

Abstract
Background & Aims:
Helicobacter pylori (H. pylori) infection is the predominant risk factor for gastric cancer. RAS protein activator like 2 (RASAL2) is considered a double-edged sword in carcinogenesis. Herein, we investigated the role of RASAL2 in response to H. pylori infection and gastric tumorigenesis.
Methods:
Bioinformatics analyses of local and public databases were applied to analyzeRASAL2 expression, signaling pathways, and clinical significance. In vitro cell culture, spheroids, patient-derived organoids, and in vivo mouse models were utilized. Molecular assays included ChIP, Co-IP, western blotting, qPCR and immunocyto/histochemistry.
Results:
H. pylori infection induced RASAL2 expression via an NF-κB-dependent mechanism where NF-kB was directly bound to the RASAL2 promoter activating its transcription. By gene silencing and ectopic overexpression, we found that RASAL2 upregulated β-catenin transcriptional activity. RASAL2 inhibited PP2A activity through direct binding with subsequent activation of the AKT/β-catenin signaling axis. Functionally, RASAL2 silencing decreased nuclear β-catenin levels and impaired tumor spheroids and organoids formation. Furthermore, the depletion of RASAL2 impaired tumor growth in gastric tumor xenograft mouse models. Clinicopathological analysis indicated that abnormal overexpression of RASAL2 correlated with poor prognosis and chemoresistance in human gastric tumors.
Conclusion:
These studies uncovered a novel signaling axis of NF-κB/RASAL2/β-catenin, providing a novel link between infection, inflammation and gastric tumorigenesis.
Keywords: RASAL2, gastric cancer, Helicobacter pylori, β-catenin, tumorigenesis
Graphical Abstract
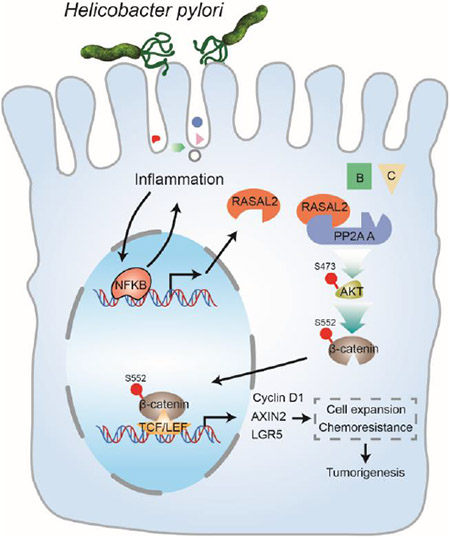
LAY SUMMARY:
Our study uncovers a signaling axis of NF-κB/RASAL2/β-catenin, providing a novel link between infection, inflammation, and gastric tumorigenesis.
Introduction
There are more than one million newly diagnosed gastric cancer cases every year, becoming the fifth most common cancer and the fourth most common cause of cancer-related death worldwide 1. The strongest risk factor for gastric cancer is H. pylori infection, classified as a class I carcinogen by the World Health Organization (WHO)2. H. pylori infection mediates activation of oncogenes and inhibition of tumor suppressor genes through genetic and epigenetic mechanisms, promoting gastric cancer development and progression3-5.
RAS proteins (HRAS, KRAS, NRAS), known as small molecular weight GTPases, play an essential role in the pathophysiological processes of human deseases6. Approximately 20% of cancer patients harbor activating mutations of the RAS gene7. Surprisingly, RAS mutations in gastric cancer are rare, accounting for 6% in TCGA cohort8 and 5% in a multicenter cohort9. Nevertheless, the RAS signaling pathway is aberrantly activated in approximately 40% of gastric cancer cases10, where H. pylori infection appears to play a critical role11. However, the mechanisms underlying the activation of RAS signaling by H. pylori remain unclear.
Two key effectors mediate the RAS signaling pathway in mammals: guanine nucleotide exchange factors (GEFs) and GTPase Activating Proteins (GAPs). As an important member of the RAS GAPs family, RAS protein activator like 2 (RASAL2) has been shown to have cell-type and context-dependent opposing functions. RASAL2 is regarded as a tumor suppressor in luminal-B breast cancer, bladder cancer, lung cancer and ovarian cancer12-16. At the same time, there are accumulating multiple lines of evidence suggesting an oncogenic role of RASAL2 in some human cancer types such as triple-negative breast cancer, colorectal cancer, and liver cancer17-19. However, the exact role of RASAL2 remains largely unknown in gastric cancer. In this study, we demonstrate previously unknown oncogenic functions of RASAL2 in gastric cancer. Our findings uncover a novel mechanism by which H. pylori infection induces RASAL2 expression to promote gastric tumorigenesis and chemoresistance. These findings suggest RASAL2 as a potential prognostic factor and molecular vulnerability in gastric cancer.
Materials and Methods
H. pylori strains
Wild-type H. pylori CagA+ strains 7.13, J166, and rodent-adapted H. pylori CagA+ strain PMSS1 were used in this study, as described previously20. Briefly, the H. pylori strains were cultured on trypticase soy agar with 5% sheep blood agar plates (BD Biosciences, Bedford, Massachusetts) at 37 °C in a humidified incubator with 10% CO2 for passaging. After passaging, H. pylori strains were cultured in Brucella broth (BB, BD Biosciences) with 10% FBS (Invitrogen Life Technologies, Carlsbad, CA) at 37 °C in a humidified incubator with 5% CO2 overnight. H. pylori 7.13 and J166 strains were used for in vitro co-culture with gastric cancer cells at a multiplicity of infection of 100:1. In addition, the PMSS1 strain was utilized to infect gastric epithelial cells of C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME) by oral gavage (1x 109 colony-forming units/mouse) for one or two weeks.
In vivo tumor stem cell frequency assay
For tumor xenograft formation, MKN45 RASAL2-knockdown or control cells were suspended in a mixture of 50% RPMI 1640 medium and 50% matrigel. For limiting dilution assay, 103, 104, 105, and 106 cells were injected subcutaneously into the flank regions of NOD/SCID mice (The Jackson Laboratory, Bar Harbor, ME). Tumor xenografts were measured twice per week for six weeks, and tumor volume was calculated following the formula: tumor volume = (length×width2)/2. The frequency of tumor initiating cells was calculated using Extreme Limiting Dilution Analysis (http://bioinf.wehi.edu.au/software/elda/).
Additional materials and methods are described in supplementary data.
Results
Integrated transcriptomics analysis identified elevated expression of RASAL2 in gastric cancer.
