

Abstract
Elastin is a long-lived extracellular matrix protein that is organized into elastic fibers that provide elasticity to the arterial wall, allowing stretch and recoil with each cardiac cycle. By forming lamellar units with smooth muscle cells, elastic fibers transduce tissue-level mechanics to cell-level changes through mechanobiological signaling. Altered amounts or assembly of elastic fibers leads to changes in arterial structure and mechanical behavior that compromise cardiovascular function. In particular, genetic mutations in the elastin gene (ELN) that reduce elastin protein levels are associated with focal arterial stenosis, or narrowing of the arterial lumen, such as that seen in supravalvular aortic stenosis and Williams–Beuren syndrome. Global reduction of Eln levels in mice allows investigation of the tissue- and cell-level arterial mechanical changes and associated alterations in smooth muscle cell phenotype that may contribute to stenosis formation. A loxP-floxed Eln allele in mice highlights cell type- and developmental origin-specific mechanobiological effects of reduced elastin amounts. Eln production is required in distinct cell types for elastic layer formation in different parts of the mouse vasculature. Eln deletion in smooth muscle cells from different developmental origins in the ascending aorta leads to characteristic patterns of vascular stenosis and neointima. Dissecting the mechanobiological signaling associated with local Eln depletion and subsequent smooth muscle cell response may help develop new therapeutic interventions for elastin-related diseases.
Keywords: extracellular matrix, mechanobiology, neointima, smooth muscle cell, vascular stenosis
INTRODUCTION
Elastic arteries expand with applied hemodynamic load and recoil to their original configuration with the transfer of stored strain energy. This “Windkessel effect” dampens the pulsatile blood flow generated by the heart during systole and efficiently translates the energy into near constant blood flow during diastole to perfuse downstream organs. Reduced arterial elasticity causes elevated blood pressure and reduced pulse dampening, which places undue strain on the heart and downstream target organs.
Arterial elasticity is conferred by elastic fibers whose elastic properties come from cross-linked elastin. Elastin evolutionarily emerges with the advent of the closed circulatory system in vertebrates (1). The production of elastin occurs in a small developmental time window, peaking in late gestation to early postnatal life, concurrent with the rise of blood pressure and flow during development (2). Once blood pressure and flow plateau after birth, new elastin production is drastically reduced whereby there is minimal elastin synthesis during adulthood (2). The existing elastic fibers are long-lived with an estimated half-life of ∼70 years (3). Once elastic fibers are compromised by aging, injury, or disease they do not fully regenerate (4).
Elastin is produced by the ELN gene in humans, with 34 exons spanning a 45 kb region in chromosome 7. ELN encodes tropoelastin (precursor to elastin) that contains alternating hydrophobic and cross-link domains (5). Once secreted to the extracellular space, tropoelastin undergoes coacervation, or self-assembly into globular aggregates based on hydrophobicity (6). The lysine residues in tropoelastin are modified by lysyl oxidases to form extensive covalent cross links within and between molecules (7). Coacervation and cross linking triggers elastin assembly into elastic fibers on a scaffold composed of microfibrils (6, 8). The assembled elastin then undergoes final cross linking to form mature elastic fibers (8).
Given the functional importance of elastic fibers, it is not surprising that altered amounts or assembly may lead to human diseases. Some of the diseases associated with ELN gene mutations feature arterial stenosis, such as supravalvular aortic stenosis (SVAS), which occurs either as an isolated condition or as part of Williams–Beuren syndrome (WBS) (9). In this review, we provide a brief overview of the role of elastin in arterial mechanics and stenosis from studies of congenital human disease and mouse models. We highlight future research areas and possible therapeutic approaches.
ELASTIN AND ARTERIAL MECHANICS
The role of elastin in arterial mechanics has been well established through studies examining arteries with different wall structures and extracellular matrix (ECM) amounts, protease applications to remove specific wall components, and genetic studies to alter elastic fiber amounts, assembly, and organization. Additional detailed reviews can be found in Refs. 10–12. In this section, we briefly review the role of elastin in arterial mechanics, with a focus on tissue- and cell-level effects of altered elastin amounts.
Tissue-Level Mechanical Behavior
The mechanical properties of the arterial wall are determined by its three main layers. The innermost layer, tunica intima, is a single layer of endothelial cells (ECs) in direct contact with circulating blood. The outermost layer, tunica adventitia, is a loose structure containing fibroblasts, progenitor cells, and a collagen-rich ECM. The middle layer, tunica media, contains layers of smooth muscle cells (SMCs) separated by circumferential sheets of elastic fibers (elastic laminae), together forming lamellar units. The number of lamellar units in the tunica media (and hence its thickness) varies along the arterial vasculature, with ∼60 units (layers) in the human aorta and <3 units in the smaller, peripheral arteries (13). Accordingly, the elastin content decreases along the length of the vasculature (14). The number of units corresponds with local vessel size, maintaining a constant wall tension per lamellar unit (15). An internal elastic lamina (IEL) separates the tunica intima and media, whereas the external elastic lamina (EEL) demarks the border between the tunica media and adventitia.
The arterial wall possesses nonlinear mechanical properties, significantly stiffening at high pressure to prevent over-distension (16). In this review, “material stiffness” and “structural stiffness” refer to the geometry-independent and geometry-dependent resistance to stretch, respectively. Material stiffness can be approximated by the slope of the circumferential stretch-stress curve, whereas structural stiffness can be approximated by the slope of the diameter-pressure curve. The nonlinear mechanical properties of the arterial wall arise mainly from elastic fibers and collagen fibers, responsible for stretch resistance at sub- and supraphysiologic pressures, respectively, as indicated by selective protease digestion experiments (16–19). In line with the elastase digestion results, genetic ablation of elastin (Eln−/−) in mice leads to reduced aortic circumferential material stiffness at (20) and below (21) physiologic pressures. Consistent with the role of elastic fibers in transferring stored strain energy, Eln−/− aorta returns only 25% of the stored energy of a control aorta under similar in vitro loading protocols. The reduction in stored energy leads to the Eln−/− aorta behaving as a viscoelastic material, rather than an elastic material (21). Alterations in the material and structural stiffness and viscoelasticity of the aorta will affect its performance as a Windkessel and the hemodynamic forces on distal organs.
Although SMCs may affect vascular tone by active contraction or relaxation, they contribute minimally to the passive mechanical behavior of large elastic arteries. The main role of SMCs in passive arterial mechanics is deposition, maintenance, and remodeling of the ECM in development and disease. In 1-wk-old mouse aorta, the most highly expressed Reactome pathways for SMCs and fibroblasts are associated with ECM synthesis, assembly, cross linking, organization, and interaction (22). Dysregulation of these ECM pathways is associated with cardiovascular diseases including atherosclerosis, aneurysms, and stenosis.
Cell-Level Mechanical Behavior
The elastic laminae and associated ECM proteins determine the physical microenvironment surrounding SMCs in the arterial wall. ECM deposition after birth serves to stress-shield the SMCs as hemodynamic forces increase (23, 24). The circumferential stress carrying capacity of isolated SMCs is ∼10–15 kPa (25) or 30% of the physiological wall stress in newborn mouse aorta (20) and 3% of the physiological wall stress in adult mouse aorta (26). If elastin is missing, reduced, or compromised, stress will be transferred to the SMCs. Although the outcomes of stress-mediated remodeling in the arterial wall are well-studied (23, 27), the SMC mechanosensors are still unknown. Candidates include integrin complexes, G protein-coupled receptors, transient receptor potential (Trp) channels, Piezo channels, and epithelial sodium channels (ENaC) (28).
Stress transfer to the SMCs stimulates phenotypic changes in the cells and remodeling of the arterial wall, typically characterized by deposition of additional ECM proteins (29). Since functional elastic fibers are not fully regenerated, the additional ECM proteins are usually collagen and proteoglycans that serve to alter the arterial mechanical behavior. Abundant collagen deposition causes fibrosis that is common to many diseases and serves to increase the material stiffness of the tissue and surrounding microenvironment. The material stiffness of the underlying microenvironment dictates differentiation of stem cells (30), and in SMCs it regulates contractility (31), migration (32), proliferation (33), and RNA expression (34). Changes in viscoelasticity, in addition to substrate material stiffness, affect stem cell differentiation (35), spreading (36), and signaling activity (37). SMCs in Eln−/− aorta have decreased expression of contractile proteins, increased proliferation and migration (38), and a decreased material stiffness (39), although it is not clear which changes are caused by the absence of soluble tropoelastin signaling compared with changes in the ECM microenvironment (40). Dissecting out these mechanisms will be important for a better understanding of SMC phenotypic changes in elastin-related diseases.
