

ABSTRACT
Diabetic nephropathy (DN) has become a major cause of end-stage renal disease, and autophagy disorder is implicated in the pathogenesis of DN. Our previous studies found that vitamin D (VD) and VDR (vitamin D receptor) played a renoprotective role by inhibiting inflammation and fibrosis. However, whether VD-VDR regulates autophagy disorders in DN remains unclear. In this study, we established a streptozotocin (STZ)-induced diabetic model in vdr knockout (vdr-KO) mice and VDR specifically overexpressed in renal proximal tubular epithelial cells (Vdr-OE) mice. Our results showed that paricalcitol (an activated vitamin D analog) or Vdr-OE could alleviate STZ-induced ALB (albumin) excretion, renal tubule injury and inflammation, while these were worsened in vdr-KO mice. Defective autophagy was observed in the kidneys of STZ mice, which was more pronounced in vdr-KO mice and could be partially restored by paricalcitol or Vdr-OE. In high glucose-induced HK-2 cells, defective autophagy and decreased PRKAA1/AMPK phosphorylation was observed, which could be partially restored by paricalcitol in a VDR-dependent manner. AMPK inhibitor abolished paricalcitol-induced autophagy activation, and AMPK activator restored the defective autophagy in high glucose-induced HK-2 cells. Furthermore, paricalcitol-mediated AMPK activation was abrogated by CAMKK2/CaMKKβ inhibition, but not by STK11/LKB1 knockout. Meanwhile, paricalcitol rescued the decreased Ca2+ concentration induced by high glucose. In conclusion, VD-VDR can restore defective autophagy in the kidney of STZ-induced diabetic mice, which could be attributed to the activation of the Ca2+-CAMKK2-AMPK pathway in renal tubular epithelial cells.
Abbreviations: ACTB/β-actin: actin beta;AGE: advanced glycation end-products;AMPK: AMP-activated protein kinase;CAMKK2/CaMKKβ: calcium-calmodulin dependent protein kinase kinase 2;CQ: chloroquine;DN: diabetic nephropathy;HG: high levels of glucose;KO: knockout;LG: low levels of glucose;MAP1LC3/LC3: microtubule associated protein 1 light chain 3;NOD2: nucleotide binding oligomerization domain containing 2;OE: overexpression;PAS: periodic acid Schiff; Pari: paricalcitol;PTECs: proximal renal tubule epithelial cells;RT: room temperature;SQSTM1/p62: sequestosome 1;STK11/LKB1: serine/threonine kinase 11;STZ: streptozotocin;TEM: transmission electron microscopy;VD: vitamin D;VDR: vitamin D receptor;WT: wild-type
KEYWORDS: AMPK, autophagy, diabetic nephropathy, renal tubular epithelial cell, vitamin D receptor
Introduction
Diabetic nephropathy (DN) is a serious microvascular complication of diabetes, and approximately 30%-40% of diabetic patients suffer from DN [1]. DN has become the primary cause of chronic kidney disease in elderly individuals, and its incidence in China has risen dramatically, making it the second leading cause of end-stage renal disease [2].
Many molecular pathways are related to the pathogenesis of DN, including the formation of advanced glycation end-products (AGE), inflammation, oxidative stress, endoplasmic reticulum stress and macroautophagy/autophagy [3–6]. Autophagy is an evolutionarily conserved homeostatic cellular process that plays an important role in degrading damaged organelles and abnormal and misfolded proteins to sustain cellular metabolism. Autophagy is regulated by many autophagy-related genes. The conversion of inactive MAP1LC3/LC3 (microtubule associated protein 1 light chain 3; LC3-I) to the active LC3-II isoform indicates autophagosome formation [7]. As LC3-I is more labile than LC3-II, the quantification of changes in LC3-II is one of the most widely used to monitor autophagy [8]. The SQSTM1/p62 (sequestosome 1) protein binds to ubiquitinated substrate cargo and targets them for degradation in the autophagy-lysosome system via interacting with LC3 [9]. Dysregulated autophagy has been observed in the proximal tubules and podocytes of streptozotocin (STZ)-induced diabetic models [10,11]. Moreover, SQSTM1 accumulates in proximal tubule epithelial cells of kidney biopsy samples from type 2 diabetes mellitus (T2DM) patients [12]. These findings indicate that autophagy is deficient in the diabetic kidney. However, the molecular mechanisms influencing autophagy in DN are not yet understood.
VDR (vitamin D receptor) is a nuclear receptor that exert a renoprotective effect through anti-inflammation, antifibrosis and inhibiting the renin-angiotensin system activities [13]. Our previous research showed that the expression of VDR is reduced in peripheral blood mononuclear cells and renal tubular epithelial cells from T2DM patients with albuminuria and negatively correlated with urine albumin-to-creatine ratio [14]. Further studies have confirmed that VDR deficiency participated in the development of DN and vitamin D (VD)-VDR played a protective role in kidney by inhibiting inflammation and fibrosis [15,16]. Although VD could suppress apoptosis of pancreatic β‑cells, prevent insulitis, increase insulin secretion and protect insulinoma cells from oxidative damage through activation of autophagy in STZ mice [17,18], the relationship between VD-VDR and autophagy in DN is not clear. In this study, we found defective autophagy in kidney of STZ-diabetic mice, which was further worsened in vdr knockout (vdr-KO) mice. However, paricalcitol treatment or VDR specifically overexpressed in renal proximal tubular epithelial cells (Vdr-OE) partially reversed the defective autophagy.
Results
VD-VDR delayed DN progression
Twelve weeks after induction, STZ-induced diabetic WT mice developed albuminuria while paricalcitol (pari) treatment blocked albuminuria in STZ-induced mice without altering blood glucose level or body weight. Meanwhile, vdr-KO mice developed more severe albuminuria than WT mice in response to STZ-induction (Figure 1A). Periodic acid Schiff (PAS) staining showed various degrees of partial dilatation of renal tubule and flattened proximal renal tubule epithelial cells (PTECs) with a reduced brush border following STZ treatment, which were further worsened by vdr-KO but partially prevented by pari treatment (Figure 1B).
Figure 1.

VD-VDR delayed diabetic nephropathy progression. (A) Body weight, blood glucose levels and albuminuria of mice during the treatment period in different treatment groups as indicated. *p < 0.05; **p < 0.01 vs WT group. #p < 0.05; ##p < 0.01 vs KO group. $p < 0.05; $$p < 0.01 vs WT+STZ group. (B) PAS staining of kidney sections. The arrows indicate examples of damaged tubules. (C) Body weight, blood glucose levels and albuminuria of mice during the treatment period in different treatment groups as indicated. **p < 0.01 vs WT group. ##p < 0.01 vs OE group. (D) PAS staining of kidney sections. The arrows indicate examples of damaged tubules. Scale bar: 50 μm. KO, knockout; OE, overexpression; pari, paricalcitol.
On the other hand, Vdr-OE reduced STZ-induced albuminuria without affecting either body weight or blood glucose level (Figure 1C). PAS staining showed various degrees of flattened proximal tubular epithelial cells in both WT+STZ and OE+STZ mice, with OE+STZ mice exhibiting partially attenuated PAS-positive staining and tubular damage. (Figure 1D).
VD-VDR alleviated renal inflammation in STZ-induced diabetic mice
Since inflammation plays an important role in the development of DN, we evaluated the effect of VD-VDR on inflammation in STZ-induced diabetic mice. ADGRE1/F4/80, a marker of macrophage infiltration, was elevated in vdr-KO mice and STZ-induced diabetic WT mice. The increase of ADGRE1 was most dramatic in KO+STZ mice, and was significantly inhibited in WT+STZ+pari group (Figure 2A). Real-time RT-PCR quantification revealed that the expression of proinflammatory cytokines (CCL2/MCP-1 and TNF/TNF-α) was markedly higher in vdr-KO mice and STZ-induced diabetic mice than that in WT mice. This increase was most robust in KO+STZ mice. Furthermore, pari partially restored the increase in proinflammatory cytokines (Figure 2B).
