

Abstract
Retinoic acid receptor responder protein 1 (RARRES1) has been identified as a novel gene for the regulation of podocyte function, and its expression is increased in glomerular disease and associated with disease progression. Increased expression of RARRES1 in podocytes leads to apoptosis through an autocrine effect. Möller-Hackbarth et al. recently found that RARRES1 expression is increased in the endothelial cells in some diseased kidneys to promote podocyte injury, likely through a paracrine effect.
Podocytes form the last layer of the glomerular filtration barrier, and its injury leads to proteinuric kidney disease. Over the last decade, multiple podocyte-specific proteins have been identified, whose mutations or dysregulation lead to the development of glomerular disease. Examples of such proteins are Wilms tumor 1, nephrin, podocin, synaptopodin, actinin-4, transient receptor potential canonical type 6, and Kruppel-like factor 15.1,2 A major function of these proteins is to maintain normal podocyte actin cytoskeleton and cellular quiescence. Recently, Chen et al. identified retinoic acid receptor responder protein 1 (RARRES1) as a novel protein that is expressed specifically in podocytes, as validated by single-cell RNA sequencing.3 RARRES1 was first identified in skin raft cultures treated with tazarotene, thus also named tazarotene-induced gene 1, and its expression is regulated by retinoic acid.4 Recent data by Chen et al. indicate that its expression is also regulated by tumor necrosis factor α.3,5 Similar to Wilms tumor 1, RARRES1 is considered to be a tumor suppressor gene, and as such, it may contribute to podocyte quiescence in adult kidneys. However, in contrast to Wilms tumor 1, whose loss of function results in podocyte injury and glomerular disease, loss of RARRES1 did not lead to any remarkable glomerular phenotype. However, its overexpression in podocytes led to apoptosis and glomerular injury,3 suggesting that a tight balance of RARRES1 is required to help maintain podocyte quiescence, but an uncontrolled expression of RARRES1 leads to cell death and demise of podocytes.3 Such findings in mouse models are well supported by the observations made in human kidney diseases, where the glomerular expression of RARRES1 is increased, correlates with the severity of renal function, and can predict disease progression.3 Both in vitro and in vivo data suggest that RARRES1 acts in an autocrine manner to cause podocyte injury in disease settings: membrane-bound RARRES1 is cleaved and is endocytosed, thereby inhibiting the intracellular atypical protein kinase, RIO kinase 1, to induce podocyte apoptosis (Figure 1).3,5
Figure 1 |. Proposed mechanisms of injury induced by retinoic acid receptor responder protein 1 (RARRES1) in glomerular cells through autocrine and/or paracrine pathways.
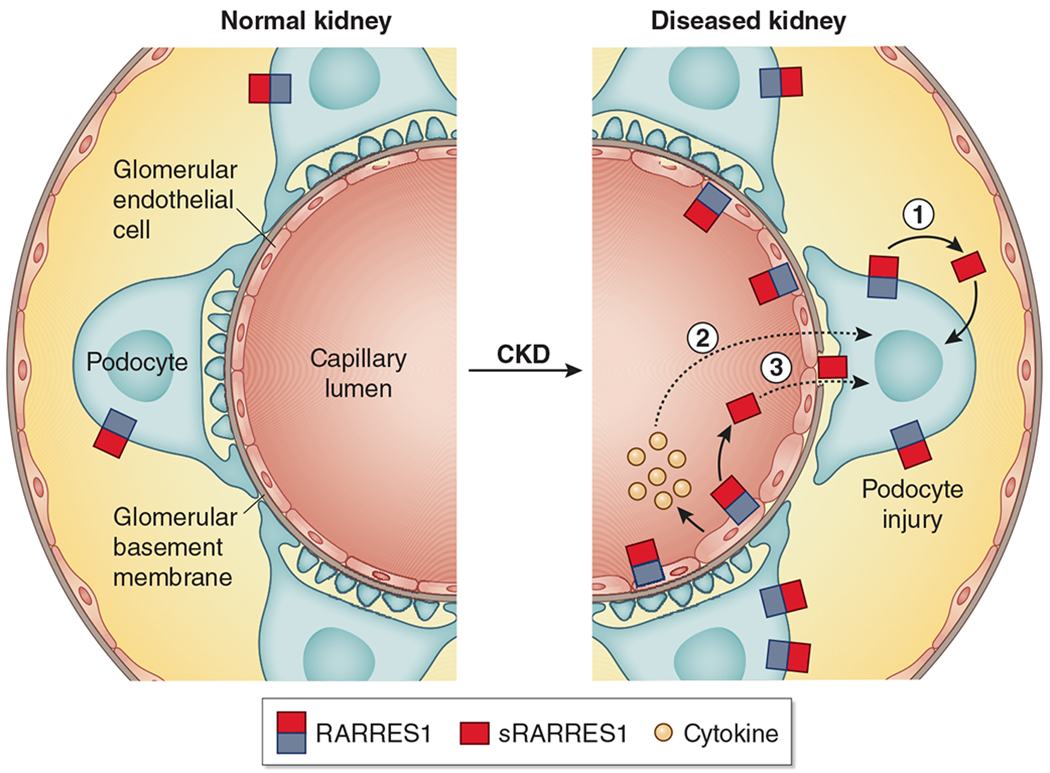
In normal kidneys, RARRES1 is expressed specifically in podocytes. In diseased kidneys, RARRES1 expression is significantly increased in podocytes, which can be cleaved into its soluble form and endocytosed by podocytes, leading to podocyte apoptosis (1); RARRES1 expression is also found to be increased in endothelial cells to promote podocyte injury through an endothelial–podocyte crosstalk, probably via cytokine released by injured endothelial cells (2) or by podocyte uptake of soluble RARRES1 (sRARRES1) generated from the endothelial cells (3). CKD, chronic kidney disease.
