

Abstract
Chondrocytes, the only cells in articular cartilage, are metabolically active and responsible for the turnover of extracellular matrix and maintenance of the tissue homeostasis. Changes in chondrocyte function can cause degradation of the matrix and loss of articular cartilage integrity, leading to development and progression of osteoarthritis (OA). These changes are exemplified by accumulated mitochondrial damage and dysfunction. Because mitochondria are the critical organelles to produce energy and play a key role in cellular processes, the approaches to assess mitochondrial function under both physiological and pathological conditions enable us to uncover the mechanisms on how dysfunction of mitochondria in chondrocytes mediates signaling pathways that are involved in disturbance of cartilage homeostasis. In this chapter, we describe the methods to evaluate mitochondrial biogenesis, activity and mitochondrial DNA (mtDNA) integrity in chondrocytes.
Keywords: Osteoarthritis, Chondrocyte, Mitochondrial biogenesis, Mitochondrial DNA, Peroxisome proliferator-activated receptor γ coactivator 1α, Mitochondrial transcription factor A
1. Introduction
Osteoarthritis (OA) is the most common form of arthritis. The central feature of OA is the progressive degeneration of articular cartilage in the synovial joints, influenced by aging, biomechanical injury, metabolic disturbance, and other factors [1, 2]. Chondrocytes, the sole cell type in cartilage, regulate extracellular matrix turnover and maintain tissue homeostasis [2]. Mitochondria are critical subcellular organelles to produce energy and are involved in regulation of many cellular processes [3]. Human OA chondrocytes exhibit mitochondrial dysfunction, manifested by decreases in mitochondrial biogenesis including reduced mitochondrial DNA content, mitochondrial mass, mitochondrial respiratory complex expression and activity and intracellular ATP levels, increases in mitochondria-mediated oxidative stress, reduced antioxidant capacity, and enhanced catabolic responses to inflammatory cytokines [4, 5].
Reduced mitochondrial biogenesis in OA chondrocytes is reflected by decreases in mitochondrial DNA content, mitochondrial mass, oxygen consumption, oxidative phosphorylation (OXPHOS), and intracellular ATP levels (Fig. 1). Decreased activity of AMP-activated protein kinase (AMPK) in OA chondrocytes is a core event compromising mitochondrial biogenesis, mediated through downregulation of the NAD+-dependent protein deacetylase SIRT1 and peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), a critical regulator of mitochondrial biogenesis [7]. PGC-1α induces expression of transcription of nuclear respiratory factors (NRF1 and NRF2) that promote mitochondrial transcription factor A (TFAM) gene expression [8], leading to an increase in transcription of mtDNA [9, 10]. Pharmacologic activation of AMPK can reverse established impairment of mitochondrial biogenesis in cultured advanced human knee OA chondrocytes via SIRT1-PGC-1α signaling [6].
Fig. 1.

Reduced mitochondrial biogenesis capacity and function in human osteoarthritis (OA) chondrocytes. (a) PGC-1α, acetyl-lysine PGC-1α, expression of mitochondrial transcriptional factor A (TFAM), nuclear respiratory factor 1 (NRF-1), and NRF-2 in normal donor and human OA chondrocyte lysates, as determined by Western blotting. (b) Expression of mitochondrial respiratory complexes I–IV and ATP synthase (complex V) in normal and OA chondrocytes, as determined by Western blotting. (c) Top, Mitochondrial DNA (mtDNA) content as determined by the mtDNA-encoded cytochrome c oxidase subunit I or II (COX1 or COX2): nucleus-encoded 18S ribosomal DNA (18S rDNA) ratio. Bottom, Mitochondrial mass, as determined by MitoTracker Green FM staining. Values are the mean ± SD and are from a representative experiment of five independent experiments performed using chondrocytes from five different pairs of normal and OA (grade III or IV) donors. Original magnification × 10. (d) Oxygen consumption rate (OCR) (top) and intracellular ATP level (bottom) in normal and OA chondrocytes. Values are the mean ± SD of 3 independent experiments performed using chondrocytes from 3 different pairs of normal and OA (grade III or IV) donors. Results in a and c are representative of experiments using chondrocytes from 5 different pairs of normal and OA (grade III or IV) donors. nDNA = nuclear DNA. (This figure is reproduced from ref. 6 with permission from John Wiley & Sons)
Given that mitochondria are the main source of reactive oxygen species (ROS), mtDNA is vulnerable to oxidative damage, which can cause mtDNA mutations, promoting mitochondrial respiratory chain dysfunction and augmenting ROS production [11]. Accumulation of mtDNA damage is observed in human OA chondrocytes [12, 13]. The most frequent mtDNA mutation in human tissues is a 4977-bp deletion (mtDNA4977), known as “the common deletion.” This mtDNA4977 deletion is prevalent in human knee OA chondrocytes, correlated with reduced expression and function of the mitochondrial-localized NAD-dependent deacetylase SIRT3 (Fig. 3). Deficiency of SIRT3 greatly increased acetylation of antioxidant enzyme manganese superoxide dismutase (MnSOD or SOD2) and the DNA repair enzyme 8-oxoguanine glycosylase (OGG) in chondrocytes, leading to decreased mitochondrial biogenesis and antioxidative oxidative stress and DNA repair capacity [13], therefore compromising mitochondrial function. Pharmacologic activation of the AMPK can eliminate preexisting mtDNA mutation in human OA chondrocytes and improved mitochondrial function via SIRT3-SOD2 and SIRT3-OGG1 signaling [13].