ELASTIN AND ARTERIAL STENOSIS
Human genetics and mouse models have clearly established elastin deficiency as the cause of some arterial stenotic diseases. In this section, we review the insights obtained from studies of human congenital diseases and mouse genetic models with reduced elastin amounts. Additional in-depth reviews can be found in Ref. 41.
Human Diseases
Supravalvular aortic stenosis and Williams–Beuren syndrome.
Supravalvular aortic stenosis (SVAS; OMIM 185500) is an autosomal dominant genetic disease that affects an estimated ∼1:20,000 live births. SVAS is characterized by congenital stenosis of the ascending aorta and other arteries, such as the pulmonary or coronary arteries while sparing the cerebral circulation, descending aorta, renal arteries, and other aortic branches (42). Histologically, the stenosis results from nonatherosclerotic thickening of the tunica media and neointimal formation (43) from SMC hyperproliferation. Increased collagen deposition and thin/disorganized elastic fibers can be observed in the tunica media. Perhaps as a compensatory mechanism, there are more elastic lamellar units in the tunica media (43).
SVAS can present as a nonsyndromic entity but is commonly associated with Williams–Beuren syndrome (WBS; OMIM 194050) that includes neurobehavioral, craniofacial, metabolic, and cardiovascular phenotypes (9). Vascular stenosis is a prominent feature of WBS and may affect the ascending aorta, aortic arch, descending aorta, peripheral pulmonary, coronary, renal, mesenteric, and intracranial arteries (9). A common feature in patients with WBS is hypertension, attributed to increased vascular stenosis and/or structural stiffness (44). The arterial stenotic features of SVAS and WBS are consequent to reduced elastin protein level. Genetically, SVAS results from loss-of-function mutations of the ELN gene, whereas WBS originates from a microdeletion of 7q11.23 that spans ∼27 genes including ELN. De novo gene alterations are more common than inherited mutations (45). Generally, there remains one functional copy of ELN in WBS or SVAS, suggesting that ELN haploinsufficiency represents the inheritance pattern.
Mouse Models of Elastin Disease
The autosomal dominant inheritance pattern of SVAS and WBS implies a role for ELN haploinsufficiency in these diseases. None of the existing mouse models recapitulate every aspect of the symptomatology of these conditions (Table 1). Some differences in human and mouse phenotypes may be partly due to the gene structure. The human ELN gene lost 2 exons during evolution and also harbors unique alternative splicing patterns (51), resulting in the 34 exons in human (52) compared with 37 in mouse. Despite the differences, the mouse models have delivered important insights into pathogenic mechanisms of arterial stenosis in these diseases.
Table 1.
Summary of genetic mouse models targeted to reduce or eliminate elastin expression
| Mouse Model | Elastin Levels and/or Localization | Cardiovascular Phenotype | Neointima and/or Stenosis | Refs. |
|---|---|---|---|---|
| Eln −/− | No elastin | Perinatal death, increased SMC proliferation migration, increased aortic thickness, and structural stiffness. | Stenosis, affecting multiple arteries. | (46) |
| Eln +/− | 60% elastin | Normal lifespan, hypertension, smaller arteries with increased structural stiffness, disorganized elastin, increased lamellar units. | No | (47) |
| WBS | 60% elastin | Normal lifespan, hypertension, smaller arteries with increased structural stiffness, but no increase in lamellar units. | No | (48) |
| hBAC-mNull | 30% elastin | Exaggeration of Eln+/− phenotype, hypertension, smaller, stiffer, thicker arteries with more lamellar units. | No | (49) |
| ElnSMHet (TaglnCre;Elnf/+) | Presumably ∼50% | Similar phenotype to Eln+/−, but not hypertensive at 1 mo of age. Normal lifespan, increased structural aortic stiffness, and lamellar units. | No | (50) |
| ElnSMKO (TaglnCre;Elnf/f) | 10%, localized to IEL | Die ∼2 wk of age. Severe cardiac hypertrophy. Fragmented IEL in ascending aorta, but intact IEL in all other arteries. | Neointima in ascending aorta (Fig. 6), focal stenosis in arch (Fig. 1). | (50) |
| ElnECKO (Tie2Cre;Elnf/f and Cdh5Cre;Elnf/f) | Presumably 100% in elastic arteries | Normal lifespan. No hypertension. Elastic arteries appear normal. Resistance arteries have thin, fragmented IEL. | No | (50) |
| ElnSHFKO (Isl1Cre;Elnf/f) | Localized to the inner part of the ascending aortic wall | Die at 2–3 mo of age. Fragmented IEL in ascending aorta. | Neointima in ascending aorta (Fig. 6), no stenosis (Fig. 1) | (22) |
| ElnCNCKO (Wnt1Cre;Elnf/f) | Localized to the IEL and outer part of the ascending aortic wall | Die at 2–3 mo of age. IEL mostly intact. | Minimal neointima in ascending aorta (Fig. 6), focal stenosis in arch (Fig. 1) | (22) |
IEL, internal elastic lamina; SMC, smooth muscle cell; WBS, Williams–Beuren syndrome
Eln−/− mice.
Eln−/− mice represent an extreme form of elastinopathy generally not observed in human patients. These mice die within 1–3 days after birth. At postnatal day (P) 0, their arteries are tortuous and diffusely stenotic (Table 1). By P1, extreme stenosis of the descending aorta as well as aortic arch arteries can be seen by angiography (53). Histologically, the aortic stenosis in Eln−/− mice results from tunica media hyperplasia (46). Prenatal examination at embryonic day (E) 18 shows patent aorta (54), indicating that the aortic stenosis progresses in a brief perinatal window. Eln−/− mice have normal blood pressure at E18, but elevated blood pressure at birth (53). Their aortic structural stiffness, however, is already elevated by E18 and remains so at birth (53, 54). It is noteworthy that the blood pressure increases occur simultaneously with stenotic remodeling of the aortic wall, whereas the changes in structural stiffness precede blood pressure elevation, implying a causal relationship. Increased blood pressure during development increases the circumferential wall stress and the requirement for stress shielding of SMCs by ECM deposition.
Mechanical characterization and gene expression analyses of Eln−/− aorta have been performed. Mechanical characterization shows decreased material stiffness (20) and increased viscoelastic behavior (21) of Eln−/− aorta compared with control that alter the mechanical microenvironment surrounding the SMCs. Decreased material stiffness would increase strain on SMCs at the same stress level, whereas increased viscoelastic behavior would increase the phase lag between stress and strain and could alter the time-dependent SMC biological response. Gene set enrichment analyses reveal upregulation of ECM-, integrin-, and syndecan-associated terms in Eln−/− compared with control aorta, including “integrin cell surface interactions,” “ECM receptor interaction,” “syndecan 1 pathway,” and “syndecan 4 pathway,” among others. Syndecan-1 and -4 are transmembrane proteoglycans that act as co-receptors for growth factors and cell-ECM and cell-cell interactions. As syndecan-1 and syndecan-4 expression in SMCs are stimulated by mechanical strain (55, 56), these pathways may mediate the transduction of altered mechanical stress and/or strain into dysregulated molecular pathways.
The SMC mechanical environment can also be altered by changes in the applied hemodynamic forces. Blood pressure lowering during development, achieved by captopril delivered in drinking water to the pregnant dam, lowers blood pressure and the resulting circumferential aortic wall stress in newborn Eln−/− mice (57). The prenatal blood pressure lowering leads to decreased wall thickness and alterations in structural and material stiffness of newborn Eln−/− aorta, independent of changes in ECM protein amounts. Furthermore, the SMCs in captopril-treated Eln−/− aorta appear more organized compared with untreated Eln−/−, suggesting a response to altered tissue- or cell-level mechanics in the presence of reduced blood pressure rather than elastin signaling. It is not clear whether prenatal blood pressure lowering with captopril may increase survival in Eln−/− mice.
SMCs from Eln−/− mice have higher proliferation rates (38), which can be reversed by rapamycin, an inhibitor of the cell growth regulator mammalian target of rapamycin (mTOR), although rapamycin treatment does not extend the lifespan of Eln−/− mice (58). Another pathway implicated in the stenosis pathogenesis is integrin β3 signaling, which is increased in the aorta of patients with SVAS and Eln−/− mice. Pharmacological inhibition of integrin β3 reduces aortic stenosis, whereas genetic inactivation of integrin β3 increases the lifespan of Eln−/− mice by ∼2 days (59). Integrin complexes are putative mechanosensors (28) and may be overexpressed or activated due to alterations in the mechanical environment of Eln−/− aorta. There is evidence that the Notch signaling pathway is also regulated directly or indirectly by mechanical forces (60). Jagged1/Notch3 signaling is upregulated in SMCs and aortic samples from patients with SVAS/WBS and Eln−/− mice, and aortic stenosis in Eln−/− mice can be mitigated by inhibition of the Jagged1/Notch3 signaling pathway (61).
