

Abstract
Background:
Cholesterol is necessary for proper neurodevelopment and neuronal health. The brain relies on neural and astrocytic de novo cholesterol synthesis. Huntington’s disease presents with altered levels of cholesterol precursors however it is unknown when the disruption in this molecular pathway occurs and whether Manganese (Mn) may alter these metabolic alterations.
Objective:
To examine the effect of Mn exposure on cholesterol biosynthesis in pre-manifest and manifest Huntington’s disease mice.
Methods:
12-week (pre-manifest) male and female and 42-week old (manifest) female YAC128Q and littermate control mice received 3 subcutaneous Mn or vehicle injections. Animals were sacrificed 24 hours after the final injection and striatum, cerebral cortex and cerebellum were collected to measure cholesterol and cholesterol precursors using LC/MS-MS.
Results:
Striatal desmosterol and cholesterol are increased in pre-manifest HD females compared to age-matched WT female mice. Striatal lanosterol, 8-DHC and desmosterol and cholesterol are reduced in manifest HD females compared to age- and sex-matched WT mice with minimal effects in the cortex and cerebellum. Mn treatment had no effect in the pre-manifest or manifest female brain except reduced lanosterol levels in the cortex of pre-manifest female mice. Neither Mn or HD altered brain cholesterol precursor levels in the pre-manifest HD or WT male mouse.
Conclusions:
Cholesterol biosynthesis is impaired in early disease stage in female HD mice only and continues throughout disease. These alterations appear largely striatal-specific. Acute systemic exposure to Mn did not significantly alter cholesterol biosynthesis in the striatum at any disease stage.
Keywords: cholesterol, sterols, Huntington’s Disease, striatum, manganese
Introduction
Cholesterol is an amphipathic sterol which participates in numerous biological functions in the brain such as neurite growth and synaptic connectivity, as well as structural functions1. Cholesterol is unable to freely cross the blood-brain barrier (BBB) and as such, the brain relies on de novo synthesis. Interestingly, the brain has a high cholesterol content compared to other organs with estimates indicating that approximately 20% of all body cholesterol is found within the brain2. Cholesterol is primarily considered a component of myelin but is also found in the plasma membrane of neurons and astrocytes. Neurons and astrocytes utilize different precursors to synthesize cholesterol: neurons contain higher levels of select cholesterol precursors such as lanosterol and 7-dehydrocholesterol (7-DHC) originating from the Kandutsch-Russel pathway whereas astrocytes convert desmosterol to cholesterol via the Bloch pathway3. This distinction is corroborated with work demonstrating tissue-specific pathways utilizing either 7-DHC or desmosterol as penultimate metabolites of cholesterol biosynthesis4.
The change in cellular composition throughout the course of select neurodegenerative diseases should be carefully considered when trying to understand select cellular contributions to disease. For instance, Huntington’s disease (HD) is associated with the progressive loss of medium spiny neurons, an increase in astrocytic infiltration and an activation within the basal ganglia throughout disease progression. Given what is known about HD pathology and the death of medium spiny neurons of the striatum, reductions in total cholesterol levels might be expected. Indeed, several human and animal model papers have identified an alteration in cholesterol biosynthesis and metabolism in HD5–10. Recent work examining global metabolomic changes in HD in mice, sheep and post-mortem human brains have indicated that sterols among other lipids are altered in HD9,11. Investigators found an elevation in the cholesterol precursors lanosterol, desmosterol, and 7-dehydrocholesterol as well as cholesterol levels in the putamen of post-mortem brains of HD patients compared to healthy controls9. The inconsistency observed in alterations of cholesterol biosynthesis in HD may be due, in part, to the role of disease stage and the activity of select cell populations within these stages.
Just as important as the consideration of specific cellular metabolic profiles with regards to a disease state is the temporal alterations of those metabolic profiles as HD progresses. For instance, in the R6/1 mouse model of HD, investigators found evidence that the aberrations in cholesterol biosynthesis and metabolism that occur in HD change with age. They observed reductions in lanosterol and lathosterol in earlier stages of disease while desmosterol was elevated at later stages12. The extent of these metabolic changes appear dependent on disease state – with later stage disease models exhibiting more exaggerated phenotypes. It is unknown whether these molecular aberrations subsist throughout disease state or rather initiate a larger cascade of pathogenic events which exacerbate this molecular phenomenon in later disease stages. Regardless, even the early molecular mechanisms responsible for these observed changes in cholesterol biosynthesis in HD are unknown.
