

Abstract
CD8 T cell memory critically contributes to long-term immunity. Both low- and high-affinity TCR signals are able to support the differentiation of memory CD8 T cells. However, it is unclear whether the requirements for memory development change when TCR signal strength is altered. To gain further insight into this question, we used a TCRβ transmembrane domain mutant model that is defective in the generation of memory in response to high-affinity ligands. Surprisingly, lowering TCR signal strength, by stimulation with low-affinity ligands, resulted in normal memory development. Restoration of memory correlated with recovery of TCR-dependent NF-κB signaling. Thus, these data provide novel evidence that the requirements for memory are qualitatively different depending on TCR signal strength. The Journal of Immunology, 2013, 191: 5797–5801.
Memory CD8 T cells provide protective immunity against intracellular pathogens and tumors. It has been assumed that high-affinity TCR ligands are required for naive CD8 T cells to acquire effector function and differentiate into long-lived memory T cells. However, there is compelling evidence that low-affinity TCR ligands support memory development (1–4). In the context of infection, T cells responding to low-affinity TCR ligands are able to differentiate into functional memory T cells (5). Furthermore, in lymphopenic conditions, T cells homeostatically proliferate (HP) and develop into memory cells in response to self-peptide–MHC complexes (low affinity), high levels of homeostatic cytokines, and low levels of inflammation (6–8). These HP memory T cells are similar to Ag-experienced (true) memory cells at clearing pathogens upon infection and generating secondary memory (9). Curiously, T cells differentiating into memory under lymphopenic conditions exhibit an expression profile that is comparable with Ag-experienced cells. However, it remains unclear whether the biochemical mechanisms required to induce the memory program are the same for T cells stimulated by high- versus low-affinity TCR ligands.
Indeed, the TCR is able to differentially signal depending on the ligand it recognizes (10–12). In line with this, recent data suggest that the TCR can selectively regulate the transduction of signals that drive T cell memory (2, 13, 14). We have previously shown that CD8 T cells bearing a point mutation in the TCRβ transmembrane domain (βTMDmut or MUT) acquire effector function but are uniquely defective in the development of memory in response to high-affinity TCR ligands (13). In this article, we show unexpectedly that lowering TCR affinity leads to a recovery in memory programming in MUT T cells. This process was dependent on NF-κB signaling, which was efficiently triggered by low- but not high-affinity TCR ligands in MUT CD8+ T cells. These findings support the idea that depending on TCR signal strength, there is differential assembly of the signaling machinery needed to generate memory.
Materials and Methods
Mice and reagents
C57BL/6, C57BL/6 Rag−/−, B6.SJL (CD45.1+), OT-1 (wild type [WT]), and MUT OT-1 TCR transgenic mice (13) were bred and maintained in accordance with University of Missouri Office of Animal Resources Animal Care and Use Committee or University of Minnesota Institutional Animal Care and Use Committee. OVA and Q4H7 peptides were from New England Peptides. rmIL-7, rmIL-15, and rmIL-12 were from Peprotech. Abs anti-CD90.1, −IL-7Rα, −CD44, −CD25, −CD122, –phospho-STAT-4, and −STAT-5 and −Bcl-2 were from BD Pharmingen. Anti-CD62L and anti-CD8α were from BD Pharmingen or Biolegend. Anti-CD45.1, −CD45.2, −CD11a (LFA-1), and −Ly6C were from eBioscience. Anti-pIκBα, −p-p65 NF-κB (Ser536), and −p-ERK1/2 were from Cell Signaling. Anti–granzyme B was from Invitrogen. Anti–α-tubulin was from Sigma. Anti-A20 (59A426) was from Millipore. Anti-Bim was from Enzo. Anti-p65 for confocal studies was from Santa Cruz. Intracellular staining was performed as described previously (13).
Flow cytometry
Cells were prepared for staining and assays as described previously (13). Frequencies and phenotype are based on the live gate (FSC/SSC) for lymphocytes and then on donor T cells (congenic marker+ CD8+). For determining phospho–NF-κB, geometric mean fluorescent intensity was normalized to a naive control. Cells were analyzed on a FACSCalibur, LSR II (BD Biosciences) and FlowJo (TreeStar) software.
Adoptive transfer and infections
Naive (1 × 103) CD8 T cells were purified from the lymph nodes of OT-1 or MUT Rag1−/− mice and transferred i.v. as performed by Hamilton et al. (7). LM-N4 and LM-Q4H7 strains were grown as described by Zehn et al. (3). All infections were performed i.v. at least 1 d after adoptive transfer of naive transgenic T cells. Listeria monocytogenes titers in the spleen were obtained as previously described (15).
