

ABSTRACT
Depolarized mitochondria can be degraded via mitophagy, a selective form of autophagy. The RAB GTPase RAB7A was recently shown to play a key role in this process. RAB7A regulates late endocytic trafficking under normal growth conditions but is translocated to the mitochondrial surface following depolarization. However, how RAB7A activity is regulated during mitophagy is not understood. Here, using a proximity-dependent biotinylation approach (miniTurbo), we identified C5orf51 as a specific interactor of GDP-locked RAB7A. C5orf51 also interacts with the RAB7A guanine nucleotide exchange factor (GEF) complex members MON1 and CCZ1. In the absence of C5orf51, localization of RAB7A on depolarized mitochondria is compromised and the protein is degraded by the proteasome. Furthermore, depletion of C5orf51 also inhibited ATG9A recruitment to depolarized mitochondria. Together, these results indicate that C5orf51 is a positive regulator of RAB7A in its shuttling between late endosomes and mitochondria to enable mitophagy.
Abbreviations: ATG9A: autophagy related 9A; Baf A1: bafilomycin A1; BioID: proximity-dependent biotin identification; CCCP: carbonyl cyanide m-chlorophenylhydrazone; CCZ1: CCZ1 homolog, vacuolar protein trafficking and biogenesis associated; DQ-BSA: dye quenched-bovine serum albumin; FYCO1: FYVE and coiled-coil domain autophagy adaptor 1; GAP: GTPase activating protein; GEF: guanine nucleotide exchange factor; KO: knockout; LRPPRC: leucine rich pentatricopeptide repeat containing; MG132: carbobenzoxy-Leu-Leu-leucinal; MON1: MON1 homolog, secretory trafficking associated; mtDNA: mitochondrial DNA; PINK1: PTEN induced kinase 1; PRKN/PARKIN: parkin RBR E3 ubiquitin protein ligase; RMC1: regulator of MON1-CCZ1; TBC1D15: TBC1 domain family member 15; TBC1D17: TBC1 domain family member 17; TOMM20: translocase of outer mitochondrial membrane 20; WDR91: WD repeat domain 91; WT: wild type.
KEYWORDS: Autophagy, C5orf51, guanine nucleotide exchange factor, mitophagy, RAB7A
Introduction
Macroautophagy (hereafter referred to as autophagy) is a conserved cellular process in eukaryotes that involves lysosome-mediated degradation of unnecessary or damaged intracellular components [1]. Autophagy was initially regarded as a nonspecific process that randomly envelops cytosolic cargo in double-membrane compartments called autophagosomes, which then fuse with lysosomes for cargo degradation and recycling. However, increasing evidence has revealed selective modes of autophagy that deliver a wide range of specific cargo to the lysosome for degradation [2]. Selective autophagy is mediated by proteins that function as autophagy cargo receptors, targeting cellular components to forming autophagosomes [3].
Mitophagy is the selective elimination of damaged mitochondria by the autophagy pathway [4]. Under pathologic conditions, damaged mitochondria initiate a series of signaling cascades on their surface, including PINK1 (PTEN induced kinase 1)-mediated phosphorylation and PRKN/PARKIN (parkin RBR E3 ubiquitin protein ligase)-mediated ubiquitination, which leads to recruitment of autophagy machinery to the mitochondria with the help of receptor proteins [5,6]. Mitophagy defects have been linked to numerous diseases, including neurodegeneration, cancer, muscle atrophy and inflammatory diseases [7–10].
Recent discoveries revealed that RAB7A is targeted to depolarized mitochondria and is required for mitophagy [11–13]. RAB7A is a RAB family GTPase that regulates membrane transport activities in its active (GTP-bound) state. Two mitochondria localized RAB7A GTPase activating proteins (GAPs), TBC1D15 and TBC1D17, are known to modulate RAB7A activity and limit mitophagy [11,14], thereby contributing to mitochondrial homeostasis. MON1 and CCZ1, previously described components of a RAB7A guanine nucleotide exchange factors (GEF) complex [15], are required for the recruitment of RAB7A to damaged mitochondria [12]. However, MON1 and CCZ1 are known to require other factors for their function on specific organelles [16]. Under normal growth conditions, RMC1/C18orf8 (regulator of MON1-CCZ1) forms a complex with MON1-CCZ1 that enables RAB7A functions in late endocytic trafficking [17]. Whether this or another GEF complex can regulate RAB7A during mitophagy is not known.
Here, we examined the RAB7A regulation network through interactome analysis using a proximity-dependent biotinylation approach, conducted under normal growth conditions and mitochondrial depolarization. Notably, C5orf51 was found to interact with a dominant negative (GDP-locked) RAB7A mutant protein under both normal and depolarized conditions, and deletion of C5orf51 attenuated RAB7A translocation to damaged mitochondria. Functional analysis revealed that C5orf51 is a positive regulator of RAB7A, necessary for the removal of depolarized mitochondria. Moreover, deletion of C5orf51 leads to rapid proteasomal degradation of RAB7A. Based on these results, we propose that C5orf51 is a regulator of mitophagy that controls RAB7A localization and stability.
Results
miniTurbo analysis of RAB7A during mitophagy
To gain insight into RAB7A function during mitophagy, we characterized RAB7A protein-protein interactions using a proximity-dependent biotinylation (BioID) approach. For this, we used the miniTurbo variant of a abortive biotin ligase, which provides high efficiency biotin labeling of proximal proteins and precise temporal control of the labeling window [18–20]. Human embryonic kidney (HEK) 293 cells stably expressing wild type RAB7A, a constitutively active (Q67L, referred to as CA) mutant or a dominant negative (T22N, referred to as DN) mutant were generated using the Flp-In T-REx site-specific recombination system. Each RAB7A open reading frame was cloned in-frame with an N-terminal 3xFlag-miniTurbo moiety (Figure 1A), expressed under the control of a tetracycline-inducible promoter. Tagged RAB7A proteins were assessed for targeting to their established intracellular compartments, as determined by immunofluorescence (Figure 1B) and by Gene Ontology cellular component analysis of biotinylated proximal proteins (Figure 1C).
Figure 1.
