

ABSTRACT
Hepatocellular carcinoma is the most frequent primary liver cancer. Macroautophagy/autophagy inhibitors have been extensively studied in cancer but, to date, none has reached efficacy in clinical trials. In this study, we demonstrated that GNS561, a new autophagy inhibitor, whose anticancer activity was previously linked to lysosomal cell death, displayed high liver tropism and potent antitumor activity against a panel of human cancer cell lines and in two hepatocellular carcinoma in vivo models. We showed that due to its lysosomotropic properties, GNS561 could reach and specifically inhibited its enzyme target, PPT1 (palmitoyl-protein thioesterase 1), resulting in lysosomal unbound Zn2+ accumulation, impairment of cathepsin activity, blockage of autophagic flux, altered location of MTOR (mechanistic target of rapamycin kinase), lysosomal membrane permeabilization, caspase activation and cell death. Accordingly, GNS561, for which a global phase 1b clinical trial in liver cancers was just successfully achieved, represents a promising new drug candidate and a hopeful therapeutic strategy in cancer treatment.
Abbreviations: ANXA5:annexin A5; ATCC: American type culture collection; BafA1: bafilomycin A1; BSA: bovine serum albumin; CASP3: caspase 3; CASP7: caspase 7; CASP8: caspase 8; CCND1: cyclin D1; CTSB: cathepsin B; CTSD: cathepsin D; CTSL: cathepsin L; CQ: chloroquine; iCCA: intrahepatic cholangiocarcinoma; DEN: diethylnitrosamine; DMEM: Dulbelcco’s modified Eagle medium; FBS: fetal bovine serum; FITC: fluorescein isothiocyanate; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; HCC: hepatocellular carcinoma; HCQ: hydroxychloroquine; HDSF: hexadecylsulfonylfluoride; IC50: mean half-maximal inhibitory concentration; LAMP: lysosomal associated membrane protein; LC3-II: phosphatidylethanolamine-conjugated form of MAP1LC3; LMP: lysosomal membrane permeabilization; MALDI: matrix assisted laser desorption ionization; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MKI67: marker of proliferation Ki-67; MTOR: mechanistic target of rapamycin kinase; MRI: magnetic resonance imaging; NH4Cl: ammonium chloride; NtBuHA: N-tert-butylhydroxylamine; PARP: poly(ADP-ribose) polymerase; PBS: phosphate-buffered saline; PPT1: palmitoyl-protein thioesterase 1; SD: standard deviation; SEM: standard error mean; vs, versus; Zn2+: zinc ion; Z-Phe: Z-Phe-Tyt(tBu)-diazomethylketone; Z-VAD-FMK: carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone.
KEYWORDS: Antitumor, autophagy, liver cancer, lysosome, mtor, PPT1
Introduction
With an estimated 782,000 deaths in 2018, hepatocellular carcinoma (HCC) stands as the most common primary liver cancer and constitutes the fourth leading cause of cancer-related death worldwide [1]. The rising incidence of HCC, the high worldwide mortality rate, and limited therapeutic options at advanced stages, make HCC a significant unmet medical need.
Autophagy-related lysosomal cell death, either alone or in connection with several other cell death pathways, has been recognized as a major target for cancer therapy [2]. Dysregulated autophagic-lysosomal activity and MTOR (mechanistic target of rapamycin kinase) signaling were shown to allow cancer cells to become resistant to the cellular stress induced by chemotherapy and targeted therapy [3]. Recently, several lysosome-specific inhibitors were shown to target PPT1 (palmitoyl-protein thioesterase 1, resulting in the modulation of protein palmitoylation and autophagy, and antitumor activity in melanoma and colon cancer models [4,5].
Chloroquine (CQ) and hydroxychloroquine (HCQ) have been used for more than 50 years to prevent and treat malarial infections and autoimmune diseases. Based on the lysosomotropic properties and the capacity for autophagy inhibition, these molecules have been proposed as active drugs in cancer [6–9]. Over 40 clinical trials have been reported to evaluate the activity of both CQ or HCQ as single agent or in combination with chemotherapy in several tumor types [6–8]. However, the required drug concentrations to inhibit autophagy were not achieved in humans, leading to inconsistent results in cancer clinical trials [5,10]. This prompted research to identify novel compounds with potent inhibitory properties against autophagy for cancer therapy.
We previously reported that GNS561 was efficient in intrahepatic cholangiocarcinoma (iCCA) by inhibiting late-stage autophagy [11]. In this study, we investigated the mechanism of action of GNS561. We identified lysosomal PPT1 as a target of GNS561. Exposure to GNS561 induced lysosomal accumulation of unbound zinc ion (Zn2+), inhibition of PPT1 and cathepsin activity, blockage of autophagic flux and MTOR displacement. Interestingly, these effects resulted in lysosomal membrane permeabilization (LMP) and caspase activation that led to cancer cell death. This mechanism was associated with dose-dependent inhibition of cancer cell proliferation and tumor growth inhibition in two HCC in vivo models. These data establish PPT1 and lysosomes as major targets to kill cancer cells and led to the development of a clinical program investigating the effects of GNS561 in patients with advanced HCC.
Results
GNS561 displays activity against human cancer cell lines and patient-derived cells
The effects of GNS561 on cell viability were investigated in a panel of human cancer cell lines, including HCC, iCCA and colon, renal, breast, prostate, lung, and ovarian carcinoma as well as acute myeloid leukemia, glioblastoma, and melanoma. As shown in Table 1, GNS561 showed potent antitumor activity ranging from 0.22 ± 0.06 µM for the most sensitive cell line (LN-18, a glioblastoma cell line) to 7.27 ± 1.71 µM for the least sensitive cell line (NIH:OVCAR3, an ovarian cancer cell line). GNS561 was at least 10-fold more effective than HCQ in tested cancer cell lines. GNS561 also displayed activity in primary HCC patient-derived cells and was on average 3-fold more potent than sorafenib, a reference drug in HCC treatment (mean half-maximal inhibitory concentration [IC50] 3.37 ± 2.40 μM for GNS561 vs 10.43 ± 4.09 μM for sorafenib).
Table 1.
In vitro activity of GNS561 and HCQ in human cancer cell lines (left, IC50 ± SD, µM) and in vitro activity of GNS561 and sorafenib in primary HCC patient-derived cells (right, IC50, µM)
| Mean IC50 ± SD (µM) |
IC50 (μM) |
|||||
|---|---|---|---|---|---|---|
| Cancer type | Cell lines | GNS561 | HCQ | Primary HCC patient-derived cells | GNS561 | sorafenib |
| Colon Carcinoma | HCT-116 | 1.22 ± 0.15 | 14.41 ± 1.5 | LI0050 | 3.54 | 9.12 |
| HT-29 | 1.35 ± 0.04 | 24.18 ± 5.14 | LI0574 | 2.41 | 8.65 | |
| Renal Cell Carcinoma | 786-O | 1.72 ± 0.17 | 21.65 ± 3.15 | LI0612 | 6.93 | 17.94 |
| CAKI-1 | 1.10 ± 0.19 | 17.69 ± 1.29 | LI0752 | 0.49 | 6.34 | |
| Ovarian Cancer | NIH:OVCAR3 | 7.27 ± 1.71 | 98.01 ± 12.75 | LI0801 | 2.07 | 5.7 |
| Melanoma | A375 | 1.2 ± 0.13 | 12.27 ± 2.8 | LI1005 | 3.16 | 14.49 |
| SK-MEL-28 | 1.81 ± 0.5 | 22.78 ± 2.65 | LI1098 | 6.95 | 10.85 | |
| Breast Cancer | MDA-MB-231 | 2.17 ± 0.14 | 14.13 ± 3.06 | LI1646 | 1.44 | 10.33 |
| Prostate Cancer | DU-145 | 1.09 ± 0.18 | 45.74 ± 0.55 | Mean | 3.37 ± 2.40 | 10.43 ± 4.09 |
| PC-3 | 2.56 ± 0.23 | 43.43 ± 6.04 | ||||
| Lung Cancer | A549 | 1.69 ± 0.34 | 14.33 ± 1.59 | |||
| NCI-H358 | 2.54 ± 0.34 | 54.07 ± 14.19 | ||||
| HCC | HepG2 | 0.47 ± 0.15 | 11.55 ± 1.52 | |||
| Huh7 | 0.88 ± 0.31 | 13.62 ± 0.71 | ||||
| Glioblastoma | LN-229 | 0.60 ± 0.24 | 10.87 ± 1.23 | |||
| LN-18 | 0.22 ± 0.06 | 5.27 ± 0.74 | ||||
| Acute Myeloid Leukemia | KG-1 | 5.86 ± 1.64 | 43.92 ± 2.76 | |||
| Mean | 1.99 ± 1.86 | 27.52 ± 23.28 | ||||
GNS561 has antitumor properties in HCC in vivo models
The whole-body tissue distribution of GNS561 was investigated in Sprague Dawley rats after repeated oral administration of GNS561 at a dose of 40 mg/kg/day for 28 days. Seven hours after the last administration, the GNS561 level was measured by mass spectrometry imaging in the liver, lung, stomach, brain, eye, salivary gland, kidney, heart, fat, muscle, testis and skin (Figure 1A). GNS561 mainly accumulated in the liver, stomach and lung as shown by the calculated organ:blood ratio (Figure 1B). Lower concentrations of GNS561 were also detected in eyes, skin, brain and testis, indicating that GNS561 crosses the blood/brain barrier and the blood/testis barrier to a limited extent (brain to blood and testis to blood ratios were 0.21 and 0.40, respectively). High liver concentrations were also highlighted in rats after repeated administration of GNS561 at the dose level of 15 and 30 mg/kg/day for 28 days and 50 mg/kg/day for 21 days (data not shown).
Figure 1.
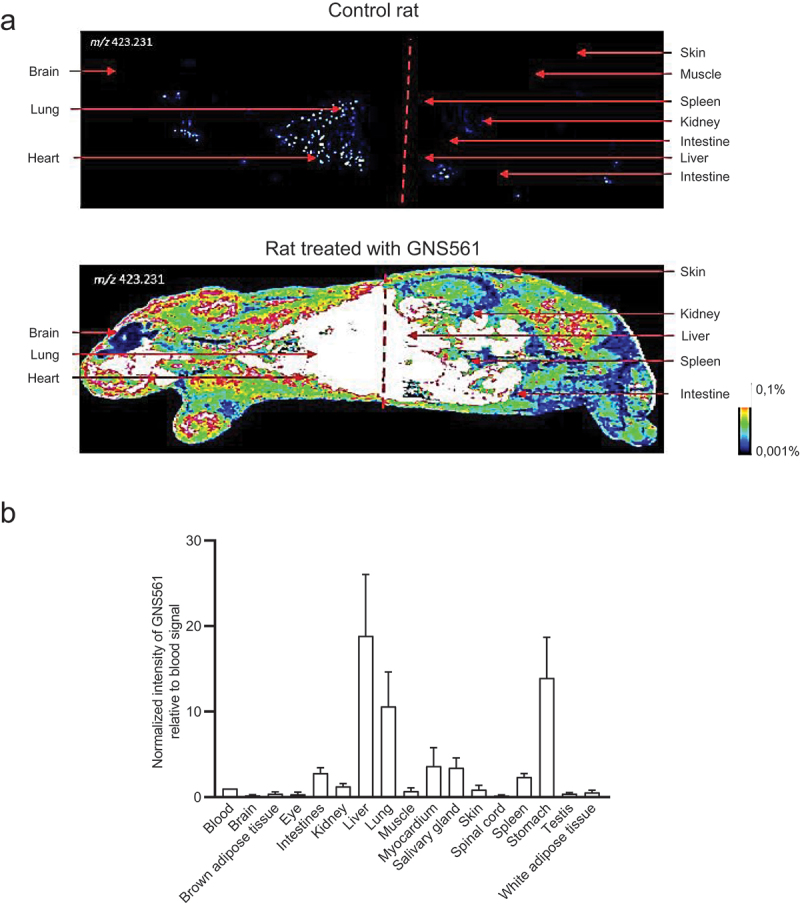
Whole body tissue distribution of GNS561. (A) Mass spectrometry imaging of a control rat (top) and a rat treated with GNS561 at a dose of 40 mg/kg/day for 28 days (bottom). (B) Normalized intensity of GNS561 relative to blood signal in several organs of GNS561-treated rats (Mean + SEM, n = 2 except for eye, n = 1). Of note, the GNS561 liver-to-blood ratio is underestimated due to liver signal saturation.
