

SUMMARY
Endoplasmic reticulum quality control (ERQC) pathways comprised of chaperones, folding enzymes, and degradation factors ensure the fidelity of ER protein folding and trafficking to downstream secretory environments. However, multiple factors including tissue-specific secretory proteomes, environmental and genetic insults, and organismal aging challenge ERQC. Thus, a key question is: ‘How do cells adapt ERQC to match the diverse, ever-changing demands encountered during normal physiology and in disease?’. The answer lies in the unfolded protein response (UPR), a signaling mechanism activated by ER stress. In mammals, the UPR comprises three signaling pathways regulated downstream of the ER membrane proteins IRE1, ATF6, and PERK. Upon activation, these UPR pathways remodel ERQC to alleviate cellular stress and restore ER function. Here, we describe how UPR signaling pathways adapt ERQC, highlighting their importance for maintaining ER function across tissues and the potential for targeting the UPR to mitigate pathologies associated with protein misfolding diseases.
Keywords: ATF6, IRE1, XBP1s, PERK, chaperone, ER-associated degradation (ERAD), protein misfolding disease, amyloid, loss-of-function disease, protein aggregation
Graphical Abstract
eTOC Blurb
Wiseman et al describe the ways in which the unfolded protein response (UPR) regulates endoplasmic reticulum (ER) quality control pathways, highlighting the importance of UPR signaling for maintaining ER function across different tissues and the potential for targeting UPR-dependent ERQC to mitigate pathologies associated with protein misfolding diseases.
INTRODUCTION
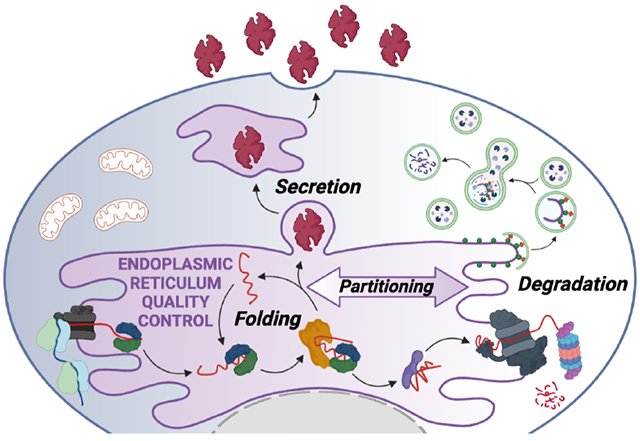
One-third of the human proteome is directed to the endoplasmic reticulum (ER) where proteins undergo folding and assembly prior to trafficking to downstream secretory environments such as lysosomes, the plasma membrane, and extracellular space (Dancourt and Barlowe, 2010). These secretory proteins are involved in nearly all aspects of cell physiology, including protein biosynthesis and turnover, as well as extracellular functions required for multicellular life. To perform these critical functions, secretory proteins must attain and maintain a folded and functional conformation. The integrity of the secretory proteome is primarily regulated by ER quality control (ERQC) pathways comprised of resident chaperones, folding enzymes, and degradation factors, as well as proteins that control transport of properly matured proteins from the ER to the Golgi (Ellgaard and Helenius, 2003). As newly synthesized proteins enter the ER lumen co-translationally, they encounter ERQC chaperones and folding enzymes that promote their maturation into native, functional conformations (Fig. 1). They are then packaged into COPII trafficking vesicles for transport to the Golgi and subsequently downstream secretory compartments. However, proteins unable to reach a properly folded conformation through interactions with ER chaperones and folding components are targeted for ER-associated degradation (ERAD) or ER-phagy for destruction by the proteasome or lysosome, respectively. Through the partitioning of nascent proteins between ER protein folding/trafficking and degradation pathways, ERQC preferentially promotes secretion of proteins in folded conformations, while actively preventing the transport of non-native proteins, thus protecting the integrity of downstream secretory environments (Fig. 1).
Figure 1. ERQC is defined by the partitioning of proteins between ER protein folding/trafficking and degradation pathway.
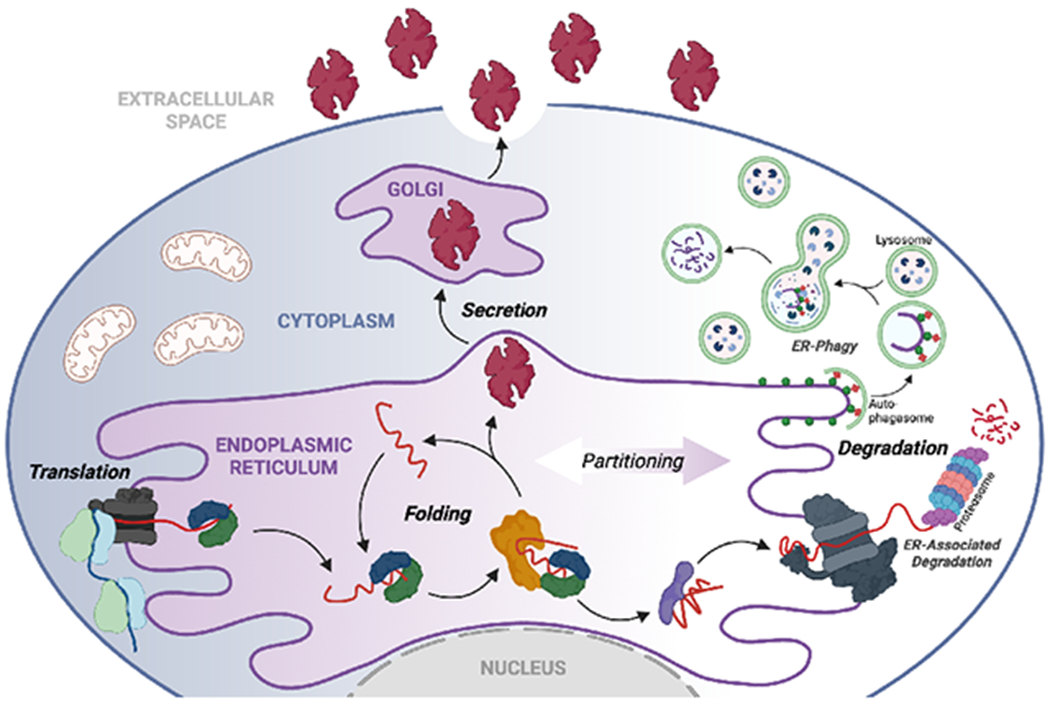
Secretory proteins are co-translationally imported into the ER through the translocon channel where they immediately engage ER chaperones and folding factors. Through these interactions, proteins are assisted in attaining their folded conformation, allowing them to be trafficked to the Golgi and subsequently to downstream secretory environments such as the extracellular space. Proteins unable to attain a folded conformation within the ER are instead recognized by degradation factors and directed towards proteasomal or lysosomal degradation through ERAD or ER-phagy, respectively. Created with BioRender.com.
Despite the efficacy of ERQC, this process is constantly challenged by environmental, genetic, behavioral, disease, or aging-related insults that increase protein misfolding in the ER and can promote downstream disruptions in the integrity and function of the secreted proteome. To protect against these insults, eukaryotes evolved the unfolded protein response (UPR), which is activated by disruptions in ER homeostasis in order to modify ERQC pathways and restore ER function. Here, we discuss how UPR signaling promotes adaptive remodeling of ERQC pathways to regulate the partitioning of secretory proteins between folding/trafficking and degradation pathways, primarily focusing on mammalian cells. Further, we will describe how dysregulation of UPR pathways can impact the secretory proteome and highlight how pharmacologic targeting of UPR-dependent ERQC regulation offers a unique opportunity to intervene in etiologically diverse diseases (vide infra).
ER Quality Control Pathways Integrate to Ensure Fidelity of Protein Maturation
The same dangers that plague protein synthesis throughout the cell also exist in the ER. However, this problem is further exacerbated in the ER by uniquely high concentrations of unfolded proteins, intracellular calcium stores used in signal transduction cascades, a redox system that catalyzes the formation of disulfide bonds, the addition of glycans to specific asparagine residues, and the need for many proteins to form multimeric complexes to reach their native state. Despite this seemingly treacherous environment, a vast number of unrelated proteins can be synthesized at high rates and mature properly in this organelle. This is made possible through a diverse complement of molecular chaperones and folding enzymes. These resident ER proteins serve to promote folding and minimize aggregation, while at the same time acting to target proteins that fail to achieve their native structure for degradation. Levels of the ER quality control machinery are tuned to the biosynthetic load of proteins entering the ER by components of the unfolded protein response (UPR), which is sufficiently versatile to adapt to wide variations in quantities and types of cargo in diverse tissues and metabolic states.
ATP-dependent chaperoning pathways of the ER
Proteins entering the ER co-translationally have the capacity to form secondary and tertiary structures almost immediately, even though the correct interactions required for reaching their native state may still be untranslated. Critical to the success of this endeavor is the abundance of chaperones the nascent polypeptide chain encounters as it emerges from the protein conducting channel or translocon. The binding of chaperones to the nascent unfolded protein protects vulnerable regions, prevents premature, non-native folding, and can dictate the fate of proteins entering the ER. The ER possesses cognates of most of the major chaperone families found in other organelles (i.e., Hsp70, Hsp90, and Hsp110), but it is also populated by ER-specific chaperones and co-factors that are critical for monitoring the processing of N-linked glycans and assisting the formation of disulfide bonds that uniquely modify nascent secretory pathway proteins in the ER.
The three most abundant molecular chaperones of the ER, GRP78/BiP, GRP94, and GRP170, consume ATP in their functional cycle, significantly increasing the energy costs of protein biosynthesis. This cycle is best understood for BiP (Fig. 2A), an Hsp70 cognate and the first ER chaperone identified in any organism (Bole et al., 1986; Haas and Wabl, 1983). Like all Hsp70 chaperones, BiP is composed of an N-terminal adenosine nucleotide binding domain, a C-terminal substate binding domain (SBD), and a linker between them, which provides allosteric coupling of the two domains. In the ATP-bound state, the lid of the SBD is open allowing a client to bind with a high on-off rate. ATP hydrolysis triggers lid closing leaving the bound client in a protected state with a low off rate. The release of ADP and rebinding of ATP are required to reopen the lid covering the unfolded client, allowing the client to escape and fold (Mayer and Gierasch, 2019; Pobre et al., 2019). BiP’s nucleotide cycle is regulated by both ER resident DnaJ-type co-chaperones (ERdjs), which increase the rate of ATP hydrolysis by BiP, and nucleotide exchange factors that promote ADP release (Fig. 2A). Early peptide binding studies indicated BiP preferred peptides of ~7-9 amino acids in length that were enriched in hydrophobic amino acids (Blond-Elguindi et al., 1993; Flynn et al., 1991), and later structural studies revealed a cleft in the SBD of Hsp70s that could accommodate short, unstructured regions of a protein (Zhu et al., 1996). Because this type of short hydrophobic sequence occurs frequently in most proteins and is usually buried upon folding, these data provided an understanding of both how BiP discriminates between unfolded and folded proteins and how it is able to interact with a vast number of sequence unrelated proteins. The association of protein disulfide isomerase (PDI) and peptidyl prolyl isomerase family members with BiP (Meunier et al., 2002) also aid in the folding of clients through this BiP-chaperoning pathway.
Figure 2. Core ERQC hubs that control the fate of nascent proteins entering the ER.