In the current issue of Kidney International, Möller-Hackbarth et al.6 reported additional salient findings on the role of RARRES1 in the pathogenesis of glomerular disease using the nephrotoxin-induced glomerulonephritis model. Consistent with the findings by Chen et al., they similarly observed that RARRES1 is expressed mostly in podocytes in the kidney and that the loss of RARRES1 expression in podocytes did not cause glomerular abnormalities at baseline.3,5 Interestingly, the podocyte-specific loss of RARRES1 did not improve the renal outcome in nephrotoxin-induced glomerulonephritis,6 whereas its loss ameliorated kidney injury in Adriamycin-induced nephropathy.3 These discrepancies may be caused by the following: (i) the differences between the nephrotoxin-induced glomerulonephritis model versus focal segmental glomerulosclerosis model induced by Adriamycin and (ii) the use of podocyte-specific knockout of RARRES1 in Möller-Hackbarth et al. versus an inducible podocyte-specific short hairpin RNA knockdown of RARRES1 in Chen et al., which may leave a residual and necessary RARRES1 function intact in podocytes.
Möller-Hackbarth et al. also found that in diseased conditions, RARRES1 expression was upregulated in endothelial cells, including glomerular endothelial cells and peritubular endothelial cells, and that the overexpression of RARRES1 specifically in endothelial cells aggravated nephrotoxin-induced glomerulonephritis, with more profound podocyte dysfunction and loss. Nonetheless, no obvious injury of endothelial cells was observed, as the amount of cluster of differentiation 31–labeled endothelial cells did not differ significantly in nephrotoxin-induced glomerulonephritis mice with or without RARRES1 overexpression. These intriguing findings raise several questions. The first question regards the mechanism of potential cell-specific regulation of RARRES1 in podocytes versus endothelial cells in normal and diseased conditions that would allow RARRES1 to be expressed primarily in podocytes (and not endothelial cells) at baseline, but to enhance its expression only in endothelial cells (but not in podocytes) in disease conditions. Thus, it would be valuable to further explore the cell-specific mechanisms of regulation of RARRES1 expression in normal and diseased conditions. Second, the cell-specific injury mechanism whereby RARRES1 overexpression in endothelial cells would be innocuous while causing significant podocyte damage needs to be clarified. It is plausible that (i) RARRES1 induces endothelial cell alterations through an autocrine effect, subsequently causing podocyte injury via a potential crosstalk, possibly involving cytokine release; and that (ii) soluble RARRES1, which is released from endothelial cells, acts on podocytes to induce injury through a paracrine effect (Figure 1). To confirm this, it would be important to determine whether endothelial cell injury occurs in these mice by providing a detailed characterization of endothelial cell injury markers.