Fig. 3.
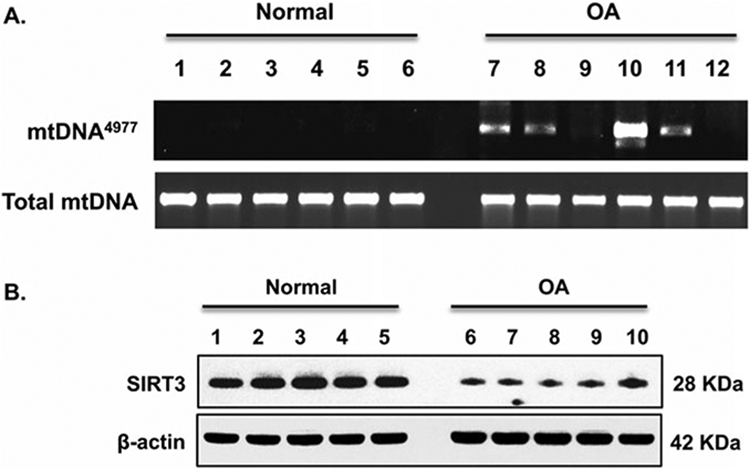
Occurrence of mtDNA4977 deletion mutation in chondrocytes associated with SIRT3 deficiency. (a) Total DNA was directly isolated from human knee cartilage from normal and OA donors. PCR analyzed mtDNA4977 deletion mutation and total mtDNA as described in the Methods. Data in A represent primary chondrocytes from six normal and six OA donors. (b) Cell lysate SIRT3 expression was examined by Western blot analysis. Data in B represent five normal and five OA donors. (This figure is reproduced from ref. 13 with permission from Elsevier)
Here we describe the methods for studying mitochondrial biogenesis, activity and mtDNA integrity in chondrocytes. These methods have allowed us to further understand critical mitochondrial function in regulation chondrocyte homeostasis and study the potential of new therapeutic approaches (e.g., activation of AMPK) for improving mitochondrial function in OA chondrocytes.
2. Materials
Prepare all the solutions using deionized water or specifically indicated. Other common materials include scalpel, nylon cell strainer, DPBS, tubes, cell culture flasks and plates, incubators, centrifuges, and tissue culture hood.
2.1. Cell Culture Medium and Reagents
Dulbecco’s Modified Eagle Medium (DMEM) containing 4.5 g/L glucose, l-glutamine, and without sodium pyruvate.
Fetal calf serum (FCS).
Streptomycin and penicillin (100×).
Type II collagenase (Worthington).
Trypsin (0.5 mg/ml).
Chondrocyte complete media: add 100 ml fetal calf serum (FCS) and 10 ml Pen/strep (100×) to 890 ml DMEM. The final complete media for chondrocyte culture is DMEM with 10% FCS, 100 μg/ml streptomycin, and 100 IU/ml penicillin.
Chondrocyte digestion media: 2 mg/ml Type II collagenase in 5% FCS DMEM.
Primary human knee chondrocytes are obtained from autopsy donors (see Note 1). The autopsy donors are graded macroscopically according to a modified Outer bridge scale [14]. Grade I represents intact cartilage surface (normal); grade II represents minimal fibrillation (OA); grades III and IV represent overt fibrillation and erosion (OA).
2.2. Mitochondrial Mass Determination
Fluorescent probe MitoTracker Green FM (see Note 2).
DMSO.
Prepare stock solutions: dissolve the lyophilized MitoTracker Green in high-grade DMSO to a final concentration of 1 mM.
Prepare working solution: dilute 1 mM MitoTracker stock solution to the final work concentration 20 nM–200 nM in growth medium.
Fluorescence microscope with Ex 490 nM and Em 516 nM.
2.3. Mitochondrial DNA Measurement
DNeasy Blood & Tissue kit (see Note 3).
Ethanol (96–100%).
Prepare buffer AW1 and buffer AW2, add the appropriate volume of ethanol (96–100%) as indicated on the bottle and shake well. The buffer is stable for at least 1 year after adding ethanol at room temperature.
Real-time PCR machine (e.g., LightCycler 480 system [Roche] 96-well).
NanoDrop spectrophotometer.
-
DNA primers.
18 s rRNA forward primer: 5′-TAGAGGGACAAGTGGCGTTC-3′
18 s rRNA reverse primer: 5′-CGCTGAGCCAGTCAGTGT-3′
mtCOXI forward primer: 5′-GGCCTGACTGGCATTGTATT-3′,
mtCOXI reverse primer: 5′-TGGCGTAGGTTTGGTCTAGG-3′,
mtCOXII forward primer: 5′-GCCGACTAAATCAAGCAACA-3′,
mtCOXII reverse primer: 5′-CAATGGGCATAAAGCTATGG-3′,
SYBR Green I Master (Roche) (see Note 4).
Prepare PCR mix of SYBR Green I Master and primers according to 20 μl standard reaction volume per well: 10 μl SYBR Green Master (2×), 1 μl forward primer (10 μM stock), 1 μl reverse primer (10 μM stock), and 3 μl PCR-grade H2O.