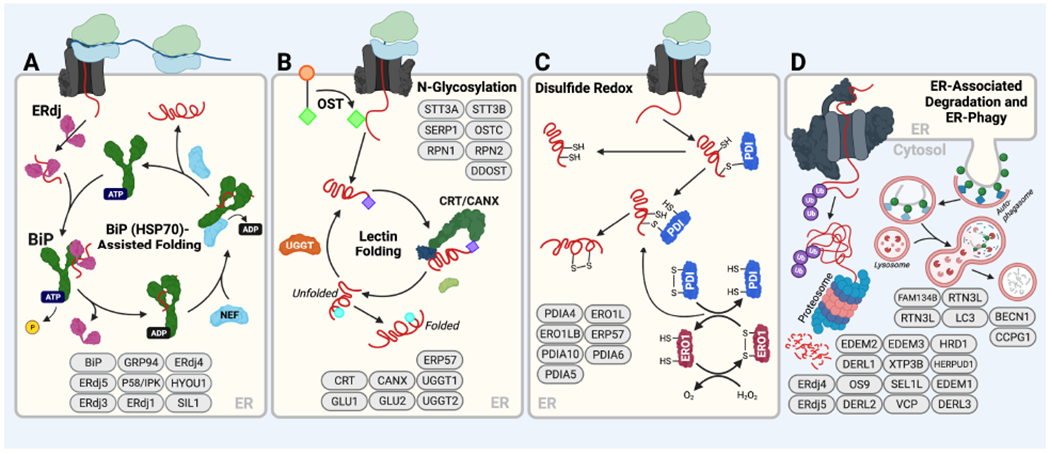
A. The BiP HSP70 chaperoning pathway. Substrates either bind directly to ATP-bound BiP or are delivered by ER-localized J-proteins (ERdjs), which stimulate BiP ATPase activity. This converts BiP to the ADP-bound conformation that has high affinity for substrates. Nucleotide exchange factors (NEFs) such as GRP170/HYOU1 then engage BiP to facilitate ADP-ATP exchange, returning BiP to the low-affinity ATP bound state and releasing the substrate for subsequent rounds of folding. Transfer of a client to ERdj family members like ERdj4 or ERdj5 can remove it from the folding cycle and transfer it for degradation. Components of the BiP chaperoning pathway are shown in ovals at the bottom of the panel. B. The Calnexin/Calreticulin lectin chaperoning pathway. Oligosaccharyl transferase (OST) appends a core glycan comprising Glc3-Man9-GlcNAc2 (green diamonds) to Asn at specific N-glycosyation site sequences (Asn-X-Ser/Thr). Two distal glucose residues are then removed from this core glycan by glucosidases, leaving a protein with a Glc-Man9-GlcNAc2 glycan (purple diamonds). Proteins possessing this singly glucosylated glycan are substrates for the lectin chaperones, calnexin (CANX) and calreticulin (CRT). Upon release from these chaperones, proteins can either fold into their native state or the final glucose of the N-glycan can be removed by glucosidases to leave a glycan comprising Man9-GlcNAc2 (blue circles) that cannot rebind CANX/CRT. This glucose-free glycan can then be either further trimmed by mannosidases that direct the protein to degradation or re-glucosylated by UGGT to allow further rounds of interactions with CANX or CRT. Select components of the Calnexin/Calreticulin lectin chaperoning pathway are shown at the bottom of the panel. C. Protein disulfide isomerase (PDI) activity in the ER. Oxidized PDIs form mixed disulfide bonds with Cys residues in substrate proteins that are then resolved by a second Cys in the substrate, creating a disulfide within the substrate protein. The resulting reduced PDI is then re-oxidized through the activity of ERO1. PDIs like ERdj5, a protein that also includes a J-domain, bind clients while in the reduced state causing the client disulfide to be transferred to ERdj5, thus serving to reduce the client for ERAD. Select components of the PDI folding pathway are shown. D. ER-associated degradation (ERAD) and ER-phagy. In ERAD, terminally misfolded proteins are directed to the ER retrotranslocon, which facilitates removal of non-native proteins from the ER to the cytosol. In the cytosol, these proteins are then ubiquitinated and subsequently degraded by the proteasome. Select components of the ERAD pathway are shown. In ER-phagy, misfolded proteins are directed to the lysosome for degradation by lysosomal hydrolases; select components are shown. Created with BioRender.com.
The two other nucleotide binding chaperones in the ER also have cognates in other organelles. GRP94, is an Hsp90 family member with a more restricted clientele than BiP, but ones that are essential for murine embryonic development (Wanderling et al., 2007). Its ability to bind and hydrolyze ATP is needed to chaperone its clients. GRP170 is an Hsp110 cognate that has chaperoning activity (Behnke and Hendershot, 2014; Park et al., 2003), but it also serves as a nucleotide exchange factor for BiP (Andréasson et al., 2010; Weitzmann et al., 2006). Unlike BiP, GRP170 prefers peptides that are enriched in aromatic residues (Behnke et al., 2016), which have a propensity to form dangerous β-sheet aggregates and therefore occur less frequently in most proteins. All three of these soluble resident ER proteins possess a C-terminal tetra-peptide sequence, KDEL, that is responsible for their retention in the ER, and as a result, their bound clients (Munro and Pelham, 1987).
BiP is an essential protein and is critically involved in most ER functions, ranging from gating the translocon through which nascent proteins enter the ER, promoting proper folding of nascent proteins, maintaining the UPR transducers in an inactive state, and targeting proteins that fail to mature properly for degradation. Its ability to participate in diverse, and even opposing, functions in the ER is made possible through its association with distinct ERdjs. Like all DnaJ family members, the ERdjs possess a highly conserved J domain through which they interact with BiP. To date, eight ERdj proteins have been identified within the ER lumen of mammalian cells, and except for ERdj7, they are expressed throughout metazoans. Four of the ERdjs also bind directly to secretory pathway clients (reviewed in (Pobre et al., 2019)), although two of these, ERdj3 (Genereux et al., 2015) and ERdj6 (Rutkowski et al., 2007), can relocalize to the extracellular space or cytosol, respectively, in response to ER stress. In the case of ERdjs that bind directly to clients, they transfer the unfolded protein to the ATP-bound form of BiP, while at the same time stimulating ATP hydrolysis by BiP, thereby inducing its tight binding to the client. The ERdj is then released due to its lower affinity for ADP-bound BiP (Fig. 2A) (Jin et al., 2008). Additional properties of these co-chaperones dictate in which ER functions BiP participates. For example, ERdj1 is a transmembrane protein that associates with the ribosome and inhibits translocation in the absence of BiP binding to the luminally oriented J domain (Blau et al., 2005). ERdj2/SEC63 is a multipass transmembrane protein and is the only ERdj protein with a clear homologue in yeast where it is essential for post-translational translocation (Brodsky et al., 1995). In mammalian cells, ERdj2/SEC63 is also a component of the translocon through its association with SEC61.
ERdj3 is homologous to the founding member of the family, E. coli DnaJ (Shen and Hendershot, 2005; Yu et al., 2000), and is the third member of the luminal ERdjs that associates with the translocon (Guo and Snapp, 2013). As the only one of the translocon-associated ERdjs that binds peptides, its localization here likely positions it to interact with clients as they enter the ER lumen, although this remains to be demonstrated. Like BiP, ERdj3 appears to prefer hydrophobic sequences, allowing it to interact with a vast number of sequence unrelated clientele.
In contrast, ERdj4 is associated with components of the retrotranslocon (Lai et al., 2012) – a membrane complex through which proteins that fail to fold are transported to the cytosol for degradation by the proteasome (Wu and Rapoport, 2018). In keeping with this, over-expression of ERdj4 accelerates the turnover of misfolded ERAD clients (Dong et al., 2008). In addition to being a DnaJ cognate, ERdj5 is also a member of the PDI family of proteins and possesses reductase activity, which is involved in breaking disulfide bonds in misfolded proteins to enable their extraction from the ER through the retrotranslocon (Ushioda et al., 2008). Both ERdj4 and ERdj5 bind the same type of aromatic-rich sequences as GRP170 (Behnke et al., 2016). ERdj6/p58IPK is both a member of the DnaJ and tetratricopeptide families. It binds clients through its TPR domains (Tao et al., 2010) and has been shown to aid in maturation of some clients (Rutkowski et al., 2007). ERdj8 governs the size of autophagic vesicles to deliver aggregates that cannot pass through the retrotranslocon to the lysosome for degradation (Yamamoto et al., 2020). ERdj7’s role in ERQC is not understood. The sub-organellar locations and additional functions of the client-binding ERdj family members can profoundly affect client outcome by either maintaining it on a pro-folding pathway or assisting in its degradation.
Calnexin/Calreticulin chaperoning pathways
The addition of an N-linked glycan complex to specific asparagine residues occurs as the polypeptide chain enters the ER and is unique to luminal portions of secretory pathway proteins (Fig. 2B) (Kornfeld and Kornfeld, 1985). Once outside the cell, these glycans will contribute to cellular homing, enhance longevity of the protein to which they are attached, and promote ligand:receptor interactions among multiple other functions. However, while still in the ER, N-linked glycans play an important role in restricting folding pathways and executing quality control of the glycoprotein maturation (Fig. 2B). Three glucose molecules are attached in tandem to the high mannose core of the glycan complex, and almost immediately after the glycan is attached, two of these glucose residues are targeted sequentially by glucosidases. Once a monoglucosylated species is produced, the nascent glycoprotein becomes a substrate for the lectin chaperones, calnexin and calreticulin (reviewed in (Molinari and Helenius, 2000)). Calnexin is a resident ER membrane protein associated with the translocon and binds glycans positioned near the membrane, whereas calreticulin is a soluble counterpart with a C-terminal KDEL sequence that interacts with glycans further from the membrane (Daniels et al., 2003). These lectin chaperones serve to retain the unfolded protein in the ER, and through their association with ERp57, a PDI family member, they assist in the oxidative folding of the client (Kozlov et al., 2010). Glucosidase removal of the remaining glucose disrupts binding of the glycoprotein from the lectin chaperones, and if the glycoprotein has completed folding it will be transported to the Golgi. If, however, portions of the protein remain unfolded, UDP-glucose:glycoprotein glucosyl transferase (UGGT1/2) binds the unfolded region of the client and reattaches a single glucose to the deglucosylated high mannose core of the glycan. The glycoprotein once again becomes a client for the lectin chaperones and is given another chance to fold (Adams et al., 2020; Maattanen et al., 2010). Recent studies have identified client specificity of the two UGGTs (Adams et al., 2020), but possible preferences in their recognition of amino acid sequences have not been determined. It is noteworthy that the inhibition of glycosylation often results in these lectin chaperone clients binding to BiP, raising the possibility that the UGGTs bind BiP-like sequences. A client will continue to interact with the lectin chaperones until they complete their folding or until the activity of mannosidases prevents reglucosylation of the glycan. At this point, the client is targeted for degradation.
Protein folding enzymes and their involvement in regulating ER quality control
In addition to the molecular chaperones and their co-factors, the ER possesses members of two types of folding enzymes that modify nascent proteins to assist in folding or degradation. The protein disulfide isomerase (PDI) family is unique to the ER, whereas two types of peptidyl-prolyl isomerases (PPIs) exist in the ER that are related to those present in the cytosol.