Changes in manganese (Mn) dependent processes and sensitivity to Mn toxicity have been reported across multiple HD model systems, with some corroborating evidence in HD patients as well13–15 . Thus, one variable which may impact the level of neuronal cholesterol biosynthesis in HD is the essential metal Mn. In the general population, adequate levels of Mn are easily obtained through diet; however, there are several occupational and environmental exposure settings, as well as patients receiving total parenteral nutrition, which may lead to excessive Mn accumulation16–18. Gross overexposure to Mn can result in ‘manganism,’ a condition which exhibits a parkinsonian-like phenotype. The cellular consequences of Mn exposure are complex, but it is known that the transition metal is a potent oxidative stressor by disrupting mitochondrial function and has been linked to the pathogenesis of several chronic diseases associated with increased oxidative stress19. For instance, Mn impairs antioxidant defense mechanisms by inhibiting glutathione-peroxidase activity20. Currently, there are no known Mn-dependent enzymes regulating central or peripheral cholesterol biosynthesis, there appears to be some Mn-responsive elements as treatment with daily Mn exposure in rats increased hippocampal cholesterol21. It remains unclear at what step during cholesterol biosynthesis that this increase occurs and the molecular mechanism(s) driving this phenomenon. In the following study, we investigated the impact of age/disease progression and Mn treatment on pre-manifest male and female HD and control mice, and a follow-up study looking at older manifest (42-weeks of age) female HD mice versus female controls. We sought to understand 1) how age impacts the alteration in CNS cholesterol biosynthesis in HD and control; and 2) if differences in Mn biology may play a role in altering HD dependent effects on CNS cholesterol biosynthesis; and 3) whether sex alters the impact of HD genotype or Mn on cholesterol biosynthesis.
Materials and Methods
Materials
Unless otherwise noted, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO). HPLC grade solvents were purchased from Thermo Fisher Scientific Inc. (Waltham, MA). All sterol standards, natural and isotopically labeled, used in this study are available from Kerafast, Inc. (Boston MA).
Animals
All animals were approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee. Animal husbandry and genotyping information has been previously described by Bichell et al14. Briefly, male and female FVB-Tg(YAC128)53Hay/J mice purchased from Jackson Laboratory (Bar Harbor, ME; stock number: 027432) were weaned at 21 days and co-housed in groups of 2–4. Hemizygous transgenic animals and wild-type (WT) littermates were used for experiments. It should be acknowledged that mice expressing normal human HTT (YAC18) were not used in this study22. Pre-manifest male and female YAC128 and WT at 12-weeks of age as well as 42-week old manifest female mice were included in the study. Mice were exposed to a 7-day Mn injection paradigm (injections on Days 1,4 and 7) previously published and known to elevate brain Mn levels23. Mice were injected with a 1% solution of MnCl2 × 4H2O in filtered MilliQ water at 50 mg/kg body weight, or with vehicle (filtered water). Twenty-four hours after the final injection, mice were sacrificed, the brain removed and cerebellum, striatum and motor cortex were dissected, flash-frozen in liquid nitrogen and stored at −80°C until later analysis. Wet tissue weight was not recorded. It should be acknowledged that sterols present in the small amount of blood remaining in the dissected tissue were incorporated into the brain sterol levels.
Sterol Extraction
The cholesterol precursors lanosterol, 7-dehydrocholesterol (7DHC), 8-dehydrocholesterol (8DHC), desmosterol as well as total cholesterol levels were measured using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) with multiple reaction monitoring (MRM). The sensitivity of the targeted lipidomic MRM analysis following the derivitizaiton of sterols to 4-phenyl-1,2,4-triazoline-3,5-dione (PTAD)24 is improved compared to previous bioanalystical analyses of sterols25,26. In order to conserve tissue sample for subsequent analyses, we first confirmed that lipid extraction from a protein lysate from a tissue homogenate27,28 provided similar extraction efficacy compared to extracting sterols from the entire tissue (Supplementary Figure 1). All sterol levels for both extraction methods are significantly correlated with no significant differences in mean sterol levels between methods with the exception of cholesterol (tissue extraction more efficient) and lanosterol (protein extraction more efficient). For validation of the extraction method, we selected brain and peripheral tissue from 5 non-experimental animals, 12 weeks of age of mixed-sex. Tissue was collected in an identical manner as in the primary study. Each tissue was split in half with each half used to measure sterols either a) from a protein lysate or b) directly from tissue. For the primary study, striatal, cortical and cerebellar tissue was homogenized in 500 μL tissue lysis buffer consisting of 120mM NaCl, 50mM HEPES and 1% IGEPAL. 10 μL of tissue lysate was reserved for protein quantification (Pierce BCA protein assay kit, cat# 23225). 10-50 μg protein were diluted with lysis buffer into a total volume of 20 μL. To each sample, 0.087 nmol d7-Chol, 0.033 nmol d7-7-DHC, 0.25 nmol 13C3-Des, 0.23 nmol 13C-Lanosterol, 0.066 nmol d7-8DHC was added. 400μL of Folch solution (2:1 Chloroform:Methanol) was added to the diluted sample and vortexed. To this mixture, 400 μL of 0.9% NaCl was added, vortexed, and centrifuged at max speed for 5 minutes. 100 μL from the lower organic layer were transferred to a LC-MS/MS compatible 96-well plate (Waters #186005837) where samples were dried under vacuum to concentrate lipids. 100 μL of 2 mg/mL 4-phenyl-1,2,4-triazoline-3,5-dione (PTAD) solution in methanol was added to each well and shaken for 30 minutes at room temperature and then immediately sealed with Easy Pierce Heat Sealing Foil (ThermoScientific AB-1720). The sealed plates were then kept in −80C until LC-MS/MS analysis.