Based on the high concentrations of GNS561 in the liver and potent in vitro activity against HCC cells, the effects of GNS561 were investigated in vivo using two liver cancer models, including the human HCC orthotopic patient-derived LI0752 xenograft BALB/c nude mouse model and the diethylnitrosamine (DEN)-induced cirrhotic immunocompetent rat model of HCC.
In the HCC patient-derived LI0752 xenograft BALB/c nude mouse model, tumor volume and weight were reduced by 37.1% and 34.4%, respectively, in mice treated with GNS561 at 50 mg/kg compared to the control (Fig. S1A and S1B). Consistently, GNS561 treatment induced a decrease in serum alpha fetoprotein levels (AFP) in a dose-dependent manner (Fig. S1C). AFP levels was also evaluated at day 7 (Fig. S1D), 21 (Fig. S1E), 28 (Fig. S1F) and 41 (Fig. S1G) days post tumor inoculation and was significantly different after GNS561 treatment compared to control at days 21 and 28 (Fig. S1E and S1F).
Because HCC often develops in cirrhotic livers in humans, we further characterized the antitumor effects of GNS561 in a DEN-induced cirrhotic rat model of HCC. Rats with already developed HCC were either treated with sorafenib at 10 mg/kg, GNS561 at 15 mg/kg, or the combination of both drugs (Fig. S2). In this model, tumor progression was significantly reduced by sorafenib (33%) and GNS561 (33%) compared to the untreated control group, and the greatest decrease in tumor progression was observed by the combination (68%) that displayed an additive effect (Figure 2A). Magnetic resonance imaging (MRI) analyses further showed a significant decrease in the mean tumor size of 6.45 ± 0.35 mm with sorafenib, 5.48 ± 1.00 mm in GNS561 and 3.83 ± 0.52 mm in the combination group compared to 9.97 ± 0.97 mm in control rats (Figure 2B). Following liver resection, the macroscopic counting of tumor nodules revealed significantly lower numbers in all treated groups compared to the control group (Figure 2C). Immunohistochemical analyses of liver tumors showed a significantly lower CCND1 (cyclin D1)-positive nuclear staining in the tumors of rats treated with GNS561 or by the combination of GNS561 with sorafenib compared to the control group (Figure 2D). GNS561 and combination treatments also significantly reduced MKI67 staining compared to the control group (Figure 2E). The effects on CCND1 and MKI67 were primarily related to GNS561 exposure, as sorafenib alone showed no statistically significant differences in CCND1 or MKI67 staining compared to the control group. Our results further showed that GNS561 and the combination treatment did not interfere with lipid or glucose metabolism, or kidney function but slightly affected some liver functions (Table S1).
Figure 2.
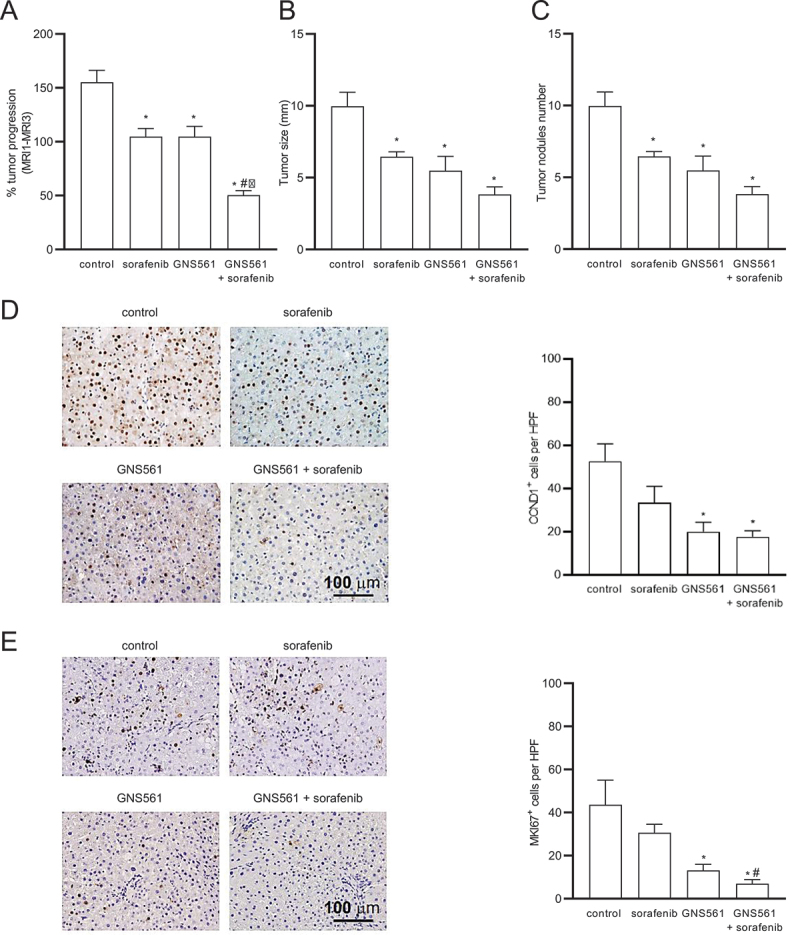
GNS561 activity in a diethylnitrosanime-induced cirrhotic rat model of hepatocellular carcinoma. (A) Tumor progression assessment by comparison of tumor size obtained by MRI 1 and MRI 3 in the control, sorafenib at 10 mg/kg, GNS561 at 15 mg/kg and combination (GNS561 + sorafenib) groups. Macroscopic examination of livers with assessments of (B) tumor size and (C) tumor nodules number at the surface of livers. (D) Representative images of nuclear CCND1 staining and quantification of CCND1-positive staining per high-power field (HPF). (E) Representative images of nuclear MKI67 staining and quantification of MKI67-positive staining per HPF. For all studies, mice n ≥ 6 per group. Data represent the mean + SEM. Comparison of means was performed by one-way ANOVA with Dunnett’s post hoc analysis. *represents significant difference compared to control, #represents significant difference compared to sorafenib, ǂrepresents significant difference compared to GNS561, at least p < 0.05.
GNS561 activates the caspase-dependent apoptosis pathway
We further wanted to characterize the antitumor effect of GNS561 and to determine whether GNS561 could trigger apoptotic cell death. To this end, ANXA5 (annexin A5)-propidium iodide analysis was performed by flow cytometry after 48 h of GNS561 exposure in HepG2 cells. Early (ANXA5+ propidium iodide− staining) and late (ANXA5+ propidium iodide+ staining) apoptosis increased in a dose-dependent manner after GNS561 exposure (Figure 3A). The induction of apoptosis was confirmed by immunodetection of PARP (poly(ADP-ribose) polymerase) cleavage in GNS561-treated cells (Figure 3B). We further examined whether GNS561-induced apoptosis was related to caspase activation. Cleaved CASP3 (caspase 3) was detected using immunoblot analysis (Figure 3C) and caspase activation was confirmed by flow cytometry (Figure 3D) and luminescence analysis (Figure 3E). After 6 h of exposure, GNS561 had no effect on CASP8 and CASP3-CASP7 activity in HepG2 cells (Figure 3E). In contrast, activation of CASP8 and CASP3-CASP7 was observed after 24 h of treatment with GNS561, and this effect was sustained at 30 h. A decrease in cell viability was concomitant with caspase activation (Figure 3E). Moreover, to confirm that GNS561-induced cell death is caspase-dependent apoptosis, pre-treatment (1 h) with the cell-permeable pan-caspase inhibitor, carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone (Z-VAD-FMK) (5 µM) was performed. Cell viability was restored in the presence of Z-VAD-FMK (Figure 3F), further confirming that GNS561 induced a caspase-dependent apoptotic cell death.
Figure 3.
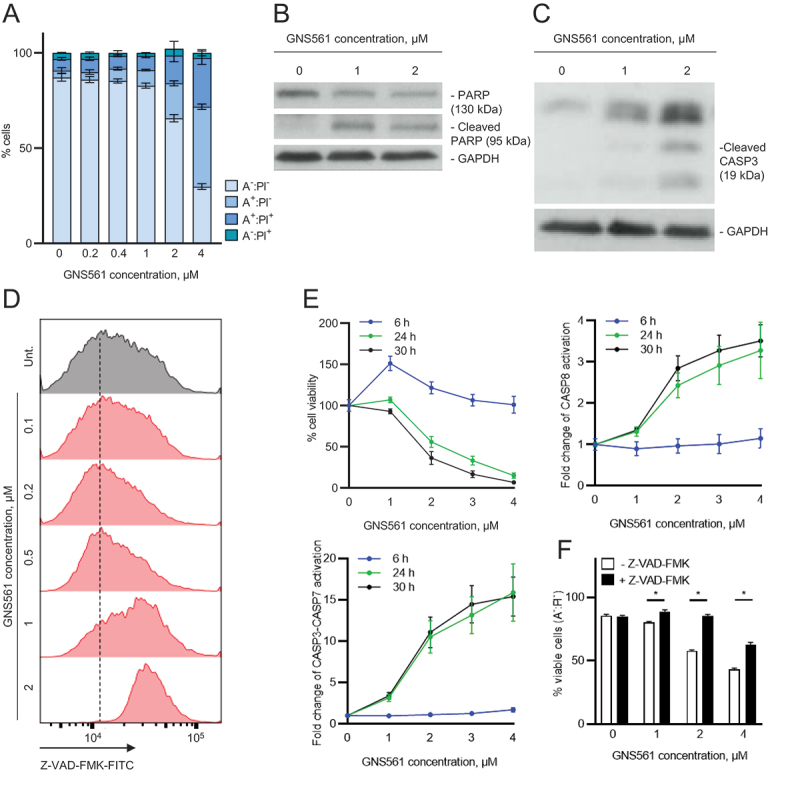
GNS561 induces apoptotic cell death in HepG2 cells in a dose and time-dependent manner through caspase activation. (A) ANXA5 (A):propidium iodide (PI) analysis by flow cytometry after 48 h of GNS561 treatment. (B) Representative immunoblotting of the cleaved and non-cleaved forms of PARP after 24 h of GNS561 treatment. (C) Representative immunoblotting of cleaved CASP3 levels after 24 h of treatment with GNS561. (D) Caspase-glow analysis by flow cytometry after 48 h of treatment with GNS561. (E) Cell viability and activation of CASP3-CASP7and CASP8 after 6, 24 and 30 h of treatment with GNS561. (F) Viable cell (A-:PI-) analysis by flow cytometry after pre-treatment with Z-VAD-FMK at 5 µM for 1 h and then treatment with Z-VAD-FMK at 5 µM and GNS561 for 48 h. For all blots, GAPDH was used as a loading control. For all studies, n ≥ 3 biological replicates. Data represent the mean + SEM. For comparison, Student t-test was used. *represents significant difference, at least p < 0.05.