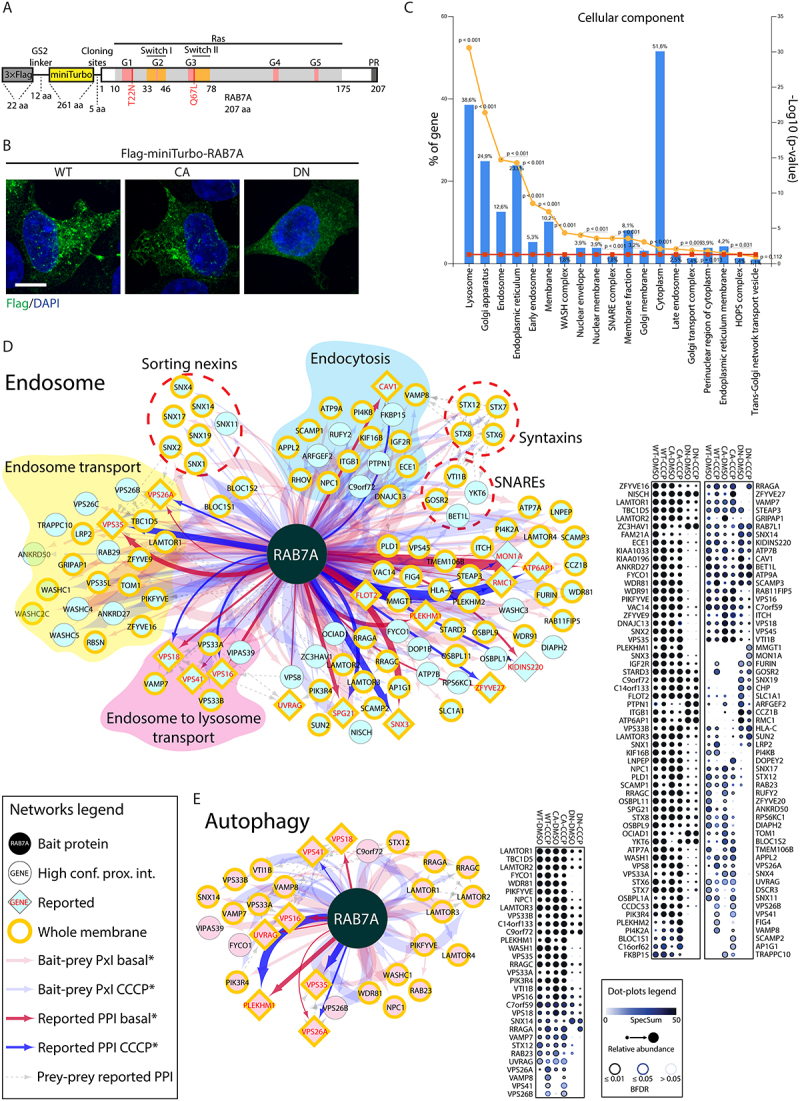
miniTurbo, a proximity-based strategy to identify RAB7A interactors.
(A) Domain architecture of Flag-miniTurbo-RAB7A constructs. G1 to G5: conserved motifs involved in guanine nucleotide binding; Switch I and II: flexible domains that substantially change their conformation upon exchange between GDP and GTP; PR: C-terminal prenylation motif. In addition to the wild-type allele, constructs were generated with a constitutively active (Q67L, referred to as CA) mutant or a dominant negative (T22N, referred to as DN) mutant of RAB7A. (B) HEK 293 T stable cell lines, in the presence of tetracycline (1.0 μg/ml), expressing Flag-miniTurbo-RAB7A WT, CA or DN respectively, were fixed and stained for Flag epitope (green) and DAPI (blue). Size bars: 10 µm. (C) FunRich software tool was used to depict the cellular component terms of the RAB7A high confidence interactors identified by miniTurbo. Left y-axis depicts the percentage of genes of the input list (all interactors) assigned to each category. Right y-axis depicts the category enrichment p-value (orange line) and significance threshold (0.05; red line). (D) RAB7A proximal interactors from endosomes. Cytoscape software was used to generate network (left) representing significance analysis of INTeractome (SAINT)-filtered putative RAB7A-prey interactions identified by miniTurbo. Each node represents a selected hit from the screen, organized by known localization and function through ToppGene analysis focused on cellular components and biological process. If known, preys were also subgrouped by subcomplex or function. Whole membrane (membrane) proteins have been highlighted. Edge thickness is proportional to peptide counts. ProHits-viz web tool was used to generate dot plot (right) displaying prey abundance across baits and prey confidence. (E) RAB7A proximal interactors in autophagy pathway, analyzed as in A.
In total, 531 high confidence RAB7A proximity interactors were identified (<1% FDR; Dataset S1). The resulting protein proximity interaction network (Figure 1D, E) depicts some of these hits clustered according to function and localization, where edge thickness is proportional to peptide count. The RAB7A interaction network is significantly enriched in endosomal and lysosomal proteins, consistent with its known roles in endocytic membrane traffic and autophagy which validates the usage of the miniTurbo technique for analysis of the RAB7A interactome. Our data also identified RAB7A proximity interactions with proteins from other organelles (based on GO annotations), including the endoplasmic reticulum membrane (133/531) (Figure S1A), Golgi membrane (144/531) (Figure S1B) and mitochondrial outer membrane (14 proteins) (Figure S1C), which is consistent with prior studies [21–23].
The RAB7A CA mutant displayed proximity interactions with known effectors of RAB7A, including the microtubule transport effector FYCO1 (FYVE and coiled-coil domain autophagy adaptor 1) [24], organelle contact site protein PDZD8 (PDZ domain containing 8) [25,26] and neuronal development effector WDR91 (WD repeat domain 91) [27] (Figure 2A). We also identified the interaction of RAB7A with GAP proteins such as TBC1D5 [28] (Figure 2A). MiniTurbo of the RAB7A DN mutant revealed a loss of interaction with known RAB7A effectors, as expected. Notably, the DN mutant displayed a selective interaction with GEF proteins MON1A, CCZ1B and RMC1. Thus, our dataset successfully identified both known effectors and regulators of RAB7A.
Figure 2.
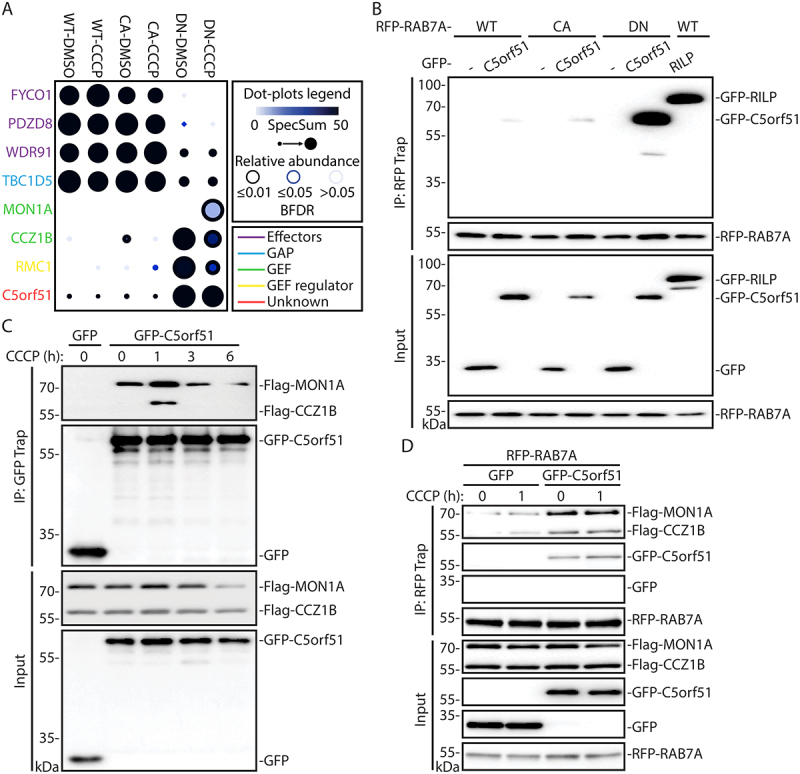
Identification of C5orf51 as a component of the MON1-CCZ1 GEF complex for RAB7A during mitophagy.
(A) ProHits-viz web tool was used to generate the dot plot view of known and novel RAB7A interactors from miniTurbo profiling, displaying prey abundance across baits and prey confidence. (B) HEK 293 T cells were cotransfected with RFP-tagged RAB7A mutants and either GFP or GFP-RILP or GFP-C5orf51. The RFP-tagged proteins were precipitated with RFP-Trap beads and the elutions were analyzed by western blotting and probed for GFP or RFP. Data are representative of 3 independent experiments. (C) Co-IPs were performed as in B using GFP-C5orf51, Flag-MON1A and Flag-CCZ1B constructs. Cells were treated with 10 μM CCCP for indicated time before collection. Lysates were precipitated with GFP-Trap beads. Data are representative of 3 independent experiments. (D) Co-IPs were performed as in B using RFP-RAB7A, GFP-C5orf51, Flag-MON1A and Flag-CCZ1B constructs. Cells were treated with 10 μM CCCP for indicated time before collection. Lysates were precipitated with RFP-Trap beads. Data are representative of 3 independent experiments.
