

ABSTRACT
Current immunotherapies for lung cancer are only effective in a subset of patients. Identifying tumor-derived factors that facilitate immunosuppression offers the opportunity to develop novel strategies to supplement and improve current therapeutics. We sought to determine whether expression of driver oncogenes in lung cancer cells affects cytokine secretion, alters the local immune environment, and influences lung tumor progression. We demonstrate that oncogenic EGFR and KRAS mutations, which are early events in lung tumourigenesis, can drive cytokine and chemokine production by cancer cells. One of the most prominent changes was in CCL5, which was rapidly induced by KRASG12V or EGFRL858R expression, through MAPK activation. Immunocompetent mice implanted with syngeneic KRAS-mutant lung cancer cells deficient in CCL5 have decreased regulatory T cells (Tregs), evidence of T cell exhaustion, and reduced lung tumor burden, indicating tumor-cell CCL5 production contributes to an immune suppressive environment in the lungs. Furthermore, high CCL5 expression correlates with poor prognosis, immunosuppressive regulatory T cells, and alteration to CD8 effector function in lung adenocarcinoma patients. Our data support targeting CCL5 or CCL5 receptors on immune suppressive cells to prevent formation of an immune suppressive tumor microenvironment that promotes lung cancer progression and immunotherapy insensitivity.
KEYWORDS: Lung cancer, CCL5, tregs, oncogenes, EGFR, kras
Introduction
Lung cancer remains the leading cause of cancer-associated mortality despite advancements in targeted therapies. Immune checkpoint inhibitors (ICIs) targeting PD-1/PD-L1 have recently been approved for non-small cell lung cancer (NSCLC) treatment.1,2 However, only 23–28% of PD-L1hi NSCLC patients demonstrate durable responses, with tumor-intrinsic and microenvironmental factors that limit overall response remaining poorly understood.2 As the main mediators of anti-tumor immunity, the presence of cytotoxic CD8+ T cell tumor-infiltrating lymphocytes (TIL), are associated with favorable responses to ICI,3 while ICI responses may be adversely affected by the presence of immune suppressive cells in the tumor.3 Cytokines are key mediators of immune effector and immune suppressor cell recruitment and activation, and cytokines can be produced by a variety of different cell types. Solid tumors can develop an immune suppressive microenvironment through recruitment and polarization of suppressive tumor associated macrophages (M2-TAMs), tumor associated neutrophils, myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs).4–6 However, the tumor cell-intrinsic signaling mechanisms that stimulate the development of immune suppressive microenvironments that facilitate tumor initiation and progression are not fully understood. Oncogenic signaling is typically thought to drive tumor progression by modifying tumor cell phenotype, such as increasing proliferation, motility, and invasion.7 However, there is evidence that activating mutations in EGFR or KRAS, which are frequent in lung adenocarcinoma (LUAD), may also enable cancer cells to alter PD-L1 expression and cytokine production in the tumor microenvironment to promote tumourigenesis.8–10 The interplay between cancer cell signaling and immune cell infiltrate is an important, yet poorly understood area of cancer biology with potential relevance for patient prognosis.11 Understanding oncogene-driven factors involved in recruitment of immune suppressive cells to the tumor microenvironment may identify novel and combination therapies to benefit patients who do not respond to current ICIs.
We investigated whether oncogenic signaling induces chemokine production by tumor cells that may recruit immune suppressive cells and create a pro-tumourigenic lung environment. Constitutive signaling by oncogenic EGFR or KRAS induced production of the cytokine CCL5. Reducing CCL5 production by lung tumor cells led to a decrease in tumor burden in immune competent mice that was associated with decreased Tregs and M2-like macrophages, and increased expression of checkpoint receptors on CD8+ T cells within the lungs. Furthermore, we found that Tregs are significantly enriched in human LUAD tumors and that high CCL5 expression correlated with Treg abundance and poor patient survival. Taken together, our findings indicate an additional key tumor extrinsic role for oncogenic signaling in immune cell recruitment and promotion of tumor development. This suggests that targeted inhibition of CCL5 activity may be a viable therapeutic strategy to block the pulmonary recruitment of immune suppressive cells in lung cancer patients.
Results
Oncogene expression drives CCL5 production
To assess whether activation of oncogenic signaling induces cytokine production during malignant transformation, we generated an in vitro model of oncogene-driven transformation. We used non-transformed NIH/3T3 mouse embryonic fibroblasts infected with lentiviral constructs expressing doxycycline (DOX)-inducible wild-type EGFR (TetO-EGFRWT), or oncogenic EGFR or KRAS mutant alleles found in lung cancer (TetO-EGFRL858R or TetO-KRASG12V). Cells expressing mutant oncogenes exhibited a transformed phenotype over time as previously described.12,13 Transgene expression following DOX administration was validated (Figure 1(a)), and expression of constitutively active mutant EGFRL858R or KRASG12V in these cells corresponded with increased phosphorylation of both AKT and ERK, which was not seen in cells expressing EGFRWT (in the absence of EGFR ligand). To determine whether cytokine or chemokine production increases as a result of oncogene induction and subsequent transformation, we quantified secretion of cytokines and chemokines using a multiplex assay (LUMINEX). Production of three chemokines (CCL5, CCL3 and CCL4) and one cytokine (IL-5) increased after oncogene induction in both EGFRL858R and KRASG12V expressing cells in comparison to EGFRWT expressing cells (Figure 1(b-c), Supplemental Figure S1(a-d)). CCL5 demonstrated the most prominent temporal increase in the context of oncogene activation compared to EGFRWT (Figure 1(c)) and was subsequently validated by ELISA (Figure 1(d)). The addition of exogenous EGF to NIH/3T3 cells over-expressing EGFRWT activated downstream signaling as indicated by increased AKT and ERK phosphorylation (Figure 1(e)) and subsequent cellular transformation.12 EGFRWT induction and stimulation with EGF also induced secretion of CCL5 (Figure 1(f)), demonstrating proto-oncogene activation was sufficient to substantially increase CCL5 production.
Figure 1.