Eln+/− mice.
Eln+/− mice recapitulate some aspects of the SVAS/WBS symptomatology, including increased blood pressure, increased structural arterial stiffness, and a higher number of elastic lamellae in the tunica media (47, 62). However, they have a normal lifespan and do not have focal arterial stenosis (Table 1). Mice with reduced elastin due to a large genetic deletion that mimics the heterozygous deletion observed in WBS (48) have hypertension and increased structural arterial stiffness, but do not have the increased number of lamellar units observed in Eln+/− mice (63). During postnatal development, Eln+/− aortas exhibit decreased diameter and increased structural stiffness before their increased blood pressure compared with controls, hinting at a causative effect (64) that is consistent with the results in Eln−/− aorta during embryonic development (65).
Eln+/− ascending aorta shows interesting developmental remodeling suggesting that it has a different homeostatic set point for circumferential wall stretch, but is able to maintain wall stress and material stiffness near normal levels, despite the observed hypertension. In particular, Eln+/− aorta has increased circumferential stretch throughout postnatal development (64) and aging (66), but circumferential stress and material stiffness are similar to control at most time points. A component-based constitutive model reveals that the stress levels may be maintained by increased stress contributions from the collagen fibers that are not due to increased collagen amounts, but could be caused by altered collagen fiber alignment (67). Evaluation of the material stiffness in aorta from a variety of species (19), including control and Eln+/− mice (23), illustrates a universal value under physiologic conditions that would maintain a constant change in stress for a given physiologic change in strain. Hawes et al. (66) found an increase in physiologic circumferential stress and material stiffness with aging in both Eln+/− and control aorta with interactions between independent variables (age, sex, and genotype), demonstrating that sex and elastin amounts affect the timeline of cardiovascular remodeling with respect to age. Pezet et al. (68) report an alternative aging process in Eln+/− aorta compared with control, consistent with our statistical interactions, however they found a significant increase in material stiffness with age specifically in Eln+/− aorta and not in control. Part of the discrepancies may be explained by differences in experimental technique, differences in equations used to calculate stress, strain, and material stiffness, and the dependence of the calculations on the wall thickness, which is challenging to measure accurately in the mouse aorta.
hBAC-mNull mice.
The phenotypic differences between Eln+/− mice and human patients with SVAS/WBS with ELN haploinsufficiency may partly be due to elastin protein doses. Elastin protein level in patients with WBS is ∼15% of control subjects, whereas Eln+/− mice have ∼50%–60% elastin level compared with Eln+/+ control mice, despite both having one intact allele (43). In an attempt to recapitulate the human condition, a humanized hBAC allele was created by insertion of the human ELN gene into a bacterial artificial chromosome (BAC) (49). The hBAC allele, when crossed into the Eln−/− (mNull) background, expresses human elastin protein at ∼30% of control elastin amounts. hBAC-mNull mice have hypertension and increased arterial structural stiffness. As would be predicted from the elastin levels, hBAC-mNull mice have a more severe phenotype than Eln+/− mice, with higher blood pressure, thicker and stiffer arteries, and more secondary cardiac hypertrophy (Table 1). Although focal aortic stenosis is not observed in hBAC-mNull mice, their aortas have decreased circumferential growth and increased axial growth, which in combination lead to normal medial cross-sectional area and a ∼70% reduction in ascending aortic lumen area compared with control mice (69).
Cell type-specific Eln knockout mice.
In the arterial wall, elastin is primarily produced from SMCs in the tunica media (70, 71), although it can also be produced by ECs in the intima (72, 73) and fibroblasts in the adventitia (74). The role of elastin in these cell types and their contribution to tissue- and cell-level arterial mechanics are beginning to be understood. A loxP-floxed allele (Elnf) allows conditional inactivation of Eln in a cell-specific manner. We summarize recent results in mice where elastin has been reduced (Het) or eliminated (knockout, KO) in SMCs (ElnSMHet; ElnSMKO), ECs (ElnECKO), and secondary heart field- and cardiac neural crest-derived (ElnSHFKO; ElnCNCKO) cells.
Smooth muscle cell-specific Eln heterozygous and knockout mice.
TaglnCre mice (75) were used to target Eln in SMCs. Similar to Eln+/− mice, ElnSMHet (TaglnCre;Elnf/+) mice are indistinguishable from controls in appearance, are fertile, and have a normal lifespan (50). ElnSMHet aortas have a slightly thicker tunica media and more circumferential elastic laminae. They also have reduced diameter and increased structural stiffness in the carotid artery, showing that Eln expression from SMCs is responsible for most of the changes in the Eln+/− arterial wall (Table 1). However, unlike Eln+/− mice, ElnSMHet mice do not exhibit systolic hypertension at 1 mo of age, suggesting a delayed increase in blood pressure or that elastin in other cell types may contribute to blood pressure regulation.
ElnSMKO mice (50) (TaglnCre;Elnf/f) are born at the expected Mendelian ratio but die prematurely by 2 wk of age. They develop severe stenosis in the aorta (Fig. 1A) due to tunica media hyperplasia and neointimal formation (Fig. 1B), similar to patients with SVAS (Table 1). ElnSMKO mice develop cardiac phenotypes attributed to the aortic stenosis, including compromised fractional shortening, cardiac enlargement, thrombosis, necrosis, and calcification in the cardiac chambers. In addition to the coarctation, the arterial vasculature in the ElnSMKO mice shows tortuosity, most prominently in the aortic arch arteries (Fig. 1A), similar to Eln−/− mice (53).
Figure 1.

Stenosis of the aorta in elastin conditional knockout mouse models. A: latex angiography in mouse mutants with Eln specifically deleted in smooth muscle cell (SMC) (ElnSMKO), secondary heart field (SHF) (ElnSHFKO), and cardiac neural crest (CNC) (ElnCNCKO). Note the presence (red arrows) and absence (white arrow) of stenosis in the different mouse models. Scale = 0.5 mm. B: histology of aortic stenosis. Verhoeff-Van Gieson staining shows neointima (red arrows) in ElnSMKO aortic stenosis, likely due to the fragmented internal elastic lamina (IEL). Tunica media hypertrophy (black arrowheads) is observed in both ElnSMKO and ElnCNCKO aorta. Black arrows point to the IEL. Scale = 50 µm. Adapted from Refs. 22 and 50.
Histologically, ElnSMKO arteries lack elastic layers in the tunica media, indicating that the medial elastic lamellae are exclusively made by SMCs (50). We performed pressure-diameter tests on newborn ElnSMKO (n = 4) ascending aorta and control (n = 6), as we have done previously on newborn (53) and late embryonic (54) Eln−/− aorta, and found no differences in outer diameter at each pressure, but an 83%–226% increase in structural stiffness at pressures between 20 and 40 mmHg (Fig. 2) using a two-tailed t test with unequal variance. Structural stiffness was calculated as the slope of the diameter-pressure curve. The systolic blood pressure in newborn mice is ∼30 mmHg (53, 57). The increases in structural stiffness in the ElnSMKO aorta are consistent with those previously observed in Eln−/− aorta (53, 54) and demonstrate mechanically induced remodeling of the aortic wall when elastin is absent in the medial layer.
Figure 2.

Aortic mechanical behavior in elastin conditional knockout mouse model. Diameter-pressure (A) and pressure-structural stiffness (B) behavior of postnatal day 1 (P1) control (Elnf/f) and ElnSMKO ascending aorta. Structural stiffness is the local slope of the diameter-pressure curve and is increased in ElnSMKO aorta between 20 and 40 mmHg. n = 6 (control) and 4 (ElnSMKO). Means ± SD. P values from two-tailed Student’s t test with unequal variance.
The EEL is largely absent in ElnSMKO arteries (50), so the EEL cannot be made by adventitial fibroblasts and/or non-Tagln expressing SMCs in the outer wall alone. The IEL is intact in the smaller elastic arteries, muscular, and resistance arteries, so ECs can make this elastic layer. However, in the ascending aorta, the IEL is fragmented. Therefore, SMCs are required for IEL formation in the ascending aorta. Furthermore, this finding reveals that the relative cellular contributions to the IEL are different in different parts of the vasculature tree (Fig. 3).