GNS561 is a lysosomotropic agent
The intracellular localization of GNS561 in HepG2 cells was visualized using GNS561D, a photoactivable analog of GNS561 containing a diazide moiety (Figure 4A). It has been noticed that before using GNS561D, we validated that this GNS561 analog retained the biological hallmarks of the parent molecule GNS561 (Table S2). GNS561D showed a punctate fluorescent signal that colocalized with the intracellular vesicle-like structure stained by LAMP1 (lysosomal associated membrane protein 1) (Figure 4B), demonstrating that GNS561 accumulated in lysosomes and is a lysosomotropic agent. Pre-treatment with ammonium chloride (NH4Cl), a weak base that rapidly increases lysosomal pH, was further used to validate the lysosomotropic character of GNS561. As shown in Figure 4B, NH4Cl pre-treatment strongly prevented lysosomal accumulation of GNS561D. Then, we investigated whether GNS561 lysosomotropism was related to its antitumor effect. For this purpose, HepG2 cells were pre-treated for 2 h with NH4Cl and then treated with GNS561 for 24 h. Although a concentration of 20 mM NH4Cl alone slightly decreased viability (Figure 4C), it significantly attenuated the larger decrease in viability induced by GNS561. These results were confirmed by pre-treatment with bafilomycin A1 (BafA1), an inhibitor of the vacuolar H+-ATPase (Fig. S3). Therefore, disrupting GNS561 lysosomal localization protected against GNS561-mediated cell death. These results suggested that GNS561 antitumor activity in HepG2 cells is caused by its lysosomotropism.
Figure 4.
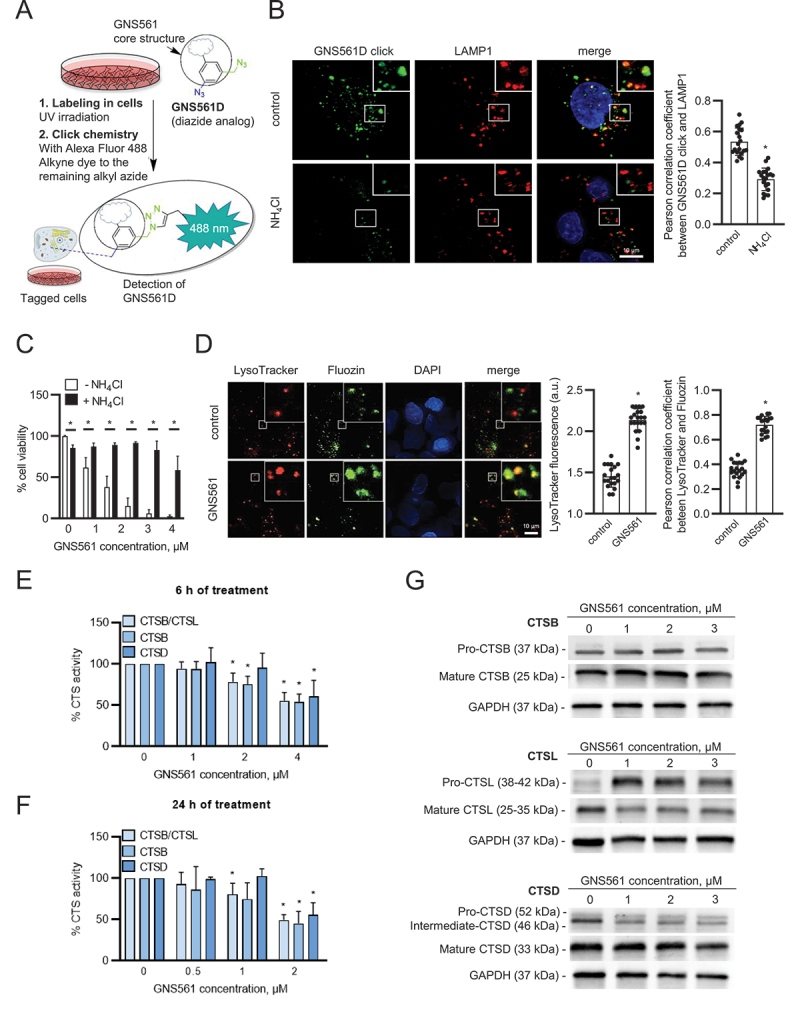
The lysosomotropic agent GNS561 modulates lysosomal functions in the HepG2 cell line. (A) Chemical labeling of GNS561D in cells. The UV-irradiation activates the aryl azide, and then the click chemistry activates the alkyl azide to create the fluorescent moiety with the dye. (B) Lysosomal localization of GNS561D after NH4Cl pre-treatment (20 mM) for 30 min and then treatment with GNS561D (10 µM) and NH4Cl (20 mM) for 90 min. (C) Cell viability after 24 h of GNS561 exposure in the presence or absence of NH4Cl (20 mM). (D) Staining of lysosomes (LysoTracker) and unbound Zn2+ (Fluozin) after GNS561 treatment (1 h, 10 µM). Quantification of LysoTracker fluorescence in arbitrary units (a.u.) (middle panel) and lysosomal unbound Zn2+ accumulation by Pearson correlation coefficient between LysoTracker and Fluozin (right panel). Fold change of peptidase activity of cysteine cathepsins (including both CTSB-CTSL), CTSB and CTSD after (E) 6 h and (F) 24 h of treatment with GNS561 calculated in comparison with the control condition. (G) Representative immunoblotting of pro-CTSB (precursor form) and mature CTSB (top), pro-CTSL (precursor form) and mature CTSL (middle) and pro-CTSD, intermediate and mature CTSD (bottom) after GNS561 treatment for 16 h. For all blots, GAPDH was used as a loading control. For all studies, n ≥ 3 biological replicates. Data represent the mean + SEM. For comparison, Student t-test was used for (B), (C) and (D), and one-way ANOVA with Dunnett’s post hoc analysis was performed for (E) and (F). *represents significant difference, at least p < 0.05.
GNS561 modulates lysosomal functions
The GNS561 lysosomotropism-dependent cell death prompted us to examine GNS561 capacity to modulate lysosomal characteristics and functions. Following continuous exposure to GNS561, staining of LysoTracker, which is a reagent allowing the identification of the lysosomal compartment, increased in HepG2 cells (Figure 4D), suggesting that GNS561 prompted a dose-dependent buildup of enlarged lysosomes. We therefore examined the enzymatic activity of three prominent lysosomal proteinases, two cysteine cathepsins, CTSB and CTSL, and aspartic CTSD. After 6 h (Figure 4E) and 24 h (Figure 4F) of treatment, GNS561 significantly impaired, in a dose-dependent manner, the enzymatic activity of cathepsins. However, this decreased activity did not relate to a direct GNS561-dependent inhibition of cathepsin activities (Fig. S4A). Based on the literature, depressed proteolytic activity of cathepsins may result from an increased Zn2+ lysosomal concentration and/or altered maturation of cathepsin precursors. Indeed, it has been described that Zn2+ may downregulate the proteolytic activity of CSTB and CTSL [12,13]. We investigated whether GNS561 modified unbound Zn2+ localization in HepG2 cells. As shown in Figure 4D, GNS561 induced a strong accumulation of unbound Zn2+ in lysosomes, as evidenced by colocalization of the fluorescent signals of Fluozin and LysoTracker in the merged images. This increase in lysosomal unbound Zn2+ could explain the decreased proteolytic activity of CTSL and CTSB.
Cathepsins are synthesized as inactive zymogens, which are converted to their mature active forms by other proteases or by autocatalytic processing [14]. As depicted in Figure 4G, GNS561 did not have an impact on CTSB maturation, while it impaired the maturation of both CTSL and CSTD (increase of precursor forms) and decreased their catalytic activity accordingly. As GNS561 induced lysosomal dysfunction, the effect of GNS561 on the autophagic process was investigated. Herein, we showed that the GNS561-induced accumulation of MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) phosphatidylethanolamine conjugate (LC3-II) was not enhanced in the presence of BafA1 (Fig. S4B and S4C), suggesting that GNS561 blocked autophagic flux.
PPT1 is a target of GNS561
Because PPT1 is critical for lysosomal function and is described to be the molecular target of CQ derivatives [4,5], we investigated whether PPT1 could be a molecular target of GNS561. First, the binding of GNS561 to recombinant PPT1 was analyzed in vitro by nano differential scanning fluorimetry using HCQ as a positive control [4]. In the presence of GNS561 and HCQ, we observed a significant dose-dependent decrease in PPT1 melting temperature (Figure 5A). Additionally, inhibition of PPT1 enzymatic activity was observed in HepG2 cells treated with GNS561 (Figure 5B). Moreover, the chemical mimetic N-tert-butylhydroxylamine (NtBuHA) attenuated autophagy inhibition associated with GNS561 at 3 µM (Figure 5C), suggesting that inhibition of PPT1 function by GNS561 induced the observed anti-autophagy effect.
Figure 5.
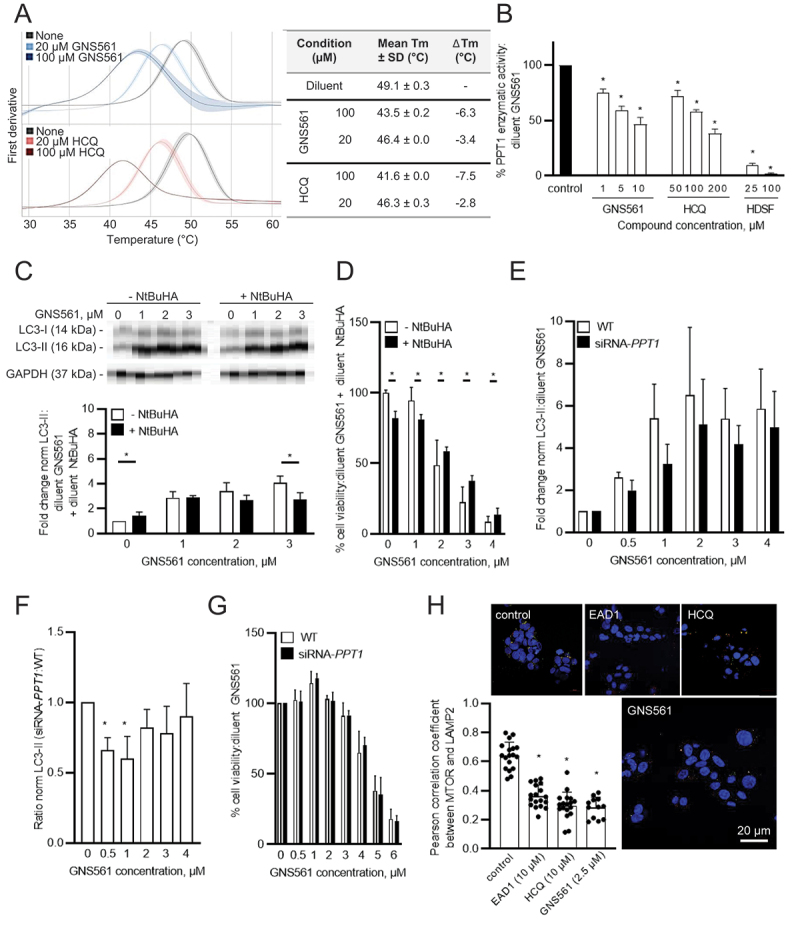
GNS561 targets PPT1. (A) Nano differential scanning fluorimetry assays comparing GNS561 + PPT1 and HCQ + PPT1 against the apo-PPT1 ligand. Data represent the mean first derivative values (solid lines) ± SEM (shaded areas) of two experiments. SD from the mean is indicated by the light-color shading around the mean-line. Tm were determined by detecting the maximum of the first derivative of the fluorescence ratios. ΔTm values of each compound condition were determined by subtracting average Tm of PPT1 (in the respective buffer) by the average Tm of the respective compound condition. (B) PPT1 enzymatic activity of HepG2 cells treated with GNS561 for 3 h. HCQ and HDSF were used as positive controls. The results were compared to the diluent of GNS561 (control condition). (C) Representative immunoblotting of LC3-II in HepG2 cells treated with GNS561 for 16 h in the presence or absence of NtBuHA (8 mM). GAPDH was used as a loading control. Fold changes of normalized LC3-II level were calculated against the control condition (diluent of GNS561 + diluent of NtBuHA). (D) Cell viability percent against the control condition (diluent of GNS561 + diluent of NtBuHA) after 24 h of treatment with GNS561 in the presence or absence of NtBuHA (8 mM). (E) Fold change of normalized LC3-II (norm LC3-II) level were calculated against the control condition (diluent of GNS561) in HepG2 cells WT or siRNA-PPT1 treated by GNS561 for 24 h. GAPDH was used as a loading control. (F) Ratio of norm LC3-II between siRNA-PPT1 and WT HepG2 cells treated by GNS561 for 24 h. (G) Cell viability percent against the control condition (diluent of GNS561) after 24 h of treatment with GNS561 of WT and siRNA-PPT1 HepG2 cells. (H) Staining of lysosomes (LAMP2, green), MTOR (red) and nucleus (4′,6-diamidino-2-phenylindole [DAPI], blue) after treatment with GNS561 and two positive controls, EAD1 and HCQ, for 16 h. Pearson correlation coefficient between MTOR and LAMP2 was represented using scatter dot plot representation. In (B), (C), (D), (E), (F) and (G), data represent the mean + SEM. For comparison, Student t-test was used for (C), (D), (E), and (G) and one-way ANOVA with Dunnett’s post hoc analysis was performed for (B), (F) and (H). For all studies except (A), n ≥ 3 biological replicates. *represents significant difference, at least p < 0.05.