We performed whole-transcriptome sequencing on 36 cases of human gastric tumor tissues and matched adjacent non-tumor tissues (Supplementary Table 1). We also enrolled a public data set of Prom1 conditional mutant mouse gastric cancer model from the Gene Expression Omnibus (GEO) (GSE40634). These two data sets were together defined as tumor-vs.-normal (TN) cohorts. In addition, we performed the whole-transcriptome sequencing on 10 cases of TFF1-knockout mouse stomach tissues with and without H. pylori infection (PMSS1 strain), as well as enrolled a public data set of H. pylori infection (H. felis CS1 strain) C57BL/6 mouse model from GEO (GSE13873) 21. These two data sets were defined as H. pylori infection (HP) cohorts. As a result, we identified 280 and 758 robust overlapping differential expression genes (DEGs) from TN cohorts and HP cohorts, respectively, by the significance levels (∣logFC∣ >1 and P < .01 for TN cohorts, and P < .05 for HP cohorts) (Figure 1A, Supplementary Table 2-5). We further obtained 56 DEGs by overlapping TN and HP cohorts (Figure 1A, Supplementary Table 6). To identify genes with significant clinical value for gastric cancer, we performed survival analysis across the 56 DEGs in TCGA cohorts. We identified 11 up-regulated DEGs and 2 down-regulated DEGs by the significance levels (∣HR∣ >1.0 and P < .05, Figure 1A). The upregulated DEGs were considered the candidate genes which were abnormally expressed in both human and mouse gastric tumors, suggesting their contribution to H. pylori infection-promoted gastric tumorigenesis.
Figure 1. Overexpression of RASAL2 in gastric cancer samples.

(A) Analyses of the whole-transcriptome sequencing datasets and two Gene Expression Omnibus (GEO) datasets from tumor-vs.-normal (TN) cohorts and H. pylori infection (HP) cohorts. Hierarchical clustering heatmap and volcano plots of significant differential expression genes (DEGs) were shown in DEGs Analysis. Overlapping TN cohorts and HP cohorts identified 56 DEGs. Survival analysis of the 56 DEGs was performed in TCGA cohort and the genes with the significance levels (∣HR∣ > 1.0 and P < .05) were identified as candidate genes. (B) Analyses of TCGA pan-cancer database displayed diverse mRNA expression patterns of RASAL2 in different cancer types; ns indicates no significance, *P < .05, **P < .01, ****P < .0001. (C) Analyses of public GEO datasets showed that the mRNA expression levels of RASAL2 were consistently elevated in gastric cancers compared to normal samples; *P < .05, **P < .01, ***P < .001, ****P < .0001. (D) The mRNA expression levels of RASAL2 were examined in the paired tumor and adjacent non-tumor tissues from the GEO datasets and our local cohort; ****P < .0001.
Among these 11 genes, we focused on the RASAL2 because: 1) abnormal overexpression of RASAL2 had a significant inverse correlation with gastric cancer patients’ prognosis (Supplementary Table 6), 2) RASAL2 is a key member of RAS GTPase-activating proteins (RAS GAPs) family17-19, and 3) RASAL2 was frequently overexpressed in the TCGA gastrointestinal cancer datasets including esophageal carcinoma (ESCA, P > .05), stomach adenocarcinoma (STAD, P < .001), colon adenocarcinoma (COAD, P < .001), and rectum adenocarcinoma (READ, P < .05) (Figure 1B). Furthermore, we have confirmed the upregulation of RASAL2 in gastric cancers by analyses of several additional databases (Figure 1C). Upregulation of RASAL2 in gastric cancers was also supported by comparing the tumor samples with the paired normal adjacent stomach samples from GEO datasets (GSE122401, GSE65801 and GSE13195), as well as our own paired samples (local dataset) (Figure 1D, all P < .001). In addition, elevated expression of Rasal2 was detected in mouse primary gastric cancer from conditional knockout mouse models of SMAD4, TP53, and CDH1 (GSE45956, P = .008) (Supplementary Figure 1A)22. These findings indicate that RASAL2 expression is significantly elevated in human and mouse gastric cancer samples.
H. pylori infection transcriptionally upregulated RASAL2 expression through NF-κB
Our results indicated that RASAL2 expression was significantly higher in TFF1-knockout mouse gastric tissues with H. pylori infection than in the untreated control mouse tissues (Supplementary Figure 1B, logFC = 0.27, P = .032). In addition, we also found that RASAL2 expression was significantly increased in an H. pylori infection of C57BL/6 mouse model21 (Supplementary Figure 1B, logFC = 0.38, P = .032). Gene set enrichment analysis (GSEA) showed that H. pylori infection-associated signature and its downstream signaling (NF-κB signaling and β-catenin signaling) were enriched in RASAL2-high expression samples, compared to RASAL2-low expression samples, in TCGA, GEO and our local cohorts (Supplementary Figure 1C). Western blots analysis showed that H. pylori infection significantly upregulated RASAL2 protein levels in human gastric cancer cell lines (Figure 2A and 2B, Supplementary Figure 2A). The qRT-PCR results confirmed that H. pylori infection induced RASAL2 mRNA overexpression (Figure 2A and 2B, Supplementary Figure 2B, all P < .05). These findings indicate that RASAL2 overexpression is mediated via a transcription mechanism.
Figure 2. H. pylori infection transcriptionally regulated RASAL2 expression through NF-κB.

(A and B) Western blot and quantitative real-time (qRT) PCR analysis of RASAL2 in AGS and STKM2 cells following H. pylori infection; *P < .05, **P < .01, ***P < .001. (C) The transcription factor binding sites were predicted by the PROMO website using a 600bp conserved segment of RASAL2 promoter. (D) A conserved sequence of two putative NF-κB binding sites (P1 and P2) with higher JASPAR scores in humans and mice. (E) Western blot and qRT-PCR analysis of RASAL2 in AGS and STKM2 cells with and without transient expression of NF-κB (p65); ***P < .001. (F and G) Chromatin immunoprecipitation (ChIP) assay using NF-κB antibody was performed, followed by qPCR applying primers covering P1 and p2 region; ns, no significance, **P < .01, ***P < .001. AGS cells with P65 transient expression (F). AGS cells with infection of the two H. pylori strains (7.13 and J166) (G). (H and I) RASAL2 promoter luciferase reporter assays were performed in AGS and STKM2 cell lines with TNFα treatment (H) or H. pylori infection (7.13 and J166, I); **P < .01, ***P < .001.