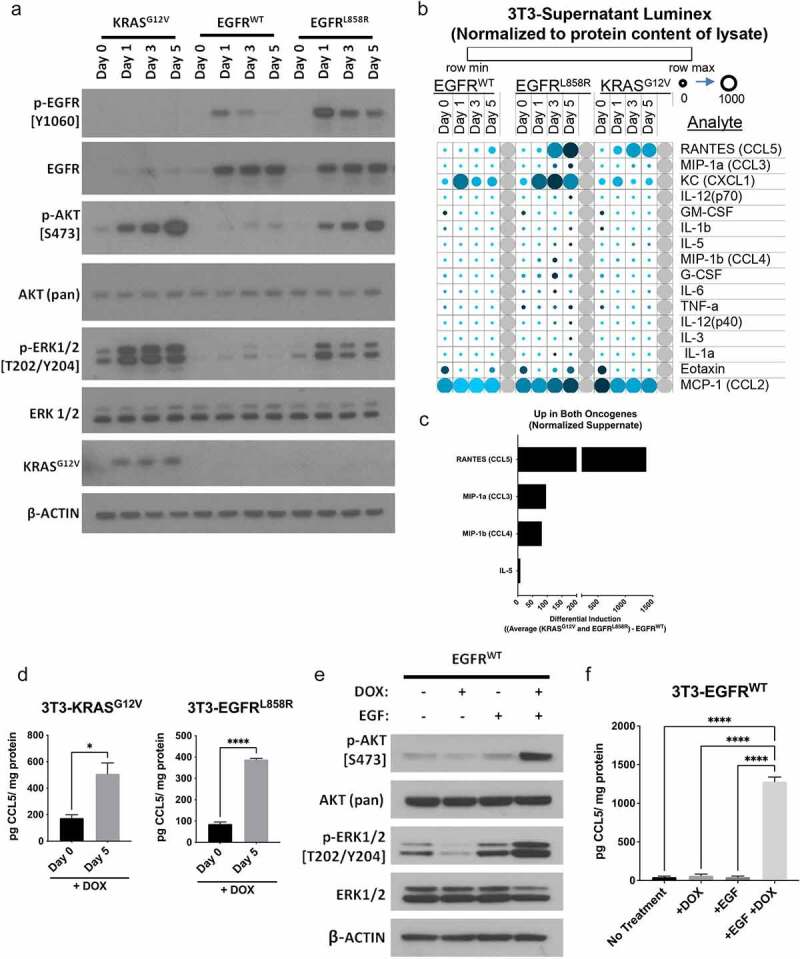
Oncogene signaling regulates cytokine production in premalignant cells. 3T3 cells were transduced with lentiviral constructs to express doxycyline (DOX) inducible wild-type EGFR, EGFRL858R, or KRASG12V. (a) Induced expression of oncogenic EGFRL858R and KRASG12V corresponded with increased phosphorylation of AKT and ERK, which was not seen with induced expression of EGFRWT. (b) Luminex assays were used to assess cytokines and chemokines produced after induction of EGFRWT, EGFRL858R, and KRASG12V. Data normalized to protein content of lysate (pg/mg) are visualized with a heatmap displaying levels of secreted protein for each cell line with increasing time post DOX treatment. Row Z-scores were calculated across all rows to show change in analyte and plotted, shown in color legend (row min-max). Value in picogram (pg) per milligram (mg) shown by circles small (0) to large (1000). (c) The analytes increased in both oncogene expressing cell lines are shown (see Supplemental Methods). (d) Secretion of CCL5 increased with 5 days of DOX treatment in 3T3 expressing KRASG12V and EGFRL858R as quantified by ELISA with DOX treatment day 0 (no treatment) and day 5. Induced expression of EGFRWT with the addition of 30 ng/mL EGF led to increased phosphorylation of AKT and ERK (e) and increased secretion of CCL5 (f). ELISA data show n = 3, with bars representing mean ± SEM, significance was determined by a Student’s T-test or an ANOVA with a post-hoc Dunnett’s test. *P < .05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001
Oncogenic MAPK/ERK signaling regulates CCL5 secretion in KRAS mutant lung cancer cells
We next assessed human lung cancer cells to confirm that oncogene signaling regulates CCL5 production in the transformed state. From gene expression microarray data for a panel of KRAS and EGFR mutant lung adenocarcinoma cell lines, we identified NCI-H23 KRASG12C as having high levels of CCL5 expression (Supplemental Figure S2). Using ELISAs, we confirmed that CCL5 was secreted at high levels by NCI-H23 cells in comparison to a cell line with low CCL5 mRNA expression, NCI-H2030 (Figure 2(a)). We used the MEK inhibitor Trametinib (TRAM) to inhibit the RAS/RAF/MAPK signaling pathway downstream of KRAS, which reduced the level of phospho-ERK and phospho-AKT (Figure 2(b)) and decreased CCL5 production by NCI-H23 cells in a dose-dependent manner (Figure 2(c)). We were next interested in whether KRAS regulates CCL5 in murine Lewis lung carcinoma (LLC) cells that also harbor a KrasG12C mutation.14 We found that transient knockdown of Kras with siRNA decreased phospho-ERK (Figure 2(d)) and decreased production of CCL5 (Figure 2(e)). Together, these findings support that oncogenic signaling through MAPK/ERK regulates CCL5 expression in human and murine lung cancer cells.
Figure 2.

Production of CCL5 is regulated by oncogenic signaling in KRAS mutant LUAD cells. (a) Expression level of secreted CCL5 in human lung adenocarcinoma cell lines H23 and H2030 as determined by ELISA. (b) Treatment of H23 cells with a MEK inhibitor (Trametinib) for 24 hours decreased phosphorylation of ERK and AKT relative to DMSO (0.1%) or untreated controls. (c) Trametinib treatment caused a dose dependent decrease in secreted CCL5 in H23 cells. Knockdown of KRAS with siRNA in LLC cells decreased ERK activation (d) and decreased production of secreted CCL5 (e). CCL5 production was quantified by ELISA. Data show n = 3, bars represent mean ± SEM, analyzed using an ANOVA with a post-hoc Dunnett’s test. *P < .05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001
Reduction of CCL5 in lung cancer cells inhibits tumor development in an immune dependent manner
To evaluate the role of tumor-derived CCL5 on lung tumor growth and the immune microenvironment in vivo, we used CRISPR-Cas9 guides to facilitate Ccl5 deletion in LLC cells, which we termed sgCCL5, and confirmed reduced CCL5 secretion by ELISA (Figure 3(a), Supplemental Figure S3a). LLC cells are syngeneic to the C57BL/6 mouse strain, providing an immune competent model with which to study the effects of tumor-derived CCL5 on immune cell recruitment to the lung and tumor microenvironment. Ccl5 deletion did not influence the proliferation of LLC cells in vitro (Figure 3(b)) or the development of lung tumors in immune deficient NRG mice (Figure 3(c)). However, immune competent C57BL/6 mice injected intravenously with sgCCL5 cells had a significantly lower tumor burden in the lungs than those injected with control guide transduced LLC cells (sgCNTRL, see supplemental methods) (Figure 3(d), Supplemental Figures S3b, and FS4–5). Taken together, these data indicate that tumor cell-derived CCL5 promotes LLC tumor growth in an immune response-dependent manner.
Figure 3.