To determine whether inhibition of PPT1 function was responsible for the antitumoral activity of GNS561, HepG2 cells were treated with GNS561 with or without NtBuHA treatment. First, we demonstrated that NtBuHA had no impact on GNS561 lysosomal localization (Fig. S5A). Then, as shown in Figure 5D, NtBuHA prevented the antitumor activity of GNS561, as evidenced by the increased viability of cells treated with NtBuHA. The same rescue effect of NtBuHA treatment was observed for HCQ used as a positive control (Fig. S5B). Thus, these results suggested that inhibition of PPT1 function by GNS561 was partially associated with its antitumoral activity and its autophagy modulation. Finally, to confirm our results, we transfected HepG2 cells with a siRNA directed against PPT1 (Fig. S6). HepG2 siRNA-PPT1 treated by GNS561 presented a trend toward a decrease in the LC3-II accumulation (Figure 5E) that was confirmed by comparing the ratio siRNA-PPT1:WT of LC3-II in HepG2 cells (Figure 5F). Indeed, a significant decrease of this ratio after GNS561 treatment at 0.5 (p = 0.008) and 1 µM (p = 0.0018) suggesting that absence of PPT1 impaired the GNS561 action on autophagy. These results confirmed that the observed attenuation of GNS561-induced autophagy modulation by NtBuHA (Figure 5C) was due to its PPT1 mimetic function and highlighted a direct link between autophagy flux inhibition by GNS561 and PPT1. Finally, the knockdown of PPT1 did not impact on the GNS561-induced viability decrease (Figure 5G), showing that antitumoral activity of GNS561 in HepG2 cells was not limited to PPT1 inhibition by GNS561 as also shown by partial rescue observed with NtBuHA treatment (Figure 5D), suggesting that antitumoral activity of GNS561 in HepG2 cells was not exclusively linked to inhibition of PPT1 function.
The results of Rebecca et al. suggest that PPT1 inhibition could result in MTOR inhibition through the displacement of MTOR from the lysosomal membrane [4,5]. Thus, we investigated the localization of MTOR after GNS561 treatment using immunofluorescence microscopy. HCQ and EAD1 were used as positive controls [15]. As shown in Figure 5H, GNS561 treatment, as well HCQ and EAD1 treatments, significantly impaired MTOR localization to the lysosomal surface. Therefore, GNS561-induced PPT1 inhibition resulted in displacement of MTOR from the lysosomal membrane and consequently likely inhibited the MTOR signaling pathway.
GNS561 induces LMP and cathepsin-dependent cell death
To characterize GNS561-induced changes in lysosomes, we analyzed LMP. To this end, we took advantage of the steady endocytic capacity of cells to load fluorescent dextran into lysosomes and the translocation of lysosomal localized dextran into the cytosol after LMP-inducing insult. Fluorescent dextran in healthy cells appeared in dense punctate structures representing intact lysosomes, whereas after LMP, a diffuse staining pattern throughout the cytoplasm is seen. After GNS561 treatment, such diffuse dextran staining was observed (Figure 6A), suggesting an induction of LMP. As seen in Figure 6B, the loss of membrane integrity, which is the hallmark of LMP, was observed by transmission electron microscopy of HepG2 cells treated with 3 µM GNS561 for 24 h. To confirm this effect, cathepsin localization was studied after GNS561 treatment. After 48 h of treatment, GNS561 decreased cathepsin staining (Figure 6C), indicating that cathepsins were released into the cytosol, thus validating LMP.
Figure 6.
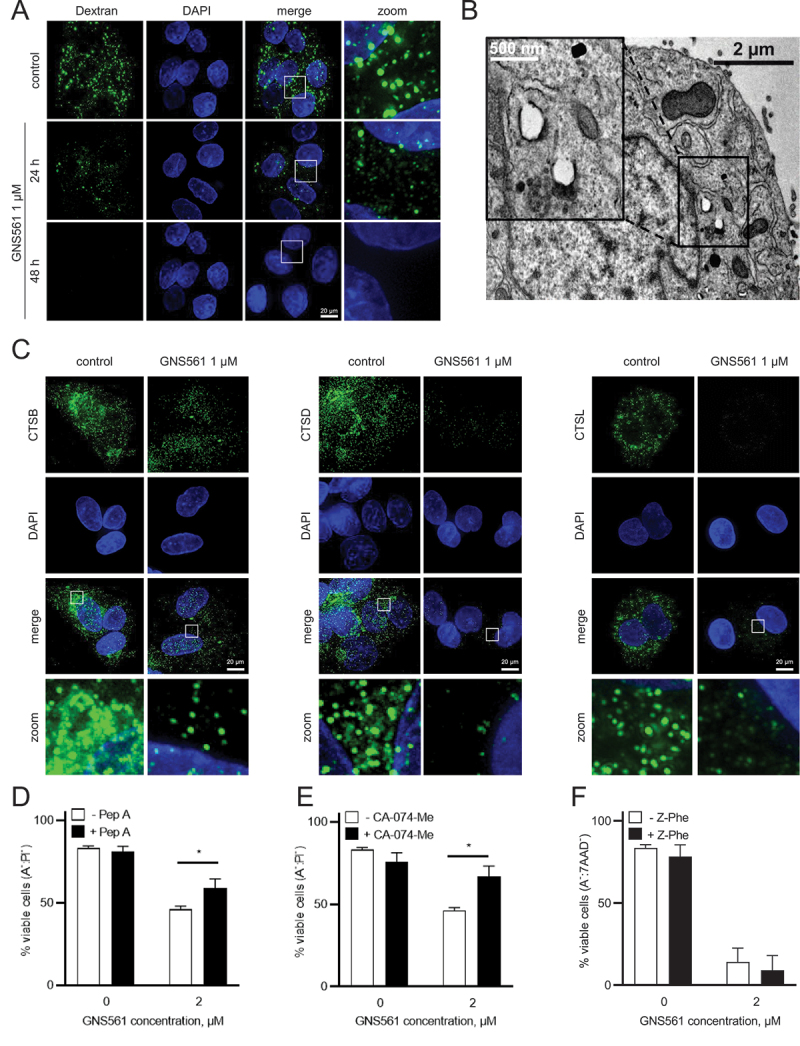
GNS561 induces lysosomal membrane permeabilization and cathepsin-dependent cell death in HepG2 cells. (A) Localization of FITC-dextran after GNS561 treatment for the indicated times. (B) Electron microscopy imaging of lysosomal membrane permeabilization (arrows) after GNS561 treatment (3 µM) for 24 h. (C) Localization of CTSB, CTSD and CTSL after GNS561 treatment for 48 h. (D) Viable cell (ANXA5 (A)−:propidium iodide (PI)−) analysis by flow cytometry of cells pre-treated or not with pepstatin A (Pep A) (5 µM) for 1 h and then treated with Pep A (5 µM) and GNS561 or with GNS561 alone for 48 h. (E) Viable cell (A−:PI−) analysis by flow cytometry of cells pre-treated or not with CA-074-Me (20 µM) for 1 h and then treated with CA-074-Me (20 µM) and GNS561 or with GNS561 alone for 48 h. (F) Viable cell (A−:7-aminoactinomycine D [7AAD]−) analysis by flow cytometry of cells pre-treated or not with Z-Phe (10 µM) for 1 h and then treated with Z-Phe (10 µM) and GNS53-61 or with GNS561 alone for 48 h. Three independent experiments were performed. Data represent the mean + SEM. For comparison, Student t-test was used. For all studies, n ≥ 3 biological replicates. Data represent the mean + SEM. For comparison, Student t-test was used. *represents significant difference, at least p < 0.05.
As cathepsin release into the cytosol after LMP may trigger cytosolic cellular death signaling [16], we evaluated the role of cathepsins in GNS561-induced cell death. To this end, HepG2 cells were pre-treated with an inhibitor of CTSD, pepstatin A, an inhibitor of CTSB, CA-074-Me, or an inhibitor of CTSL, Z-Phe-Tyt(tBu)-diazomethylketone (Z-Phe). Cell viability was partially rescued with an inhibitor of CTSD or CTSB (Figures 6D and 6E) but no rescue was observed with an inhibitor of CTSL (Figure 6F), suggesting that the GNS561-induced apoptotic pathway is at least partially CTSD and CTSB-dependent.
Discussion
Taken together, our study reports a specific antitumoral mechanism of action of GNS561 in HCC highlighting the key role of PPT1 in cancer (Figure 7). As reported, PPT1 overexpression [4] and autophagy activation [17] represent two key mechanisms involved in the cell survival and tumor growth in HCC. Here, we showed that GNS561 treatment in HCC leads to (1) PPT1 inhibition, (2) autophagy inhibition, (3) tumor cell apoptosis and (4) tumor regression. More specifically, we showed that GNS561 compound localizes in lysosomes where it binds and inhibits PPT1 leading to lysosomal unbound Zn2+ accumulation, impairment of cathepsin activity, autophagic flux inhibition, altered location of MTOR and lysosomal membrane permeabilization. Finally, all these events induce caspase activation and tumor cell apoptosis (Figure 7).
Figure 7.