Magnetic bead enrichment
Single-cell suspensions were generated from the spleens and lymph nodes of noninfected and LM-Q4H7–infected CD45.1+ mice that were previously adoptively transferred with WT and MUT CD45.2+ donor CD8 T cells. Total cells were stained with CD45.2-FITC Abs, then incubated with MACS anti-FITC microbeads (Miltenyi Biotech) and passed over magnetic MACS LS Columns (Miltenyi Biotech) to enrich for CD45.2+ WT or MUT CD8 donor T cells following a protocol similar to Akue et al. (16). Bound and unbound fractions from individual uninfected and LM-Q4H7–infected mice were stained and analyzed by flow cytometry for detection and phenotype of CD45.2+ WT or MUT CD8 T cells. No CD45.2+ cells were detected in the unbound fraction.
Immunoblot analysis
A total of 2–5 × 106 T cells were stimulated with OVA- or Q4H7-tetramer, and anti-CD28 were subjected to immunoblot as described by Daniels et al. (12) for p-IκBα signaling and p-ERK1/2 and A20 blots. Densitometry of the immunoblots was obtained using ImageJ.
Confocal microscopy
Microscopy was performed and analyzed as described by Teixeiro et al. (13).
Statistical analysis
For statistical analysis, two-tailed unpaired Student t test was applied using GraphPad Prism software. Significance was set at p < 0.05.
Results and Discussion
Memory-defective T cells recover memory development in response to low-affinity TCR ligands
In previous studies, we reported that OT-1 MUT cells are defective in memory development upon infection with L. monocytogenes expressing the high-affinity ligand OVA (LM-OVA) (13). Interestingly, T cells responding to low-affinity ligands are able to support memory development in the context of infection (5). To test whether the requirements for memory change depending on TCR signal strength, we determined whether MUT T cells differentiate into memory in response to low-affinity TCR ligands. For this, we chose a L. monocytogenes strain that expresses Q4H7, an OVA peptide variant that exhibits very low affinity for the OT-1 TCR (12) and induces memory differentiation of OT-I T cells (5, 17). WT and MUT CD8 T cells were adoptively transferred into congenic hosts and challenged with LM-OVA (strong) or LM-Q4H7 (weak). As expected, challenge with LM-OVA led to a marked decrease in the frequency of MUT CD8 T cells developing into memory and in the ability of MUT memory cells to respond to rechallenge (Fig. 1A, 1B, lower panels) (13). Surprisingly, in response to LM-Q4H7 infection, MUT T cells generated memory T cells at frequencies comparable with WT (Fig. 1B, upper panel). We confirmed this using magnetic bead enrichment procedures (Supplemental Fig. 1A) (16). WT and MUT cells reached similar frequencies and numbers at the peak (day 6) and memory points of the LM-Q4H7 immune response. The frequencies and number of T cells in the LM-Q4H7 response were substantially higher than in uninfected control mice (transferred in parallel with equal number of naive WT and MUT donor T cells). Furthermore, in response to LM-Q4H7, both WT and MUT displayed an effector (day 6) and memory (day 32) phenotype clearly distinct from matched noninfected controls where donor cells remained naive (Supplemental Fig. 1B). Importantly, MUT memory T cells primed with the low-affinity TCR ligand underwent robust secondary responses (to LM-OVA), similar to WT cells. This is in stark contrast with MUT cells primed with the high-affinity ligand OVA (Fig. 1A, 1B, lower panels), where MUT memory generation is defective. Hence, these data indicate that MUT cells are unexpectedly able to generate functional memory T cells in response to low-affinity TCR ligands.
FIGURE 1.
