

Abstract
Monoclonal antibodies (mAbs) with neutralizing activity against SARS-CoV-2 have demonstrated clinical benefit in cases of mild to moderate SARS-CoV-2 infection, substantially reducing the risk for hospitalization and severe disease1–4. Treatment generally requires the administration of high doses of these mAbs with limited efficacy in preventing disease complications or mortality among hospitalized COVID-19 patients5. Here we report the development and evaluation of Fc-optimized anti-SARS-CoV-2 mAbs with superior potency to prevent or treat COVID-19 disease. In several animal models of COVID-19 disease6,7, we demonstrate that selective engagement of activating FcγRs results in improved efficacy in both preventing and treating disease-induced weight loss and mortality, significantly reducing the dose required to confer full protection upon SARS-CoV-2 challenge and treatment of pre-infected animals. Our results highlight the importance of FcγR pathways in driving antibody-mediated antiviral immunity, while excluding any pathogenic or disease-enhancing effects of FcγR engagement of anti-SARS-CoV-2 antibodies upon infection. These findings have important implications for the development of Fc-engineered mAbs with optimal Fc effector function and improved clinical efficacy against COVID-19 disease.
Several neutralizing mAbs targeting the SARS-CoV-2 Spike have entered clinical testing over the past months, yielding FDA approval of two mAb cocktails –casirivimab and imdevimab and bamlanivimab and etesevimab– for the treatment of mild to moderate COVID-19 patients4. In phase II/III studies, these and other mAbs that are currently awaiting regulatory approval demonstrated clear therapeutic benefit in mild to moderate COVID-19 cases, reducing the risk for hospitalization by over 80%1–3. These results are in stark contrast to the findings from phase III trials (e.g. ACTIV-3, NCT04501978) that assessed the therapeutic activity of these mAbs in hospitalized COVID-19 patients. In all cases, none of the tested mAbs offered any therapeutic benefit, even when administered at exceedingly high doses or in combination with remdesivir5.
The antiviral activity of IgG antibodies is the outcome of Fab-mediated virus neutralization coupled with the capacity of the Fc domain to mediate effector functions through interactions with Fcγ receptors (FcγRs) expressed on effector leukocytes8. FcγR engagement mediates pleiotropic functions, including the clearance of viral particles9, the cytotoxic elimination of virus-infected cells10, as well as the induction of antiviral T-cell responses11. Several reports have independently demonstrated in well-defined in vivo models of SARS-CoV-2 infection that the antiviral activity of neutralizing anti-SARS-CoV-2 antibodies depends on Fc-FcγR interactions12–15. Additionally, mechanistic studies determined that these protective effects are primarily mediated by CCR2+ monocytes, as well as cytotoxic CD8+ T cells that infiltrate the lung and confer antiviral activities12. Despite these findings, no studies have explored whether optimization of neutralizing anti-SARS-CoV-2 mAbs for enhanced FcγR binding could improve their therapeutic activity, especially in the setting of severe COVID-19. Indeed, maximizing the capacity of neutralizing anti-SARS-CoV-2 mAbs to engage and activate the appropriate FcγR pathways is expected to lower the mAb dose required for the treatment of mild to moderate COVID-19, as well as improve their activity in hospitalized patients.
Currently, most mAbs in clinical use or development are expressed as human IgG1, which despite its affinity for activating FcγRs also exhibits binding to the inhibitory FcγRIIb, thereby limiting protective Fc effector activities11. Additionally, due to presumptive safety concerns over the capacity of antibodies to exacerbate disease through ADE (antibody-dependent enhancement) mechanisms8, several clinical mAbs (etesevimab, AZD8895, and AZD1061) have been engineered to lack FcγR binding activity. However, numerous in vivo studies in animal models failed to provide evidence for ADE12–16, and therapeutic administration of high doses of convalescent plasma or neutralizing anti-SARS-CoV-2 mAbs in COVID-19 patients has not been associated with worse disease outcomes1–3,5,17. Likewise, comparable safety profiles were evident in clinical trials of neutralizing mAbs with intact or diminished Fc effector function3,18,19.
Hamster FcγRs and IgG Fc domain activity
To assess the role of FcγRs in the mAb-mediated protection and develop mAbs with superior therapeutic potency, we selected well-established small animal SARS-CoV-2 infection models that recapitulate the clinical features of human COVID-196,7,20. One of these models involves the use of Syrian hamsters (Mesocricetus auratus), a species that sustains productive SARS-CoV-2 replication and exhibits evidence of severe disease upon challenge6. However, a major obstacle in the study of human IgG Fc effector activity is the substantial interspecies differences in the affinity of human IgG antibodies for FcγRs expressed by rodent species, such as hamsters21. We therefore cloned the four classes of hamster FcγRs and characterized their affinity for human, hamster, and mouse IgG subclasses and Fc variants (Fig. 1a–b, Extended Data Fig. 1). Comparative analysis of hamster FcγRs revealed substantial sequence homology to mouse FcγRs, with three hamster FcγRs (FcγRI, FcγRIII, FcγRIV) corresponding to activating FcγRs, whereas FcγRIIb represents the sole inhibitory FcγR.
Fig. 1: Contribution of Fc effector function to the protective activity of neutralizing anti-SARS-CoV-2 mAbs in hamster infection models.
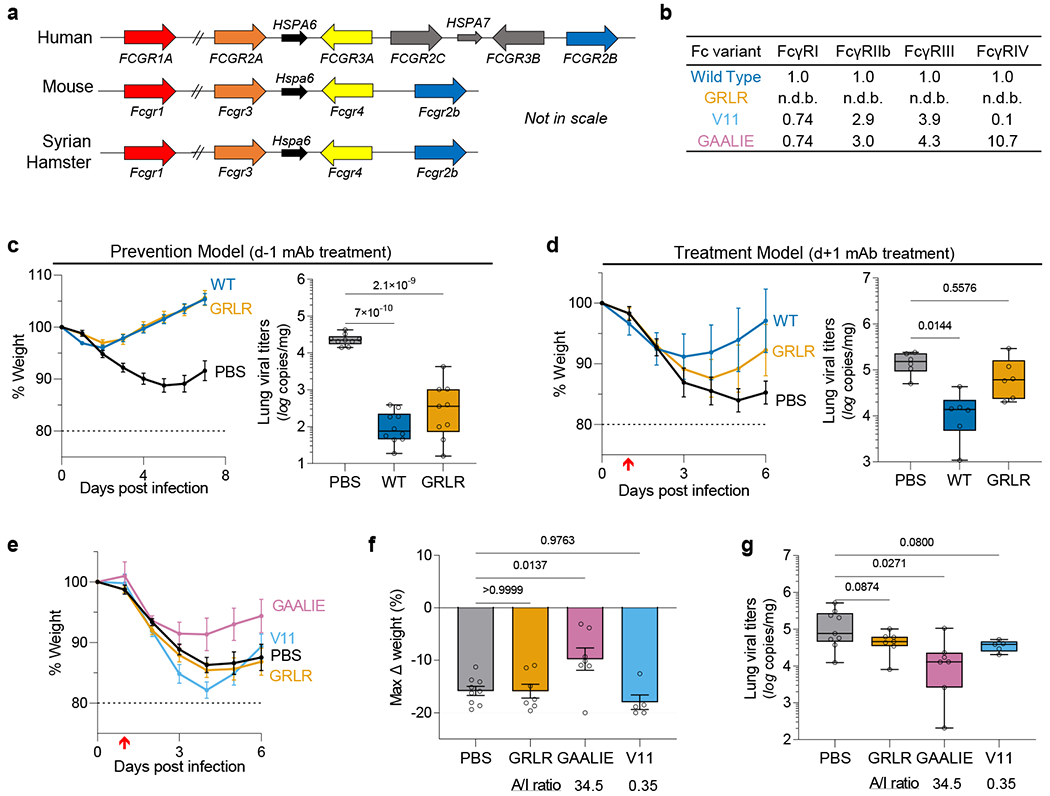
a, Overview of the FcγR locus organization in humans, mice, and Syrian hamsters. b, Fc variants of human IgG1 were evaluated for their affinity for hamster FcγRs. Numbers indicate the fold-change in affinity compared to wild-type human IgG1. n.d.b., no detectable binding. c, d, Wild-type and FcR null (GRLR) variants of REGN mAb cocktail (c) or S309 mAb (d) were administered i.v. (5 mg/kg) to Syrian hamsters one day before (prevention model, c) or after (therapy model, d) i.n. challenge with SARS-CoV-2 (NYC isolate, 105 pfu) (n=9 hamsters per group for PBS and GRLR-treated, n=10 for wild-type from two independent experiments for c and n=6 hamsters per group from two independent experiments for d). Hamsters were monitored for weight loss (left; mean ± s.e.m.) and lung viral titers (right, analyzed on day 7 (c) or 6 (d) post-infection) were compared between treatment groups by one-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons). P values are indicated. e-g, SARS-CoV-2-infected hamsters (105 pfu, NYC isolate) were treated on day 1 post-infection with Fc variants of the REGN mAb cocktail (5 mg/kg, i.v.) exhibiting differential hamster FcγR binding affinity and A/I ratio (calculated based on FcγRIV/FcγRIIb affinity). Weight loss (e, plotted over time (mean ± s.e.m.) or f, as max change) and lung viral titers (g, assessed on day 6 post-infection) were compared by one-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons). P values are indicated. n=9 hamsters per group for PBS-treated, n=7 for GRLR and GAALIE, and n=5 for V11 from two independent experiments. Red arrow indicates time point of mAb treatment post-infection. Boxes and whiskers represent the median, quartiles, and range (minimum to maximum).
To assess the contribution of Fc-FcγR interactions to mAb-mediated protection, we selected neutralizing mAbs in clinical use or development, including casirivimab and imdevimab (REGN cocktail22) and S309/VIR-7831 (Vir23) and expressed them as human IgG1 or as Fc variants with defined affinity for hamster FcγRs. In agreement with recent reports12, we observed that when mAbs are administered prophylactically, Fc effector function has minimal contribution to the mAb antiviral activity (Fig. 1c). By contrast, in the therapy setting, wild-type, but not FcR null (GRLR) variants are able to suppress lung viremia and prevent weight loss (Fig. 1d). Since previous studies in mouse models of influenza and HIV-1 infection support a key role for FcγRIV in the mAb-mediated protection10,24, we compared the in vivo therapeutic activity of two Fc domain variants –GAALIE and V11– that exhibit differential hamster FcγRIV binding activity, but comparable affinity for the other hamster FcγRs (Fig. 1b). Whereas the FcγRIV-enhanced variant (GAALIE) demonstrates potent antiviral activity, no therapeutic activity is evident for V11, which exhibits minimal affinity for hamster FcγRIV (Fig. 1e–g).
