

Abstract
Since the discovery of insulin 100 years ago, our knowledge and understanding of diabetes have grown exponentially. Specifically, with regards to the genetics underlying diabetes risk, our discoveries have paralleled developments in our understanding of the human genome and our ability to study genomics at scale; these advancements in genetics have both accompanied and led to those in diabetes treatment. This review will explore the timeline and history of gene discovery and how this has coincided with progress in the fields of genomics. Examples of genetic causes of monogenic diabetes are presented and the continuing expansion of allelic series in these genes and the challenges these now cause for diagnostic interpretation along with opportunities for patient stratification are discussed.
Keywords: diabetes, pancreas, genomics, b-cell development
Introduction
It was in the 1930s, after the discovery and first use of insulin, that Harold Percival Himsworth first made the distinction between type 1 and type 2 diabetes (Bryder & Harper 2013). However, it was not until the 1970s, following decades of observation, that both Stefan Fajans and Robert Tattersall extended this classification to recognize the existence of diabetes subtypes inherited in an autosomal dominant manner (Tattersall 1974, Tattersall & Fajans 1975) (Fig. 1). This dominantly inherited form of diabetes was originally reported as ‘mild familial diabetes with dominant inheritance’, and subsequently ‘maturity-onset diabetes of the young (MODY)’, to distinguish it from juvenile onset type 1 diabetes and indicate its similarities in clinical presentation to maturity-onset diabetes (now known as type 2) (Tattersall & Fajans 1975). In recent years, we have seen a gradual move toward using the broader term ‘monogenic diabetes’ to encompass other genetic types of diabetes, such as neonatal or syndromic diabetes. Fueled by steady improvements in our understanding of the human genome, and coupled with technological advancement, we have seen the careful dissection of the genetic causes of these monogenic forms of diabetes. This has provided numerous insights into the machinery responsible for making and maintaining functional insulin-secreting pancreatic β-cells (Fig. 2). In this review, we focused on how these discoveries have shaped our understanding of insulin secretion across the phenotypic spectrum of diabetes and provided a model for precision medicine in diabetes.
Figure 1.
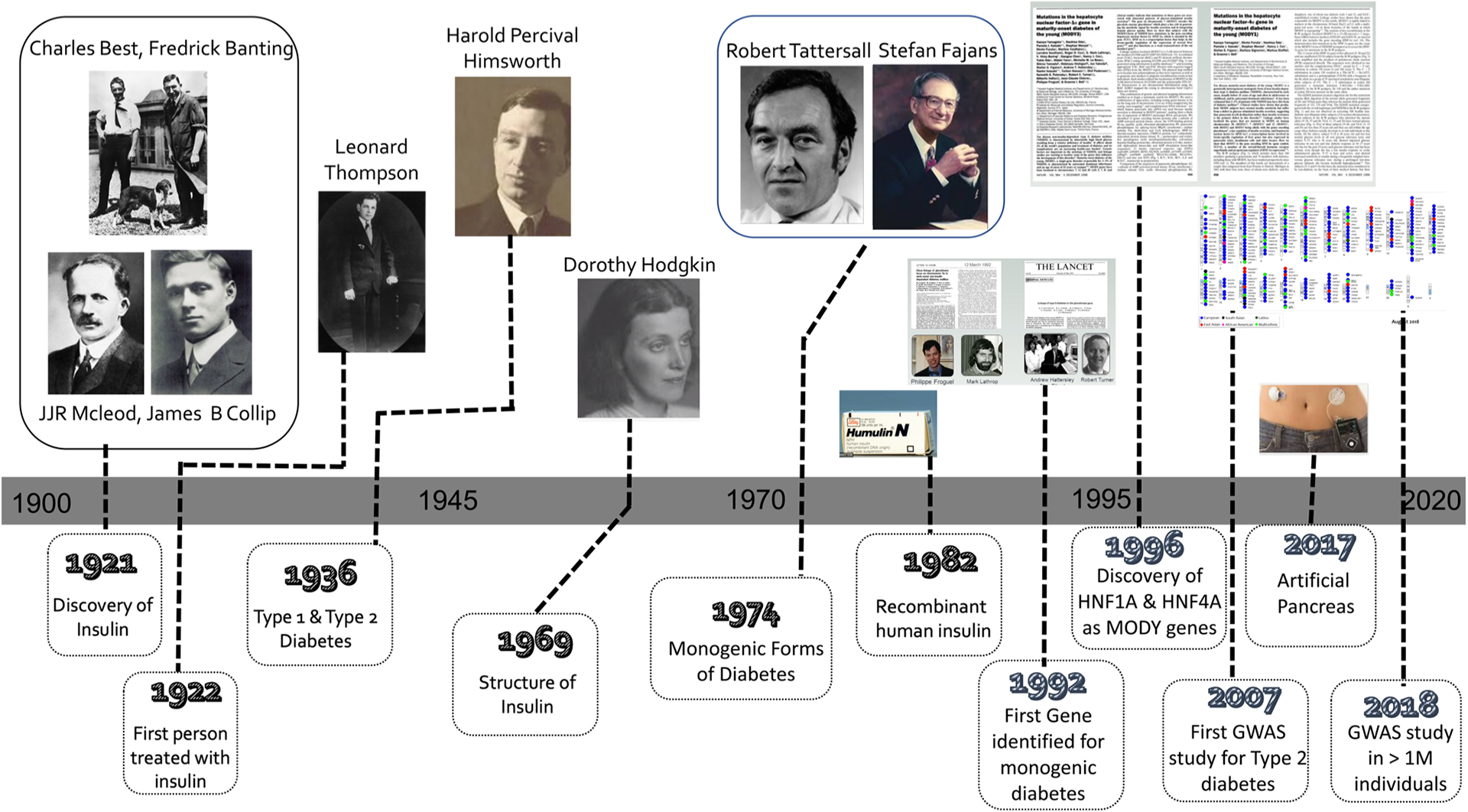
Timeline indicating key genetic discoveries in the context of our understanding of the hormone insulin and its use therapeutically.
Figure 2.
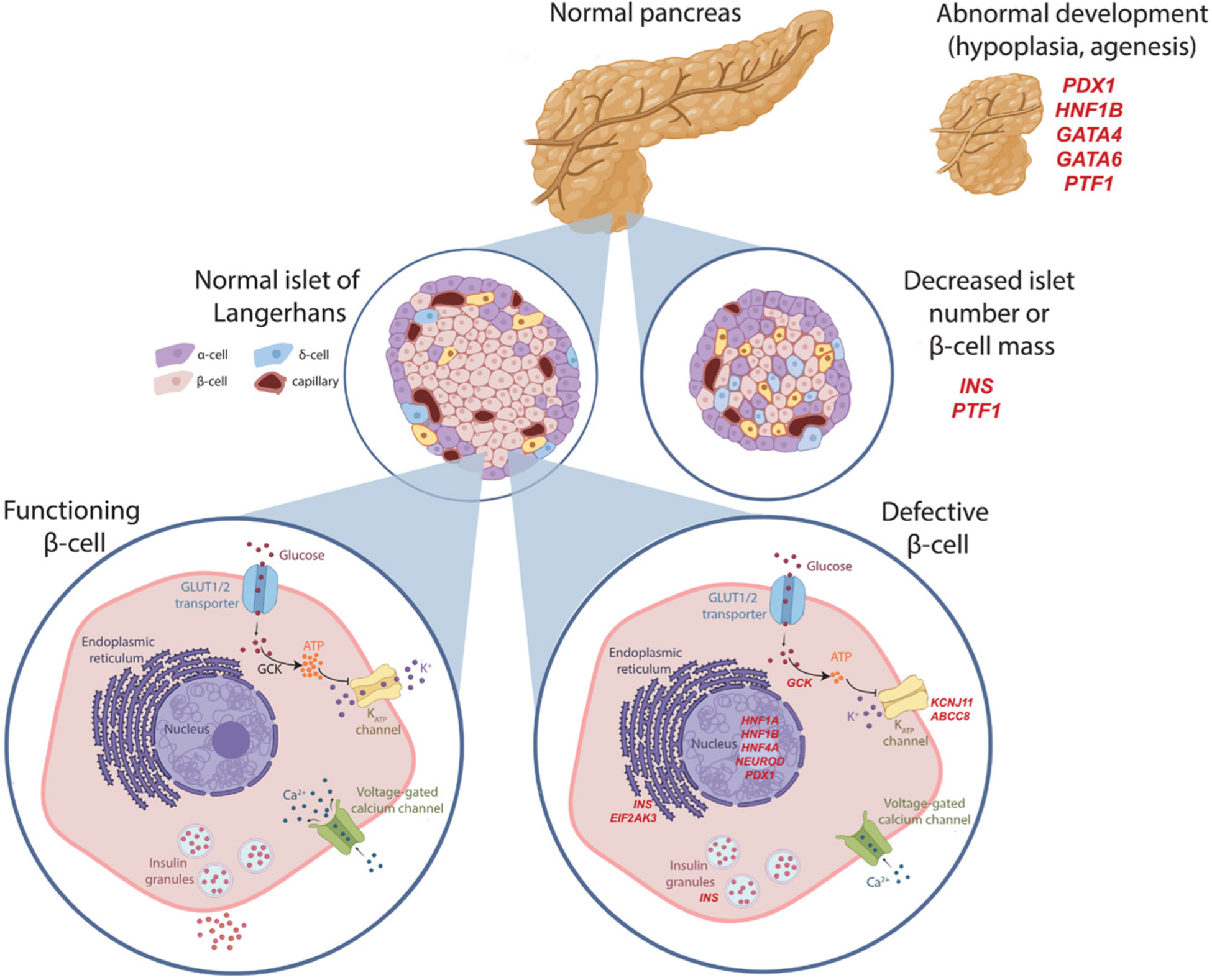
Schematic representation of the pancreatic β-cell illustrating the three main mechanisms by which monogenic diabetes arises. Gene defects of selected representative genes at each stage indicated in red italics. Defect in pancreas/β-cell development leading to pancreatic agenesis or hypoplasia (PDX1, HNF1B, GATA4, GATA6, PTF1A), decreased β-cell mass or proliferations (INS, PTF1A) or β-cell dysfunction, including glucose sensing (GCK), ATP responsiveness (ABCC8, KCNJ11), ER stress (INS, EIF2AK3), and transcriptional regulation (HNF1A, HNF1B, HNF4A, NEUROD1, PDX1). Created with BioRender.com.