Figure 2.
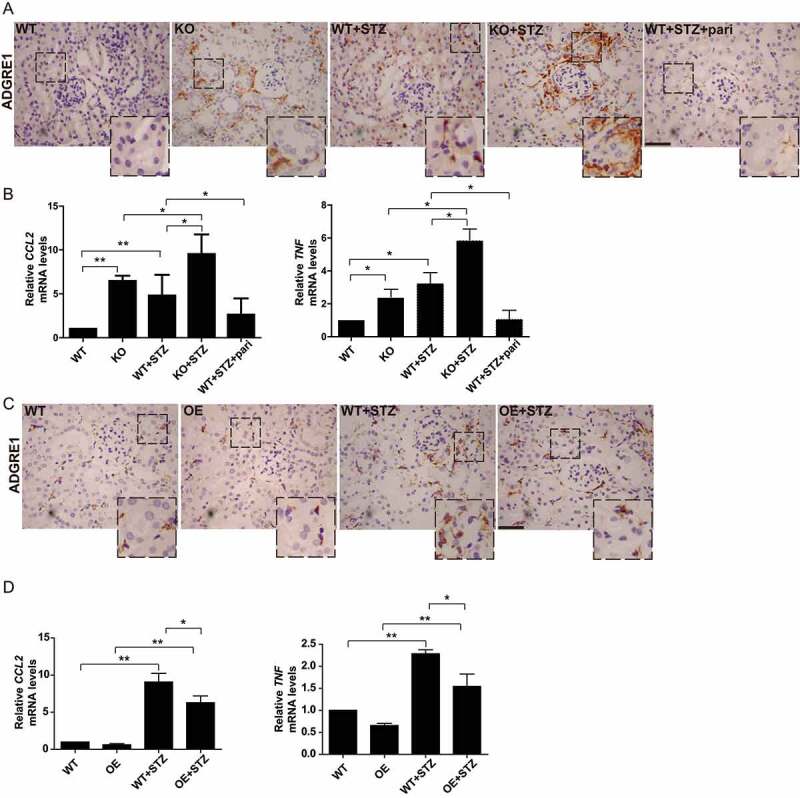
VD-VDR alleviated renal inflammation in STZ-induced diabetic mice. (A) Immunohistochemical staining for ADGRE1 in the kidneys of mice from each group as indicated. ADGRE1-positive cells are stained brown, and nuclei are stained blue. Scale bar: 100 μm. (B) Real-time quantitative PCR analysis of CCL2 and TNF expression in the kidneys of mice from each group as indicated. *p < 0.05, **p < 0.01. (C) Immunohistochemical staining for ADGRE1 in the kidneys of mice from each group as indicated. Scale bar: 100 μm. (D): Real-time quantitative PCR analysis of CCL2 and TNF expression in the kidneys of mice from each group as indicated. *p < 0.05, **p < 0.01.
Compared with that in WT and Vdr-OE mice, the expression of ADGRE1, CCL2 and TNF were all increased in STZ-induced diabetic WT mice. VDR overexpression apparently reduced inflammation infiltration, as OE+STZ mice showed lower expression of inflammation factors than WT+STZ mice (Figure 2C,D).
VD-VDR relieved abnormal autophagosome accumulation in the diabetic mice kidney
Next, we utilized transmission electron microscopy (TEM) to examine changes in the kidney of diabetic mice. More autophagic vacuoles in renal tubular epithelial cells were found from WT+STZ mice than WT mice alone, and the greatest number of autophagic vacuoles was found in diabetic vdr-KO mice (Figure 3A,D). To further confirm these findings, we examined the expression of LC3, a key marker of autophagy. As shown in Figure 3B and 3E, LC3-II was increased in mice following STZ treatment, and this effect was more pronounced in the KO+STZ group, while pari treatment restored STZ-induced LC3 changes. To further verify, we assessed the autophagosomes by immunofluorescence staining. The number of LC3 puncta was increased in STZ mice, and such increase was more obvious in KO+STZ mice, while decreased LC3 puncta was observed in pari treatment (Figure 3C,F). To explore whether the increase in the number of autophagy vacuoles and LC3 indicates autophagic activation or impaired autophagic degradation, autophagy substrate SQSTM1 was examined. As shown in Figure 3B and 3E, higher SQSTM1 expression was observed in STZ-induced diabetic mice than WT mice, indicating defective autophagy in STZ-treated mice, which is consistent with published data [11]. Pari partially restored defective autophagy, as it reduced SQSTM1 expression in STZ-induced diabetic mice. In addition, vdr-KO mice showed higher level of autophagy defect than control mice after STZ treatment, while no SQSTM1 aggregation was observed in OE+STZ mice. Our data demonstrate that autophagy in STZ-induced diabetic kidneys is defective and can be partially restored by VD-VDR.
Figure 3.
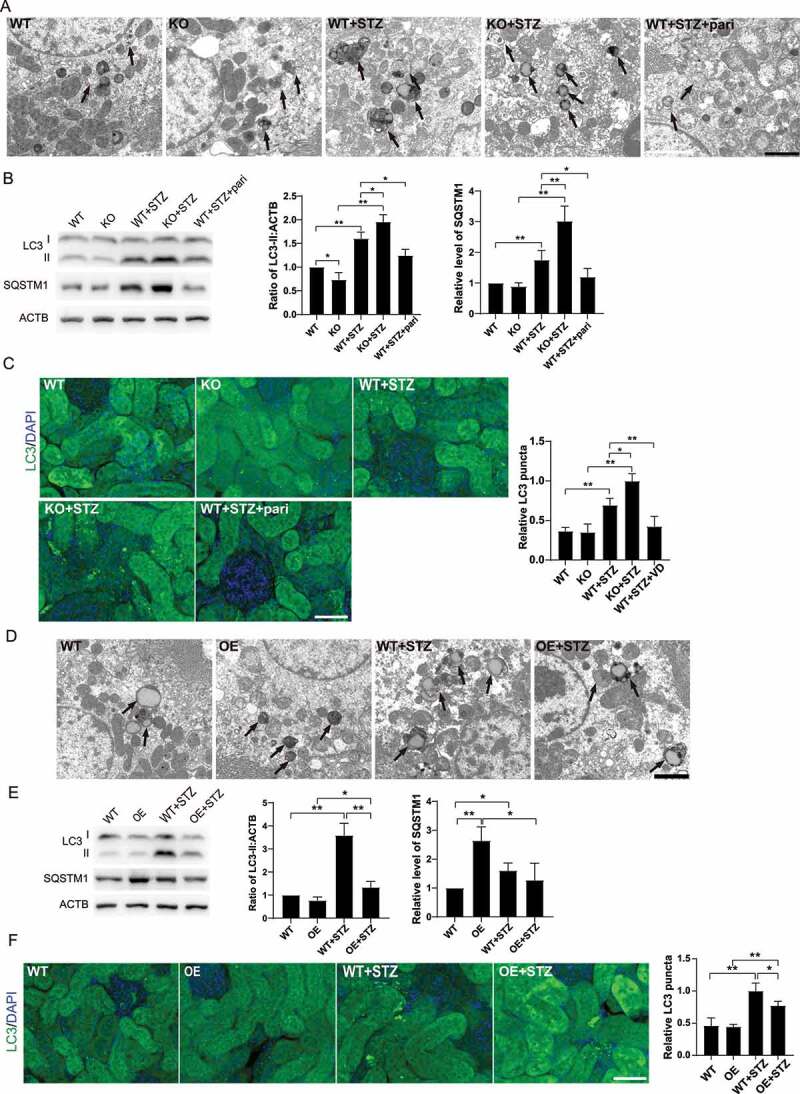
VD-VDR relieved the abnormal autophagosome accumulation in STZ-induced diabetic mice kidney. (A) TEM of proximal tubule epithelial cells from mice of each group as indicated. The arrows indicate autophagic vacuoles. Scale bar: 2 μm. (B) Western blot analysis of LC3 and SQSTM1 in the renal cortex of mice from each group as indicated. ACTB/β-actin was used as the loading control. *p < 0.05, **p < 0.01. (C) Immunofluorescence analysis of LC3 puncta in proximal tubule epithelial cells from mice of each group as indicated. Scale bar: 50 μm. (D) TEM of proximal tubule epithelial cells from mice of each group as indicated. The arrows indicate autophagic vacuoles. Scale bar: 2 μm. (E) Western blot analysis of LC3 and SQSTM1 in the renal cortex of mice from each group as indicated. (F) Immunofluorescence analysis of LC3 puncta in proximal tubule epithelial cells from miceof each group as indicated. Scale bar: 50 μm. ACTB was used as the loading control. *p < 0.05, **p < 0.01.