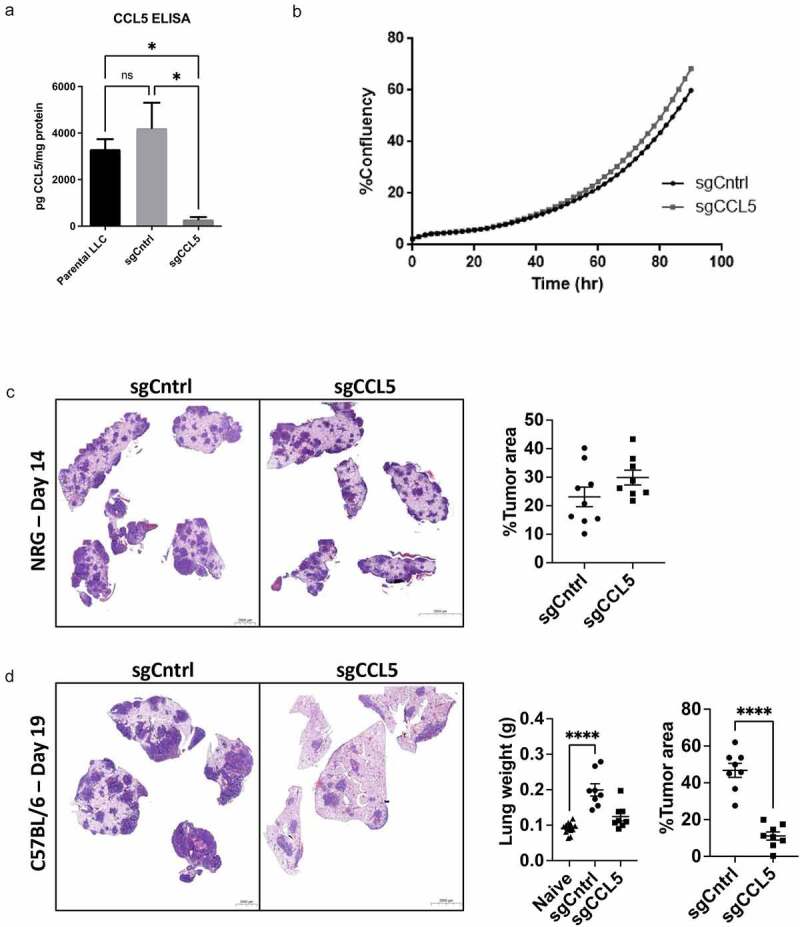
Ccl5 deletion in lung cancer cells reduces tumor burden. LLC cells were infected with lentiviral particles containing CRISPR/cas9 guide targeting Ccl5 (sgCCL5) or a scramble sequence (sgCNTRL). (a) CCL5 production was quantified by ELISA. Data show n = 3, significance by students t-test, bars indicate mean ± SEM. (b) Ccl5 deletion did not alter the in vitro growth rate of LLC cells determined by real-time cell confluence tracking by Incucyte as % confluency over time. (c) Day 14 after immune deficient NRG mice injected i.v. with Ccl5 deletion cells did not display a significant difference in lung tumor burden. Data show n = 8-9 , bars indicate mean ± SEM. (d) Day 19 after immune competent C57BL/6 mice injected i.v. with Ccl5 deletion cells had reduced tumor burden in the lungs. Data show n = 8, bars indicate mean ± SEM. Tumor burden was quantified as the fraction of tumor area/total lung area from 5 step sections 150 µm apart. Scale bars represent 2000-5000 µm. Significance from Graphpad Prism was determined using an ANOVA with a post-hoc Tukey’s test. If data did not have normal distribution a Kruskal-Wallis test was performed. *P < .05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001
Reduction of tumor-derived CCL5 prevents the formation of an immune suppressive microenvironment
CCL5 can be chemotactic for several different immune cell types.15,16 We used flow cytometry to quantify innate and adaptive immune cell populations in the lungs of C57BL/6 mice influenced by LLC tumors and by the reduction of tumor-derived CCL5. Firstly, the lungs of mice bearing sgCntrl LLC tumors had increased monocyte-derived macrophages (M0) relative to the lungs of tumor-free mice, and these tumor-associated macrophages were trending toward a decrease in the lungs of mice injected with sgCCL5 LLC cells (Figure 4(a)). There was no change in the frequency of natural killer cells (NK), alveolar macrophages, dendritic cells (DC), eosinophils (Eo), or myeloid-derived suppressor cells/neutrophils (MDSC/Neu) between lungs injected with LLC-sgCCL5 and LLC-sgCntrl cells (Figure 4(b-c), Supplemental Figure S6a-c). We quantified the expression of phenotypic markers for classically activated (M1) or alternatively activated immunosuppressive (M2) macrophages. While the frequency of macrophages was unchanged when compared to sgCNTRL, mice injected with LLC-sgCCL5 had, increased frequency of MHCII+ macrophages and fewer M2-like macrophages (CD206+MHCII−) than mice injected with LLC-sgCntrl cells (Figure 4(d-f)). There was no alteration to M1-like macrophages (CD206−MHCII+) (Figure 4(g)).
Figure 4.
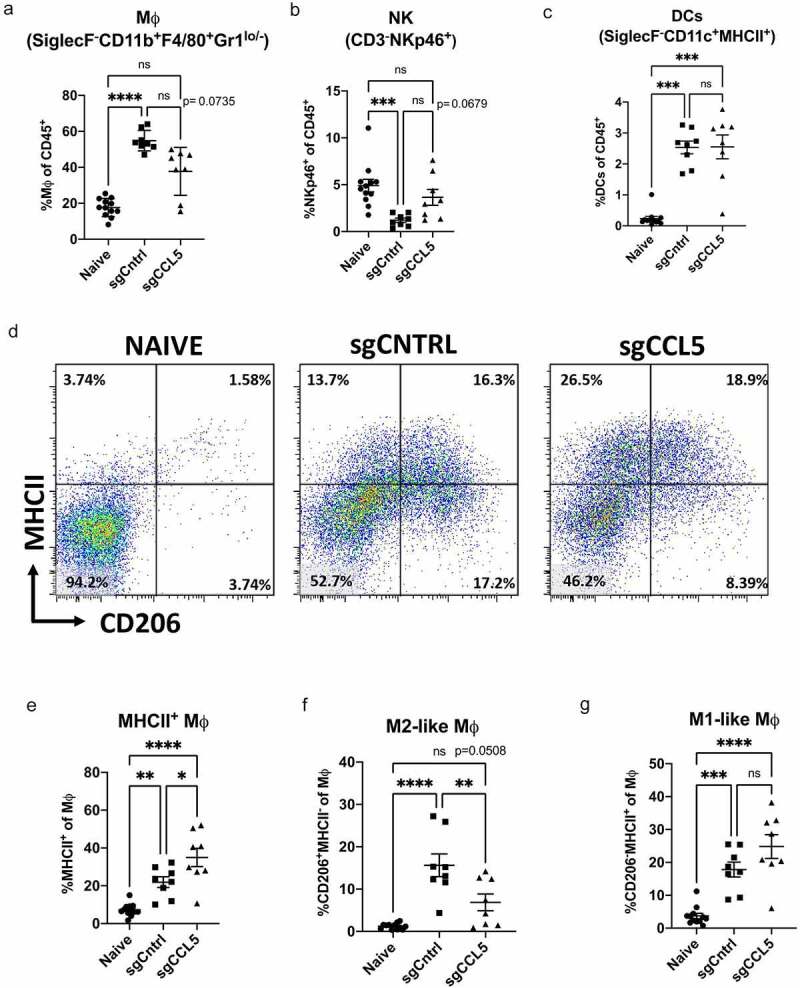
Tumor derived CCL5 alters innate immune suppressive cells. Innate immune cell populations within the lungs of C57BL/6 mice injected i.v. with Ccl5 deletion LLC cells, LLC scramble control cells, and tumor naïve mice were quantified by flow cytometry on day 19 post injection (see supplemental methods for more details). There was a trend toward a reduction in frequency of SiglecF−CD11b+F4/80+ macrophages (M0,total) (a) of mice injected with Ccl5 deletion cells (sgCCL5) compared to the scramble controls (sgCNTRL). Additionally, there was reduced frequency of NKp46+ (NK) cells in the sgCNTRL and increased (DC) cells in both sgCNTRL and sgCCL5 compared to naïve mice (b-c). Representative flow gates for staining of MHC-II and CD206 out of M0 populations shown (d). The frequency of MHCII+ M0 was increased, and CD206+ MHCII− M0 decreased in the sgCCL5 when compared to sgCNTRL (e-f). M0 that were CD206- MHCII+ were increased in both sgCNTRL and sgCCL5 when compared to naïve (g). Data shown n = 8–12, bars indicate mean ± SEM, analyzed using an ANOVA with a post-hoc Tukey’s test. If data did not have normal distribution a Kruskal-Wallis test was performed. *P < .05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001
Lungs of mice injected with sgCtrl LLC tumor cells exhibited decreased CD19 + B cells and CD4+ helper T cells, no change in CD8 + T cells, and increased immune suppressive Tregs compared to lungs from naïve tumor-free mice (Figure 5(a-d), Supplemental Figure 7). Injection of sgCCL5 tumor cells resulted in increased frequency of B cells, trend toward an increase in CD8+ T cells, no change in CD4+ cells and decreased Tregs compared to mice with sgCntrl tumors (Figure 5(a-d)).