SARS-CoV-2 in FcγR humanized mice
Although these findings support the importance of Fc-FcγR interactions in the mAb-mediated protection against SARS-CoV-2 infection, their translational relevance is limited, given the structural complexity of the FcγR family and the divergence of these receptors between humans and other mammalian species21. To address this problem, we have previously developed a mouse strain in which only human FcγRs are expressed in a pattern that recapitulates as faithfully as possible the expression pattern of human tissues25. Despite differences in the absolute FcγR expression levels between humans and FcγR-humanized mice (e.g. the expression of FcγRI on neutrophils and FcγRIIb on monocytes), this strain represents a suitable platform for studies on human IgG antibody function with translational relevance to humans. Human FcγR expression among the various leukocyte populations is stable and does not differ between young and old mice (Extended Data Fig. 2). Infection of old (>15 weeks old), but not young (7 weeks), FcγR humanized mice with the mouse-adapted SARS-CoV-2 strain MA107 results in challenge dose-dependent weight loss and mortality (Fig. 2a–b). Recent studies determined that on day 2 post-infection, SARS-CoV-2MA10-infected mice exhibit peak lung viral titers, accompanied by severely compromised pulmonary function and extensive lung histopathologic damage7. Histological evaluation of lungs from SARS-CoV-2MA10-infected FcγR humanized mice (>15 weeks old) revealed multifocal areas of interstitial pneumonia, extensive inflammatory cell infiltration, as well as occasional necrotic and proteinaceous material and hyalinized membranes within affected alveoli (Extended Data Fig. 3). Such histopathological findings are consistent with those observed in well-established animal models of SARS-CoV-2 infection used extensively in previous studies and resemble the lung pathology of human COVID-197,12–14. Although it is not classified as a variant of concern (VOC), the SARS-CoV-2MA10 strain contains several mutations near the epitopes targeted by mAbs currently in clinical use or development. As a result, several of these mAbs exhibit significantly reduced neutralizing activity against SARS-CoV-2MA10, which is comparable to that observed for VOCs, like B1.351 (Extended Data Fig. 4).
Fig. 2: Fc-FcγR interactions are required for the therapeutic activity of neutralizing anti-SARS-CoV-2 mAbs in mouse infection models.
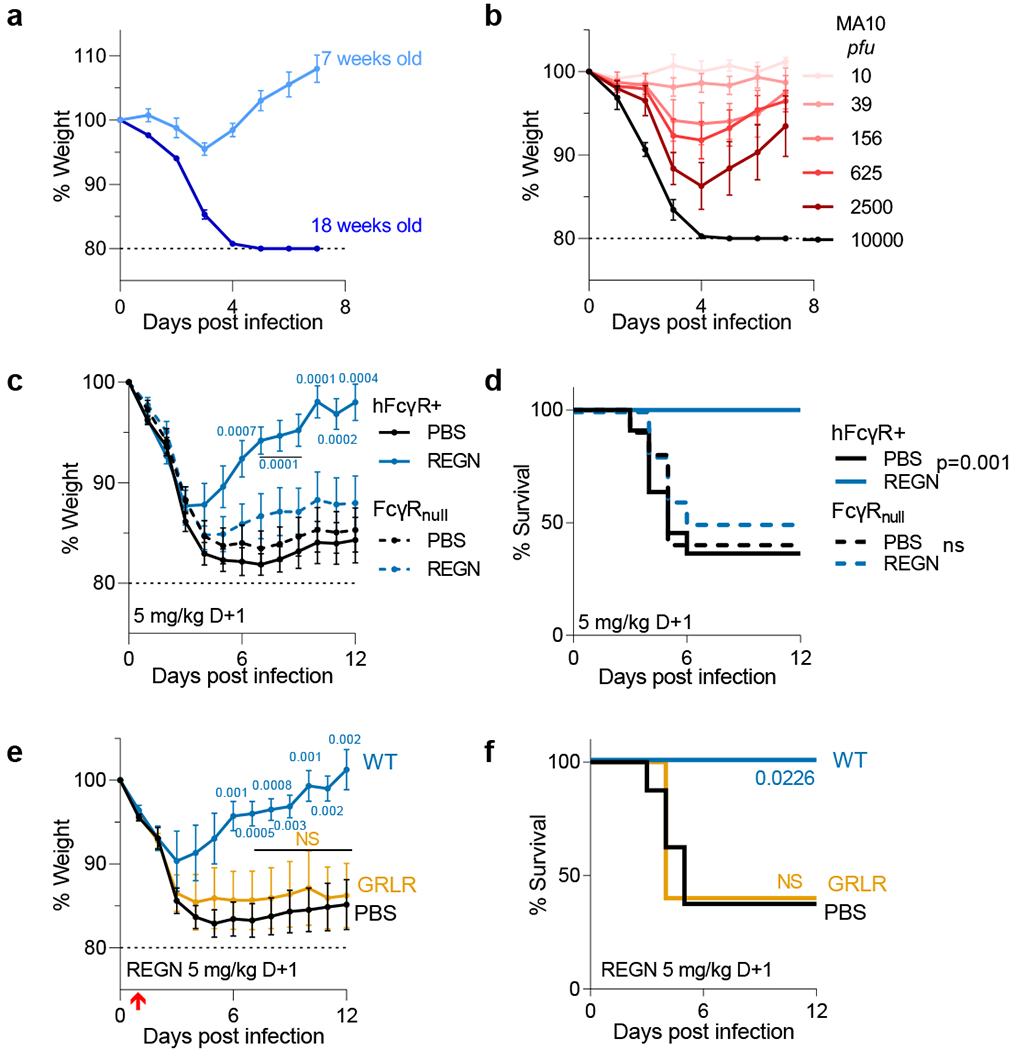
a, b, FcγR humanized mice were infected with mouse-adapted SARS-CoV-2 (MA10 strain, 104 pfu, i.n.) and weight loss (mean ± s.e.m.) was compared in (a) young (7 weeks old; n=5) and older (18 weeks old; n=4) mice, as well as in (b) mice (16–19 weeks old) challenged with the indicated inoculum dose. n=5 mice per group; n=4 for 104 and 10 pfu dose groups from two independent experiments. c, d, The therapeutic activity of REGN mAb cocktail (expressed as human IgG1 and administered at 5 mg/kg one day post-infection) was evaluated in FcγR humanized and FcγR deficient (FcγRnull) mouse strains challenged with SARS-CoV-2 (MA10 strain, 104 pfu i.n.). n= total of 10 mice per group for FcγRnull and n= total of 11 (PBS) and n=12 (REGN) mice per group for FcγR humanized mice from two independent experiments. e, f, SARS-CoV-2-infected FcγR humanized mice (MA10 strain, 104 pfu i.n.) were treated with wild-type human IgG1 or GRLR variants of REGN mAb cocktail one day post-infection. n= total of 8 (PBS), 5 (WT), and 6 (GRLR) mice per group from two independent experiments. Weight loss (c, e; mean ± s.e.m.) and survival curves (d, f) were compared to the corresponding PBS-treated group by two-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons) and log-rank (Mantel–Cox) test, respectively. P values are indicated. NS, not significant. Red arrow indicates time point of mAb treatment post-infection.
IgG Fc effector function in mAb therapy
In a model of mAb-mediated therapy, we observed that the REGN mAb cocktail (expressed as wild-type human IgG1) confers full protection of FcγR humanized mice when administered at 5 mg/kg one day after lethal challenge with SARS-CoV-2MA10 (Fig. 2c–f). By contrast, no therapeutic activity is evident in mice lacking FcγRs (FcγRnull) or when mAbs are expressed as variants (GRLR) with minimal affinity for human FcγRs, highlighting the importance of Fc effector function in the therapeutic activity of neutralizing mAbs (Fig. 2c–f). To determine the mechanisms by which human FcγRs contribute to the mAb-mediated protection, REGN cocktail mAbs were expressed as human IgG1 Fc variants11 and exhibit differential affinities for the various human FcγRs (Fig. 3a). As outlined in Figure 3b, we assessed the therapeutic activity of Fc variants of the REGN mAb cocktail at a dose (1 mg/kg) which wild-type human IgG1 confers minimal protection (Fig. 3c). Consistent with a protective role for activating FcγRs, Fc variants enhanced for either FcγRIIa (GA) or FcγRI and FcγRIII (ALIE) show a trend for improved therapeutic potency over wild-type IgG1, whereas maximal therapeutic activity was evident only for the GAALIE variant, which is enhanced for both FcγRIIa and FcγRIII and has reduced affinity for the inhibitory FcγRIIb (Fig. 3d–e). These findings suggest that synergy between the two activating FcγRs, FcγRIIa and FcγRIII likely accounts for the therapeutic activity of the GAALIE variant, which achieves the same degree of protection as wild-type IgG1, but at a 5-times lower dose. Additionally, the observed differences in the in vivo therapeutic activity among Fc variants could not be attributed to differences in their in vitro neutralization activity, antigen binding specificity, or in vivo half-life (Extended Data Fig. 5–6).
Fig. 3: Selective engagement of activating FcγRs improves the therapeutic activity of anti-SARS-CoV-2 mAbs.
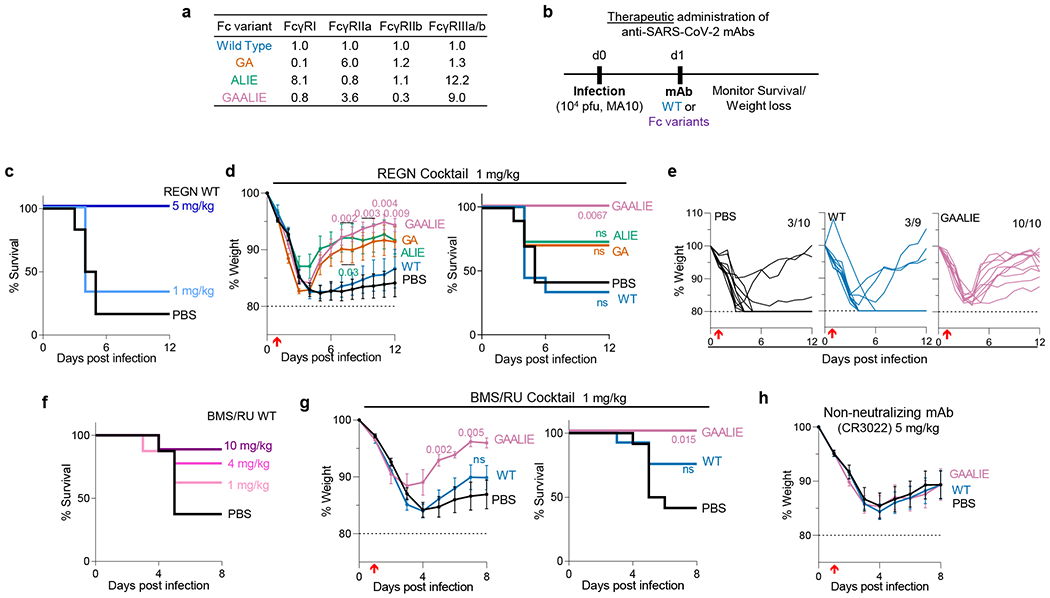
a, Human IgG1 Fc variants with differential affinity for specific classes of human FcγRs were generated for anti-SARS-CoV-2 mAbs. Numbers indicate the fold-change in affinity compared to wild-type human IgG1. b-g, Following the experimental strategy in panel b, SARS-CoV-2-infected FcγR humanized mice were treated (i.v.) at the indicated dose with REGN (c-e) or BMS/RU (f-g) mAb cocktail, or the non-neutralizing mAb CR3022 (h), expressed as wild-type human IgG1 or as Fc variants with differential affinity for human FcγRs. Weight loss (mean ± s.e.m.) (h and d, g, left panels; e, curves from individual mice) and survival curves (c, f and d, g, right panels) of antibody-treated mice were compared with the corresponding PBS-treated group by two-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons) and log-rank (Mantel–Cox) test, respectively. P values are indicated. NS, not significant. c, n=6 mice per group from two independent experiments; d, e, n= total of 10 mice per group (for PBS, GAALIE, GA groups), n=9 for WT, and n=11 for ALIE from four independent experiments; f, n= total of 8 (PBS and 1 mg/kg) or 9 (10 mg/kg and 4 mg/kg groups) mice per group from two independent experiments; g, n= total of 7 (GAALIE) or 12 (PBS, WT) mice per group from three independent experiments. h, n= total of 8 (GAALIE) or 10 (PBS, WT) mice per group from two independent experiments. Red arrow indicates time point of mAb treatment post-infection.