Most peptide bonds form in the trans configuration during protein synthesis with the exception of bonds between proline and the preceding amino acid, which can occur in either the cis or trans conformation. However, in the native structure for a given protein, the conformational state of this bond is usually defined specifically as either cis or trans. The isomerization of this bond is catalyzed by PPIs, which exist in two structurally distinct classes in the ER; cyclophilins (Cyps), and FK506-binding proteins (FKBPs) (Schiene-Fischer, 2015). Both classes of PPIs have been detected in protein complexes with BiP and the lectin chaperones. In vitro studies revealed that CypB accelerates folding of the CH1 domain of immunoglobulin (Ig) heavy chains (Feige et al., 2009) and the covalent assembly of Ig heavy and light chains (Jansen et al., 2012). While the isomerization of peptidyl-prolyl bonds is critical to protein folding, it is reasonable to assume that isomerase activity could play a role in unfolding domains to aid in the degradation of a client that fails to mature properly, although this remains to be shown.
The ER possesses an oxidizing environment that is similar to the extracellular space. To prepare secretory pathway proteins for this eventuality, disulfide bonds are formed in proteins between juxtaposed cysteines; a reaction that is catalyzed by PDI family members (Fig. 2C). More than 20 resident ER PDI family members have been identified that catalyze disulfide formation, isomerization, or reduction (Bulleid and Ellgaard, 2011). PDIs are characterized by having at least one thioredoxin (Trx)-like domain, which contains a CXXC active site. The redox status of their active site determines whether they will primarily act to catalyze disulfide bond formation and stabilize folded states or serve as a reductase to break non-native disulfide bonds or reduce folded regions so a client that ultimately fails to reach its native state can be retrotranslocated to the cytosol for degradation. Both the Hsp70 and lectin chaperones have a family member associated, PDI and ERp57 respectively, that assist in the oxidative folding of clients.
ER degradation factors and pathway
Proteins that fail to mature properly in the ER must be identified and removed from this organelle through mechanisms such as ER-associated degradation (ERAD; Fig. 2D). This involves recognition of the protein as misfolded, transferring it to a membrane channel (retrotranslocon) for transport to the cytosol, ubiquitination as the client emerges at the cytosolic face of this channel, extraction from the ER, and finally degradation by the 26S proteasome (reviewed in (Wu and Rapoport, 2018)). Misfolding of a single domain or region of a protein requires that the entire protein be degraded, even if other portions have folded properly. Misfolding can occur in luminal, transmembrane, and even cytosolic portions of secretory pathway clients, which can require some unique components of the ERAD machinery, but most basic steps are the same. This process is best understood for glycosylated proteins that utilize the lectin chaperones (Sun and Brodsky, 2019). The N-linked glycan, which is the basis of lectin chaperone recognition, upon further modification is also important for ERAD. A terminal mannose on the glycan of a misfolded protein is eventually trimmed by a mannosidase, which prevents reglucosylation and exposes an underlying 1-6-linked mannose residue. This modified glycan is recognized by another resident ER lectin, Yos9/OS9, which associates with luminal portions of the retrotranslocon (Sun and Brodsky, 2019). The interaction of ERdj5 with EDEM and OS9 (Maattanen et al., 2010; Riemer et al., 2009) provides a reductase to dismantle folded regions stabilized with disulfide bonds allowing the misfolded glycoprotein to travel through the retrotranslocon.
In the case of BiP substrates, BiP co-factors play a role in dictating the fate of clients in the ER. ERdj3 is associated with the translocon where it is positioned to bind nascent proteins entering the ER and protect vulnerable regions giving them a chance to fold (Guo and Snapp, 2013). ERdj6 binds nascent proteins through its TPR domains and seems also to be a pro-folding BiP co-factor (Rutkowski et al., 2007). On the other hand, ERdj4 and ERdj5 assist in degradation. While the details remain incompletely understood, ERdj4 enhances degradation of bound clients via its association with the retrotranslocon (Lai et al., 2012), whereas ERdj5 acts to reduce disulfides in misfolded BiP clients similar to its function in glycoprotein unfolding (Ushioda et al., 2008). GRP170 over-expression enhances the degradation of clients, and studies have implicated both its chaperone (Buck et al., 2013) and NEF activity (Inoue and Tsai, 2016). GRP170, ERdj4, and ERdj5 have been shown to bind identical peptides rich in aromatic residues that are prone to forming aggregates (Behnke et al., 2016). While these peptides are well-tolerated when rapidly buried by native folding, their prolonged exposure represents a danger and signals the need for degradation. How clients are transferred from one ERdj to another has not been well-delineated. Although at least for some substrates, BiP does not continuously cycle (Vanhove et al., 2001), whereas ERdjs are released upon a productive interaction with BiP (Jin et al., 2008).
Apart from ERAD, misfolding prone ER proteins can also be targeted for degradation through alternative mechanisms such as ER-phagy, which uses ER-specific factors to intersect with downstream components of conventional autophagic vesicle production components. In this process, fragments of the ER are directed to lysosomes for degradation through different mechanisms including micro-ER-phagy (direct envelopment of an ER fragment by the lysosome) or macro-ER-phagy (incorporation of an ER piece into autophagasomes for targeting to the lysosome) (Chino and Mizushima, 2020; Ferro-Novick et al., 2021). This allows for both reductions in total ER volume and degradation of misfolded or aggregated ER proteins that are resistant to ERAD. Specific receptors of ER-phagy, including those involved in ERQC such as FAM134B and RTN3L, have been identified over the past 5-6 years, and it is already clear that this pathway represents an important mechanism to remove terminally misfolded proteins from the ER (Chino and Mizushima, 2020; Ferro-Novick et al., 2021). Numerous misfolding-prone proteins that are resistant to ERAD, including pro-collagen and destabilized insulin variants, have been shown to be removed through ER-phagy (Cunningham et al., 2019; Forrester et al., 2019; Omari et al., 2018). While there remains much to learn about the mechanism and regulation of ER-phagy dependent ERQC, the evolution of this alternative pathway further highlights the critical importance of degrading terminally misfolded proteins within the ER to prevent their potentially toxic accumulation. For additional reviews on this topic please see (Chino and Mizushima, 2020; Ferro-Novick et al., 2021) and (Gubas and Dikic in this issue).
Partitioning of substrates between ER protein folding and degradation pathways dictates ERQC
Ensuring fidelity of the secretory proteome is as dependent on chaperone-assisted folding and assembly reactions as it is on the capacity of the ERAD machinery to rapidly detect and destroy those nascent proteins that do not properly mature (Fig. 1). This process is complicated by the fact that nascent unfolded polypeptide chains entering the ER have many of the same features as proteins that do not manage to mature properly, including exposed hydrophobic patches or aggregation-prone regions. Furthermore, the presence of components of both folding and degradation systems in the ER implies the possibility for competition between these two outcomes. This is averted in part through distinct sub-organellar localization of some components of each system and the ability to reprogram their individual levels to meet changing demands of protein biosynthesis.
Ensuring sufficient time to allow nascent proteins to mature, while at the same time ridding the ER of proteins that cannot achieve their native state, is the essence of ERQC. It is not fully understood how the balance between folding attempts and degradation for individual proteins is set, although several aspects of this process have been identified. The client itself participates in this decision, as proteins that cannot mature have dramatically different half-lives. For instance, an unassembled Ig heavy chain has a half-live of >12 hrs, whereas an lg light chain that cannot fold properly is removed from the ER within ~1 hr. In this case, the exposure of recognition sites for the pro-degradation chaperones, ERdj4/ERdj5/Grp170, appears to contribute. Reduction of an oxidized but improperly folded domain exposes these sites in the light chain, whereas the unstructured, first constant (CH1) domain of the unassembled heavy chain is devoid of them (Behnke et al., 2016). In other cases, the relative affinity of pro-folding chaperones and co-factors are likely to affect the accessibility of pro-degradation components with the client. The critical aspect of glycoprotein quality control is a competition between reglucosylation and removal of two terminal mannose residues, reactions that are catalyzed by distinct enzymes (Fig. 2B) (Adams et al., 2020; Maattanen et al., 2010). Over-expression experiments with various components of either pro-folding or pro-degradation pathways reveal this balance is affected by alterations in the concentrations of these opposing components. A signal transduction pathway referred to as the unfolded protein response (UPR) can reset the relative concentrations of components of both pathways in response to changes in the secretory capacity of the cell or alterations in the extracellular environment that adversely affect ER homeostasis.
UPR-dependent regulation of ERQC
ERQC is constantly challenged by environmental, behavioral, genetic, or aging-related insults that impair the ability of proteins to attain natively folded, functional conformations within the ER. These types of insults are commonly referred to as ER stress and have the potential to disrupt the integrity, and thus function, of proteins localized throughout the secretory pathway and the extracellular milieu. To protect the secretory proteome from ER stress, ERQC pathways are actively remodeled to ensure proper folding of secreted proteins and prevent the trafficking of non-native proteins to downstream secretory compartments where they could impair protein function or promote toxic protein aggregation. This is achieved through the unfolded protein response (UPR) – the predominant stress-responsive signaling pathway activated by a disruption in ER homeostasis (Hetz et al., 2020; Preissler and Ron, 2019). The induction of the UPR is best understood to occur in response to insults that lead to the accumulation of unfolded proteins within the ER lumen, although other disruptors of ER function like lipid stress are also potent inducers of the response (Hetz et al., 2020; Preissler and Ron, 2019). The UPR regulates diverse aspects of cellular physiology in response to ER stress (e.g., metabolism, mitochondrial function, apoptosis), but here, we will limit our focus to its role in maintaining ER homeostasis. For reviews on UPR-dependent regulation of these other aspects of cellular physiology please see (Hetz and Papa, 2018; Lebeaupin et al., 2018; Rainbolt et al., 2014; Rutkowski, 2019).
The Mammalian UPR
In mammals, the UPR comprises three signaling pathways emanating downstream of the ER transmembrane proteins IRE1, PERK, and ATF6 (Fig. 3). These three proteins can be differentially activated in response to ER stress and other types of ER insults (e.g., lipid disequilibrium) through diverse mechanisms, providing an opportunity to sensitively adapt ER function to various types of pathologic insult. We briefly describe the activation mechanism for UPR signaling pathways here. For more detailed description of the mechanisms of UPR pathway activation, please refer to (Hetz et al., 2020; Preissler and Ron, 2019).
Figure 3. Mammalian UPR signaling pathways involved in adapting ERQC following an acute ER insult.