Figure 5.

CCL5 deletion induces increased adaptive suppressor response and decreased CD8 T cell exhaustion. Adaptive immune cell populations were quantified from the tumor bearing lungs of sgCCL5, sgCNTRL and tumor free naïve. The sgCCL5 had higher CD19+ B cells, CD3+ CD8+ T cells, and no change to CD3+ CD4+ T cells when compared to sgCNTRL (a-c). In the CD4+ T cell populations, sgCCL5 had decreased CD25+ FOXP3+ (Tregs), decreased CD62L− CD44+ (TEM), and increased CD62L+ CD44− (Tnaive) compared to sgCNTRL (d-g). The CD8+ T cells in sgCCL5 had increased frequency of CD62L+ CD44+ (TCM) and significantly decreased PD1hi EOMEShi TBETlo CD8+ TEM than the sgCNTRL (h-k). Data shown n = 8–12, bars indicate mean ± SEM, analyzed using an ANOVA with a post-hoc Tukey’s test. If data did not have normal distribution a Kruskal-Wallis test was performed. *P < .05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001
We further characterized the pulmonary CD4+ and CD8+ T cell effector and memory populations. Mice with sgCtrl LLC tumors had decreased naïve CD4+ and CD8 + T cells (naïve T; Figure 5(e,h)), increased central memory-like CD4 + T cells (TCM; Figure 5(f)), and increased effector, memory-like17,18 CD4+ and CD8 + T cells (TEM; Figure 5(g-j)) in the lungs. CCL5 knockout partially rescued the tumor-induced changes in CD4 + T cell populations (Figure 5(e-g)), although naïve CD8 + T cells remained lower, central memory CD8 + T cells increased, and effector memory CD8+ were not significantly affected by loss of CCL5 in the LLC tumor cells (Figure 5(h-j)). Interestingly, while PD-1hi EOMEShi TBETdim CD8+ TEM cells indicative of T cell dysfunction or exhaustion were present in the lungs of mice with sgCtrl tumors, exhausted T cells were significantly less abundant in mice injected with sgCCL5 tumor cells (Figure 5(k), Supplemental Figure 8).19 Together, our in vivo data suggest CCL5 secreted by LLC tumor cells creates a pro-tumourigenic, immune suppressed lung environment that includes M2-like macrophages, Tregs, and exhausted CD8+ TEM cells. CCL5 knockout in LLC tumor cells reduces the formation of this immune suppressed lung environment, leading to reduced tumor growth in immune competent mice.
High CCL5 expression correlates with poor survival and suppression of effector immune response in lung cancer patients
We next assessed the clinical impact of CCL5 expression on recruitment of immune cells during LUAD development. To do this, we investigated the composition of tumor-infiltrating immune cells in LUAD patients using clinical gene expression data and determined cell types enriched in tumor bearing regions of the lung compared to matched adjacent normal lung tissue to identify immune cells most likely influenced by tumor cell-derived factors. We utilized two independent LUAD datasets for this purpose, one previously generated in-house (BCCRC) and one from The Cancer Genome Atlas (TCGA) (Supplemental Table 1). We identified hematopoietic cell types within LUAD tumors at varying abundance by CIBERSORT analysis20 and found a clear distinction in the immune cell repertoire of tumors compared to normal lung (Figure 6(a), Supplemental Figure S9 A-B).
Figure 6.

High CCL5 transcriptomic expression in LUAD patients correlates with lower survival and suppression of effector response in tumors. (a) CIBERSORT analysis of BCCRC microarray data from 83 patients showing matched tumor and normal biopsies and cell subsets organized by -high to -low fold change tumor/normal (T/N) in the heatmap. (b) Frequency of Tregs in TCGA and BCCRC samples are significantly increased in tumor compared to normal tissues. (c) CIBERSORT Treg relative cell subset frequency in BCCRC LUAD samples plotted by stage of tumor development relative to matched normal lung tissue. (d) Violin plot showing CCL5 (RNAseq) expression separated by median (low (n = 251) and high (n = 251)) value for RAS-MAPK-ERK signature score in TCGA LUAD (see supplemental methods). (e) Bar graph of exhaustion linked genes in the CD8 T cells from CIBERSORTX for low (n = 127) and high (n = 127) tertiles of CCL5 expression in TCGA LUAD patients. (f) High expression of CCL5 (ID 1555759_a_at, split by median) was associated with poor outcomes in LUAD patients using the online program KM plotter. CIBERSORT is plotted as a box plot with whiskers representing min to max values. Significance for CIBERSORT, and RAS-MAPK-ERK signature score data by Mann-Whitney. Survival curves were compared using a log-rank (Mantel-cox) statistical test, *P < .05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001
Consistent differences in several immune cell types were noted across tumor and normal lung regions in both datasets. We observed increased abundance of Tregs, macrophages (M0), Tfh, M1 macrophages, and plasma cells in tumor regions of the lung (Figure 6(a)).21 Tregs were the only immune cell type that was present in tumors and not detectable in adjacent normal tissues in both datasets, with the largest tumor/normal relative cell abundance (Figure 6(a-b), Supplemental Figure S9A). The relative Treg abundance derived from CIBERSORT was validated by FOXP3 IHC staining on a subset of BCCRC tumors (Supplemental Figure S10A-B), which showed a significant correlation between values from the two methods (one-way Pearson correlation coefficient test, r = 0.4586, *p = .0321). The abundance of Tregs assessed by CIBERSORT or by IHC in tumor tissues was elevated in early stage (I/II) patients and was consistent across early stage and late stage (III/IV) patients, suggesting Treg recruitment occurs early during lung tumourigenesis (Figure 6(c), Supplemental Figure S10 C-D). FOXP3 expression correlated with Treg abundance prediction by CIBERSORT, and we also found that FOXP3 expression correlated with CCL5 in LUAD tumors (Supplemental Figure S10E-G). Overall, there was a strong correlation between CCL5 and CCR5 expression in LUAD samples, supporting the functional link between CCL5 and CCR5 in immune cell recruitment (Supplemental Figure S10 H). Finally, we found CCL5 expression was higher in patients with a gene expression signature score that corresponds to high RAS-MAPK-ERK activity (Figure 6(d), Supplemental Figure S11A), which is predicted for tumors with activating mutations in EGFR, KRAS and other genes that drive signaling through this pathway (see supplemental methods).