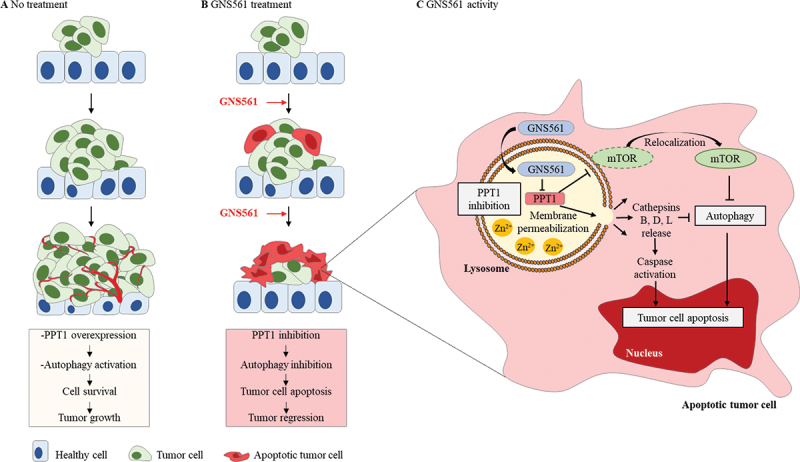
Schematic representation of molecular and cellular mechanisms involved in the antitumoral activity of GNS561. (A) Schematic illustration showing the stages of untreated tumor progression where autophagy activation and overexpression of PPT1 have been singled out in cell survival and tumor growth. (B) GNS561 compound localizes in lysosomes where it binds and inhibits PPT1 resulting in lysosomal unbound Zn2+ accumulation, impairment of cathepsin activity, autophagic flux inhibition, alters location of MTOR and leads to lysosomal membrane permeabilization. Finally, all these events induce caspase activation and tumor cell apoptosis.
Rapidly dividing and invasive cancer cells are strongly dependent on effective lysosomal functions. Lysosomes are acidic and catabolic organelles that are responsible for the disposal and recycling of used and damaged macromolecules and organelles, as well as the assimilation of extracellular materials incorporated into the cell by endocytosis, autophagy, and phagocytosis. Increased autophagic flux and changes in lysosomal compartments in cancer cells have been shown to promote invasion, proliferation, tumor growth, angiogenesis, and drug resistance. Consistently, lysosomal changes are expected to sensitize cells to lysosome-targeting anticancer drugs [18]. Many steps in the autophagy pathway represent potentially druggable targets and several clinical trials have aimed to block autophagy by inhibiting lysosomal functions using CQ and HCQ. Unfortunately, CQ and HCQ failed to demonstrate consistent antitumor effects possibly due to subeffective anticancer concentrations in humans, even with high doses. Drug screening led us to identify GNS561 as a lead compound that displays lysosomotropism and significantly higher antiproliferative effects in human cancer cells compared to HCQ.
We previously reported that GNS561 yielded antiproliferative activity in iCCA, inhibited late-stage autophagy, and induced a dose-dependent enlargement of lysosomes [11]. Based on these preliminary results, we further investigated the cellular mechanisms by which GNS561 may lead to lysosomal changes and death in cancer cells. In this study, we confirmed that GNS561 antitumor properties are strongly dependent on its lysosomotropic properties. In accordance with the hypothesis proposed in our previous study [11], we showed here that GNS561 induced a dose-dependent increase in the number of enlarged lysosomes and LMP leading to cytosolic cathepsin release, caspase activation, and apoptotic cell death. These observations confirm prior reports that highlight the capability of lysosomotropic agents to cause lysosomal stress and lysosomal enlargement [19]. Further investigations are needed to identify the upstream signals that initiate LMP in GNS561-treated cells.
PPT1 plays a central role in the control of cellular autophagy by enabling the degradation and intracellular trafficking of membrane-bound proteins. PPT1 is highly expressed in several cancer cell lines as well as in advanced stage cancers in patients [4]. Indeed, it was reported that PPT1 expression was higher in tumor vs normal tissue from patients suffering of gastric, renal, lung or liver cancers [4]. Moreover, survival analysis revealed that the high tumor expression of PPT1 was correlated with shorter overall survival more specifically in renal and hepatocellular cancer [4]. Recent data have shown that lysosome-specific inhibitors targeting PPT1 can modulate protein palmitoylation and display antitumor activity in melanoma and colon cancer models [5]. Our data showed that PPT1 acts as a molecular target of GNS561. GNS561 bound to PPT1 and inhibited its activity in cells. It is to highlight that as showed in studies of Amaravadi team [4,5], a partial inhibition of PPT1 activity (25% at 1 µM GNS561, 3 h) led to stronger cell viability effects at later time points (more than 50% at 1 µM GNS561, 72 h). Treatment with the chemical mimetic NtBuHA or PPT1 silencing affected the GNS561-associated autophagic flux inhibition highlighting that the mechanism of action of GNS561 on tumor cells is specifically associated with PPT1. Moreover, siRNA-PPT1 did not impact the GNS561-induced viability decrease, confirming that the antitumoral activity of GNS561 is not exclusively link to inhibition of PPT1 function by GNS561 as shown by partial rescue observed with NtBuHA treatment. Taken together, we identified PPT1 as a target of GNS561 but further studies are required to precise others potential mechanisms associated to the antitumor activity of GNS561.
Moreover, we observed that GNS561 modified the intracellular localization of MTOR. This is in accordance with previous studies showing that inhibition of PPT1 may displace the MTOR protein from the lysosomal membrane as a result of the inhibition of V-ATPase-Ragulator-RAG GTPase interactions [4,5]. It was also described that lysosomal MTOR localization brings it in close vicinity to its main regulator, RHEB (Ras homolog, mTORC1 binding), and that as a result, the MTOR-RHEB interaction can activate MTOR kinase activity leading to the phosphorylation of downstream effectors [20]. Consistently, we hypothesized that GNS561-induced PPT1 inhibition led to MTOR signaling pathway inhibition. Further investigations on downstream targets of MTOR as S6 and 4E-BP1 kinases are currently ongoing to fully validate this hypothesis.
As previously observed in iCCA [11], we showed here that GNS561 induced a significant decrease in the enzymatic activity of cathepsins. This decreased activity is unlikely due to a direct inhibition of CTSL, CTSB and CTSD by GNS561 but rather could be the consequence of both impairment of CTSL and CTSD maturation and lysosomal unbound Zn2+ accumulation. As cathepsin activity is optimal in acidic pH [21], we could also speculate that GNS561 may negatively influence the proteolytic activity of cathepsins by inducing an increase in lysosomal pH via PPT1 inhibition. In fact, other authors have shown that PPT1 deficiency in ppt1/cln1−/− mice disrupted the delivery of the ATP6V0A1 (ATPase H+ transporting V0 subunit a1) to the lysosomal membrane, leading to a dysregulation of lysosomal acidification [22]. The authors suggested that S-palmitoylation by PPT1 may play a critical role in the trafficking of ATP6V0A1 to the lysosomal membrane and in lysosomal pH regulation.
Based on prior studies, GNS561 was neither a zinc ionophore nor a zinc chelator (data not shown), unlike CQ [23]. However, our hypothesis that GNS561-induced PPT1 inhibition could lead to lysosomal deacidification could also explain the observed lysosomal unbound Zn2+ accumulation after GNS561 treatment. In fact, as lysosomal pH is mainly regulated by cation/anion movement across the lysosomal membrane, it was suggested that a proton motive force was required to mediate unbound Zn2+ efflux [12].
In summary, GNS561-induced PPT1 inhibition may lead to two main mechanisms inducing cancer cell death. One is related to lysosomal deacidification, which induces lysosomal unbound Zn2+ accumulation, a decrease in the enzymatic activity of cathepsins, inhibition of autophagic flux, lysosomal swelling, LMP, cathepsin release, and caspase-dependent apoptosis. The other is linked to prevention of the interaction between v-ATPase and the Ragulator complex, blockage of MTOR lysosomal recruitment, impairment of MTOR-RHEB interaction and finally the inhibition of MTOR signaling pathway. Thus, by targeting PPT1, GNS561 acts as a regulator of autophagy and MTOR, two major processes that drive cancer aggressiveness. Finally, as lysosomes and autophagy are associated with adaptive mechanisms of resistance to MTOR inhibition [24], GNS561 can disable MTOR function and downregulate adaptive mechanisms of resistance.
An extensive preclinical program has been conducted to evaluate the antitumor activity, pharmacological properties and toxicology of GNS561. Our data showed that GNS561 displays antiproliferative effects in several human cancer cells (cell lines and primary patient-derived cells) and that GNS561 was more potent than HCQ. Analysis of the whole-body tissue distribution of GNS561 in rats after repeated oral dosing of GNS561 showed that GNS561 was mainly concentrated in the liver, stomach and lung. The data are consistent with the basic lipophilic nature of GNS561 (pKa1 = 9.4, pKa2 = 7.6, LogD 7,4 = 2.52 and tPSA = 39.7) and with studies showing that basic lipophilic drugs show high lysosomal tropism and high uptake in lysosome-profuse tissues, such as the liver and the lung [25,26]. As GNS561 had a high liver tropism, the effect of GNS561 on tumor growth in vivo was evaluated using two liver cancer models: one orthotopic human liver cancer xenograft mouse model (with an HCC patient-derived cell line, LI0752) and one DEN-induced cirrhotic rat model with HCC. These studies showed that GNS561 administered by oral gavage was well tolerated up to the doses of 50 mg/kg/day for 6 days in mice and up to 15 mg/kg/day for 6 weeks in rats and induced significant antitumor growth activity that was either comparable to or higher than sorafenib. In addition, in a DEN-induced cirrhotic rat model with HCC, the combination of GNS561 with sorafenib exerted an additive effect in controlling tumor progression and cell proliferation. This result was in accordance with a study completed by Shimizu et al. showing that sorafenib increased autophagy in some HCC cells, which leads to resistance and that combination of sorafenib with an autophagy inhibitor significantly increase the suppression of tumor growth in vivo [27]. Further studies are required to investigate GNS561 mechanism of action in vivo and particularly to confirm the engagement of PPT1 in GNS561 antitumor efficacy in vivo.
Furthermore, instead of that observed with CQ and HCQ [28], the distribution of GNS561 into the central nervous system was limited. Inactivating PPT1 mutations have long been known to induce infantile neuronal cerebral lipofuscinosis and induce retinopathy during childhood [29]. Germline PPT1 mutations were shown to selectively affect the central nervous system, with no effects in other tissues. Prior clinical experience using CQ and HCQ showed that retinopathy was one of the major toxicities in patients [30]. Authors have suggested that novel PPT1 inhibitors may take advantage of not crossing the blood-brain barrier to avoid retinal toxicity [4]. Interestingly, our data shown that the disposition of GNS561 displays limited penetration into the brain in rats, consistent with the lack of neurological and retinal toxicity observed in the current phase 1b/2a clinical trial of GNS561 [31–33].
In brief, our findings strengthen the importance of PPT1 and lysosomes as cancer targets. Moreover, we reported that through the inhibition of PPT1, GNS561 represents an innovative therapeutic strategy in cancer as we recently showed proof of concept in a clinical study targeting HCC and iCCA cancers [33]. Recently, it was shown that PPT1 inhibition by CQ derivatives or genetic PPT1 inhibition increases the antitumor activity of anti-PDCD1 (programmed cell death 1) antibody in melanoma by M2 to M1 phenotype switching in macrophages and a reduction in myeloid-derived suppressor cells in the tumor [34]. As such, GNS561 represents a promising new candidate for drug development in HCC either alone or in combination with other drugs, such as immune checkpoint inhibitors.