VD restored defective autophagy induced by high glucose in HK-2 cells
Next, we explored the effect of pari on autophagy and inflammation in DN in vitro. We treated HK-2 cells with high levels of glucose (HG, 30 mM). As shown in Figure 4A, the expression of LC3-II and SQSTM1 was increased under high glucose conditions, and further increased when treated with chloroquine (CQ), an autophagy inhibitor which leads to inhibition of both fusion of autophagosome with lysosome and lysosomal protein degradation. Furthermore, we used tf-LC3 to investigate the autolysosome maturation process. As mCherry is more stable than GFP in the acidic environment of lysosome, the normal maturation of autolysosomes is characterized with increased red-only puncta. In contrast, colocalization of GFP and mCherry puncta would indicate disruption of autophagic flux which presented with yellow puncta. High glucose induced colocalization of mCherry and GFP, which was more obvious when treated with CQ (Figure 4C). These results further indicated that high glucose impaired autophagic flux. On the other hand, paricalcitol led to decreased expression of LC3-II and SQSTM1, and increased red-only puncta when compared with HG group, indicating efficient autophagic flux (Figure 4B,C).
Figure 4.

VD-VDR restored defective autophagy induced by high glucose in HK-2 cells. (A) Expression of LC3 and SQSTM1 in HK-2 cells treated with low levels of glucose (LG, 5 mM), high levels of glucose (HG, 30 mM) with/without chloroquine (CQ, 40 μM) for 3 days. ACTB was used as the loading control. n = 3; a representative image is shown. *p < 0.05, **p < 0.01. (B) Expression of LC3 and SQSTM1 in HK-2 cells treated with LG, HG with/without paricalcitol (pari, 0.5 ng/ml) for 3 days. ACTB was used as the loading control. n = 3; a representative image is shown. *p < 0.05, **p < 0.01. (C) HK-2 cells were transfected with tfLC3, and then treated with LG, HG, HG+CQ or HG+pari. The autophagosomes were shown as yellow puncta with both mcherry (red) and GFP (green) labels, autolysosome were shown as red only puncta because after fusion with lysosomes, GFP loses its fluorescence in acidic PH. DAPI (blue) was used to stain nuclei. At least 10 microscopy fields were assessed in each experiment. The images are representative of three independent experiments. Scale bar, 5 μm. *p < 0.05, **p < 0.01. (D) Expression of CCL2, TNF and IL6 mRNA levels in HK-2 cells post treated with LG, HG, HG+CQ or HG+pari. n = 3. *p < 0.05, **p < 0.01.
To investigate whether defective autophagy contributes to high glucose-induced inflammation, real-time quantitative PCR was used to detect the mRNA levels of proinflammatory factors (CCL2, TNF, IL6). Results showed CQ aggravated inflammation induced by high glucose, while paricalcitol decreased the expression of proinflammatory factors (Figure 4D), which indicated that high glucose-induced inflammation in HK-2 cells was partly due to defective autophagy, and paricalcitol could restore autophagic flux and reduce inflammation.
VD restored defective autophagic flux via activation of AMPK pathway
It has been reported that autophagy changes induced by high glucose were closely related to AMP-activated protein kinase (AMPK) [19]. To explore the molecular mechanisms under which paricalcitol induces autophagy in HK-2 cells, we investigated the status of AMPK-ULK1 kinase network. As showed in Figure 5A, phosphorylation of PRKAA1/AMPKα1 was decreased under high glucose conditions, which was reversed with paricalcitol treatment. Paricalcitol-induced autophagy activation was abolished in the presence of PRKAA1 inhibitor, compound C (Figure 5A). In contrast, metformin, a PRKAA1 activator, decreased levels of LC3-II, SQSTM1 and inflammation induced by high glucose (Figure 5B,C). These results suggested that AMPK activation represents a key mechanism underlying paricalcitol-induced activation of autophagy and inhibition of inflammation under high glucose conditions.
Figure 5.
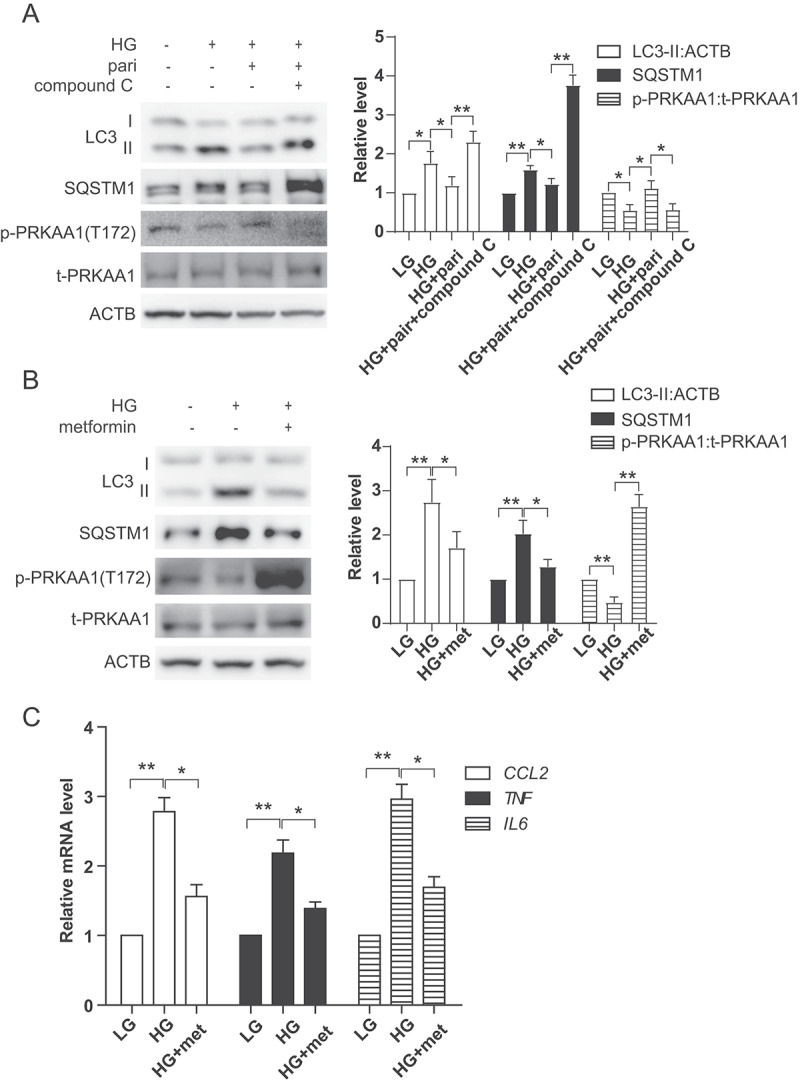
VD restored defective autophagic flux via activation of AMPK pathway. (A) Expression of LC3, SQSTM1, p-PRKAA1 and t- PRKAA1 in HK-2 cells treated with LG, HG, HG+pari or HG+pari+compound C (10 μM, pretreated 1 h) for 3 days. ACTB was used as the loading control. *p < 0.05, **p < 0.01. (B) Expression of LC3, SQSTM1, p-PRKAA1 and t-PRKAA1 in HK-2 cells treated with LG, HG and HG+metformin (met, 2 mM, pretreated 1 h) for 3 days. ACTB was used as the loading control. *p < 0.05, **p < 0.01. (C) Expression of CCL2, TNF and IL6 mRNA levels in HK-2 cells post LG, HG or HG+ metformin. n = 3. *p < 0.05, **p < 0.01.