Recognition of autosomal dominantly inherited diabetes
Maturity-onset diabetes of the young (MODY) is defined as autosomal, dominantly inherited diabetes which is characterized by non-insulin dependence, with an early age of onset (at least one affected family member with an onset before 25 years of age) and pancreatic β-cell dysfunction. In the 1950s, Fajans and Conn were attempting to develop a method for earlier detection of diabetes; whilst on this quest, they discovered ‘unsuspected diabetes’ in about 20% of subjects who had a close relative with diabetes. They noted that not only was the unsuspected ‘chemical diabetes’ distinct from juvenile-onset diabetes (now known as type 1 diabetes) but also that the inheritance was different (Fajans & Conn 1959).
Given what we now know, it is particularly fascinating that the early descriptions of MODY in the literature indicate that subjects were often treated with sulfonylureas to maintain their glucose levels (Fajans & Brown 1993). Even before the various subtypes of MODY were genetically dissected, it was apparent that there were families with distinct clinical phenotypes. Some cases were characterized by years of mild fasting hyperglycemia, whereas others progressed to persistent fasting hyperglycemia, often preceded by varying degrees of glucose intolerance (Fajans 1987, 1990). Of course, this now fits perfectly with our understanding of the differences in the underlying pathophysiology of glucokinase (GCK) vs transcription factor (HNF1A/HNF4A/HNF1B) MODY.
At the time of writing this article, the number of genetic subtypes causing MODY differs depending on the source of information. Online Mendelian inheritance in man (OMIM) lists 14 genetic subtypes; however, not all of these meet the American College of Medical Genetics criteria and a shorter list of 10 subtypes is more broadly acknowledged (Zhang et al. 2021). While there are multiple genetic causes, most cases in the clinical setting are due to mutations in three of these genes (HNF1A, HNF4A, GCK) which can collectively account for up to 95% of all MODY cases (Shields et al. 2010).
Subtypes of monogenic diabetes
Although we still use the name maturity-onset diabetes of the young (MODY), the field has moved in recent years to rebrand these conditions to the broader monogenic diabetes and to use the names of the genes responsible, for example, ‘glucokinase diabetes’ or ‘transcription factor diabetes’ (Ellard et al. 2008, McDonald & Ellard 2013). This shift in naming convention reflects our broadening understanding of the wide phenotypic spectrum which can result from a given gene mutation, leading to blurred boundaries between historic non-genetic nomenclature which relied on the age of diagnosis (e.g. MODY vs neonatal diabetes). As we unravel the genetic causes for monogenic diabetes (Fig. 1), the rational for this shift in thinking will become apparent along with the emerging themes of monogenic forms of pancreatic β-cell dysfunction – largely arising from either an intrinsic defect in β-cell function (e.g. GCK, KCNJ11), pancreas development (e.g. PDX1, HNF1A) or β-cell proliferation and mass (e.g. INS). This review will focus on the most common genes connected with monogenic diabetes while also highlighting a selection of less commonly seen genes to illustrate additional pathologic mechanisms.
Hepatocyte nuclear factor 4 alpha (HNF4A-MODY, MODY1)
In 1992, a locus on chromosome 20p containing HNF4A was identified by linkage analysis in a large pedigree, known as the RW pedigree (Bowden et al. 1992). However, it was not until 1996 that back-to-back publications reported the positional cloning of the genes responsible for both MODY1 (HNF4A) and MODY3 (HNF1A). Prior to this discovery, both transcription factors had recognized roles in the liver but these studies unearthed a previously unknown role in the regulation of pancreatic β-cell development and function (Yamagata et al. 1996a,b).
HNF4A belongs to a group of hepatocyte nuclear transcription factors, some of which are important in regulating insulin secretion (including HNF1A and HNF1B discussed below) (Yamagata et al. 1996a). As the name suggests, these transcription factors were originally described in the liver but they also function within the pancreatic β-cell to directly regulate the gene expression important for both pancreatic islet-cell development as well as glucose metabolism, including the insulin gene itself (Yamagata et al. 1996a).
Patients with heterozygous loss-of-function mutations in HNF4A have a progressive decline in β-cell function, with most carriers developing diabetes in adolescence or early adulthood. Because HNF4A also has a role in cholesterol and fatty acid metabolism, individuals with HNF4A-MODY may have unique changes in cholesterol and triglyceride levels (Pearson et al. 2005). Importantly, a diagnosis of HNF4A-MODY has implications for treatment as patients can often be successfully treated with oral sulfonylureas rather than insulin (Pearson et al. 2003, Hattersley et al. 2018).
Glucokinase (GCK-MODY, MODY2)
Shortly after the reports of linkage on chromosome 20p, linkage to chromosome 7p, where the gene for the key glycolytic enzyme glucokinase (GCK) resides, was described in both French and UK pedigrees in 1992 (Froguel et al. 1992, Hattersley et al. 1992) (Fig. 1). The demonstration of linkage was quickly followed by characterization of the GCK human gene and the detection of diabetes-causing mutations (Stoffel et al. 1992). GCK is a member of the hexokinase family of enzymes and is largely expressed in hepatocytes and pancreatic β-cells where it is responsible for catalyzing the transfer of phosphate from ATP to glucose, generating glucose-6-phosphate (Magnuson et al. 1989). This catalytic transfer is the first, rate-limiting step in glucose metabolism, and as such GCK is often considered the glucose sensor of the β-cell (Bedoya et al. 1986). A single inactivating mutation in GCK impairs that glucose sensor, as well as the β-cell ability to respond to increasing glucose concentrations, ultimately leading to MODY. Thus, hyperglycemia in individuals with GCK-MODY is essentially caused by increasing the glucose set point that is necessary to induce insulin secretion (Velho et al. 1992). To date, in vitro studies have indicated mutation-specific variation in the severity of the functional defect; however, in the clinical setting, heterozygous inactivating mutations result in strikingly similar phenotypes, irrespective of the specific mutation (Stride et al. 2002). This observation is explained by compensation by the WT allele, which is post-translationally upregulated in response to the elevated circulating glucose levels (Gloyn et al. 2004c). In contrast, activating GCK mutations lead to over-secretion of insulin and hypoglycemia, and given the lack of compensation by the WT allele, the phenotype is more varied (Glaser et al. 1998, Gloyn 2003, Cuesta-Muñoz et al. 2004).
Patients with GCK-MODY have stable mild fasting hyperglycemia throughout their lifespan; however, due to the mild degree of hyperglycemia, and the low incidence of micro- or macrovascular complications, individuals with GCK-MODY typically do not require pharmacological treatment (Steele et al. 2014). The exception to this is during pregnancy, where glucose-lowering medications may be considered in the case of increased fetal growth observed on ultrasound (Chakera et al. 2015).
Hepatocyte nuclear factor 1 alpha (HNF1A-MODY, MODY3)
Positional cloning of the linkage signal on chromosome 12p, which had been identified in multiple families with monogenic diabetes, identified another liver transcription factor gene, hepatocyte nuclear factor 1 alpha (HNF1A), as a major cause of monogenic diabetes (Yamagata et al. 1996b). Like HNF4A, HNF1A functions as an important transcriptional regulator of insulin and other genes involved in islet-cell development and glucose metabolism. HNF1A is also a key transcriptional regulator of the sodium-glucose transporter in the kidney (SGLT2), therefore, patients with HNF1A-MODY have a low renal threshold for glucose (Pontoglio et al. 2000).
Functional studies have demonstrated that protein-truncating mutations result in haploinsufficiency (Harries et al. 2004), whilst missense variants lead to loss of function through a variety of mechanisms (Althari et al. 2020). More recently, our appreciation for the role of HNF1A in MODY has been expanded, as homozygous hypomorphic variants have also been identified as a cause of MODY (Misra et al. 2020). These observations fit with a growing body of evidence demonstrating that genetic variation across the functional severity spectrum corresponds to phenotype (Althari et al. 2020).
HNF1A-MODY is characterized by normal glucose tolerance early in childhood, with progressive hyperglycemia developing in adolescence or adulthood. Because of the progressive nature of HNF1A-MODY, these individuals are at risk for microvascular complications and do require glucose-lowering therapy (Lee et al. 1999, Hattersley et al. 2018). Importantly, as with a diagnosis of HNF4A-MODY, patients with HNF1A-MODY can usually be treated with oral sulfonylureas rather than insulin (Pearson et al. 2003, Hattersley et al. 2018).