2.4. Mitochondrial DNA Mutation Detection
Platinum Taq DNA polymerase high fidelity.
-
Primers: for detection of deletion mtDNA4977 mutant: L8150L + H13650, WT: L3304 + H3836).
L8150L: 5′ CCGGGGGTATACTACGGTCA 3′.
H13650: 5′ GGGGAAGCGAGGTTGACCTG 3′.
L3304: 5′ AACATACCCATGGCCAACCT 3′.
H3836: 5′ GGCAGGAGTAATCAGAGGTG 3′.
PCR reaction setup: 5 μl 10× high Fidelity PCR buffer, 2 μl 50 mM MgSO4, 1 μl 10 mM dNTP mix, 1 μl forward primer, 1 μl reverse primer, 1 μl DNA, 0.2 μl Taq, 38.8 μl nuclease free water.
2.5. SDS/Western Blot Reagents
RIPA buffer: 150 mM NaCl, 5 mM EDTA pH 8.0, 50 mM Tris, pH 8.0, NP-40, 1%, 0.5% sodium deoxycholate, 0.1% SDS, 2 mM sodium vanadate and 1 × protease inhibitor cocktail.
-
Pierce BCA protein assay
Albumin (BSA) standards dilution: prepare a series of BSA standard stock solution and conduct a series of two-fold serial dilutions to generate 0.0625, 0.125, 0.25, 0.5, 1 and 2 mg/ml protein standards.
BCA Working Reagent: mix 50 parts of BCA reagent A with 1 part of BCA reagent B (Reagent A: B, 50:1).
-
Antibodies
Primary antibody: anti-PGC-1α, anti-TFAM, anti-NRF1, anti-NRF2, anti-OXPHOS, anti-SOD2, anti-acetylated SOD2, anti-OGG1, anti-acetylated OGG1, anti-acetyl lysine, and anti-β-Actin. Secondary antibody: donkey anti-mouse IgG (H + L) HRP, donkey anti-rabbit IgG (H + L) HRP.
SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific): working solution made by mixing equal parts of detection reagents 1 and 2 in the kit. Use 0.125 ml working solution per cm2 of membrane.
4–20% SDS-PAGE gradient gel (see Note 5)
SDS-PAGE running buffer: 0.025 M Tris–HCI, PH8.3, 0.192 M glycine, 0.1% SDS (see Note 6).
Western blot transfer buffer: 0.025 M Tris–HCI, 0.192 M glycine, 20% methanol (see Note 6).
Western blot wash buffer (TBST): 20 mM Tris–HCI and 150 mM NaCl, pH 7.4 containing 0.05% Tween-20 (see Note 6).
PVDF transfer membranes.
Blocking/antibody diluent solution: 5% milk or 5% BSA in TBST.
2.6. Assessment of Acetylation Levels of PGC-1a
Pierce classic IP kit for PGC-1α immunoprecipitation (Thermo Fisher Scientific).
Primary antibody anti-PGC-1α for IP (Santa Cruz Biotechnology), anti-acetyl lysine antibody (Cell Signaling).
2.7. siRNA Transfection Reagent
X-tremeGene siRNA transfection reagent (see Note 7).
TFAM siRNA and control siRNA (Thermo Fisher Scientific).
Opti-MEM® I Reduced-Serum Medium.
2.8. SIRT3 Deacetylation Activity Assay
SIRT3 activity assay kit (Abcam).
96-well plate—black wall
Microplate reader capable of measuring fluorescence at Ex/Em = 340–360/440–460 nm.
Orbital shaker.
2.9. Oxygen Consumption Rate (OCR) Measurement
Agilent Seahorse XFe/XF analyzers.
XFe 96 FluxPax (containing sensor cartridge, cell culture microplates, and calibrant solution. Agilent Tech).
XF 1 M Glucose solution (Agilent Tech).
XF DMEM medium (Agilent Tech). The glucose is added into Agilent DMEM to make final concentration of 4500 mg/l, since the basal medium does not contain glucose. Adjust pH to 7.4.
Seahorse XF cell Mito Stress test kit (Agilent Tech).
Prepare oligomycin, FCCP, and rotenone/antimycin A into the final concentration of 2.5 μM, 1 μM, and 0.5 μM per well (optimal concentrations for human OA chondrocytes).
2.10. Intracellular ATP Measurement
- ATP colorimetric/fluorometric assay kit (Biovision) (see Note 8).
- (a) Buffer preparation: warm ATP assay buffer to room temperature before use.
- (b) ATP converter: dissolve ATP converter in 220 μl ATP assay buffer. Aliquot and store at −20 °C.
- (c) Developer Mix: Dissolve in 220 μl ATP Assay Buffer. Aliquot and store at −20 °C.
- (d) ATP Standard: Dissolve in 100 μl of distilled water to generate 10 mM stock solution. Keep on ice while in use. Store at −20 °C.
- (e) Sample preparation: Lyse 1 × 106 cells in 100 μl ATP Assay Buffer and deproteinize using the Kit (BioVision) or 10 kDa Spin Column (Millipore).
- (f) Standard Curve Preparation: dilute 10 μl of the ATP Standard with 90 μl of dH2O to generate 1 mM ATP standard, mix well.
96-well plate with flat bottom
Multi-well spectrophotometer (OD 570 nM).