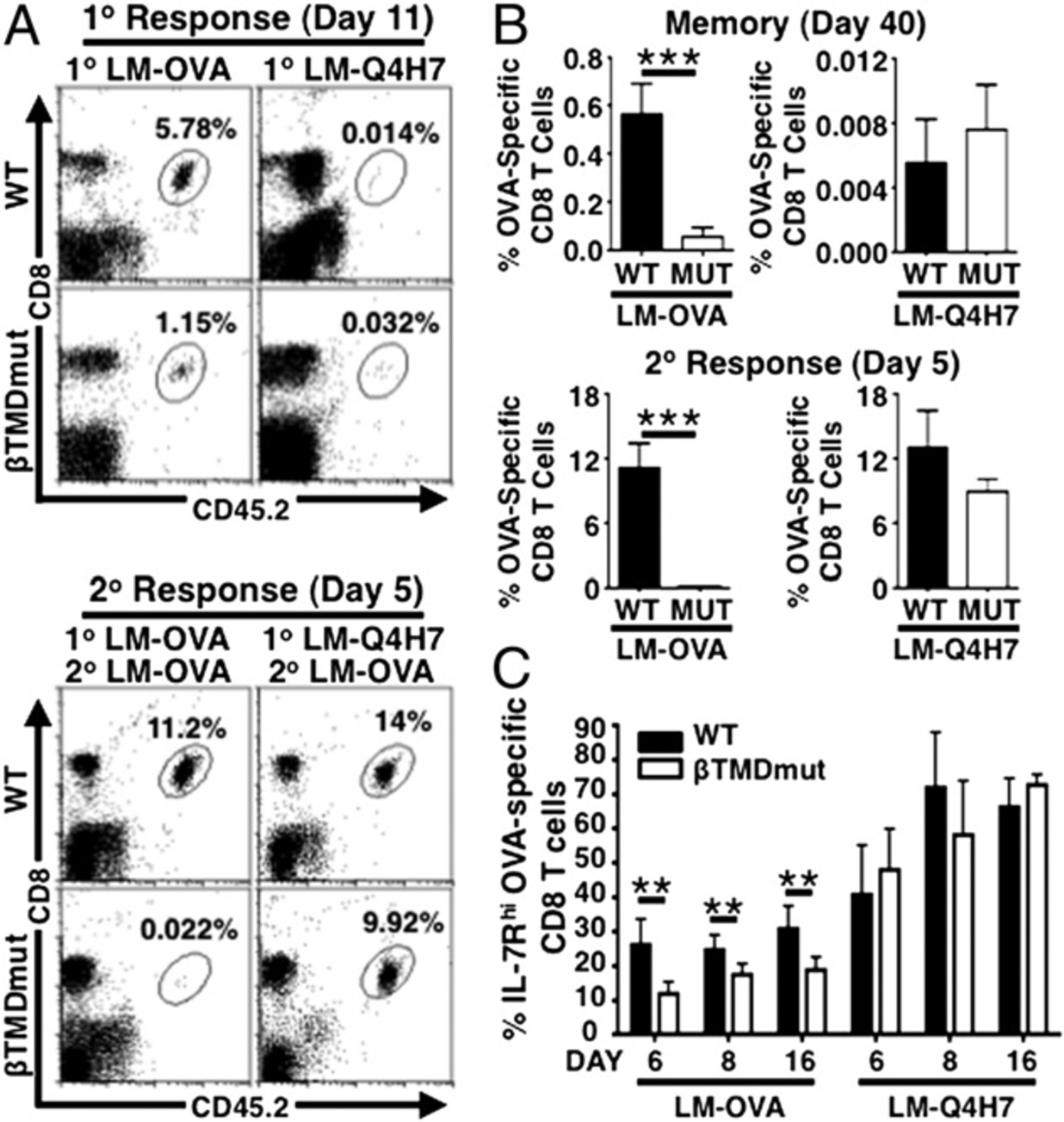
MUT memory defect is restored in response to low-affinity TCR ligands. (A and B) Naive T cells were transferred into B6.CD45.1+ hosts and challenged with 1 × 103 CFU LM-OVA or LM-Q4H7. (A) Representative plots show frequencies of OVA-specific CD8 T cells in blood at day 11 of 1° response (upper panel) and day 5 of 2° response (lower panel). (B) Graphs show frequencies of OVA-specific cells in blood at day 40 of 1° response (upper panel) and day 5 of 2° response (lower panel, mean ± SD) in experiments analogous to (A). (C) Graph shows frequencies of IL-7Rhi cells in blood determined by flow cytometry (mean ± SD). Data are representative of n ≥ 3 independent experiments, n ≥ 3 mice per group. **p ≤ 0.005, ***p ≤ 0.001.
Next, we measured the frequency of T cells differentiating into IL-7Rhi memory precursors. MUT cells exhibited a significantly lower frequency of IL-7Rhi memory precursors during the immune response to LM-OVA. In contrast, weak (low-affinity) TCR signals overcame this MUT defect, because frequencies of MUT IL-7Rhi memory precursors were restored to WT levels in response to LM-Q4H7 (Fig. 1C). These data suggest that mutant T cells are defective in memory programming in response to strong TCR ligands but recover the ability to program memory in response to weak TCR signals.