Figure 3.
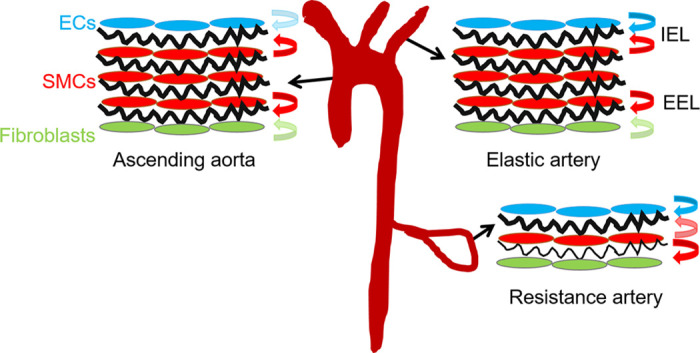
Cellular contributions to the internal (IEL) and external (EEL) elastic laminae differs throughout the vascular tree. In the ascending aorta, the IEL is primarily made by smooth muscle cells (SMCs). In other elastic arteries it is made by both endothelial cells (ECs) and SMCs, while in the resistance arteries it is mainly made by ECs. Cellular contributions are shown by colored arrows, with minor contributions shown in lighter colors. In all arteries, SMCs are the main cell type contributing to the EEL, however there is a minor contribution from fibroblasts in the ascending aorta and elastic arteries. Elastic laminae are shown in black.
The fragmented IEL in the ascending aorta is associated with neointimal formation in ElnSMKO mice (50). The IEL serves as both an attachment substrate for and a physical barrier between SMCs and ECs. Myoendothelial junctions between ECs and SMCs through gaps in the IEL serve as direct (76) and indirect (77) communication pathways between cell types. Increased gaps due to fragmentation may allow increased signaling between these cell types that contribute to disease (78). We performed ligand receptor analysis on our previously published single-cell RNA-sequencing data of ElnSMKO ascending aorta (22) following the methods of Farbehi et al. (79). Cell-cell interaction networks were constructed with weighted edges reflecting expression fold changes of ligands and receptors in source and target populations. Ligand-receptor interactions were derived from a curated map of human ligand-receptor pairs with mouse-specific weights added after reference to the STRING database (80). Results show a complex web of cell-cell interactions with ECs receiving the largest number of inbound signals and neutrophils sending the highest number of outbound signals (Fig. 4), demonstrating that abundant cross talk between cell types occurs in the aortic wall that may play a role in homeostasis and disease.
Figure 4.
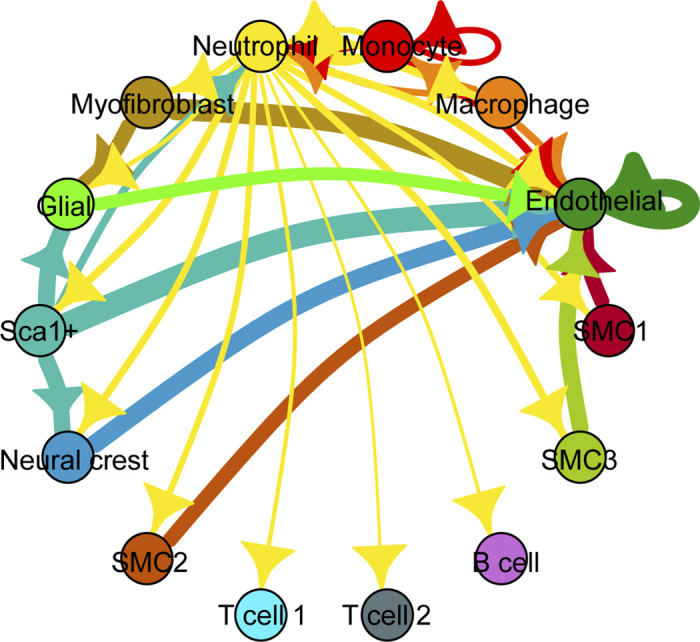
Network diagram of significant cell-cell interactions via ligand-receptor pairs expressed in cell clusters (79) determined from previously published single-cell RNA-sequencing data of ElnSMKO ascending aorta (22). Arrows and color indicate direction (ligand to receptor) and thickness indicates the sum of the weighted paths between cell clusters.
Fragmentation of the IEL may also facilitate migration of SMCs into the subendothelial space. Migration of SMCs through IEL gaps has been observed in genetic and mechanical models of neointimal formation (81) and atherosclerosis (82) in mice. Migration of immune cells from the blood across the EC layer and into the aortic wall may also be facilitated by IEL gaps. ElnSMKO aorta has almost three times more monocytes in the aortic wall than controls (22). In humans, it has been hypothesized that differences in IEL discontinuity may explain differences in atherosclerosis and stenosis susceptibility of different arteries (83, 84). Additional work is needed to understand how pore size of the IEL microenvironment contributes to alterations in cellular communication and migration in stenosis.
The neointima in ElnSMKO ascending aorta is composed of SMCs that are luminal to the IEL. Single-cell RNA-sequencing of ElnSMKO and control ascending aortas showed three populations of SMCs, termed SMC1, -2 and -3, respectively (22). SMC1 includes the mature SMCs, as this group expresses the highest levels of contractile SMC marker genes. SMC2 has the lowest level of SMC marker genes and increased expression of myofibroblast marker genes. SMC3 has the highest level of genes involved in cell proliferation. Identification of unique gene signatures for each group showed that SMC2 and SMC3, which express Ptch1 whereas SMC1 does not, are localized to the neointima in ElnSMKO aorta. When comparing the major SMC1 cluster between ElnSMKO and control aorta, 147 upregulated and 184 downregulated genes are identified. The most upregulated gene is Igfbp2, which was also identified in our previous microarray analysis of Eln−/− versus control aorta (85). Nine of the 25 most differentially regulated genes from the previous microarray analysis overlapped with the single-cell analysis, validating this approach. Reactome pathway analysis shows “Regulation of IGF transport and uptake by IGFBPs” to be highly upregulated in SMC1 ElnSMKO compared with control, implying that targeting this pathway may have therapeutic benefit for stenosis prevention. Igfbp2 has a heparin binding domain sequence that interacts with proteoglycans (86) and an RGD sequence that binds select integrins (87), indicating a role for Igfbp2 for sequestering IGF in the ECM that may be mechanosensitive.
Based on our interest in linking tissue- and cell-level arterial mechanics in this review, we re-examined our previously published single-cell RNA-sequencing data (22) for expression of putative mechanosensors (28). We had previously found no changes in β integrin subunits in ElnSMKO cell clusters compared with control and significantly increased expression of Piezo2 in ElnSMKO SMC1 compared with control (22). Violin plots of Piezo1 and -2 show high expression of Piezo1 in ECs that is unchanged by elastin amounts in SMCs and high expression of Piezo2 in SMC and myofibroblast clusters for ElnSMKO only (Fig. 5). Piezo1 and -2 are mechanically activated cation channels (88). Piezo1 senses mechanical stimuli in multiple organ systems, including flow-induced shear stress in ECs, however the role of Piezo2 is less well studied (89). Piezo2, in combination with Piezo1, is involved in baroreceptor pressure sensing in mice (90), and both Piezos have genetic associations with heart failure, thoracic aortic aneurysm rupture, and stenosis (91). The single-cell RNA-sequencing results identify both specific cell types (i.e., SMC2 and SMC3) and pathways (i.e., IGF or Piezo signaling) that merit further research and eventually may be targeted to prevent neointima formation and stenosis in SVAS/WBS.
Figure 5.

Violin plots of Piezo1 (A) and -2 (B) expression in different cell clusters for postnatal day 8 (P8) control (Elnf/f or Elnf/+) and ElnSMKO ascending aorta determined from previously published single-cell RNA-sequencing data (22).
Endothelial cell-specific Eln knockout mice.
Both Tie2Cre (92) and Cdh5Cre (93) were used to inactivate Eln in ECs, with similar results. ElnECKO (Tie2Cre;Elnf/f or Cdh5Cre;Elnf/f) mice are grossly normal, fertile, and have a normal lifespan (Table 1). In contrast to the ElnSMKO model, elastin deletion in ECs does not lead to aortic stenosis. In ElnECKO mice, the overall vascular architecture and IEL are indistinguishable from controls in the aorta, medium-sized elastic, and muscular arteries. However, the IEL in resistance arteries, such as second-order mesenteric arteries, is fragmented, indicating that ECs are the main contributor to the IEL in these arteries (Fig. 3) (50). It is not known why IEL fragmentation in the ascending aorta is associated with neointima and stenosis, whereas IEL fragmentation in the second-order mesenterics is not. The results suggest that additional factors beyond IEL integrity, such as unique SMC phenotype or specific hemodynamic forces in the ascending aorta, play a role in stenosis.