Importantly, treatment of SARS-CoV-2MA10-infected mice one day post-infection with the GAALIE variant was not associated with enhanced disease not only when administered at a low dose (1 mg/kg), but also at a much higher dose (40 mg/kg) (Extended Data Fig. 7a). At this dose, which is typically used in the clinical setting, FcR null variants (GRLR) also exhibit full protective activity comparable to the GAALIE variants, suggesting that Fc-independent protection could be achieved once neutralizing mAbs are administered at sufficiently high doses, as has been documented previously for other viral pathogens24. Likewise, treatment of SARS-CoV-2-infected mice two days post-infection, which coincides with the peak of lung viral replication7, has no disease-enhancing effects and neither GAALIE nor GRLR variants offer any therapeutic benefit, confirming recent reports that defined that the therapeutic window for neutralizing mAbs is limited to 24 hours post-infection26 (Extended Data Fig. 7b).
Similar results were obtained when we assessed the in vivo therapeutic activity of another neutralizing anti-SARS-CoV-2 mAb cocktail (C135+C144; BMS/RU27) currently in clinical development27. When BMS-RU mAbs are administered to FcγR humanized mice at 1 mg/kg, only GAALIE variants, but not wild-type human IgG1 confer protective activity and prevent disease-induced weight loss (Fig. 3f–g). In contrast to neutralizing anti-SARS-CoV-2 mAbs, Fc-engineering of the non-neutralizing anti-RBD mAb, CR3022, does not result in improved therapeutic activity, as both CR3022-GAALIE and CR3022-WT failed to protect mice from lethal SARS-CoV-2 challenge (Fig. 3h).
Fc domain activity in mAb prophylaxis
Our findings in hamsters suggest that when neutralizing mAbs are administered prophylactically, Fc-FcγR interactions are not critical for their antiviral activity (Fig. 1c). However, given the substantial interspecies differences in FcγR biology between hamsters and humans, we assessed the contribution of activating FcγR engagement to the mAb-mediated prophylaxis of SARS-CoV-2MA10-challenged FcγR humanized mice (Fig. 4a). When administered at a dose where wild-type human IgG1 exhibits no protective activity (0.5 mg/kg) (Fig. 4b), GAALIE variants of the REGN mAb confer full protection against lethal SARS-CoV-2 challenge, suggesting that selective activating FcγR engagement could improve the efficacy of neutralizing mAbs both at the therapeutic as well as at the prophylactic setting (Fig. 4c).
Fig. 4: Prophylactic activity of anti-SARS-CoV-2 mAbs is enhanced by selective engagement of activating FcγRs.
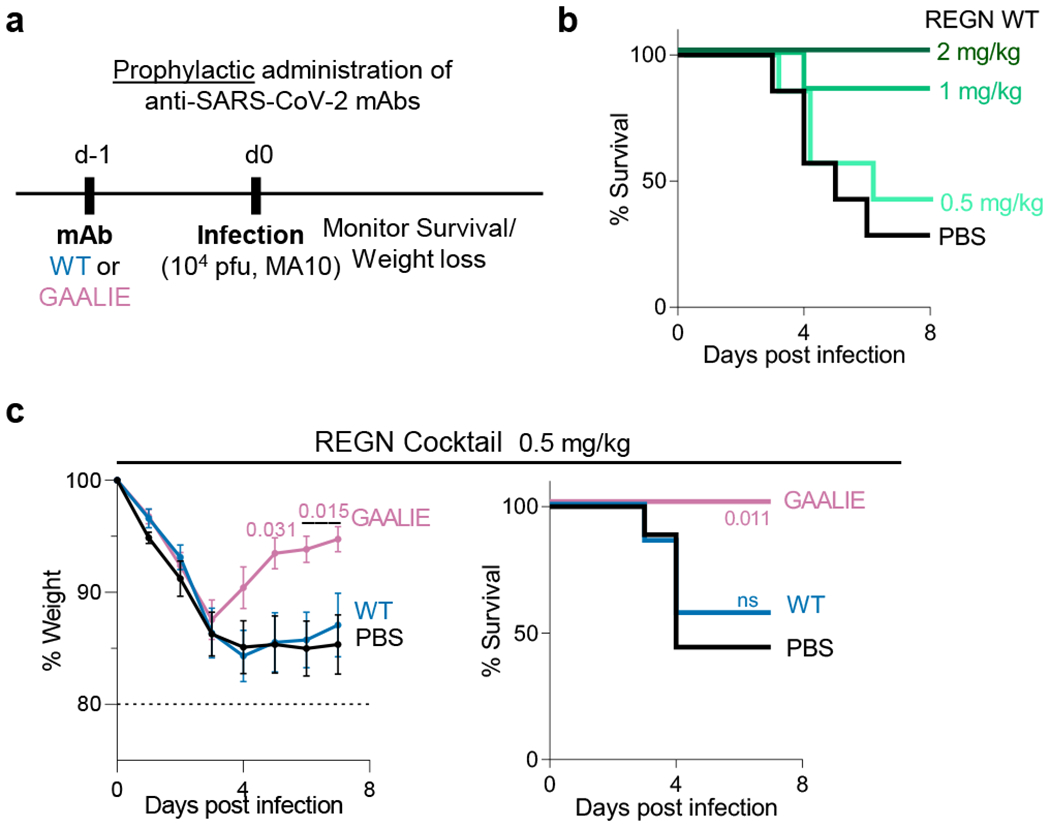
In a model of mAb-mediated prophylaxis of SARS-CoV-2 infection (a), the activity of wild-type and GAALIE variants of the REGN mAb cocktail was assessed. FcγR humanized mice were treated (i.v.) at the indicated dose with REGN mAb cocktail expressed as wild-type human IgG1 or as GAALIE variant one day prior to challenge with SARS-CoV-2 (MA10, 104 pfu i.n.). Weight loss (mean ± s.e.m.) (c, left panel) and survival curves (b and c, right panel) of antibody-treated mice were compared with the PBS-treated group by two-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons) and log-rank (Mantel–Cox) test, respectively. P values are indicated. NS, not significant. b, n= total of 7 mice per group (n=6 mice/group for 2 mg/kg) from two independent experiments; c, n= total of 7 (WT) or 9 (PBS, GAALIE) mice per group from three independent experiments.
Discussion and concluding remarks
To maximize the translational relevance of our findings, the present study focused on neutralizing anti-SARS-CoV-2 mAbs that are currently in clinical use or development and assessed their in vivo protective activity in mouse strains that recapitulate the unique complexity of human FcγRs25. Challenge of FcγR humanized mice with SARS-CoV-2MA10 results in accelerated weight loss and significant mortality, resembling the clinical manifestations of severe COVID-19. However, such rapid symptomatic disease development limits the therapeutic window for mAb treatment and does not allow to fully unravel the mechanisms of COVID-19 disease pathogenesis and the potential immunopathologies due to host immune-related responses. Despite such limitation, in the selected experimental setting, we observed that in models of mAb-mediated prophylaxis or early treatment, Fc engineering for selective binding to activating FcγRs results in approximately 5-fold reduction in the mAb dose required to achieve full therapeutic benefit; a finding consistent with our recent in vivo evaluation studies on Fc-optimized anti-influenza virus mAbs11. Consistent with previous studies on other viral pathogens, including HIV-128, influenza24, and Ebola29, our findings suggest that FcγR, but not complement pathways confer mAb-mediated protection, as mAbs lack protective activity in mice deficient for all classes of FcγRs (Fig. 2c–d). Moreover, Fc variants like GAALIE, which exhibit diminished C1q binding activity (Extended Data Fig. 8), demonstrate improved therapeutic efficacy compared to wild-type IgG1.
Analysis of IgG Fc glycan characteristics in hospitalized COVID-19 patients has previously revealed an association of disease severity with elevated levels of afucosylated glycoforms30,31, which exhibit increased affinity for the activating FcγRIII11. Although these results suggest a potential pathogenic effect for FcγRIII pathways, severe COVID-19 patients are also characterized by delayed induction of neutralizing antibody responses yet elevated antibody levels32, suggesting that any disease-enhancing effects of afucosylated antibodies might also be related to their poor neutralizing activity33. However, in our study, we failed to observe any pathogenic or disease-enhancing effects for anti-SARS-CoV-2 mAbs engineered for enhanced binding to activating FcγRs, irrespective of their neutralizing potency (Fig. 3). It is therefore likely that susceptibility to severe disease is not entirely driven by the FcγRIII binding activity of anti-SARS-CoV-2 IgGs, but rather by multiple, complex immunological determinants.
Despite the successful deployment of highly effective vaccines, neutralizing anti-SARS-CoV-2 mAbs are expected to continue to play an important role in virus containment efforts, as well as in the clinical management of COVID-19, especially in high-risk populations and immunocompromised individuals. Although regulatory approval of anti-SARS-CoV-2 mAb cocktails has been based on their remarkable clinical efficacy in mild to moderate COVID-19 patients1–3, none of the currently approved mAbs take full advantage of the potential of an IgG molecule to mediate protective antiviral Fc effector functions. Our findings provide a paradigm for the development of mAb-based therapeutics with improved potency and superior therapeutic efficacy against COVID-19 through selective engagement of specific FcγR pathways.