The highly active miniTurbo enzyme [20] also allowed us to characterize RAB7A interactions in response to carbonyl cyanide m-chlorophenyl hydrazone (CCCP) treatment, which causes mitochondrial depolarization and subsequent mitophagy [29,30]. Under CCCP treatment, the interaction between RMC1 and the DN mutant of RAB7A was weakened, suggesting that the RMC1-containing GEF complex does not regulate RAB7A activity during mitophagy (Figure 2A). Notably, however, we observed increased proximity labeling of C5orf51 with the DN mutant of RAB7A. Our findings are consistent with Yamano et al, who identified C5orf51 as a candidate interacting partner of RAB7A (T22N) during mitophagy [12]. For these reasons, C5orf51 was a highly ranked candidate for our subsequent functional studies.
Identification of C5orf51 as a component of the MON1-CCZ1 GEF complex for RAB7A during mitophagy
The C5orf51-RAB7A interaction detected by miniTurbo was validated via immunoprecipitation experiments performed using HEK 293T cells expressing GFP-C5orf51 and RFP-RAB7A (Figure 2B). RILP, a known RAB7A effector, was used as a positive control. Consistent with the RAB7A miniTurbo results, C5orf51 was observed to interact with RAB7A, with a distinct preference for the DN mutant (Figure 2B). We also examined this interaction by expressing Flag-C5orf51 in cells alone. Immunoprecipitation of Flag-C5orf51 revealed its interaction with endogenous RAB7A, and this interaction increased upon CCCP treatment (Figure S2A).
Next, we examined a potential interaction between C5orf51 and the MON1-CCZ1 complex, a RAB7A GEF [15]. We performed co-immunoprecipitation experiments using HEK 293T cells transiently expressing GFP-C5orf51 and Flag-MON1A/Flag-CCZ1B. By treating the cells with CCCP, we found that the interaction between C5orf51 and MON1-CCZ1 was enhanced and peaked shortly after mitochondrial depolarization (Figure 2C). To test whether C5orf51 interacts with either MON1 or CCZ1, we performed the immunoprecipitation assay using C5orf51 co-transfected individually with MON1 or CCZ1. We observed that C5orf51 has a weak interaction with CCZ1 but not with MON1 when the proteins are expressed individually (Figure S2B).
To determine whether C5orf51 regulates GEF complex binding with RAB7A, GFP-C5orf51, RFP-RAB7A and Flag-MON1A/Flag-CCZ1B were co-transfected into HEK 293T cells. An increased interaction between the MON1-CCZ1 complex and RAB7A in C5orf51 expressing cells was observed (Figure 2D and Figure S2C). Notably, CCCP treatment further enhanced the interaction between C5orf51 and RAB7A (Figure 2D and Figure S2D).
To investigate if the RAB7A-GEF interaction depends on C5orf51, RFP-RAB7A and HA-MON1A/Flag-CCZ1B were co-transfected into wild type or C5orf51 knockout HEK 293T cells. Our results suggested that C5orf51 was not necessary for RAB7A-GEF interaction (Figure S2E). However, knockdown of MON1-CCZ1 could effectively weaken the interaction between RAB7A and C5orf51 (Figure S2F). Collectively, these data suggest that C5orf51 interacts with GDP-bound RAB7A and promotes its interaction with the MON1-CCZ1 GEF complex after mitochondrial depolarization.
C5orf51 is required for clearance of depolarized mitochondria
To determine whether C5orf51 is required for mitophagy, we performed mitochondria clearance assays with longer CCCP treatment times, as previously described [31,32]. These assays were performed with a HeLa cell line stably expressing GFP-PRKN, a protein that is essential for mitophagy [30,33]. The cells were transfected with constructs expressing two distinct CRISPR-Cas9 sgRNA directed against C5orf51 [34]. Western blot analyses indicated that C5orf51 protein expression was reduced to negligible levels in the respective cell pools, and single cells were isolated and expanded to generate clonal knockout populations (Figure S3A).
We first examined mitochondrial degradation through immunofluorescence analysis of mitochondrial DNA (mtDNA) (Figure S3B). At the early timepoint of CCCP treatment (3 h), the aggregation of damaged mitochondria and the translocation of PRKN were at very similar levels in control or C5orf51 deficient cells (Figure 3A). After 24 h of CCCP treatment, most control cells displayed significant clearance of mitochondria, as indicated by the absence of mtDNA, Mito-DsRed and GFP-PRKN. In the small fraction of control cells that retained mitochondria, these organelles tended to be aggregated, a precursor to mitophagy [29]. However, in C5orf51 deficient cells, the clearance of mitochondria was impaired, despite colocalization with GFP-PRKN and aggregation of these organelles (Figure 3A-C), a phenotype also displayed in RAB7A deficient cells (Figure S3C-E).
Figure 3.
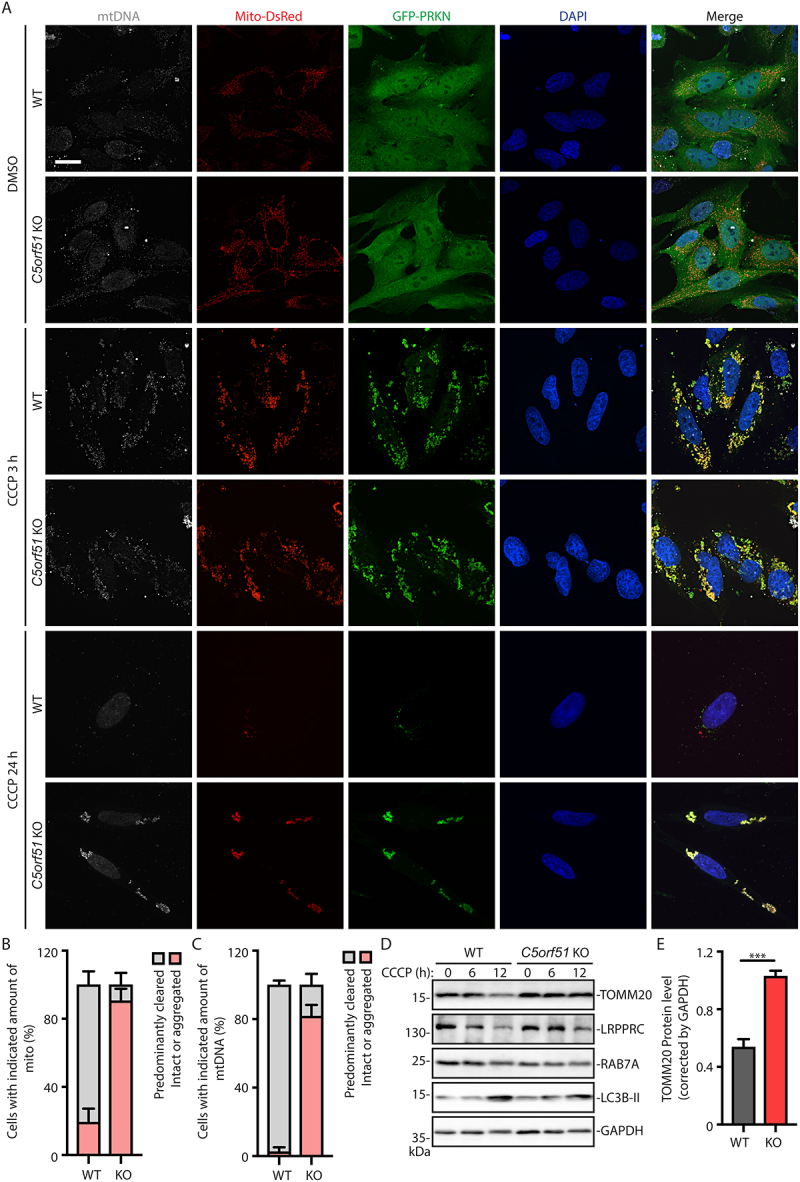
C5orf51 is required for clearance of depolarized mitochondria.