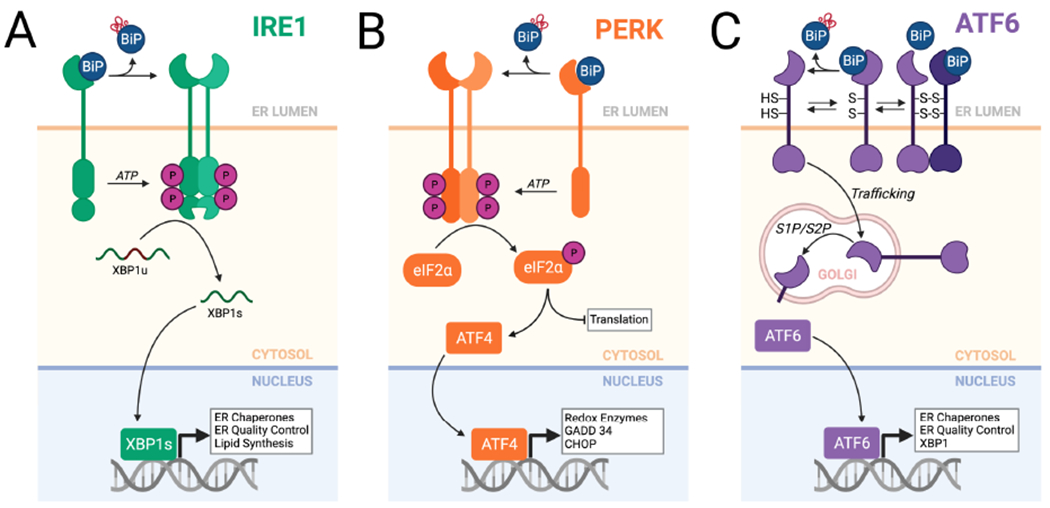
A. The IRE1/XBP1s signaling arm of the UPR. In response to ER stress, IRE1 is activated through a process involving dissociation of BiP from the IRE1 luminal domain. This leads to IRE1 oligomerization and subsequent activation of the IRE1 cytosolic kinase domain to promote autophosphorylation and allosteric activation of the IRE1 RNase. The activated IRE1 RNase adapts ERQC primarily through the non-canonical splicing of XBP1 mRNA, resulting in the production of the active transcription factor XBP1s. XBP1s activates expression of numerous genes involved in ERQC pathways including ER chaperones and degradation factors. B. The PERK arm of the UPR. In response to ER stress, BiP dissociates from the PERK luminal domain, allowing dimerization and autophosphorylation of the cytosolic kinase domain. This leads to selective PERK-dependent phosphorylation of eIF2α, which promotes both translational attenuation and selective activation of stress-responsive transcription factors such as ATF4. C. The ATF6 arm of the UPR. ATF6 is maintained in the ER as monomers and dimers, possessing intra- and inter-molecular disulfide bonds, that are bound to the ER chaperone BiP. ER stress promotes reduction of ATF6 disulfides and BiP dissociation, increasing populations of reduced ATF6 monomers that traffic to the Golgi where they are processed by site 1 (S1P) and site 2 (S2P) proteases. This releases the active ATF6 bZIP transcription factor domain, which localizes to the nucleus and induces expression of stress-responsive genes primarily involved in ERQC. Created with BioRender.com.
IRE1 is the most evolutionarily conserved arm of the UPR with homologues found in all eukaryotes (Hetz et al., 2020; Preissler and Ron, 2019). It consists of a luminal ER stress sensing domain and a cytoplasmic effector domain that possesses kinase and RNA endonuclease (RNase) activities. IRE1 is maintained in an inactive, monomeric conformation in the absence of stress through the binding of BiP to the luminal domain (Fig. 3A). Like the ERdj-mediated recruitment of BiP to unfolded clients in the ER, the association of BiP with IRE1 is supported by two luminal ERdj family members; ERdj2/SEC63 (Li et al., 2020) and ERdj4 (Amin-Wetzel et al., 2017). In response to the accumulation of unfolded proteins within the ER, IRE1 is activated through a process involving BiP dissociation and potentially direct binding of misfolded protein to the IRE1 luminal domain (Hetz et al., 2020; Preissler and Ron, 2019). This facilitates IRE1 oligomerization, autophosphorylation, and subsequent allosteric activation of the cytosolic RNase domain (Hetz et al., 2020; Preissler and Ron, 2019). Once activated, IRE1 promotes signaling through three distinct mechanisms. Most prominently, IRE1 is involved in the non-canonical splicing of XBP1 mRNA through its endonuclease activity, excising 26 bases that result in a frameshift when translating the spliced XBP1 (XBP1s) transcript (Fig. 3A) (Hetz et al., 2020; Preissler and Ron, 2019). IRE1-dependent XBP1 splicing is predicted to be mediated through a process involving translational pausing during the ribosomal synthesis of XBP1 (Kanda et al., 2016; Plumb et al., 2015; Yanagitani et al., 2011). This exposes a hydrophobic sequence that engages SRP, directing the nascent polypeptide:ribosome complex to the SEC61 translocon. Since IRE1 is associated with the translocon, this is predicted to be the site of IRE1-dependent XBP1 splicing (Kanda et al., 2016; Plumb et al., 2015; Yanagitani et al., 2011). However, the vast molar excess of SEC61 compared to IRE1 argues more studies are needed on this point, as most XBP1 transcripts would seemingly be targeted to translocons devoid of IRE1. XBP1s encodes an active bZIP transcription factor that regulates expression of many genes that are involved in diverse biological pathways including lipid regulation, gluconeogenesis, and ERQC (Lee et al., 2003; Shoulders et al., 2013; Yamamoto et al., 2007; Yamamoto et al., 2004). IREV’s RNase can also promote the promiscuous degradation of mRNAs and miRNAs through a mechanism termed regulated IRE1-dependent decay (RIDD). RIDD is implicated in reducing the quantity of proteins entering the ER during conditions of stress, as well as the regulation of diverse biological activities including lysosomal flux (Bae et al., 2019; Hetz et al., 2020; Preissler and Ron, 2019). Active, phosphorylated IRE1 can also bind to TRAF2 to promote JNK signaling in response to severe or chronic ER insults (Hetz and Papa, 2018; Hetz et al., 2020; Preissler and Ron, 2019), resulting in activation of apoptotic programs. Thus, IRE1 can signal through three different mechanisms to promote adaptation or apoptosis in response to varying degrees and extents of ER stress.
PERK is also a transmembrane protein comprising a luminal ER stress-sensing domain and a cytosolic kinase domain. Like IRE1, PERK is maintained in an inactive conformation through the binding of BiP to its luminal domain (Fig. 3B). In response to ER stress, BiP dissociates from PERK, allowing dimerization and subsequent autophosphorylation (Hetz et al., 2020; Preissler and Ron, 2019). Activated PERK primarily functions as a kinase that selectively phosphorylates the α subunit of eukaryotic initiation factor 2 (eIF2α). Phosphorylated eIF2α inhibits the GTP exchange activity of the initiation factor eIF2B, thereby blocking cap-dependent translation initiation in response to ER stress (Hetz et al., 2020; Preissler and Ron, 2019). Despite this global reduction in protein translation, eIF2α phosphorylation also leads to the selective translation of a subset of transcripts containing small upstream open reading frames (uORFs) in their 5’ untranslated regions (UTRs) that inhibit their correct initiation during normal protein synthesis (Hetz et al., 2020; Preissler and Ron, 2019). Genes translationally regulated downstream of PERK include two transcription factors, ATF4 and CHOP, which induce genes involved in redox regulation, amino acid synthesis, and mitochondrial protein homeostasis (Han et al., 2013; Harding et al., 2003). ATF4 and CHOP also control the expression of PPP1R15A/GADD34, a phosphatase regulatory subunit that de-phosphorylates eIF2α to restore protein translation following ER insults (Ma and Hendershot, 2003; Novoa et al., 2001). While PERK-dependent translational attenuation and transcriptional signaling is adaptive in response to acute ER stress, chronic PERK activation promotes apoptotic signaling through multiple mechanisms, including the upregulation of pro-apoptotic factors downstream of CHOP (Hetz and Papa, 2018). Three additional eIF2α kinases, apart from PERK, can similarly promote eIF2α phosphorylation and subsequent downstream translational and transcriptional signaling in response to other types of stress as part of the integrated stress response (Costa-Mattioli and Walter, 2020), potentially explaining the less ER-centric functional outputs of PERK signaling.
ATF6 is activated in response to ER stress through a mechanism distinct from both IRE1 and PERK (Fig. 3C). ATF6 is a transmembrane protein comprising an ER stress-sensing luminal domain and a cytosolic domain encoding a bZIP transcription factor. In the absence of ER stress, ATF6 is retained within the ER through a mechanism involving the binding of BiP to its luminal domain and intra- and intermolecular disulfides that stabilize ER-localized ATF6 (Hetz et al., 2020; Preissler and Ron, 2019). Following an acute ER insult, BiP dissociates from this luminal domain and ATF6 disulfides are reduced through the activities of PDIs, increasing the population of ATF6 monomers that traffic to the Golgi where they are proteolytically processed by site 1 protease (S1P) and site 2 protease (S2P). This releases the cytosolic, active ATF6 transcription factor domain, which localizes to the nucleus. Unlike IRE1 and PERK, which more broadly affect cellular processes, ATF6 primarily induces expression of genes involved in ERQC (Adachi et al., 2008; Okada et al., 2002; Shoulders et al., 2013; Yamamoto et al., 2007; Yamamoto et al., 2004).
XBP1s and ATF6 differentially remodel the composition and activity of ERQC pathways
The role of individual arms of the UPR in regulating ERQC pathways has been determined through a combination of genetically disrupted cells, providing information that can be affected by the acquisition of compensatory mechanisms, and the selective activation of individual transducers in the absence of ER stress (Adachi et al., 2008; Lee et al., 2003; Okada et al., 2002; Shoulders et al., 2013; Yamamoto et al., 2007; Yamamoto et al., 2004). These studies reveal that the UPR-associated transcription factors XBP1s and ATF6 are primarily responsible for regulating the composition and activity of ERQC pathways involved in the partitioning of secretory proteins between folding/trafficking and degradation. XBP1s and ATF6 form homo- or hetero-dimers that bind to specific sequences localized in the promoter regions of UPR target genes (Yamamoto et al., 2004). Stress-independent activation of XBP1s and/or the active transcription factor domain of ATF6 to physiological levels achieved during ER stress in cell culture models demonstrate that these transcription factors induce overlapping, but distinct, sets of ERQC genes (Fig. 4) (Shoulders et al., 2013). Consistent with its conservation throughout evolution, XBP1s regulates expression of nearly all ERQC genes to some extent, including those involved in ER targeting/import, folding, glycosylation, disulfide formation, degradation (via both ERAD and ER-phagy), and trafficking (Lee et al., 2003; Molinari, 2021; Ren et al., 2021; Shoulders et al., 2013; Yamamoto et al., 2007; Yamamoto et al., 2004). In contrast, ATF6 regulates a more select group of ERQC factors, including chaperones such as BiP, GRP94, and CRT, as well as folding factors such as PDIA4 (Adachi et al., 2008; Okada et al., 2002; Shoulders et al., 2013; Yamamoto et al., 2007; Yamamoto et al., 2004). These chaperones and folding factors can also be induced by XBP1s in cell culture models, albeit less potently, suggesting that ATF6 is the primary transcription factor responsible for their regulation in many cells (Shoulders et al., 2013). XBP1s and ATF6 can also heterodimerize to enhance expression of select ERQC factors, including ERAD factors such as HERP, enabling these transcription factors to sensitively adapt ERQC to diverse types of pathologic insults (Shoulders et al., 2013; Vidal et al., 2021; Yamamoto et al., 2007).
Figure 4. XBPIs and/or ATF6 activation promote distinct remodeling of ERQC pathways.
