, 8112S) overnight. After washing, the sections were incubated with GTVision detection system (Dako, GK500705) for 1 h, and then were stained with the DAB kit (Maixin Biological, DAB-0031). Images were obtained using a Nikon Eclipse Ni microscope (Nikon Instruments Inc., USA) with NIS-Elements D software, and positive cells were quantified using ImagePro Plus software. The staining score was blindly calculated by different individuals using the staining intensity multiplied by the cell percentage (+) %*1 + (++) %*2 + (+++) %*3. + indicates a weak staining signal; ++ indicates a medium staining signal; +++, indicates a strong staining signal.
Immunofluorescence
Immunofluorescence was performed essentially as previously reported [46]. In brief, cells were fixed with 4% paraformaldehyde and permeabilized with 100% methanol. The permeabilized cells were incubated with appropriate primary antibodies at 4°C overnight. After PBS wash, cells were incubated with CF488-conjugated goat-anti-mouse IgG (Biotium, 20018) or CF568-conjugated goat-anti-rabbit IgG (Biotium, 20103) at room temperature for 1 h. Nuclei were revealed by Hoechst 33342 (Sigma, B2261) staining. Fluorescence images were collected using a confocal laser scanning microscope (Zeiss LSM880+Airyscan, Germany). To label LDs, fixed samples were washed in 60% isopropanol (Aladdin, I112023) for 30 s and 60% Oil Red O (Sigma, O0625) solution (5 mg/ml in isopropanol) for 1.5 min and then washed in 60% isopropanol for an additional 30 s. Images were acquired using confocal microscope. LD numbers were quantified by blind counting.
Statistical analysis
Student’s t-test was used for statistical analysis with GraphPad Prism 5.0 software. Pearson’s correlation test was used to analyze the correlation between the levels of IHC staining. P < 0.05 is considered to be statistically significant.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81970509, 81974436, 81901613, 81800559, 82001663, 31900661), National Science and Technology Major Project (2018ZX10723203), Natural Science Foundation of Guangdong Province (2017A030310252, 2020A1515011299), China Postdoctoral Science Foundation Grant (2019M653189), the Fundamental Research Funds for the Central Universities (20ykzd03, 20ykpy28, 19ykpy26, 19ykzd06), Young Elite Scientists Sponsorship Program by China Association for Science and Technology (No.2020-QNRC1-03), and the Program for Guangdong Introducing Innovative and Enterpreneurial Teams (2019ZT08Y485).
Funding Statement
This work was supported by the Fundamental Research Funds for the Central Universities [20ykzd03]; Fundamental Research Funds for the Central Universities [20ykpy28]; Fundamental Research Funds for the Central Universities [19ykzd06]; Fundamental Research Funds for the Central Universities [19ykpy26]; Natural Science Foundation of Guangdong Province [2020A1515011299]; National Natural Science Foundation of China [81800559]; Natural Science Foundation of Guangdong Province (CN) [2017A030310252]; National Natural Science Foundation of China [81974436]; young elite scientists sponsorship program by china association for science and technology [No.2020-QNRC1-03]; National Natural Science Foundation of China [81901613]; National Natural Science Foundation of China [81970509]; National Natural Science Foundation of China [82001663]; National Natural Science Foundation of China [31900661]; National Science and Technology Major Project [2018ZX10723203]; program for guangdong introducing innovative and enterpreneurial teams [2019ZT08Y485]; Postdoctoral Research Foundation of China [2019M653189].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Eslam M, Sanyal AJ, George J, et al. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020. Feb 7;158(7):1999–2014.e1. [DOI] [PubMed] [Google Scholar]
- [2].Friedman SL, Neuschwander-Tetri BA, Rinella M, et al. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018. Jul;24(7):908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Musso G, Cassader M, Gambino R.. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov. 2016. Apr;15(4):249–274. [DOI] [PubMed] [Google Scholar]
- [4].Martin S, Parton RG.. Lipid droplets: a unified view of a dynamic organelle. Nat Rev Mol Cell Biol. 2006. May;7(5):373–378. [DOI] [PubMed] [Google Scholar]
- [5].Yasuda M, Tanaka Y, Kume S, et al. Fatty acids are novel nutrient factors to regulate mTORC1 lysosomal localization and apoptosis in podocytes. Biochim Biophys Acta. 2014. Jul;1842(7):1097–1108. [DOI] [PubMed] [Google Scholar]
- [6].Chakrabarti P, English T, Shi J, et al. Mammalian target of rapamycin complex 1 suppresses lipolysis, stimulates lipogenesis, and promotes fat storage. Diabetes. 2010. Apr;59(4):775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Le Bacquer O, Petroulakis E, Paglialunga S, et al. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J Clin Invest. 2007. Feb;117(2):387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Um SH, Frigerio F, Watanabe M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004. Sep 9;431(7005):200–205. [DOI] [PubMed] [Google Scholar]
- [9].Yecies JL, Zhang HH, Menon S, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011. Jul 6;14(1):21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Luo X, Li H, Ma L, et al. Expression of STING Is Increased in Liver Tissues From Patients With NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology. 2018. Dec;155(6):1971–1984 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cho C-S, Park H-W, Ho A, et al. Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1–mediated p62/sequestosome 1 phosphorylation. Hepatology. 2018. Oct;68(4):1331–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yu Y, Liu Y, An W, et al. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest. 2019. Feb 1;129(2):546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Qiao JT, Cui C, Qing L, et al. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism. 2018. Apr;81:13–24. [DOI] [PubMed] [Google Scholar]