LC-MS/MS Analysis and Sterol Quantification
The addition of PTAD allows for the MRM analysis of Lan, 8-DHC, 7-DHC, and Des as described previously24,29 by monitoring the loss of the PTAD-moiety from the parent ion. Briefly, derivatized sterol samples (10 uL injections) were analyzed on an UPLC C18 column (Acquity UPLC BEH C18, 1.7 um, 2.1 mm x 50 mm) with 100% methanol (0.1%v/v acetic acid) mobile phase at a flow rate of 500 uL/min and runtime of 1.2 min. A TSQ Quantum Ultra tandem mass spectrometer (ThermoFisher) was used for MS detections, and data were acquired with a Finnigan Xcalibur software package. MRMs of the PTAD derivatives were acquired in the positive ion mode using atmospheric pressure chemical ionization (APCI). MS parameters were optimized for the 7-DHC-PTAD adduct and were as follows: auxiliary nitrogen gas pressure at 55 psi and sheath gas pressure at 60 psi; discharge current at 22 µA and vaporizer temperature at 342°C. Collision induced dissociation (CID) was optimized at 12 eV under 1.0 mTorr of argon. The monitored transitions included: 7-DHC 560→365, d7-7-DHC 567→372, 8-DHC 558→363, d7-8-DHC 565→370, Des 592→365, Lan 634→602,13C3-Des 595→368, and 13C3-Lan 637→605. Cholesterol and d7-Cholesterol were monitored using pseudo-MRMS of 369→369 and 376→376, respectively, during the same analytical run. Sterol levels were then analytically determined based on response to their respective isotopically labeled internal standard and normalized to total protein content determined using the Pierce BCA protein assay kit (Thermo Scientific).
Statistics
Data are shown as the mean ± s.e.m. Statistical significance among treatment groups was determined using 2-way ANOVA with Holm-Sidak’s multiple comparisons post-hoc test after confirmation of a statistically significant (p<0.05) main effect or interaction. Data was determined to be normally distributed upon visual inspection of Q-Q plots for each sterol30,31.
Results
To begin investigating the role of disease progression on cholesterol metabolism in HD we compared vehicle-treated pre-manifest (12wk) and manifest (42wk) HD model and control females. We identified several age x HD interactions in striatal sterol levels (Supplementary Table 1). There was a non-significant trend in elevation in striatal 8DHC and desmosterol levels in HD compared to WT pre-manifest females (Figure 1A); however, this elevation was significant in cholesterol (Figure 1A). Interestingly, manifest HD female mice have a significant 20%, 35%, 25% and 22% reduction in the levels of lanosterol, 8DHC, desmosterol and cholesterol, respectively, in the striatum compared to manifest WT female mice (Figure 1A). The effect of age on HD sterol levels was not present in cortical or cerebellar tissues with the exception of 8DHC in the cortex which showed a 50% reduction in HD compared to WT manifest females (Figure 1B). Desmosterol in the cerebellum showed a 32% reduction with age in HD (Figure 1C).
Figure 1.
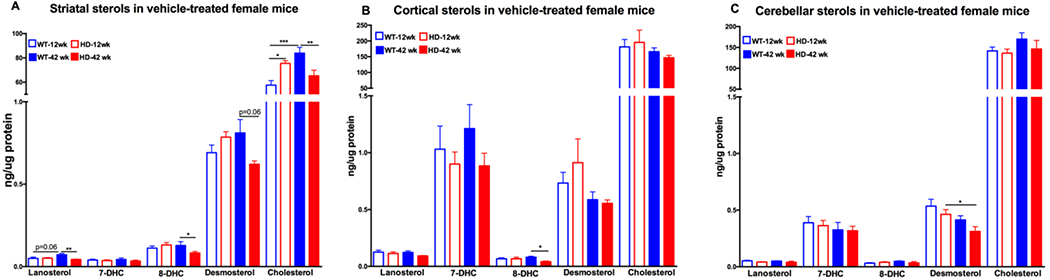
Sterols in female 12 week old and 42 week old vehicle-treated wild-type (WT) and YAC128 (HD) mice in the A) striatum, B) cortex, and C) cerebellum. 12 week old WT mice in empty blue (n=10), 12 week old HD mice in empty red (n=8), 42 week old WT mice in solid blue (n=10) and 42 week old HD mice in solid red bars (n=10). Data represents the mean ± s.e.m. An asterisk (*) indicates a p-value <0.05, ** indicates p-value <0.01, *** indicates p-value <0.001.
We next tested whether systemic subcutaneous Mn treatment would alter brain sterols in pre-manifest and manifest HD females. We found that cortical levels of lanosterol, desmosterol and cholesterol were approximately 30% reduced in both WT and HD 12-week old female animals with Mn exposure (Figure 2B), yet resulted in a significant elevation in cerebellar desmosterol in WT females (Figure 2C). In manifest females, Mn treatment resulted in a 40% reduction in striatal lanosterol levels in WT mice (Figure 3A) without any effect in the cortex or cerebellum (Figure 3B and C). As Mn treatment had minimal effects on brain sterol levels in female mice, we examined Mn-treated animals only and observed the same interactions between age and HD in the striatum, cortex and cerebellum (Supplementary Table 2; Supplemental Figure 2 A,B and C) as seen in the vehicle-treated female pre-manifest and manifest mice (Figure 1A, B and C). Specifically, in pre-manifest females, striatal 8DHC, desmosterol and cholesterol were elevated in Mn-exposed HD compared to Mn-exposed WT mice (Supplementary Figure Figure 2A) without any effect of age or HD in the cortex or cerebellum (Supplementary Figure 2B and C). In manifest Mn-treated females, there were reductions in desmosterol and cholesterol (Supplemental Figure 2A) and reductions in 7DHC in the cerebellum in HD compared to WT (Supplemental Figure 2C). No HD related changes were observed in manifest females in the cortex.