Methods
Viruses, Cell Lines, and Animals
A P1 stock of the SARS-CoV-2 MA10 strain7 was amplified in VeroE6 cells obtained from the ATCC that were engineered to stably express TMPRSS2 (VeroE6TMPRSS2). To generate a P2 working stock, VeroE6TMPRSS2 cells were infected at a multiplicity of infection (MOI) of 0.1 plaque forming units (pfu)/cell and incubated at 37°C for 4 days. The virus-containing supernatant was subsequently harvested, clarified by centrifugation (3,000g; 10 min), and filtered using a disposable vacuum filter system with a 0.22 μm membrane. Virus stock titers were measured by standard plaque assay on Huh-7.5 cells that stably express ACE2 and TMPRSS2 (Huh-7.5ACE2/TMPRSS2). Briefly, 500 μl of serial 10-fold virus dilutions in Opti-MEM were used to infect 4×105 cells seeded the day prior into wells of a 6-well plate. After 90 min adsorption, the virus inoculum was removed, and cells were overlayed with DMEM containing 10% FBS with 1.2% microcrystalline cellulose (Avicel). Cells were incubated for four days at 33°C, followed by fixation with 7% formaldehyde and crystal violet staining for plaque enumeration. To confirm virus identity and evaluate for unwanted mutations that were acquired during the amplification process, RNA from virus stocks was purified using TRIzol Reagent (ThermoFisher). Briefly, 200 μl of each virus stock was added to 800 μl TRIzol Reagent, followed by 200 μl chloroform, which was then centrifuged at 12,000g for 5 min. The upper aqueous phase was moved to a new tube, mixed with an equal volume of isopropanol, and then added to an RNeasy Mini Kit column (Qiagen) to be further purified following the manufacturer’s instructions. Viral stocks were subsequently confirmed via next generation sequencing using libraries for Illumina MiSeq.
The SARS-CoV-2 NYC isolate was obtained from a patient’s saliva generously provided by Agnès Viale (Memorial Sloan Kettering Cancer Center) and amplified in Caco-2 cells. This P1 virus was used to generate a P2 working stock by infecting Caco-2 cells at a MOI of 0.05 pfu/cell. Cells were incubated at 37°C for 6 days before harvesting virus-containing supernatant as described above. Similarly, virus stock titers were determined by plaque assay performed on Huh-7.5ACE2/TMPRSS2 cells. All SARS-CoV-2 experiments were carried out in biosafety level 3 (BSL-3) containment in compliance with institutional and federal guidelines.
VeroE6 cells (ATCC, CRL-1586), Caco-2 cells (ATCC, HTB-37), and Huh-7.5 hepatoma cells (described in 34) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 1% nonessential amino acids (NEAA) and 10% fetal bovine serum (FBS). 293T cells (ATCC, CRL-3216) and HT1080ACE2 (kindly provided by Dr. Paul Bieniasz, Rockefeller University) were maintained in DMEM supplemented with 10% FBS, 50 U/ml penicillin and 50 μg/ml streptomycin (ThermoFisher). All cell lines were maintained at 37°C at 5% CO2. Expi293F cells (ThermoFisher) were maintained at 37°C, 8% CO2 in Expi293 expression medium (ThermoFisher) supplemented with 10 U/ml penicillin and 10 μg/ml streptomycin. Cell lines were not authenticated after purchase. All cell lines have been tested negative for mycoplasma contamination.
In vivo experiments were approved by the Rockefeller University Institutional Animal Care and Use Committee in compliance with federal laws and institutional guidelines. Hamsters and mice were maintained at the Comparative Bioscience Center at the Rockefeller University at a controlled ambient temperature (20–25°C) and humidity (30–70%) environment with 12-h dark/light cycle. Golden Syrian hamsters were purchased from Charles River laboratories (strain code 049) and maintained in compliance with USDA regulations. FcγR knockout (mFcγRα−/−; Fcgr1−/−) and FcγR humanized mice (mFcγRα−/−, Fcgr1−/−, hFcγRI+, hFcγRIIaR131+, hFcγRIIb+, hFcγRIIIaF158+, and hFcγRIIIb+) were generated in the C57BL/6 background and characterized in previous studies11,25.
Cloning, Expression, and Purification of Recombinant Proteins
Human IgG1 Fc domain variants were generated by site-directed mutagenesis using specific primers as previously described9 and recombinant IgG antibodies were expressed and purified using previously described protocols11. Purity was assessed by SDS–PAGE followed by SafeStain blue staining (ThermoFisher), as well as by size exclusion chromatography (SEC) using a Superdex 200 Increase 10/300GL column (GE Healthcare) on an Äkta Pure 25 HPLC system (data analyzed using Unicorn v.6.3 software). All antibody preparations were more than 94% pure and endotoxin levels were less than 0.05 EU/mg, as measured by the limulus amebocyte lysate assay.
The two plasmid-based HIV/NanoLuc-SARS-CoV-2 pseudovirus system was kindly provided by Dr. Paul Bieniasz (described in 35). The S gene was modified by site-directed mutagenesis to introduce the amino acid changes present in the MA10 or the B.1.351 strains7. SARS-CoV-2WT, SARS-CoV-2MA10 and SARS-CoV-2B.1.351 pseudovirus particles were generated by transfection of the two plasmid-based system to 293T cells using X-tremeGENE HP DNA transfection reagent (Sigma).
Golden Syrian hamster FcγR cDNA sequences were identified based on the current Mesocricetus auratus genome assembly (MesAur1.0) and recent transcriptomic data36. To validate sequences, hamster FcγR sequences were amplified and sequenced (Genewiz) from Syrian hamster spleen cDNA (Zyagen) using the following primers: hamster FcγRI (5’-GGC GGA CAA GTG GTA AAT GTC AC-3’, 5’-CGG ACA CAT CAT TGC TTC AGA CTT ACT AAG-3’), hamster FcγRII (5’-CTG CTG GGA CAC ATG ATC TCC-3’, 5’-TTA AAT GTG GTT CTG GTA ATC ATG CTC TG-3’), hamster FcγRIII (5’-GAG TCT GGA GAC ACA GAT GTT TCA G-3’, 5’-CGA CGT CAT TTG TCC CGA GG-3’), hamster FcγRIV (5’-AAT GGG TGA GGG TGC TTG AG-3’, 5’-GAG GGA ATG TTG GGG ACA GG-3’). To identify the ectodomain, transmembrane, and cytoplasmic domains, Syrian hamster FcγR protein sequences were then aligned against annotated mouse FcγR protein sequences (Uniprot). Soluble Syrian hamster FcγR ectodomains were generated by transient transfection as described above for mAbs. Syrian hamster FcγR expression plasmids were generated encoding a secretion signal peptide, the predicted Syrian hamster FcγR ectodomain, and a C-terminal Strep or His tag. Following transfection of Expi293 cells, recombinant FcγRs were purified from cell-free supernatants by affinity purification using Strep-Tactin Superflow Plus resin (Qiagen) or Ni-NTA Agarose (Qiagen), per manufacturer’s instructions. Purified proteins were dialyzed into PBS and assessed for purity by SDS-PAGE gel electrophoresis followed by SafeStain blue staining. Monomeric FcγR ectodomains were fractionated from aggregates by size exclusion chromatography on an Äkta Pure system using a Superdex 200 Increase 10/300 GL column (GE Healthcare).
Surface Plasmon Resonance
All experiments were performed with a Biacore T200 SPR system (GE Healthcare) at 25 °C in HBS-EP+ buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3.4 mM EDTA, 0.005% (v/v) surfactant P20). IgG antibodies were immobilized on Series S Protein G sensor chip (GE Healthcare) at a density of 2,000 response units (RU). Serial dilutions of recombinant soluble hamster FcγR ectodomains were injected to the flow cells at 20 μl/min, with the concentration ranging from 1000 to 15.625 nM (1:2 successive dilutions). Association time was 60 s followed by a 900-s dissociation step. At the end of each cycle, sensor surface was regenerated with a glycine HCl buffer (10 mM, pH 2.0; 50 μl/min, 30 s). Background binding to blank immobilized flow cells was subtracted, and affinity constants were calculated using BIAcore T200 evaluation software v.2.0 (GE Healthcare) using the 1:1 Langmuir binding model.
Neutralization Assay
Neutralization activity of IgG1 Fc domain variants was measured as previously described35. Briefly, HT1080ACE2 cells were seeded in 96 U-well black plates 24 h prior to infection with SARS-CoV-2WT, SARS-CoV-2MA10 or SARS-CoV-2B.1.351 pseudoviruses. Pseudovirus particles were pre-incubated with mAbs (four-fold serially diluted starting at 10 μg/ml) for 1 h at 37°C and then added to a monolayer of HT1080ACE2 cells. Following a 48-h incubation at 37°C, cells were carefully washed with PBS and lysed with Luciferase Cell Culture Lysis reagent (Promega) for 15 min. Nano luciferase activity was detected by adding Nano-Glo Luciferase Assay System (Promega) and measured by SpectraMax Plus spectrophotometer (Molecular Devices), using 0.5 s integration time. Data were collected and analyzed using SoftMax Pro v.7.0.2 software (Molecular Devices). Relative luciferase units were normalized to those derived from cells infected with the relevant SARS-CoV-2 pseudoviruses in the absence of mAbs.
Anti-SARS-CoV-2 RBD ELISA
Recombinant SARS-CoV-2 RBD was immobilized (1 μg/ml) into high-binding 96-well microtitre plates (Nunc) and after overnight incubation at 4 °C, plates were blocked with PBS plus 2% (w/v) BSA for 2 h. After blocking, plates were incubated for 1 h with serially diluted IgG antibodies or serum samples (1:3 consecutive dilutions in PBS starting at 100 ng/ml), followed by HRP-conjugated goat anti-human IgG (1 h; 1:5,000; Jackson Immunoresearch). Plates were developed using the TMB two-component peroxidase substrate kit (KPL) and reactions were stopped with the addition of 1 M phosphoric acid. Absorbance at 450 nm was immediately recorded using a SpectraMax Plus spectrophotometer (Molecular Devices) and background absorbance from negative control samples was subtracted. Data were collected and analyzed using SoftMax Pro v.7.0.2 software (Molecular Devices).
C1q Binding Assay
Human and mouse C1q binding to IgG1 Fc variants was assessed following previously described protocols37. Briefly, antibodies were serially diluted (100–0.046 μg/ml) in PBS and coated overnight (4 °C) onto high-binding 96-well microtiter plates. After washing with PBS+0.05% Tween-20, plates were blocked with 2% BSA. Normal mouse or human serum (3%) was added and incubated for 60 min with gentle shaking. For the detection of C1q binding, monoclonal mouse anti-C1q antibody (JL-1, Abcam) was added at 0.5 μg/ml, HPR-conjugated goat anti-mouse IgG was used at a dilution of 1:5000, and plates were developed and analyzed as described above.