The inability of MUT cells to differentiate into memory against strong TCR ligands could not be attributed to altered apoptosis because WT and MUT exhibited similar levels of the regulators of contraction Bim and Bcl-2 (Supplemental Fig. 1C) (18). In addition, we did not find defects in IL-12R, IL-7R, and IL-15R signaling in MUT cells as measured by levels of phosphorylation of STAT-4 (as a readout for IL-12R signaling) and STAT-5 (as a readout of IL-7R and IL-15R signaling; Supplemental Fig. 1D). Thus, these data demonstrate that increased apoptosis or impaired cytokine signal input cannot account for the defective mutant memory phenotype. Rather, MUT cells are defective in the TCR signals that regulate memory programming in response to high-affinity TCR ligands. Overall, lowering TCR signal strength leads to a recovery in mutant memory differentiation. These findings unexpectedly reveal that the signals necessary for programming memory are qualitatively different depending on TCR signal strength.
Memory-defective T cells recover memory development in lymphopenic conditions
In lymphopenic conditions, T cell memory development also occurs in response to low-affinity TCR ligands but in the context of IL-7, IL-15, and low levels of inflammation (6, 19). To determine whether MUT cells were able to differentiate into HP memory, we adoptively transferred WT or MUT OT-1 CD8 T cells into sublethally irradiated B6 hosts. MUT T cells underwent normal homeostatic proliferation and acquired a memory-like phenotype, similar to the results obtained in the LM-Q4H7 response (Fig. 2A, 2B). Equal numbers of WT and MUT HP-memory cells were adoptively transferred into naive congenic hosts and challenged with LM-OVA to test HP memory cell function. Importantly, MUT HP memory T cells were able to expand and clear LM-OVA in the spleen comparable with WT and the true-memory control (Fig. 2C, 2D). Thus, MUT T cells also recovered memory development in response to lymphopenia. These data further support that memory development is differentially regulated depending on TCR signal strength, although it is possible that high doses of homeostatic cytokines also contribute to the TCR-dependent recovery of memory in these conditions.
FIGURE 2.
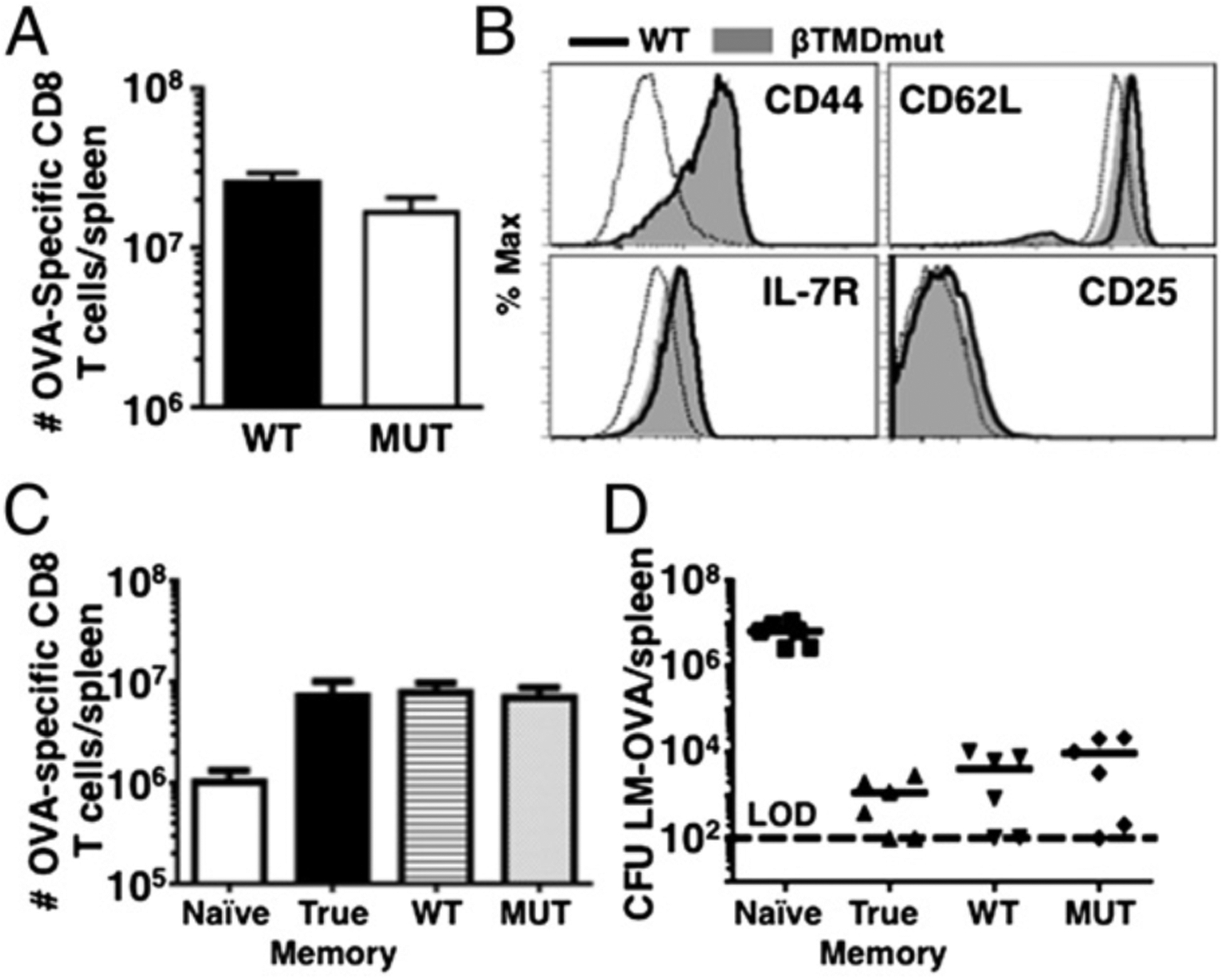
Memory-deficient MUT T cells recover memory development in response to lymphopenia. A total of 1–3 × 106 naive CD45.2+ T cells were transferred into sublethally irradiated hosts and allowed to differentiate into HP memory. (A) Representative graphs show total cell number at day 28 (mean ± SD). (B) Memory phenotype determined at day 28. Dashed line represents naive control. (C) A total of 3 × 105 HP memory cells from (A) were transferred to B6.CD45.1+ and challenged with 1 × 105 CFU LM-OVA. Graphs show total cell numbers (means ± SD) (C) and LM-OVA titers (D) at day 5 p.i. Data are representative of n ≥ 3 independent experiments, n ≥ 3 mice per experiment.