Figure 2.
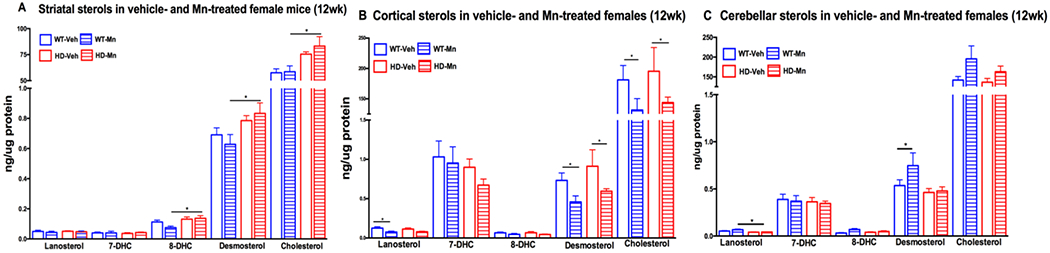
Sterols in female 12 week old vehicle-treated and Mn-treated wild-type (WT) and YAC128 (HD) mice in the A) striatum, B) cortex, and C) cerebellum. Vehicle-treated WT mice in empty blue (n=9), Mn-treated WT in striped blue (n=10), vehicle-treated HD in empty red (n=8) and Mn-treated HD in striped red bars (n=9). For clarity, vehicle data from Figure 1 are included.
Figure 3.
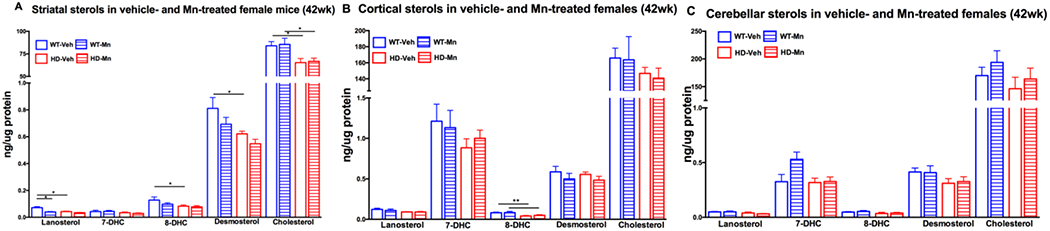
Sterols in female 42 week old vehicle-treated and Mn-treated wild-type (WT) and YAC128 (HD) mice in the A) striatum, B) cortex, and C) cerebellum. Vehicle-treated WT mice in empty blue (n=10), Mn-treated WT in striped blue (n=7), vehicle-treated HD in empty red (n=10) and Mn-treated HD in striped red bars (n=10). For clarity, vehicle data from Figure 1 are included.
In order to determine whether the HD effect on brain sterols was sex dependent, we investigated the effect of HD and Mn on pre-manifest male mice. Unlike pre-manifest female mice, there were no effects of HD or Mn in the striatum (Figure 4A) or cerebellum (Figure 4C), however there was a reduction in cortical 7DHC in HD with Mn treatment in 12-week old male mice (Figure 4B).
Figure 4.
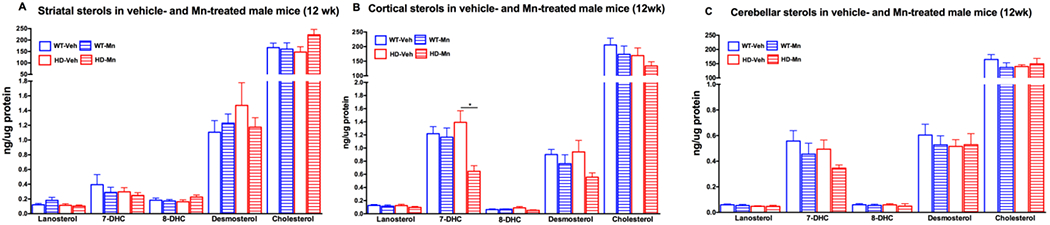
Sterols in male 12 week old vehicle- and Mn-treated wild-type (WT) and YAC128 (HD) mice in the A) striatum, B) cortex, and C) cerebellum. Vehicle-treated WT mice in empty blue (n=9), Mn-treated WT in striped blue (n=10), vehicle-treated HD in empty red (n=8) and Mn-treated HD in striped red bars (n=9). Data represents the mean ± s.e.m. An asterisk (*) indicates a p-value <0.05.
Discussion
Huntington’s disease (HD) results in the preferential loss of medium spiny neurons within the basal ganglia of the brain with noticeable changes also observed in the cortex and hypothalamus32,33. Areas such as the cerebellum and hippocampus remain relatively unaffected33. Although it is clear that the mutant Huntingtin (HTT) gene results in the development of HD, the molecular pathophysiology of the disease remains unclear – impeding the development of genetic, pharmacokinetic or environmental modifiers of disease that delay or prevent symptom onset. Post-mortem studies in HD patients show that several aspects of metabolism are altered in HD compared to non-HD controls, including lipid and sterol metabolism in particular. Kreilaus and colleagues found that the putamen of HD patients had significantly elevated levels of cholesterol precursors such as lanosterol and desmosterol as well as total cholesterol levels9. However, human data gives us little insight into when these metabolic alterations occur throughout the pathophysiology of HD, how they might change over the course of disease and potential interventions which may correct select metabolic deficiencies. For instance, we recently found that systemic Mn treatment in these mice corrects elevations in striatal urea cycle metabolites14. As such, we sought here to investigate the impact of acute manganese (Mn) exposure on alterations in brain cholesterol biosynthesis in pre-manifest and manifest stages of HD given the reported changes in cholesterol metabolism in HD models and the potential for changes in Mn levels to impact metabolic pathways.