Materials and methods
Reagents and antibodies
BafA1 (254,134–5 G), NH4Cl (254,134–5 G), E-64 (3132), CA-074 (C57325 MG), Dextran-FITC (FD10S), pepstatin A (P531825 MG), dithiothreitol (D0632), DEN (N0756), almond β-glucosidase (G4511), 4-methylumbelliferone (M1381), NtBuHA (479,675), CA-074-Me (C57325 MG) and HCQ (H0915) were obtained from Sigma-Aldrich. EAD1 was purchased from Selleckchem (SE-S8576). The fluorogenic substrates Z-Phe-Arg-7-amido-4-methylcoumarin (03–321501), Z-Arg-Arg-7-amido-4-methylcoumarin (219,392) and (7-methoxycoumarin-4-yl)acetyl-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys(Dnp)-D-Arg-NH2 (219,360) were supplied by Merck. Mammalian Cell Lysis Buffer (GE Healthcare, 28–941279), 4-methylumbelliferyl-β-D-6-thiopalmitoyl-glucoside (Moscerdam, EM06650), Z-VAD-FMK (Bio Techne, FMK001), sorafenib (Santa Cruz Biotechnology, Sc-357,801), hexadecanesulfonyl fluoride (HDSF; Santa Cruz Biotechnology, sc-221,708), human PPT1 (OriGene Technologies, TP721098), DC661 (Vagdevi Innoscience), Triton X-100 (Dutscher, 091584B) and cOmplete™ Protease Inhibitor Cocktail (Roche, 4,693,132,001) were used. Fluozin-3 (F24195), LysoTracker Deep Red (L12492), CaspGLOW™ Fluorescein Active Staining Kit (88–700342), Annexin V/PI kit (V13242) were purchased from Thermo Fisher Scientific.
PPT1 Silencer® Select Pre-designed siRNA (4,392,420, ID s11017), Opti-MEM™ I Reduced Serum Medium (11,058,021) Lipofectamine™ RNAiMAX Transfection Reagent (13,778,075) were obtained from Life Technologies. Z-Phe was purchase from Cayman Chemical (27,877).
For immunoblotting assays, rabbit anti-LC3-II (Sigma-Aldrich, L7543; 1:3,000), mouse anti- glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Abnova, H00002597-M01; 1:5,000), rabbit anti-PARP (GeneTex, GTX100573; 1:1,500), rabbit anti-cleaved CASP3 (Asp175; Cell Signaling Technology, 9661; 1:200), goat anti-CTSD (Santa Cruz Biotechnology, Sc-6486; 1:200), rabbit anti-CTSB (Santa Cruz Biotechnology, sc-13,985; 1:200), goat anti-CTSL (Santa Cruz Biotechnology, sc-6498; 1:200), mouse anti-PPT1 (Invitrogen, MA526,471; 1:2,000), goat anti-rabbit (Santa Cruz Biotechnology, sc-2004; 1:5,000), donkey anti-goat (Santa Cruz Biotechnology, sc-2020; 1:5,000), goat anti-mouse (Jackson ImmunoResearch, 115–035003; 1:40,000), goat anti-rabbit (Sigma-Aldrich, AP307P; 1:25,000) and goat anti-rabbit (Jackson ImmunoResearch, 111–035003; 1:40,000) antibodies were used.
For immunofluorescence assays, anti-LAMP1 (Cell Signaling Technology, 9091S; 1:200), anti-lysosomal associated membrane protein 2 (LAMP2) (Developmental Studies Hybridoma Bank, AB_528129; 1:100), anti-MTOR (Cell Signaling Technology, 2972; 1:250), anti-CTSB (Abcam, ab58802; 1:200), anti-CTSD (Abcam, ab75852; 1:200), anti-CTSL (Abcam, ab133641; 1:200), Alexa Fluor 594 conjugate (Life Technologies, A-11072, A-11032; 1:500), Alexa Fluor 488 conjugate (Life Technologies, A-11017, A-11008; 1:500), Alexa Fluor 647 conjugate (Life Technologies, A-21447; 1:500) and Alexa Fluor 546 conjugate (Life Technologies, A-11003; 1:1,000) antibodies were used. Antibodies were diluted in blocking solution (5% bovine serum albumin [BSA; Dutscher, 871,001], 0.1% Tween-20 [Biosolve, 20,452,335] in TBS [1 M Tris HCl, pH 7.4, 5 M NaCl dissolved in pure water] or 5% fetal bovine serum [FBS] in PBS [Dutscher, L0615500]).
For immunohistochemical analysis, anti-MKI67/Ki67 (Thermo Fisher Scientific, MA514,520; 1:150) and anti-CCND1/cyclin D1 (Abcam, ab134175; 1:200) antibodies were used.
Cell lines and cell culture
Huh7 (HCC, JCRB Cell Bank, JCRB0403), HCT-116 (colorectal carcinoma, ATCC, CCL-247), A549 (lung cancer, Sigma-Aldrich, 86,012,804), LN-18 (glioblastoma, ATCC, CRL-2610), LN-229 (glioblastoma, ATCC, CRL-2611), MDA-MB-231 (breast cancer, ATCC, HTB-26) and A375 (malignant melanoma, ATCC, CRL-1619) cell lines were cultured in DMEM (Dutscher, L0103500). KG-1 cell line (acute myeloid leukemia; ATCC, CCL-246) was maintained in Iscove medium (Dutscher, L0191500). CAKI-1 (renal adenocarcinoma, NCI, 0507829), 786-O (renal adenocarcinoma, NCI, 05007648), DU-145 (prostate cancer, ATCC, HTB-81), PC-3 (prostate cancer, Sigma-Aldrich, 90,112,714), NCI-H358 (lung cancer, Sigma-Aldrich, 95,111,733) and NIH:OVCAR3 (ovarian adenocarcinoma, ATCC, HTB-161) cell lines were maintained in RPMI 1640 medium (Dutscher, L0498500). HepG2 (hepatocellular carcinoma, ATCC, HB-8065), HT-29 (colorectal carcinoma, ATCC, HTB-38) and SK-MEL-28 (malignant melanoma, ATCC, HTB-72) cell lines were cultured in DMEM low glucose (Thermo Fisher Scientific, 21,885,025), Mc Coy’s medium (Dutscher, L0210500), DMEM: Nutrient Mixture F-12 (Thermo Fisher Scientific, 31,765–027) and MEM (Dutscher, L0416500) respectively.
Cell lines were tested for Mycoplasma at each thawing and were used at a number of passage lower than 20. All cell lines were maintained in medium containing 1% penicillin-streptomycin (Dutscher, P0607100) and 10% FBS (GE Healthcare, SV30160.03 C), except NIH:OVCAR3 and KG-1 cell lines that were cultured in medium supplemented 20% FBS. In NIH:OVCAR3 medium, 0.01 mg/mL insulin (Actrapid® Penfill®, Novo Nordisk) was also added.
Cell viability assay
Cell viability was assessed using the CellTiter-Glo Luminescent Cell Viability Assay following the manufacturer’s protocol (Promega, G7573). Briefly, cells were plated in a 96-well tissue culture plate in 90 µL of medium. Twenty-four hours after plating, cells were treated with 10 µL of increasing concentrations of GNS561 or with GNS561 vehicle and were incubated for 72 h. At the end of the treatment, 100 µL of CellTiter-Glo solution was added to each well; cells were briefly shaken and then were incubated at room temperature (RT) for 10 min to allow stabilization of the luminescent signal. The luminescence was recorded using an Infinite F200 Pro plate reader (Tecan, Männedorf, Switzerland) and cell viability was expressed as a percentage of the values obtained from the negative control cells (vehicle treated cells). The half-maximal inhibitory concentration (IC50) was evaluated using a nonlinear regression curve in Prism 8.4.3 software (GraphPad Software, La Jolla, CA, USA). Each concentration was tested in triplicate. Mean IC50 was calculated as the average of three independent experiments.
Flow cytometry for ANXA5/propidium iodide assay
Cells were treated as indicated in the figures. Cell media containing floating cells were recovered and pelleted by centrifugation at 300 × g. Cells were trypsinized and recovered in medium and pelleted by centrifugation at 300 × g. Both cell pellets were combined and washed twice with ice cold PBS. ANXA5 and propidium iodide or ANXA5 and 7-aminoactinomycine D staining was performed according to the manufacturer’s protocol. Cells were analyzed immediately using flow cytometry and data were recorded on a BD Accuri C6 (BD Biosciences, San Jose, CA, USA) or an Attune NxT flow cytometer (Thermo Fisher Scientific) and processed using Cell Quest or BD FACSDiva softwares (BD Biosciences) or FlowJo software (FLOWJO, USA).
Flow cytometry detection of caspase activity
For these experiments the manufacturer’s protocol of CaspGLOW Fluorescein Active Staining Kit was followed. In brief, cells were collected, washed with PBS and stained with Z-VAD-FMK-FITC. Cells were kept on ice and analyzed immediately using flow cytometry and data were recorded on a BD Accuri C6 (BD Biosciences) and processed using BD Cell Quest (BD Biosciences) and FlowJo softwares.
Luminescence detection of caspase activity
The activity of CASP3-CASP7 and CASP8 was measured using the Caspase-Glo 3/7 Assay (Promega, G8092) and Caspase-Glo 8 Assay (Promega, G8202) following the manufacturer’s protocol. Briefly, HepG2 cells were plated in a 96-well plate (7,500 cells per well) in 90 µL of medium. Twenty-four hours after plating, cells were treated with 10 µL of GNS561 (1–4 µM) or GNS561 vehicle and incubated for 6, 24 and 30 h. At the end of the treatment, 100 µL of Caspase-Glo 3/7 or Caspase-Glo 8 reagent were added to each well and cells were incubated for 1 h at room temperature. Then, luminescence was measured by an Infinite F200 Pro plate reader. Fold change of activation of CASP3-CASP7and CASP8 was determined by comparing the luminescence in the treated groups with the luminescence observed in the negative control wells (vehicle-treated cells), with the luminescence of blank wells subtracted. At each time point, in parallel with the activation of CASP3-CASP7 and CASP8, cell viability was also investigated using CellTiter-Glo Luminescent Cell Viability Assay. Each GNS561 concentration was tested in triplicate in three independent experiments.
Z-VAD-FMK, CA-074-Me, pepstatin A, NtBuHA, BafA1, NH4Cl and GNS561 treatments
On the day of the treatment, cells were treated with warmed media and specified concentrations of Z-VAD-FMK (5 µM, pretreatment for 1 h), CA-074-Me (20 µM, pretreatment for 1 h), pepstatin A (5 µM, pretreatment for 1 h), NtBuHA (8 mM), BafA1 (200 nM, pretreatment for 2 h), NH4Cl (20 mM, pretreatment for 2 h) and GNS561 (concentrations and time as indicated on the figures) or vehicle were added. Cells were treated for the times indicated and processed for several analyses as indicated. Each condition was tested in triplicate, and three independent experiments were performed.
Chemical labeling of GNS561D in cells
GNS561D, is a GNS561 analogue, synthetized with one additional azide group (-N3) directly linked to an aromatic group of GNS561 structure (meta-1, “aryl azide”) and with a second alkyl azide group (-CH2-N3) linked on the same aromatic group (meta-2, “alkyl aryl”). Cells were cultured at ~80% confluence and were treated with 10 μM GNS561D, the photoactivable analogue of GNS561 (with an aryl and an alkyl azide moieties), for 90 min. Cell were fixed with formaldehyde (2% in PBS, 12 min) prior to permeabilization (Triton X-100, 0.1% in PBS, 5 min) and washed three times with 1% BSA-PBS. Fluorescent signal was recorded after that GNS561D was immobilized on its target site by UV-crosslinking, through reaction with the aryl azide specie, and then GNS561D was labeled with the green-fluorescent Alexa Fluor 488 alkyne dye by click-chemistry, through reaction with the alkyl azide specie, in the presence of copper (Figure 4A).
The click reaction cocktail was prepared from Click-iT EdU Imaging kits (Life Technologies, C10337) according to the manufacturer’s protocol. Briefly, mixing 430 μL of 1 × Click-iT reaction buffer with 20 μL of CuSO4 solution, 1.2 μL Alexa Fluor azide, 50 μL reaction buffer additive (sodium ascorbate) to reach a final volume of 500 μL. Cover-slips were incubated with the click reaction cocktail in the dark at room temperature for 30 min, then washed three times with PBS. Immunofluorescence was then performed as indicated.