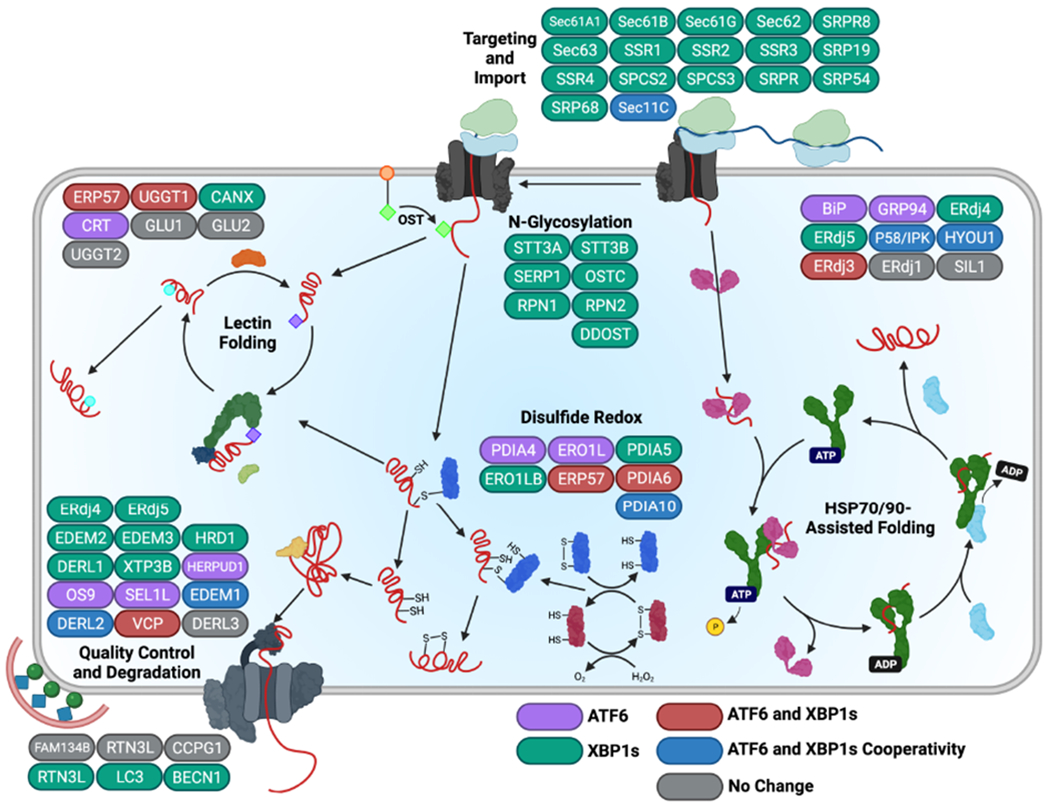
XBPIs and ATF6 induce expression of specific, but overlapping, sets of ERQC factors that differentially impact ER function. ERQC genes primarily up-regulated by XBP1s are shown in green, while those primarily induced by ATF6 are depicted in purple. Genes targeted by both XBP1s and ATF6 to similar extents are shown in red, whereas genes regulated cooperatively by ATF6 and XBP1s activation are in blue. Image was adapted from (Shoulders et al., 2013) where the individual expression of ERQC factors induced by XBP1s and/or ATF6 activation in the absence of ER stress was measured in HEK293 cells. Created with BioRender.com.
The differential remodeling of ERQC afforded by XBP1s and/or ATF6 activation can distinctly influence partitioning of proteins between ER protein folding, trafficking and degradation pathways (Plate and Wiseman, 2017). This appears to be largely dependent on the specific dependencies of ER substrates to various ERQC pathways. One example of this is the differential sensitivity of the model ERAD substrate null-Hong Kong α1-antitrypsin (NHK-A1AT) to XBP1s- or ATF6-dependent ERQC remodeling. Stress-independent activation of XBP1s, but not ATF6, enhances NHK-A1AT degradation (Shoulders et al., 2013). However, an NHK-A1AT variant lacking the three N-linked glycosylation sites (NHK-A1ATQQQ) shows preferential increases in degradation following activation of ATF6, relative to XBP1s. The differential sensitivity of glycosylated NHK-A1AT and non-glycosylated NHK-A1ATQQQ to ERQC remodeling induced by XBP1s or ATF6 activation reflects the differential dependence of their degradation on ERAD components (Adams et al., 2019; Sun and Brodsky, 2019; van der Goot et al., 2018). For example, XBP1s regulates expression of numerous ERAD factors critical for degradation of glycosylated proteins, such as the mannosidases EDEM2 and EDEM3 and the lectin XTP3B (Fig. 3) (Shoulders et al., 2013), likely contributing to the increased degradation of NHK-A1AT observed in this condition.
Since XBP1s or ATF6 activation broadly regulate the composition of multiple pathways, their activation can influence ERQC in distinct ways. For example, ATF6-dependent reductions in the secretion of destabilized immunoglobulin light chains (LCs) involves increased interactions with ATF6-regulated ER chaperones that retain this protein in the ER lumen (Plate et al., 2019). Alternatively, ATF6-dependent increases in the degradation of the non-secreted, highly-destabilized TTR variant D18G appears to involve both enhanced activity of both ERAD and ER-phagy, evident by the inability for inhibitors of either pathway independent to impair ATF6-dependent degradation of this ER client (Chen et al., 2014). This latter point reflects the fact that UPR signaling coordinates regulation of both ERAD and ER-phagy, highlighting how UPR activation integrates remodeling of pathways to enhance ERQC in response to stress. While it can be difficult to predict how XBP1s- or ATF6-dependent adaptive remodeling of ERQC will influence the folding, trafficking, and/or degradation of a given substrate, it is clear the ability to access distinct ER environments through the independent or combined activation of these two transcription factors offers a unique opportunity to tune ERQC during conditions of ER stress.
PERK-dependent regulation of the secretory proteome
While XBP1s and ATF6 are often viewed as the primary mechanisms by which cells regulate ERQC, PERK signaling also has an important role in regulating ERQC for a wide range of secretory proteins. One mechanism by which PERK regulates ERQC is through controlling the ‘load’ of newly synthesized proteins entering the ER environment downstream of eIF2α phosphorylation-dependent translation attenuation (Fig. 3B) (Harding et al., 2001; Scheuner et al., 2001; Scheuner et al., 2005). This reduction in newly synthesized proteins lessen the burden on ERQC factors, freeing them to engage non-native proteins that accumulate in the ER during stress. Apart from regulating protein folding load, PERK signaling in tissues such as pancreatic beta cells has also been suggested to contribute to the regulation of ERQC by controlling the expression and activity of select ER chaperones such as BiP and PDIA4, as well as components of ER-phagy, including the receptor CCPG1 and multiple ATGs (Adamson et al., 2016; Molinari, 2021; Sowers et al., 2018).
The importance of PERK in regulating ERQC is evident as deficiencies in PERK signaling increase the misfolding and/or aggregation of numerous secretory proteins, including proinsulin in pancreatic beta cells (Harding et al., 2012; Sowers et al., 2018), collagen in chondrocytes (Hisanaga et al., 2018), and mutant rhodopsin in the retina (Athanasiou et al., 2017). Intriguingly, deficiencies in PERK signaling also increase secretion of destabilized variants of amyloidogenic proteins such as TTR in non-native conformations that accelerate their accumulation as toxic oligomers in extracellular environments (Romine and Wiseman, 2019). This demonstrates that reduced PERK signaling can also impair ERQC-dependent regulation of downstream secretory environments. While the specific mechanisms by which PERK protects the ER and downstream environments from the accumulation of misfolded/aggregated proteins remains to be further defined, it is clear that PERK signaling is an important factor in adapting ERQC or preventing its overload, especially for secretory proteins highly expressed in certain tissues.
Tissue-specific challenges met by the UPR
The UPR plays a critical role in several developmental programs as well as in maintaining homeostasis in response to cyclic changes in the secretory capacity of various organs. We highlight a few of these here.
UPR-dependent regulation of B cell secretory capacity
The terminal differentiation of Ig-producing B cells requires a massive metamorphosis of quiescent B cells, which are nearly devoid of ER, into plasma cells that are packed with ER and capable of synthesizing a thousand antibody molecules per second (Hendershot and Sitia, 2004). In the case of IgM-secreting plasma cells, this requires the formation of 100 disulfide bonds, the addition of 50 N-linked glycans, and the assembly of 21 protein subunits to produce each IgM pentamer (Hendershot and Sitia, 2004; Muller et al., 2013). This cyto-architectural make-over begins immediately after the B cell is triggered by an antigen or mitogen and is initiated by the IRE1/XBP1s axis (Shaffer et al., 2004) in what is referred to as a preparatory UPR. This drives sequential waves of expansion of the secretory pathway membrane system, and fills the ER with the chaperones, folding enzymes, and ERAD components that will be required to produce proteins at this scale, even before the synthesis of antibody subunits begins (van Anken et al., 2003). Genetic ablation of XBP1 does not affect normal B cell development, but in its absence, stimulated B cells cannot undergo terminal differentiation and antibody synthesis is profoundly impaired (Reimold et al., 2001). Although ATF6 is activated during terminal B cell differentiation, ex vivo studies with LPS-stimulated ATF6 null B cells reveals it is dispensable for up-regulation of chaperones like BiP and GRP94 that are primarily regulated by ATF6 in other cell types, and Ig synthesis is not compromised (Aragon et al., 2012). This reveals the presence of tissue-specific functions for the various UPR transducers. PERK is dispensable plasma cell differentiation (Gass et al., 2008), and in fact is actively suppressed (Ma et al., 2010) by IRE1/XBP1s-dependent up-regulation of UFBP1 (Zhu et al., 2019). The dispensation of this braking system on translation during early UPR activation allows unfettered antibody production by the short-lived plasma cells.
UPR-dependent regulation of insulin secretion in pancreatic islets
Pancreatic β islet cells face a different type of challenge. They can produce up to a million pro-insulin molecules in a minute in response to glucose stimulation (Schuit et al., 1991), but unlike plasma cells their need to do so is intermittent resulting in vast changes in the demands on the ERQC system. In response to signals to induce insulin production the UPR is activated. The IRE1/XBP1s axis is critical for the up-regulation of multiple ERQC components including BiP and PDIs (Lee et al., 2011), which chaperone insulin. Consequently, disruption of XBP1 in β cells in a murine model and cell lines results in pro-insulin retention in the cells, β cell death, and elevated blood glucose levels (Lee et al., 2011). Unlike plasma cells, the PERK arm of the response is also critical to β cell homeostasis. PERK null mice develop a diabetic phenotype soon after birth due to β cell loss (Harding et al., 2001), and neonatal mice expressing a mutation in eIF2α that prevents it from being phosphorylated have a deficiency in pancreatic β cells (Scheuner et al., 2001), underscoring the importance of protein synthesis attenuation to moderate the biosynthetic burden in these cells. In addition, activation of the ATF4 transcription factor downstream of eIF2α phosphorylation up-regulates targets necessary for recovery from stress, including those involved in amino acid transport and protection from oxidative stress (Back et al., 2009; Harding et al., 2000). In keeping with the PERK/eIF2α axis serving critical functions in maintaining homeostasis in the pancreas, mutations in PERK are a cause of Wolcott-Rallison syndrome (Delepine et al., 2000), which is a rare autosomal recessive disorder characterized by insulin-dependent diabetes developing in neonates. The ability of XBP1s to assume a prominent role in regulating ER chaperones in plasma cells and β islet cells, one that that is attributed to ATF6 in other cells, reveals the plasticity of the UPR in distinct physiological settings.