Quantification of Serum IgG Levels
Blood was collected into microvette serum gel tubes (Sarstedt) and serum was fractionated by centrifugation (10,000g, 5 min). IgG levels were determined by ELISA following previously described protocols11.
In vivo SARS-CoV-2 Infection Models
All animal infection experiments were performed at the Comparative Bioscience Center of the Rockefeller University in animal biosafety level 3 (ABSL-3) containment in compliance with institutional and federal guidelines. Hamsters (males; 6-8 weeks old) were anaesthetized with isoflurane (3%) in a VetFlo high-flow vaporizer followed by an intraperitoneal injection of a ketamine (150 mg/kg) and xylazine (10 mg/kg) mixture. Hamsters were challenged intranasally with 105 pfu SARS-CoV-2 (NYC isolate, 100 μl viral inoculum). Mice (males or females; 6-7 or 16-22 weeks old) were anaesthetized with a ketamine (75 mg/kg) and xylazine (15 mg/kg) mixture (administered intraperitoneally) prior to challenge with SARS-CoV-2 (MA10 strain, 104 pfu in 30 μl, intranasally).
No statistical method was used to predetermine sample size. Based on preliminary studies that determined experimental variation in survival following infection and mAb treatment, we have performed power calculations and determined that at least n=6 animals/group is sufficient to detect differences among experimental groups (powered at 80% for 5% significance level; survival assessed by log-rank (Mantel-Cox) test).
After infection, animals were monitored daily and humanely euthanized by CO2 asphyxiation at endpoints authorized by the Rockefeller University Institutional Animal Care and Use Committee, including any of the following: marked lethargy or inactivity, severe respiratory distress or labored breathing, inability to ambulate, and weight loss of greater than 20% of baseline. Animals were randomized based on age, gender, and weight. Before treatment, we ensured that the mean weight, gender, and age were comparable among the various treatment groups. The treatment groups were not blinded to the personnel involved in monitoring animal survival and weight upon challenge. For antibody-mediated prophylaxis, antibodies were administered intravenously one day before virus challenge, whereas for antibody-mediated therapy, antibodies were administered one day after infection. Antibody dose was calculated as mg/kg.
Histological Analysis
Lungs from euthanized mice were instilled with 10% neutral buffered formalin and fixed overnight by submersion in 10% formalin. Fixed tissues were embedded in paraffin, sectioned at 4 μm thickness, and stained with hematoxylin and eosin. Sections of lung were microscopically evaluated by a board-certified veterinary anatomic pathologist and representative images were captured with an Olympus BX45 light microscope using an SC30 camera with the cellSens Dimension software.
Determination of Lung Viral Titers
Hamsters were euthanized at the indicated timepoints following infection and lung weights were recorded. Lungs were lysed in Trizol (ThermoFisher) and dissociated in gentle MACS M tubes using the gentleMACS Octo Dissociator (Miltenyi Biotec). Samples were transferred into Phasemaker tubes (ThermoFisher) and chloroform was added (200 μl chloroform/1 ml TRIzol). After vigorous shake, tubes were rested for 5 min and then centrifuged for 15 min at 12,000g at 4°C. The aqueous phase containing the RNA was transferred into a new tube and RNA extraction was performed by using RNeasy mini kit (Qiagen). SARS-CoV-2 lung titers were determined by qRT-PCR assay using TaqMan Fast Virus 1-Step Master Mix and specifically designed primers and a TaqMan probe that bind a conserved region in the nucleocapsid gene of SARS-CoV-2 (2019-nCoV_N1-F :5’-GAC CCC AAA ATC AGC GAA AT-3’. 2019-nCoV_N1-R: 5’-TCT GGT TAC TGC CAG TTG AAT CTG-3’. 2019-nCoV_N1-P: 5’-FAM-ACC CCG CAT TAC GTT TGG TGG ACC-BHQ1-BHQ1–3’). qPCR was performed using an Applied Biosystems QuantStudio 6 Flex cycler using the following parameters: 50°C for 5 min, 95°C for 20 s followed by 40 cycles of 95°C for 3 s, and 30 s at 60°C. Signal from unknown samples was compared to a known standard curve (obtained through BEI Resources, NIAID, NR-52358) and viral titers were expressed as RNA copies/mg tissue.
Flow Cytometry
After lysis of red blood cells (RBC lysis buffer; Biolegend), cells were resuspended in PBS containing 0.5% (w/v) BSA and 5 mM EDTA and labelled with the following fluorescently labelled antibodies (all used at 1:200 dilution unless otherwise stated): anti-human FcγRIIa (clone IV.3)-FITC, anti-human FcγRIIb (clone 2B6)-Dylight650 (used at 5 μg/ml), anti-B220-AlexaFluor700, anti-Gr1-BrilliantViolet421, anti-CD8β-BrilliantViolet510, anti-human FcγRI (clone 10.1)-BrilliantViolet605, anti-CD3-BrilliantViolet650 (used at 1:100 dilution), anti-CD11b-BrilliantViolet711, anti-CD4-BrilliantViolet785, anti-human FcγRIIIa/b (clone 3G8)-PE, and anti-NK1.1-PE/Cy7. Relevant isotype control antibodies were used and included: mouse IgG1 isotype control-Dylight650 (used at 5 μg/ml), mouse IgG2b kappa isotype control-FITC, mouse IgG1 kappa isotype control-PE, mouse IgG1 kappa isotype control-BrilliantViolet605. Samples that were stained with isotype control antibodies were also blocked with unlabeled anti-FcγR antibodies as follow: anti-human FcγRI (clone 10.1), anti-human FcγRIIa (clone IV.3), anti-human FcγRIIb (clone 2B6), and anti-human FcγRIIIa/b (clone 3G8) (used at 10 μg/ml and incubated for 5 min prior to staining with fluorescently labelled antibodies). Samples were analyzed on an Attune NxT flow cytometer (ThermoFisher) using Attune NxT software v3.1.2 and data were analyzed using FlowJo (v10.7) software.
Statistical Analysis
Raw data for all main and extended data figures are included in the manuscript as source files. Results from multiple experiments are presented as mean ± s.e.m. One- or two-way ANOVA was used to test for differences in the mean values of quantitative variables, and where statistically significant effects were found, post hoc analysis using Bonferroni (adjusted for multiple comparisons) test was performed. Statistical differences between survival rates were analyzed by comparing Kaplan–Meier curves using the log-rank (Mantel–Cox) test. Data were collected and analyzed with Microsoft Excel and GraphPad Prism v.9.1 software (GraphPad) and p < 0.05 were considered to be statistically significant.
Extended Data
Extended Data Fig. 1: Cloning and characterization of the IgG binding activity of hamster FcγRs.
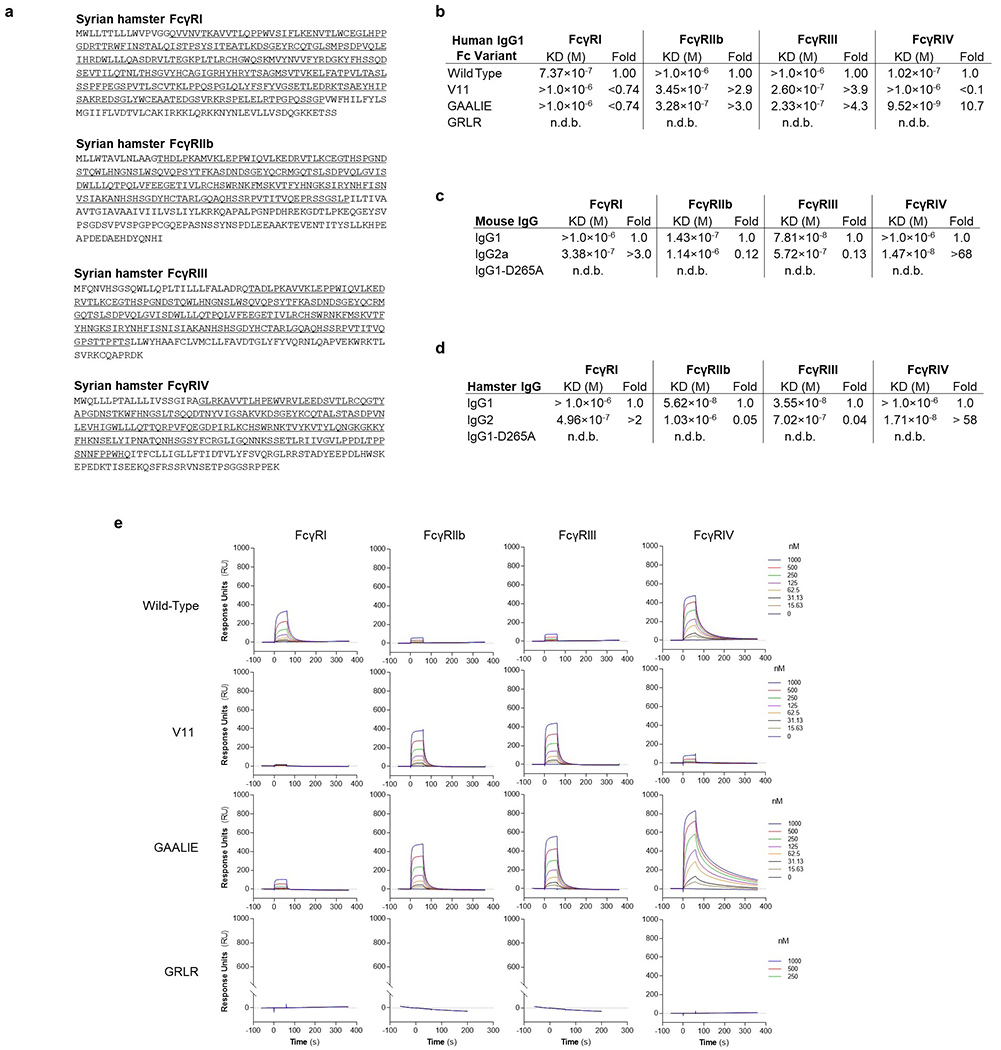
a, Syrian hamster FcγRs were cloned, and their sequences were determined. The FcγR ectodomains are underlined. b-e, The affinity of human IgG1 and Fc variants (b, e, SPR sensorgrams), as well as of mouse (c) and hamster (d) IgG subclass variants for the various classes of hamster FcγRs was determined by surface plasmon resonance (SPR), using soluble hamster FcγR ectodomains. n.d.b., no detectable binding.
Extended Data Fig. 2: Comparison of the FcγR expression levels in the various effector leukocyte populations between young and older FcγR humanized mice.