3. Methods
3.1. Primary Human Knee Chondrocyte Cell Preparation and Culture as Reported [15, 16]
Human knee chondrocytes are isolated from autopsy donors that are graded as described in Subheading 2.1, item 8.
Before the chondrocyte isolation, the cartilage surfaces of autopsy donor are rinsed with saline first and cartilage sections are cut at around 5 mm apart by scalpel vertically from the cartilage surface onto the subchondral bone. These strips are washed with DMEM (see Note 9).
After discarding the supernatant wash, the cartilage slices are cut into 3 mm × 3 mm pieces and digested with double volume of digestion media (from Subheading 2.1, item 7) on a gyratory shaker at 37 °C overnight.
The next day, the digested mixture is filtered through 100 μm nylon cell strainer and human chondrocytes are pelleted by centrifuge and washed using complete DMEM. And then the cells are resuspended in compete DMEM media and seeded into the culture flasks (3 × 106 cells/T75 flask). The cells are incubated at 37 °C in a humidified gas mixture containing 5% CO2 balanced with air and ready for use upon confluency (see Note 10).
Unless indicated, chondrocytes are plated at 3 × 105 cells per well in 12 well plates or 7.5 × 105 cells per well in 6-well plates.
3.2. Protein Expression of PGC-1α, TFAM, NRF1, NRF2, SIRT3, SOD2, OGG1, Acetylated SOD2, Acetylated OGG1, and OXPHOS
3.2.1. Cell Lysate Preparation
Human chondrocyte cells are harvested after desired treatment and centrifuged at 500 × g, 4 °C for 5 min, and are washed once with PBS.
The cell pellets are resuspended in RIPA buffer with 2 mM sodium vanadate and protease inhibitor cocktail (1 × 106 cells in 100 μl) and sonicated for 5 s, and then lysed for half an hour on ice.
The mixture of lysed cells is centrifuged at 12,000 × g, 4 °C for 10 min and the supernatant is the final protein extract.
3.2.2. Protein Concentration Determined by BCA
Dilute the protein supernatant from Subheading 3.2.1 if needed.
BCA Working Reagent is mixed as described in Subheading 2.5, item 2.
Microplate procedure: pipet 5 μl standards or samples into 100 μl BCA working solution and mix the plate thoroughly.
Cover the plate and incubate for 30 min at RT and measure the absorbance at 562 nM on a plate reader. Determine the protein concentration for each sample based on the BSA standard curve.
Prepare protein extracts of cell lysates with loading buffer and heat at 95 °C for 5 min.
3.2.3. Protein Separation and Membrane Transfer
Centrifuge the heated samples at 3000 × g for 30 s to bring down the condensate.
Load the samples (15–20 μg) to 4–20% SDS-PAGE gradient gels, and electrophoreses them at constant 80 volts (V) until the samples enter the gel and then continue at 150 V till the dye reaches the bottom of the gel.
Prepare the Polyvinylidene difluoride (PVDF) membrane and quickly immerse in methanol for 30 s, after rinsing in water, put the membrane in transfer buffer until use.
Following electrophoresis, prepare the wet-sandwich transfer cassette and run at constant current 250 mA for 90 min in cold room or ice box; or use semi-dry transfer at 15 V for 25 min.
3.2.4. Western Blotting
After transfer, rinse the membrane with TBST and incubate in blocking solution for 30 min to 1 h at RT.
Incubate the membrane with appropriate primary antibodies (OXPHOS diluted at 1:300 in 5% milk. Others are diluted at 1:1000 in 5% BSA) on a shaker at 4 °C overnight.
The next day, wash the membrane thoroughly with TBST 3 times (5 min each time), and then add secondary antibody (1:3000 in 5% milk) for 1 h at RT.
Wash the membrane 3 times with TBST and incubate the membrane with working solution from Subheading 2.5, item 4 for 1 min at room temperature.
Place the membrane in a plastic sheet protector and place it in a film cassette. Place the film on top of the membrane. Vary the exposure time to achieve optimal results. Figures 1a, b and 3b are examples of Western blot showing that PGC-1α, NRF-1, NRF-2, OXPHOS, and SIRT3 decreased in OA chondrocytes.
3.3. Assessment of Acetylation Levels of PGC-1α
3.3.1. Immunoprecipitation with PGC-1α Antibody Using Pierce Classic IP Kit (See Note 11)
Human chondrocytes are cultured in 6-well plate with and without treatment of testing agents for desired time. After removing the culture media, the cells are washed once with cold DPBS.
Add 400 μl ice cold IP lysis/wash buffer into the cells and incubate on ice for 5 min with periodic mixing.
Transfer the lysate to a microcentrifuge tube and incubate on ice for another 20 min, then centrifuge at ~13,000 × g for 10 min to pellet the cell debris.
Transfer supernatant to a new tube and determine protein concentration as described in Subheading 3.2. The cell lysate is ready for preclear by control agarose resin.
Prepare enough control agarose resin slurry (80 μl for 1 mg lysate), add the slurry into a spin column and wash once with PBS, centrifuge, and discard the flow-through.
Add 1 mg lysate to the column containing the resin and incubate at 4 °C for 1 h with gentle end-over-end mixing.
Centrifuge column at 1000 × g for 1 min. Discard the column containing the resin and save the flow-through, which will be cell lysate for next step.
Determine the concentration of cell lysate from step 7 as described in Subheading 3.2.2.