- [14].Wang X, Yu W, Nawaz A, et al. Palmitate induced insulin resistance by PKCtheta-dependent activation of mTOR/S6K pathway in C2C12 myotubes. Exp Clin Endocrinol Diabetes. 2010. Oct;118(9):657–661. [DOI] [PubMed] [Google Scholar]
- [15].Duran A, Amanchy R, Linares JF, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011. Oct 7;44(1):134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wu J, Sun L, Chen X, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013. Feb 15;339(6121):826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sun L, Wu J, Du F, et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013. Feb 15;339(6121):786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mao Y, Luo W, Zhang L, et al. STING-IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler Thromb Vasc Biol. 2017. May;37(5):920–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sancak Y, Bar-Peled L, Zoncu R, et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010. Apr 16;141(2):290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008. Jun 13;320(5882):1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Matsumoto G, Wada K, Okuno M, et al. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011. Oct 21;44(2):279–289. [DOI] [PubMed] [Google Scholar]
- [22].Prabakaran T, Bodda C, Krapp C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. 2018. Apr 13;37(8):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bodur C, Kazyken D, Huang K, et al. The IKK-related kinase TBK1 activates mTORC1 directly in response to growth factors and innate immune agonists. EMBO J. 2018. Jan 4;37(1):19–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007. Sep-Oct;3(5):452–460. [DOI] [PubMed] [Google Scholar]
- [25].Tsai T-H, Chen E, Li L, et al. The constitutive lipid droplet protein PLIN2 regulates autophagy in liver. Autophagy. 2017. Jul 3;13(7):1130–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schott MB, Weller SG, Schulze RJ, et al. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J Cell Biol. 2019. Oct 7;218(10):3320–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu D, Wu H, Wang C, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 2019. Sep;26(9):1735–1749. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Adams LA, Lymp JF, St Sauver J, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005. Jul;129(1):113–121. . [DOI] [PubMed] [Google Scholar]
- [29].Cai X, Chiu Y-H, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell. 2014. Apr 24;54(2):289–296. [DOI] [PubMed] [Google Scholar]
- [30].Ahn J, Barber GN. Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr Opin Immunol. 2014. Dec;31:121–126. [DOI] [PubMed] [Google Scholar]
- [31].Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017. Mar 9;168(6):960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li Y-N, Su Y. Remdesivir attenuates high fat diet (HFD)-induced NAFLD by regulating hepatocyte dyslipidemia and inflammation via the suppression of STING. Biochem Biophys Res Commun. 2020. May 28;526(2):381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cooper JM, Ou Y-H, McMillan EA, et al. TBK1 provides context-selective support of the activated AKT/mTOR pathway in lung cancer. Cancer Res. 2017. Sep 15;77(18):5077–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hasan M, Gonugunta VK, Dobbs N, et al. Chronic innate immune activation of TBK1 suppresses mTORC1 activity and dysregulates cellular metabolism. Proc Natl Acad Sci U S A. 2017. Jan 24;114(4):746–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kim JK, Jung Y, Wang J, et al. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia. 2013. Sep;15(9):1064–1074. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Antonia RJ, Castillo J, Herring LE, et al. TBK1 limits mTORC1 by promoting phosphorylation of raptor Ser877. Sci Rep. 2019. Sep 17;9(1):13470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Porstmann T, Santos CR, Griffiths B, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008. Sep;8(3):224–236. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zechner R, Zimmermann R, Eichmann TO, et al. FAT SIGNALS–lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012. Mar 7;15(3):279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gui X, Yang H, Li T, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019. Mar;567(7747):262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ding H, Ge G, Tseng Y, et al. Hepatic autophagy fluctuates during the development of non-alcoholic fatty liver disease. Ann Hepatol. 2020;19(5):516–522. Sep–Oct. [DOI] [PubMed] [Google Scholar]
- [41].Liu K-P, Zhou D, Ouyang D-Y, et al. LC3B-II deacetylation by histone deacetylase 6 is involved in serum-starvation-induced autophagic degradation. Biochem Biophys Res Commun. 2013. Nov 29;441(4):970–975. [DOI] [PubMed] [Google Scholar]
- [42].Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010. Aug;24(8):3052–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012. Jun 12;5(228):ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhou J, Tan S-H, Nicolas V, et al. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013. Apr;23(4):508–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mederacke I, Dapito DH, Affo S, et al. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc. 2015. Feb;10(2):305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Liu K, Zhang C, Li B, et al. Mutual stabilization between TRIM9 short isoform and MKK6 potentiates p38 signaling to synergistically suppress glioblastoma progression. Cell Rep. 2018. Apr 17;23(3):838–851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.