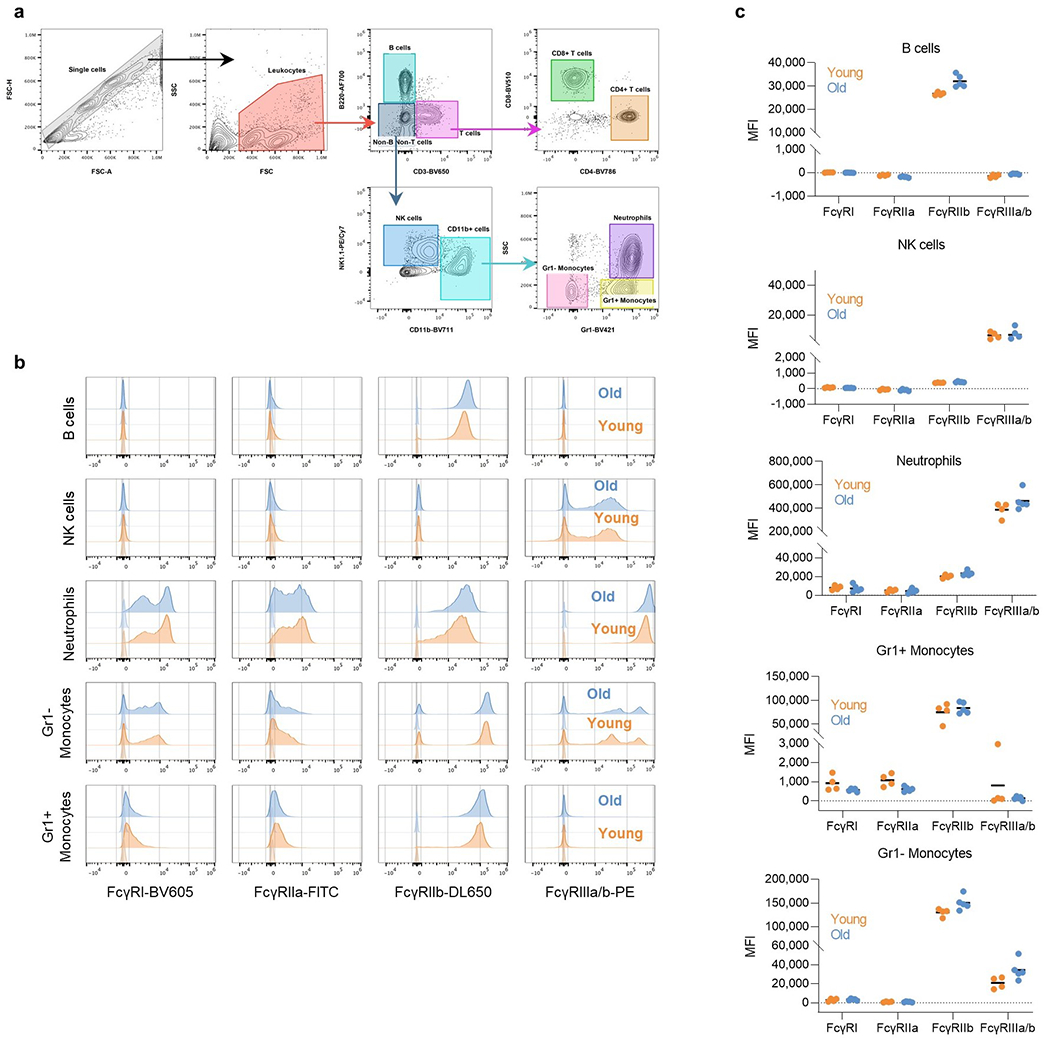
FcγR expression was assessed by flow cytometry in peripheral blood leukocyte populations from young (6-7 weeks old; orange) and older (17 weeks old; blue) FcγR humanized mice. a, Gating strategy to identify the various leukocyte populations, b, Representative histogram overlay plots of FcγR expression in young and older FcγR humanized mice. Corresponding isotype controls are indicated in lighter shade. c, Quantitation of FcγR expression (MFI, median fluorescence intensity subtracted from the respective isotype control) in various leukocyte populations. Results are from 4 or 5 mice per group for young and older mice, respectively.
Extended Data Fig. 3: Histopathological analysis of lung tissue from SARS-CoV-2-infected FcγR humanized mice.
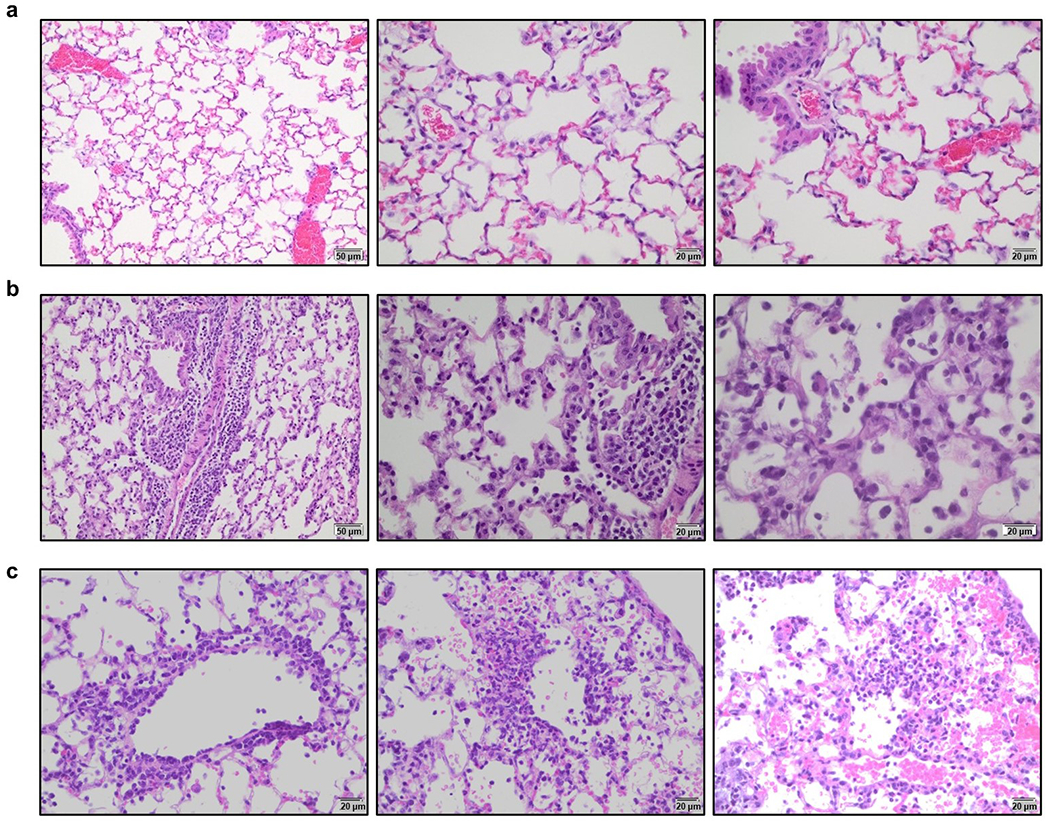
Lungs from SARS-CoV-2-infected (MA10 strain, 104 pfu, i.n.) FcγR humanized mice (16-22 weeks old) were harvested on day 4 post-infection and evaluated histologically to assess the pathological changes associated with SARS-CoV-2 infection. a, Uninfected mice were characterized by clear alveolar spaces and absence of inflammatory cell infiltrates (low magnification (left panel, 200x); high magnification (center and right panels, 400x). b, In contrast, SARS-CoV-2 infection was associated with perivascular and peribronchial mononuclear leukocyte infiltration (200x left; 400x center panel), as well as the presence of macrophages and neutrophils in alveoli and necrotic cellular debris in alveolar spaces (600x, right panel). c, In addition, SARS-CoV-2-infected mice exhibited perivenular mixed neutrophilic, histiocytic, and lymphocytic inflammation, reactive endothelium and extravasation of leukocytes (left panel, 400x), as well as foci of interstitial neutrophilic and macrophage inflammation with hemorrhage and single cell necrosis (center and right panels, 400x). Images are representative of one uninfected and six infected mice.
Extended Data Fig. 4: In vitro neutralization activity of anti-SARS-CoV-2 mAbs against SARS-CoV-2 strains.
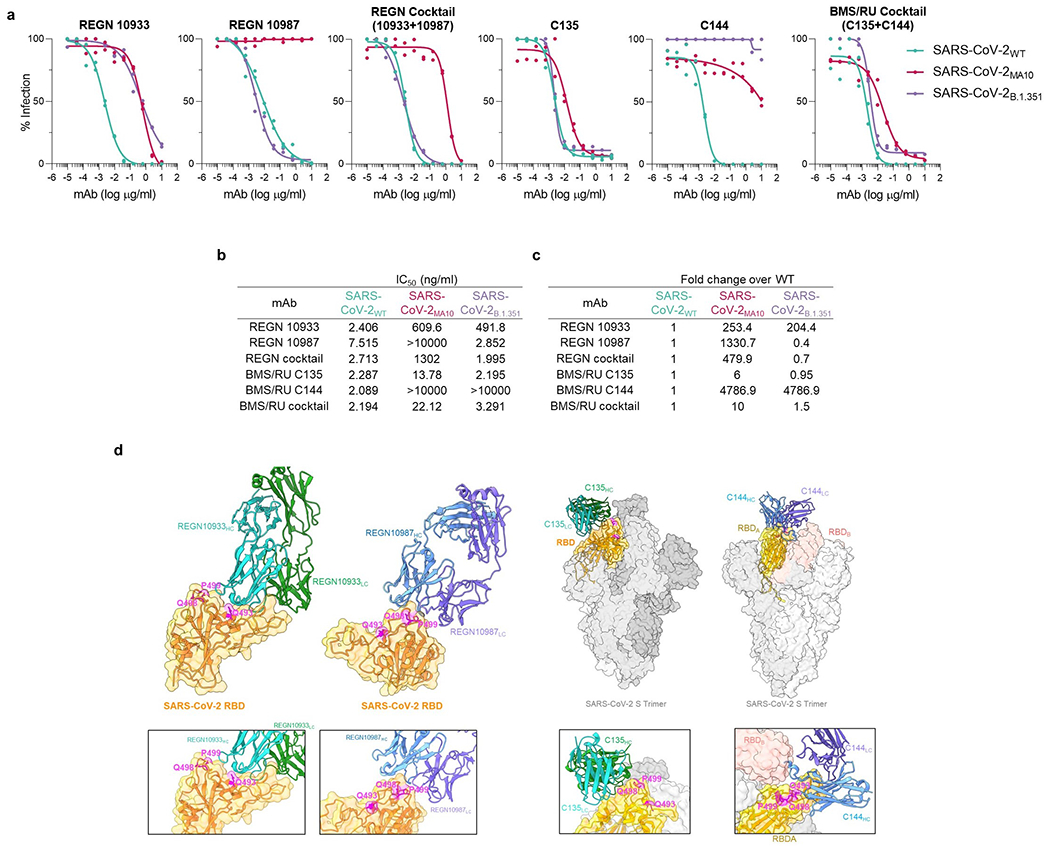
Neutralization activity of REGN and BMS/RU mAbs (individual mAbs or as a cocktail) against SARS-CoV-2 pseudotyped reporter viruses was measured by in vitro neutralization assay. (a) In vitro neutralization curves and (b) IC50 values of REGN (upper panel) and BMS/RU (lower panel) against SARS-CoV-2 WT, MA10, or B.1.351. (c) Fold change of SARS-CoV-2MA10 and SARS-CoV-2B.1.351 IC50 was calculated over WT. n= 1 experiment performed in duplicates. (d) Cryo-EM structures of REGN10933 and REGN10987 complexed with SARS-CoV-2 (PDB: 6XDG) or C135 (PDB: 7K8Z) and C144 (PDB: 7K90) complexed with the SARS-CoV-2 spike trimer. Residues within the SARS-CoV-2WT RBD that are mutated in the SARS-CoV-2MA10 strain (Q493K, Q498Y, P499T) are indicated in magenta.