MUT T cells recover NF-κB signaling in response to low-affinity TCR signals
Next, we investigated the mechanism responsible for the recovery of MUT memory with weak TCR ligands (low affinity). MUT βTMD TCRs are impaired in transducing NF-κB signals in response to strong TCR ligands (13, 20, 21). Thus, we tested whether the MUT TCR could activate NF-κB signaling in response to low-affinity ligands. As previously described (13), upon OVA stimulation in vitro, MUT cells were defective in translocating the p65–NF-κB subunit to the nucleus (Fig. 3A). This correlated with a defect in the induction of p-IκBα, an upstream regulator of NF-κB signals (13, 22) (Fig. 3B). Remarkably, in response to the low-affinity ligand Q4H7, a similar percentage of WT and MUT cells translocated NF-κB to the nucleus, although the percentage observed was lower than upon OVA stimulation (Fig. 3A). Interestingly, WT and MUT cells stimulated with Q4H7 exhibited induction of p-IκBα levels similar to WT OVA stimulation (Fig. 3B). Together with the confocal data, this indicates that, on a per-cell basis, the NF-κB signal input is higher upon Q4H7 than OVA stimulation. Furthermore, MUT cells were impaired in keeping the balance between degradation and expression of the NF-κB target A20 upon OVA but not Q4H7 stimulation (23) (Fig. 3B). A20 is also a negative regulator of NF-κB signal, and we observed A20 degradation (A20p37 cleaved form) (24). This suggests that the MUT defect occurs upstream of A20. Indeed, we have previously described that MUT cells are defective in the translocation of protein kinase C θ (PKCθ) to the membrane, an event that is required to assemble the IκB kinase complex with the Carma1/Bcl10/Malt1 (CBM) signalosome (13, 25). Of note, there was not an overall enhancement in other TCR signaling pathways in response to Q4H7, as indicated by the levels of ERK phosphorylation (Fig. 3B). Finally, we determined whether the early NF-κB signaling defect observed in MUT cells in vitro would occur and persist in vivo in the context of infection. We monitored NF-κB signaling in vivo by measuring the IκB kinase complex–mediated phosphorylation of the NF-κB subunit p65 (p-NF-κB) (26). Notably, MUT T cells were impaired in the induction of p-NF-κB in the immune response to LM-OVA. Conversely, MUT cells recovered p-NF-κB levels in vivo when challenged with LM-Q4H7 (Supplemental Fig. 2). Taken together, these data show that TCR-dependent NF-κB signaling can be differentially regulated depending on TCR signal strength.
FIGURE 3.