Pancreatic duodenal homeobox factor 1 (PDX1-MODY, MODY4)
Pancreatic duodenal homeobox factor 1 (PDX1), previously referred to as insulin promoter factor 1 (IPF1), is yet another transcription factor involved in monogenic diabetes. Located on chromosome 13, PDX1 plays a central role in determining cell fate in embryonic pancreatic development, and also in the differentiation, maturation, and function of pancreatic β-cells (Stoffel et al. 1995, Inoue et al. 1996). In 1997, Stoffers and colleagues shed light on this critical transcription factor first by demonstrating that homozygous loss resulted in pancreatic agenesis and neonatal diabetes (NDM) (Stoffers et al. 1997b) and secondly, that heterozygous carriers of these mutations had defective insulin secretion, consistent with previously described MODY phenotypes (Stoffers et al. 1997a).
PDX1-MODY is characterized by often having a slightly later age of onset than other forms of MODY (Stoffers et al. 1997a). Some patients have only mildly impaired glucose tolerance while others have fulminant diabetes requiring daily insulin injections. Although early reports suggested that variants in PDX1 were associated with increased risk of diabetes due to impaired PDX1-mediated INS expression (Ahlgren et al. 1998), larger genome-wide studies have not substantiated these early findings (Edghill et al. 2011). Since there is variation in phenotype within PDX1-MODY, the treatment must be tailored to the individual patient; options range from dietary changes alone to sulfonylureas or insulin (Hattersley et al. 2018, Delvecchio et al. 2020).
Hepatocyte nuclear factor 1 beta (HNF1B-MODY, MODY5)
HNF1B is closely related to HNF1A and in fact actually dimerizes with it (Mendel et al. 1991). Unsurprisingly, it plays a similarly important function in pancreatic embryonic development but unlike HNF1A, it also has a vital role in nephron embryonic development (Horikawa et al. 1997). This leads to a wide spectrum of phenotypes which may include diabetes or kidney disease alone or a combination of the two. Kidney disease, typically renal cysts, is somewhat unique to HNF1B-MODY, and so should be expected in young patients presenting with both diabetes and kidney disease. HNF1B-MODY can also be complicated by urogenital tract malformations, hyperuricemia, and gout (Firdous et al. 2018). Interestingly, patients with HNF1B-MODY display both defects in insulin secretion and insulin sensitivity (Pearson et al. 2004).
Neurogenic differentiation-1 (NEUROD1-MODY, MODY6)
Neurogenic differentiation-1 (NEUROD1, also known as BETA2) is a helix-loop-helix (HLH) transcription factor that functions as a regulatory switch for pancreatic and neuronal development. One important role of NEUROD1 is in directly regulating insulin gene (INS) expression by binding to a critical E-box motif on the INS promoter (Malecki et al. 1999). NEUROD1 also has important roles in activating other genes encoding components of the insulin secretion pathway within the β-cell, including ABCC8, GCK, and PAX6 (Kim et al. 2002, Marsich et al. 2003, Moates et al. 2003).
In 2001, Kristinsson and colleagues confirmed that heterozygous NEUROD1 mutations are a rare cause of MODY (Kristinsson et al. 2001), while homozygous loss-of-function mutations lead to NDM and neurological impairment (Rubio-Cabezas et al. 2010). NEUROD1-MODY can be diagnosed throughout the lifespan, as early as childhood and into the seventh decade most likely reflecting the incomplete penetrance of some variants and requirement for additional environmental or genetic risk factors. As with many of the other MODY subtypes, clinical presentation and course can be quite variable even within a single family (Delvecchio et al. 2020); approximately 20% of patients require insulin therapy, whilst others may respond well to dietary modifications alone or in combination with oral glucose-lowering agents.
Genetic causes of monogenic diabetes outside the β-cell
Mutations in CEL, encoding carboxy-ester lipase, cause young-onset diabetes coupled with exocrine pancreas dysfunction (CEL-MODY), which is characterized by low fecal elastase levels (Raeder et al. 2006). It is notable that the disease-causing mutations are caused by alterations in the variable number of tandem repeats (VNTR) region rather than in the protein-coding sequence (Raeder et al. 2006). This variety of monogenic diabetes is unusual as it does not appear to directly involve defects in the β-cell rather it involves the acinar cells of the pancreas and may involve defects in mitogen-activating protein kinase (MAPK) signaling (Ræder et al. 2014). This atypical monogenetic diabetes may prove to be a paradigm for other types of diabetes as multiple recent studies have suggested a role for the exocrine pancreas in mediating genetic association signals for both type 1 and type 2 diabetes (Ng et al. 2019, Chiou et al. 2021).
Monogenic diabetes diagnosed in the first 6 months of life
Many of the early strides in identifying genetic diabetes, particularly in describing key MODY genes, relied on chromosomal linkage analysis. In 2001, the Human Genome Project published the first complete sequence for the human genome, and with that, sparked a new era in genetic discoveries (Lander et al. 2001). As our genetic capabilities continued to progress, so did our understanding of genetic regulation of insulin secretion and related disorders, specifically in regards to neonatal diabetes mellitus (NDM). NDM was first described in 1852 by J F Kitselle and is often complicated by poor in utero growth and neonatal failure to thrive (Shield 2000). NDM is a heterogeneous disease defined as diabetes with onset during the first 6 months of life with a monogenic cause identifiable in up to 85% of cases (Letourneau & Greeley 2018). NDM exists on a spectrum, with mild forms being transient (TNDM) and resolving spontaneously by 18 months of life, permanent forms (PNDM) which do not resolve, and syndromic forms that may be accompanied by a number of other complications. It is worth noting that many of the genes described above in causing MODY can also be related to NDM, with a strong genotype-phenotype correlation. Of the >80% of NDM cases with a known genetic cause, more than 18 causative genes have been described in the literature but many of these are very rare and only seen in a small number of families (Delépine et al. 2000, Gloyn et al. 2004c, Babenko et al. 2006, Støy et al. 2007, Flanagan et al. 2014, De Franco et al. 2015, 2017, 2019, 2020a, Johnson et al. 2017, De Franco 2020). In this review, we highlight the most common varieties and those which have informed our current thinking on β-cell biology and the continuums of risk in diabetes.
ATP-sensitive potassium channel (KATP) defects (KATP-NDM)
Mutations in pancreatic KATP channel genes are the most common known causes of NDM. These channels, composed of four SUR1 subunits (encoded by ABCC8) and four Kir6.2 subunits (encoded by KCNJ11), are critical for regulating membrane excitability and insulin secretion by the β-cell (Shimomura & Maejima 2017). At low plasma glucose concentration, KATP channels are normally open, the cell membrane is hyperpolarized, and voltage-dependent calcium channels (VDCCs) are closed, thus inhibiting insulin secretion. Glucose metabolism increases the intracellular (ATP)/(ADP) ratio, closing KATP channels, leading to membrane depolarization, calcium influx through VDCCs, and triggering insulin release.
The role of these two KATP channel subunits in NDM was first appreciated in 2004 when heterozygous activating mutations in the KCNJ11 gene were reported as a major cause of permanent neonatal diabetes (Gloyn et al. 2004b). A number of additional causative mutations were subsequently identified in KCNJ11 (Gloyn et al. 2004a, Sagen et al. 2004, Yorifuji et al. 2005). The physiologic role of these specific mutations was further confirmed by co-expressing mutated Kir6.2 and demonstrating a significant decrease in response to ATP by the KATP channel (Gloyn et al. 2004a,b, Tammaro et al. 2005). In 2005, it was further demonstrated that distinct mutations in KCNJ11 can also lead to transient, or remitting, diabetes, suggesting that certain abnormalities within this channel can lead to a fluctuating phenotype (Gloyn et al. 2005). Shortly after, it was also demonstrated that some patients with particular KCNJ11 mutations have diabetes complicated by additional neurological features, a syndrome which has been termed developmental delay, epilepsy and neonatal diabetes (DEND) (Gloyn et al. 2006). Around this same time, Babenko et al. identified seven novel activating mutations in ABCC8, which also lead to NDM (Babenko et al. 2006). In direct contrast to the activating mutations leading to NDM, inactivating mutations in these same genes result in reduced KATP channel activity, β-cell hyperexcitability, and excessive insulin secretion (Kane et al. 1996, Nichols et al. 1996, Thomas et al. 1996, Dunne et al. 1997, Huopio et al. 2000).
The discovery linking the KATP channel mutations to neonatal diabetes was critical for patient care – prior to this knowledge, individuals with neonatal diabetes required lifelong treatment with insulin for survival. However, since this pathology is directly due to a failure of the KATP channels to close in response to ATP generated by glucose metabolism, the majority of patients with this variety of diabetes are responsive to oral sulfonylureas, which act on SUR1 to close the channel in an ATP-independent manner restoring insulin secretion (Gloyn et al. 2004a, Sagen et al. 2004). This discovery has enabled many affected patients to discontinue insulin use with great success, and often achieving better glycemic control with sulfonylureas than with their prior insulin therapy. More recent studies have shown that the majority of these patients still had excellent glycemic control at 10 year follow-up, confirming sulfonylureas as a safe and effective approach over the long-term (Bowman et al. 2018). This responsiveness to oral medication, along with a strong genotype-phenotype correlation (Flanagan et al. 2006) which can guide medical decision-making and family counseling, makes an early molecular diagnosis for NDM imperative.