Add 5 μg PGC-1α primary antibody into 500 μg cell lysate in a microcentrifuge tube and dilute the antibody–lysate solution to 300–600 μl with IP Lysis/Wash Buffer. Incubate with gentle rocking overnight at 4 °C to form antibody–sample complex.
Gently swirl the bottle of Pierce Protein A/G Agarose to obtain an even suspension. Using a cut pipette tip, add 50 μl of the resin slurry into a Pierce Spin Column. Place column into a microcentrifuge tube and centrifuge at 1000 × g for 1 min. Discard the flow-through.
Wash resin twice with 100 μL of cold IP Lysis/Wash Buffer. Discard the flow-through after each wash.
Gently tap the bottom of the spin column on a paper towel to remove excess liquid and insert the bottom plug.
Add the antibody–sample complex to Protein A/G plus Agarose in the spin column. Attach the screw cap and incubate column with gentle end-over-end mixing or shaking for 1–2 h.
Remove bottom plug, loosen the screw cap, and place the column in a collection tube. Centrifuge column and save the flow-through. Do not discard flow-through until confirming that the IP has been successful.
Remove the screw cap, place the column into a new collection tube, add 200 μl of IP Lysis/Wash Buffer, and centrifuge.
Wash the resin three times with 200 μl IP Lysis/Wash Buffer and centrifuge after each wash.
Wash the resin once with 100 μl of 1× Conditioning Buffer (diluted from 100× with ultrapure water).
Sample-buffer elution: place the spin column containing the resin into a new collection tube and add 50 μl of 2× loading buffer. Keep the column unplugged and in the collection tube, and incubate at 100 °C for 5–10 min. Centrifuge to collect eluate. Allow the sample to cool to room temperature before applying to the SDS-PAGE gel.
3.3.2. Western Blot with Anti-Acetyl Lysine Antibody
Western blot is performed with anti-acetyl-lysine as described in Subheading 3.2. As depicted in Fig. 1a, acetylation level of PGC-1α increases in OA chondrocytes.
3.4. Mitochondrial Mass by MitoTracker Green FM Staining
Before staining, all the reagents are warmed up to room temperature and working solution is prepared as described in Subheading 2.2 (see Note 12).
Human chondrocytes are seeded into 6 well plates with the complete medium and treated with appropriate compound for desired time.
When the cells reach the desired confluency and treatment, replace the growth medium with prewarmed (37 °C) staining solution (see Note 13) and place the plates back in an incubator (see Note 14).
After incubation for 30 min, staining solution is replaced with the prewarmed media, and fluorescence stained cells are visualized under a fluorescence microscope (see Note 15). The example shown in Fig. 1c depicts that mitochondrial mass is much less in OA compared to normal chondrocytes.
3.5. mtDNA Content by Real-Time PCR
3.5.1. Total DNA Is Isolated from Chondrocytes Using DNeasy Blood & Tissue Kit
Buffer AW1 and buffer AW2 are prepared as described in Subheading 2.3.
Centrifuge the appropriate number of cells (maximum 5 × 106) for 5 min at 300 × g. Resuspend the pellet in 200 μl PBS, add 20 μl proteinase K.
Add 200 μl Buffer AL (without added ethanol), mix thoroughly by vortexing for 5–10 s, and incubate at 56 °C for 10 min.
Add 200 μl ethanol (100% ethanol) to the sample and mix thoroughly by vortexing to generate a homogeneous solution.
Pipet the mixture from last step into the DNeasy Mini spin column placed in a 2 ml collection tube. Centrifuge at 6000 × g for 1 min. Discard the flow-through and collection tube.
Place the column in a new 2 ml collection tube, add 500 μl buffer AW1, centrifuge at 6000 × g for 1 min. Discard the flow-through and collection tube.
Place the column in a new 2 ml collection tube, add 500 μl buffer AW2, centrifuge at 20,000 × g for 3 min. Discard the flow-through and collection tube (see Note 16).
Place the column in a clean 1.5 ml or 2 ml Eppendorf tube and pipet 200 μl Buffer AE onto dry DNeasy membrane. Incubate at RT for 1 min, then centrifuge for 1 min at 6000 × g to elute.
3.5.2. DNA Concentration Measured by NanoDrop
Turn on the computer attached to the NanoDrop.
Wash the NanoDrop pedestal using purified water.
Open the NanoDrop software and click on the ‘Nucleic Acid’ button. Add water to the pedestal and wait for the machine to initialize. When it’s done, life the upper arm and dry the pedestal with a wipe.
Add 2 μl water or elution buffer to blank the NanoDrop. Dry the pedestal with wipe once done (see Note 17).
Add 2 μl sample from Subheading 3.5.1 to the pedestal and click the ‘measure’ button for measurement, and then export the data to excel.
Clean the pedestal when finishing all the measurement.
3.5.3. Real-Time PCR
Pipet 15 μl PCR mix from Subheading 2.3, item 7 into each well.
Add 5 μl DNA template Mitochondrial DNA (0.1 ng) or nuclear DNA (10 ng) into each well. Seal the plates and centrifuge at 1500 × g for 2 min to bring the mixture to the bottom.
Load the plate into the Real-time PCR machine (e.g., LightCycler 480 instrument) and start the quantitative real-time PCR program: initial denaturation at 94 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing/extension and fluorescence reading at 60 °C for 1 min.