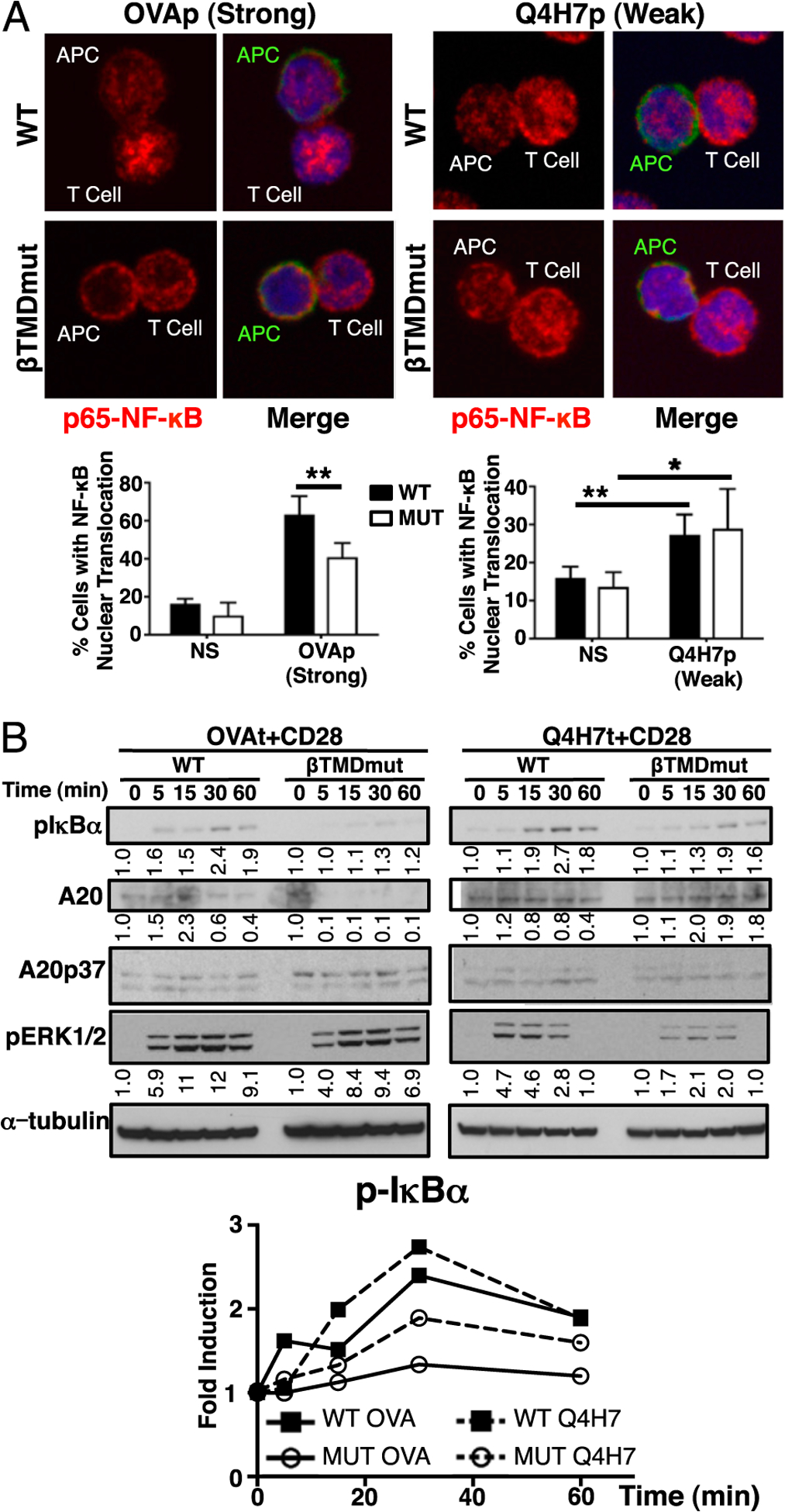
MUT T cells recover NF-κB signaling in response to low-affinity ligands. (A) Nuclear translocation of p65–NF-κB determined by confocal microscopy: CD45.1 (green), NF-κB (red), nucleus (blue). Images are representative of n ≥ 50 conjugates from n ≥ 3 independent experiments. Graphs show the mean percentage ± SD of conjugated T cells that exhibit p65 in the nucleus over unstimulated cell levels. Original magnification ×60. (B) Phosphorylation of IκBα, ERK1/2, and expression of A20 in response to high- and low-affinity TCR ligands was determined in WT or MUT T cells stimulated with Kb-OVA or -Q4H7 tetramers and anti-CD28 Abs by immuno-blot. All immunoblots correspond to the same representative experiment. A representative experiment of n = 4 is shown. Graph and numbers in the blot show relative induction of p-IκBα, A20, p-ERK1/2 levels over unstimulated (time 0), normalized to α-tubulin (loading control). *p ≤ 0.05, **p ≤ 0.005.