Lysosome staining using Lysotracker
LysoTracker Deep Red was added to the cells at the same time as GNS561 at 1:10,000.
Cathepsin activity assay in cell lysate
Twenty-four hours after HepG2 cell plating, the cells were treated with GNS561 (1, 2 and 4 µM) for 6 and 24 h. Treatment with vehicle was used as a baseline for cathepsin activity control. Cell lysates (1 µg of total protein) were pre-incubated with acetate buffer (0.1 M sodium acetate, pH 5.5, 10 mM DTT, 2 mM EDTA, and 0.01% Brij35 (Merck, 101,894) or with citrate buffer (0.1 M sodium citrate, pH 4.0, 2 mM EDTA, 0.01% Brij35) prior to measurement of the respective CTSB-CTSL (including both cathepsins) and CTSB activities and CTSD activity as previously reported [35]. The peptidase activity of CTSB, CTSB-CTSL and CTSD were determined fluorometrically with a fluorescence reader (Gemini spectrofluorometer, Molecular Devices, San José, CA, USA) using respectively, the synthetic substrates Z-Arg-Arg-7-amido-4-methylcoumarin (excitation wavelength: 350 nm; emission wavelength: 460 nm), Z-Phe-Arg-7-amido-4-methylcoumarin (excitation wavelength: 350 nm; emission wavelength: 460 nm) and 7-methoxycoumarin-4-yl)acetyl-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys(Dnp)-D-Arg-NH2 (excitation wavelength: 325 nm; emission wavelength: 390 nm) [36]. The synthetic protease inhibitors E-64 (pan-inhibitor of cysteine cathepsins), CA-074 (specific inhibitor of CTSB) and pepstatin A (inhibitor of CTSD) were used as controls to confirm the detection of specific activities. Slopes of the enzymatic activities were calculated with the software SoftMax Pro (Molecular Devices). For each experiment and tested condition, fold change of the cathepsin activity was determined by comparing the slope of the enzymatic activity in treated conditions to the slope of the enzymatic activity in the vehicle condition. Three independent experiments were performed.
Unbound zinc staining using Fluozin
Fluozin-3 was added for 30 min to live cells at 1:1,000.
Western blotting
In brief, cells were lysed with Mammalian Cell Lysis Buffer. cOmplete™ Protease Inhibitor Cocktail was added extemporaneously to the lysis buffer. Ten to twenty micrograms of protein from each cell lysate was separated on a 15% or 4–15% SDS-PAGE gel, transferred to a PVDF membrane, and blotted with antibodies. For all blots, GAPDH immunoblotting was used as a loading control. All the experiments were repeated at least three times. Representative autoradiograms are shown.
Autophagy assay
The autophagy pathway was studied as performed previously [11]. Twenty-four hours after HepG2 or Huh7 cell plating, the cells were treated with GNS561 (0.5, 1 and 2 µM for HepG2 cell line and 0.4, 0.8 and 1.6 µM for Huh7 cell line) for 24 h. Treatment with vehicle was used as a baseline for autophagic flux control. In specified conditions, BafA1 (100 nM) was added for the last 2 h of treatment. The autophagic flux was calculated as the ratio between the LC3-II level normalized against GAPDH level (Norm LC3-II) with or without BafA1.
Nano differential scanning fluorimetry measurements
Nano differential scanning fluorimetry was used to measure thermal stability of PPT1 in absence (0 µM ligand) and presence of the ligands, GNS561 and HCQ (used at 20 µM and 100 µM). Recombinant PPT1 was used at 3.6 µM in 1x PBS pH 7.4, 0.05% Tween-20. In order to facilitate comparability, the assay buffer was supplied with 1% DMSO in experiments using the DMSO-solved ligand GNS561. For each condition, a duplicate experiment was prepared and measured in standard capillaries (NanoTemper Technologies GmbH, München, Germany, PR-C002) at 40% sensitivity, temperature range from 20–95°C, and a heating speed of 1°C/min on a Prometheus NT.48 instrument (NanoTemper Technologies GmbH). PPT1 unfolding was measured by detecting the temperature-dependent change in tryptophan/tyrosine fluorescence at emission wavelengths of 350 nm and 330 nm, respectively. Melting temperatures (Tm) were determined by detecting the maximum of the first derivative of the fluorescence ratios (F350/F330). For this, an 8th order polynomial fit was applied to the transition region (PR.StabilityAnalysis_x64_1.0.3.10009, NanoTemper Technologies). For determination of the influence of ligands on PPT1 stability, ΔTm values of each compound condition were determined by subtracting average Tm of PPT1 (in the respective buffer) by the average Tm of the respective compound condition. The standard deviation (SD) of the Tm of each condition was calculated from two replicates (n = 2). ΔTm > 6*SD of PPT1 (0.3°C) were considered as significant.
PPT1 enzymatic assay
HepG2 cell line was treated with GNS561 (1, 5 and 10 µM), HCQ (50, 100 and 200 µM) and HDSF (25 and 100 µM) for 3 h. The cells were lysed in 0.5% Triton X-100 with cOmplete™ Protease Inhibitor Cocktail. The cell lysates were used a source of PPT1 and PPT1 activity was assayed using 4-methylumbelliferyl-β-D-6-thiopalmitoyl-glucoside as reported [37]. Reaction mixtures contained 5 µL of cell lysate + 5 µL 0.5% Triton X-100 with cOmplete™ Protease Inhibitor Cocktail + 20 μL of substrate preparation (0.5 mM substrate, 1.5 mM dithiothreitol, 0.1 U almond β-glucosidase (Sigma-Aldrich, G4511), 0.2% Triton X-100 and McIlvain’s phosphate citrate buffer (0.2 M Na2HPO4, 0.1 M citric acid, pH 4). After 1 h incubation at 37°C, the reaction was stopped by adding 200 µl of 0.5 M glycine-NaOH, pH 10.5 buffer. The amount of the released fluorescent product 4-methylumbelliferone was determined by fluorometry at 358 and 448 nm for the excitation and emission wavelengths, respectively. Infantile subtype of ceroid lipofuscinosis fibroblasts which contain PPT1 mutations and normal fibroblasts were used as control. 4-methylumbelliferone diluted in 0.5 M glycine-NaOH, pH 10.5 buffer was used to do a standard curve and to calculate the enzymatic activity of PPT1.
Small interference RNA
Knockdown of PPT1 expression was performed using PPT1 Silencer® Select Pre-designed siRNA following the manufacturer’s protocol and the target sequence: sense (5ʹ-GGAGAGAGCUCUCACAUCUtt-3ʹ) and antisense (5ʹ-AGAUGUGAGAGCUCUCUCCtg-3ʹ). HepG2 cells (250,000 cell per well in 6-well tissue culture plates) were cultivated during 24 h. Cultivated cells were treated with 10 nM of siRNA-PPT1 in Opti-MEM™ I Reduced Serum Medium and using 7.5 µL per well of Lipofectamine™ RNAiMAX Transfection Reagent according to the manufacturer’s instructions during 6 h. After medium change, cells were further cultivated for 48 h before GNS561 treatment for an additional 24 h. The efficiency of siRNA interference of PPT1 was monitored by immunoblotting assays using the mouse anti-PPT1 antibody (Fig. S6).
Lysosomal membrane permeabilization assay
HepG2 cells were plated 24 h prior to the experiments and then treated as indicated. Then, cells were treated with Dextran-FITC at 1 mg/mL for 1 h in cell medium. Cells where then fixed with formaldehyde (2% in PBS, 12 min) and analyzed by fluorescence microscopy.
Cell imaging
For immunofluorescence, HepG2 cells were blocked with 2% BSA or 10% FBS supplemented with 0.2% Tween-20 in PBS (blocking buffer) for 20 min at room temperature. Cover-slips were incubated with 50 to 100 µL of diluted primary antibodies in blocking buffer 1 h at room temperature. Cover-slips were then washed three times with blocking buffer and incubated as described above with the appropriate secondary antibodies for 1 h. Cover-slips were washed three times with PBS and mounted using Mowiol (Sigma-Aldrich, 81,381) or Vectashield Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI; 1/500, VECTOR Labs, H-1200). Fluorescence images were acquired using a Deltavision real-time microscope (Applied Precision, Rača, Slovakia) with 60×/1.4NA and 100×/1.4NA objectives or using a LSM 800 Airyscan confocal microscope (Carl Zeiss, Oberkochen, Germany) with a 63X oil objective. A typical z-stack of a field contained cells in the range of 3–10. Pearson correlation coefficients were determined using the ImageJ plugin Coloc 2 for each individual cell. The background (no cell) was set to 0 and Pearson correlation coefficients were calculated above that threshold. In immunofluorescence quantifications, one point represents one cell for Figures 4B and 4D, or one field for Figure 5H and S5A.
For electron microscopy, HepG2 cells were fixed at least for 1 h with glutaraldehyde 2.5% in 0.1 M sodium cacodylate buffer (Sigma-Aldrich, 70,114). For resin embedding, cells were washed three times with a mixture of 0.2 M saccharose (Sigma-Aldrich, 1,076,870,250), 0.1 M sodium cacodylate. Cells were then post-fixed for 1 h with 1% OsO4 diluted in 0.2 M potassium hexa-cyanoferrate (III) (Sigma-Aldrich, P8131), 0.1 M sodium cacodylate solution. After three 10 min washes with distilled water, the cells were gradually dehydrated with ethanol by successive 10 min baths in 30, 50, 70, 96, 100, and 100% ethanol. Substitution was achieved by successively placing the cells in 25, 50, and 75% Epon (Fisher Scientific, 50–980448) solutions for 15 min. HepG2 cells were placed for 1 h in 100% Epon solution and in fresh Epon 100% overnight under vacuum at room temperature. Polymerization occurred with cells in 100% fresh Epon for 72 h at 60°C. All solutions used above were 0.2 µm filtered. Ultrathin 70-nm sections were cut using a UC7 ultramicrotome (Leica, Wetzlar, Germany) and placed on HR25 300 mesh copper-rhodium grids (TAAB, GM033). Sections were contrasted according to Reynolds [38]. Electron micrographs were obtained on a Morgagni 268D (FEI Company, Hillsboro, Or, USA) transmission electron microscope operated at 80 keV.
Whole body rat distribution of GNS561 by mass spectrometry imaging
Sprague Dawley male rats (N = 2) have been dosed orally with GNS561 at 40 mg/kg/day for 28 days with a single administration and were sacrificed 7 h after the last administration. One rat received water and used as a control animal. After the sacrifice, whole body rats were shaved and their legs and tails sawed off. They were individually embedded in CMC 3% and fast-frozen in a dry ice/hexane mix then stored at −80°C before sample preparation for matrix assisted laser desorption ionization (MALDI) analysis. For each animal, 20 μm tissue sections through the sagittal section plan were performed on tape (Leica, 14,041,739,651) in a cryomacrotome cryostat (Leica, CM3600) at −20°C. Tissue sections on tape were cryodesiccated 24 h then stored at −80°C until use. Low resolution optical images of each slide were acquired using a standard office type scanner (Hewlett-Packard, Palo Alto, CA, USA).
Prior to starting the MALDI matrix deposit, sections on tape were stuck on double conductive tape (XYZ-Axis Electrically Conductive Tape 9713, 3 M) on MALDI target (Bruker, Billerica, MA, USA). 2,5-Dihydroxybenzoic acid solution was prepared at 40 mg/mL in methanol/H2O + 0.2% trifluoroacetic acid (1:1 v:v) for matrix deposition in the positive ion mode study. TM sprayer (HTX Imaging, Chapel Hill, NC, USA) was used for spraying the MALDI matrices over the tissue sections. Deuterated GNS561 was added to the MALDI matrix at 3 µM and used as an internal standard.