INS-PNDM
It was really only a matter of time before mutations in the insulin gene (INS) were discovered as a cause of diabetes. The search had been on for many years but the first descriptions were in patients with hyperproinsulinemia and not diabetes (Gruppuso et al. 1984). The reason for this, as often is the case, is the site and consequence of the mutation. In 2007 a team led by Graeme Bell, who had cloned the gene back in the early 1980s (Bell et al. 1980), combined linkage analysis with a candidate gene approach to identify first diabetes-causing INS mutation, and subsequently identified nine additional mutations in a total of 16 probands with NDM (Støy et al. 2007, Hodish et al. 2010). In this cohort, subjects had a median age of diabetes presentation at 9 weeks and were often complicated by ketoacidosis. Due to the mutation being within the insulin gene itself, predicted to prevent normal folding and secretion, these individuals are not eligible for treatment with sulfonylureas, which acts on the secretory pathway rather than the insulin protein itself. It has been observed that mice with similarly misfolded insulin proteins may actually have increased β-cell death due to extreme endoplasmic reticulum (ER) stress (Støy et al. 2007). Large-scale screening of patients with PNDM identified INS mutations as one of the most common causes of this disease, secondly only to KATP channel mutations (Edghill et al. 2008). A recent study by Balboa et al. demonstrated β-like cells generated from in vitro differentiated induced pluripotent stem cells (iPSCs) from individuals carrying INS mutations have diminished β-cell proliferation, leading to a decreased β-cell mass, likely playing a role in diabetes pathogenesis (Balboa et al. 2018). Recessively inherited INS mutations have also been shown to cause PNDM due to a contrasting pathogenic mechanism of decreased insulin biosynthesis through a variety of regulatory mechanisms (Garin et al. 2010).
GCK-PNDM
Glucokinase, discussed in detail above as a MODY-causing gene when it presents as a heterozygous mutation, can also cause permanent NDM when presenting with homozygous or compound heterozygous mutations (Njølstad et al. 2001, 2003). These individuals have similar traits to those with KATP-related NDM, including intrauterine growth restriction, low birth weights, and insulin requirements shortly after birth (Njølstad et al. 2003). Not all homozygous or compound heterozygous inactivating GCK mutations cause NDM though. A relatively recent study demonstrated the importance of protein stability as a major contributor to the severity of inactivating GCK mutations. This became apparent through the study of a homozygous GCK mutation which presented as diabetes in young-adolescence rather than at birth (Raimondo et al. 2014).
PDX1-PNDM
PDX1 is another MODY-causing gene that is also a rare, but important, cause of permanent neonatal diabetes. PDX1 is critical for pancreatic development (Jonsson et al. 1994), and some individuals with PDX1 mutations have little or no development of functional pancreatic islets (Stoffers et al. 1997b). Although most cases are confirmed to have pancreatic hypoplasia or agenesis, there are some reports in patients with a normally formed pancreas that may still be affected by permanent NDM due to a PDX1 mutation (De Franco et al. 2013). By regulating the expression of a number of important islet transcripts (insulin, glucokinase, somatostatin), PDX1 serves as a master regulator of gene expression within the islet (Hui & Perfetti 2002). Additional genes important for pancreatic development, including NKX2–2, GATA4, GATA6, GLIS3, RFX6, and PTF1A, have also been identified as rare causes of NDM (Wang et al. 2004, Senée et al. 2006, Doyle & Sussel 2007, Chen et al. 2008, Fukuda et al. 2008, Smith et al. 2010, Allen et al. 2011, Carrasco et al. 2012, Stanescu et al. 2015, Xuan & Sussel 2016).
GLIS3-PNDM
Gli-similar 3 (GLIS3) is a Krüppel-like zinc finger transcription factor that is important during cell specification and patterning during pancreatic development. GLIS3 interacts with other important β-cell transcription factors including PDX1 and NEUROD1 and binds directly to the insulin promoter to positively regulate gene expression. In mice, deletion of both copies of Glis3 leads to neonatal diabetes (Kang et al. 2009, Yang et al. 2009). GLIS3 is also important in humans, as homozygous mutations in GLIS3 are responsible for a syndrome characterized by neonatal diabetes, congenital hypothyroidism, polycystic kidneys, and facial anomalies (Senée et al. 2006). At least one patient with compound heterozygous GLIS3 mutations has been described; this patient has isolated NDM but none of the other features (Dimitri et al. 2011). Genome-wide association studies (GWAS) have also demonstrated variants at the GLIS3 locus which are associated with both type 1 and type 2 diabetes (Aylward et al. 2018).
Syndromic monogenic diabetes
Neonatal diabetes has also been identified as a component of a number of multi-system syndromes, many of which have identified genetic causes. The majority of these syndromes are rare, and because of variable presentation, are at risk for late or missed diagnosis. Despite the heterogeneous nature of these conditions, the genetic dissection of these conditions and molecular understanding of the underlying pathophysiology make it apparent that there are some shared clinical features and biological mechanisms. For example, a large number of patients with NDM also have neurological symptoms (e.g. epilepsy, developmental delay) (De Franco et al. 2015, 2020b). Neurons and β-cells have similar electrophysiology and signaling mechanisms, making this frequent association between defects in glucose homeostasis and neurology unsurprising (De Franco et al. 2020a,b). A majority of the genes implicated in these syndromes are transcription factors with key roles in the development of both cell types (ex. PTF1A, NEUROD1, MNX1, and NKX2–2).
Other causative gene mutations are related to ER stress, either due to mutations in important checkpoints in controlling the ER stress response, or due to mutations that directly result in protein-misfolding, and subsequently, lead to ER stress (ex. EIF2AK3, SLC19A2, IER3IP1, WFS1, TRMT10A, PPP1R15B, EIF2S3) (De Franco et al. 2020b). Clinical presentation with diabetes can be diagnosed beyond the neonatal period well into adolescence for some of these etiologies. One example of NDM caused by ER stress is Wolcott–Rallinson syndrome (WRS); the most common cause of syndromic NDM in areas with high consanguinity (Rubio-Cabezas et al. 2009). It is an autosomal recessive condition and is often accompanied by skeletal dysplasia, poor growth, and liver dysfunction, in addition to NDM. Homozygous or compound heterozygous mutations in eukaryotic translation initiation factor 2-α kinase 3 (EIF2AK3), a critical component of the ER stress response, are responsible for WRS (Delépine et al. 2000).
Blurring the boundaries of NDM genes in MODY
As with PDX1, where mutations were first described in neonatal diabetes (NDM), additional genes which are major causes of NDM have been reported as the cause of diabetes outside of infancy, consistent with MODY. These include those encoding the ATP-sensitive potassium channel subunits (ABCC8 and KCNJ11). In 2012, Bonnefond and colleagues used whole-exome sequencing (WES) to study some of the 30% of French-MODY patients without a genetic diagnosis. This analysis identified a KCNJ11 mutation, specifically, p.E227K, as the cause of MODY in a single-family (Bonnefond et al. 2012). In that same year, Bowman and colleagues found four novel ABCC8 mutations within a cohort of sulfonylurea responsive MODY patients (Bowman et al. 2012). The term MIDY (mutant INS-gene induced diabetes of youth) has also been used to describe patients with INS mutations presenting outside the neonatal period (Liu et al. 2010, 2012).
Expanding the spectrum of the role of monogenic diabetes genes in type 2 diabetes (T2D) risk
The early type 2 diabetes (T2D) genetics literature is plagued by both poorly designed and executed genetic association studies (Siontis et al. 2010). With a few exceptions, most earlier findings failed to replicate when expanded to large, well-powered studies. Of the early studies which have stood the test of time, it is notable that two of three such studies include genes (PPARG, KCNJ11, and TCF7L2) involved in monogenic forms of diabetes (Altshuler et al. 2000, Gloyn et al. 2003, Grant et al. 2006). In the late 2000s, people began to explore the utility of genome-wide association studies (GWAS) for understanding complex diseases and traits. GWAS compare multiple individual genomes to identify associations between specific genetic changes and a given disease or trait. International efforts for both type 1 and type 2 diabetes have been terrifically successful in uncovering hundreds of association signals. As has been the case for most complex traits, the vast majority of GWAS signals for both T1D and T2D exert their impact through non-coding variants with a presumed regulatory impact (Mahajan et al. 2018). A consistent finding has also been the identification of signals in or close to genes with known roles in monogenic diabetes, such as HNF1A and KCNJ11. A number of these also have coding variants across the frequency spectrum, which are associated with altered risk for T2D: sometimes they are specific to a population (e.g. p.E508K in HNF1A) (SIGMA Type 2 Diabetes Consortium et al. 2014) whilst others are common in the population (e.g. p.E23K in KCNJ11) (Gloyn et al. 2003). The field has also uncovered common regulatory variants near monogenic diabetes genes which likely have an impact in a tissue and developmental time-specific manner (e.g. HNF4A) (Kathiresan et al. 2009). These findings bring into focus a continuum between normal function and complete loss of function in these genes – an allelic spectrum – where the relationship between the specific variant and subsequent disease presentation is not necessarily straightforward (Althari et al. 2020). The genetic characterization of large Biobanks (e.g. UKBioBank, BioMe) will facilitate the studies required to fully understand both the relationship between genetic variation in monogenic diabetes genes and clinical phenotype as well as the impact of common genetic variation (e.g. T2D genetic risk scores) on altering the presentation of variants in these genes. Genetics has certainly delivered precision medicine for monogenic forms of diabetes; the challenge for the next decade is extending this to all types of diabetes.