To assess whether CCL5 expression in patients is associated with signatures of immune suppressive cells and exhausted T cells as observed in our mouse model, we stratified the TCGA LUAD tumor samples by CCL5 transcript levels into low- (n = 127) and high- (n = 127) expressing groups and determined immune cell content using CIBERSORT. We then used CIBERSORTX, a program that infers gene expression profiles of immune cell subsets identified by CIBERSORT, to determine gene expression differences between immune cell subsets in low vs high CCL5 expressing patients. Consistent with findings in our mouse models, Tregs, CD8 + T cells, and memory activated CD4+ T cells were significantly increased in TCGA LUAD patients with high CCL5 expression (Supplemental Figure S12A-D). The macrophage populations did not align with the mouse data as subset frequency of M1 were significantly increased, and M2 decreased in the CCL5 high patients (Supplemental Figure S12E-F). Supporting the clinical relevance of CCL5 expression and T cell response, CCL5 was most strongly correlated with CD8 + T cell abundance in LUAD patients (r = 0.6068, Supplemental Table 3–4, Supplemental Figure S12 G). While CD8 T cells are associated with an anti-tumor immune response, CD8 T cells in CCL5 high LUAD patients had increased expression of exhaustion-linked transcription factors and inhibitory receptor genes including EOMES, TBX21 (encoding Tbet), PDCD1 (encoding PD1), LAG3, BTLA, TIGIT, TOX, and CD244 (Figure 6(e)). There was an increase in CD8 T cell expression of cytokine and cytotoxic linked genes (IFNG, GZMA and GZMB) in the CCL5 high LUAD patients (Supplemental Figure S12 H). Furthermore, high CCL5 expression was strongly associated with poor survival in lung cancer patients, more so than smoking status and gender (figure 6(f), Supplemental Figure S13 A-B), highlighting the importance of CCL5 in promoting lung cancer progression and the potential use of CCL5 expression for LUAD prognosis. Our clinical data indicate that high CCL5 expression in LUAD patient lung tumors is associated with increased Tregs and CD8 checkpoint receptor expression, which, when taken with our in vitro and in vivo data, identify oncogene-induced expression of CCL5 by lung tumor cells as an important contributor to a pro-tumourigenic, immunosuppressive lung microenvironment.
Discussion
Mutations in oncogenes are generally thought to drive tumourigenesis through cancer cell intrinsic mechanisms such as increasing proliferation and suppressing apoptosis. We found that oncogenic signaling can also impact tumourigenesis by influencing the tumor immune microenvironment. Secretion of the chemokine CCL5 was found to be driven by mutant EGFR and KRAS through increased MAPK/ERK signaling, and tumor-derived CCL5 was important for optimal tumor growth in immunocompetent mice in vivo. Ccl5 deletion in an orthotopic mouse model of lung cancer led to decreased pulmonary Tregs, M2-like macrophages, and decreased PD-1hi EOMEShi TBETdim CD8+ TEM cells compared to CCL5 producing lung tumors. In human LUAD patients, Tregs were significantly increased in tumors but not detectable in adjacent normal lung tissues, and Treg levels correlated with expression of CCL5 and its receptor CCR5. High CCL5 expression in LUAD was associated with an increase in Tregs, expression of CD8 T cell exhaustion markers, and poor survival. Therefore, we conclude that CCL5 produced by oncogene expressing lung cancer cells contributes to the formation of an immune suppressive, tumor-permissive environment within the lungs that may be associated with worse therapeutic outcome.
The development of an immune suppressive microenvironment is central to the ability of a tumor to avoid detection and elimination by the immune system. CCL5 has previously been shown to be elevated in the bronchoalveolar lavage (BAL) fluid of mice with oncogene-induced tumors,8 but the effect of CCL5 on oncogenic tumor formation and the immune environment within the lungs was not clear. We found that Ccl5 deletion by use of the CRISPR-Cas9 system reduced tumor formation in the lungs of immune competent mice, which was associated with lower levels of immunosuppressive Tregs, M2-like macrophages, and decreased exhaustion in CD8+ TEM compared to control tumors. While a tumor cell intrinsic role for CCL5 in autocrine regulation of proliferation and invasion in lung cancer cells has been reported by others,22–24 we did not see any effect of Ccl5 deletion on LLC growth in vitro or on tumor formation in the lungs of immune deficient mice. Our data therefore indicate a cell extrinsic role for tumor-derived CCL5 that contributes to recruitment of M2-like macrophages and immune suppressive Tregs to promote lung tumor growth and inhibit CD8+ TEM cells. Importantly, CCL5 levels were also significantly correlated with increased Tregs and RAS-MAPK-ERK activity in human LUAD patient tumors as well as an exhausted CD8 profile, indicating the clinical relevance of these experimental findings. While the macrophage findings did not align between mouse models and human tumor data, we speculate that this disconnect could be from the ex-vivo isolated and polarized macrophages used in the CIBERSORT M1/M2 signatures, which may not translate to an in situ clinical setting.
We found that KRAS or EGFR-induced transformation of NIH-3T3 cells, or EGF induced signaling in NIH-3T3 cells over-expressing EGFRWT, significantly increased CCL5 production. In human lung cancer cells, we found that CCL5 secretion was reduced by blocking oncogenic signaling through the MAPK/ERK pathway. CCL5 expression can be regulated by the transcription factor AP-1, which is regulated downstream of the KRAS/MAPK/ERK signaling pathway.25,26 Zhu et al22 reported an increase in CCL5 signaling during KRAS driven transformation mediated through TBK1/IKKε, which is also activated by KRAS signaling in LUAD.27 Not all lung cancer cell lines with oncogenic KRAS and EGFR mutations expressed high levels of CCL5, which may be due to the lack of selective pressure to maintain CCL5 expression after prolonged culturing in the absence of immune cells. Furthermore, cross talk with other signaling pathways may play a role as CCL5 expression in lung cancer cells can also be regulated by the signaling of growth factors and pro-inflammatory cytokines (e.g. TNFα, IL-1β, IL-6) through NF-κB,22,28,29 STAT3,30 and RUNX3.31 Multiple regulators of CCL5 under different contexts present a clinical challenge to targeting CCL5 production through inhibition of oncogenic signaling.32 Our results indicate that while oncogenic signaling is an important driver of CCL5 production by lung cancer cells, the importance and influence of CCL5 on the immune microenvironment and tumor development may not be limited to EGFR and KRAS driven lung cancer.
Tumor derived CCL5 may be acting directly upon Tregs or indirectly through other immune or stromal cells. Elevated levels of Tregs have been reported in both oncogene (mutant EGFR and KRAS)8,33 and carcinogen-induced (NNK, via KRAS mutation)34 mouse models of lung cancer. Multiple chemokine signaling axes have been shown to induce Treg recruitment to tumors35 and metastatic target organs.4,36 The CCL5/CCR5 axis was associated with the recruitment and immunosuppressive function of Tregs in pancreatic cancer37,38 and colorectal cancer.39 We found that CCL5 and CCR5 correlated with FOXP3 expression and Treg abundance in LUAD patients, which was consistent with the role of CCL5 we observed in the animal tumor model. Furthermore, depletion of Tregs is known to decrease tumor burden,34,40 highlighting their importance in lung tumor development. Previous research has shown that LUAD are enriched for Tregs41,42 and that Tregs are elevated in the blood of NSCLC patients compared to healthy controls.41,43,44 High levels of tumor infiltrating Tregs are commonly associated with poor patient outcomes.45–47 Using CIBERSORT to assess Treg frequency in LUAD patient samples, we observed a significant accumulation of Tregs in tumors relative to matched normal lung tissue. We found Tregs were elevated in early-stage disease and did not observe a significant increase in Tregs with stage in our cohorts, in contrast to Erfani et al43 who reported that Tregs increase with lung cancer stage. Our findings indicate that Treg populations are recruited early in LUAD development, and our pre-clinical data and clinical correlates indicate Treg recruitment is at least partly driven by CCL5.