Using bioinformatics analysis of the RASAL2 gene and promoter, we found a highly conserved 600bp DNA sequence in human and mouse, containing the transcription start site (TSS). Analysis of the transcription factor (TF) binding sites, using the conserved regions, in the PROMO website23 (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) identified a total of 46 human and 28 mouse TF binding regions. Interestingly, three often overlapping binding regions were NF-κB or RelA (Figure 2C). We focused on the two putative NF-κB binding regions (P1 and P2), which have higher JASPAR scores (http://jaspar.genereg.net/) (Figure 2D)24. To confirm the computational analysis, we examined whether NF-κB regulated RASAL2 expression in our gastric cancer models. Indeed, activation of NF-κB by TNF-α significantly induced RASAL2 mRNA and protein expression levels in AGS, SNU-1, and STKM2 cells. Conversely, an NF-κB inhibitor, BAY 11-7082, repressed RASAL2 expression in MKN45 and MKN28 cells (Supplementary Figure 2C and 2D). Moreover, transient overexpression of p65 induced upregulation of RASAL2 expression in gastric cancer cell lines (Figure 2E, Supplementary Figure 2E). Using chromatin immunoprecipitation (ChIP) assay, the results demonstrated direct binding of p65 on two RASAL2 promoter regions after transient overexpression, compared with the control groups (Figure 2F, Supplementary Figure 2F). H. pylori infection induced significant recruitment of p65 to the RASAL2 promoter regions, compared with control groups (Figure 2G, Supplementary Figure 2G, all P < .01). To confirm whether H. pylori infection activated RASAL2 promoter, gastric cancer cells were transfected with RASAL2-promoter-luciferase reporter plasmid and then treated with TNF-α or infected with H. pylori. The results confirmed that both TNF-α treatment and H. pylori infection upregulated RASAL2 promoter activity (Figure 2H and 2I, all P < .01). Consequently, western blots analysis showed that H. pylori infection induced activation of NF-κB (phosphorylation on Ser536) with upregulation of RASAL2 in human gastric cancer cell lines (Figure 2A and 2B, Supplementary Figure 2A). Taken together, these multiple lines of evidence demonstrated that H. pylori infection upregulated RASAL2 via activation of NF-κB in gastric cancer cells.
H. pylori infection activates AKT/β-catenin signaling axis via RASAL2
To elucidate the downstream molecular mechanism regulated by RASAL2 in gastric cancer, we performed GSEA in TCGA and several GEO gastric cancer datasets by comparing RASAL2-high-expressed cohorts with RASAL2-low-expressed cohorts. Intriguingly, several crucial oncogenic gene sets, including epithelial-mesenchymal transition (EMT), TNF-α/NF-κB, TGFβ, Hedgehog, and Wnt/β-catenin, were consistently enriched across all the datasets as shown in the heatmap (Supplementary Figure 3A). The Wnt/β-catenin signaling pathway plays an important role in organ development, cellular expansion, and maintenance of stem cell homeostasis for all mammal species25. As expected, the Wnt/β-catenin gene set (NES = 1.923, FDR = 0.004), including CTNNB1 and its targets MYC and AXIN2 (all P < .001), was significantly enriched in RASAL2-high tumors (Supplementary Figure 3B and 3C). Pearson’s correlation test further supported the strong correlations between RASAL2 expression and CTNNB1 and its downstream targets (Supplementary Figure 3D, all P < .01).
To determine the causal relationship between RASAL2 and β-catenin, we performed RASAL2 silencing or transient overexpression in multiple gastric cancer cell lines. RASAL2 knockdown using siRNA strikingly decreased β-catenin activity/phosphorylation (Ser552) (Supplementary Figure 4A), whereas RASAL2 overexpression activated β-catenin (Supplementary Figure 4B), compared with respective control groups. AKT is a direct upstream regulator of β-catenin phosphorylation (S552). Therefore, we investigated whether RASAL2 activated β-catenin via AKT. Consistently, RASAL2 positively regulated phosphorylation of AKT in gastric cancer cell lines (Supplementary Figures 4A and 4B). Based on previous studies showing that H. pylori infection can trigger AKT/β-catenin signaling26, we investigated whether H. pylori infection activated this signaling axis in a RASAL2-dependent manner. Our results indicated that phosphorylations of AKT and β-catenin induced by H. pylori-infection were abrogated by RASAL2 knockdown (Supplementary Figure 4C). Furthermore, RASAL2-overexpression- or H. pylori infection-induced activation of AKT/β-catenin axis was abolished by treatment of AKT inhibitor (MK2206) (Supplementary Figures 4D and 4E). These results suggested that H. pylori infection activated the AKT/ β-catenin signaling axis via induction of RASAL2.
H. pylori infection induced β-catenin transcriptional activity in a RASAL2-dependent manner
Given that RASAL2 played a critical role in H. pylori infection-induced active phosphorylation of β-catenin, we questioned whether RASAL2 regulated the transcriptional activity and downstream targets of the β-catenin transcription complex. Therefore, the TOP/FOP flash luciferase assays were performed in gastric cancer cells with RASAL2 silencing. RASAL2 knockdown significantly repressed TOP-flash reporter activity (P < .01), compared with CTRL siRNA, while mutant FOP-flash reporter activity was unchanged (a negative control) (Figure 3A, Supplementary Figure 5A). H. pylori infection induced the TOP-flash reporter activities (P < .05); however, this increase was abrogated by RASAL2 knockdown (Figure 3B, Supplementary Figure 5B). On the other hand, RASAL2 overexpression enhanced TOP-flash reporter activity in AGS and SNU-1 cells (Figure 3C, all P < .01). Consistent with these findings, RASAL2 silencing downregulated the protein and mRNA expression levels of β-catenin downstream targets, such as AXIN2, cyclin D1 (CCND1), and LGR5 (Figure 3D, Supplementary Figure 5C and 5D), whereas RASAL2 overexpression upregulated their expression (Figure 3E, Supplementary Figure 5E). In line with these findings, immunofluorescence (IF) and nuclear/cytoplasmic protein extraction assay results showed a decrease in β-catenin nuclear localization, following knockdown of RASAL2 (Figure 3F and 3G), whereas RASAL2 overexpression promoted nuclear accumulation of β-catenin (Supplementary Figure 5F). These results demonstrated that H. pylori-induced RASAL2 promoted nuclear accumulation and transcriptional activity of β-catenin.
Figure 3. H. pylori infection induced β-catenin transcriptional activity in a RASAL2-dependent manner.

(A - C) β-catenin luciferase reporter assays. TOP-flash contains wild-type TCF binding sites. FOP-flash, containing mutated TCF binding sites (negative control); *P < .05, **P < .01, ***P < .001. (A) Gastric cancer cells were transfected with two independent RASAL2 siRNAs (#1, or #2) or scramble siRNA (CTRL). (B) 72h after transfection of RASAL2 siRNAs (#1, or #2), gastric cancer cells were infected with H. pylori strains for 6h. (C) Gastric cancer cells were transfected with the indicated mounts (0.25μg or 0.5μg) of pLV-RASAL2 expression vector or empty vector control (CTRL). (D and E) Western blots and qRT-PCR analysis of β-catenin targets, including AXIN2, Cyclin D1, and LGR5; *P < .05, ***P < .001. Gastric cancer cell lines were transfected with RASAL2 siRNA (#1) or scramble siRNA control (CTRL) (D). Gastric cancer cell lines were transfected with the indicated mounts (0.25μg or 0.5μg) of pLV-RASAL2 expression plasmid or empty vector (CTRL) (E). (F and G) MKN28 cells were transfected with RASAL2 siRNA (#1) or scrambled siRNA (CTRL). Immunofluorescence staining (scale bars, 20μm) for RASAL2 (red) and β-catenin (green) was performed. Representative images are shown (F). Nuclear and cytoplasmic protein extraction and Western blots of RASAL2, p-β-catenin (S552) and β-catenin were performed (G).