VD regulated AMPK in a VDR-dependent manner
As vitamin D exerts its most biological effects through VDR, we used HK-2 VDR KO cells to investigate whether VD activates AMPK via VDR. Results showed that paricalcitol increased PRKAA1 and ULK1 phosphorylation, which was decreased under high glucose conditions. However, such effect was totally abolished in HK-2 VDR KO cells (Figure 6A). Furthermore, metformin activated PRKAA1 and ULK1 under high glucose conditions in HK-2 WT cells, but this effect of metformin was still observed in HK-2 VDR KO cells (Figure 6B). These results indicated that VD activated AMPK in a VDR-dependent manner.
Figure 6.
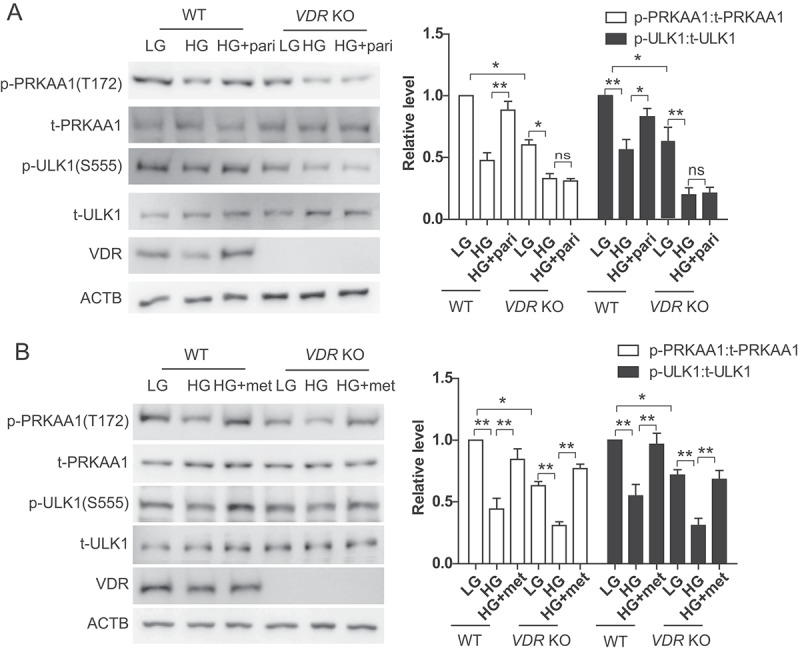
VD regulated AMPK in a VDR-dependent manner. (A) Expression of p- PRKAA1, t- PRKAA1, p-ULK1, t-ULK1 and VDR in HK-2 WT or HK-2 VDR KO cells treated with LG, HG or HG+pari for 3 days. ACTB was used as the loading control. *p < 0.05, **p < 0.01, ns: no significant. (B) Expression of p- PRKAA1, t- PRKAA1, p-ULK1, t-ULK1 and VDR in HK-2 WT or HK-2 VDR KO cells treated with LG, HG or HG+metformin (HG+met) for 3 days. ACTB was used as the loading control. *p < 0.05, **p < 0.01.
VD regulated AMPK via the Ca2+-CAMKK2 pathway
AMPK is activated by phosphorylation via upstream kinase, mainly including STK11/LKB1 (serine/threonine kinase 11) and CAMKK2/CaMKKβ (calcium-calmodulin dependent protein kinase kinase 2) [20]. To determine whether AMPK kinase is responsible for paricalcitol-mediated AMPK activation, STO-609 (a selective CAMKK inhibitor), CAMKK2 siRNA and HK-2 STK11 KO cells were used. As showed in Figure 7A and 7B, co-treatment with STO-609 or CAMKK2 siRNA completely abrogated paricalcitol-stimulated PRKAA1 and ULK1 phosphorylation. However, paricalcitol was able to activate PRKAA1 and ULK1 in HK-2 STK11 KO cells (Figure 7C). These results suggested that paricalcitol actived AMPK through CAMKK2, but not STK11. Furthermore, CAMKK2 is mainly regulated by intracellular Ca2+ concentration. To elucidate the involvement of Ca2+ signaling in the paricalcitol-mediated effect, we assessed changes in the intracellular Ca2+ concentration in response to paricalcitol treatment, using a fluorescent Ca2+ probe, Fluo 4AM. As shown in Figure 7D, Ca2+ concertration decreased when HK2 cells were exposed to high glucose, and this decrease was rescued by paricalcitol.
Figure 7.
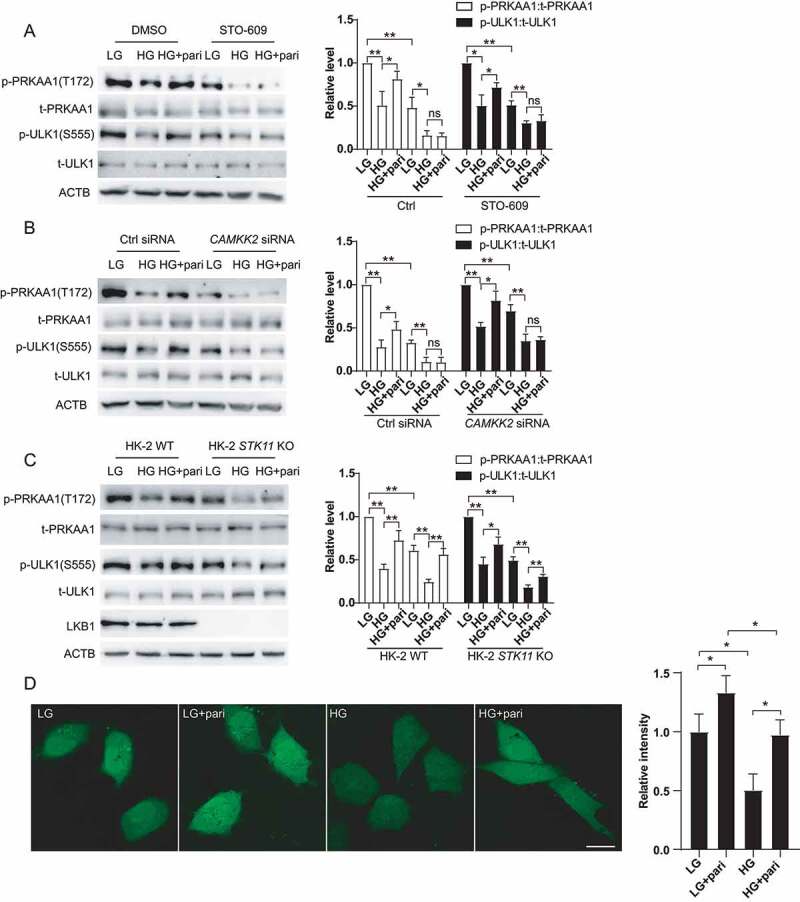
VD regulated AMPK via Ca2+-CAMMK2. (A) Expression of p-PRKAA1, t-PRKAA1, p-ULK1 and t-ULK1 in HK-2 cells treated with LG, HG, HG+pari, STO-609, STO-609+ HG or STO-609+ HG+pari for 72 h. ACTB was used as the loading control. *p < 0.05, **p < 0.01, ns: no significant. (B) Expression of p-PRKAA1, t-PRKAA1, p-ULK1 and t-ULK1 in HK-2 CAMKK2-KD by siRNA cells treated with LG, HG, or HG+pari for 72 h. *p < 0.05, **p < 0.01, ns: no significant. (C) Expression of p-PRKAA1, t-PRKAA1, p-ULK1 and t-ULK1 in HK-2 WT cells or HK-2 LKB1 KO cells treated with LG, HG or HG+pari for 24 h. ACTB was used as the loading control. *p < 0.05, **p < 0.01. (D) Fluo-4 AM analysis of the Ca2+ concentration in HK-2 cells treated with LG, HG or HG+pari for 24 h. *p < 0.05.