(A) Wildtype and C5orf51 KO HeLa cells stably expressing GFP-PRKN and Mito-DsRed were treated with 20 μM CCCP for the indicated times and subjected to immunostaining with DNA antibody. Size bars: 20 μm. (B) Histogram of the Mito-DsRed signal quantification results (24 h of CCCP treatment) for A. Error bars represent SEM from 3 independent experiments, with >100 cells analyzed per replicate. (C) Histogram of the mtDNA signal quantification results (24 h of CCCP treatment) for A. Error bars represent SEM from 3 independent experiments, with >100 cells analyzed per replicate. (D) Wildtype and C5orf51 KO HeLa cells stably expressing GFP-PRKN and Mito-DsRed were treated with 20 μM CCCP for the indicated time and total cell lysates were analyzed by immunoblotting. (E) Quantification of the TOMM20 (12 h of CCCP treatment) for D. Error bars represent SEM from 3 independent experiments. Statistical differences were determined by Student’s t-test. ***p < 0.001.
We also examined mitochondrial clearance after CCCP treatment using western blot analysis. In control cells, the mitochondrial outer membrane protein TOMM20 (translocase of outer mitochondria membrane 20) and matrix protein LRPPRC (leucine rich pentatricopeptide repeat containing) were efficiently degraded after 12 h of CCCP treatment (Figure 3D, E). However, in C5orf51 deficient cells, protein levels of TOMM20 and LRPPRC remained unaltered after CCCP treatment (Figure 3D, E). These results indicate that C5orf51 plays an important role in mitophagy.
Late endocytic trafficking and lysosome activity is normal in C5orf51 deficient cells
RAB7A regulates late endocytic trafficking to lysosomes [35]. To determine whether C5orf51 is also involved in late endocytic trafficking events, we used DQ-BSA (dye quenched-bovine serum albumin) as a probe to examine endocytic trafficking to degradative lysosomes [36]. In control cells we observed a robust fluorescent signal that was inhibited by bafilomycin A1 (Baf A1), an inhibitor of the vacuolar-type proton-translocating ATPase that is known to block lysosomal degradation [37] (Figure S3F, G). RAB7A knockout cells displayed very weak DQ-BSA fluorescence (Figure S3F, G). However, the fluorescence intensity of C5orf51 deficient cells was similar to control cells (Figure S3F, G). Similarly, overexpression of GFP-C5orf51 had no effect on the DQ-BSA fluorescence, compared with cells expressed GFP (Figure S3H, I). This data indicates that late endocytic trafficking and lysosomal activity is not affected by C5orf51 expression. Thus, we conclude that C5orf51 promotes mitophagy by a mechanism independent of lysosome activity.
C5orf51 is required for RAB7A translocation to depolarized mitochondria
Active Rab7A predominantly localizes to late endosomes and lysosomes [22]. However, during mitophagy a portion of RAB7A is recruited to damaged mitochondria [12,13]. To examine this RAB7A translocation event, we visualized the localization of endogenous RAB7A (detected by antibody staining) during mitophagy. Consistent with previous findings [12,13], we observed clear recruitment of RAB7A to puncta containing the mitochondrial marker Mito-DsRed (Figure S4A).
Under normal growth conditions, GFP-PRKN localized throughout the cytosol and RAB7A did not colocalize with mitochondria (Figure 4A). CCCP treatment induced PRKN recruitment to mitochondria in both control and C5orf51 deficient HeLa cells (Figure 4A, B), indicating effective depolarization of these organelles. In control cells, we observed a significant translocation of endogenous RAB7A to the mitochondria, consistent with previous studies [12]. However, this translocation was impaired in C5orf51 deficient cells (Figure 4A, C). These findings indicate that C5orf51 is required for the translocation of RAB7A to mitochondria during mitophagy.
Figure 4.
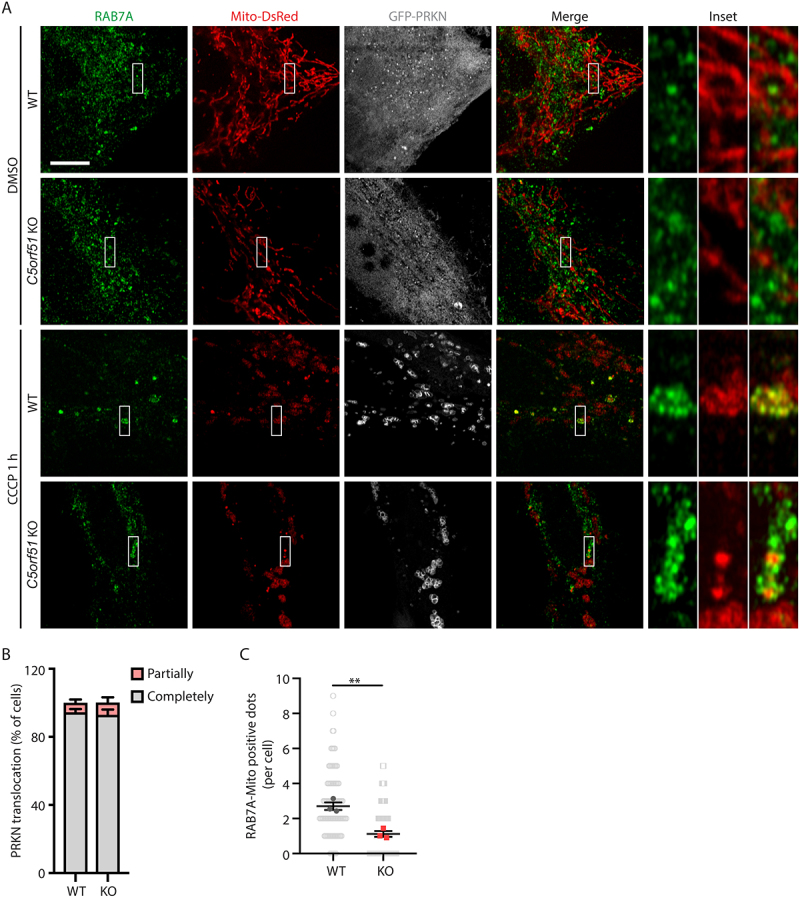
C5orf51 is required for RAB7A translocation to depolarized mitochondria.
(A) Wildtype and C5orf51 KO HeLa cells stably expressing GFP-PRKN and Mito-DsRed were treated with DMSO or CCCP for 1 h and subjected to immunostaining with RAB7A antibody. Size bars: 10 μm. (B) Histogram of the PRKN translocation results for A. Error bars represent SEM from 3 independent experiments, with >50 cells analyzed per replicate. (C) Number of RAB7A-mitochondria positive dots in each cell was counted for A. Each point representing the number of RAB7A-mitochondria positive dots in a cell. Error bars represent SEM from 3 independent experiments, with >30 cells analyzed per replicate. Statistical differences were determined by Student’s t-test. ***p < 0.001.