RASAL2 promoted AKT phosphorylation via interaction with and inactivation of PP2A
Previous studies illustrated that RASAL2, a key member of the RAS GTPase-activating protein family, could positively or negatively regulate the RAS signaling pathway, depending on the cell context or stimulus13, 27. In our studies, RASAL2 silencing or overexpression did not affect KRAS protein level or downstream MEK signaling in both KRAS mutant (MT) and wild-type (WT) gastric cancer cell lines (Supplementary Figure 6A). We observed that RASAL2 overexpression induced activation of AKT and β-catenin in AGS cells. Of note, KRAS depletion did not abrogate RASAL2-induced activation of AKT and β-catenin in AGS cells (Supplementary Figure 6B), suggesting that RASAL2 utilizes a RAS-independent pathway to promote AKT activation. We found that RASAL2 expression negatively correlated with genes (PPP2CA and PPP2CB) encoding protein phosphatase 2A catalytic (PP2Ac) subunit across four GEO cohorts (Supplementary Figure 6C). This finding prompted us to investigate the relationship between RASAL2 and PP2A.
PP2A can negatively regulate AKT activity, whereas inactivation of its catalytic subunit (PP2Ac) by phosphorylation at Y307 activates AKT signaling28, 29. We found that RASAL2 depletion inhibited the PP2A phosphorylation and increased the active form of PP2A (P < .01), with subsequent inactivation of AKT (Figure 4A and 4B). On the contrary, overexpression of RASAL2 had opposite effects (Figure 4C and 4D, all P < .05). We also found that H. pylori infection promoted phosphorylation of PP2A (inactive) and activated AKT/β-catenin axis. Notably, RASAL2 silencing abolished these H. pylori infection-induced changes (Figure 4E and 4F, Supplementary Figure 6D and 6E). Importantly, the treatment with a PP2A inhibitor, Okadaic Acid (OA), abrogated the repressive effects of RASAL2 depletion on PP2A/AKT/β-catenin cascades in gastric cancer cells (Figure 4G). These results supported the notion that RASAL2 activated AKT by promoting the inactivation of PP2A. Using immunoprecipitation, we detected a novel protein-protein interaction between RASAL2 and PP2AA (Figure 4H, Supplementary Figure 6F). These findings were further supported by the proximity ligation assays (PLA) showing the physical proximity of RASAL2 and PP2AA (Figure 4I). Consistent with these findings, immunfluoresence analysis demonstrated co-localization of these two proteins (Supplementary Figure 6G-6I). Collectively, these results indicated that RASAL2 could activate AKT signaling by directly binding to and inactivating PP2A.
Figure 4. RASAL2 enhanced AKT phosphorylation through inhibition of protein phosphatase 2A (PP2A) activity.

(A,C and E) Western blots for RASAL2, p-PP2Ac (Y307), total PP2Ac, p-AKT (Ser473), and total AKT were performed in gastric cancer cell lines. (A) Gastric cancer cells with RASAL2 depletion, (C) Gastric cancer cells with RASAL2 overexpression, (E) Gastric cancer cells with RASAL2 depletion followed by H. pylori infection. (B,D and F) PP2A Phosphatase immunoprecipitation assays. The active PP2A form, PP2Ac, was specifically pulled down from whole-cell lysates, according to the manufacturer's instructions; *P < .05, **P < .01, ***P < .001. (B) Gastric cancer cell lines with RASAL2 depletion, (D) RASAL2 overexpression, and (F) RASAL2 depletion followed by H. pylori infection. (G) Western blot in AGS and STKM2 cell lines, following RASAL2 silencing and PP2A inhibitor (Okadaic acid, OA, 100nM) treatment. (H) Immunoprecipitation (IP) assay using antibody against RASAL2 or PP2AA in MKN45 cells. IgG was used as a negative control. Western blots of RASAL2 and PP2AA were performed. (I) Proximity ligation assays (PLA) in MKN45 and STKM2 cells were performed by using anti-RASAL2 and anti-PP2AA antibodies (scale bars, 10μm). PBS (CTRL) and single antibody only was used as negative controls. Red dots represent close relationship between two proteins.
RASAL2 depletion inhibited expansion of cancer cells in vitro and suppressed tumor formation in vivo
The contribution of the Wnt/β-catenin signaling axis to cell survival and expansion has been widely reported in several human malignancies30, 31, including gastric cancer32. Pearson’s correlation analyses revealed that both RASAL2 (Figure 5A, P < .01) and CTNNB1 (Supplementary Figure 7A, P < .01) expression levels correlated with single sample GSEA (ssGSEA)33 scores for two verified stem cell signatures (Supplementary Table 7), namely Stem Cell Gene Set (SCGS)_Smith and SCGS_Benporath_Sox234, 35. Consistently, GSEA demonstrated enrichments of several genes/pathways known to promote self-renewal and expansion properties in patients with high RASAL2 expression across four databases (Figure 5B). Following these findings, we first tested the functions of RASAL2 on cell expansion stem-like properties utilizing spheroids derived from MKN28 and MKN45 cells (Supplementary Figure 7B). RASAL2 knockdown remarkably decreased the number (P < .05) and size (P < .01) of spheroids, as compared to control groups (Figure 5C and 5D, Supplementary Figure 7C and 7D). The spheroids with RASAL2 knockdown displayed less nuclear and more cytosolic staining of β-catenin than in control cells, indicating nuclear export of β-catenin in the spheroids (Figure 5E, Supplementary Figure 7E-7H, P < .001). Consistent with these observations, the knockdown of RASAL2 decreased the number (P < .05) and size (P < .01) of the organoids from human gastric cancer cells (Figure 5F and 5G, Supplementary Figure 7I). Immunofluorescence staining demonstrated similar results as observed in spheroids, with decreased numbers of cells positive for nuclear β-catenin (P < .01) and Ki-67 (P < .001) (Figure 5H, Supplementary Figure 7J). To test the role of RASAL2 in spheroid/tumor formation, we used a series dilution assay of MKN45 tumor cells with or without RASAL2 knockdown in vitro and in vivo. RASAL2 silencing in MKN45 cells significantly suppressed spheroid formation capacity (P < .0001, Supplementary Figure 8A-8C). Furthermore, the tumor xenograft results showed a significant impairment of tumor initiation capacity from 1 of 3,656 cells (control) to 1 of 57,086 cells (RASAL2 knockdown) (P < .001, Figure 6A-6C). Western blots confirmed down-regulation of PP2A/AKT/β-catenin signaling axis in the RASAL2 knockdown cells, consistent with suppression of tumor growth (Figure 6D). These data indicated that RASAL2 played a critical role in promoting stem-like expansion properties of cancer cells in gastric tumorigenesis.