CCL5 is an inflammatory chemokine that can recruit a variety of immune cell types in response to injury and infection including T cells (CD4+, CD8+), monocytes, macrophages, eosinophils, and dendritic cells.48 Therefore, the effect of CCL5 on the immune environment varies in different cancer models and tissue types. We found mouse models with tumor cell Ccl5 deletion had reduced populations of PD-1hi EOMEShi TBETlo CD8+ TEM cells when compared to Ccl5 expressing LLC controls. CD8+ cells can become dysfunctional or “exhausted” in lung tumors by prolonged antigen exposure, interaction with suppressive immune cells or suppressive cytokines, or direct engagement with checkpoint ligands on tumor cells.49,50 Decreased IL-7 signaling, high expression of checkpoint receptor proteins (i.e. PD-1, LAG3), and increased EOMES expression are markers of exhaustion in T cells.19,49,51,52 Similarly, in LUAD we found that high CCL5 expression was associated with increased CD8+ T cells with higher levels of exhaustion markers (EOMES, PD-1, BTLA, CD244, LAG-3 and TIGIT) and high expression of TBET, cytokines, and cytotoxic markers (TBX21, IFNg, GZMA, and GZMB). The combination of expression of these markers indicates CIBERSORTX detected subsets of CD8 T cells (exhausted, activated, and memory) that are increased in the CCL5 high LUAD patients. The data indicates the signature of exhaustion from CD8 + T cells by CIBERSORT occurs when patients have higher vs. lower CCL5; therefore, we posit levels of CCL5 could be an indicator of influx of immunosuppressive cells and exhaustion. Previous studies indicated CCL5 produced in the TME polarizes macrophages and MDSCs to an immunosuppressive phenotype thereby inhibiting CD8 + T cells and leading to exhaustion.53 Based off our mouse model data, we posit increased CCL5 leads to recruitment of immunosuppressive cells (i.e. Tregs, M2-Tumor associated macrophages, and more) that are known to induce exhaustion of CD8 + T cells. Future experiments would involve depleting CCL5 expression by tumor cells or myeloid populations to help determine if CCL5 produced by lung tumor cells vs. other myeloid cells contributes more significantly to exhaustion of CD8 + T cells. Further analysis using CIBERSORTX incorporating a gene signature matrix from isolated tumor infiltrating exhausted CD8 T cells would enable better assessment of exhausted CD8+ T cells from LUAD patient tumors.
In early-stage LUAD, Moran et al.54,55 reported that CCL5 expression was a predictor of survival associated with favorable active lymphocytic response. Conversely in another study, CCL5 and CCR5 were associated with invasion and increased risk of mortality in LUAD patients, although it did not examine the immune microenvironment.23 We found high CCL5 expression correlated with poor survival in LUAD patients, in addition to higher levels of Tregs and markers of T cell exhaustion. Although CCL5 may be capable of recruiting anti-tumourigenic immune cells in some contexts, our data highlight a role for CCL5 in facilitating an immune suppressive environment that facilitates CD8+ T cell exhaustion, lung cancer development and progression.
Conclusion
Our findings indicate that oncogenic signaling regulates CCL5 to promote lung tumor growth in vivo through the formation of an immune suppressive lung environment. Since oncogenic mutations are an early event in lung tumourigenesis, oncogene-driven CCL5 production may shape the immune microenvironment from an early stage and drive alternate priming of an anti-tumor immune response. However, additional study is necessary to understand the different functional impacts of tumor cell-derived CCL5 on immunosuppressive cells and antigen presenting cells. Our data implicate tumor production of CCL5 as a potential biomarker for an immunosuppressive microenvironment with CD8 + T cell exhaustion that may be susceptible to checkpoint inhibitors or immunotherapies targeting Treg function. Furthermore, neutralization of tumor-derived CCL5 by monoclonal antibody or inhibition of CCR5 by the CCR5 inhibitor maraviroc may reduce immunosuppression in the lung cancer microenvironment. Overall, this work provides evidence that oncogenic signaling directly influences the tumor immune environment to promote cancer development, offering potential avenues for diagnosis and therapy.
Supplementary Material
Acknowledgments
BC Cancer patient tumours were collected and profiled as part of a previous collaboration with Dr. Adi Gazdar, the Early Detection Research Network (EDRN) and the Canary Foundation. We thank technicians at the BC Cancer ARC for their technical support. This work was funded by the Canadian Institutes of Health Research (CIHR; MOP-142313; WWL and KLB), the CURE Foundation and the Cancer Research Society (21441; KLB), the BC Cancer Foundation and the Terry Fox Research Institute. ESM was funded by a Four-Year Fellowship from the University of British Columbia (UBC). RAC was funded by a Cordula and Gunter Paetzold Fellowship and a Four-Year Fellowship from UBC. FJ and BJW were supported by a CIHR Doctoral Research Award, JLC was funded by a Walter C Koerner Fellowship from UBC, and ECH was funded by the Li Tze Fong Memorial Fellowship and a Four-Year Fellowship from UBC. KLB is a Michael Smith Foundation for Health Research (MSFHR) Scholar. WWL is a MSFHR Scholar and CIHR New Investigator.
Funding Statement
This work was supported by the Canadian Institutes of Health Research [MOP-142313]; CURE Foundation and the Cancer Research Society [21441]; [Terry Fox Research Institute New Investigator Award]; BC Cancer Foundation.
Materials and Methods
Cell lines and reagents
A detailed description of the cell lines and reagents used as well as LUMINEX, ELISA, Western blotting, and Incucyte methodologies can be found in the Supplemental Methods.
Mice
C57BL/6 mice were obtained from Jackson Laboratories. NRG mice were bred in-house at the BC Cancer Research Centre’s (BCCRC) Animal Resource Centre. Mice were housed under specific pathogen-free conditions at the BCCRC. All housing, care, and experimental conditions were followed in accordance with the guidelines of the University of British Columbia Animal Care Committee and the Canadian Council for Animal Care. Detailed descriptions of methodologies for mouse i.v. injection of tumour cells, flow cytometry, histology and tumour burden quantification can be found in the Supplemental Methods.
Patient samples
Details of patient biopsy samples are summarized in Supplemental Table 1. Samples from BC Cancer (BCCRC) were collected with written patient consent, following protocols approved by BC Cancer and University of British Columbia (H04-60060). These samples were profiled as part of a collaboration with the Early Detection Research Network (EDRN) and the Canary Foundation (55). The Cancer Genome Atlas (TCGA) dataset of lung adenocarcinoma (LUAD) samples was collected with informed patient consent through the National Cancer Institute (NCI) and National Human Genome Research Institute (NHGRI) with approval from the local Institutional Review Boards (IRBs). Normal lung tissue samples are patient-matched and derived from adjacent non-tumour tissue. Detailed descriptions for the array and RNAseq expression analyses, CIBERSORT, CIBERSORTX, histology, heatmaps and survival analyses can be found in the Supplemental Methods.