Secondary heart field and cardiac neural crest origin Eln knockout mice.
SMCs in the ascending aorta and aortic arch originate from two embryonic sources: the secondary heart field (SHF) and the cardiac neural crest (CNC). SHF-derived cells populate the outer (abluminal) portion of the tunica media, whereas cells from the CNC generally constitute the inner (luminal) portion (94). The availability of SHF- and CNC-specific Cre drivers (Isl1Cre and Wnt1Cre, respectively) allows interrogation of the contribution of elastin production from SMCs of these embryonic origins to aortic stenosis. Isl1Cre also targets the ECs. Both ElnSHFKO (Isl1Cre;Elnf/f) and ElnCNCKO (Wnt1Cre;Elnf/f) mice have premature mortality around 2–3 mo of age (Table 1). However, the phenotype of IEL fragmentation and neointimal formation is different in these models. In all ElnSHFKO mice, IEL in the ascending aorta is fragmented, which is associated with neointima. Lineage tracing shows that Isl1Cre mostly targets SMCs in the abluminal aspect of the ascending aorta and there are scattered SMCs underneath the IEL that are labeled by the Isl1Cre lineage. Cells from the Isl1Cre lineage populate most of the neointima (Fig. 6). Due to their localization, it is possible that the sub-IEL SMCs derived from the SHF lineage contribute disproportionately to the neointima, but additional genetic or molecular tools are needed to specifically target this cell population. The fragmented IEL in ElnSHFKO mice could be due to lack of elastin contributions from the sub-IEL SMCs and/or lack of the minor elastin contributions expected from ECs (Fig. 3), since Isl1Cre also targets ECs. The effect of elastin contributions from these different cell populations is underscored by the fact that the majority (∼2/3) of ElnCNCKO mice with lack of elastin production in the luminal portion of the ascending aortic wall have an intact IEL and no neointima, whereas one-third develop neointima, but the burden is less than that of ElnSHFKO mice (Fig. 6). ElnCNCKO but not ElnSHFKO mice develop aortic coarctation due to tunica media hyperplasia (Fig. 1) (22), suggesting that SMCs from these embryonic origins have different propensities to form aortic coarctation and neointima.
Figure 6.

Neointima in the ascending aorta of elastin conditional knockout mouse models as demonstrated by VVG staining (ElnSMKO and ElnCNCKO) and lineage tracing with the ROSA26mT/mG allele (ElnSHFKO) (22). Internal elastic lamina (IEL) demarcated by dashed lines. Neointimal burden indicated by double arrows. Note the younger age of ElnSMKO mice. Scale = 50 µm.
SUMMARY AND FUTURE DIRECTIONS
We have summarized the role of elastin in arterial mechanics and stenosis from studies of human congenital disease and genetic animal models. Elastin plays unique roles in multiple dimensions—from the Windkessel effect, to tissue- and cell-level arterial mechanics, to regulating interactions between SMCs and ECs. Elastic fiber synthesis and assembly in the arterial wall is an intricately orchestrated process with respect to time and space. We reviewed the spatial specificity of elastogenesis across the arterial wall and throughout the arterial tree. Recent evidence with conditional elastin knockout mouse models demonstrates that different transmural and arterial tree locations have distinct cell type requirements for elastogenesis and that loss of elastin in one cell type may not be compensated for by elastin produced in other cell types. Therefore, tissue engineering efforts to create elastic fibers in the arterial wall will have to account for this added layer of complexity.
The formation of the IEL exemplifies this complexity, with varying contributions of SMCs and ECs depending on the artery type (Fig. 3). While separating the SMCs and ECs, the fenestrated IEL also allows communication between the two cell types, such as through the myoendothelial junctions (95). Ultrastructural changes in the fenestrae can be associated with pathological conditions (96). Not only can SMCs move across the fenestrae (81), it is conceivable that breaks in the IEL cause aberrant SMC-EC interactions that affect pathogenesis and disease progression (97). Immune cells can also move across the IEL. There are increased percentages of monocytes, macrophages, and neutrophils in the wall (22) and extensive cross talk between immune cells and other cell clusters (Fig. 4) in ElnSMKO ascending aorta with a fragmented IEL. As immune cells are known to express elastolytic enzymes, how these infiltrating immune cells and signaling events facilitate elastin destruction, ECM remodeling, and changes in tissue- and cell-level mechanical properties remains to be understood.
Elucidating the pathways affected by IEL fragmentation may require in vitro cell coculture models that can faithfully recreate the physical and mechanical microenvironment. These models may also help tease out SMC mechanosensing pathways that play a role in SVAS/WBS. Altering circumferential stress in SMCs within a composite tissue is much more challenging experimentally than altering fluid-induced shear stress across ECs or stretch of SMCs attached to a deformable monolayer. Most mechanosensors are activated by specific modes (28) (i.e., osmotic pressure, flow, negative or positive pressure, local indentation, or stretch), hence it is critical that relevant mechanical stimuli are applied to the SMCs. Although circumferential stretch is likely an appropriate short-term mechanical stimulus for SMCs in resistance arteries that contract or relax to modulate pressure and blood flow, circumferential wall stress may be a more relevant mechanical stimulus for SMCs in large, elastic arteries with abundant ECM and long-term remodeling that leads to arterial stenosis.
An additional area of future research is modulating the temporal expression of elastin in arterial development and disease. Elastin is normally expressed during a short window during late gestation and early postnatal life. Emerging evidence reveals that elastin expression may be reactivated postnatally in vascular diseases (98), although the process can be abnormal, perhaps due to lack of orchestrated elastic fiber assembly and cross-linking machinery. Knowledge of this process will help improve therapeutic options, such as minoxidil treatment, which induces elastin expression postnatally and improves arterial mechanics in aging and/or Eln+/− mice (99, 100). Current knowledge of elastin vascular biology stems from human genetics and mouse mutants with constitutive deletions (Table 1), whereby the individual never experiences normal amounts of elastin during development. With the availability of inducible conditional knockout mouse models, manipulating the temporal expression of elastin will provide valuable information on the role, if any, of elastin in vascular pathogenesis and aging. Such understanding may guide therapeutic interventions to prevent disease progression in arterial stenosis caused by reduced elastin amounts, including SVAS/WBS.
GRANTS
This study was partially funded by National Science Foundation Grant 1662434 (to J. E. Wagenseil) and National Heart, Lung, and Blood Institute (NHLBI) Grants R56 HL152420 (to J. E. Wagenseil). C-J. Lin was supported by NHLBI Grants T32HL007081 and T32HL125241.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C-J.L. and J.E.W. conceived and designed research; C-J.L., A.J.C., and J.E.W. performed experiments; C-J.L., A.J.C., and J.E.W. analyzed data; C-J.L. and J.E.W. interpreted results of experiments; C-J.L. and J.E.W. prepared figures; C-J.L. and J.E.W. drafted manuscript; C-J.L. and J.E.W. edited and revised manuscript; C-J.L., A.J.C., and J.E.W. approved final version of manuscript.