Figure 5. RASAL2 depletion represses gastric cancer cell expansion.

(A) Pearson’s correlation analyses between RASAL2 mRNA level and single-sample gene set enrichment analysis (ssGSEA) scores for two verified stem cell signatures, namely Stem cell gene set (SCGS)_Smith and SCGS_Benporath_Sox2, in TCGA cohort. (B) GSEA was employed in the TCGA, GEO and local cohorts, using stem cell relative signatures from GO biological processes items. The CSC markers, YAP1, NES, CTNNB1 (β-catenin), NF1, NOTCH1, and WNT5A, were analyzed across the datasets. (C) Spheroids (scale bars, 100μm) derived from MKN28 cells with stable knockdown of RASAL2 (shRASAL2-#1) displayed significantly smaller spheroids as compared to the scrambled shRNA cells (CTRL). (D) The quantification of sphere size and the number was expressed as the mean ± SD of 3 independent fields; *P < .05, **P < .01, ***P < .001. (E) Representative immunofluorescent images (scale bars, 25μm) of β-catenin (green) in spheroids; nuclei were stained with DAPI (blue). White arrows indicate nuclear β-catenin staining. The quantification of nuclear β-catenin fluorescence is shown as the mean ± SD of 3 independent fields; ***P < .001. (F) Human organoids (scale bars, 100μm) from gastric cancer tissues were stable knocked down of RASAL2 (lentivirus shRASAL2) or control (CTRL), showing that knockdown of RASAL2 inhibited organoids growth. (G) The quantification of organoids size and number was expressed as the mean ± SD of 5 independent fields; *P < .05, **P < .01. (H) Representative immunofluorescence images (scale bars, 50μm) of β-catenin (green) and RASAL2 (red) in organoids; nuclei were stained with DAPI (blue). White arrows indicate nuclear β-catenin staining. The quantification of nuclear β-catenin fluorescence is shown as the mean ± SD of 3 independent fields; **P < .01.
Figure 6. In vivo studies of RASAL2 in gastric tumorigenesis.

(A and B) MKN45 cells with/without RASAL2 knockdown were serially diluted and xenografted into NOD/SCID mice subcutaneously. (A) shows tumor the cell numbers injected and frequency of tumor formation at day 42. (B) displays the probability estimates calculated with Extreme Limiting Dilution Analysis (ELDA) software (http://bioinf.wehi.edu.au/software/elda/). A significant difference in tumor formation capacity was observed between the control and sh-RASAL2 groups. (C) Tumor growth curves for subcutaneous tumor xenografts with shRNA knockdown or control (n = 6 per 1 million cells dose group); *P < .05. (D) Western blots for RASAL2 and its downstream signaling genes for xenograft tumors with/without RASAL2 knockdown (from 1 million cells dose group). (E-G) Immunohistochemistry staining (scale bars, 100μm) of RASAL2 (E), qRT-PCR of Rasal2 (F), and Western blots for RASAL2 (G) and its downstream targets were performed in H. pylori-infected mouse stomach tissues using mouse-adapted H. pylori strain, PMSS1, for 1 week (PMSS1-1W) or 2 weeks (PMSS1-2W) infection. *P < .05, **P < .01, ***P < .001. (H-J) Hematoxylin and eosin (H&E) staining (H, scale bars, 100μm), qRT-PCR of Rasal2 (I), and Western blots for RASAL2 and its downstream targets (J) were performed in TFF1-knockout mouse neoplastic gastric tissues. WT, wide-type mouse; LGD, low-grade dysplasia; HGD, High-grade dysplasia. *P < .05, ***P < .001.
H. pylori infection induced RASAL2 expression in gastric tissues and in primary gastric tumors in mouse models
To further investigate the role of RASAL2 in H. pylori infection-induced gastric tumorigenesis, we utilized an in vivo mouse model for infection with PMSS1. The mouse adapted H. pylori strain. After one week or two weeks of infection, infected mice showed upregulation of RASAL2 at the mRNA and protein levels as well as activation of AKT/β-catenin axis in gastric tissues, compared to the untreated control mice (Figure 6E-6G, Supplementary Figure 8D). We next analyzed stomach tissues from the TFF1-knockout gastric cancer mouse model (Figure 6H), which developed gastric tumors with activation of NF-κB pathway 36, 37. We also detected high levels of RASAL2 and its downstream signaling targets in neoplastic lesions, with a progressive increase from low grade dysplasia (LGD) to high grade dysplasia (HGD)/adenocarcinoma tissues (Figure 6I and 6J). Our data strongly indicate that RASAL2 plays a critical role in gastric tumorigenesis in mouse models.
Elevated RASAL2 correlated with β-catenin expression and predicted poor prognosis and chemoresistance of patients with gastric cancer.
Following our data in mice, we evaluated the clinical significance of RASAL2 in gastric cancer. We examined RASAL2 protein expression using immunohistochemistry (IHC) staining in three tumor tissue microarrays (TMA) with 365 evaluable gastric cancer cases and one adjacent non-tumor TMA with 124 evaluable cases. The clinicopathological characteristics were listed in Supplementary Table 8. Representative RASAL2 positive and negative images are shown in Figure 7A. RASAL2 positive expression was observed in 57.0% (208/365) of primary gastric cancer, which was significantly higher than in adjacent non-tumor tissues (37.9%, 47/124, P < .001, Figure 7B). In addition, IHC staining of β-catenin was performed in one of the three tumor TMA with 119 evaluable cases. Spearman’s correlation analysis indicated a strong correlation between RASAL2 and β-catenin (Figure 7C, R = 0.47, P < .001, Supplementary Figure 9A). There was no significant correlation between RASAL2 level and clinicopathological features in local or GSE66229 cohorts (Supplementary Table 9 and 10), suggesting that RASAL2 may be an inherent early event in gastric tumorigenesis. Kaplan-Meier survival analysis revealed that RASAL2-positive patients had much worse overall survival (OS, Hazard Ratio, HR = 1.8, 95% CI: 1.3-2.4, P < .001) and recurrence-free survival (RFS, HR = 2.1, 95% CI: 1.4-3.0, P < .001), as compared with RASAL2-negative patients (Figure 7D). Similar results were obtained from analyses of TCGA data and a panel of GEO cohorts (Figure 7D, Supplementary Figure 9B, all P < .05). Furthermore, analysis of our data identified RASAL2 as one of the independent prognostic factors for shorter OS in gastric cancer by multivariate analysis with Cox’s proportional hazards regression model (Supplementary Table 11, HR = 1.8, 95% CI: 1.3-2.4, P < .001). These results were in line with the results from the GSE66229 cohort (Supplementary Table 12, HR = 1.7, 95% CI: 1.2-2.5, P = .004).