Extended Data Fig. 5: In vitro neutralization activity and antigenic specificity of Fc variants of anti-SARS-CoV-2 mAbs.
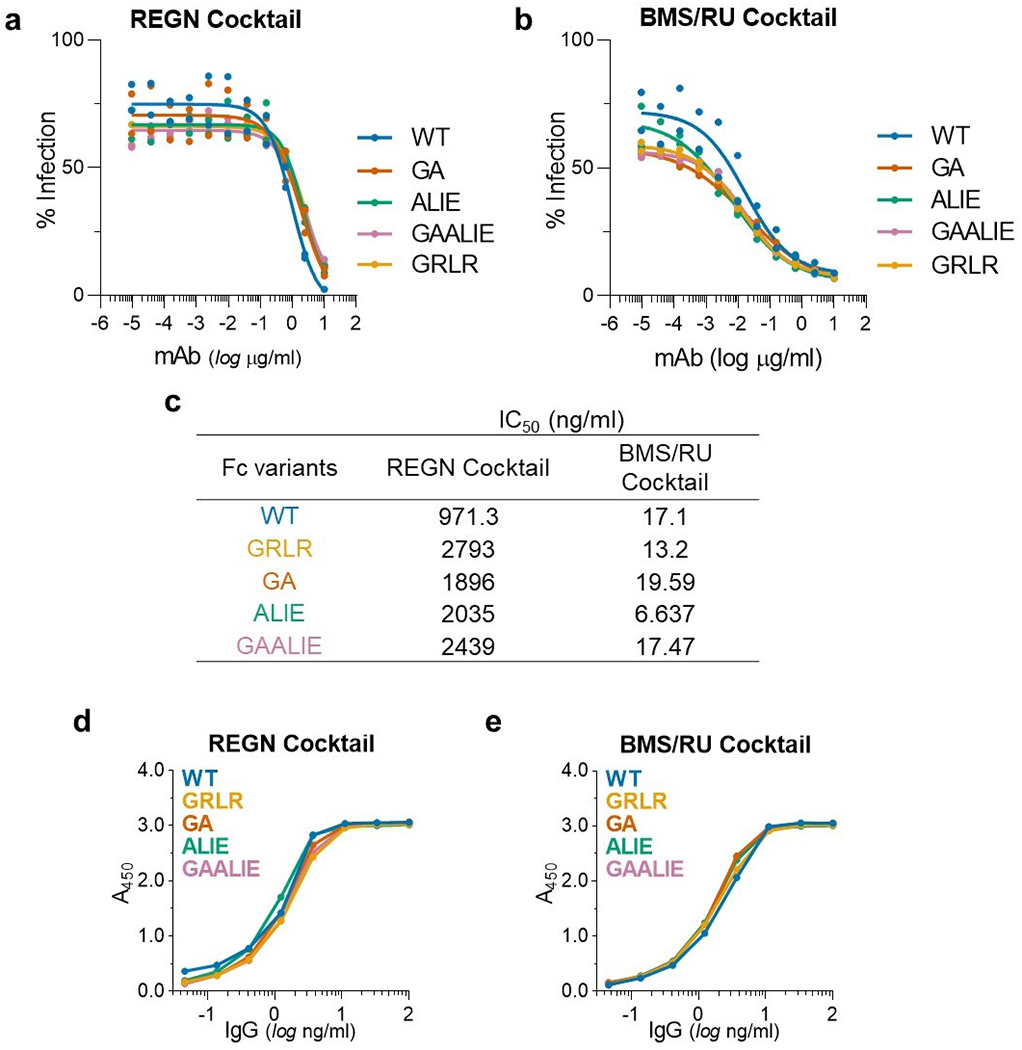
To confirm that changes in the Fc domain have no effect on the neutralization activity and Fab-mediated functions of anti-SARS-CoV-2 mAb, Fc domain variants were characterized in (a-c) in vitro neutralization assays using SARS-CoV-2MA10 pseudotyped reporter viruses and (d, e) in ELISA assays using SARS-CoV-2 RBD. n= 1 experiment performed in duplicates. (a, d) REGN and (b, e) BMS/RU mAb cocktails were expressed as Fc variants and their in vitro neutralization activity (a, b, c, IC50 values) and (d, e) antigenic specificity was compared among Fc variants.
Extended Data Fig. 6: In vitro and in vivo stability of Fc variants of anti-SARS-CoV-2 mAbs.
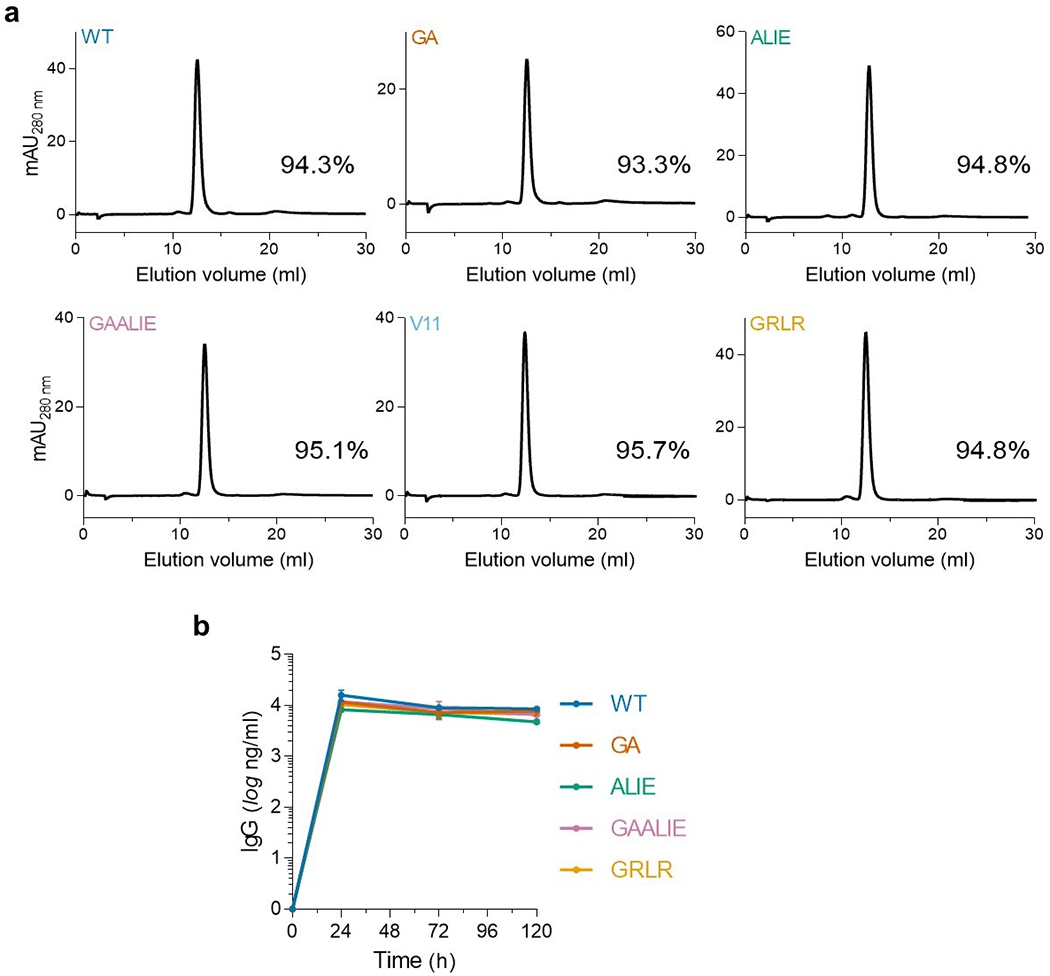
(a) Size-exclusion chromatography (SEC) analysis of Fc variants to determine aggregate formation among Fc domain variants. The SEC elution profiles and abundance (percentage) of monomeric IgG is indicated for the different Fc variants. (b) Fc variants of the REGN mAb cocktail were administered (i.v.; 50 μg) to FcγR humanized mice and antibody serum levels were determined by ELISA at various time points after antibody administration. n= total of 3 mice per group from two independent experiments. Data are mean ± s.e.m.
Extended Data Fig. 7: High-dose treatment of SARS-CoV-2-infected FcγR humanized mice with anti-SARS-CoV-2 mAbs Fc variants enhanced for activating FcγR binding is not associated with enhanced disease.
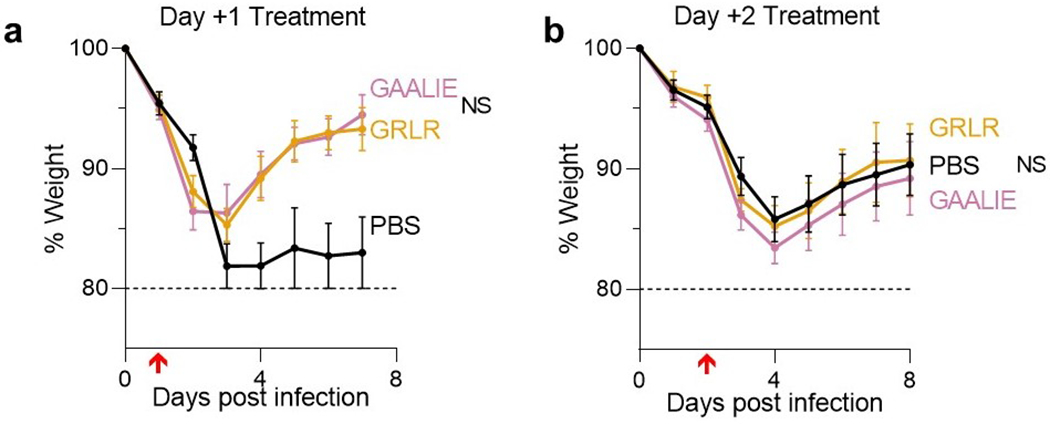
(a) Following the experimental strategy in Fig. 3b, SARS-CoV-2-infected (MA10, 104 pfu, i.n.) FcγR humanized mice (n=3 for PBS and n=5 for mAb-treated groups) were treated (i.v.) with 40 mg/kg REGN mAb cocktail expressed as Fc variants with diminished (GRLR) or enhanced activating FcγR binding (GAALIE). (b) SARS-CoV-2-infected mice were treated with 10 mg/kg BMS/RU mAb cocktail expressed as Fc variants GRLR or GAALIE on day 2 post infection. n= total of 10 mice per group for PBS, n=9 for GAALIE, and n=6 for GRLR from two independent experiments Weight loss (mean ± s.e.m.) was compared between GRLR and GAALIE-treated groups by two-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons). NS, not significant. Red arrow indicates time point of mAb treatment post-infection.