Statistics
For the comparison of immune cell populations by CIBERSORT analysis, a Mann–Whitney statistical test was performed. A one-tailed Pearson correlation coefficient (r) was used to assess the correlation between immunohistochemistry (IHC) and CIBERSORT data and RNAseq data. Survival curves were compared using a log-rank (Mantel-cox) statistical test. Remaining data were analyzed using a one-way ANOVA and correction for multiple comparisons was made with a Tukey’s test to compare each value to a single control. If data did not have normal distribution a Kruskal–Wallis test was performed. Statistical significance is denoted as p < .05 designated by (*), p < .01 as (**), p <.001 as (***), and p <.0001 as (****). Incucyte cell proliferation analysis was performed using Microsoft Excel (Microsoft). All other data were analyzed using GraphPad Prism software (GraphPad Software, Inc). Unless otherwise stated, all graphs are reported as mean ± standard error of the mean (SEM).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Govindan R, Szczesna A, Ahn MJ, Schneider CP, Gonzalez Mella PF, Barlesi F, Han B, Ganea DE, Von Pawel J, Vladimirov V, et al. Phase III Trial of Ipilimumab Combined With Paclitaxel and Carboplatin in Advanced Squamous Non-Small-Cell Lung Cancer. J Clin Oncol. 2017;35(30):3449–12. doi: 10.1200/JCO.2016.71.7629. [DOI] [PubMed] [Google Scholar]
- 2.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016;375(19):1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 3.Hegde PS, Karanikas V, S. E.. The Where the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin Cancer Res. 2016;22(8):1865–1874. doi: 10.1158/1078-0432.CCR-15-1507. [DOI] [PubMed] [Google Scholar]
- 4.Halvorsen EC, Mahmoud SM, Bennewith KL. Emerging roles of regulatory T cells in tumour progression and metastasis. Cancer Metastasis Rev. 2014;33(4):1025–1041. doi: 10.1007/s10555-014-9529-x. [DOI] [PubMed] [Google Scholar]
- 5.Conway EM, Pikor LA, Kung SH, Hamilton MJ, Lam S, Lam WL, Bennewith KL. Macrophages, Inflammation, and Lung Cancer. Am J Respir Crit Care Med. 2016;193(2):116–130. doi: 10.1164/rccm.201508-1545CI. [DOI] [PubMed] [Google Scholar]
- 6.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumours. Cancer Discov. 2013;3(12):1355–1363. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, Matsumoto K, Takayama K, Takamori S, Kage M, et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol. 2014;25(10):1935–1940. doi: 10.1093/annonc/mdu242. [DOI] [PubMed] [Google Scholar]
- 10.Chen N, Fang W, Lin Z, Peng P, Wang J, Zhan J, Hong S, Huang J, Liu L, Sheng J, et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol Immunother. 2017;66(9):1175–1187. doi: 10.1007/s00262-017-2005-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wellenstein Md, de Visser KE. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumour Immune Landscape. Immunity. 2018;48(3):399–416. doi: 10.1016/j.immuni.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2(11):e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tong JH, Lung RW, Sin FM, Law PP, Kang W, Chan AW, Ma BB, Mak TW, Ng SS, To KF, et al. Characterization of rare transforming KRAS mutations in sporadic colorectal cancer. Cancer Biol Ther. 2014;15(6):768–776. doi: 10.4161/cbt.28550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agalioti T, Giannou AD, Krontira AC, Kanellakis NI, Kati D, Vreka M, Pepe M, Spella M, Lilis I, Zazara DE, et al. Mutant KRAS promotes malignant pleural effusion formation. Nat Commun. 2017;8(1):15205. doi: 10.1038/ncomms15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang S, Zhong M, Wang C, Xu Y, Gao WQ, Zhang Y. CCL5-deficiency enhances intratumoural infiltration of CD8(+) T cells in colorectal cancer. Cell Death Dis. 2018;9(7):766. doi: 10.1038/s41419-018-0796-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Araujo JM, Gomez AC, Aguilar A, Salgado R, Balko JM, Bravo L, Doimi F, Bretel D, Morante Z, Flores C, et al. Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer. Sci Rep. 2018;8(1):4899. doi: 10.1038/s41598-018-23099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin MD, Badovinac VP. Defining Memory CD8 T Cell. Front Immunol. 2018;9:2692. doi: 10.3389/fimmu.2018.02692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bengsch B, Ohtani T, Khan O, Setty M, Manne S, O’Brien S, Gherardini PF, Herati RS, Huang AC, Chang K-M, et al. Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity. 2018;48(5):1029–45 e5. doi: 10.1016/j.immuni.2018.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, et al. Defining ‘T cell exhaustion’. Nat Rev Immunol. 2019;19(11):665–674. doi: 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng KW, Marshall EA, Enfield KS, Martin SD, Milne K, Pewarchuk ME, Abraham N, Lam WL. Somatic mutation-associated T follicular helper cell elevation in lung adenocarcinoma. Oncoimmunology. 2018;7(12):e1504728. doi: 10.1080/2162402X.2018.1504728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, Moody SE, Shen RR, Schinzel AC, Thai TC, et al. Inhibition of KRAS-Driven Tumorigenicity by Interruption of an Autocrine Cytokine Circuit. Cancer Discov. 2014;4(4):452–465. doi: 10.1158/2159-8290.CD-13-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borczuk AC, Papanikolaou N, Toonkel RL, Sole M, Gorenstein LA, Ginsburg ME, Sonett JR, Friedman RA, Powell CA. Lung adenocarcinoma invasion in TGFbetaRII-deficient cells is mediated by CCL5/RANTES. Oncogene. 2008;27(4):557–564. doi: 10.1038/sj.onc.1210662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang CY, Fong YC, Lee CY, Chen MY, Tsai HC, Hsu HC, Tang C-H. CCL5 increases lung cancer migration via PI3K, Akt and NF-kappaB pathways. Biochem Pharmacol. 2009;77(5):794–803. doi: 10.1016/j.bcp.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Roebuck KA, Carpenter LR, Lakshminarayanan V, Page SM, Moy JN, Thomas LL. Stimulus-specific regulation of chemokine expression involves differential activation of the redox-responsive transcription factors AP-1 and NF-kappaB. J Leukoc Biol. 1999;65(3):291–298. doi: 10.1002/jlb.65.3.291. [DOI] [PubMed] [Google Scholar]
- 26.Henriquet C, Gougat C, Combes A, Lazennec G, Mathieu M. Differential regulation of RANTES and IL-8 expression in lung adenocarcinoma cells. Lung Cancer. 2007;56(2):167–174. doi: 10.1016/j.lungcan.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ayad O, Stark JM, Fiedler MM, Menendez IY, Ryan MA, Wong HR. The heat shock response inhibits RANTES gene expression in cultured human lung epithelium. J Immunol. 1998;161:2594–2599. [PubMed] [Google Scholar]
- 29.Wickremasinghe MI, Thomas LH, O’Kane CM, Uddin J, Friedland JS. Transcriptional mechanisms regulating alveolar epithelial cell-specific CCL5 secretion in pulmonary tuberculosis. J Biol Chem. 2004;279(26):27199–27210. doi: 10.1074/jbc.M403107200. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Yang X, Tsai Y, Yang L, Chuang KH, Keng PC, Lee SO, Chen Y. IL-6 Mediates Macrophage Infiltration after Irradiation via Up-regulation of CCL2/CCL5 in Non-small Cell Lung Cancer. Radiat Res. 2017;187(1):50–59. doi: 10.1667/RR14503.1. [DOI] [PubMed] [Google Scholar]