Figure 7. Elevated expression of RASAL2 predicted chemoresistance and poor prognosis in human samples.

(A) Representative immunohistochemistry staining (scale bars, 100μm) of RASAL2 in human gastric tumors and adjacent non-tumor tissues. (B) Percentages of RASAL2 negative and positive staining in tumor (n=365) and non-tumor tissues (n=124). (C) Spearman’s correlation between IHC staining scores of RASAL2 and β-catenin in tumor slides of 119 patients. (D) Kaplan-Meier survival analysis for RASAL2 staining in the local cohort (left for OS and middle for RFS) or mRNA levels (right) in TCGA cohorts. (E) Kaplan-Meier survival analysis for patients with and without adjuvant chemotherapy (ACT) in RASAL2 positive or negative staining cases in the local cohort. (F) Cox regression model for an adjusted hazard ratio of the patients with ACT in RASAL2 positive and negative staining cases, compared with the patients without ACT. (G) Gross morphology of tumors in NOD/SCID mice model using MKN45 cells with RASAL2 knockdown or combination with CDDP treatment. Mice were dosed with CDDP (2 mg/kg/3 day, ip) (n = 6 tumors per group). (H) Tumor growth curve over time in each group. Data are shown as the mean ± SEM. (I) Relative tumor volume at the end of treatment. Boxes in the graph indicate the median with interquartile range. ns indicates no significance. **P < .01.
Given the results obtained from spheroids, organoids and tumor xenograft models, showing RASAL2 ability to promote cell expansion, we tested whether RASAL2 promoted chemoresistance. We used our cohort of 324 patients with available adjuvant chemotherapy treatment (ACT) information (all regimens were platinum and fluorouracil-based chemotherapy). Our data showed a survival benefit in patients with ACT, compared with those without ACT (Supplementary Figure 9C, P < .001). A stratified analysis demonstrated a significant survival benefit in RASAL2-negative patients who received ACT (HR = 0.31, 95% CI: 0.18-0.52, P < .001), compared to RASAL2-positive patients (Figure 7E). This was further confirmed by adjusting to other clinicopathological features in the Cox regression model (Figure 7F, Supplementary Figure 9D). These results suggest that RASAL2-positive gastric cancer cells/patients may be more resistant to ACT. To confirm this intriguing clinical observation in a lab setting, we performed cytotoxicity assays. We found that RASAL2 silencing significantly sensitized gastric cancer cells to the treatments of cisplatin (CDDP), oxaliplatin (OXA), and docetaxel (DTX) as indicated by reductions in IC50 values (Supplementary Figure 10A). Using CDDP-resistant AGS cell models38, we detected higher RASAL2 levels and active AKT/β-catenin signaling in these cells than in parental cells (Supplementary Figure 10B). Notably, RASAL2 silencing abrogated chemoresistance and reversed the above signaling axis in the CDDP-resistant cells (Supplementary Figure 10C and 10D). An in vivo tumor xenograft model of RASAL2 knockdown combined with CDDP treatment was employed to confirm the in vitro finding from cytotoxicity assays (Supplementary Figure 10E). The combination of RASAL2 silencing and CDDP inhibited 40.9% of tumor growth, compared to the shRASAL2 cells without CDDP, whereas CDDP didn’t significantly inhibit the tumor growth of knockdown control cells (tumor inhibition rate is 15.9%) (Figure 7G-7I). In addition, western blots confirmed that RASAL2 knockdown suppressed the AKT/β-catenin signaling axis, possibly contributing to the inhibition of gastric tumor growth (Supplementary Figure 10F and 10G). These results suggest synergistic anti-tumor effects of RASAL2 silencing and CDDP treatment in gastric cancer.
Discussion
In this study, we investigated the molecular mechanisms underlying H. pylori infection-driven gastric tumorigenesis. We report direct transcription up-regulation of RASAL2 via NF-κB in response to H. pylori infection. The results illustrate the role of RASAL2 in promoting tumorigenic cells’ expansion via activation of the AKT/β-catenin signaling axis. Our findings provide a novel link between H. pylori infection, inflammation, and cell signaling in gastric tumorigenesis.
H. pylori is classified as a class I carcinogen2 and considered the main risk factor for two-thirds of non-cardia gastric cancer39, 40. We performed whole-transcriptome analysis of gastric tumors to screen novel candidate genes and signaling pathways in H. pylori-infected gastric cancers. Analyses of sequencing data identified RASAL2 as one of the most upregulated genes in H. pylori-infected mouse stomach tissues. We found that H. pylori-infection induces the endogenous RASAL2 expression in vitro and in vivo, suggesting an oncogenic role for RASAL2 in H. pylori-infection-promoted tumorigenesis. Although we characterized transcriptional regulation of RASAL2 by NF-kB in response to H. pylori, we can not exclude a post-transcriptional regulatory mechanism. Our finding of changes in protein levels at early time points preceding the increase in mRNA suggests this additional possibility. In fact, earlier studies reported negative regulation of RASAL2 by miR-203 and miR-136 17 41, raising the possibility that these miRNAs might be silenced in response to H. pylori. Alternatively, H. pylori signaling may play a role in mediating RASAL2 protein stability. Nevertheless, our findings confirm the role of inflammation in mediating transcriptional regulation of RASAL2 where active NF-κB, induced by H. pylori42, can directly bind to and activate RASAL2 transcription. The TFF1-knockout mouse model of gastric neoplasia, characterized by NF-kB activation, demonstrated high levels of Rasal2. Therefore, it is reasonable to assume that high expression levels of RASAL2 and activation of its downstream signaling can accompany conditions of pro-inflammatory NF-κB signaling in gastic tissues and tumors, regardless of the H. pylori status at cancer diagnosis.
RASAL2 is a RAS GAPs family member with reports of opposing functions as a tumor suppressor or oncogene, depending on the cell type and exogenous stimulus12, 17, 43. Our analysis of the TCGA Pan-cancer dataset revealed a diverse expression pattern of RASAL2 in different cancer types (Figure 1B). Interestingly, consistent with our study, the upregulation of RASAL2 was present in stomach adenocarcinoma, as well as other digestive system tumors such as hepatocellular carcinoma19, colon adenocarcinoma18, and rectal adenocarcinoma27. In our hands, RASAL2 silencing had no effect on KRAS expression or the downstream MEK signaling. On the other hand, we found that RASAL2 can directly bind to and inactivate PP2A, subsequently activating the AKT/β-catenin signaling axis. These results, including whole-transcriptome sequencing of human samples and in vitro and in vivo mouse models, support this novel role of RASAL2 signaling in H. pylori-related gastric tumorigenesis.