Based upon the previous work investigating the impact of HD on cholesterol metabolism, we examined the role of disease stage and Mn treatment on cholesterol biosynthesis in HD pre-manifest and manifest female mice (manifest male mice were unavailable). Pre-manifest (12wk old) HD female mice have elevations in 8-dehydrocholesterol (8-DHC), desmosterol and cholesterol compared to WT mice (Figure 1A). Manifest (42wk old) HD females have significantly reduced striatal lanosterol, 8-DHC, desmosterol and cholesterol compared to WT animals (Figure 1A). It is noteworthy that these changes are occurring selectively in the striatum and are not present in the cortex or cerebellum (Figure 1B and C) with the exception of 8-DHC. Our findings are corroborated in a more advanced mouse model of HD, where both female and male R6/1 mice showed reductions in cholesterol precursors at early ages and increases at later time points12. Yet another model, the Q175 knock-in mouse, has a slight reduction in cholesterol precursors at a young age which is exacerbated over time34. Previously it has been shown that the 10 month YAC128 striatum has lower levels of lanosterol, desmosterol and cholesterol compared to their wild-type control which is confirmed in this report, but in that same study it was shown that lanosterol levels were reduced at 2 months which we were unable to recapitulate5. Despite the conflicting findings regarding the impact of disease progression and sex on HD cholesterol biosynthesis, it is clear that age alters cholesterol biosynthesis. Further, the additional sterols analyzed (7-DHC and 8-DHC, in particular) as well as the reported similarities in cholesterol levels in manifest animals provides a better understanding of sterol biology in rodent models of HD. Finally, it is important to note that the transcriptional regulation of cholesterol biosynthesis is markedly changed in HD models of disease. Specifically, the sterol regulatory elemenat-binding protein (SREBP) is significantly reduced in HD models providing a possible mechanism as to how sterol levels in HD models is lower than their matched control35.
The effect of Mn treatment on cholesterol metabolism in pre-manifest and manifest female mice is largely localized to non-striatal tissue. The only observed effect of Mn exposure was seen in the cortex of young females with reduced lanosterol, desmosterol and cholesterol (Figure 2B). The localization of Mn effects to the cortex is unexpected as subcutaneous Mn administration in rodents results in equitable increases in Mn levels across brain regions36. However, imaging studies in humans with various occupational Mn exposures show that the cerebral cortex is particularly vulnerable to the neurotoxic effects of Mn37 and in non-human primates, Mn injections led to protein aggregation in several cortical layers38. Mn-induced reductions in lanosterol, desmosterol and cholesterol in young mice may be an initiating molecular event which leads to the development of mutant HTT aggregation both in the cortex and striatum. Given the neurotoxicity of Mn at these doses and the potential for Mn-induced aggregation, it is clear that any use of Mn as an HD therapeutic would require sub-toxic administration.
Cholesterol is synthesized differently by neurons and astrocytes: neurons favoring the Kandutsch-Russel pathway which converts the precursors lanosterol, 7-dehydrocholesterol, and lathosterol into cholesterol while astrocytes favor the Bloch pathway that utilizes desmosterol to synthesize cholesterol39. Thus, our observation that the astrocytic cholesterol precursor, desmosterol, is increased in pre-symptomatic striatal HD may be partially explained by increases in astrocytic numbers and activation in HD compared to non-disease states. The reduction in desmosterol with age has been observed in several tissues including the brain40,41 although it is unclear why this occurs in the cortex and cerebellum, but not in the striatum of WT animals. Another consideration is that astrocytic activation is positively correlated with disease state progression, however, astrocytic dysfunction has also been shown in several in vivo and in vitro models of HD42. An increase in astrocytic dysfunction could explain the reduction in desmosterol production in our symptomatic HD females32.
We also found that lanosterol, the first mammalian sterol synthesized in cholesterol biosynthesis, was unchanged in pre-symptomatic HD mice but reduced in symptomatic HD mice. Lanosterol has been shown to reduce protein aggregation43–45 in rodent cataract models as well as reduce cytotoxicity46. Zhao et al found that treatment with exogenous lanosterol dissolved ‘amyloid-like’ crystallin aggregates46. It is possible that reduced striatal lanosterol production due to neuronal loss in HD in the later stages of disease promotes mutant Huntingtin aggregation and thereby further exacerbates to a loss of medium spiny neurons (MSNs). YAC128 mice accumulate mHTT aggregates well before 42-weeks of age47 which correlates with the reduction in striatal lanosterol levels. The potential to reduce HTT aggregation with lanosterol administration is worthy of investigation and has direct clinical implications.