Fold changes of mtDNA-encoded cytochrome c oxidase subunit I (COXI) and COXII are calculated by 2-ΔΔct methods. 18 s rDNA is used as internal standard. Ratio of COXI or COXII DNA copies to 18S rDNA represents relative mitochondrial copy number. The examples shown in Figs. 1c and 2b demonstrate that mtDNA COXI and COXII are significantly decreased in OA chondrocytes.
Fig. 2.

Decreased mitochondrial biogenesis with knockdown of mitochondrial transcriptional factor A (TFAM). (a) Expression of mitochondrial respiratory complexes I–IV and ATP synthase (complex V) in normal and OA chondrocytes transfected with TFAM siRNA or control siRNA, as determined by Western blotting. Results are representative of 5 independent experiments using five different pairs of normal donors and donors with OA. (b) and (c) Mitochondrial DNA (mtDNA) content (b) and mitochondrial mass (c) in human knee chondrocytes (passage 1) from both normal donors and donors with osteoarthritis (OA) (grade IV) transfected with TFAM siRNA and control (Ctrl) siRNA, as determined by the mtDNA-encoded cytochrome c oxidase subunit I or II (COX1 or COX2): nucleus-encoded 18S ribosomal DNA (18S rDNA) ratio. Values are the mean ± SD and are representative of three independent experiments performed using chondrocytes from three different donors. (This figure is reproduced from ref. 6 with permission from John Wiley & Sons)
3.6. OCR Measurement
Prepare XF medium (from Subheading 2.9, item 3).
Plate 5 × 103 cells into each well/Agilent 96-well cell culture microplate with regular culture medium (see Note 18).
Incubate cells in the 5% CO2 incubator overnight to allow the cells to adhere to the plate.
Add 200 μl sterile water into the utility plate, immerse sensor into water, and place the module into 37 °C oven (non-CO2) overnight (see Note 19).
On the day of experiment, aspirate water and replenish with prewarm XF calibration solution for at least 1 h.
Wash the cells with XF medium twice to remove buffered medium. Add 180 μl of XF medium into each well and place plate in 37 °C oven (non-CO2) for at least 45 min to 1 h prior to the assay.
Load 20 μl of 2.5 μM oligomycin (from Subheading 2.9, item 6) into “A” position of cartridge, 22 μl of 1 μM FCCP into “B” position of cartridge, and 25 μl of 0.5 μM rotenone/antimycin A into port “C” position of cartridge.
Place the cartridge with calibration fluid plate into instrument tray of Seahorse analyzer and click “Continue.” It takes 15–30 min to complete.
When prompted, replace the calibration plate with the cell culture plate then click start. The example in Fig. 1d shows that OA chondrocytes have significantly lower oxygen consumption rate.
3.7. ATP Measurement
Assay buffer, ATP converter, developer mix, ATP standard, and sample are prepared as described in Subheading 2.10.
Add appropriate volume of sample (2–50 μl; from Subheading 2.10, item 1(e)) to a 96-well plate. Adjust the volume to 50 μl/well with ATP Assay Buffer (from Subheading 2.10, item 1(a); see Note 20).
Add 0, 2, 4, 6, 8, and 10 μl of ATP Standard solution (see Subheading 2.10, item 1(f)) into a series of wells and adjust volume to 50 μl/well with ATP Assay Buffer to generate 0, 2, 4, 6, 8, 10 nmol/well of ATP Standard.
-
Reaction Mix: Mix enough reagent for the number of samples and standards to be performed: For each well, prepare a total 50 μl Reaction Mix:
Colorimetric
assay (μl)Fluorometric
assay (μl)ATP assay buffer 44 45.8 ATP probe 2 0.2 ATP converter (from Subheading 2.10, item 1(b)) 2 2 Developer (from Subheading 2.10, item 1(c)) 2 2 Mix well. Add 50 μl of the Reaction Mix to each well containing the ATP Standard and test samples.
Measurement: Incubate at room temperature for 30 min, protected from light. Measure absorbance (OD 570 nm) or fluorescence (Ex/Em = 535/587 nm) in a microplate reader. The signals are stable for 2 h.
-
Calculation: Correct background by subtracting the value derived from the 0 ATP standards from all readings. If the background control reading is significant, subtract the background control reading from sample reading. Plot the Standard Curve. Apply ATP sample readings to the standard curve to get B nmol of ATP in the sample well.
where: B is ATP amount in the reaction well from standard curve (nmol).V is the sample volume added into the sample well (μl).
D is the dilution factor.
The example shown in Fig. 1d illustrates that OA chondrocytes contain lower levels of ATP compared to normal healthy chondrocytes (Fig. 1d).
3.8. Knockdown of TFAM in Human Knee Articular Chondrocytes
3.8.1. Cell Preparation Before Transfection
Cells are seeded in 6-well plate (2 ml media/well) the day before and reach 70% confluence on transfection day.
3.8.2. Transfection (Single Well of 6-Well Plate as an Example)
Dilute 10 μl X-tremeGENE siRNA transfection reagent with 100 μl serum/antibiotics-free Opti-MEM I Medium.
Dilute 2 μg siRNA with 100 μl serum/antibiotics-free Opti-MEM I Medium.
Mix the transfection reagent and siRNA complex by pipetting up and down and incubate for 15–20 min at RT (see Note 21).