REFERENCES
- 1.Sage H, Gray WR. Studies on the evolution of elastin. I. Phylogenetic distribution. Comp Biochem Physiol B 64: 313–327, 1979. doi: 10.1016/0305-0491(79)90277-3. [DOI] [PubMed] [Google Scholar]
- 2.Kelleher CM, McLean SE, Mecham RP. Vascular extracellular matrix and aortic development. Curr Top Dev Biol 62: 153–188, 2004. doi: 10.1016/S0070-2153(04)62006-0. [DOI] [PubMed] [Google Scholar]
- 3.Shapiro SD, Endicott SK, Province MA, Pierce JA, Campbell EJ. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of d-aspartate and nuclear weapons-related radiocarbon. J Clin Invest 87: 1828–1834, 1991. doi: 10.1172/JCI115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandberg LB, Soskel NT, Leslie JG. Elastin structure, biosynthesis, and relation to disease states. N Engl J Med 304: 566–579, 1981. doi: 10.1056/NEJM198103053041004. [DOI] [PubMed] [Google Scholar]
- 5.Tamburro AM, Bochicchio B, Pepe A. The dissection of human tropoelastin: from the molecular structure to the self-assembly to the elasticity mechanism. Pathol Biol (Paris) 53: 383–389, 2005. doi: 10.1016/j.patbio.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 6.Cox BA, Starcher BC, Urry DW. Communication: coacervation of tropoelastin results in fiber formation. J Biol Chem 249: 997–998, 1974. [PubMed] [Google Scholar]
- 7.Smith-Mungo LI, Kagan HM. Lysyl oxidase: properties, regulation and multiple functions in biology. Matrix Biol 16: 387–398, 1998. doi: 10.1016/s0945-053x(98)90012-9. [DOI] [PubMed] [Google Scholar]
- 8.Ozsvar J, Yang C, Cain SA, Baldock C, Tarakanova A, Weiss AS. Tropoelastin and elastin assembly. Front Bioeng Biotechnol 9: 643110, 2021. doi: 10.3389/fbioe.2021.643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pober BR. Williams-Beuren syndrome. N Engl J Med 362: 239–252, 2010. [Erratum in N Engl J Med 362: 2142, 2010]. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- 10.Cocciolone AJ, Hawes JZ, Staiculescu MC, Johnson EO, Murshed M, Wagenseil JE. Elastin, arterial mechanics, and cardiovascular disease. Am J Physiol Heart Circ Physiol 315: H189–H205, 2018. doi: 10.1152/ajpheart.00087.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giudici A, Wilkinson IB, Khir AW. Review of the techniques used for investigating the role elastin and collagen play in arterial wall mechanics. IEEE Rev Biomed Eng 14: 256–269, 2021. doi: 10.1109/RBME.2020.3005448. [DOI] [PubMed] [Google Scholar]
- 12.Yanagisawa H, Wagenseil J. Elastic fibers and biomechanics of the aorta: insights from mouse studies. Matrix Biol 85–86: 160–172, 2020. doi: 10.1016/j.matbio.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolinsky H. Comparison of medial growth of human thoracic and abdominal aortas. Circ Res 27: 531–538, 1970. doi: 10.1161/01.res.27.4.531. [DOI] [PubMed] [Google Scholar]
- 14.Burton AC. Relation of structure to function of the tissues of the wall of blood vessels. Physiol Rev 34: 619–642, 1954. doi: 10.1152/physrev.1954.34.4.619. [DOI] [PubMed] [Google Scholar]
- 15.Wolinsky H, Glagov S. A lamellar unit of aortic medial structure and function in mammals. Circ Res 20: 99–111, 1967. doi: 10.1161/01.res.20.1.99. [DOI] [PubMed] [Google Scholar]
- 16.Roach MR, Burton AC. The reason for the shape of the distensibility curves of arteries. Can J Biochem Physiol 35: 681–690, 1957. [PubMed] [Google Scholar]
- 17.Dobrin PB, Canfield TR. Elastase, collagenase, and the biaxial elastic properties of dog carotid artery. Am J Physiol Heart Circ Physiol 247: H124–H131, 1984. doi: 10.1152/ajpheart.1984.247.1.H124. [DOI] [PubMed] [Google Scholar]
- 18.Fonck E, Prod'hom G, Roy S, Augsburger L, Rufenacht DA, Stergiopulos N. Effect of elastin degradation on carotid wall mechanics as assessed by a constituent-based biomechanical model. Am J Physiol Heart Circ Physiol 292: H2754–H2763, 2007. doi: 10.1152/ajpheart.01108.2006. [DOI] [PubMed] [Google Scholar]
- 19.Shadwick RE. Mechanical design in arteries. J Exp Biol 202: 3305–3313, 1999. doi: 10.1242/jeb.202.23.3305. [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Cocciolone AJ, Staiculescu MC, Mecham RP, Wagenseil JE. Passive biaxial mechanical behavior of newborn mouse aorta with and without elastin. J Mech Behav Biomed Mater 126: 105021, 2022. doi: 10.1016/j.jmbbm.2021.105021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J, Staiculescu MC, Cocciolone AJ, Yanagisawa H, Mecham RP, Wagenseil JE. Crosslinked elastic fibers are necessary for low energy loss in the ascending aorta. J Biomech 61: 199–207, 2017. doi: 10.1016/j.jbiomech.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin CJ, Hunkins BM, Roth RA, Lin CY, Wagenseil JE, Mecham RP. Vascular smooth muscle cell subpopulations and neointimal formation in mouse models of elastin insufficiency. Arterioscler Thromb Vasc Biol 41: 2890–2905, 2021. doi: 10.1161/ATVBAHA.120.315681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev 89: 957–989, 2009. doi: 10.1152/physrev.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murtada SI, Kawamura Y, Li G, Schwartz MA, Tellides G, Humphrey JD. Developmental origins of mechanical homeostasis in the aorta. Dev Dyn 250: 629–639, 2021. doi: 10.1002/dvdy.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steucke KE, Win Z, Stemler TR, Walsh EE, Hall JL, Pw. A. Empirically determined vascular smooth muscle cell mechano-adaptation law. J Biomech Eng 139: 0710051–0710059, 2017. doi: 10.1115/1.4036454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bellini C, Bersi MR, Caulk AW, Ferruzzi J, Milewicz DM, Ramirez F, Rifkin DB, Tellides G, Yanagisawa H, Humphrey JD. Comparison of 10 murine models reveals a distinct biomechanical phenotype in thoracic aortic aneurysms. J R Soc Interface 14: 20161036, 2017. doi: 10.1098/rsif.2016.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fridez P, Zulliger M, Bobard F, Montorzi G, Miyazaki H, Hayashi K, Stergiopulos N. Geometrical, functional, and histomorphometric adaptation of rat carotid artery in induced hypertension. J Biomech 36: 671–680, 2003. doi: 10.1016/s0021-9290(02)00445-1. [DOI] [PubMed] [Google Scholar]
- 28.Drummond HA. What evolutionary evidence implies about the identity of the mechanoelectrical couplers in vascular smooth muscle cells. Physiology (Bethesda) 36: 292–306, 2021. doi: 10.1152/physiol.00008.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wanjare M, Agarwal N, Gerecht S. Biomechanical strain induces elastin and collagen production in human pluripotent stem cell-derived vascular smooth muscle cells. Am J Physiol Cell Physiol 309: C271–C281, 2015. doi: 10.1152/ajpcell.00366.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell 126: 677–689, 2006. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 31.Steucke KE, Tracy PV, Hald ES, Hall JL, Alford PW. Vascular smooth muscle cell functional contractility depends on extracellular mechanical properties. J Biomech 48: 3044–3051, 2015. doi: 10.1016/j.jbiomech.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rickel AP, Sanyour HJ, Leyda NA, Hong Z. Extracellular matrix proteins and substrate stiffness synergistically regulate vascular smooth muscle cell migration and cortical cytoskeleton organization. ACS Appl Bio Mater 3: 2360–2369, 2020. doi: 10.1021/acsabm.0c00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown XQ, Bartolak-Suki E, Williams C, Walker ML, Weaver VM, Wong JY. Effect of substrate stiffness and PDGF on the behavior of vascular smooth muscle cells: implications for atherosclerosis. J Cell Physiol 225: 115–122, 2010. doi: 10.1002/jcp.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu CK, Xu T, Assoian RK, Rader DJ. Mining the stiffness-sensitive transcriptome in human vascular smooth muscle cells identifies long noncoding RNA stiffness regulators. Arterioscler Thromb Vasc Biol 38: 164–173, 2018. doi: 10.1161/ATVBAHA.117.310237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee HP, Lippens E, Duda GN, Mooney DJ. Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat Mater 15: 326–334, 2016. doi: 10.1038/nmat4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaudhuri O, Gu L, Darnell M, Klumpers D, Bencherif SA, Weaver JC, Huebsch N, Mooney DJ. Substrate stress relaxation regulates cell spreading. Nat Commun 6: 6364, 2015. doi: 10.1038/ncomms7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cameron AR, Frith JE, Gomez GA, Yap AS, Cooper-White JJ. The effect of time-dependent deformation of viscoelastic hydrogels on myogenic induction and Rac1 activity in mesenchymal stem cells. Biomaterials 35: 1857–1868, 2014. doi: 10.1016/j.biomaterials.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 38.Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY. A critical role for elastin signaling in vascular morphogenesis and disease. Development 130: 411–423, 2003. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- 39.Espinosa MG, Gardner WS, Bennett L, Sather B, Yanagisawa H, Wagenseil JE. The effects of elastic fiber protein insufficiency and treatment on the modulus of arterial smooth muscle cells. J Biomech Eng 136: 021030, 2014. doi: 10.1115/1.4026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karnik SK, Wythe JD, Sorensen L, Brooke BS, Urness LD, Li DY. Elastin induces myofibrillogenesis via a specific domain, VGVAPG. Matrix Biol 22: 409–425, 2003. doi: 10.1016/S0945-053X(03)00076-3. [DOI] [PubMed] [Google Scholar]