VD-VDR actived AMPK in STZ-induced diabetic mice
To further confirm the effect of VD-VDR on AMPK activation under high glucose environment, we detected phosphorylation level of AMPK-ULK1 in STZ-induced diabetic mice. As Figure 8 showed, PRKAA1 and ULK1 phosphorylation levels were decreased in STZ-induced diabetic mice, and this change was more pronounced in KO+STZ mice. Furthermore, paricalcitol or VDR overexpression promoted PRKAA1 and ULK1 phosphorylation, even though there was no statistic difference between WT+STZ groups and OE+STZ groups (Figure 8A,C). We also assessed the level of p-PRKAA1 by immunofluorescence staining, which showed similar trend as it in western blotting (Figure 8B,D). Combined with results in Figure 3E, we speculated that in the STZ-induced diabetic kidneys, VDR required activated by VD to effectively phosphorylate PRKAA1 and then regulate autophagy (Figures 3E and 8A,B). In summary, our in vivo results indicated that VD-VDR restored defective autophagy in kidney of STZ-induced diabetic mice by activating AMPK-ULK1 pathway, which was roughly consistent with the results in vitro.
Figure 8.
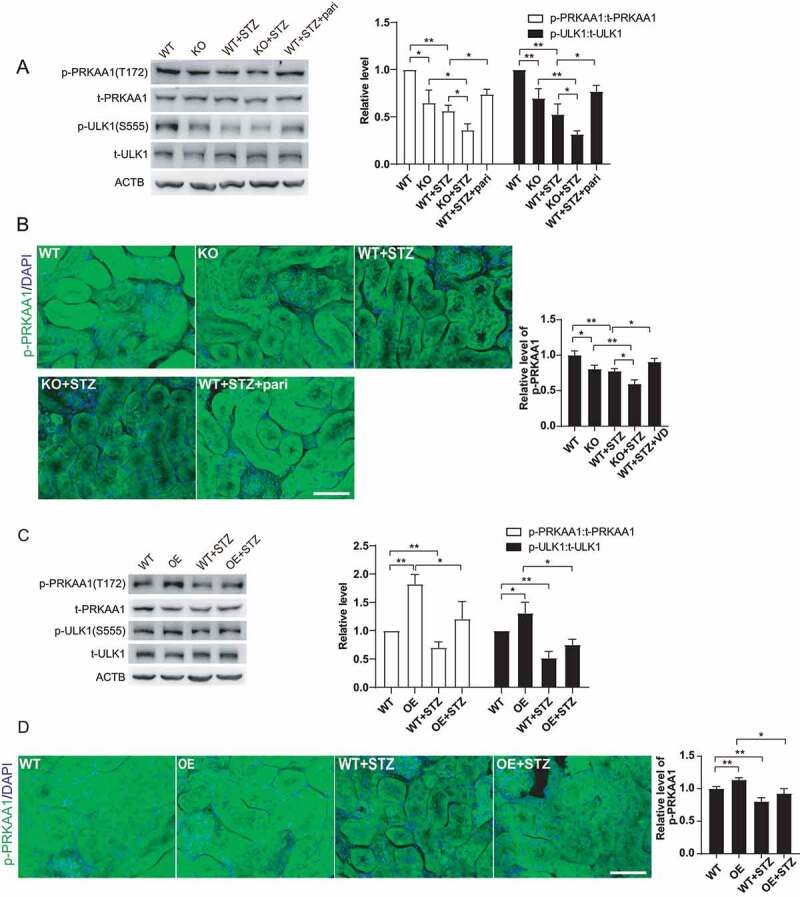
VD-VDR actived AMPK in STZ-induced diabetic mice. (A) Western blot analysis of p- PRKAA1, t- PRKAA1, p-ULK1 and t-ULK1 in the renal cortex of mice from each group as indicated. ACTB was used as the loading control. *p < 0.05, **p < 0.01. (B) Immunofluorescence analysis of p-PRKAA1 in proximal tubule epithelial cells from mice of each group as indicated. Scale bar: 50 μm. *p < 0.05, **p < 0.01. (C) Western blot analysis of p-PRKAA1, t-PRKAA1, p-ULK1 and t-ULK1 in the renal cortex of mice from each group as indicated. ACTB was used as the loading control. *p < 0.05, **p < 0.01. (D) Immunofluorescence analysis of p- PRKAA1 in proximal tubule epithelial cells from mice of each group as indicated. Scale bar: 50 μm. *p < 0.05, **p < 0.01.
Discussion
In the present study, we established an STZ-induced diabetic model in vdr-KO or Vdr-OE mice to investigate the role of vitamin D-VDR in inflammation and autophagy in DN. We found that vdr-KO led to more severely defective autophagy and increased inflammation level in STZ-induced mice than controls. However, paricalcitol or VDR overexpression restored defective autophagy and reduced inflammation caused by STZ-induced diabetes. Further, we report for the first time that paricalcitol ameliorated high glucose induced autophagy deficiency and inflammation in HK-2 cells partially via the Ca2+-CAMKK2-AMPK pathway.
Type 2 diabetic nephropathy is characterized by chronic, low-grade inflammation, which exacerbates the progression of DN [21]. Studies have suggested that autophagy participates in kidney inflammation. In kidney tubular cells, autophagy suppresses inflammation through removing damaged and malfunctioning mitochondria and damaged lysosomes, and inhibiting damage-associated molecular patterns released by cellular protection and damage-associated molecular patterns degradation [22]. AGEs are endocytosed by kidney proximal tubules for lysosomal degradation to suppress AGE-induced inflammation [23]. In PTECs, both high-glucose conditions and AGE overload gradually blunt autophagic flux, while in DN autophagy promotes AGEs degradation by increasing lysosomal biogenesis and function [24]. Thus, impaired AGE degradation due to defective autophagy in DN activates inflammation and thus further promotes DN. We observed in this study that inflammation was worsened when autophagy was blunted by CQ and alleviated when autophagy was restored by paricalcitol or metformin, indicating that defective autophagy at least partially contributes to high glucose-induced inflammation in DN. VDR plays an anti-inflammatory role in DN, but the mechanism of this function is not clear. Our previous study indicated that VDR exerts anti-inflammatory effects in high glucose-treated THP-1 cells via PTPN2 [25]. Zhang et al. also reported that 1,25(OH)2D3 blocks high glucose-induced CCL2 expression in mesangial cells by blunting NF-κB activation [26]. NOD2 (nucleotide binding oligomerization domain containing 2), which can stimulate host immune response, is known to interact with and recruit ATG16L1 to bacterial entry site, which triggering autophagy to eliminate intracellular bacteria [27]. In primary human monocytic and epithelial cells, vitamin D3 induces the expression of NOD2, and VDR promotes NOD2 gene transcription through its vitamin D response elements (VDRES) [28]. Thus, VD-VDR may regulate inflammation by increasing vesicle elongation via NOD2. As VD-VDR both regulate kidney inflammation and autophagy in diabetic mice, we propose that the anti-inflammatory effect of VD-VDR is related to their effect on autophagy, which requires further study.