UPR-dependent regulation of liver ERQC
Another highly secretory tissue is the liver. Unlike plasma cells and pancreatic islets, which primarily synthesize a single protein, the liver is responsible for secreting numerous highly abundant serum proteins, all of which have distinct structures. Further, the liver is a highly metabolic organ that responds to diverse nutritional and metabolic cues that adapt overall tissue function. This combination of high metabolic activity and a structurally distinct, highly-expressed secretory proteome presents a unique challenge to UPR-dependent regulation of ERQC in the liver. To confront this challenge, the hepatic UPR integrates both transcriptional ERQC remodeling with global regulation of tissue metabolism and function. For example, apart from regulating ERQC pathway composition, XBP1s also plays key roles in regulating hepatic gluconeogenesis and lipid accumulation, tightly linking ER function to overall tissue activity (Herrema et al., 2016; Zhou et al., 2011). Unsurprisingly, the liver is highly sensitive to alterations in the activity of specific UPR signaling pathways. Notably, whole body Xbp1 knockout is embryonic lethal in mice, owing to liver hypoplasia and apoptosis (Reimold et al., 2000). Further, liver-specific knockout of Ire1, Xbp1 or Atf6 lead to pathologic liver dysfunctions including inflammation, hepatic steatosis, and impaired glucose regulation in mice challenged with various insults such as ER stress or high fat diet (Lebeaupin et al., 2018; Rutkowski, 2019). Imbalanced UPR-dependent regulation of ERQC in the liver can also lead to disruptions in ER and secretory proteostasis. For example, age-related imbalances in hepatic UPR signaling in the liver have been suggested to contribute to the toxic aggregation and distal deposition of the amyloidogenic protein transthyretin (TTR) in mouse models of TTR amyloid disease (Buxbaum et al., 2012; Giadone et al., 2020; Romine and Wiseman, 2020). This suggests that dysregulation of hepatic UPR signaling could directly challenge the ability of UPR pathways to regulate ERQC, thus leading to disruptions in downstream secretory environments and the extracellular space. While there is still much to learn about UPR-dependent regulation of ERQC in the liver, it is evident from the work of many investigators that the UPR plays a central role in coordinating multiple physiologic aspects of liver function in response to diverse types of organismal challenges.
Targeting UPR-dependent ERQC regulation in protein misfolding diseases
Numerous human protein misfolding diseases are associated with failures of ERQC induced by genetic, environmental, or aging-related insults. For example, the aberrant secretion of destabilized, aggregation-prone proteins can promote the toxic extracellular aggregation of proteins implicated in etiologically diverse amyloid diseases including both transthyretin (TTR) and light chain amyloidoses, as well as Alzheimer disease (Kelly, 2020; Plate and Wiseman, 2017). In contrast, premature targeting of secretory proteins to degradation pathways can reduce their activity and contribute to the pathogenesis of loss-of-function protein misfolding disorders including Gaucher disease and idiopathic epilepsy (Kelly, 2020; Plate and Wiseman, 2017). The potential to promote adaptive remodeling of ERQC pathways through activation of UPR signaling pathways, most notably IRE1/XBP1s and ATF6, suggests that these pathways could be therapeutically accessed to alter the folding, degradation, and/or trafficking of proteins implicated in protein misfolding diseases. Here, we discuss evidence demonstrating the potential for improving ERQC of destabilized, disease-associated proteins through arm-selective activation of IRE1/XBP1s or ATF6 signaling. Further, we describe pharmacologic approaches to selectively activate these adaptive UPR signaling pathways, specifically focusing on their potential to improve ERQC in the context of disease.
Stress-independent XBP1s or ATF6 activation improves ERQC of disease-associated proteins
The potential to differentially regulate expression of ERQC factors through the independent or combined activation of ATF6 and XBP1s provides a highly sensitive mechanism to adapt ERQC to mitigate pathologic imbalances in the folding, trafficking, and degradation of destabilized proteins associated with protein misfolding diseases. For example, stress-independent ATF6 activation selectively reduces secretion, and subsequent aggregation, of destabilized, aggregation-prone variants of disease-associated proteins including transthyretin (TTR), Ig light chains (LC), and α1-antitrypsin (A1AT) (Chen et al., 2014; Cooley et al., 2014; Plate et al., 2019; Shoulders et al., 2013; Smith et al., 2011). Similarly, XBP1s over-expression enhances ERAD of destabilized amyloid precursor protein (APP) to reduce accumulation the APP cleavage product Aβ in the extracellular environment (Kaneko et al., 2010). Further, ATF6- or XBP1s-dependent remodeling of ERQC pathways can increase secretion of proteins such as mutant GABAA receptors that are prematurely targeted to ER degradation in loss-of-function protein misfolding diseases like idiopathic epilepsy (Fu et al., 2018). Despite the ability for increased XBP1s or ATF6 activity to correct pathologic imbalances in ERQC for destabilized proteins, the independent activation of these UPR-associated transcription factors does not appear to globally influence the folding, trafficking, or degradation of the endogenous secretory proteome or stable variants of disease-associated proteins (Shoulders et al., 2013). This suggests that pharmacologically accessing IRE1/XBP1s or ATF6 signaling offers a unique opportunity to improve outcomes across many different protein misfolding diseases, without globally impacting secretion of endogenous tissue proteomes.
Pharmacologically Targeting IRE1/XBP1s or ATF6 to enhance ERQC efficiency
Over the past 10 years, numerous compounds have been developed that selectively activate adaptive IRE1/XBP1s or ATF6 signaling (reviewed in (Grandjean and Wiseman, 2020; Marciniak et al., 2021)). These compounds have offered new opportunities to probe the therapeutic potential for pharmacologic targeting of UPR signaling pathways to mitigate pathologic imbalances in cellular function associated with etiologically diverse diseases including ischemia/reperfusion (Blackwood et al., 2019; Kudo et al., 2008; Oida et al., 2010; Prachasilchai et al., 2009), obesity-related diabetes (Madhavan et al., 2020), and, most relevant for this discussion, protein misfolding diseases (Plate and Wiseman, 2017). The ATF6 activating compound AA147 preferentially activates the ATF6 signaling arm of the UPR through a mechanism involving compound metabolic activation and covalent modification of a subset of ER PDIs (Paxman et al., 2018; Plate et al., 2016). Through this mechanism, AA147 selectively decreases secretion and subsequent extracellular aggregation of destabilized variants of amyloidogenic proteins including TTR and Ig LC, without significantly impacting secretion of stable, non-amyloidogenic variants of these proteins (Plate et al., 2016), mirroring results observed upon genetic ATF6 activation (Cooley et al., 2014; Plate et al., 2019; Shoulders et al., 2013). This reduction in amyloidogenic protein secretion can be attributed to AA147-dependent ERQC remodeling, although, in the case of amyloidogenic LCs, this is mediated more through AA147-dependent PDI modification rather than ATF6 activation (Rius et al., 2021). Another compound, BiX, which induces expression of BiP through an ATF6-dependent mechanism (Kudo et al., 2008), increases surface expression of mutant GABAA receptors (Fu et al., 2018), further demonstrating the potential for pharmacologic targeting ATF6 to improve outcomes in the context of protein misfolding diseases.
IRE1/XBP1s also represents an attractive target to adapt ERQC for destabilized, disease-associated proteins. Recently, compound IXA4 was identified to selectively activate the IRE1/XBP1s signaling arm of the UPR, without significantly inducing pathologic IRE1 activities such as RIDD or TRAF2-mediated JNK signaling (Grandjean et al., 2020). Importantly, this compound induced adaptive, XBP1s-dependent remodeling of ERQC pathways, highlighting its potential to influence secretion of disease-associated proteins (Grandjean et al., 2020). Consistent with this, IXA4-dependent IRE1 activation increased degradation of destabilized APP variants, thereby reducing the accumulation of toxic Aβ in conditioned media and decreasing APP-associated toxicity (Grandjean et al., 2020), mimicking results observed with XBP1s overexpression (Kaneko et al., 2010). IXA4-dependent IRE1 activation also improved glucose stimulated insulin secretion in pancreatic islets isolated from obese mice (Madhavan et al., 2020), further demonstrating the potential for this compound to improve ERQC in the context of disease.
Concluding Remarks
A vast array of cell surface and secreted proteins are critical to the complex processes that form the basis of mammalian life. To ensure the fidelity of this secretome, a dedicated ERQC system has evolved which both aids and monitors their maturation allowing only properly folded and assembled proteins to be expressed in the extracellular space, while simultaneously identifying those that fail and targeting their degradation. The UPR serves to adjust the ERQC components to ever changing metabolic demands encountered by various tissues in response to internal and external cues. The essential nature of ERQC and the contribution of the UPR’s various components in maintaining it are dramatically revealed when disruptions in this process occur leading to protein folding diseases. While we have learned a lot about the myriad ways in which the UPR regulates ERQC, many outstanding questions still exist that need to be addressed before we can fully understand the pathologic impact and therapeutic potential for UPR-dependent ERQC remodeling in health and disease. For example, the structural features that define a specific client protein’s sensitivity to distinct ERQC pathways and how this is influenced by activation of different UPR signaling pathways remains to be established. A better understanding of this relationship will enable efforts to develop pharmacologic approaches that selectively enhance ERQC outcomes for specific ER clients. In addition, more work needs to be directed toward defining the tissue-specific impact of UPR activation on ERQC capacity. Along these same lines, it is important to better understand how UPR-dependent ERQC regulation integrates with other UPR activities to globally adapt tissue-specific physiology during conditions of stress. This type of understanding is critical to avoid potential toxicities when developing pharmacologic strategies to enhance UPR-dependent ERQC remodeling for protein misfolding diseases. However, while we still have a lot to learn regarding how UPR signaling integrates to influence ERQC pathway composition and function, our current understanding of these two systems lays the groundwork for the further development of new therapeutic interventions for treating ER-based protein folding diseases.
Acknowledgements.
We would like to thank Evan Powers (Scripps Research) for critical reading of this manuscript. Funding for this work was provided by the National Institutes of Health (DK123038 and DK107604 to RLW), Alex’s Lemonade Stand Foundation ALSF 182026010 to LMH, and the American Lebanese Syrian Associated Charities of St. Jude to LMH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests. RLW is an inventor on patents for IRE1/XBP1s and ATF6 activating compounds and is a scientific advisory board member and shareholder for Protego Biopharma who has licensed UPR activating compound for translational development. No other conflicts are identified.