Outlook
Moving forward, our expanding knowledge surrounding the genetic changes underlying both monogenic and polygenic forms of diabetes will continue to advance our potential for personalized medicine through both precision diagnostics and therapeutics. These genetic breakthroughs, in combination with clinical biomarkers, will allow us to refine our classification and diagnosis of diabetes subtypes and provide evidence-based care to determine optimal management and decrease morbidity and mortality associated with these conditions. The importance of molecular diagnosis of MODY and NDM is quite apparent on its own; however, the groundwork laid in understanding them will continue to provide insight and guidance for more complex forms of diabetes as well.
Closing remarks
The careful dissection of the genetic etiology of monogenic forms of diabetes has created a framework of potential mechanisms to understand how common genetic variants could exert their effect and lead to increased risk for complex forms of diabetes such as type 1 and type 2 diabetes. There are now documented examples of single-gene defects causing alterations in glucose sensing, insulin secretion, pancreas development and β-cell mass, and for each of these examples of how these same mechanisms influence type 1 and type 2 diabetes. As we mark the centenary of the discovery of insulin, it is remarkable to see the speed at which its discovery was translated into changes in clinical practice. With the exception of the remarkable efforts with the COVID19 vaccinations, this is not often witnessed. Progress in understanding the genetic basis of pancreatic islet-cell dysfunction in diabetes has also had an incredible century but we have some way to go before we have the same impact as Banting, Best, Collip, and Mcleod.
Acknowledgement
The authors thank their many colleagues in the field of diabetes genetics for their contributions to our understanding of the genetic basis of diabetes and ask their forgiveness for not being able to ‘shine a light’ in this brief review on all the wonderful work that has been done over the past 100 years.
Funding
A L G is a Wellcome Senior Fellow in Basic Biomedical Science. A L G is funded by the Wellcome (095101, 200837) and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (U01-DK105535, U01-DK085545, UM1DK126185), the Stanford Diabetes Research Center (NIDDK award P30DK116074). J M I is supported by NIDDK 1K12DK122550-01.
Footnotes
Declaration of interest
A L G declares that her spouse is an employee of Genentech and hold stock options in Roche. J K declares that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
References
- Ahlgren U, Jonsson J, Jonsson L, Simu K & Edlund H 1998. beta-Cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes and Development 12 1763–1768. ( 10.1101/gad.12.12.1763) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen HL, Flanagan SE, Shaw-Smith C, De Franco E, Akerman I, Caswell R, International Pancreatic Agenesis Consortium, Ferrer J, Hattersley AT & Ellard S & Ellard S 2011. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nature Genetics 44 20–22. ( 10.1038/ng.1035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althari S, Najmi LA, Bennett AJ, Aukrust I, Rundle JK, Colclough K, Molnes J, Kaci A, Nawaz S, van der Lugt T, et al. 2020. Unsupervised clustering of missense variants in HNF1A using multidimensional functional data aids clinical interpretation. American Journal of Human Genetics 107 670–682. ( 10.1016/j.ajhg.2020.08.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, et al. 2000. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nature Genetics 26 76–80. ( 10.1038/79216) [DOI] [PubMed] [Google Scholar]
- Aylward A, Chiou J, Okino ML, Kadakia N & Gaulton KJ 2018. Shared genetic risk contributes to type 1 and type 2 diabetes etiology. Human Molecular Genetics [In press]. ( 10.1093/hmg/ddy314) [DOI] [PubMed] [Google Scholar]
- Babenko AP, Polak M, Cavé H, Busiah K, Czernichow P, Scharfmann R, Bryan J, Aguilar-Bryan L, Vaxillaire M & Froguel P 2006. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. New England Journal of Medicine 355 456–466. ( 10.1056/NEJMoa055068) [DOI] [PubMed] [Google Scholar]
- Balboa D, Saarimäki-Vire J, Borshagovski D, Survila M, Lindholm P, Galli E, Eurola S, Ustinov J, Grym H, Huopio H, et al. 2018. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. eLife 7 e38519. ( 10.7554/eLife.38519) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedoya FJ, Wilson JM, Ghosh AK, Finegold D & Matschinsky FM 1986. The glucokinase glucose sensor in human pancreatic islet tissue. Diabetes 35 61–67. ( 10.2337/diab.35.1.61) [DOI] [PubMed] [Google Scholar]
- Bell GI, Pictet RL, Rutter WJ, Cordell B, Tischer E & Goodman HM 1980. Sequence of the human insulin gene. Nature 284 26–32. ( 10.1038/284026a0) [DOI] [PubMed] [Google Scholar]
- Bonnefond A, Philippe J, Durand E, Dechaume A, Huyvaert M, Montagne L, Marre M, Balkau B, Fajardy I, Vambergue A, et al. 2012. Whole-exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS ONE 7 e37423. ( 10.1371/journal.pone.0037423) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden DW, Akots G, Rothschild CB, Falls KF, Sheehy MJ, Hayward C, Mackie A, Baird J, Brock D & Antonarakis SE 1992. Linkage analysis of maturity-onset diabetes of the young (MODY): genetic heterogeneity and nonpenetrance. American Journal of Human Genetics 50 607–618. [PMC free article] [PubMed] [Google Scholar]
- Bowman P, Flanagan SE, Edghill EL, Damhuis A, Shepherd MH, Paisey R, Hattersley AT & Ellard S 2012. Heterozygous ABCC8 mutations are a cause of MODY. Diabetologia 55 123–127. ( 10.1007/s00125-011-2319-x) [DOI] [PubMed] [Google Scholar]
- Bowman P, Sulen Å, Barbetti F, Beltrand J, Svalastoga P, Codner E, Tessmann EH, Juliusson PB, Skrivarhaug T, Pearson ER, et al. 2018. Effectiveness and safety of long-term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: an international cohort study. Lancet: Diabetes and Endocrinology 6 637–646. ( 10.1016/S2213-8587(18)30106-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryder L & Harper C 2013. Commentary: more than ‘tentative opinions’: Harry Himsworth and defining diabetes. International Journal of Epidemiology 42 1599–1600. ( 10.1093/ije/dyt200) [DOI] [PubMed] [Google Scholar]
- Carrasco M, Delgado I, Soria B, Martín F & Rojas A 2012. GATA4 and GATA6 control mouse pancreas organogenesis. Journal of Clinical Investigation 122 3504–3515. ( 10.1172/JCI63240) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakera AJ, Steele AM, Gloyn AL, Shepherd MH, Shields B, Ellard S & Hattersley AT 2015. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care 38 1383–1392. ( 10.2337/dc14-2769) [DOI] [PubMed] [Google Scholar]
- Chen R, Hussain K, Al-Ali M, Dattani MT, Hindmarsh P, Jones PM & Marsh P 2008. Neonatal and late-onset diabetes mellitus caused by failure of pancreatic development: report of 4 more cases and a review of the literature. Pediatrics 121 e1541–e1547. ( 10.1542/peds.2007-3543) [DOI] [PubMed] [Google Scholar]
- Chiou J, Geusz RJ, Okino M-L, Han JY, Miller M, Benaglio P, Huang S, Korgaonkar K, Heller S, Kleger A, et al. 2021. Large-scale genetic association and single cell accessible chromatin mapping defines cell type-specific mechanisms of type 1 diabetes risk. bioRxiv [In press]. ( 10.1101/2021.01.13.426472) [DOI] [Google Scholar]
- Cuesta-Muñoz AL, Huopio H, Otonkoski T, Gomez-Zumaquero JM, Näntö-Salonen K, Rahier J, López-Enriquez S, García-Gimeno MA, Sanz P, Soriguer FC, et al. 2004. Severe persistent hyperinsulinemic hypoglycemia due to a de novo glucokinase mutation. Diabetes 53 2164–2168. ( 10.2337/diabetes.53.8.2164) [DOI] [PubMed] [Google Scholar]
- De Franco E 2020. From biology to genes and back again: gene discovery for monogenic forms of beta-cell dysfunction in diabetes. Journal of Molecular Biology 432 1535–1550. ( 10.1016/j.jmb.2019.08.016) [DOI] [PubMed] [Google Scholar]