Our previous data indicated that the βTMD has a role in the assembly of the PKCθ–CBM complex in the membrane (13, 21). It has been proposed that digitization of NF-κB signaling occurs upstream or at the level of the TCR such that only strong TCR ligands will generate efficient signal to induce gene expression (27). Instead of this, the data presented in this article suggest that even weak TCR signals can produce efficient NF-κB signaling that persists during the immune response. It is possible that the βTMD plays a role in integrating the antigenic signal input to determine which cells acquire productive NF-κB signaling required to generate memory. Alternatively, other TCR-dependent downstream mechanisms that control NF-κB induction or act together with NF-κB signals to drive memory programming may be differentially regulated by high- and low-affinity TCR ligands. It is still unclear how the TCR regulates early signaling events upstream of the PKCθ–CBM complex. Thus, defining whether the mechanisms that differentially regulate memory through the TCR lie upstream or downstream of PKCθ–CBM is an exciting area that needs further investigation.
Several studies have explored the impact of TCR signal strength on T cell memory development. It has been shown that changing the dose of high-affinity ligands influences T cell memory (5, 14, 28, 29); in contrast, changing TCR affinity does not (5, 14, 28, 29). Thus, it is possible that the quantity of TCR signal alone is not sufficient to define a T cell’s ability to differentiate into memory. The results presented in this article provide evidence that weak TCR signals can support memory in conditions where strong TCR signals fail. Hence, we favor the hypothesis that high- and low-affinity ligands signal qualitatively different to promote memory programming. Intriguingly, this implies that the diversity of signals generated from the TCR that control T cell memory might be more complex than previously appreciated. This may also contribute to the diversity of the CD8 memory pool (30, 31).
In summary, the results from this study fit a model where T cell memory programming is qualitatively different depending on TCR signal strength. This offers the possibility of specifically targeting TCR antigenic signals to improve memory generation in vaccine regimens. Thus, the βTMD mutant model rises as an ideal platform to provide deeper insight into the structural and molecular mechanisms that can enable the manipulation of T cell memory.
Supplementary Material
Acknowledgments
We thank M. Bevan and D. Zehn for providing Listeria strains. We thank Cody Cunningham for technical assistance.
This work was supported by a University of Missouri Life Sciences fellowship (to K.M.K.), the Leukemia and Lymphoma Society Career Development Program (to S.E.H.), and grants from the University of Missouri Mission Enhancement Fund and University of Missouri Research Board (to E.T. and M.A.D.).
Abbreviations used in this article:
- CBM
Carma1/Bcl10/Malt1
- HP
homeostatically proliferate
- LM-OVA
Listeria monocytogenes expressing the high-affinity ligand OVA
- PKCθ
protein kinase C θ
- p-NF-κB
phosphorylation of the NF-κB subunit p65
- βTMDmut or MUT
mutation in the TCRβ transmembrane domain
- WT
wild type
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.de Visser KE, Cordaro TA, Kioussis D, Haanen JB, Schumacher TN, and Kruisbeek AM. 2000. Tracing and characterization of the low-avidity self-specific T cell repertoire. Eur. J. Immunol 30: 1458–1468. [DOI] [PubMed] [Google Scholar]
- 2.Smith-Garvin JE, Burns JC, Gohil M, Zou T, Kim JS, Maltzman JS, Wherry EJ, Koretzky GA, and Jordan MS. 2010. T-cell receptor signals direct the composition and function of the memory CD8+ T-cell pool. Blood 116: 5548–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zehn D, Turner MJ, Lefrançois L, and Bevan MJ. 2010. Lack of original antigenic sin in recall CD8(+) T cell responses. J. Immunol 184: 6320–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kieper WC, and Jameson SC. 1999. Homeostatic expansion and phenotypic conversion of naı¨ve T cells in response to self peptide/MHC ligands. Proc. Natl. Acad. Sci. USA 96: 13306–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zehn D, Lee SY, and Bevan MJ. 2009. Complete but curtailed T-cell response to very low-affinity antigen. Nature 458: 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldrath AW, and Bevan MJ. 1999. Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity 11: 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamilton SE, Wolkers MC, Schoenberger SP, and Jameson SC. 2006. The generation of protective memory-like CD8+ T cells during homeostatic proliferation requires CD4+ T cells. Nat. Immunol 7: 475–481. [DOI] [PubMed] [Google Scholar]