Thus, the major discrepancy between Chen et al. and Möller-Hackbarth et al. lies in the glomerular cell type responsible for the increased RARRES1 expression in the diseased kidneys. Both studies relied in part on the immunostaining methods that led Chen et al. to conclude that RARRES1 expression was increased mostly in podocytes,3 whereas the current study by Möller-Hackbarth et al. shows RARRES1 expression to be largely limited to endothelial cells.6 Such discrepancy could be caused by the difference in the detection of cleaved and uncleaved RARRES1 by immunostaining methods, utilizing the currently available antibodies that recognize both intact and soluble forms of RARRES1. Thus, positive staining of RARRES1 in the cells could detect both locally synthesized RARRES1 and the endocytosed RARRES1 synthesized by neighborhood cells.3 In the current study, the authors have demonstrated that mRNA expression of RARRES1 was upregulated in endothelial cells in the kidney of nephrotoxin-induced glomerulonephritis mice by using RNAscope. Future studies are required to further confirm different glomerular diseases and by using more sensitive methods, such as single-cell RNA sequencing of diseased mice. The proliferation of parietal epithelial cells is a major contributor of crescent glomerulonephritis, and previous studies showed that retinoic acid was able to reduce crescent formation in nephrotoxin-induced glomerulonephritis mice.7 Therefore, it would be interesting to determine whether the effects of retinoic acid in parietal epithelial cells depend on RARRES1 expression. Understanding of the role of RARRES1 in different glomerular cell types will help to develop a more effective therapy for glomerulonephritis, potentially by combining retinoic acid with RARRES1 inhibitors.
Mechanistically, Möller-Hackbarth et al.6 showed that RARRES1 overexpression in endothelial cells induces podocyte injury by activation of nuclear factor-κB signaling pathway via receptor tyrosine kinase Axl. However, it is unclear whether this pathway is activated by RARRES1 in endothelial cells, podocytes, or both. As the study by Chen et al. indicated that RARRES1 induces podocyte apoptosis through interaction with RIO kinase 1, RARRES1 likely activates multiple downstream pathways in a cell type–specific manner.
Overall, this study further validates the critical role of RARRES1 in glomerular disease and suggests that RARRES1 may be activated in endothelial cells in diseased conditions and act in an autocrine and paracrine manner to activate multiple downstream pathways in glomerular cells, as summarized in Figure 1. Therefore, Rarres1 might be a potential new biomarker of glomerular cell injury and a novel therapeutic target for glomerular disease.
ACKNOWLEDGMENTS
AC is supported by grants from National Natural Science Foundation of China (81800637) and Hunan Natural Science Foundation Outstanding Youth Science Fund Project (2021JJ10075); JCH is supported by Veterans Affairs Merit Award IBX000345C, National Institutes of Health (NIH) 1R01DK078897, NIH 1R01DK088541, and NIH P01DK56492; and KL is supported by NIH R01DK117913.
Footnotes
DISCLOSURE
All the authors declared no competing interests.
REFERENCES
- 1.Kopp JB, Anders HJ, Susztak K, et al. Podocytopathies. Nat Rev Dis Primers. 2020;6:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mallipattu SK, Liu RJ, Zheng F, et al. Kruppel-like factor 15 (KLF15) is a key regulator of podocyte differentiation. J Biol Chem. 2012;287:19122–19135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen AQ, Feng Y, Lai H, et al. Soluble RARRES1 induces podocyte apoptosis to promote glomerular disease progression. J Clin Invest. 2020;130:5523–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagpal S, Patel S, Asano AT, et al. Tazarotene-induced gene 1 (TIG1), a novel retinoic acid receptor-responsive gene in skin. J Invest Dermatol. 1996;106:269–274. [DOI] [PubMed] [Google Scholar]
- 5.Chen AQ, Liu Y, Lu Y, et al. Disparate roles of retinoid acid signaling molecules in kidney disease. Am J Physiol Renal Physiol. 2021;320:F683–F692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Möller-Hackbarth K, Dabaghie D, Charrin E, et al. Retinoic acid receptor responder1 promotes development of glomerular diseases via the Nuclear Factor-κB signaling pathway. Kidney Int. 2021;100:809–823. [DOI] [PubMed] [Google Scholar]
- 7.Dai Y, Chen AQ, Liu RJ, et al. Retinoic acid improves nephrotoxic serum-induced glomerulonephritis through activation of podocyte retinoic acid receptor α. Kidney Int. 2017;92:1444–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]