- De Franco E, Shaw-Smith C, Flanagan SE, Edghill EL, Wolf J, Otte V, Ebinger F, Varthakavi P, Vasanthi T, Edvardsson S, et al. 2013. Biallelic PDX1 (insulin promoter factor 1) mutations causing neonatal diabetes without exocrine pancreatic insufficiency. Diabetic Medicine 30 e197–e200. ( 10.1111/dme.12122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franco E, Flanagan SE, Houghton JA, Allen HL, Mackay DJ, Temple IK, Ellard S & Hattersley AT 2015. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet 386 957–963. ( 10.1016/S0140-6736(15)60098-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franco E, Flanagan SE, Yagi T, Abreu D, Mahadevan J, Johnson MB, Jones G, Acosta F, Mulaudzi M, Lek N, et al. 2017. Dominant ER stress-inducing WFS1 mutations underlie a genetic syndrome of neonatal/infancy-onset diabetes, congenital sensorineural deafness, and congenital cataracts. Diabetes 66 2044–2053. ( 10.2337/db16-1296) [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franco E, Watson RA, Weninger WJ, Wong CC, Flanagan SE, Caswell R, Green A, Tudor C, Lelliott CJ, Geyer SH, et al. 2019. A specific CNOT1 mutation results in a novel syndrome of pancreatic agenesis and holoprosencephaly through impaired pancreatic and neurological development. American Journal of Human Genetics 104 985–989. ( 10.1016/j.ajhg.2019.03.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franco E, Caswell R, Johnson MB, Wakeling MN, Zung A, Chí Dũng VC, Bích Ngọc CT, Goonetilleke R, Vivanco Jury M, El-Khateeb M, et al. 2020a. De novo mutations in EIF2B1 affecting eIF2 signaling cause neonatal/early onset diabetes and transient hepatic dysfunction. Diabetes 69 477–483. ( 10.2337/db19-1029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franco E, Lytrivi M, Ibrahim H, Montaser H, Wakeling MN, Fantuzzi F, Patel K, Demarez C, Cai Y, Igoillo-Esteve M, et al. 2020b. YIPF5 mutations cause neonatal diabetes and microcephaly through endoplasmic reticulum stress. Journal of Clinical Investigation 130 6338–6353. ( 10.1172/JCI141455) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delépine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM & Julier C 2000. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nature Genetics 25 406–409. ( 10.1038/78085) [DOI] [PubMed] [Google Scholar]
- Delvecchio M, Pastore C & Giordano P 2020. Treatment options for MODY patients: a systematic review of literature. Diabetes Therapy 11 1667–1685. ( 10.1007/s13300-020-00864-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitri P, Warner JT, Minton JA, Patch AM, Ellard S, Hattersley AT, Barr S, Hawkes D, Wales JK & Gregory JW 2011. Novel GLIS3 mutations demonstrate an extended multisystem phenotype. European Journal of Endocrinology 164 437–443. ( 10.1530/EJE-10-0893) [DOI] [PubMed] [Google Scholar]
- Doyle MJ & Sussel L 2007. Nkx2.2 regulates beta-cell function in the mature islet. Diabetes 56 1999–2007. ( 10.2337/db06-1766) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne MJ, Kane C, Shepherd RM, Sanchez JA, James RF, Johnson PR, Aynsley-Green A, Lu S, Clement JP, Lindley KJ, et al. 1997. Familial persistent hyperinsulinemic hypoglycemia of infancy and mutations in the sulfonylurea receptor. New England Journal of Medicine 336 703–706. ( 10.1056/NEJM199703063361005) [DOI] [PubMed] [Google Scholar]
- Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, Shepherd MH, Hussain K, Kapoor RR, Malecki M, et al. 2008. Insulin mutation screening in 1044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes 57 1034–1042. ( 10.2337/db07-1405) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edghill EL, Khamis A, Weedon MN, Walker M, Hitman GA, McCarthy MI, Owen KR, Ellard S, T Hattersley A & Frayling TM 2011. Sequencing PDX1 (insulin promoter factor 1) in 1788 UK individuals found 5% had a low frequency coding variant, but these variants are not associated with Type 2 diabetes. Diabetic Medicine 28 681–684. ( 10.1111/j.1464-5491.2011.03269.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellard S, Bellanné-Chantelot C, Hattersley AT & European Molecular Genetics Quality Network (EMQN) 2008. Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia 51 546–553. ( 10.1007/s00125-008-0942-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajans SS 1987. MODY – a model for understanding the pathogeneses and natural history of Type II diabetes. Hormone and Metabolic Research 19 591–599. ( 10.1055/s-2007-1011888) [DOI] [PubMed] [Google Scholar]
- Fajans SS 1990. Scope and heterogeneous nature of MODY. Diabetes Care 13 49–64. ( 10.2337/diacare.13.1.49) [DOI] [PubMed] [Google Scholar]
- Fajans SS & Brown MB 1993. Administration of sulfonylureas can increase glucose-induced insulin secretion for decades in patients With maturity-onset diabetes of the young. Diabetes Care 16 1254–1261. ( 10.2337/diacare.16.9.1254) [DOI] [PubMed] [Google Scholar]
- Fajans SS & Conn JW 1959. The early recognition of diabetes mellitus. Annals of the New York Academy of Sciences 82 208–218. ( 10.1111/j.1749-6632.1959.tb44901.x) [DOI] [PubMed] [Google Scholar]
- Firdous P, Nissar K, Ali S, Ganai BA, Shabir U, Hassan T & Masoodi SR 2018. Genetic testing of maturity-onset diabetes of the young current status and future perspectives. Frontiers in Endocrinology 9 253. ( 10.3389/fendo.2018.00253) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan SE, Edghill EL, Gloyn AL, Ellard S & Hattersley AT 2006. Mutations in KCNJ11, which encodes Kir6.2, are a common cause of diabetes diagnosed in the first 6 months of life, with the phenotype determined by genotype. Diabetologia 49 1190–1197. ( 10.1007/s00125-006-0246-z) [DOI] [PubMed] [Google Scholar]
- Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, McDonald TJ, Rajala H, Ramelius A, Barton J, et al. 2014. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nature Genetics 46 812–814. ( 10.1038/ng.3040) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froguel P, Vaxillaire M, Sun F, Velho G, Zouali H, Butel MO, Lesage S, Vionnet N, Clément K & Fougerousse F 1992. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature 356 162–164. ( 10.1038/356162a0) [DOI] [PubMed] [Google Scholar]
- Fukuda A, Kawaguchi Y, Furuyama K, Kodama S, Horiguchi M, Kuhara T, Kawaguchi M, Terao M, Doi R, Wright CVE, et al. 2008. Reduction of Ptf1a gene dosage causes pancreatic hypoplasia and diabetes in mice. Diabetes 57 2421–2431. ( 10.2337/db07-1558) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garin I, Edghill EL, Akerman I, Rubio-Cabezas O, Rica I, Locke JM, Maestro MA, Alshaikh A, Bundak R, del Castillo G, et al. 2010. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. PNAS 107 3105–3110. ( 10.1073/pnas.0910533107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, Stanley CA, Thornton PS, Permutt MA, Matschinsky FM, et al. 1998. Familial hyperinsulinism caused by an activating glucokinase mutation. New England Journal of Medicine 338 226–230. ( 10.1056/NEJM199801223380404) [DOI] [PubMed] [Google Scholar]
- Gloyn AL 2003. Glucokinase (GCK) mutations in hyper- and hypoglycemia: maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemia of infancy. Human Mutation 22 353–362. ( 10.1002/humu.10277) [DOI] [PubMed] [Google Scholar]
- Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, et al. 2003. Large-scale association studies of variants in genes encoding the pancreatic β-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 52 568–572. ( 10.2337/diabetes.52.2.568) [DOI] [PubMed] [Google Scholar]
- Gloyn AL, Cummings EA, Edghill EL, Harries LW, Scott R, Costa T, Temple IK, Hattersley AT & Ellard S 2004a. Permanent neonatal diabetes due to paternal germline mosaicism for an activating mutation of the KCNJ11 gene encoding the Kir6.2 subunit of the beta-cell potassium adenosine triphosphate channel. Journal of Clinical Endocrinology and Metabolism 89 3932–3935. ( 10.1210/jc.2004-0568) [DOI] [PubMed] [Google Scholar]
- Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JMCL, Molnes J, et al. 2004b. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. New England Journal of Medicine 350 1838–1849. ( 10.1056/NEJMoa032922) [DOI] [PubMed] [Google Scholar]
- Gloyn AL, Odili S, Buettger C, Njolstad PR, Shiota C, Magnuson MA & Matschinsky FM 2004c. Glucokinase and the regulation of blood sugar. Glucokinase and Glycemic Disease: From Basics to Novel Therapeutics 16 92–109. ( 10.1159/000079009) [DOI] [Google Scholar]
- Gloyn AL, Reimann F, Girard C, Edghill EL, Proks P, Pearson ER, Temple IK, Mackay DJG, Shield JPH, Freedenberg D, et al. 2005. Relapsing diabetes can result from moderately activating mutations in KCNJ11. Human Molecular Genetics 14 925–934. ( 10.1093/hmg/ddi086) [DOI] [PubMed] [Google Scholar]
- Gloyn AL, Diatloff-Zito C, Edghill EL, Bellanné-Chantelot C, Nivot S, Coutant R, Ellard S, Hattersley AT & Robert JJ 2006. KCNJ11 activating mutations are associated with developmental delay, epilepsy and neonatal diabetes syndrome and other neurological features. European Journal of Human Genetics 14 824–830. ( 10.1038/sj.ejhg.5201629) [DOI] [PubMed] [Google Scholar]
- Grant SFA, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, et al. 2006. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature Genetics 38 320–323. ( 10.1038/ng1732) [DOI] [PubMed] [Google Scholar]
- Gruppuso PA, Gorden P, Kahn CR, Cornblath M, Zeller WP & Schwartz R 1984. Familial hyperproinsulinemia due to a proposed defect in conversion of proinsulin to insulin. New England Journal of Medicine 311 629–634. ( 10.1056/NEJM198409063111003) [DOI] [PubMed] [Google Scholar]