Many studies have reported a link between autophagy and DN, and that vitamin D3 and its analogs induce autophagy in many cell types. Autophagy deficiency or insufficiency in renal cells, including podocytes, mesangial cells, endothelial cells and tubular cells, contributes to the pathogenesis of DN [29]. Cellular stress and excess nutrition induced by metabolic dysfunction in diabetes are implicated in autophagy impairment through alterations in the nutrient-sensing pathways, including AMPK, mammalian target of rapamycin complex1 and Sirt1 in renal cells [30]. Adaptive autophagy exerts renoprotective effects and autophagy impairment is involved in the pathogenesis of DN [31]. Autophagy degradation was defective in diabetic mice and patients as SQSTM1 was accumulated [11,12], which was consistent with our study. Other than triggering autophagy by upregulating BECN1/Beclin 1 and downregulating MTORC1 and its kinase activity [32,33], vitamin D promotes the formation of autophagosomes via cathelicidin, which is a target gene of VDR [34]. Meanwhile, vitamin D induces Ca2+-dependent autophagy, which depends on the Ca2+-CAMKK2-dependent activation of AMPK [35,36]. Study indicated that vitamin D could stimulate autophagy by downregulating MTOR gene expression in DN [33], but the mechanisms required more direct and in-depth evidence and research. In our study, vdr-KO, Vdr-OE, pharmacological intervention, and gene knockdown were used to explore the mechanism of VD-VDR on autophagy regulating, and results indicated VD-VDR may regulates autophagy in DN through activating CAMKK2, leading to AMPK phosphorylation. Moreover, results in HK-2 cells suggested that VD-VDR mainly regulated AMPK activity in high glucose environment and restored it to near basal level, without causing overactivation of AMPK. AMPK induces autophagy by activating ULK1 or inhibiting MTORC1 activity, while study reported that loss of AMPK or ULK1 resulted in aberrant accumulation of SQSTM1 [37], and AICAR (an AMPK activator) restored downregulated autophagy degradation induced by high glucose [38]. These indicated AMPK may regulate autophagy by regulating autophagy degradation whose mechanism needs further study.
Calcium is involved in various cellular and organ functions, such as neurotransmitter release, intracellular signal transduction, cardiac contractility, muscle contraction, and bone metabolism. Studies have demonstrated that vitamin D increases cytoplasmic Ca2+ level via many aspects. Vitamin D upregulates calcium absorption through transient receptor potential vanilloid calcium channel-6 by forming 1,25(OH)2D3-VDR-RXR complex [39], increases expression of calcium-binding proteins and/or their transcripts, e.g., calbindin-D9k, calbindin-D28k, parvalbumin, calmodulin, and sorcin [40,41], and increase the solvent drag-induced duodenal calcium transport, which is probably mediated by pathways involving 1,25D3-MARRS, PI3K, PKC, and MEK [42]. Moreover, VDR modulates sarcoendoplasmic reticulum Ca2+-ATPase activity, leading toenhanced calcium tunneling across the duodenal cytoplasm [43]. In MCF-7 breast cancer cells, 1,25(OH)2D3 and EB1089 increase intracellular Ca2+, which is required for vitamin D compounds-induced autophagosome formation, and Ca2+-induced autophagy is dependent on CAMKK2 [35]. In db/db mice, calcitriol reduces hepatic triglyceride accumulation and glucose output through increasing cytosolic calcium Ca2+ and activation of the Ca2+-CAMKK2-AMPK pathway [44]. Although the mechanism of how VD regulating Ca2+ is not examined in our study, these findings support our proposed mechanism that vitamin D-VDR regulate autophagy and inhibit inflammation partially through activation of Ca2+-CAMKK2-AMPK pathway.
Taken together, our results demonstrated defective autophagy in the kidneys of STZ-induced diabetic mice, and that vitamin D-VDR partially restored defective autophagy and reduced inflammation. Mechanistic studies demonstrated that AMPK was involved in defective autophagy and inflammation induced by high glucose, and vitamin D-stimulated AMPK phosphorylation was due to CAMKK2 activation in response to increased intracellular Ca2+ concentration. Our results illuminate a new mechanism for the renoprotective effect of vitamin D-VDR in DN.
Materials and methods
Reagents and antibodies
HK-2 cells were from the American Type Culture Collection (CRL-2190). DMEM was obtained from HyClone (SH30023.01). Fetal bovine serum was obtained from Gibco (FBS-CBT). STZ was acquired from Sigma (S0130), chloroquine (CQ, S6999), compound C (S7306), metformin (S5958) and STO-609 (S8274) were acquired from Selleck. Complete protease cocktail was obtained from Roche (5892970001). The following antibodies were used in the experiments: anti-VDR (Santa Cruz Biotechnology, sc-13133), anti-LC3B (Cell Signaling Technology, 2775, 3868), anti-SQSTM1/p62 (Sigma-Aldrich, P0067), anti-ADGRE1/F4/80 (BD Biosciences, 565409), anti-p-PRKAA1/p-AMPKα1 (Cell Signaling Technology, 2535), anti-t-PRKAA1/AMPKα1 (Cell Signaling Technology, 2795), anti-p-ULK1 (Cell Signaling Technology, 5869), anti-t-ULK1 (Cell Signaling Technology, 8054), and anti-STK11/LKB1 (Cell Signaling Technology, 3050). Cy™2 AffiniPure Goat Anti-Rabbit (111–225-144), Peroxidase AffiniPure Goat Anti-Mouse (115–035-003), and Peroxidase AffiniPure Goat Anti-Rabbit (111–035-144) were from Jackson ImmunoResearch Laboratories.
Mice and treatment
vdr knockout (vdr-KO), VDR specifically overexpressed in kidney proximal tubular epithelial cells (Vdr-OE) and wild-type (WT) control mice were constructed in cooperation with the Model Animal Research Center of Nanjing University. The establishment of vdr-KO and Vdr-OE models was confirmed by immunohistochemistry, western blotting and real-time quantitative PCR analyses (Figure S1). To induce diabetic mellitus, 8-week-old male mice received intraperitoneal injections (i.p.) of either sodium citrate (pH 4.5, NC group) or 50 mg/kg STZ (dissolved in sodium citrate, pH 4.5, DN group) for 5 consecutive days. One week later, mice with a casual blood glucose level above 16.7 mmol/L were used for subsequent experiments. vdr-KO mice and their WT counterparts were randomly separated into the WT, KO, WT+STZ, KO+STZ, and WT+STZ+pari groups, and all mice received an i.p. injection of 0.4 µg/kg paricalcitol (an activated vitamin D analog, pari; a present from professor Yan Chun Li, Chicago university) or PBS (Hyclone, SH30256.01) three times a week. Vdr-OE mice and their WT counterparts were randomly separated into the WT, OE, WT+STZ, and OE+STZ groups. Mice were sacrificed 12 weeks after diabetes induction, and their blood, serum and kidneys were harvested. Ethical approval was obtained from the Third Xiangya Hospital of Central South University.
Cell culture and treatment
HK-2 cells were cultured in DMEM with F12 (1:1) supplemented with 10% fetal bovine serum. HK-2 VDR KO cells and HK-2 STK11 KO were generated by using the CRISPR-Cas9 system. The sgRNA sequence for VDR KO is 5ʹ-ACGTTCCGGTCAAAGTCTCC-3ʹ. The sgRNA sequence for STK11 KO is 5ʹ-CAGGTGTCGTCCGCCGCGAA-3ʹ. To evaluate the effect of high-glucose conditions on autophagy, HK-2 cells were incubated for up to 72 h in DMEM containing either low levels of glucose (5 mM, LG), high levels of glucose (30 mM, HG) or CQ (40 μM) with/without paricalcitol (0.5 ng/ml) treatment. To evaluate the effect of AMP activated kinase (AMPK) pathway on high glucose induced autophagy, HK-2 cells were treated with compound C (an AMPK inhibitor; 10 μM, pretreated 1 h) or metformin (an AMPK activator, met; 2 mM, pretreated 1 h). To explore the molecular mechanisms by which paricalcitol activates AMPK, STO-609 (a selective CAMKK2 inhibitor) (10 μM, pretreated 1 h), CAMKK2 siRNA and HK-2 STK11 KO cells were used.