- 8.Surh CD, and Sprent J. 2008. Homeostasis of naive and memory T cells. Immunity 29: 848–862. [DOI] [PubMed] [Google Scholar]
- 9.Cheung KP, Yang E, and Goldrath AW. 2009. Memory-like CD8+ T cells generated during homeostatic proliferation defer to antigen-experienced memory cells. J. Immunol 183: 3364–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards LJ, and Evavold BD. 2011. T cell recognition of weak ligands: roles of signaling, receptor number, and affinity. Immunol. Res 50: 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corse E, Gottschalk RA, and Allison JP. 2011. Strength of TCR-peptide/MHC interactions and in vivo T cell responses. Journal of immunology (Baltimore, Md: 1950) 186: 5039–5045. [DOI] [PubMed] [Google Scholar]
- 12.Daniels MA, Teixeiro E, Gill J, Hausmann B, Roubaty D, Holmberg K, Werlen G, Holländer GA, Gascoigne NRJ, and Palmer E. 2006. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature 444: 724–729. [DOI] [PubMed] [Google Scholar]
- 13.Teixeiro E, Daniels MA, Hamilton SE, Schrum AG, Bragado R, Jameson SC, and Palmer E. 2009. Different T cell receptor signals determine CD8+ memory versus effector development. Science 323: 502–505. [DOI] [PubMed] [Google Scholar]
- 14.Leignadier J, and Labrecque N. 2010. Epitope density influences CD8+ memory T cell differentiation. PLoS ONE 5: e13740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamilton SE, Badovinac VP, Khanolkar A, and Harty JT. 2006. Listeriolysin O-deficient Listeria monocytogenes as a vaccine delivery vehicle: antigen-specific CD8 T cell priming and protective immunity. J. Immunol 177: 4012–4020. [DOI] [PubMed] [Google Scholar]
- 16.Akue AD, Lee JY, and Jameson SC. 2012. Derivation and maintenance of virtual memory CD8 T cells. J. Immunol 188: 2516–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King CG, Koehli S, Hausmann B, Schmaler M, Zehn D, and Palmer E. 2012. T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity 37: 709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurtulus S, Tripathi P, Opferman JT, and Hildeman DA. 2010. Contracting the ‘mus cells’—does down-sizing suit us for diving into the memory pool? Immunol. Rev 236: 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamilton SE, and Jameson SC. 2008. The nature of the lymphopenic environment dictates protective function of homeostatic-memory CD8+ T cells. Proc. Natl. Acad. Sci. USA 105: 18484–18489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teixeiro E, García-Sahuquillo A, Alarcón B, and Bragado R. 1999. Apoptosis-resistant T cells have a deficiency in NF-kappaB-mediated induction of Fas ligand transcription. Eur. J. Immunol 29: 745–754. [DOI] [PubMed] [Google Scholar]
- 21.Teixeiro E, Daniels MA, Hausmann B, Schrum AG, Naeher D, Luescher I, Thome M, Bragado R, and Palmer E. 2004. T cell division and death are segregated by mutation of TCRbeta chain constant domains. Immunity 21: 515–526. [DOI] [PubMed] [Google Scholar]
- 22.Vallabhapurapu S, and Karin M. 2009. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol 27: 693–733. [DOI] [PubMed] [Google Scholar]
- 23.Krikos A, Laherty CD, and Dixit VM. 1992. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J. Biol. Chem 267: 17971–17976. [PubMed] [Google Scholar]
- 24.Düwel M, Welteke V, Oeckinghaus A, Baens M, Kloo B, Ferch U, Darnay BG, Ruland J, Marynen P, and Krappmann D. 2009. A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J. Immunol 182: 7718–7728. [DOI] [PubMed] [Google Scholar]
- 25.Paul S, and Schaefer BC. 2013. A new look at T cell receptor signaling to nuclear factor-κB. Trends Immunol. 34: 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayden MS, and Ghosh S. 2008. Shared principles in NF-kappaB signaling. Cell 132: 344–362. [DOI] [PubMed] [Google Scholar]
- 27.Kingeter LM, Paul S, Maynard SK, Cartwright NG, and Schaefer BC. 2010. Cutting edge: TCR ligation triggers digital activation of NF-kappaB. J. Immunol 185: 4520–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prlic M, Hernandez-Hoyos G, and Bevan MJ. 2006. Duration of the initial TCR stimulus controls the magnitude but not functionality of the CD8+ T cell response. J. Exp. Med 203: 2135–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corse E, Gottschalk RA, Krogsgaard M, and Allison JP. 2010. Attenuated T cell responses to a high-potency ligand in vivo. PLoS Biol. 8: e1000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jameson SC, and Masopust D. 2009. Diversity in T cell memory: an embarrassment of riches. Immunity 31: 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerlach C, Rohr JC, Perié L, van Rooij N, van Heijst JW, Velds A, Urbanus J, Naik SH, Jacobs H, Beltman JB, et al. 2013. Heterogeneous differentiation patterns of individual CD8+ T cells. Science 340: 635–639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.