Sections were analyzed by MALDI imaging. MALDI images were obtained using a 7 T MALDI-FTICR (Solarix, Bruker) equipped with a SmartBeam II laser used with a repetition rate of 1000 Hz, in positive ion mode. Mass spectra were acquired with a full scan mode of acquisition within the m/z 100–1,000 range at 650 µm of spatial resolution. The mass spectrum obtained for each position of the images corresponds to the averaged mass spectra of 300 consecutive laser shots on the same location. Prior to each data acquisition, external calibration was performed using endogenous compounds well known and MALDI matrix ions. FTMS Control 2.0 and FlexImaging 4.1 software packages (Bruker) were used to control the mass spectrometer and set imaging parameters. Multimaging™ 1.1 software (ImaBiotech, Loos, France) was used to create the molecular distributions of GNS561 normalized by the internal standard.
Animal treatment
The animals were checked daily for clinical signs, effects of tumor growth and any other abnormal effects. For experiments involving the mouse model (performed in CrownBio [United Kingdom] facilities), the protocol and any amendment(s) or procedures involving the care and use of animals were reviewed and approved by the Institutional Animal Care and Use Committee of CrownBio prior to experimentation, and during the study, the care and use of animals was conducted in accordance with the regulations of the Association for Assessment and Accreditation of Laboratory Animal Care. For the rat model, all animals received humane care in accordance with the Guidelines on the Humane Treatment of Laboratory Animals (Directive 2010/63/EU), and experiments were approved by the animal Ethics Committee: GIN Ethics Committee No.004.
DEN-induced cirrhotic rat model of HCC
Thirty 6-week-old Fischer 344 male rats (Charles River Laboratories) were housed in the animal facility of “Plateforme de Haute Technologie Animale” (Jean Roget, University of Grenoble-Alpes, France). Rats were kept in individually ventilated cage systems at constant temperature and humidity with 2–3 animals in each cage having free access to food (standard diet) and water during the entire study period. Rats were treated weekly with intra-peritoneal injections of 50 mg/kg of DEN, which were diluted in olive oil in order to obtain a fully developed HCC on a cirrhotic liver after 14 weeks [39]. Rats were randomized in 4 different groups and treated during six weeks by i) sorafenib (n = 8), ii) GNS561 (n = 8), iii) combination of GNS561 and sorafenib (n = 6) or iv) rested untreated (control, n = 8), as specified in Fig. S2. Treatments of GNS561 (15 mg/kg/day), sorafenib (10 mg/kg/day) and combination (GNS561+ sorafenib) were administered by oral gavage for a period of six weeks. The nutritional state was monitored by daily weighing of rats and protein-rich nutrition was added to the standard food in every cage where a loss of weight was observed. The food intake per cage was monitored during the last 6 weeks of the experiment. Food was withheld for 3–4 h before the animals were sacrificed.
Imaging study was conducted on a 4.7 Tesla MR Imaging system (BioSpec 47/40 USR, Bruker). All rats were subjected to 3 MRI scans: MRI1 was performed before randomization, MRI2 was performed after 3 weeks of treatment and MRI3 after 6 weeks of treatment. MRI analysis was done by an investigator who was blinded of treatment allocation.
After the third MRI scan, all rats were euthanized with vena cava blood sampling for hematological and biochemical analyses. Each liver was weighed, the diameter of the five largest tumors was measured and the number of tumors larger than 1 mm on the surface of the liver was counted, all in a blinded manner. Tumor proliferation was studied by using anti-MKI67 and anti-CCND1 antibodies.
Statistical analysis
Statistical analyses were performed using Prism 8.4.3 software. For datasets with normal distribution, multiple comparisons were performed using one-way ANOVA with Dunnett’s or Tukey’s post hoc analysis. The parametric Student t-test was used to compare two groups of data with normal distribution. Data are presented as the mean values ± standard error mean (SEM) or SD unless stated otherwise. Statistical significance was defined as a p-value < 0.05, which *represents significant difference, at least p < 0.05.
Supplementary Material
Acknowledgments
The authors are very grateful to Dr. Sebastian Müller and Dr. Raphaël Rodriguez from Curie Institute for mechanistic analysis, Pr. Thierry Levade and Dr. Nathalie Andrieu from CRCT for the PPT1 enzymatic assay, Keerthi Kurma and Seyedeh Tayebeh Ahmad Pour from the Institute for Advanced Biosciences for technical support during animal experiments and Dr. François Autelitano, Dr. Marie Guillemot and Philippe Fabre for Zn2+ localization analysis.
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Disclosure statement
SB, EB, FB, ER, SM, MR, MN, JT, JC, CD, GJ, CS, CA and PH are employees of Genoscience Pharma. SB, ER, FB, CD, CS, CA and PH are shareholders of Genoscience Pharma. SB, FB, JC and PH are co-inventors of a pending patent. The other authors declare that they have no conflicts of interest to report.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- [2].Aits S, Jäättelä M.. Lysosomal cell death at a glance. J Cell Sci. 2013;126(9):1905–1912. [DOI] [PubMed] [Google Scholar]
- [3].Klempner SJ, Myers AP, Cantley LC, et al. What a tangled web we weave: emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov. 2013;3(12):1345–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rebecca VW, Nicastri MC, Fennelly C, et al. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov. 2019;9(2):220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rebecca VW, Nicastri MC, McLaughlin N, et al. A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov. 2017;7(11):1266–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shi -T-T, et al. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother Pharmacol. 2017;79(2):287–294. [DOI] [PubMed] [Google Scholar]
- [7].Verbaanderd C, Maes H, Schaaf MB, et al. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience. 2017;11:781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Manic G, Obrist F, Kroemer G, et al. Chloroquine and hydroxychloroquine for cancer therapy. Mol Cell Oncol. 2014;1(1):e29911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pérez-Hernández M, Arias A, Martínez-García D, et al. Targeting autophagy for cancer treatment and tumor chemosensitization. Cancers (Basel). 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Plantone D, Koudriavtseva T. Current and future use of chloroquine and hydroxychloroquine in infectious, immune, neoplastic, and neurological diseases: a mini-review. Clin Drug Investig. 2018;38(8):653–671. [DOI] [PubMed] [Google Scholar]
- [11].Brun S, Bassissi F, Serdjebi C, et al. GNS561, a new lysosomotropic small molecule, for the treatment of intrahepatic cholangiocarcinoma. Invest New Drugs. 2019;37(6):1135–1145. [DOI] [PubMed] [Google Scholar]
- [12].Lockwood TD. Lysosomal metal, redox and proton cycles influencing the CysHis cathepsin reaction. Metallomics. 2013;5(2):110–124. [DOI] [PubMed] [Google Scholar]
- [13].Lockwood TD. Biguanide is a modifiable pharmacophore for recruitment of endogenous Zn2+ to inhibit cysteinyl cathepsins: review and implications. Biometals. 2019;32(4):575–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Turk V, Stoka V, Vasiljeva O, et al. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta. 2012;1824:68–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sironi J, Aranda E, Nordstrøm LU, et al. Lysosome membrane permeabilization and disruption of the molecular target of rapamycin (mTOR)-lysosome interaction are associated with the inhibition of lung cancer cell proliferation by a chloroquinoline analog. Mol Pharmacol. 2019;95(1):127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Oberle C, Huai J, Reinheckel T, et al. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 2010;17(7):1167–1178. [DOI] [PubMed] [Google Scholar]
- [17].Yang S, Yang L, Li X, et al. New insights into autophagy in hepatocellular carcinoma: mechanisms and therapeutic strategies. Am J Cancer Res. 2019;9:1329–1353. [PMC free article] [PubMed] [Google Scholar]
- [18].Kallunki T, Olsen OD, Jäättelä M, et al. Cancer-associated lysosomal changes: friends or foes? Oncogene. 2013;32(16):1995–2004. [DOI] [PubMed] [Google Scholar]
- [19].Wang F, Gómez-Sintes R, Boya P, et al. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19(12):918–931. [DOI] [PubMed] [Google Scholar]
- [20].Carroll B, Maetzel D, Maddocks OD, et al. Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. eLife. 2016;5:e11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Turk B, Dolenc I, Lenarčič B, et al. Acidic pH as a physiological regulator of human cathepsin L activity. Eur J Biochem. 2001;259(3):926–932. [DOI] [PubMed] [Google Scholar]
- [22].Bagh MB, Peng S, Chandra G, et al. Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nat Commun. 2017;8(1):14612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Xue J, Moyer A, Peng B, et al. Chloroquine is a zinc ionophore. PLoS One. 2014;9(10):e109180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xie X, White EP, Mehnert JM, et al. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS One. 2013;8(1):e55096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nadanaciva S, Lu S, Gebhard DF, et al. A high content screening assay for identifying lysosomotropic compounds. Toxicol in Vitro. 2011;25(3):715–723. [DOI] [PubMed] [Google Scholar]
- [26].Daniel WA, Wójcikowski J. Contribution of lysosomal trapping to the total tissue uptake of psychotropic drugs. Toxicol Pharmacol. 1997;80:62–68. [DOI] [PubMed] [Google Scholar]
- [27].Shimizu S, Takehara T, Hikita H, et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer. 2012;131(3):548–557. [DOI] [PubMed] [Google Scholar]
- [28].Harder BG, Blomquist MR, Wang J, et al. Developments in blood-brain barrier penetrance and drug repurposing for improved treatment of glioblastoma. Front Oncol. 2018;8:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Metelitsina TI, Waggoner DJ, Grassi MA. Batten disease caused by a novel mutation in the PPT1 gene. Retin Cases Brief Rep. 2016;10(3):211–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Marmor MF, Kellner U, Lai TYY, et al. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology. 2011;118(2):415–422. [DOI] [PubMed] [Google Scholar]
- [31].Eide DJ. The SLC39 family of metal ion transporters. Pflugers Arch. 2004;447(5):796–800. [DOI] [PubMed] [Google Scholar]
- [32].ClinicalTrials.gov Study of GNS561 in patients with liver cancer.
- [33].Harding JJ, Awada A, Decaens T, et al. First-in-human phase I, pharmacokinetic (PK), and pharmacodynamic (PD) study of oral GNS561, a palmitoyl-protein thioesterase 1 (PPT1) inhibitor, in patients with primary and secondary liver malignancies. JCO. 2021;39(15_suppl):e16175–e16175. [Google Scholar]
- [34].Sharma G, Ojha R, Noguera-Ortega E, et al. PPT1 inhibition enhances the antitumor activity of anti-PD-1 antibody in melanoma. JCI Insight. 2020;5(17). DOI: 10.1172/jci.insight.133225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bestion E, Jilkova ZM, Mège J-L, et al. GNS561 acts as a potent anti-fibrotic and pro-fibrolytic agent in liver fibrosis through TGF-β1 inhibition. Ther Adv Chronic Dis. 2020;11:2040622320942042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Galibert M, Wartenberg M, Lecaille F, et al. Substrate-derived triazolo- and azapeptides as inhibitors of cathepsins K and S. Eur J Med Chem. 2018;144:201–210. [DOI] [PubMed] [Google Scholar]
- [37].van Diggelen OP, Keulemans JLM, Winchester B, et al. A rapid fluorogenic palmitoyl-protein thioesterase assay: pre- and postnatal diagnosis of INCL. Mol Genet Metab. 1999;66(4):240–244. [DOI] [PubMed] [Google Scholar]
- [38].Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol. 1963;17(1):208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jilkova ZM, Kuyucu AZ, Kurma K, et al. Combination of AKT inhibitor ARQ 092 and sorafenib potentiates inhibition of tumor progression in cirrhotic rat model of hepatocellular carcinoma. Oncotarget. 2018;9(13):11145–11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.