Western blot analysis
Total protein was isolated from the kidney cortex or model cells as previously described (30). Protein concentrations were measured using a BCA protein assay kit (Pierce, 23227). Total protein was separated by SDS/PAGE and electrotransferred to PVDF membranes (Millipore, IPVH00010). The resulting membranes were blocked with PBST (PBS+0.1% Triton X-100 [Sigma-Aldrich, T8787]) containing 5% nonfat milk for 1 h before they were incubated with primary antibodies at 4°C overnight. After being washed three times with PBST, the membranes were further incubated with HRP-conjugated goat anti-mouse or anti-rabbit IgG antibodies at room temperature (RT) for 1 h. Finally, the protein expression levels were measured using chemiluminescent staining reagent kits (SuperSignal West Femto, 34095), and images of stained proteins were captured using Image Scanner. The intensities of bands on the images were quantified with ImageJ software.
Periodic acid Schiff staining
Kidney tissues were immediately preserved in 4% neutral buffered formalin, embedded in paraffin after dehydration through a graded alcohol series, cut into 4-μm sections and stained with PAS (Solarbio, G1281) for histological analysis.
Immunohistochemical staining
VDR and ADGRE1 levels in the formalin-fixed, paraffin-embedded tissues were evaluated using immunohistochemical staining. Briefly, the tissue sections (5-μm thickness) were deparaffinized and rehydrated, and microwave antigen retrieval was conducted in citrate buffer. After cooling, endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 10 min at RT. Then, the slides were blocked with 5% BSA for 1 h at RT and incubated overnight at 4°C with primary antibodies (VDR: 1:100; ADGRE1: 1:200). After washing in PBS, the sections were incubated with secondary antibody for 1 h at RT. After subsequent washes with PBS, DAB was applied to visualize the indicated proteins, followed by counterstaining with hematoxylin to identify cellular nuclei.
Real-time quantitative PCR
Total RNA was extracted from the renal cortex using TRIzol reagent (Invitrogen, 15596–018). cDNA was synthesized using a reverse transcription kit (ReverTra Ace qPCR RT Kit; Thermo Scientific, AB1453B) according to the manufacturer’s instructions. Real-time quantitative PCR was performed using SYBR Green PCR Master Mix (Thermo Scientific, K0251) on an Applied Biosystems 7300 Sequence Detection System. PCR primers were designed using Oligo 6.0 software and synthesized by Shanghai Sangon. PCR was performed with oligonucleotide primers (R&D Systems) specifically designed to amplify mouse CCL2 (forward primer: 5-CACTCACCTGCTGCTACTCA-3 and reverse primer: 5-CTTCTTGGGGTCAGCACAGA-3), mouse TNF (forward primer: 5-TTCTATGGCCCAGACCCTCA-3 and reverse primer: 5- TGTCTTTGAGATCCATGCCGT-3), human CCL2 (forward primer: 5-AGCAGCAAGTGTCCCAAAGA-3 and reverse primer: 5-CGGAGTTTGGGTTTGCTTGT-3), human TNF (forward primer: 5-CCCAGGCAGTCAGATCATCT-3 and reverse primer: 5-AGGACCTGGGAGTAGATGAGG-3), and human IL6 (forward primer: 5-GACAGCCACTCACCTCTTCA-3 and reverse primer: 5-GCCTCTTTGCTGCTTTCACA-3). The relative amounts of the mRNAs were expressed as 2−ΔΔCT.
Transmission electron microscopy
The kidney tissue specimens were sliced into 1x1x3 mm3 in size and double-fixed in 2.5% glutaraldehyde solution (Alfa Aesar, A17876) with phosphate buffer (pH 7.3, 2.28 g NaH2PO4, 29.01 g Na2HPO4.12H2O, with ddH2O volume up to 500 ml) and shipped overnight at ambient temperature to the TEM laboratory at the Pathology Department of Xiangya Hospital, Changsha, Hunan, where specimens were processed as followed. In sample preparations, the samples were washed three times at 10-min intervals with phosphate buffer. Incubated for 1 h in 1% osmium tetroxide and washed three times at 10-min intervals with phosphate buffer. Dehydration of the samples were carried out at room temperature in a graded series of 50%, 70%, and 90% acetone at 10-min intervals for each step followed by 100% acetone twice at 15-min intervals. Sample resin (Electone Microscopy Sciences, GE14120) soaking and embedding process was done with the specimens in 1:1 mix of acetone:resin for 12 h and 100% resin to polymerize overnight at 37°C. Sample resin solidifying process was the specimens 100% resin to polymerize overnight at 37°C and then 12 h at 60°C. 50- to 100-nm ultrathin sections of specimens were made with an ultramicrotome and a diamond knife. After 3% uranyl acetate and lead nitrate double staining, the specimens were examined and photographed on a Hitachi HT-7700 electron microscope.
Immunofluorescence staining
LC3 and p-PRKAA1/AMPKα1 levels in the formalin-fixed, paraffin-embedded tissues were evaluated using immunofluorescence staining. Briefly, the tissue sections (5-μm thickness) were deparaffinized and rehydrated, and microwave antigen retrieval was conducted in citrate buffer. After cooling, endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 10 min at RT. Then, the slides were blocked with 5% BSA for 1 h at RT and incubated overnight at 4°C with primary antibodies (1:100). After washing in PBS, the sections were incubated with Alexa Fluor-conjugated goat anti-rabbit secondary antibody for 1 h at RT. After subsequent washes with PBS, the sections were then stained with DAPI to visualize nuclei. Confocal microscopy was performed using a Leica TCS SP laser scanning microscope (Leica Microsystems, Exton, Pa.) fitted with a 100× Leica objective (Planaprochromatic; 1.4 numerical aperture) and connected to Leica image software.
Statistical analysis
Data were analyzed using SPSS16.0 statistical software and are expressed as the mean ± standard deviation. Qualitative data are representatives of at least 3 experiments. Statistical differences in multiple groups were determined by multiple comparisons with ANOVA followed by Tukey’s post-hoc tests. Student’s t-test was used to analyze the differences between different 2 groups. P < 0.05 indicated statistical significance.
Supplementary Material
Acknowledgments
This study was funded by the National Natural Science Foundation of China (No.81870498, No. 82000696 and No. 82070759) and the science and technology innovation Program of Hunan Province (No. 2020RC2071).