Next, we examined the localization of C5orf51. The commercially available antibodies to C5orf51 we used for western immunoblotting were found to be unsuitable for immunofluorescence of fixed cells, since a specific signal was not detectable in WT cells compared to C5orf51 KO cells (Figure S4B). As an alternative strategy to visualize C5orf51, we performed live cell imaging using fluorescently labeled C5orf51. We observed that GFP-C5orf51 and RFP-C5orf51 were diffusely localized throughout the cytosol of cells and did not translocate to mitochondria following treatment of cells with CCCP (Figure S4C, D). We conclude that either C5orf51 acts on RAB7A within cytosol or that available reagents are insufficient to detect translocation of C5orf51 during mitophagy.
C5orf51 is required for ATG9A vesicle recruitment to depolarized mitochondria
We also examined ATG9A vesicle assembly on damaged mitochondria, an early step of autophagosome biogenesis during mitophagy that requires RAB7A [11,12]. Under normal growth conditions, ATG9A was mainly localized to the Golgi apparatus in both control and C5orf51 deficient cells, as expected [11,12]. After CCCP treatment, we observed that a fraction of ATG9A was relocated from Golgi to mitochondria in control cells (Figure S4E, F). However, the relocation of ATG9A was significantly impaired and ATG9A retained its association with the Golgi in C5orf51 deficient cells (Figure S4E, F). These findings suggest that C5orf51 promotes mitophagy through RAB7A-mediated translocation of ATG9A vesicles.
C5orf51 promotes stability of RAB7A during mitophagy
RAB7A protein levels were reduced in C5orf51 deficient cells, compared with control cells, and RAB7A protein was dramatically reduced following CCCP treatment in C5orf51 deficient cells (Figure 5A, B). However, RAB7A mRNA levels were similar in control and C5orf51 KO cells, and the RAB7A transcript increased following CCCP treatment (Figure 5C). In addition, expression of Flag-C5orf51 in C5orf51 KO cells could rescue the decrease of RAB7A protein (Figure S5A). RAB7A protein level displayed no difference in MON1-CCZ1 knockdown cells compared with control cells (Figure S5B, C). These results suggested that C5orf51 can specifically act to stabilize the RAB7A protein. Consistent with this possibility, treatment with the proteasome inhibitor carbobenzoxy-Leu-Leu-leucinal (MG132) treatment partially restored RAB7A protein levels in C5orf51 deficient cells under CCCP treatment conditions, while Baf A1 treatment had no effect (Figure 5D). Thus, C5orf51 inhibits the proteasomal degradation of RAB7A during mitophagy.
Figure 5.
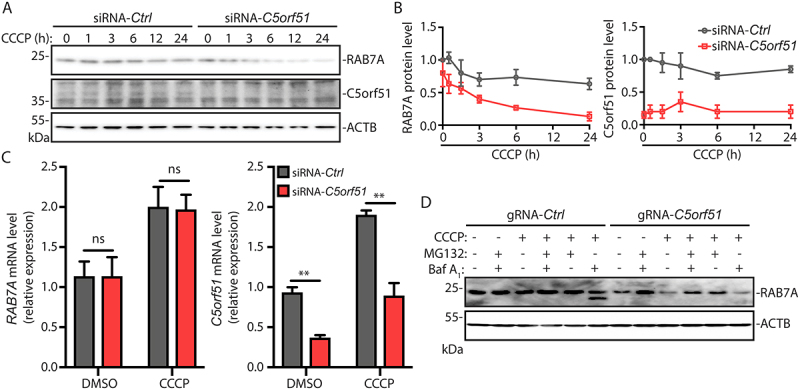
C5orf51 promotes stability of RAB7A during mitophagy.
(A) siRNA treated HEK293T cells were treated with CCCP for the indicated time and total cell lysates were analyzed by immunoblotting, probing for RAB7A, C5orf51 or ACTB. (B) RAB7A and C5orf51 protein level were analyzed for A. The RAB7A or C5orf51 protein densities were calculated using Image Lab 6.0.1 software (Bio-Rad) compared with ACTB and normalized to untreated control cells. Error bars represent SEM from 3 independent experiments. (C) RAB7A and C5orf51 mRNA levels (12 h of DMSO or CCCP treatment) were analyzed by qPCR in siRNA treated HEK293T cells. Error bars represent SEM from 3 independent experiments. Statistical differences were determined by Student’s t-test. **p < 0.01; ns, not significant. (D) MG132 (5 μM) or Baf A1 (0.1 μM) treated HeLa cells were depolarized with CCCP for 24 h. Cell lysates were immunoblotted for total RAB7A and ACTB.
Discussion
Recent studies have shown that RAB7A and its regulators are critical for mitophagy in vertebrates [12,13]. The recruitment of RAB7A to mitochondria is regulated by MON1-CCZ1, components of the RAB7A GEF complex [12]. However, structural analysis revealed that MON1-CCZ1 exists as a heterotetramer, and the GEF complex requires additional factors to coordinate its membrane recruitment [16]. Our studies identify C5orf51 as a novel component of the MON1-CCZ1 complex required for RAB7A translocation to mitochondria during depolarization of this organelle. We also showed that C5orf51 controls the stability of RAB7A following mitochondrial depolarization. Thus, C5orf51 may also act as a chaperone in the RAB7A activation cycle, similar to the way RABIF stabilizes RAB10 [38]. The mechanisms by which C5orf51 promotes RAB7A translocation and stability will be an important topic for future studies.
RMC1 was previously shown to regulate lysosome function [17]. However, we show that C5orf51 does not impact lysosome function under normal growth conditions (Figure S3 F-I). These findings highlight the specificity of C5orf51 to control RAB7A function during mitophagy but not endocytic traffic under normal growth conditions and imply specific modes of regulation of these degradation pathways. Furthermore, our miniTurbo studies identified many other RAB7A-proximal proteins lacking functional annotations. It will be important to determine how these interacting proteins contribute to RAB7A function in different cellular processes and organelle sites.
As the RAB7A gene is implicated in multiple diseases [39–41], mitophagy could represent an important therapeutic target. For example, mitochondrial integrity is disrupted in Charcot-Marie Tooth syndrome type 2B, a disease associated with RAB7A mutations that affects peripheral neurons [42]. It is also noteworthy that C5orf51 was identified as a potential susceptibility gene for amyloid-β accumulation in Alzheimer’s Disease using an integrated analysis of human genome wide association study (GWAS) data and mouse transcriptomic analysis [43]. Future work on C5orf51 in relevant animal models will help to define the molecular basis of RAB7A regulation and its role in disease pathogenesis.
Materials and methods
Cell culture and transfection
HeLa and HEK 293T were obtained from the American Type Culture Collection (HeLa, CCL-2; HEK 293T, CRL-11268). All cells used were authenticated and tested negative for mycoplasma by ATCC and The Hospital for Sick Children Biobank. Cell cultures were maintained in growth medium (DMEM, high glucose (HyClone, 319–005-CL), supplemented with 10% fetal bovine serum (FBS; Wisent, 080–450)) at 37°C in 5% CO2. HeLa cells stably expressing GFP-PRKN and Mito-DsRed (GPRM2) were generously provided by Dr. Richard J Youle (NIH, NINDS) and previously described [44,45]. RAB7A−/- and C5orf51−/- cells were generated by the CRISPR-Cas9 technique following the protocol adapted from the Feng Zhang lab [46]. Briefly, cells were seeded in 6-well tissue culture plates and transfected with control Cas9 plasmid and indicated Cas9 plasmids separately. After 24 h, cells were trypsinized and resuspended in growth media containing puromycin. After puromycin selection, remaining cells were expanded, and the knockout efficiency was examined by sequencing or western blots.