Add the complex directly to the cells and incubate until assay for gene knockdown is performed (72 h) (see Note 22).
Levels of TFAM expression is examined by SDS-PAGE/Western blot analysis. Figure 2 demonstrates that knocking down TFAM expression (A) results in lower OXPHOS that is indicated by reduced expression subunits of mitochondrial respiratory complexes (A), and reduced mitochondrial mass (Fig. 2c) and DNA content (Fig. 2b).
3.9. mtDNA Mutation Detection by Real-Time PCR
Total DNA isolation from chondrocytes and DNA concentration measurement are as described as 3.5.
The PCR reaction (from Subheading 2.4, item 3) is carried out at 94 °C for 15 s, 58 °C for 15 s, and 72 °C for 40 s per cycle for total 35 cycles.
Products are separated by electrophoresis on a 1.5% agarose gel. The example in Fig. 3a shows that OA chondrocytes have a higher occurrence rate for mtDNA4977 deletion mutation.
3.10. SIRT3 Deacetylase Activity Assay (See Note 23)
5 × 105 chondrocytes are cultured in 6-well plate with and without treatment of testing agents for desired time. After removing the culture media, the cells are washed once with cold DPBS. The cells are collected in 500 μl cold DPBS using a cell scraper and are then transferred into 1.5 ml Eppendorf tube.
After centrifugation at 200 × g for 10 min at 4 °C, the cell pellets are resuspended with fresh cold 30 μl DPBS and are dispersed using a micro tube homogenizer for 5–10 s on ice.
The cell lysates are centrifuged at 12500 × g for 10 min at 4 °C. The supernatant is collected for SIRT3 activity assay.
The fluorometric SIRT3 activity assay kit is used to measures the enzymatic activity of SIRT3 by the basic principle of changing a SIRT3 reaction into the activity of the peptidase. In this kit, fluorophore and quencher are coupled to amino terminal and carboxyl terminal of substrate peptide, respectively, and before reaction of deacetylase, the fluorescence cannot be emitted. However, if SIRT3 performs deacetylation, substrate peptide will be cut by the action of peptidase added simultaneously, quencher will separate from fluorophore, and fluorescence will be emitted. Deacetylase enzyme activity is measured by measuring this fluorescence intensity.
Follow the manufacturer’s assay protocol exactly to quantify SIRT3 activity (see Note 24). The example illustrated in Fig. 4 shows that SIRT3 activity decreases in OA chondrocytes.
Fig. 4.
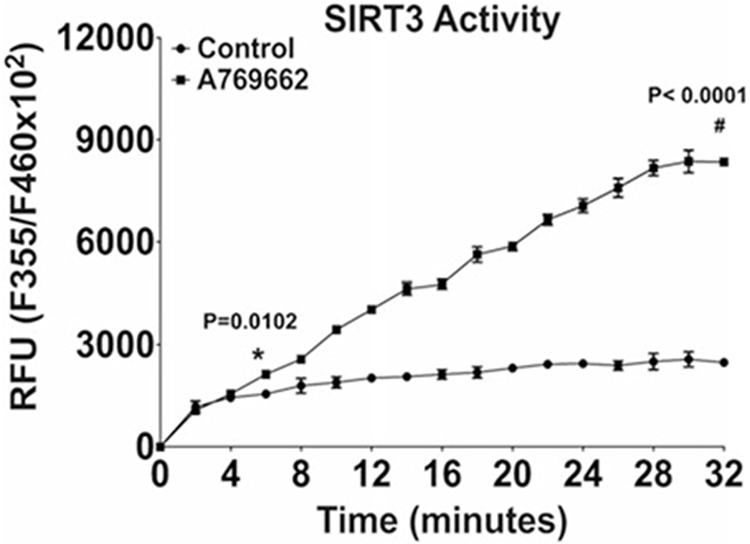
Pharmacologic activation of AMPK significantly improved SIRT3 activity in OA chondrocytes. Cultured primary human knee OA chondrocytes were treated with AMPK pharmacological activator A-769662 (0.125 mM) for 2 h, and the SIRT3 activity was measured. (This figure is reproduced from ref. 13 with permission from Elsevier)
Acknowledgments
This work was supported by Department of Veterans Affairs Merit Review grant 1I01BX002234 and NIH grant AR1067966 to R.L-B.
Footnotes
Human chondrocytes isolated from deidentified human knee donors are performed in compliance with institutional IRB reviewed and approved human subjects’ protocols.
The kit is stable for at least 6 months if stored in ≤−20 °C upon arrival and protect from light.
All the buffers in this kit should be stored dry at room temperature (15–25 °C) and are stable for 1 year. Other equipment or reagent needed: Vortexer, ethanol (96–100%), microcentrifuge tubes (1.5 ml or 2 ml), thermomixer, shaking water bath, or rocking platform for heating at 56 °C, PBS, pH 7.2 (50 mM potassium phosphate, 150 mM NaCl).
Keep the master away from light and store the kit at −15 to −25 °C until expiration date.
4–20% Mini-PROTEAN® TGX™ Precast Protein Gels are bought from Bio-Rad or self-made in house.
10 × stock of the buffers are prepared and dilute into 1× using DI H2O on use day. Do not reuse all these 1× buffers.
X-tremeGene siRNA transfection reagent is stored at 2–8 °C. Do not freeze. Serum-free culture medium without supplements or Opti-MEM I medium is recommended for diluting the siRNA or the reagent.