Funding Statement
This work was supported by the National Natural Science Foundation of China [81870498]; The science and technology innovation Program of Hunan Province [2020RC2071]; National Natural Science Foundation of China [82070759]; National Natural Science Foundation of China [82000696].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Adler AI, Stevens RJ, Manley SE, et al. Development and progression of nephropathy in type 2 diabetes: the United Kingdom prospective diabetes study (UKPDS 64) [J]. Kidney Int. 2003;63(1):225–232. [DOI] [PubMed] [Google Scholar]
- [2].Liu ZH. Nephrology in china [J]. Nat Rev Nephrol. 2013;9(9):523–528. [DOI] [PubMed] [Google Scholar]
- [3].Du P, Fan B, Han H, et al. NOD2 promotes renal injury by exacerbating inflammation and podocyte insulin resistance in diabetic nephropathy [J]. Kidney Int. 2013;84(2):265–276. [DOI] [PubMed] [Google Scholar]
- [4].Forbes JM, Coughlan MT, Cooper ME.. Oxidative stress as a major culprit in kidney disease in diabetes [J]. Diabetes. 2008;57(6):1446–1454. [DOI] [PubMed] [Google Scholar]
- [5].Kume S, Thomas MC, Koya D. Nutrient sensing, autophagy, and diabetic nephropathy [J]. Diabetes. 2012;61(1):23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Calcutt NA, Cooper ME, Kern TS, et al. Therapies for hyperglycaemia-induced diabetic complications: from animal models to clinical trials [J]. Nat Rev Drug Discov. 2009;8(5):417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology [J]. Physiol Rev. 2010;90(4):1383–1435. [DOI] [PubMed] [Google Scholar]
- [8].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) [J]. Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death [J]. J Cell Biol. 2005;171(4):603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Barbosa Junior ADEA, Zhou H, Hultenschmidt D, et al. Inhibition of cellular autophagy in proximal tubular cells of the kidney in streptozotocin-diabetic and uninephrectomized rats [J]. Virchows Arch B Cell Pathol Incl Mol Pathol. 1992;61(6):359–366. [DOI] [PubMed] [Google Scholar]
- [11].Vallon V, Rose M, Gerasimova M, et al. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus [J]. Am J Physiol Renal Physiol. 2013;304(2):F156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yamahara K, Kume S, Koya D, et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions [J]. J Am Soc Nephrol. 2013;24(11):1769–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang Y, Kong J, Deb DK, et al. Vitamin D receptor attenuates renal fibrosis by suppressing the renin-angiotensin system [J]. J Am Soc Nephrol. 2010;21(6):966–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yi B, Huang J, Zhang W, et al. Vitamin D receptor down-regulation is associated with severity of Albuminuria in type 2 diabetes patients [J]. J Clin Endocrinol Metab. 2016;101(11):4395–4404. [DOI] [PubMed] [Google Scholar]
- [15].Li A, Zhang H, Han H, et al. LC3 promotes the nuclear translocation of the vitamin D receptor and decreases fibrogenic gene expression in proximal renal tubules [J]. Metabolism. 2019;98:95–103. [DOI] [PubMed] [Google Scholar]
- [16].Zhang Z, Sun L, Wang Y, et al. Renoprotective role of the vitamin D receptor in diabetic nephropathy [J]. Kidney Int. 2008;73(2):163–171. [DOI] [PubMed] [Google Scholar]
- [17].He D, Wang Y, Liu R, et al. 1,25(OH)2D3 activates Autophagy to protect against oxidative damage of INS-1 pancreatic beta cells. Biol Pharm Bull. 2019;42(4):561–567. [DOI] [PubMed] [Google Scholar]
- [18].Wang Y, He D, Ni C, et al. Vitamin D induces autophagy of pancreatic beta-cells and enhances insulin secretion [J]. Mol Med Rep. 2016;14(3):2644–2650. [DOI] [PubMed] [Google Scholar]
- [19].Shen Z, Chen Q, Jin T, et al. Theaflavin 3,3ʹ-digallate reverses the downregulation of connexin 43 and autophagy induced by high glucose via AMPK activation in cardiomyocytes [J]. J Cell Physiol. 2019;234(10):17999–18016. [DOI] [PubMed] [Google Scholar]
- [20].Carling D, Mayer FV, Sanders MJ, et al. AMP-activated protein kinase: nature’s energy sensor [J]. Nat Chem Biol. 2011;7(8):512–518. [DOI] [PubMed] [Google Scholar]
- [21].Lin M, Yiu WH, Li RX, et al. The TLR4 antagonist CRX-526 protects against advanced diabetic nephropathy [J]. Kidney Int. 2013;83(5):887–900. [DOI] [PubMed] [Google Scholar]
- [22].Kimura T, Isaka Y, Yoshimori T. Autophagy and kidney inflammation [J]. Autophagy. 2017;13(6):997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tan AL, Forbes JM, Cooper ME. AGE, RAGE, and ROS in diabetic nephropathy [J]. Semin Nephrol. 2007;27(2):130–143. [DOI] [PubMed] [Google Scholar]
- [24].Takahashi A, Takabatake Y, Kimura T, et al. Autophagy inhibits the accumulation of advanced glycation end products by promoting Lysosomal biogenesis and function in the kidney proximal tubules [J]. Diabetes. 2017;66(5):1359–1372. [DOI] [PubMed] [Google Scholar]
- [25].Zheng L, Zhang W, Li A, et al. PTPN2 downregulation is associated with albuminuria and vitamin D receptor deficiency in type 2 diabetes mellitus [J]. J Diabetes Res. 2018;2018:3984797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang Z, Yuan W, Sun L, et al. 1,25-Dihydroxyvitamin D3 targeting of NF-kappaB suppresses high glucose-induced MCP-1 expression in mesangial cells [J]. Kidney Int. 2007;72(2):193–201. [DOI] [PubMed] [Google Scholar]
- [27].Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry [J]. Nat Immunol. 2010;11(1):55–62. [DOI] [PubMed] [Google Scholar]
- [28].Wang TT, Dabbas B, Laperriere D, et al. Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-defensin beta2 innate immune pathway defective in Crohn disease [J]. J Biol Chem. 2010;285(4):2227–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ding Y, Choi ME. Autophagy in diabetic nephropathy [J]. J Endocrinol. 2015;224(1):R15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kume S, Koya D, Uzu T, et al. Role of nutrient-sensing signals in the pathogenesis of diabetic nephropathy [J]. Biomed Res Int. 2014;2014:315494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yang D, Livingston MJ, Liu Z, et al. Autophagy in diabetic kidney disease: regulation, pathological role and therapeutic potential [J]. Cell Mol Life Sci. 2018;75(4):669–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang J, Lian H, Zhao Y, et al. Vitamin D3 induces autophagy of human myeloid leukemia cells [J]. J Biol Chem. 2008;283(37):25596–25605. [DOI] [PubMed] [Google Scholar]
- [33].Khodir SA, Samaka RM, Ameen O. Autophagy and mTOR pathways mediate the potential renoprotective effects of vitamin D on diabetic nephropathy [J]. Int J Nephrol. 2020;2020:7941861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yuk JM, Shin DM, Lee HM, et al. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin [J]. Cell Host Microbe. 2009;6(3):231–243. [DOI] [PubMed] [Google Scholar]
- [35].Hoyer-Hansen M, Bastholm L, Szyniarowski P, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2 [J]. Mol Cell. 2007;25(2):193–205. [DOI] [PubMed] [Google Scholar]
- [36].Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling [J]. Cancer Cell. 2004;6(1):91–99. [DOI] [PubMed] [Google Scholar]
- [37].Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy [J]. Science. 2011;331(6016):456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bai X, Yang X, Jia X, et al. CAV1-CAVIN1-LC3B-mediated autophagy regulates high glucose-stimulated LDL transcytosis [J]. Autophagy. 2020;16(6):1111–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Meyer MB, Watanuki M, Kim S, et al. The human transient receptor potential vanilloid type 6 distal promoter contains multiple vitamin D receptor binding sites that mediate activation by 1,25-dihydroxyvitamin D3 in intestinal cells [J]. Mol Endocrinol. 2006;20(6):1447–1461. [DOI] [PubMed] [Google Scholar]
- [40].Schroder B, Schlumbohm C, Kaune R, et al. Role of calbindin-D9k in buffering cytosolic free Ca2+ ions in pig duodenal enterocytes [J]. J Physiol. 1996;492(Pt 3):715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Timmermans JA, Bindels RJ, Van Os CH. Stimulation of plasma membrane Ca2+ pump by calbindin-D28k and calmodulin is additive in EGTA-free solutions [J]. J Nutr. 1995;125(7Suppl):1981S–6S. [DOI] [PubMed] [Google Scholar]
- [42].Tudpor K, Teerapornpuntakit J, Jantarajit W, et al. 1,25-dihydroxyvitamin D(3) rapidly stimulates the solvent drag-induced paracellular calcium transport in the duodenum of female rats [J]. J Physiol Sci. 2008;58(5):297–307. [DOI] [PubMed] [Google Scholar]
- [43].Zhao G, Simpson RU. Interaction between vitamin D receptor with caveolin-3 and regulation by 1,25-dihydroxyvitamin D3 in adult rat cardiomyocytes [J]. J Steroid Biochem Mol Biol. 2010;121(1–2):159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cheng S, So WY, Zhang D, et al. Calcitriol reduces hepatic triglyceride accumulation and glucose output through Ca2+/CaMKKβ/AMPK activation under insulin-resistant conditions in type 2 diabetes mellitus [J]. Curr Mol Med. 2016;16(8):747–758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.