There are accumulating lines of evidence suggesting that extrinsic stimuli from the tumor microenvironment induce and maintain stem-like cell properties such as self-renewal and expansion in a subset of tumor cells44. Of note, several models have been proposed to elucidate how H. pylori-infection and inflammation enrich stem-like properties 45, 46. The Wnt/β-catenin signaling and the downstream gene, CDX1, have been reported to convert H. pylori-infected gastric epithelial cells into tissue stem-like progenitor cells47. The Wnt/β-catenin signaling maintains LGR5+/AXIN2+ cells in organoids and spheroids cultures to promote expansion and growth of gastrointestinal cancers31, 48, 49. Consistent with our observation of activation of β-catenin, we detected up-regulation of LGR5 and AXIN2, two important Wnt/β-catenin downstream targets, widely accepted as markers of stem cells in the intestine50 and stomach51. Our findings present a novel mechanism by which H. pylori-induced RASAL2 enriches cell expansion, tumor development, and chemoresistance properties, likely through upregulating the β-catenin signaling pathway.
Chemoresistance, a major clinical challenge leading to tumor recurrence and poor clinical outcome, is defined as the ability of cancer cells to survive and expand while escaping or coping with chemotherapeutics52. Although targeted therapy and immunotherapy approaches have revolutionized the treatment in many cancer types53, 54, the results in gastric cancer remain largely disappointing55. A key factor in chemoresistance is the presence of treatment refractory cancer cells with intrinsic or acquired stem-like cell survival and expansion properties56, 57. Our clinicopathological analysis indicated that RASAL2-positive gastric cancer patients had worse overall survival (OS) and recurrence-free survival (RFS) than RASAL2-negative patients. Furthermore, RASAL2-negative patients had more survival benefits from adjuvant chemotherapy (ACT) than RASAL2-positive patients, suggesting that RASAL2-positive patients are more resistant to ACT. Given our findings showing the role of RASAL2 in promoting tumor development in xenograft models and cellular expansion of spheroids and organoids, strategies targeting RASAL2 could eliminate RASAL2high treatment refractory cancer cells. Our data support this concept showing the efficacy of RASAL2 knockdown in sensitizing gastric cancer cells to chemotherapeutics.
In summary, our findings demonstrated, for the first time, that RASAL2 plays a critical role in H. pylori infection-induced gastric tumorigenesis. The NF-kB/RASAL2/PP2A/AKT/β-catenin signaling cascade promotes tumorigenic cell properties. High expression of RASAL2 may contribute to chemoresistance and predict poor prognosis of gastric cancer patients. These findings call for the development of RASAL2 inhibitors as a novel strategy for treating cancer patients as single agents or in combination with chemotherapeutics.
Supplementary Material
WHAT YOU NEED TO KNOW:
BACKGROUND AND CONTEXT:
H. pylori infection is the main risk factor for gastric cancer. This study provides new insights into the mechanisms underlying H. pylori infection-induced gastric tumorigenesis and chemoresistance.
NEW FINDINGS:
Overexpression of RASAL2 is common in human and mouse gastric cancers. H. pylori infection induced RASAL2 expression via activation of NF-κB binding to the RASAL2 promoter. RASAL2 mediated cancer cells’ expansion by interacting and inhibiting of PP2A leading to activation of the AKT/β-catenin signaling.
LIMITATIONS:
This study did not directly address the potential therapeutic significance of RASAL2 inhibitors.
IMPACT:
Our findings call for the development of RASAL2 inhibitors as a novel strategy for treating gastric cancer patients as single agents or in combination with chemotherapeutics.
Acknowledgments:
The authors would like to thank Changyin Feng of the Department of Pathology for assistance with assessing of human gastric tumor pathology.
Grant support:
This study was supported by grants from the U.S. National Institutes of Health (R01CA249949) and the U.S. Department of Veterans Affairs (1IK6BX003787 and I01BX001179). The use of shared resources was supported by the Sylvester Comprehensive Cancer Center (P30CA240139). This work’s content is solely the responsibility of the authors. It does not necessarily represent the official views of the Department of Veterans Affairs, National Institutes of Health, or the University of Miami.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors declare that they have no competing interests.
References
- 1.Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021;71:209–249. [DOI] [PubMed] [Google Scholar]
- 2.Infection with Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum 1994;61:177–240. [PMC free article] [PubMed] [Google Scholar]
- 3.Kiga K, Mimuro H, Suzuki M, et al. Epigenetic silencing of miR-210 increases the proliferation of gastric epithelium during chronic Helicobacter pylori infection. Nat Commun 2014;5:4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peterson AJ, Menheniott TR, O'Connor L, et al. Helicobacter pylori infection promotes methylation and silencing of trefoil factor 2, leading to gastric tumor development in mice and humans. Gastroenterology 2010;139:2005–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niwa T, Tsukamoto T, Toyoda T, et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res 2010;70:1430–40. [DOI] [PubMed] [Google Scholar]
- 6.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 2008;9:517–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prior IA, Hood FE, Hartley JL. The Frequency of Ras Mutations in Cancer. Cancer Res 2020;80:2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hewitt LC, Saito Y, Wang T, et al. KRAS status is related to histological phenotype in gastric cancer: results from a large multicentre study. Gastric Cancer 2019;22:1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng N, Goh LK, Wang H, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 2012;61:673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hatakeyama M Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe 2014;15:306–16. [DOI] [PubMed] [Google Scholar]
- 12.McLaughlin SK, Olsen SN, Dake B, et al. The RasGAP gene, RASAL2, is a tumor and metastasis suppressor. Cancer Cell 2013;24:365–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olsen SN, Wronski A, Castano Z, et al. Loss of RasGAP Tumor Suppressors Underlies the Aggressive Nature of Luminal B Breast Cancers. Cancer Discov 2017;7:202–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hui K, Gao Y, Huang J, et al. RASAL2, a RAS GTPase-activating protein, inhibits stemness and epithelial-mesenchymal transition via MAPK/SOX2 pathway in bladder cancer. Cell Death Dis 2017;8:e2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li N, Li S. RASAL2 promotes lung cancer metastasis through epithelial-mesenchymal transition. Biochem Biophys Res Commun 2014;455:358–62. [DOI] [PubMed] [Google Scholar]