The entire kit is protected from light and stored at −20 °C. ATP probe, ATP converter and developer mix are dissolved and aliquoted to store in −20 °C. They should be used within 2 months.
Use best aseptic practice to keep sample sterile.
Media is replaced every 3–4 days until use, and it usually takes 2–3 weeks for the chondrocytes to reach confluency in primary culture. Only passage 1 chondrocytes are used in all the experiments.
Perform all steps at 4 °C, unless otherwise indicated and all the resin centrifugation steps for 30–60 s at low speed (i.e., 1000 × g). A lysate preclearing step to reduce nonspecific binding to Protein A/G agarose beads is optional.
Allow the product to warm to room temperature. After dissolving the vial, aliquot the stock solution if needed and store them at ≤ −20 °C and protect from light, avoid multiple rethaw.
Optimize the dye concentration and try to keep it as low as possible. It will cause potential artifacts or toxicity at higher dose.
The incubation time varies and needs to be optimized. 15–45 min incubation is generally sufficient.
The cells are visualized right after staining, but the stained cells can be fixed for later analysis. Briefly, the cells are washed with prewarmed growth medium and then fixed with 2–4% formaldehyde growth medium for 15 min at 37 °C. After fixation, the cells are rinsed several times and kept in the medium until use.
At the last wash step, remove the column carefully to avoid any contamination of column from the flow-through ethanol. If it occurs, just repeat the centrifugation again in a new collection tube.
Use the same buffer as samples to blank the instrument. For example, if the sample is eluted in water, then use water for blank.
This is the optimal cell density to perform this experiment in our hand. It needs to optimize the cell density before performing this experiment.
Seal the sensor in the same container to prevent dry out and keep sterile.
Use fresh sample or snap frozen sample. Optimize the sample amount or dilution to make sure the reading is in the standard curve range.
Diluted siRNA should be combined with diluted transfection reagent within 5 min.
Removal of growth medium is not necessary.
All procedures are performed on ice or at 4 °C.
The reaction is initiated and the fluorescence intensity is measured by mixing simultaneously fluorescence-labeled acetylated peptide (the substrate), SIRT3, NAD and the developer. Since the reaction is not stopped, it is necessary to measure fluorescence intensity at regular intervals after the reaction is initiated, and to determine reaction velocity.
References
- 1.Felson DT, Lawrence RC, Dieppe PA, Hirsch R, Helmick CG, Jordan JM et al. (2000) Osteoarthritis: new insights. Part 1: the disease and its risk factors. Ann Intern Med 133:635–646 [DOI] [PubMed] [Google Scholar]
- 2.Loeser RF, Goldring SR, Scanzello CR, Goldring MB (2012) Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 64 (6):1697–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408:239–247 [DOI] [PubMed] [Google Scholar]
- 4.Blanco FJ, Rego I, Ruiz-Romero C (2011) The role of mitochondria in osteoarthritis. Nat Rev Rheumatol 7(3):161–169 [DOI] [PubMed] [Google Scholar]
- 5.Blanco FJ, López-Armada MJ, Maneiro E (2004) Mitochondrial dysfunction in osteoarthritis. Mitochondrion 4(5–6):715–728 [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Zhao X, Lotz M, Terkeltaub R, Liu-Bryan R(2015) Mitochondrial biogenesis is impaired in osteoarthritic chondrocytes but reversible via peroxisome proliferator-activated receptor-γ coactivator 1α. Arthritis Rheumatol 67(8):2141–2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez-Marcos PJ, Auwerx J (2011) Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93 (4):884–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piantadosi CA, Suliman HB (2006) Mitochondrial transcription factor a induction by redox activation of nuclear respiratory factor 1. J Biol Chem 281(1):324–333 [DOI] [PubMed] [Google Scholar]
- 9.Scarpulla RC, Vega RB, Kelly DP (2012) Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab 23 (9):459–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jornayvaz FR, Shulman GI (2010) Regulation of mitochondrial biogenesis. Essays Biochem 47:69–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev MF (2009) Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res 37(8):2539–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang MC, Hung SC, Chen WY, Chen TL, Lee CF, Lee HC, Wang KL, Chiou CC, Wei YH (2005) Accumulation of mitochondrial DNA with 4977-bp deletion in knee cartilage—an association with idiopathic osteoarthritis. Osteoarthr Cartil 13(11):1004–1011 [DOI] [PubMed] [Google Scholar]
- 13.Chen L-Y, Wang Y, Terkeltaub R, Liu-Bryan R (2018) Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthr Cartil 26(11):1539–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamada K, Healey R, Amiel D, Lotz M, Coutts R (2002) Subchondral bone of the human knee joint in aging and osteoarthritis. Osteoarthr Cartil 10:360–369 [DOI] [PubMed] [Google Scholar]
- 15.Blanco FJ, Ochs RL, Schwarz H, Lotz M (1995) Chondrocyte apoptosis induced by nitric oxide. Am J Pathol 146(1):75–85 [PMC free article] [PubMed] [Google Scholar]
- 16.Akasaki Y, Alvarez-Garcia O, Saito M, Caramés B, Iwamoto Y, Lotz MK (2014) FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis Rheumatol 66(12):3349–3358 [DOI] [PMC free article] [PubMed] [Google Scholar]