Extended Data Fig. 8: Mouse and human C1q binding of Fc domain variants of IgG1.
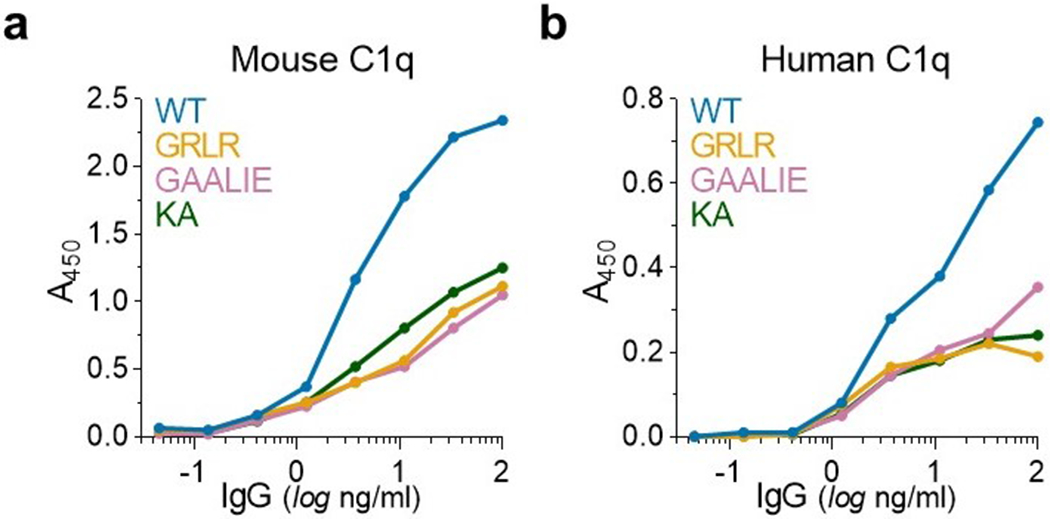
The capacity of IgG1 Fc domain variants to interact with mouse (a) and human (b) C1q was assessed by ELISA (n= 1 experiment performed in duplicates). The KA (K322A) variant, which has previously described as a complement-deficient mutant28, was included as control.
Acknowledgements
We would like to thank Patrick Smith, Emily Lam, Ruben Peraza, Luke Smith, Adriana Martin Mozqueda, and Tiffany Martinez (Rockefeller University) for technical assistance and for maintaining and genotyping the mouse colonies used in this study and all the members of the Laboratory of the Molecular Genetics and Immunology (Rockefeller University) for discussions. We also thank Sara Borghi (Rockefeller University) for providing the recombinant SARS-CoV-2 RBD, Paul Bieniasz (Rockefeller University) for providing the HT1080ACE2 cell line and plasmids for generating pseudoviruses, Agnès Viale (Memorial Sloan Kettering Cancer Center) for providing the SARS-CoV-2 NYC clinical isolate, Alison Ashbrook and Gaitree McNab (Rockefeller University) for advice on handling BSL-3 agents and processing of tissues from infected animals, David Martinez (University of North Carolina at Chapel Hill) for technical assistance, and Sebastian Carrasco (Memorial Sloan Kettering Cancer Center/Weill Cornell Medicine) and staff from the Laboratory Comparative Pathology for histopathology support (with funding from NIH Core Grant P30CA008748). Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases (R01AI137276, R01AI157155, U19AI111825, and R01AI145870) and the Bill and Melinda Gates Foundation (INV-023152). This work was in part made possible with funding from the G. Harold and Leila Y. Mathers Charitable Foundation, the Bawd Foundation, and Fast Grants (www.fastgrants.org), a part of Emergent Ventures at the Mercatus Center, George Mason University. We acknowledge support from the Rockefeller University and Vir Biotechnology Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Competing interests statement
S.B. and J.V.R. are inventors on a patent (WO2019125846A1) describing the GAALIE variant and its use in therapeutic mAbs. C.M.R. and J.V.R. are SAB members of Vir Biotechnology with equity interests.
Data availability
Raw data for all main and extended data figures are included in the manuscript as source files.
References
- 1.Chen P et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N Engl J Med 384, 229–237, doi: 10.1056/NEJMoa2029849 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weinreich DM et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid-19. N Engl J Med 384, 238–251, doi: 10.1056/NEJMoa2035002 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gottlieb RL et al. Effect of Bamlanivimab as Monotherapy or in Combination With Etesevimab on Viral Load in Patients With Mild to Moderate COVID-19: A Randomized Clinical Trial. JAMA 325, 632–644, doi: 10.1001/jama.2021.0202 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor PC et al. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat Rev Immunol, doi: 10.1038/s41577-021-00542-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lundgren JD et al. A Neutralizing Monoclonal Antibody for Hospitalized Patients with Covid-19. N Engl J Med 384, 905–914, doi: 10.1056/NEJMoa2033130 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sia SF et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature, doi: 10.1038/s41586-020-2342-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leist SR et al. A Mouse-Adapted SARS-CoV-2 Induces Acute Lung Injury and Mortality in Standard Laboratory Mice. Cell 183, 1070–1085.e1012, doi: 10.1016/j.cell.2020.09.050 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bournazos S, Gupta A & Ravetch JV The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol 20, 633–643, doi: 10.1038/s41577-020-00410-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bournazos S et al. Broadly Neutralizing Anti-HIV-1 Antibodies Require Fc Effector Functions for In Vivo Activity. Cell 158, 1243–1253, doi: 10.1016/j.cell.2014.08.023 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu CL et al. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science 352, 1001–1004, doi: 10.1126/science.aaf1279 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bournazos S, Corti D, Virgin HW & Ravetch JV Fc-optimized antibodies elicit CD8 immunity to viral respiratory infection. Nature 588, 485–490, doi: 10.1038/s41586-020-2838-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Winkler ES et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell 184, 1804–1820.e1816, doi: 10.1016/j.cell.2021.02.026 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suryadevara N et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell 184, 2316–2331.e2315, doi: 10.1016/j.cell.2021.03.029 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schäfer A et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J Exp Med 218, doi: 10.1084/jem.20201993 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ullah I et al. Live imaging of SARS-CoV-2 infection in mice reveals neutralizing antibodies require Fc function for optimal efficacy. bioRxiv, doi: 10.1101/2021.03.22.436337 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li D et al. The functions of SARS-CoV-2 neutralizing and infection-enhancing antibodies in vitro and in mice and nonhuman primates. bioRxiv, doi: 10.1101/2020.12.31.424729 (2021). [DOI] [Google Scholar]
- 17.Joyner M et al. Early Safety Indicators of COVID-19 Convalescent Plasma in 5,000 Patients. medRxiv, 2020.2005.2012.20099879, doi: 10.1101/2020.05.12.20099879 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dougan M et al. Bamlanivimab plus Etesevimab in Mild or Moderate Covid-19. N Engl J Med, doi: 10.1056/NEJMoa2102685 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu X et al. Tolerability, Safety, Pharmacokinetics, and Immunogenicity of a Novel SARS-CoV-2 Neutralizing Antibody, Etesevimab, in Chinese Healthy Adults: a Randomized, Double-Blind, Placebo-Controlled, First-in-Human Phase 1 Study. Antimicrob Agents Chemother 65, e0035021, doi: 10.1128/AAC.00350-21 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cleary SJ et al. Animal models of mechanisms of SARS-CoV-2 infection and COVID-19 pathology. Br J Pharmacol, doi: 10.1111/bph.15143 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bournazos S IgG Fc Receptors: Evolutionary Considerations. Curr Top Microbiol Immunol, doi: 10.1007/82_2019_149 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Baum A et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science 370, 1110–1115, doi: 10.1126/science.abe2402 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinto D et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583, 290–295, doi: 10.1038/s41586-020-2349-y (2020). [DOI] [PubMed] [Google Scholar]
- 24.DiLillo DJ, Tan GS, Palese P & Ravetch JV Broadly neutralizing hemagglutinin stalk-specific antibodies require FcgammaR interactions for protection against influenza virus in vivo. Nat Med 20, 143–151, doi: 10.1038/nm.3443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith P, DiLillo DJ, Bournazos S, Li F & Ravetch JV Mouse model recapitulating human Fcgamma receptor structural and functional diversity. Proc Natl Acad Sci U S A 109, 6181–6186, doi: 10.1073/pnas.1203954109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez DR et al. Prevention and therapy of SARS-CoV-2 and the B.1.351 variant in mice. Cell Rep 36, 109450, doi: 10.1016/j.celrep.2021.109450 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature 592, 616–622, doi: 10.1038/s41586-021-03324-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hessell AJ et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449, 101–104, doi: 10.1038/nature06106 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Bournazos S, DiLillo DJ, Goff AJ, Glass PJ & Ravetch JV Differential requirements for FcγR engagement by protective antibodies against Ebola virus. Proc Natl Acad Sci U S A 116, 20054–20062, doi: 10.1073/pnas.1911842116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakraborty S et al. Proinflammatory IgG Fc structures in patients with severe COVID-19. Nat Immunol 22, 67–73, doi: 10.1038/s41590-020-00828-7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larsen MD et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science 371, doi: 10.1126/science.abc8378 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lucas C et al. Delayed production of neutralizing antibodies correlates with fatal COVID-19. Nat Med, doi: 10.1038/s41591-021-01355-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atyeo C et al. Dissecting strategies to tune the therapeutic potential of SARS-CoV-2-specific monoclonal antibody CR3022. JCI Insight 6, doi: 10.1172/jci.insight.143129 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blight KJ, McKeating JA & Rice CM Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76, 13001–13014, doi: 10.1128/jvi.76.24.13001-13014.2002 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt F et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J Exp Med 217, doi: 10.1084/jem.20201181 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCann KE, Sinkiewicz DM, Norvelle A & Huhman KL De novo assembly, annotation, and characterization of the whole brain transcriptome of male and female Syrian hamsters. Sci Rep 7, 40472, doi: 10.1038/srep40472 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pietzsch J et al. A mouse model for HIV-1 entry. Proc Natl Acad Sci U S A 109, 15859–15864, doi: 10.1073/pnas.1213409109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw data for all main and extended data figures are included in the manuscript as source files.