Additionally, our investigation in pre-manifest male mice found no impact of HD or Mn treatment (with the exception of cortical 7DHC) in the striatum, cortex or cerebellum (Figure 4A,B,C). The molecular mechanism(s) responsible for the sex-specificity of the effect of HD and Mn on sterol synthesis is unclear, but it is well known that there are sex differences in lipid metabolism and cholesterol biosynthesis in humans and mice48. One potential mechanism by which this phenomenon may occur is through estrogen signaling. A meta-analysis by Mumford and colleagues found evidence that rising blood estradiol levels lower total cholesterol 49. Furthermore, differences in estrogen receptor (ER) signaling alters reverse cholesterol transport (RCT)48 which provides a mechanism by which sex hormones impact brain sterol levels; however, this work was done in peripheral tissues and has not examined the effect of ER signaling in the central nervous system (CNS). However, it is possible that the observed changes in female neural cholesterol levels could be driven by sex hormones. It also should be noted that this study did not monitor the estrous cyclicity of the female mice. Given what is known about rodent estrous and the age of reproductive senescence, we likely have a heterogenous population of both pre-manifest and manifest females in regards to reproductive stage50,51.
Future studies should investigate the mechanistic basis of changes in cholesterol metabolites in HD and Mn exposure. Elevations in total cholesterol in HD have been recently connected to impairments in the enzyme CYP46A1 – the primary enzyme which metabolizes cholesterol into 24-hydroxycholesterol (24-OHC) 52. 24-OHC, an oxysterol that can cross the BBB, is the primary means by which cholesterol leaves the brain5. Future work examining the alterations in cholesterol biosynthesis in HD should also address the downstream metabolites of cholesterol including oxysterols and steroid hormones in order to better understand the specific regulatory elements in cholesterol metabolism that are altered in HD. Lastly, our tissue collection method should be optimized prior to follow up studies to eliminate the potential for blood sterols to contaminate the levels of brain sterols.
Overall, our work here provides several additional insights into understanding the pathophysiology of Huntington’s disease, in particular, the role that cholesterol and its precursors in the brain may have on HD pathology. To our knowledge, this is the first observation of sex differences in cholesterol biosynthesis in the context of HD and provides evidence that future work on lipid metabolism in HD should be conducted in both sexes. Furthermore, our data provide a framework upon which to begin understanding the contributions of various cell types (neurons vs astrocytes) on the pathology of HD and how these contributions might change throughout the course of disease. Additionally, our observations that Mn reduced cortical lanosterol levels in HD mice coupled with the ability of lanosterol to dissolve aggregates might be a novel molecular mechanism to explore for future HD therapeutics.
Supplementary Material
Acknowledgements
This work was supported by NIH NIEHS R01 ES016931 (ABB). Stipend support for ACP and PAW was provided by NIH NIEHS 5T32ES007028.
Footnotes
Conflict of Interest
The authors have no conflict of interest to report.
References
- 1.Surguchov A, Hannan AJ, Fernandez-Checa JC, Arenas F, Garcia-Ruiz C. Intracellular Cholesterol Trafficking and Impact in Neurodegeneration. Front Mol Neurosci Intracell Cholest Traffick Impact Neurodegener Front Mol Neurosci. 2017;10(10). doi: 10.3389/fnmol.2017.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Björkhem I, Meaney S, Fogelman AM. Brain Cholesterol: Long Secret Life behind a Barrier. Arterioscler Thromb Vasc Biol. 2004;24(5):806–815. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- 3.Nieweg K, Schaller H, Pfrieger FW. Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J Neurochem. 2009;109(1):125–134. doi: 10.1111/j.1471-4159.2009.05917.x. [DOI] [PubMed] [Google Scholar]
- 4.Mitsche MA, McDonald JG, Hobbs HH, Cohen JC. Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell-type specific pathways. Elife. 2015;4(JUNE 2015):1–21. doi: 10.7554/eLife.07999.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valenza M, Leoni V, Tarditi A, et al. Progressive dysfunction of the cholesterol biosynthesis pathway in the R6/2 mouse model of Huntington’s disease. Neurobiol Dis. 2007;28(1):133–142. doi: 10.1016/j.nbd.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Shankaran M, Di Paolo E, Leoni V, et al. Early and brain region-specific decrease of de novo cholesterol biosynthesis in Huntington’s disease: A cross-validation study in Q175 knock-in mice. Neurobiol Dis. 2017;98:66–76. doi: 10.1016/j.nbd.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 7.Leoni V, Caccia C. Study of cholesterol metabolism in Huntington′s disease. Biochem Biophys Res Commun. 2014;446(3):697–701. doi: 10.1016/j.bbrc.2014.01.188. [DOI] [PubMed] [Google Scholar]