For plasmid transfection, Xtreme Gene 9 (Roche, 6365787001) and GeneJuice (Millipore, 709674) transfection reagents were used as per manufacturer’s protocols. For siRNA-mediated knockdown, cells were seeded at a concentration of 2.5 × 104 cells per well 24 h before use. Cells were transfected with 100 nM siRNA using Lipofectamine RNAiMax (Invitrogen, LS13778150) as recommended by the manufacturer for 48 h.
Microscopy and analysis
Immunostaining was conducted as previously described [47]. Briefly, cells were permeabilized and blocked in PBS (Wisent, 311–010-CL) containing 0.2% saponin (Calbiochem, 558255) or Triton X-100 (Bio-Rad, 1610407) and 10% normal goat serum (Wisent, 053–150) for 30 min. Subsequently cells were incubated for 1 h with primary antibodies in SS-PBS. Cells were washed three times with PBS and incubated with secondary Alexa Fluor-conjugated antibodies for 1 h. Cells were washed three times with PBS, mounted in fluorescence mounting medium (Dako, S302380-2). Unless otherwise indicated, cells were imaged using a Quorum spinning disk microscope with a 63× oil immersion objective (Leica DMIRE2 inverted fluorescence microscope equipped with a Hamamatsu Back-Thinned EM-CCD camera or Hamamatsu CMOS FL-400 camera, spinning disk confocal scan head) and Volocity 6.3 acquisition software (Improvision). In Super-resolution imaging, cells were visualized using a Zeiss LSM 880 confocal microscope (Zeiss) equipped with an Airyscan detector, and a 63× oil immersion objective (1.4 numerical aperture; Zeiss). Images were acquired in the Airyscan mode by ZEN Black software (Zeiss) and processed with ImageJ 1.48 software (National Institutes of Health, Bethesda, MD). Confocal images were analyzed with Volocity 6.3 software and Fiji. Each image was processed uniformly, then assembled in Adobe Illustrator for labeling.
Co-immunoprecipitation and immunoblot analysis
For in vivo Co-IP experiments, HEK 293T cells were seeded into 10-cm diameter tissue culture dishes at approximately 10% confluency and grown at 37°C in 5% CO2. After 24 h or 48 h, cells were cotransfected with epitope-tagged plasmids harboring the candidate host target and either the effector of interest or a control plasmid (6 μg total DNA) for 48 h or 24 h, respectively. Cells were washed in PBS and lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) supplemented with 1 mM phenylmethyl sulfonyl fluoride, 5 mM NaF, 5 mM NaVO4, 10 μg ml−1 aprotinin, 10 μg ml−1 leupeptin and 1 μM pepstatin A. Cell lysates were incubated for 3 h with either GFP-Trap (Chromotek, gta-20), RFP-Trap resin (Chromotek, rta-20) or Flag affinity gel (Sigma-Aldrich, F2426) and washed three times in lysis buffer. Elutions were performed using 100 μl of 1× SDS-PAGE loading buffer and boiling for 10 min.
Cell lysates and proteins eluted from Co-IPs were resolved by 12% or 10% SDS-PAGE, transferred to polyvinylidenedifluoride membrane (Bio-Rad, 1620177). Blocking was performed with 5% skimmed milk. Then the membranes were probed with antigen-specific primary antibodies. For all antibody-based analyses, HRP-conjugated secondary antibodies were used. Detection was performed using the Clarity Western ECL Substrate (Bio-Rad, 1705060) or SuperSignal West Femto Maximum Sensitivity Substrate (ThermoFisher Scientific, PI34095).
Quantitative PCR with reverse transcription analysis
RNA extractions were performed using the RNeasy Mini Kit (QIAGEN, 74104), and complementary DNA was subsequently generated using the iScript cDNA Synthesis Kit (Bio-Rad, 1708841). Quantitative PCR analysis was performed on the QuantStudio 3 Real-time PCR system (ThermoFisher Scientific, QuantStudio™ 3–96-Well 0.2 mL Block) using the SsoFast EvaGreen Supermix Kit (Bio-Rad, 1725201).
Biotin-streptavidin affinity purification and mass spectrometry
miniTurbo sample processing
MiniTurbo was conducted as described previously [48], utilizing a shorter biotin labeling time of 30 min. Briefly, MiniTurbo lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.1% SDS, protease inhibitor cocktail (Sigma-Aldrich, P8340), turbonuclease (Sigma-Aldrich, T4330)) was added to frozen cell pellets, incubated with gentle agitation at 4°C for 1 h, briefly sonicated and centrifuged at 16,000 × g for 30 min at 4°C. Supernatants were incubated with 30 µL streptavidin-sepharose beads (GE Healthcare, 17–5113-01, Lot-A 10269523, Lot-B 10264650) for 3 h, at 4°C with gentle agitation. Beads were washed with NH4HCO3 (50 mM) prior to overnight digestion with MS grade, TPCK-treated trypsin (1 µg; Promega, V5111) at 37°C. The next morning, additional trypsin (0.5 µg) was added, and beads incubated for an additional 2 h at 37°C. Supernatant was collected and beads rinsed with NH4HCO3 (50 mM). Pooled supernatant and washes were lyophilized. Samples were reconstituted in HCOOH (0.1%), de-salted on C18 columns and lyophilized.
Liquid chromatography – mass spectrometry
Samples were reconstituted in HCOOH (0.1%), loaded on a pre-column (C18 Acclaim PepMapTM 100, 75 µm × 2 cm, 3 µm, 100 Å; ThermoFisher Scientific, 164535) and separated on an analytical column (C18 Acclaim PepMapTM RSLC, 75 µm × 50 cm, 3 µm, 100 Å; ThermoFisher Scientific, 164570) via high performance liquid chromatography (HPLC) over a 120-min reversed-phase gradient (5–30% CH3CN in 0.1% HCOOH) running at 250 nl/min on an EASY-nLC1200 pump in-line with a Q-Exactive HF mass spectrometer (ThermoFisher Scientific, 0726090) operated in positive ion mode ESI. An MS1 ion scan was performed at 60,000 FWHM followed by MS/MS scans (HCD, 15,000 FWHM) of the twenty most intense parent ions (minimum ion count of 1000 for activation). Dynamic exclusion (within 10 ppm) was set for 5 s.
LC-MS data processing
Raw files (.raw) were converted to .mzML format using Proteowizard (v3.0.19311), then searched using X!Tandem (v2013.06.15.1) and Comet (2014.02 rev. 2) against Human RefSeqV104 (containing 36,113 entries). Search parameters specified a parent MS tolerance of 15 ppm and an MS/MS fragment ion tolerance of 0.4 Da, with up to two missed cleavages allowed for trypsin. No fixed modifications were set. Deamidation (NQ), oxidation (M), acetylation (protein N-term) and diglycine (K) were set as variable modifications. Search results were processed through the trans-proteomic pipeline (TPP v4.7), and proteins to which ≥2 unique peptides were assigned, and with an iProphet probability ≥0.9, were considered to be high confidence identifications. For statistical analysis, a Bayesian FDR was assigned to identified proteins using SAINT (v2.5.0; 18 BirA*Flag-only controls compressed to 4).