REFERENCES
- Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, and Mori K (2008). ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct 33, 75–89. [DOI] [PubMed] [Google Scholar]
- Adams BM, Canniff NP, Guay KP, Larsen ISB, and Hebert DN (2020). Quantitative glycoproteomics reveals cellular substrate selectivity of the ER protein quality control sensors UGGT1 and UGGT2. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams BM, Oster ME, and Hebert DN (2019). Protein Quality Control in the Endoplasmic Reticulum. Protein J 38, 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson B, Norman TM, Jost M, Cho MY, Nunez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. (2016). A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin-Wetzel N, Saunders RA, Kamphuis MJ, Rato C, Preissler S, Harding HP, and Ron D (2017). A J-Protein Co-chaperone Recruits BiP to Monomerize IRE1 and Repress the Unfolded Protein Response. Cell 171, 1625–1637 e1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andréasson C, Rampelt H, Fiaux J, Druffel-Augustin S, and Bukau B (2010). The endoplasmic reticulum Grp170 acts as a nucleotide exchange factor of Hsp70 via a mechanism similar to that of the cytosolic Hsp110. J Biol Chem 285, 12445–12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragon IV, Barrington RA, Jackowski S, Mori K, and Brewer JW (2012). The specialized unfolded protein response of B lymphocytes: ATF6alpha-independent development of antibody-secreting B cells. Mol Immunol 51, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanasiou D, Aguila M, Bellingham J, Kanuga N, Adamson P, and Cheetham ME (2017). The role of the ER stress-response protein PERK in rhodopsin retinitis pigmentosa. Hum Mol Genet 26, 4896–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, Gildersleeve RD, Pennathur S, and Kaufman RJ (2009). Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab 10, 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae D, Moore KA, Mella JM, Hayashi SY, and Hollien J (2019). Degradation of Blos1 mRNA by IRE1 repositions lysosomes and protects cells from stress. J Cell Biol 218, 1118–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke J, and Hendershot LM (2014). The large hsp70 grp170 binds to unfolded protein substrates in vivo with a regulation distinct from conventional hsp70s. J Biol. Chem 289, 2899–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke J, Mann MJ, Scruggs FL, Feige MJ, and Hendershot LM (2016). Members of the Hsp70 Family Recognize Distinct Types of Sequences to Execute ER Quality Control. Mol. Cell 63, 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood EA, Azizi K, Thuerauf DJ, Paxman RJ, Plate L, Kelly JW, Wiseman RL, and Glembotski CC (2019). Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat Commun 10, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau M, Mullapudi S, Becker T, Dudek J, Zimmermann R, Penczek PA, and Beckmann R (2005). ERj1p uses a universal ribosomal adaptor site to coordinate the 80S ribosome at the membrane. Nat. Struct. Mol. Biol 12, 1015–1016. [DOI] [PubMed] [Google Scholar]
- Blond-Elguindi S, Cwirla SE, Dower WJ, Lipshutz RJ, Sprang SR, Sambrook JF, and Gething MJ (1993). Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell 75, 717–728. [DOI] [PubMed] [Google Scholar]
- Bole DG, Hendershot LM, and Kearney JF (1986). Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J. Cell. Biol 102, 1558–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL, Goeckeler J, and Schekman R (1995). BiP and Sec63p are required for both co- and posttranslational protein translocation into the yeast endoplasmic reticulum. Proc Natl Acad Sci U S A 92, 9643–9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck TM, Plavchak L, Roy A, Donnelly BF, Kashlan OB, Kleyman TR, Subramanya AR, and Brodsky JL (2013). The Lhs1/GRP170 chaperones facilitate the endoplasmic reticulum-associated degradation of the epithelial sodium channel. J Biol. Chem 288, 18366–18380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulleid NJ, and Ellgaard L (2011). Multiple ways to make disulfides. Trends Biochem Sci 36, 485–492. [DOI] [PubMed] [Google Scholar]
- Buxbaum JN, Tagoe C, Gallo G, Walker JR, Kurian S, and Salomon DR. (2012). Why are some amyloidoses systemic? Does hepatic “chaperoning at a distance” prevent cardiac deposition in a transgenic model of human senile systemic (transthyretin) amyloidosis? FASEB J 26, 2283–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Genereux JC, Qu S, Hulleman JD, Shoulders MD, and Wiseman RL (2014). ATF6 activation reduces the secretion and extracellular aggregation of destabilized variants of an amyloidogenic protein. Chem Biol 21, 1564–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chino H, and Mizushima N (2020). ER-Phagy: Quality Control and Turnover of Endoplasmic Reticulum. Trends Cell Biol 30, 384–398. [DOI] [PubMed] [Google Scholar]
- Cooley CB, Ryno LM, Plate L, Morgan GJ, Hulleman JD, Kelly JW, and Wiseman RL (2014). Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain. Proc Natl Acad Sci U S A 111, 13046–13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CN, Williams JM, Knupp J, Arunagiri A, Arvan P, and Tsai B (2019). Cells Deploy a Two-Pronged Strategy to Rectify Misfolded Proinsulin Aggregates. Mol Cell 75, 442–456 e444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancourt J, and Barlowe C (2010). Protein sorting receptors in the early secretory pathway. Annu Rev Biochem 79, 777–802. [DOI] [PubMed] [Google Scholar]
- Daniels R, Kurowski B, Johnson AE, and Hebert DN (2003). N-linked glycans direct the cotranslational folding pathway of influenza hemagglutinin. Mol. Cell 2003. Jan. ; 11 (1):79. −90 11, 79–90. [DOI] [PubMed] [Google Scholar]
- Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, and Julier C (2000). EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet 25, 406–409. [DOI] [PubMed] [Google Scholar]
- Dong M, Bridges JP, Apsley K, Xu Y, and Weaver TE (2008). ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol Biol Cell 19, 2620–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, and Helenius A (2003). Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol 4, 181–191. [DOI] [PubMed] [Google Scholar]
- Feige MJ, Groscurth S, Marcinowski M, Shimizu Y, Kessler H, Hendershot LM, and Buchner J (2009). An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol. Cell 34, 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro-Novick S, Reggiori F, and Brodsky JL (2021). ER-Phagy, ER Homeostasis, and ER Quality Control: Implications for Disease. Trends Biochem Sci 46, 630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn GC, Pohl J, Flocco MT, and Rothman JE (1991). Peptide-binding specificity of the molecular chaperone BiP. Nature 353, 726–730. [DOI] [PubMed] [Google Scholar]
- Forrester A, De Leonibus C, Grumati P, Fasana E, Piemontese M, Staiano L, Fregno I, Raimondi A, Marazza A, Bruno G, et al. (2019). A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YL, Han DY, Wang YJ, Di XJ, Yu HB, and Mu TW (2018). Remodeling the endoplasmic reticulum proteostasis network restores proteostasis of pathogenic GABAA receptors. PLoS One 13, e0207948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass JN, Jiang HY, Wek RC, and Brewer JW (2008). The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Mol. Immunol 45, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genereux JC, Qu S, Zhou M, Ryno LM, Wang S, Shoulders MD, Kaufman RJ, Lasmezas CI, Kelly JW, and Wiseman RL (2015). Unfolded protein response-induced ERdj3 secretion links ER stress to extracellular proteostasis. EMBO J 34, 4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giadone RM, Liberti DC, Matte TM, Rosarda JD, Torres-Arancivia C, Ghosh S, Diedrich JK, Pankow S, Skvir N, Jean JC, et al. (2020). Expression of Amyloidogenic Transthyretin Drives Hepatic Proteostasis Remodeling in an Induced Pluripotent Stem Cell Model of Systemic Amyloid Disease. Stem Cell Reports 15, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean JMD, Madhavan A, Cech L, Seguinot BO, Paxman RJ, Smith E, Scampavia L, Powers ET, Cooley CB, Plate L, et al. (2020). Pharmacologic IRE1/XBP1s activation confers targeted ER proteostasis reprogramming. Nat Chem Biol 16, 1052–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean JMD, and Wiseman RL (2020). Small molecule strategies to harness the unfolded protein response: where do we go from here? J Biol Chem 295, 15692–15711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, and Snapp EL (2013). ERdj3 regulates BiP occupancy in living cells. J Cell Sci 126, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas IG, and Wabl M (1983). Immunoglobulin heavy chain binding protein. Nature 306, 387–389. [DOI] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, and Ron D (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, and Ron D (2001). Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol Cell 7, 1153–1163. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zyryanova AF, and Ron D (2012). Uncoupling proteostasis and development in vitro with a small molecule inhibitor of the pancreatic endoplasmic reticulum kinase, PERK. J Biol Chem 287, 44338–44344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot L, and Sitia R (2004). Antibody Synthesis and Assembly. In Molecular Biology of B cells, Alt FW, Honjo T, and Neuberger MS, eds. (Elsevier Science Press; ), pp. 261–273. [Google Scholar]
- Herrema H, Zhou Y, Zhang D, Lee J, Salazar Hernandez MA, Shulman GI, and Ozcan U (2016). XBP1s Is an Anti-lipogenic Protein. J Biol Chem 291, 17394–17404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, and Papa FR (2018). The Unfolded Protein Response and Cell Fate Control. Mol Cell 69, 169–181. [DOI] [PubMed] [Google Scholar]
- Hetz C, Zhang K, and Kaufman RJ. (2020). Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol 21, 421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisanaga S, Miyake M, Taniuchi S, Oyadomari M, Morimoto M, Sato R, Hirose J, Mizuta H, and Oyadomari S (2018). PERK-mediated translational control is required for collagen secretion in chondrocytes. Sci Rep 8, 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, and Tsai B (2016). The Grp170 nucleotide exchange factor executes a key role during ERAD of cellular misfolded clients. Mol. Biol. Cell 27, 1650–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen G, Maattanen P, Denisov AY, Scarffe L, Schade B, Balghi H, Dejgaard K, Chen LY, Muller WJ, Gehring K, et al. (2012). An interaction map of endoplasmic reticulum chaperones and foldases. Mol Cell Proteomics 11, 710–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Awad W, Petrova K, and Hendershot LM (2008). Regulated release of ERdj3 from unfolded proteins by BiP. EMBO J 27, 2873–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda S, Yanagitani K, Yokota Y, Esaki Y, and Kohno K (2016). Autonomous translational pausing is required for XBP1u mRNA recruitment to the ER via the SRP pathway. Proc Natl Acad Sci U S A 113, E5886–E5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Koike H, Saito R, Kitamura Y, Okuma Y, and Nomura Y (2010). Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J Neurosci 30, 3924–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JW (2020). Pharmacologic Approaches for Adapting Proteostasis in the Secretory Pathway to Ameliorate Protein Conformational Diseases. Cold Spring Harb Perspect Biol 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld R, and Kornfeld S (1985). Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem 54, 631–664. [DOI] [PubMed] [Google Scholar]
- Kozlov G, Maattanen P, Thomas DY, and Gehring K (2010). A structural overview of the PDI family of proteins. FEBS J 277, 3924–3936. [DOI] [PubMed] [Google Scholar]
- Kudo T, Kanemoto S, Hara H, Morimoto N, Morihara T, Kimura R, Tabira T, Imaizumi K, and Takeda M (2008). A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ 15, 364–375. [DOI] [PubMed] [Google Scholar]
- Lai CW, Otero JH, Hendershot LM, and Snapp E (2012). ERdj4 protein is a soluble endoplasmic reticulum (ER) DnaJ family protein that interacts with ER-associated degradation machinery. J Biol. Chem 287, 7969–7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeaupin C, Vallee D, Hazari Y, Hetz C, Chevet E, and Bailly-Maitre B (2018). Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol 69, 927–947. [DOI] [PubMed] [Google Scholar]
- Lee AH, Heidtman K, Hotamisligil GS, and Glimcher LH (2011). Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc Natl Acad Sci U S A 108, 8885–8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, and Glimcher LH (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23, 7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Sun S, Appathurai S, Sundaram A, Plumb R, and Mariappan M (2020). A Molecular Mechanism for Turning Off IRE1alpha Signaling during Endoplasmic Reticulum Stress. Cell Rep 33, 108563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, and Hendershot LM (2003). Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem 278, 34864–34873. [DOI] [PubMed] [Google Scholar]