- 8.Leoni V, Mariotti C, Nanetti L, et al. Whole body cholesterol metabolism is impaired in Huntington’s disease. Neurosci Lett. 2011;494(3):245–249. doi: 10.1016/j.neulet.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 9.Kreilaus F, Spiro AS, McLean CA, Garner B, Jenner AM. Evidence for altered cholesterol metabolism in Huntington’s disease post mortem brain tissue. Neuropathol Appl Neurobiol. 2016;42(6):535–546. doi: 10.1111/nan.12286. [DOI] [PubMed] [Google Scholar]
- 10.Valenza M, Leoni V, Karasinska JM, et al. Cholesterol Defect Is Marked across Multiple Rodent Models of Huntington’s Disease and Is Manifest in Astrocytes. J Neurosci. 2010. doi: 10.1523/JNEUROSCI.0917-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patassini S, Begley P, Reid SJ, et al. Identification of elevated urea as a severe, ubiquitous metabolic defect in the brain of patients with Huntington’s disease. Biochem Biophys Res Commun. 2015;468(1–2):161–166. doi: 10.1016/j.bbrc.2015.10.140. [DOI] [PubMed] [Google Scholar]
- 12.Kreilaus F, Spiro AS, Hannan AJ, Garner B, Jenner AM. Brain Cholesterol Synthesis and Metabolism is Progressively Disturbed in the R6/1 Mouse Model of Huntington’s Disease: A Targeted GC-MS/MS Sterol Analysis. J Huntingtons Dis. 2015;4(4):305–318. doi: 10.3233/JHD-150170. [DOI] [PubMed] [Google Scholar]
- 13.Bryan MR, Bowman AB. Manganese and the Insulin-IGF Signaling Network in Huntington’s Disease and Other Neurodegenerative Disorders. Adv Neurobiol. 2017;18:113–142. doi: 10.1007/978-3-319-60189-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bichell TJV, Wegrzynowicz M, Tipps KG, et al. Reduced bioavailable manganese causes striatal urea cycle pathology in Huntington’s disease mouse model. Biochim Biophys Acta - Mol Basis Dis. 2017;1863(6):1596–1604. doi: 10.1016/j.bbadis.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horning KJ, Caito SW, Tipps KG, Bowman AB, Aschner M. Manganese Is Essential for Neuronal Health. Annu Rev Nutr. 2015;35(1):71–108. doi: 10.1146/annurev-nutr-071714-034419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aschner JL, Anderson A, Slaughter JC, et al. Neuroimaging identifies increased manganese deposition in infants receiving parenteral nutrition. Am J Clin Nutr. 2015;102(6):1482–1489. doi: 10.3945/ajcn.115.116285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reiss B, Simpson CD, Baker MG, Stover B, Sheppard L, Seixas NS. Hair Manganese as an Exposure Biomarker among Welders. Ann Occup Hyg. 2016;60(2):139–149. doi: 10.1093/annhyg/mev064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ou SY, Luo HL, Mailman RB, et al. Effect of manganese on neural endocrine hormones in serum of welders and smelters. J Trace Elem Med Biol. 2018;50:1–7. doi: 10.1016/j.jtemb.2018.05.018. [DOI] [PubMed] [Google Scholar]
- 19.Bu S-Y, Choi M-K. Daily Manganese Intake Status and Its Relationship with Oxidative Stress Biomarkers under Different Body Mass Index Categories in Korean Adults. Clin Nutr Res. 2012;1(1):30–36. doi: 10.7762/cnr.2012.1.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liccione JJ, Maines MD. Selective vulnerability of glutathione metabolism and cellular defense mechanisms in rat striatum to manganese. J Pharmacol Exp Ther. 1988;247(1):156–161. http://www.ncbi.nlm.nih.gov/pubmed/2902211. Accessed June 18, 2018. [PubMed] [Google Scholar]
- 21.Şentürk ÜK, Öneŕ G. The effect of manganese-induced hypercholesterolemia on learning in rats. Biol Trace Elem Res. 1996;51(3):249–257. doi: 10.1007/BF02784079. [DOI] [PubMed] [Google Scholar]
- 22.Hodgson JG, Agopyan N, Gutekunst CA, et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999. doi: 10.1016/S0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 23.Dodd CA, Ward DL, Klein BG. Basal Ganglia Accumulation and Motor Assessment Following Manganese Chloride Exposure in the C57BL/6 Mouse. Int J Toxicol. 2005;24(6):389–397. doi: 10.1080/10915810500366500. [DOI] [PubMed] [Google Scholar]
- 24.Korade Z, Kim HYH, Tallman KA, et al. The Effect of Small Molecules on Sterol Homeostasis: Measuring 7-Dehydrocholesterol in Dhcr7-Deficient Neuro2a Cells and Human Fibroblasts. J Med Chem. 2016;59(3):1102–1115. doi: 10.1021/acs.jmedchem.5b01696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dietschy JM, Turley SD. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Lutjohann D, Breuer O, Ahlborg G, et al. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci. 1996. doi: 10.1073/pnas.93.18.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu L, Korade Z, Rosado DA, Liu W, Lamberson CR, Porter NA. An oxysterol biomarker for 7-dehydrocholesterol oxidation in cell/mouse models for Smith-Lemli-Opitz syndrome. J Lipid Res. 2011. doi: 10.1194/jlr.M014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu L, Mirnics K, Bowman AB, et al. DHCEO accumulation is a critical mediator of pathophysiology in a Smith-Lemli-Opitz syndrome model. Neurobiol Dis. 2012. doi: 10.1016/j.nbd.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu W, Xu L, Lamberson C, Haas D, Korade Z, Porter NA. A highly sensitive method for analysis of 7-dehydrocholesterol for the study of smith-lemli-opitz syndrome. J Lipid Res. 2014;55(2). doi: 10.1194/jlr.D043877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilk MB, Gnanadesikan R. Probability Plotting Methods for the Analysis of Data. Biometrika. 1968. doi: 10.2307/2334448. [DOI] [PubMed] [Google Scholar]