- Harries LW, Hattersley AT & Ellard S 2004. Messenger RNA transcripts of the hepatocyte nuclear factor-1alpha gene containing premature termination codons are subject to nonsense-mediated decay. Diabetes 53 500–504. ( 10.2337/diabetes.53.2.500) [DOI] [PubMed] [Google Scholar]
- Hattersley AT, Turner RC, Patel P, O’Rahilly S, Hattersley AT, Patel P, Wainscoat JS, Permutt MA, Tanazawa Y, Chin KC, et al. 1992. Linkage of type 2 diabetes to the glucokinase gene. Lancet 339 1307–1310. ( 10.1016/0140-6736(92)91958-B) [DOI] [PubMed] [Google Scholar]
- Hattersley AT, Greeley SAW, Polak M, Rubio-Cabezas O, Njølstad PR, Mlynarski W, Castano L, Carlsson A, Raile K, Chi DV, et al. 2018. ISPAD Clinical Practice Consensus Guidelines 2018: the diagnosis and management of monogenic diabetes in children and adolescents. Pediatric Diabetes 19 (Supplement 27) 47–63. ( 10.1111/pedi.12772) [DOI] [PubMed] [Google Scholar]
- Hodish I, Liu M, Rajpal G, Larkin D, Holz RW, Adams A, Liu L & Arvan P 2010. Misfolded proinsulin affects bystander proinsulin in neonatal diabetes. Journal of Biological Chemistry 285 685–694. ( 10.1074/jbc.M109.038042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, Lindner T, Yamagata K, Ogata M, Tomonaga O, et al. 1997. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nature Genetics 17 384–385. ( 10.1038/ng1297-384) [DOI] [PubMed] [Google Scholar]
- Hui H & Perfetti R 2002. Pancreas duodenum homeobox-1 regulates pancreas development during embryogenesis and islet cell function in adulthood. European Journal of Endocrinology 146 129–141. ( 10.1530/eje.0.1460129) [DOI] [PubMed] [Google Scholar]
- Huopio H, Reimann F, Ashfield R, Komulainen J, Lenko HL, Rahier J, Vauhkonen I, Kere J, Laakso M, Ashcroft F, et al. 2000. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. Journal of Clinical Investigation 106 897–906. ( 10.1172/JCI9804) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Riggs AC, Tanizawa Y, Ueda K, Kuwano A, Liu L, Donis-Keller H & Permutt MA 1996. Isolation, characterization, and chromosomal mapping of the human insulin promoter factor 1 (IPF-1) gene. Diabetes 45 789–794. ( 10.2337/diab.45.6.789) [DOI] [PubMed] [Google Scholar]
- Johnson MB, De Franco E, Lango-Allen H, Al Senani A, Elbarbary N, Siklar Z, Berberoglu M, Imane Z, Haghighi A, Razavi Z, et al. 2017. Recessively inherited LRBA mutations cause autoimmunity presenting as neonatal diabetes. Diabetes 66 2316–2322. ( 10.2337/db17-0040) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson J, Carlsson L, Edlund T & Edlund H 1994. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 371 606–609. ( 10.1038/371606a0) [DOI] [PubMed] [Google Scholar]
- Kane C, Shepherd RM, Squires PE, Johnson PR, James RF, Milla PJ, Aynsley-Green A, Lindley KJ & Dunne MJ 1996. Loss of functional KATP channels in pancreatic beta-cells causes persistent hyperinsulinemic hypoglycemia of infancy. Nature Medicine 2 1344–1347. ( 10.1038/nm1296-1344) [DOI] [PubMed] [Google Scholar]
- Kang HS, Kim YS, ZeRuth G, Beak JY, Gerrish K, Kilic G, Sosa-Pineda B, Jensen J, Pierreux CE, Lemaigre FP, et al. 2009. Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Molecular and Cellular Biology 29 6366–6379. ( 10.1128/MCB.01259-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y, Tanaka T, et al. 2009. Common variants at 30 loci contribute to polygenic dyslipidemia. Nature Genetics 41 56–65. ( 10.1038/ng.291) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Seghers V, Cho JH, Kang Y, Kim S, Ryu Y, Baek K, Aguilar-Bryan L, Lee YD, Bryan J, et al. 2002. Transactivation of the mouse sulfonylurea receptor I gene by BETA2/NeuroD. Molecular Endocrinology 16 1097–1107. ( 10.1210/mend.16.5.0934) [DOI] [PubMed] [Google Scholar]
- Kristinsson SY, Thorolfsdottir ET, Talseth B, Steingrimsson E, Thorsson AV, Helgason T, Hreidarsson AB & Arngrimsson R 2001. MODY in Iceland is associated with mutations in HNF-1α and a novel mutation in NeuroD1. Diabetologia 44 2098–2103. ( 10.1007/s001250100016) [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409 860–921. ( 10.1038/35057062) [DOI] [PubMed] [Google Scholar]
- Lee BC, Appleton M, Shore AC, Tooke JE & Hattersley AT 1999. Impaired maximum microvascular hyperaemia in patients with MODY 3 (hepatocyte nuclear factor-1alpha gene mutations). Diabetic Medicine 16 731–735. ( 10.1046/j.1464-5491.1999.00129.x) [DOI] [PubMed] [Google Scholar]
- Letourneau LR & Greeley SAW 2018. Congenital forms of diabetes: the beta-cell and beyond. Current Opinion in Genetics and Development 50 25–34. ( 10.1016/j.gde.2018.01.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hodish I, Haataja L, Lara-Lemus R, Rajpal G, Wright J & Arvan P 2010. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends in Endocrinology and Metabolism 21 652–659. ( 10.1016/j.tem.2010.07.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Lara-Lemus R, Shan SO, Wright J, Haataja L, Barbetti F, Guo H, Larkin D & Arvan P 2012. Impaired cleavage of preproinsulin signal peptide linked to autosomal-dominant diabetes. Diabetes 61 828–837. ( 10.2337/db11-0878) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson MA, Andreone TL, Printz RL, Koch S & Granner DK 1989. Rat glucokinase gene: structure and regulation by insulin. PNAS 86 4838–4842. ( 10.1073/pnas.86.13.4838) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. 2018. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nature Genetics 50 1505–1513. ( 10.1038/s41588-018-0241-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, Saad M, Warram JH, Montminy M & Krolewski AS 1999. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nature Genetics 23 323–328. ( 10.1038/15500) [DOI] [PubMed] [Google Scholar]
- Marsich E, Vetere A, Di Piazza M, Tell G & Paoletti S 2003. The PAX6 gene is activated by the basic helix-loop-helix transcription factor NeuroD/BETA2. Biochemical Journal 376 707–715. ( 10.1042/BJ20031021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald TJ & Ellard S 2013. Maturity onset diabetes of the young: identification and diagnosis. Annals of Clinical Biochemistry 50 403–415. ( 10.1177/0004563213483458) [DOI] [PubMed] [Google Scholar]
- Mendel DB, Hansen LP, Graves MK, Conley PB & Crabtree GR 1991. HNF-1 alpha and HNF-1 beta (vHNF-1) share dimerization and homeo domains, but not activation domains, and form heterodimers in vitro. Genes and Development 5 1042–1056. ( 10.1101/gad.5.6.1042) [DOI] [PubMed] [Google Scholar]
- Misra S, Hassanali N, Bennett AJ, Juszczak A, Caswell R, Colclough K, Valabhji J, Ellard S, Oliver NS & Gloyn AL 2020. Homozygous hypomorphic HNF1A alleles are a novel cause of young-onset diabetes and result in sulfonylurea-sensitive diabetes. Diabetes Care 43 909–912. ( 10.2337/dc19-1843) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moates JM, Nanda S, Cissell MA, Tsai MJ & Stein R 2003. BETA2 activates transcription from the upstream glucokinase gene promoter in islet beta-cells and gut endocrine cells. Diabetes 52 403–408. ( 10.2337/diabetes.52.2.403) [DOI] [PubMed] [Google Scholar]
- Ng NHJ, Willems SM, Fernandez J, Fine RS, Wheeler E, Wessel J, Kitajima H, Marenne G, Rundle JK, Sim X, et al. 2019. Tissue-specific alteration of metabolic pathways influences glycemic regulation. bioRxiv [In press] 790618. ( 10.1101/790618) [DOI] [Google Scholar]
- Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP, Gonzalez G, Aguilar-Bryan L, Permutt MA & Bryan J 1996. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science 272 1785–1787. ( 10.1126/science.272.5269.1785) [DOI] [PubMed] [Google Scholar]
- Njølstad PR, Søvik O, Cuesta-Muñoz A, Bjørkhaug L, Massa O, Barbetti F, Undlien DE, Shiota C, Magnuson MA, Molven A, et al. 2001. Neonatal diabetes mellitus due to complete glucokinase deficiency. New England Journal of Medicine 344 1588–1592. ( 10.1056/NEJM200105243442104) [DOI] [PubMed] [Google Scholar]
- Njølstad PR, Sagen JV, Bjørkhaug L, Odili S, Shehadeh N, Bakry D, Sarici SU, Alpay F, Molnes J, Molven A, et al. 2003. Permanent neonatal diabetes caused by glucokinase deficiency: inborn error of the glucose-insulin signaling pathway. Diabetes 52 2854–2860. ( 10.2337/diabetes.52.11.2854) [DOI] [PubMed] [Google Scholar]
- Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM & Hattersley AT 2003. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 362 1275–1281. ( 10.1016/S0140-6736(03)14571-0) [DOI] [PubMed] [Google Scholar]
- Pearson ER, Badman MK, Lockwood CR, Clark PM, Ellard S, Bingham C & Hattersley AT 2004. Contrasting diabetes phenotypes associated with hepatocyte nuclear factor-1alpha and -1beta mutations. Diabetes Care 27 1102–1107. ( 10.2337/diacare.27.5.1102) [DOI] [PubMed] [Google Scholar]