DQ-Red BSA loading of live cells
Cells seeded in eight-well coverglass chambers were incubated for 3 h in low serum medium (1% FBS) containing DQ-Red BSA (0.1 mg/ml; Invitrogen, D12051), washed with PBS, and incubated in low-serum medium for 3 h [36]. Cells were examined by spinning disk confocal microscopy.
Plasmids, siRNA and primers
The following constructs were used: GFP-RILP [49], pcDNA5-FRT/TO-Flag-MON1A [50], pcDNA5-FRT/TO-Flag-CCZ1B [50], and GFP-PRKN [51]. The following constructs was constructed by inverse PCR mutagenesis: pmRFP-C3-RAB7, pmRFP-C3-RAB7(Q67L), pmRFP-C3-RAB7(T22N) and pEGFP-C1-C5orf51. pSpCas9(BB)-2A-Puro (pX459) was purchased from Addgene (48139; deposited by Feng Zhang lab).
The following pairs of oligos for gRNA were used: RAB7A (#1: 5’ CACCGAGGCGTTCCAGACGATTGCA 3’ and 5’ AAACTGCAATCGTCTGGAACGCCTC 3’, #2: 5’ CACCGGTGCATTCCGTGCAATCGTC 3’ and 5’ AAACGACGATTGCACGGAATGCACC 3’) and C5orf51 (#1: 5’ CACCGTCTTCTAGAACTTTACACAC 3’ and 5’ AAACGTGTGTAAAGTTCTAGAAGAC 3’, #2: 5’ CACCGAGCTACTGGAATGTCTCTCT 3’ and 5’ AAACAGAGAGACATTCCAGTAGCTC 3’) The following siRNAs were purchased from Sigma-Aldrich: RAB7A (00104375), C5orf51 (00369178), MON1A (00019772), MON1B (00177957) and CCZ1/CCZ1B (00243195). Quantitative PCR primer sets: RAB7A (5’ TGATGGTGGATGACAGGCTAGT 3’ and 5’ CGAGAGACTGGAACCGTTCCT 3’), C5orf51 (5’ GCCGCAGTCTCTAGTGTGG 3’ and 5’ CAGCTTCGCTAAGTTGCTGT 3’), MON1A (5’ GGAGGCACTTTCCAGCACTATG 3’ and 5’ CGCACGAATACTACCTTGTAGCC 3’), MON1B (5’ GTGGCTGAGAAGGAGACACTAC 3’ and 5’ CGCAGGAGTTTGGTCACTACCA 3’), CCZ1/CCZ1B (5’ TTGCCGAAGACTGGACAGCATC 3’ and 5’ TGTGCTCTTCTCGGCGAGATTC 3’), ACTB (5’ TCAGAAGGATTCCTATGTGGGCGA 3’ and 5’ CACACGCAGCTCATTGTAGAAGGT 3’) and GAPDH (5’ GGAGCGAGATCCCTCCAAAAT 3’ and 5’ GGCTGTTGTCATACTTCTCATGG 3’).
Reagents
Primary antibodies: mouse monoclonal anti-Flag (clone M2; Sigma-Aldrich, F3165), mouse monoclonal anti-HA (clone 16B12; Covance, MMS-101 R), mouse monoclonal anti-GFP (clone 3E6; Molecular Probes, A11120) and rat monoclonal anti-RFP (clone 5F8; Chromotek, 5f8), mouse monoclonal anti-ACTB/β-actin (clone AC-15; Sigma-Aldrich, A5441), rabbit monoclonal anti-RAB7 (clone D95F2; Cell Signaling Technology, 9367), mouse polyclonal anti-C5orf51 (Abnova, H00285636-B01P), rabbit polyclonal anti-TOMM20 (Santa Cruz Biotechnology, sc11415), mouse monoclonal anti-TOMM20 (clone F10; Santa Cruz Biotechnology, sc11764), rabbit polyclonal anti-LRPPRC (Novus, NBP1-83349), mouse monoclonal anti-DNA (clone AC-30-10; Progen, 61014). HRP-conjugated secondary antibodies: peroxidase-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch, 111–035-144), peroxidase-conjugated goat anti-mouse IgG (Jackson ImmunoResearch, 115–035-146) and peroxidase-conjugated goat anti-rat IgG (Jackson ImmunoResearch, 112–035-003). Fluorescence-dye conjugated secondary antibodies: Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen, A11029), Alexa Fluor 568-conjugated goat anti-mouse IgG (Invitrogen, A11031), Cy5-conjugated goat anti-mouse IgG (Jackson ImmunoResearch, 115–175-166), Alexa Fluor 568-conjugated goat anti-rabbit IgG (Invitrogen, A11036), Alexa Fluor 488-conjugated goat anti-chicken IgG (Invitrogen, A11034). Cell nuclei were stained using 4,6-diamidino-2-phenylindole (DAPI; Invitrogen, D1306) at a concentration of 2 µg/ml. Drugs: Carbonyl cyanide 3-chlorophenylhydrazone (CCCP; Sigma-Aldrich, C2759); MG132 (Sigma-Aldrich, M7449); bafilomycin A1 (Baf A1; Cayman Chemical, 11038).
Statistics
Error bars represent SEM shown in the figures. The P values were calculated with GraphPad Prism v.8.0 using a two-way analysis of variance (ANOVA) with Bonferroni post hoc test, unless otherwise indicated. Where specified, a two-tailed, two-sample, equal-variance Student’s t-test was used. P < 0.05 was determined to be statistically significant. ***P < 0.001; **P value between 0.001 and 0.01; and *P value between 0.01 and 0.05 in the associated figures. Where applicable, not significant represents a comparison that is not statistically significant (P > 0.05).
Supplementary Material
Acknowledgments
J.H.B. holds the Pitblado Chair in Cell Biology. Infrastructure for the Brumell Laboratory was provided by a John Evans Leadership Fund grant from the Canadian Foundation for Innovation and the Ontario Innovation Trust. We thank Paul Paroutis for help with confocal microscopy and Andrew Sydor for critically reading the manuscript. This work was supported by operating grants from the Canadian Institutes of Health Research (FDN#154329 to J.H.B, MOP#119289 to B.R. and PJT156196 to P.K.K). B.R. holds a Canada Research Chair in Proteomics and Molecular Medicine.
Funding Statement
This work was supported by the Canadian Institutes of Health Research [FDN154329]; Canadian Institutes of Health Research [PJT156196]; Canadian Institutes of Health Research [MOP#119289].
Disclosure statement
The authors declare no competing interest.