- 31.Kim HJ, Park J, Lee SK, Kim KR, Park KK, Chung WY. Loss of RUNX3 expression promotes cancer-associated bone destruction by regulating CCL5, CCL19 and CXCL11 in non-small cell lung cancer. J Pathol. 2015;237(4):520–531. doi: 10.1002/path.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Culley FJ, Pennycook AM, Tregoning JS, Dodd JS, Walzl G, Wells TN, Hussell T, Openshaw PJM. Role of CCL5 (RANTES) in viral lung disease. J Virol. 2006;80(16):8151–8157. doi: 10.1128/JVI.00496-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Busch SE, Hanke ML, Kargl J, Metz HE, MacPherson D, Houghton AM. Lung Cancer Subtypes Generate Unique Immune Responses. J Immunol. 2016;197(11):4493–4503. doi: 10.4049/jimmunol.1600576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Granville CA, Memmott RM, Balogh A, Mariotti J, Kawabata S, Han W, LoPiccolo J, Foley J, Liewehr DJ, Steinberg SM, et al. A central role for Foxp3+ regulatory T cells in K-Ras-driven lung tumourigenesis. PLoS One. 2009;4(3):e5061. doi: 10.1371/journal.pone.0005061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mailloux AW, Young MR. Regulatory T-cell trafficking: from thymic development to tumour-induced immune suppression. Crit Rev Immunol. 2010;30(5):435–447. doi: 10.1615/CritRevImmunol.v30.i5.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halvorsen EC, Hamilton MJ, Young A, Wadsworth BJ, LePard NE, Lee HN, Firmino N, Collier JL, Bennewith KL. Maraviroc decreases CCL8-mediated migration of CCR5+regulatory T cells and reduces metastatic tumor growth in the lungs. Oncoimmunology. 2016;5(6):e1150398. doi: 10.1080/2162402X.2016.1150398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, Eberlein TJ, Hsieh C-S, Linehan DC. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumour growth in a murine model of pancreatic cancer. J Immunol. 2009;182(3):1746–1755. doi: 10.4049/jimmunol.182.3.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Lang M, Zhao T, Feng X, Zheng C, Huang C, Hao J, Dong J, Luo L, Li X, et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3+Treg cells in pancreatic ductal adenocarcinoma. Oncogene. 2017;36(21):3048–3058. doi: 10.1038/onc.2016.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang LY, Lin YC, Mahalingam J, Huang CT, Chen TW, Kang CW, Peng H-M, Chu -Y-Y, Chiang J-M, Dutta A, et al. Tumor-Derived Chemokine CCL5 Enhances TGF-β–Mediated Killing of CD8+ T Cells in Colon Cancer by T-Regulatory Cells. Cancer Res. 2012;72(5):1092–1102. doi: 10.1158/0008-5472.CAN-11-2493. [DOI] [PubMed] [Google Scholar]
- 40.Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, DuPage M, Tammela T, Kerper N, Farago A, et al. Regulatory T Cells in Tumour-Associated Tertiary Lymphoid Structures Suppress Anti-tumour T Cell Responses. Immunity. 2015;43(3):579–590. doi: 10.1016/j.immuni.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phillips JD, Knab LM, Blatner NR, Haghi L, DeCamp MM, Meyerson SL, Heiferman MJ, Heiferman JR, Gounari F, Bentrem DJ, et al. Preferential expansion of pro-inflammatory Tregs in human non-small cell lung cancer. Cancer Immunol Immunother. 2015;64(9):1185–1191. doi: 10.1007/s00262-015-1725-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Black CC, Turk MJ, Dragnev K, Rigas JR. Adenocarcinoma contains more immune tolerance regulatory t-cell lymphocytes (versus squamous carcinoma) in non-small-cell lung cancer. Lung. 2013;191(3):265–270. doi: 10.1007/s00408-013-9455-7. [DOI] [PubMed] [Google Scholar]
- 43.Erfani N, Mehrabadi SM, Ghayumi MA, Haghshenas MR, Mojtahedi Z, Ghaderi A, Amani D. Increase of regulatory T cells in metastatic stage and CTLA-4 over expression in lymphocytes of patients with non-small cell lung cancer (NSCLC). Lung Cancer. 2012;77(2):306–311. doi: 10.1016/j.lungcan.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 44.Kotsakis A, Koinis F, Katsarou A, Gioulbasani M, Aggouraki D, Kentepozidis N, Georgoulias V, Vetsika E-K. Prognostic value of circulating regulatory T cell subsets in untreated non-small cell lung cancer patients. Sci Rep. 2016;6(1):39247. doi: 10.1038/srep39247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Callaghan DS, Rexhepaj E, Gately K, Coate L, Delaney D, O’Donnell DM, Kay E, O’Connell F, Gallagher WM, O’Byrne KJ, et al. Tumour islet Foxp3 + T-cell infiltration predicts poor outcome in nonsmall cell lung cancer. Eur Respir J. 2015;46(6):1762–1772. doi: 10.1183/13993003.00176-2014. [DOI] [PubMed] [Google Scholar]
- 46.Shimizu K, Nakata M, Hirami Y, Yukawa T, Maeda A, Tanemoto K. Tumor-Infiltrating Foxp3+ Regulatory T Cells are Correlated with Cyclooxygenase-2 Expression and are Associated with Recurrence in Resected Non-small Cell Lung Cancer. J Thoracic Oncol: Official Publ Int Assoc for the Study of Lung Cancer. 2010;5(5):585–590. doi: 10.1097/JTO.0b013e3181d60fd7. [DOI] [PubMed] [Google Scholar]
- 47.Tao H, Mimura Y, Aoe K, Kobayashi S, Yamamoto H, Matsuda E, Okabe K, Matsumoto T, Sugi K, Ueoka H, et al. Prognostic potential of FOXP3 expression in non-small cell lung cancer cells combined with tumor-infiltrating regulatory T cells. Lung Cancer. 2012;75(1):95–101. doi: 10.1016/j.lungcan.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 48.Aldinucci D, Casagrande N. Inhibition of the CCL5/CCR5 Axis against the Progression of Gastric Cancer. Int J Mol Sci. 2018;19(5):5. doi: 10.3390/ijms19051477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):3449–3457. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 51.Thommen DS, Schumacher TN, Cell T. Dysfunction in Cancer. Cancer Cell. 2018;33(4):547–562. doi: 10.1016/j.ccell.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He Y, Yu H, Rozeboom L, Rivard CJ, Ellison K, Dziadziuszko R, Suda K, Ren S, Wu C, Hou L, et al. LAG-3 Protein Expression in Non-Small Cell Lung Cancer and Its Relationship with PD-1/PD-L1 and Tumour-Infiltrating Lymphocytes. J Thorac Oncol. 2017;12(5):814–823. doi: 10.1016/j.jtho.2017.01.019. [DOI] [PubMed] [Google Scholar]
- 53.Aldinucci D, Borghese C, The CN. CCL5/CCR5 Axis in Cancer Progression. Cancers (Basel). 2020. Jul;12(7):1765. doi: 10.3390/cancers12071765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moran CJ, Arenberg DA, Huang CC, Giordano TJ, Thomas DG, Misek DE, Chen G, Iannettoni MD, Orringer MB, Hanash S, et al. RANTES expression is a predictor of survival in stage I lung adenocarcinoma. Clin Cancer Res. 2002;8(12):3803–3812. [PubMed] [Google Scholar]
- 55.Girard L, Rodriguez-Canales J, Behrens C, Thompson DM, Botros IW, Tang H, Xie Y, Rekhtman N, Travis WD, Wistuba II, et al. An Expression Signature as an Aid to the Histologic Classification of Non-Small Cell Lung Cancer. Clin Cancer Res. 2016;22(19):4880–4889. doi: 10.1158/1078-0432.CCR-15-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.