- Pearson ER, Pruhova S, Tack CJ, Johansen A, Castleden HA, Lumb PJ, Wierzbicki AS, Clark PM, Lebl J, Pedersen O, et al. 2005. Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia 48 878–885. ( 10.1007/s00125-005-1738-y) [DOI] [PubMed] [Google Scholar]
- Pontoglio M, Prié D, Cheret C, Doyen A, Leroy C, Froguel P, Velho G, Yaniv M & Friedlander G 2000. HNF1alpha controls renal glucose reabsorption in mouse and man. EMBO Reports 1 359–365. ( 10.1093/embo-reports/kvd071) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raeder H, Johansson S, Holm PI, Haldorsen IS, Mas E, Sbarra V, Nermoen I, Eide SA, Grevle L, Bjørkhaug L, et al. 2006. Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nature Genetics 38 54–62. ( 10.1038/ng1708) [DOI] [PubMed] [Google Scholar]
- Ræder H, McAllister FE, Tjora E, Bhatt S, Haldorsen I, Hu J, Willems SM, Vesterhus M, El Ouaamari A, Liu M, et al. 2014. Carboxyl-ester lipase maturity-onset diabetes of the young is associated with development of pancreatic cysts and upregulated MAPK signaling in secretin-stimulated duodenal fluid. Diabetes 63 259–269. ( 10.2337/db13-1012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondo A, Chakera AJ, Thomsen SK, Colclough K, Barrett A, De Franco E, Chatelas A, Demirbilek H, Akcay T, Alawneh H, et al. 2014. Phenotypic severity of homozygous GCK mutations causing neonatal or childhood-onset diabetes is primarily mediated through effects on protein stability. Human Molecular Genetics 23 6432–6440. ( 10.1093/hmg/ddu360) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Patch AM, Minton JAL, Flanagan SE, Edghill EL, Hussain K, Balafrej A, Deeb A, Buchanan CR, Jefferson IG, et al. 2009. Wolcott-Rallison syndrome is the most common genetic cause of permanent neonatal diabetes in consanguineous families. Journal of Clinical Endocrinology and Metabolism 94 4162–4170. ( 10.1210/jc.2009-1137) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Minton JAL, Kantor I, Williams D, Ellard S & Hattersley AT 2010. Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes 59 2326–2331. ( 10.2337/db10-0011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagen JV, Raeder H, Hathout E, Shehadeh N, Gudmundsson K, Baevre H, Abuelo D, Phornphutkul C, Molnes J, Bell GI, et al. 2004. Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes 53 2713–2718. ( 10.2337/diabetes.53.10.2713) [DOI] [PubMed] [Google Scholar]
- Senée V, Chelala C, Duchatelet S, Feng D, Blanc H, Cossec JC, Charon C, Nicolino M, Boileau P, Cavener DR, et al. 2006. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nature Genetics 38 682–687. ( 10.1038/ng1802) [DOI] [PubMed] [Google Scholar]
- Shield JPH 2000. Neonatal diabetes: new insights into aetiology and implications. Hormone Research 53 (Supplement 1) 7–11. ( 10.1159/000053198) [DOI] [PubMed] [Google Scholar]
- Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT & Ellard S 2010. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia 53 2504–2508. ( 10.1007/s00125-010-1799-4) [DOI] [PubMed] [Google Scholar]
- Shimomura K & Maejima Y 2017. KATP channel mutations and neonatal diabetes. Internal Medicine 56 2387–2393. ( 10.2169/internalmedicine.8454-16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIGMA Type 2 Diabetes Consortium, Estrada K, Aukrust I, Bjørkhaug L, Burtt NP, Mercader JM, García-Ortiz H, Huerta-Chagoya A, Moreno-Macías H, Walford G, et al. 2014. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 311 2305–2314. ( 10.1001/jama.2014.6511) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siontis KCM, Patsopoulos NA & Ioannidis JPA 2010. Replication of past candidate loci for common diseases and phenotypes in 100 genome-wide association studies. European Journal of Human Genetics 18 832–837. ( 10.1038/ejhg.2010.26) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SB, Qu HQ, Taleb N, Kishimoto NY, Scheel DW, Lu Y, Patch AM, Grabs R, Wang J, Lynn FC, et al. 2010. Rfx6 directs islet formation and insulin production in mice and humans. Nature 463 775–780. ( 10.1038/nature08748) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanescu DE, Hughes N, Patel P & De Leon DD 2015. A novel mutation in GATA6 causes pancreatic agenesis. Pediatric Diabetes 16 67–70. ( 10.1111/pedi.12111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S & Hattersley AT 2014. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA 311 279–286. ( 10.1001/jama.2013.283980) [DOI] [PubMed] [Google Scholar]
- Stoffel M, Froguel P, Takeda J, Zouali H, Vionnet N, Nishi S, Weber IT, Harrison RW, Pilkis SJ & Lesage S 1992. Human glucokinase gene: isolation, characterization, and identification of two missense mutations linked to early-onset non-insulin-dependent (type 2) diabetes mellitus. PNAS 89 7698–7702. ( 10.1073/pnas.89.16.7698) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffel M, Stein R, Wright CV, Espinosa R, Le Beau MM & Bell GI 1995. Localization of human homeodomain transcription factor insulin promoter factor 1 (IPF1) to chromosome band 13q12.1. Genomics 28 125–126. ( 10.1006/geno.1995.1120) [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Ferrer J, Clarke WL & Habener JF 1997a. Early-onset type-ll diabetes mellitus (MODY4) linked to Ipf1. Nature Genetics 17 138–139. ( 10.1038/ng1097-138) [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL & Habener JF 1997b. Pancreatic agenesis attributable to a single nucleotide deletion in the human Ipf1 gene coding sequence. Nature Genetics 15 106–110. ( 10.1038/ng0197-106) [DOI] [PubMed] [Google Scholar]
- Støy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, et al. 2007. Insulin gene mutations as a cause of permanent neonatal diabetes. PNAS 104 15040–15044. ( 10.1073/pnas.0707291104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stride A, Vaxillaire M, Tuomi T, Barbetti F, Njølstad PR, Hansen T, Costa A, Conget I, Pedersen O, Søvik O, et al. 2002. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia 45 427–435. ( 10.1007/s00125-001-0770-9) [DOI] [PubMed] [Google Scholar]
- Tammaro P, Girard C, Molnes J, Njølstad PR & Ashcroft FM 2005. Kir6.2 mutations causing neonatal diabetes provide new insights into Kir6.2-SUR1 interactions. EMBO Journal 24 2318–2330. ( 10.1038/sj.emboj.7600715) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattersall RB 1974. Mild familial diabetes with dominant inheritance. Quarterly Journal of Medicine 43 339–357. [PubMed] [Google Scholar]
- Tattersall RB & Fajans SS 1975. A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes 24 44–53. ( 10.2337/diab.24.1.44) [DOI] [PubMed] [Google Scholar]
- Thomas P, Ye Y & Lightner E 1996. Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Human Molecular Genetics 5 1809–1812. ( 10.1093/hmg/5.11.1809) [DOI] [PubMed] [Google Scholar]
- Velho G, Froguel P, Clement K, Pueyo ME, Rakotoambinina B, Zouali H, Passa P, Cohen D & Robert JJ 1992. Primary pancreatic beta-cell secretory defect caused by mutations in glucokinase gene in kindreds of maturity onset diabetes of the young. Lancet 340 444–448. ( 10.1016/0140-6736(92)91768-4) [DOI] [PubMed] [Google Scholar]
- Wang J, Elghazi L, Parker SE, Kizilocak H, Asano M, Sussel L & Sosa-Pineda B 2004. The concerted activities of Pax4 and Nkx2.2 are essential to initiate pancreatic beta-cell differentiation. Developmental Biology 266 178–189. ( 10.1016/j.ydbio.2003.10.018) [DOI] [PubMed] [Google Scholar]
- Xuan S & Sussel L 2016. GATA4 and GATA6 regulate pancreatic endoderm identity through inhibition of hedgehog signaling. Development 143 780–786. ( 10.1242/dev.127217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, Fajans SS, Signorini S, Stoffel M & Bell GI 1996a. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature 384 458–460. ( 10.1038/384458a0) [DOI] [PubMed] [Google Scholar]
- Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, Southam L, Cox RD, Lathrop GM, Boriraj VV, et al. 1996b. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature 384 455–458. ( 10.1038/384455a0) [DOI] [PubMed] [Google Scholar]
- Yang Y, Chang BHJ, Samson SL, Li MV & Chan L 2009. The Krüppel-like zinc finger protein Glis3 directly and indirectly activates insulin gene transcription. Nucleic Acids Research 37 2529–2538. ( 10.1093/nar/gkp122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorifuji T, Nagashima K, Kurokawa K, Kawai M, Oishi M, Akazawa Y, Hosokawa M, Yamada Y, Inagaki N & Nakahata T 2005. The C42R mutation in the Kir6.2 (KCNJ11) gene as a cause of transient neonatal diabetes, childhood diabetes, or later-onset, apparently type 2 diabetes mellitus. The Journal of Clinical Endocrinology and Metabolism 90 3174–3178. ( 10.1210/jc.2005-0096) [DOI] [PubMed] [Google Scholar]
- Zhang H, Colclough K, Gloyn AL & Pollin TI 2021. Monogenic diabetes: a gateway to precision medicine in diabetes. Journal of Clinical Investigation 131 e142244. ( 10.1172/JCI142244) [DOI] [PMC free article] [PubMed] [Google Scholar]