- 41.Milewicz DM, Urban Z, Boyd C. Genetic disorders of the elastic fiber system. Matrix Biol 19: 471–480, 2000. doi: 10.1016/S0945-053X(00)00099-8. [DOI] [PubMed] [Google Scholar]
- 42.Merla G, Brunetti-Pierri N, Piccolo P, Micale L, Loviglio MN. Supravalvular aortic stenosis: elastin arteriopathy. Circ Cardiovasc Genet 5: 692–696, 2012. doi: 10.1161/CIRCGENETICS.112.962860. [DOI] [PubMed] [Google Scholar]
- 43.Urban Z, Riazi S, Seidl TL, Katahira J, Smoot LB, Chitayat D, Boyd CD, Hinek A. Connection between elastin haploinsufficiency and increased cell proliferation in patients with supravalvular aortic stenosis and Williams-Beuren syndrome. Am J Hum Genet 71: 30–44, 2002. doi: 10.1086/341035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pober BR, Johnson M, Urban Z. Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome. J Clin Invest 118: 1606–1615, 2008. doi: 10.1172/JCI35309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ashkenas J. Williams syndrome starts making sense. Am J Hum Genet 59: 756–761, 1996. [PMC free article] [PubMed] [Google Scholar]
- 46.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature 393: 276–280, 1998. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 47.Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest 102: 1783–1787, 1998. doi: 10.1172/JCI4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li HH, Roy M, Kuscuoglu U, Spencer CM, Halm B, Harrison KC, Bayle JH, Splendore A, Ding F, Meltzer LA, Wright E, Paylor R, Deisseroth K, Francke U. Induced chromosome deletions cause hypersociability and other features of Williams-Beuren syndrome in mice. EMBO Mol Med 1: 50–65, 2009. doi: 10.1002/emmm.200900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hirano E, Knutsen RH, Sugitani H, Ciliberto CH, Mecham RP. Functional rescue of elastin insufficiency in mice by the human elastin gene: implications for mouse models of human disease. Circ Res 101: 523–531, 2007. doi: 10.1161/CIRCRESAHA.107.153510. [DOI] [PubMed] [Google Scholar]
- 50.Lin CJ, Staiculescu MC, Hawes JZ, Cocciolone AJ, Hunkins BM, Roth RA, Lin CY, Mecham RP, Wagenseil JE. Heterogeneous cellular contributions to elastic laminae formation in arterial wall development. Circ Res 125: 1006–1018, 2019. doi: 10.1161/CIRCRESAHA.119.315348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fazio MJ, Olsen DR, Kauh EA, Baldwin CT, Indik Z, Ornstein-Goldstein N, Yeh H, Rosenbloom J, Uitto J. Cloning of full-length elastin cDNAs from a human skin fibroblast recombinant cDNA library: further elucidation of alternative splicing utilizing exon-specific oligonucleotides. J Invest Dermatol 91: 458–464, 1988. doi: 10.1111/1523-1747.ep12476591. [DOI] [PubMed] [Google Scholar]
- 52.Szabo Z, Levi-Minzi SA, Christiano AM, Struminger C, Stoneking M, Batzer MA, Boyd CD. Sequential loss of two neighboring exons of the tropoelastin gene during primate evolution. J Mol Evol 49: 664–671, 1999. doi: 10.1007/pl00006587. [DOI] [PubMed] [Google Scholar]
- 53.Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A, Mecham RP. Reduced vessel elasticity alters cardiovascular structure and function in newborn mice. Circ Res 104: 1217–1224, 2009. doi: 10.1161/CIRCRESAHA.108.192054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A, Mecham RP. The importance of elastin to aortic development in mice. Am J Physiol Heart Circ Physiol 299: H257–H264, 2010. doi: 10.1152/ajpheart.00194.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Julien MA, Haller CA, Wang P, Wen J, Chaikof EL. Mechanical strain induces a persistent upregulation of syndecan-1 expression in smooth muscle cells. J Cell Physiol 211: 167–173, 2007. doi: 10.1002/jcp.20927. [DOI] [PubMed] [Google Scholar]
- 56.Julien MA, Wang P, Haller CA, Wen J, Chaikof EL. Mechanical strain regulates syndecan-4 expression and shedding in smooth muscle cells through differential activation of MAP kinase signaling pathways. Am J Physiol Cell Physiol 292: C517–C525, 2007. doi: 10.1152/ajpcell.00093.2006. [DOI] [PubMed] [Google Scholar]
- 57.Kim J, Cocciolone AJ, Staiculescu MC, Mecham RP, Wagenseil JE. Captopril treatment during development alleviates mechanically induced aortic remodeling in newborn elastin knockout mice. Biomech Model Mechanobiol 19: 99–112, 2020. doi: 10.1007/s10237-019-01198-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li W, Li Q, Qin L, Ali R, Qyang Y, Tassabehji M, Pober BR, Sessa WC, Giordano FJ, Tellides G. Rapamycin inhibits smooth muscle cell proliferation and obstructive arteriopathy attributable to elastin deficiency. Arterioscler Thromb Vasc Biol 33: 1028–1035, 2013. doi: 10.1161/ATVBAHA.112.300407. [DOI] [PubMed] [Google Scholar]
- 59.Misra A, Sheikh AQ, Kumar A, Luo J, Zhang J, Hinton RB, Smoot L, Kaplan P, Urban Z, Qyang Y, Tellides G, Greif DM. Integrin β3 inhibition is a therapeutic strategy for supravalvular aortic stenosis. J Exp Med 213: 451–463, 2016. doi: 10.1084/jem.20150688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stassen O, Ristori T, Sahlgren CM. Notch in mechanotransduction—from molecular mechanosensitivity to tissue mechanostasis. J Cell Sci 133: jcs250738, 2020. doi: 10.1242/jcs.250738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dave JM, Chakraborty R, Ntokou A, Saito J, Saddouk FZ, Feng Z, Misra A, Tellides G, Riemer RK, Urban Z, Kinnear C, Ellis J, Mital S, Mecham R, Martin KA, Greif DM. JAGGED1/NOTCH3 activation promotes aortic hypermuscularization and stenosis in elastin deficiency. J Clin Invest 132: e142338, 2022. doi: 10.1172/JCI142338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, Mecham RP. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest 112: 1419–1428, 2003. doi: 10.1172/JCI19028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goergen CJ, Li HH, Francke U, Taylor CA. Induced chromosome deletion in a Williams-Beuren syndrome mouse model causes cardiovascular abnormalities. J Vasc Res 48: 119–129, 2011. doi: 10.1159/000316808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Le VP, Knutsen RH, Mecham RP, Wagenseil JE. Decreased aortic diameter and compliance precedes blood pressure increases in postnatal development of elastin-insufficient mice. Am J Physiol Heart Circ Physiol 301: H221–H229, 2011. doi: 10.1152/ajpheart.00119.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wagenseil JE, Mecham RP. Elastin in large artery stiffness and hypertension. J Cardiovasc Transl Res 5: 264–273, 2012. doi: 10.1007/s12265-012-9349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hawes JZ, Cocciolone A, Cui AH, Griffin DB, Staiculescu MC, Mecham RP, Wagenseil JE. Elastin haploinsufficiency in mice has divergent effects on arterial remodeling with aging depending on sex. Am J Physiol Heart Circ Physiol 319: H1398–H1408, 2020. doi: 10.1152/ajpheart.00517.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng JK, Stoilov I, Mecham RP, Wagenseil JE. A fiber-based constitutive model predicts changes in amount and organization of matrix proteins with development and disease in the mouse aorta. Biomech Model Mechanobiol 12: 497–510, 2013. doi: 10.1007/s10237-012-0420-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pezet M, Jacob MP, Escoubet B, Gheduzzi D, Tillet E, Perret P, Huber P, Quaglino D, Vranckx R, Li DY, Starcher B, Boyle WA, Mecham RP, Faury G. Elastin haploinsufficiency induces alternative aging processes in the aorta. Rejuvenation Res 11: 97–112, 2008. doi: 10.1089/rej.2007.0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiao Y, Li G, Korneva A, Caulk AW, Qin L, Bersi MR, Li Q, Li W, Mecham RP, Humphrey JD, Tellides G. Deficient circumferential growth is the primary determinant of aortic obstruction attributable to partial elastin deficiency. Arterioscler Thromb Vasc Biol 37: 930–941, 2017. doi: 10.1161/ATVBAHA.117.309079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gadson PF Jr, Rossignol C, McCoy J, Rosenquist TH. Expression of elastin, smooth muscle alpha-actin, and c-jun as a function of the embryonic lineage of vascular smooth muscle cells. In vitro Cell Dev Biol Anim 29A: 773–781, 1993. doi: 10.1007/BF02634344. [DOI] [PubMed] [Google Scholar]