- Ma Y, Shimizu Y, Mann MJ, Jin Y, and Hendershot LM (2010). Plasma cell differentiation initiates a limited ER stress response by specifically suppressing the PERK-dependent branch of the unfolded protein response. Cell Stress. Chaperones 15, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maattanen P, Gehring K, Bergeron JJ, and Thomas DY (2010). Protein quality control in the ER: the recognition of misfolded proteins. Semin. Cell Dev. Biol 21, 500–511. [DOI] [PubMed] [Google Scholar]
- Madhavan A, Kok BP, Grandjean JMD, Albert V, Sukiasyan A, Rius B, Powers ET, Galmozzi A, Saez E, and Wiseman RL (2020). Pharmacologic IRE1/XBP1s Activation Promotes Systemic Adaptive Remodeling in Obesity. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Chambers JE, and Ron D (2021). Pharmacological targeting of endoplasmic reticulum stress in disease. Nat Rev Drug Discov. [DOI] [PubMed] [Google Scholar]
- Mayer MP, and Gierasch LM (2019). Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J Biol Chem 294, 2085–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier L, Usherwood YK, Chung KT, and Hendershot LM (2002). A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol Biol Cell 13, 4456–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari M (2021). ER-phagy responses in yeast, plants, and mammalian cells and their crosstalk with UPR and ERAD. Dev Cell 56, 949–966. [DOI] [PubMed] [Google Scholar]
- Molinari M, and Helenius A (2000). Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science 288, 331–333. [DOI] [PubMed] [Google Scholar]
- Muller R, Grawert MA, Kern T, Madl T, Peschek J, Sattler M, Groll M, and Buchner J (2013). High-resolution structures of the IgM Fc domains reveal principles of its hexamer formation. Proc Natl Acad Sci U S A 110, 10183–10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, and Pelham HR (1987). A C-terminal signal prevents secretion of luminal ER proteins. Cell 48, 899–907. [DOI] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, and Ron D (2001). Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 153, 1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oida Y, Hamanaka J, Hyakkoku K, Shimazawa M, Kudo T, Imaizumi K, Yasuda T, and Hara H (2010). Post-treatment of a BiP inducer prevents cell death after middle cerebral artery occlusion in mice. Neurosci Lett 484, 43–46. [DOI] [PubMed] [Google Scholar]
- Okada T, Yoshida H, Akazawa R, Negishi M, and Mori K (2002). Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J 366, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omari S, Makareeva E, Roberts-Pilgrim A, Mirigian L, Jarnik M, Ott C, Lippincott-Schwartz J, and Leikin S (2018). Noncanonical autophagy at ER exit sites regulates procollagen turnover. Proc Natl Acad Sci U S A 115, E10099–E10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Easton DP, Chen X, MacDonald IJ, Wang XY, and Subjeck JR (2003). The chaperoning properties of mouse grp170, a member of the third family of hsp70 related proteins. Biochemistry 42, 14893–14902. [DOI] [PubMed] [Google Scholar]
- Paxman R, Plate L, Blackwood EA, Glembotski C, Powers ET, Wiseman RL, and Kelly JW (2018). Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate L, Cooley CB, Chen JJ, Paxman RJ, Gallagher CM, Madoux F, Genereux JC, Dobbs W, Garza D, Spicer TP, et al. (2016). Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate L, Rius B, Nguyen B, Genereux JC, Kelly JW, and Wiseman RL (2019). Quantitative Interactome Proteomics Reveals a Molecular Basis for ATF6-Dependent Regulation of a Destabilized Amyloidogenic Protein. Cell Chem Biol 26, 913–925 e914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate L, and Wiseman RL (2017). Regulating Secretory Proteostasis through the Unfolded Protein Response: From Function to Therapy. Trends Cell Biol 27, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumb R, Zhang ZR, Appathurai S, and Mariappan M (2015). A functional link between the co-translational protein translocation pathway and the UPR. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pobre KFR, Poet GJ, and Hendershot LM (2019). The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem 294, 2098–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prachasilchai W, Sonoda H, Yokota-Ikeda N, Ito K, Kudo T, Imaizumi K, and Ikeda M (2009). The protective effect of a newly developed molecular chaperone-inducer against mouse ischemic acute kidney injury. J Pharmacol Sci 109, 311–314. [DOI] [PubMed] [Google Scholar]
- Preissler S, and Ron D (2019). Early Events in the Endoplasmic Reticulum Unfolded Protein Response. Cold Spring Harb Perspect Biol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainbolt TK, Saunders JM, and Wiseman RL (2014). Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol Metab 25, 528–537. [DOI] [PubMed] [Google Scholar]
- Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, Horton HF, Scott A, Orkin SH, Byrne MC, et al. (2000). An essential role in liver development for transcription factor XBP-1. Genes Dev 14, 152–157. [PMC free article] [PubMed] [Google Scholar]
- Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, and Glimcher LH (2001). Plasma cell differentiation requires the transcription factor XBP-1. Nature 412, 300–307. [DOI] [PubMed] [Google Scholar]
- Ren H, Zhai W, Lu X, and Wang G (2021). The Cross-Links of Endoplasmic Reticulum Stress, Autophagy, and Neurodegeneration in Parkinson’s Disease. Front Aging Neurosci 13, 691881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemer J, Appenzeller-Herzog C, Johansson L, Bodenmiller B, Hartmann-Petersen R, and Ellgaard L (2009). A luminal flavoprotein in endoplasmic reticulum-associated degradation. Proc Natl Acad Sci U S A 106, 14831–14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rius B, Mesgarzadeh JS, Romine IC, Paxman RJ, Kelly JW, and Wiseman RL (2021). Pharmacologic targeting of plasma cell endoplasmic reticulum proteostasis to reduce amyloidogenic light chain secretion. Blood Adv 5, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romine IC, and Wiseman RL (2019). PERK Signaling Regulates Extracellular Proteostasis of an Amyloidogenic Protein During Endoplasmic Reticulum Stress. Sci Rep 9, 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romine IC, and Wiseman RL (2020). Starting at the beginning: endoplasmic reticulum proteostasis and systemic amyloid disease. Biochem J 477, 1721–1732. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT (2019). Liver function and dysfunction - a unique window into the physiological reach of ER stress and the unfolded protein response. FEBS J 286, 356–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Kang SW, Goodman AG, Garrison JL, Taunton J, Katze MG, Kaufman RJ, and Hegde RS (2007). The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol. Biol. Cell 18, 3681–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, and Kaufman RJ (2001). Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7, 1165–1176. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Vander Mierde D, Song B, Flamez D, Creemers JW, Tsukamoto K, Ribick M, Schuit FC, and Kaufman RJ (2005). Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med 11, 757–764. [DOI] [PubMed] [Google Scholar]
- Schiene-Fischer C (2015). Multidomain Peptidyl Prolyl cis/trans Isomerases. Biochim Biophys Acta 1850, 2005–2016. [DOI] [PubMed] [Google Scholar]
- Schuit FC, Kiekens R, and Pipeleers DG (1991). Measuring the balance between insulin synthesis and insulin release. Biochem Biophys Res Commun 178, 1182–1187. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, et al. (2004). XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21, 81–93. [DOI] [PubMed] [Google Scholar]
- Shen Y, and Hendershot LM (2005). ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP’s interactions with unfolded substrates. Mol. Biol. Cell 16, 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR 3rd, Su AI, Kelly JW, and Wiseman RL (2013). Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 3, 1279–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Granell S, Salcedo-Sicilia L, Baldini G, Egea G, Teckman JH, and Baldini G (2011). Activating transcription factor 6 limits intracellular accumulation of mutant alpha(1)-antitrypsin Z and mitochondrial damage in hepatoma cells. J Biol Chem 286, 41563–41577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowers CR, Wang R, Bourne RA, McGrath BC, Hu J, Bevilacqua SC, Paton JC, Paton AW, Collardeau-Frachon S, Nicolino M, et al. (2018). The protein kinase PERK/EIF2AK3 regulates proinsulin processing not via protein synthesis but by controlling endoplasmic reticulum chaperones. J Biol Chem 293, 5134–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, and Brodsky JL (2019). Protein quality control in the secretory pathway. J Cell Biol 218, 3171–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Petrova K, Ron D, and Sha B (2010). Crystal structure of P58(IPK) TPR fragment reveals the mechanism for its molecular chaperone activity in UPR. J Mol Biol 397, 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, and Nagata K (2008). ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321, 569–572. [DOI] [PubMed] [Google Scholar]
- van Anken E, Romijn EP, Maggioni C, Mezghrani A, Sitia R, Braakman I, and Heck AJ (2003). Sequential waves of functionally related proteins are expressed when B cells prepare for antibody secretion. Immunity 18, 243–253. [DOI] [PubMed] [Google Scholar]
- van der Goot AT, Pearce MMP, Leto DE, Shaler TA, and Kopito RR (2018). Redundant and Antagonistic Roles of XTP3B and OS9 in Decoding Glycan and Non-glycan Degrons in ER-Associated Degradation. Mol Cell 70, 516–530 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhove M, Usherwood Y-K, and Hendershot LM (2001). Unassembled Ig heavy chains do not cycle from BiP in vivo, but require light chains to trigger their release. Immunity 15, 105–114. [DOI] [PubMed] [Google Scholar]
- Vidal RL, Sepulveda D, Troncoso-Escudero P, Garcia-Huerta P, Gonzalez C, Plate L, Jerez C, Canovas J, Rivera CA, Castillo V, et al. (2021). Enforced dimerization between XBP1s and ATF6f enhances the protective effects of the UPR in models of neurodegeneration. Mol Ther 29, 1862–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanderling S, Simen BB, Ostrovsky O, Ahmed NT, Vogen SM, Gidalevitz T, and Argon Y (2007). GRP94 is essential for mesoderm induction and muscle development because it regulates insulin-like growth factor secretion. Mol Biol Cell 18, 3764–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzmann A, Volkmer J, and Zimmermann R (2006). The nucleotide exchange factor activity of Grp170 may explain the non-lethal phenotype of loss of Sil1 function in man and mouse. FEBS Lett 580, 5237–5240. [DOI] [PubMed] [Google Scholar]
- Wu X, and Rapoport TA (2018). Mechanistic insights into ER-associated protein degradation. Curr Opin Cell Biol 53, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, and Mori K (2007). Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell 13, 365–376. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, and Mori K (2004). Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem 136, 343–350. [DOI] [PubMed] [Google Scholar]
- Yamamoto YH, Kasai A, Omori H, Takino T, Sugihara M, Umemoto T, Hamasaki M, Hatta T, Natsume T, Morimoto RI, et al. (2020). ERdj8 governs the size of autophagosomes during the formation process. J Cell Biol 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagitani K, Kimata Y, Kadokura H, and Kohno K (2011). Translational pausing ensures membrane targeting and cytoplasmic splicing of XBP1u mRNA. Science 331, 586–589. [DOI] [PubMed] [Google Scholar]
- Yu M, Haslam RH, and Haslam DB (2000). HEDJ, an Hsp40 co-chaperone localized to the endoplasmic reticulum of human cells. J. Biol. Chem 275, 24984–24992. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Lee J, Reno CM, Sun C, Park SW, Chung J, Lee J, Fisher SJ, White MF, Biddinger SB, et al. (2011). Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat Med 17, 356–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Bhatt B, Sivaprakasam S, Cai Y, Liu S, Kodeboyina SK, Patel N, Savage NM, Sharma A, Kaufman RJ, et al. (2019). Ufbp1 promotes plasma cell development and ER expansion by modulating distinct branches of UPR. Nat Commun 10, 1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, and Hendrickson WA (1996). Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272, 1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]