Data availability statement
High confidence RAB7A proximity interactors (<1% FDR) are listed in Dataset S1. All raw mass spectrometry data have been deposited in the MassIVE repository , as accession ID MSV000086719.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Yang Z, Klionsky DJ.. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010. Sep;12(9):814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Abdrakhmanov A, Gogvadze V, Zhivotovsky B.. To eat or to die: deciphering selective forms of autophagy. Trends Biochem Sci. 2020. Apr;45(4):347–364. [DOI] [PubMed] [Google Scholar]
- [3].Behrends C, Fulda S. Receptor proteins in selective autophagy. Int J Cell Biol. 2012;2012:673290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011. Jan;12(1):9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018. Feb 19;28(4):R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Padman BS, Nguyen TN, Uoselis L, et al. LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat Commun. 2019. Jan 24;10(1):408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Palikaras K, Tavernarakis N. Mitophagy in neurodegeneration and aging. Front Genet. 2012;3:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gkikas I, Palikaras K, Tavernarakis N. The role of mitophagy in innate immunity. Front Immunol. 2018;9:1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Borgia D, Malena A, Spinazzi M, et al. Increased mitophagy in the skeletal muscle of spinal and bulbar muscular atrophy patients. Hum Mol Genet. 2017. Mar 15;26(6):1087–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vara-Perez M, Felipe-Abrio B, Agostinis P. Mitophagy in cancer: a tale of adaptation. Cells. 2019. May 22;8(5):493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jimenez-Orgaz A, Kvainickas A, Nagele H, et al. Control of RAB7 activity and localization through the retromer-TBC1D5 complex enables RAB7-dependent mitophagy. EMBO J. 2018. Jan 17;37(2):235–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yamano K, Wang C, Sarraf SA, et al. Endosomal Rab cycles regulate Parkin-mediated mitophagy. Elife. 2018. Jan 23;7:e31326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Heo JM, Ordureau A, Swarup S, et al. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci Adv. 2018. Nov;4(11):eaav0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yamano K, Fogel AI, Wang C, et al. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife. 2014. Feb 25;3:e01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nordmann M, Cabrera M, Perz A, et al. The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr Biol. 2010. Sep 28;20(18):1654–1659. [DOI] [PubMed] [Google Scholar]
- [16].Kiontke S, Langemeyer L, Kuhlee A, et al. Architecture and mechanism of the late endosomal Rab7-like Ypt7 guanine nucleotide exchange factor complex Mon1-Ccz1. Nat Commun. 2017. Jan 4;8(1):14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vaites LP, Paulo JA, Huttlin EL, et al. Systematic analysis of human cells lacking ATG8 proteins uncovers roles for GABARAPs and the CCZ1/MON1 regulator C18orf8/RMC1 in macroautophagic and selective autophagic flux. Mol Cell Biol. 2018. Jan 1;38(1). DOI: 10.1128/MCB.00392-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Roux KJ, Kim DI, Burke B. BioID: a screen for protein-protein interactions. Curr Protoc Protein Sci. 2013. Nov 5;74(1):19 23 1–19 23 14. [DOI] [PubMed] [Google Scholar]
- [19].D’Costa VM, Coyaud E, Boddy KC, et al. BioID screen of Salmonella type 3 secreted effectors reveals host factors involved in vacuole positioning and stability during infection. Nat Microbiol. 2019. Dec;4(12):2511–2522. [DOI] [PubMed] [Google Scholar]
- [20].Branon TC, Bosch JA, Sanchez AD, et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol. 2018. Oct;36(9):880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Raiborg C, Wenzel EM, Pedersen NM, et al. Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature. 2015. Apr 9;520(7546):234–238. [DOI] [PubMed] [Google Scholar]
- [22].Bucci C, Thomsen P, Nicoziani P, et al. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000. Feb;11(2):467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wong YC, Ysselstein D, Krainc D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature. 2018. Feb 15;554(7692):382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pankiv S, Alemu EA, Brech A, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010. Jan 25;188(2):253–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guillen-Samander A, Bian X, De Camilli P. PDZD8 mediates a Rab7-dependent interaction of the ER with late endosomes and lysosomes. Proc Natl Acad Sci U S A. 2019. Nov 5;116(45):22619–22623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Elbaz-Alon Y, Guo Y, Segev N, et al. PDZD8 interacts with Protrudin and Rab7 at ER-late endosome membrane contact sites associated with mitochondria. Nat Commun. 2020. Jul 20;11(1):3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu K, Xing R, Jian Y, et al. WDR91 is a Rab7 effector required for neuronal development. J Cell Biol. 2017. Oct 2;216(10):3307–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Seaman MN, Harbour ME, Tattersall D, et al. Membrane recruitment of the cargo-selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-GAP TBC1D5. J Cell Sci. 2009. Jul 15;122(Pt 14):2371–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010. Jan 5;107(1):378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Narendra D, Tanaka A, Suen DF, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008. Dec 1;183(5):795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Villa E, Proics E, Rubio-Patino C, et al. Parkin-independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep. 2017. Sep 19;20(12):2846–2859. [DOI] [PubMed] [Google Scholar]
- [32].Correia-Melo C, Ichim G, Tait SW, et al. Depletion of mitochondria in mammalian cells through enforced mitophagy. Nat Protoc. 2017. Jan;12(1):183–194. [DOI] [PubMed] [Google Scholar]
- [33].Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015. Jan 21;85(2):257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang XH, Tee LY, Wang XG, et al. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids. 2015. Nov 17;4:e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Vanlandingham PA, Ceresa BP. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J Biol Chem. 2009. May 1;284(18):12110–12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Marwaha R, Sharma M. DQ-Red BSA trafficking assay in cultured cells to assess cargo delivery to lysosomes. Biol Protoc. 2017. Oct 5;7(19):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yoshimori T, Yamamoto A, Moriyama Y, et al. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991. Sep 15;266(26):17707–17712. [PubMed] [Google Scholar]
- [38].Gulbranson DR, Davis EM, Demmitt BA, et al. RABIF/MSS4 is a Rab-stabilizing holdase chaperone required for GLUT4 exocytosis. Proc Natl Acad Sci U S A. 2017. Sep 26;114(39):E8224–E8233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang M, Chen L, Wang S, et al. Rab7: roles in membrane trafficking and disease. Biosci Rep. 2009. Jun;29(3):193–209. [DOI] [PubMed] [Google Scholar]
- [40].Verhoeven K, De Jonghe P, Coen K, et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet. 2003. Mar;72(3):722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Guerra F, Bucci C. Multiple roles of the small GTPase Rab7. Cells. 2016. Aug 18;5(3):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cioni JM, Lin JQ, Holtermann AV, et al. Late endosomes act as mRNA translation platforms and sustain mitochondria in Axons. Cell. 2019. Jan 10;176(1–2):56–72 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yamaguchi-Kabata Y, Morihara T, Ohara T, et al. Integrated analysis of human genetic association study and mouse transcriptome suggests LBH and SHF genes as novel susceptible genes for amyloid-beta accumulation in Alzheimer’s disease. Hum Genet. 2018. Jul;137(6–7):521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang Y, Serricchio M, Jauregui M, et al. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy. 2015. Apr 3;11(4):595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hasson SA, Kane LA, Yamano K, et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature. 2013. Dec 12;504(7479):291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013. Nov;8(11):2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Czuczman MA, Fattouh R, van Rijn JM, et al. Listeria monocytogenes exploits efferocytosis to promote cell-to-cell spread. Nature. 2014. May 8;509(7499):230–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gupta GD, Coyaud E, Goncalves J, et al. A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell. 2015. Dec 3;163(6):1484–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Harrison RE, Bucci C, Vieira OV, et al. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol. 2003. Sep;23(18):6494–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kinchen JM, Ravichandran KS. Identification of two evolutionarily conserved genes regulating processing of engulfed apoptotic cells. Nature. 2010. Apr 1;464(7289):778–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang Y, Nartiss Y, Steipe B, et al. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy. 2012. Oct;8(10):1462–1476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High confidence RAB7A proximity interactors (<1% FDR) are listed in Dataset S1. All raw mass spectrometry data have been deposited in the MassIVE repository , as accession ID MSV000086719.