- 71.Hungerford JE, Owens GK, Argraves WS, Little CD. Development of the aortic vessel wall as defined by vascular smooth muscle and extracellular matrix markers. Dev Biol 178: 375–392, 1996. doi: 10.1006/dbio.1996.0225. [DOI] [PubMed] [Google Scholar]
- 72.Cantor JO, Keller S, Parshley MS, Darnule TV, Darnule AT, Cerreta JM, Turino GM, Mandl I. Synthesis of crosslinked elastin by an endothelial cell culture. Biochem Biophys Res Commun 95: 1381–1386, 1980. doi: 10.1016/s0006-291x(80)80050-7. [DOI] [PubMed] [Google Scholar]
- 73.Mecham RP, Madaras J, McDonald JA, Ryan U. Elastin production by cultured calf pulmonary artery endothelial cells. J Cell Physiol 116: 282–288, 1983. doi: 10.1002/jcp.1041160304. [DOI] [PubMed] [Google Scholar]
- 74.Ruckman JL, Luvalle PA, Hill KE, Giro MG, Davidson JM. Phenotypic stability and variation in cells of the porcine aorta: collagen and elastin production. Matrix Biol 14: 135–145, 1994. doi: 10.1016/0945-053x(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 75.Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc Natl Acad Sci USA 99: 7142–7147, 2002. doi: 10.1073/pnas.102650499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ Res 86: 341–346, 2000. doi: 10.1161/01.RES.86.3.341. [DOI] [PubMed] [Google Scholar]
- 77.Little TL, Xia J, Duling BR. Dye tracers define differential endothelial and smooth muscle coupling patterns within the arteriolar wall. Circ Res 76: 498–504, 1995. doi: 10.1161/01.res.76.3.498. [DOI] [PubMed] [Google Scholar]
- 78.Halabi CM, Kozel BA. Vascular elastic fiber heterogeneity in health and disease. Curr Opin Hematol 27: 190–196, 2020. doi: 10.1097/MOH.0000000000000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JW, Nordon RE, Harvey RP. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife 8: e43882, 2019. doi: 10.7554/eLife.43882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45: D362–D368, 2017. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lu YW, Lowery AM, Sun LY, Singer HA, Dai G, Adam AP, Vincent PA, Schwarz JJ. Endothelial myocyte enhancer factor 2c inhibits migration of smooth muscle cells through fenestrations in the internal elastic lamina. Arterioscler Thromb Vasc Biol 37: 1380–1390, 2017. doi: 10.1161/ATVBAHA.117.309180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jones GT, Jiang F, McCormick SP, Dusting GJ. Elastic lamina defects are an early feature of aortic lesions in the apolipoprotein E knockout mouse. J Vasc Res 42: 237–246, 2005. doi: 10.1159/000085553. [DOI] [PubMed] [Google Scholar]
- 83.Sims FH. Discontinuities in the internal elastic lamina: a comparison of coronary and internal mammary arteries. Artery 13: 127–143, 1985. [PubMed] [Google Scholar]
- 84.Worssam MD, Jorgensen HF. Mechanisms of vascular smooth muscle cell investment and phenotypic diversification in vascular diseases. Biochem Soc Trans 49: 2101–2111, 2021. doi: 10.1042/BST20210138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Staiculescu MC, Cocciolone AJ, Procknow JD, Kim J, Wagenseil JE. Comparative gene array analyses of severe elastic fiber defects in late embryonic and newborn mouse aorta. Physiol Genomics 50: 988–1001, 2018. doi: 10.1152/physiolgenomics.00080.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Russo VC, Bach LA, Fosang AJ, Baker NL, Werther GA. Insulin-like growth factor binding protein-2 binds to cell surface proteoglycans in the rat brain olfactory bulb. Endocrinology 138: 4858–4867, 1997. doi: 10.1210/endo.138.11.5472. [DOI] [PubMed] [Google Scholar]
- 87.Kapp TG, Rechenmacher F, Neubauer S, Maltsev OV, Cavalcanti-Adam EA, Zarka R, Reuning U, Notni J, Wester HJ, Mas-Moruno C, Spatz J, Geiger B, Kessler H. A comprehensive evaluation of the activity and selectivity profile of ligands for RGD-binding integrins. Sci Rep 7: 39805, 2017. doi: 10.1038/srep39805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330: 55–60, 2010. doi: 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beech DJ, Kalli AC. Force sensing by Piezo channels in cardiovascular health and disease. Arterioscler Thromb Vasc Biol 39: 2228–2239, 2019. doi: 10.1161/ATVBAHA.119.313348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zeng WZ, Marshall KL, Min S, Daou I, Chapleau MW, Abboud FM, Liberles SD, Patapoutian A. PIEZOs mediate neuronal sensing of blood pressure and the baroreceptor reflex. Science 362: 464–467, 2018. doi: 10.1126/science.aau6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Staley JR, Blackshaw J, Kamat MA, Ellis S, Surendran P, Sun BB, Paul DS, Freitag D, Burgess S, Danesh J, Young R, Butterworth AS. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics 32: 3207–3209, 2016. doi: 10.1093/bioinformatics/btw373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol 230: 230–242, 2001. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 93.Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, Carmeliet P, Iruela-Arispe ML. VE-cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn 235: 759–767, 2006. doi: 10.1002/dvdy.20643. [DOI] [PubMed] [Google Scholar]
- 94.Sawada H, Rateri DL, Moorleghen JJ, Majesky MW, Daugherty A. Smooth muscle cells derived from second heart field and cardiac neural crest reside in spatially distinct domains in the media of the ascending aorta-brief report. Arterioscler Thromb Vasc Biol 37: 1722–1726, 2017. doi: 10.1161/ATVBAHA.117.309599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Straub AC, Zeigler AC, Isakson BE. The myoendothelial junction: connections that deliver the message. Physiology (Bethesda) 29: 242–249, 2014. doi: 10.1152/physiol.00042.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kwon HM, Sangiorgi G, Spagnoli LG, Miyauchi K, Holmes DR Jr, Schwartz RS, Lerman A. Experimental hypercholesterolemia induces ultrastructural changes in the internal elastic lamina of porcine coronary arteries. Atherosclerosis 139: 283–289, 1998. doi: 10.1016/S0021-9150(98)00081-1. [DOI] [PubMed] [Google Scholar]
- 97.Lilly B. We have contact: endothelial cell-smooth muscle cell interactions. Physiology (Bethesda) 29: 234–241, 2014. doi: 10.1152/physiol.00047.2013. [DOI] [PubMed] [Google Scholar]
- 98.Krettek A, Sukhova GK, Libby P. Elastogenesis in human arterial disease: a role for macrophages in disordered elastin synthesis. Arterioscler Thromb Vasc Biol 23: 582–587, 2003. doi: 10.1161/01.ATV.0000064372.78561.A5. [DOI] [PubMed] [Google Scholar]
- 99.Fhayli W, Boyer M, Ghandour Z, Jacob MP, Andrieu JP, Starcher BC, Esteve E, Faury G. Chronic administration of minoxidil protects elastic fibers and stimulates their neosynthesis with improvement of the aorta mechanics in mice. Cell Signal 62: 109333, 2019. doi: 10.1016/j.cellsig.2019.05.018. [DOI] [PubMed] [Google Scholar]
- 100.Knutsen R, Beeman SC, Broekelmann TJ, Liu D, Tsang KM, Kovacs A, Ye L, Danback J, Watson A, Wardlaw A, Wagenseil J, Garbow JR, Shoykhet M, Kozel BA. Minoxidil improves vascular compliance, restores cerebral blood flow and alters extracellular matrix gene expression in a model of chronic vascular stiffness. Am J Physiol Heart Circ Physiol 315: H18–H32, 2018. doi: 10.1152/ajpheart.00683.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]