- 31.Thode HC. Testing for Normality. 10th ed. New York, NY: Mercel Dekker Inc; 2002. https://books.google.com/books?id=gbegXB4SdosC&printsec=frontcover&source=gbs_ge_summary_r&cad=0#v=onepage&q=Q-Q&f=false. [Google Scholar]
- 32.Waldvogel HJ, Kim EH, Tippett LJ, Vonsattel JPG, Faull RLM. The neuropathology of Huntington’s disease. Curr Top Behav Neurosci. 2014;22:33–80. doi: 10.1007/7854_2014_354. [DOI] [PubMed] [Google Scholar]
- 33.Walker FO. Huntington’s disease. Lancet. 2007;369(9557):218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 34.Shankaran M, Di Paolo E, Leoni V, et al. Early and brain region-specific decrease of de novo cholesterol biosynthesis in Huntington’s disease: A cross-validation study in Q175 knock-in mice. Neurobiol Dis. 2017;98:66–76. doi: 10.1016/j.nbd.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 35.Valenza M Dysfunction of the Cholesterol Biosynthetic Pathway in Huntington’s Disease. J Neurosci. 2005. doi: 10.1523/JNEUROSCI.3355-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reaney SH, Bench G, Smith DR. Brain accumulation and toxicity of Mn(II) and Mn(III) exposures. Toxicol Sci. 2006;93(1):114–124. doi: 10.1093/toxsci/kfl028. [DOI] [PubMed] [Google Scholar]
- 37.Long Z, Jiang YM, Li XR, et al. Vulnerability of welders to manganese exposure - A neuroimaging study. Neurotoxicology. 2014;45:285–292. doi: 10.1016/j.neuro.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verina T, Schneider JS, Guilarte TR. Manganese exposure induces α-synuclein aggregation in the frontal cortex of non-human primates. Toxicol Lett. 2013;217(3):177–183. doi: 10.1016/j.toxlet.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell. 2015;6(4):254–264. doi: 10.1007/s13238-014-0131-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smiljanic K, Vanmierlo T, Mladenovic Djordjevic A, et al. Cholesterol metabolism changes under long-term dietary restrictions while the cholesterol homeostasis remains unaffected in the cortex and hippocampus of aging rats. Age (Omaha). 2014;36(3):1303–1314. doi: 10.1007/s11357-014-9654-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bourre JM, Clément M, Gérard D, Legrand R, Chaudière J. Precursors for cholesterol synthesis (7-dehydrocholesterol, 7-dehydrodesmosterol, and desmosterol): cholesterol/7-dehydrocholesterol ratio as an index of development and aging in PNS but not in CNS. J Neurochem. 1990;54(4):1196–1199. http://www.ncbi.nlm.nih.gov/pubmed/2156015. Accessed July 10, 2018. [DOI] [PubMed] [Google Scholar]
- 42.Tong X, Ao Y, Faas GC, et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci. 2014;17(5):694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Upadhyay A, Amanullah A, Mishra R, Kumar A, Mishra A. Lanosterol Suppresses the Aggregation and Cytotoxicity of Misfolded Proteins Linked with Neurodegenerative Diseases. Mol Neurobiol. 2018. doi: 10.1007/s12035-016-0377-2. [DOI] [PubMed] [Google Scholar]
- 44.Kang H, Yang Z, Zhou R. Lanosterol Disrupts Aggregation of Human γd-Crystallin by Binding to the Hydrophobic Dimerization Interface. J Am Chem Soc. 2018. doi: 10.1021/jacs.8b03065. [DOI] [PubMed] [Google Scholar]
- 45.Yang X, Chen XJ, Yang Z, et al. Synthesis, Evaluation, and Structure-Activity Relationship Study of Lanosterol Derivatives to Reverse Mutant-Crystallin-Induced Protein Aggregation. Journal of Medicinal Chemistry. 2018. [DOI] [PubMed] [Google Scholar]
- 46.Zhao L, Chen X-J, Zhu J, et al. Lanosterol reverses protein aggregation in cataracts. Nature. 2015;523(7562):607–611. doi: 10.1038/nature14650. [DOI] [PubMed] [Google Scholar]
- 47.Osmand AP, Bichell TJ, Bowman AB, Bates GP. Embryonic Mutant Huntingtin Aggregate Formation in Mouse Models of Huntington’s Disease. J Huntingtons Dis. 2016;5:343–346. doi: 10.3233/JHD-160217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palmisano BT, Zhu L, Eckel RH, Stafford JM. Sex differences in lipid and lipoprotein metabolism. Molecular Metabolism. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mumford SL, Dasharathy S, Pollack AZ, Schisterman EF. Variations in lipid levels according to menstrual cycle phase: Clinical implications. Clin Lipidol. 2011. doi: 10.2217/clp.11.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finch CE, Mobbs CV., Felicio LS, Nelson JF. Ovarian and steroidal influences on neuroendocrine aging processes in female rodents. Endocr Rev. 1984. doi: 10.1210/edrv-5-4-467. [DOI] [PubMed] [Google Scholar]
- 51.Brinton RD. Minireview: Translational animal models of human menopause: Challenges and emerging opportunities. Endocrinology. 2012. doi: 10.1210/en.2012-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boussicault L, Alves S, Lamazière A, et al. CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain. 2016;139(3):953–970. doi: 10.1093/brain/awv384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.