- 16.Huang Y, Zhao M, Xu H, et al. RASAL2 down-regulation in ovarian cancer promotes epithelial-mesenchymal transition and metastasis. Oncotarget 2014;5:6734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng M, Bao Y, Li Z, et al. RASAL2 activates RAC1 to promote triple-negative breast cancer progression. J Clin Invest 2014;124:5291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan Y, Tong JHM, Lung RWM, et al. RASAL2 promotes tumor progression through LATS2/YAP1 axis of hippo signaling pathway in colorectal cancer. Mol Cancer 2018;17:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang JF, Zhao HP, Wang ZF, et al. Upregulation of RASAL2 promotes proliferation and metastasis, and is targeted by miR-203 in hepatocellular carcinoma. Mol Med Rep 2017;15:2720–2726. [DOI] [PubMed] [Google Scholar]
- 20.Zhu S, Soutto M, Chen Z, et al. Helicobacter pylori-induced cell death is counteracted by NF-kappaB-mediated transcription of DARPP-32. Gut 2017;66:761–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sayi A, Kohler E, Hitzler I, et al. The CD4+ T cell-mediated IFN-gamma response to Helicobacter infection is essential for clearance and determines gastric cancer risk. J Immunol 2009;182:7085–101. [DOI] [PubMed] [Google Scholar]
- 22.Park JW, Jang SH, Park DM, et al. Cooperativity of E-cadherin and Smad4 loss to promote diffuse-type gastric adenocarcinoma and metastasis. Mol Cancer Res 2014;12:1088–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Messeguer X, Escudero R, Farre D, et al. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002;18:333–4. [DOI] [PubMed] [Google Scholar]
- 24.Fornes O, Castro-Mondragon JA, Khan A, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 2020;48:D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koni M, Pinnaro V, Brizzi MF. The Wnt Signalling Pathway: A Tailored Target in Cancer. Int J Mol Sci 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song X, Xin N, Wang W, et al. Wnt/beta-catenin, an oncogenic pathway targeted by H. pylori in gastric carcinogenesis. Oncotarget 2015;6:35579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang W, Lu Y, Li X, et al. IPO5 promotes the proliferation and tumourigenicity of colorectal cancer cells by mediating RASAL2 nuclear transportation. J Exp Clin Cancer Res 2019;38:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 1992;257:1261–4. [DOI] [PubMed] [Google Scholar]
- 29.Seshacharyulu P, Pandey P, Datta K, et al. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett 2013;335:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wend P, Holland JD, Ziebold U, et al. Wnt signaling in stem and cancer stem cells. Semin Cell Dev Biol 2010;21:855–63. [DOI] [PubMed] [Google Scholar]
- 31.Hua F, Shang S, Yang YW, et al. TRIB3 Interacts With beta-Catenin and TCF4 to Increase Stem Cell Features of Colorectal Cancer Stem Cells and Tumorigenesis. Gastroenterology 2019;156:708–721 e15. [DOI] [PubMed] [Google Scholar]
- 32.Chi HC, Tsai CY, Wang CS, et al. DOCK6 promotes chemo- and radioresistance of gastric cancer by modulating WNT/beta-catenin signaling and cancer stem cell traits. Oncogene 2020;39:5933–5949. [DOI] [PubMed] [Google Scholar]
- 33.Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ben-Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 2008;40:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith BA, Balanis NG, Nanjundiah A, et al. A Human Adult Stem Cell Signature Marks Aggressive Variants across Epithelial Cancers. Cell Rep 2018;24:3353–3366 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soutto M, Peng D, Katsha A, et al. Activation of beta-catenin signalling by TFF1 loss promotes cell proliferation and gastric tumorigenesis. Gut 2015;64:1028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lefebvre O, Chenard MP, Masson R, et al. Gastric mucosa abnormalities and tumorigenesis in mice lacking the pS2 trefoil protein. Science 1996;274:259–62. [DOI] [PubMed] [Google Scholar]
- 38.Zhu S, Chen Z, Wang L, et al. A Combination of SAHA and Quinacrine Is Effective in Inducing Cancer Cell Death in Upper Gastrointestinal Cancers. Clin Cancer Res 2018;24:1905–1916. [DOI] [PubMed] [Google Scholar]
- 39.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 40.Plummer M, Franceschi S, Vignat J, et al. Global burden of gastric cancer attributable to Helicobacter pylori. Int J Cancer 2015;136:487–90. [DOI] [PubMed] [Google Scholar]
- 41.Yan M, Li X, Tong D, et al. miR-136 suppresses tumor invasion and metastasis by targeting RASAL2 in triple-negative breast cancer. Oncol Rep 2016;36:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng C, Ouyang Y, Lu N, et al. The NF-kappaB Signaling Pathway, the Microbiota, and Gastrointestinal Tumorigenesis: Recent Advances. Front Immunol 2020;11:1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou B, Zhu W, Jiang X, et al. RASAL2 Plays Inconsistent Roles in Different Cancers. Front Oncol 2019;9:1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nassar D, Blanpain C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu Rev Pathol 2016;11:47–76. [DOI] [PubMed] [Google Scholar]
- 45.Amieva M, Peek RM Jr. Pathobiology of Helicobacter pylori-Induced Gastric Cancer. Gastroenterology 2016;150:64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giannakis M, Chen SL, Karam SM, et al. Helicobacter pylori evolution during progression from chronic atrophic gastritis to gastric cancer and its impact on gastric stem cells. Proc Natl Acad Sci U S A 2008;105:4358–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujii Y, Yoshihashi K, Suzuki H, et al. CDX1 confers intestinal phenotype on gastric epithelial cells via induction of stemness-associated reprogramming factors SALL4 and KLF5. Proc Natl Acad Sci U S A 2012;109:20584–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho YH, Ro EJ, Yoon JS, et al. 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/beta-catenin pathway activation. Nat Commun 2020;11:5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai C, Zhu X. The Wnt/beta-catenin pathway regulates self-renewal of cancer stem-like cells in human gastric cancer. Mol Med Rep 2012;5:1191–6. [DOI] [PubMed] [Google Scholar]
- 50.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449:1003–7. [DOI] [PubMed] [Google Scholar]
- 51.Barker N, Huch M, Kujala P, et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 2010;6:25–36. [DOI] [PubMed] [Google Scholar]
- 52.Mansoori B, Mohammadi A, Davudian S, et al. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv Pharm Bull 2017;7:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nikolaou M, Pavlopoulou A, Georgakilas AG, et al. The challenge of drug resistance in cancer treatment: a current overview. Clin Exp Metastasis 2018;35:309–318. [DOI] [PubMed] [Google Scholar]
- 54.Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature 2019;575:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marin JJG, Perez-Silva L, Macias RIR, et al. Molecular Bases of Mechanisms Accounting for Drug Resistance in Gastric Adenocarcinoma. Cancers (Basel) 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nunes T, Hamdan D, Leboeuf C, et al. Targeting Cancer Stem Cells to Overcome Chemoresistance. Int J Mol Sci 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phi LTH, Sari IN, Yang YG, et al. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int 2018;2018:5416923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.