

Abstract
Background and Aims:
Colonic eosinophilia, an enigmatic finding often referred to as eosinophilic colitis (EoC), is a poorly understood condition. Whether EoC is a distinct disease or a colonic manifestation of eosinophilic gastrointestinal diseases (EGIDs) or inflammatory bowel disease (IBD) is undetermined.
Methods:
Subjects with EoC (n=27) and controls (normal [NL, n=20], Crohn disease [CD, n=14]) were enrolled across sites associated with the Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR). EoC was diagnosed as colonic eosinophilia (ascending ≥100, descending ≥85, sigmoid ≥65 eosinophils/high-power field) with related symptoms. Colon biopsies were subjected to RNA sequencing. Associations between gene expression and histologic features were analyzed with Spearman correlation; operational pathways and cellular constituents were computationally derived.
Results:
We identified 987 differentially expressed genes (EoC transcriptome) between EoC and NL (>1.5-fold change, P < .05). Colonic eosinophil count correlated with 31% of EoC transcriptome, most notably with CCL11 and CLC (r=0.78 and 0.77, P < .001). Among EoC and other EGIDs, there was minimal transcriptomic overlap; and minimal evidence of a strong allergic type 2 immune response compared with other EGIDs. Decreased cell-cycle and increased apoptosis in EoC compared with NL were identified by functional enrichment analysis and immunostaining using Ki-67 and cleaved caspase-3. Pericryptal circumferential eosinophil collars were associated with the EoC transcriptome (P < .001). EoC transcriptome-based scores were reversible with disease remission and differentiated EoC from IBD, even after controlling for colonic eosinophil levels (P < .0001).
Conclusion:
We established EoC transcriptomic profiles, identified mechanistic pathways, and integrated findings with parallel IBD and EGID data. These findings establish EoC as a distinct disease compared with other EGIDs and IBD, thereby providing a basis for improving diagnosis and treatment.
Keywords: colitis, eosinophil, eosinophilic colitis, inflammatory bowel disease, transcriptome
Graphical Abstract
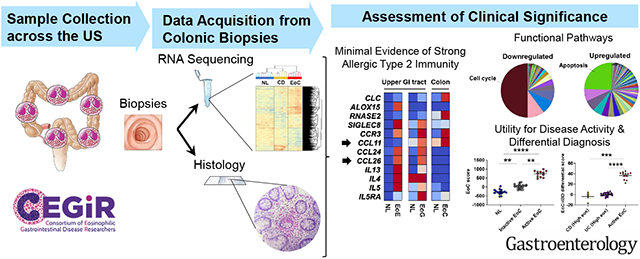
Lay summary:
We established a distinct transcriptomic profile in EoC, identified functional molecular pathways, and assessed its clinical significance.
INTRODUCTION
Eosinophilic gastrointestinal diseases (EGIDs) are clinicopathologically characterized by marked eosinophilic infiltration of the gastrointestinal (GI) tract with related symptoms and are classified according to the site of infiltration: eosinophilic esophagitis (EoE), eosinophilic gastritis (EoG), eosinophilic duodenitis (EoD), eosinophilic gastroenteritis (EGE), and eosinophilic colitis (EoC).1 Among EGIDs, EoC represents the least frequent manifestation (1.6–2.1 per 100,000 persons) and least well-understood disorder;2-4 however, patients with EoC have a higher disease burden of symptoms and comorbidities than patients with EoE, the most common EGID.5 Due to a lack of agreed-upon diagnostic criteria, EoC is currently defined as a clinicopathologic disorder that primarily affects the colon with eosinophil-rich inflammation in the absence of known causes of eosinophilia.6, 7 This diagnostic definition is problematic because there are numerous more common diseases associated with colonic eosinophilia, most notably inflammatory bowel disease (IBD), and the relationship between IBD and eosinophilic infiltration in GI biopsies is unclear. Further knowledge of EoC characteristics and pathogenesis may lead to consensus criteria for diagnosis and to reduced disease burden.
With regard to the molecular causes of EGIDs, substantial progress has been made using whole-genome transcript expression profiling (transcriptome) of tissue biopsies from patients with EoE,8-12 and, more recently, from patients with EoG.13-15 Cumulative evidence has elucidated specific molecular, cellular, and immune mechanisms involved in EoE and EoG pathogenesis,16, 17 including overproduction of type 2 cytokines (e.g., IL-5, IL-13) and IL-13–induced gene products (e.g., CCL26/eotaxin-3, CAPN14).18, 19 In contrast, EoC pathogenesis remains poorly understood due to EoC’s relative rarity and challenging differential diagnosis.
The differential diagnosis for increased eosinophil density in colonic mucosa is clinically problematic because colonic eosinophils are present during homeostasis unlike esophageal eosinophils; the eosinophil level is highest in the ascending colon, tapers to lower levels in the recto-sigmoid colon,20, 21 and increases during inflammation in many conditions.16 As eosinophil-rich inflammation is not exclusive to EoC, primary EoC is a diagnosis that can be made only after all other known causes for increased colonic mucosal eosinophils have been eliminated.6, 7, 16 Distinguishing EoC from other causes of GI eosinophilia (e.g., hypereosinophilic syndrome, IBD, infection, and autoimmune disorders) is important because the therapeutic strategy may substantially differ.6, 7, 16 If EoC were similar to other EGIDs, elimination diets and anti–type 2 cytokine therapy would be appropriate therapies; conversely, if EoC were similar to IBD, distinct anti-inflammatory and/or biologics (e.g., anti–TNF) therapy would be preferred. The lack of a way to accurately differentiate these colonic states is increasingly recognized as a clinical conundrum.22
Herein, we aimed to answer three fundamental questions in the field: (1) Is EoC a distinct disease entity?; (2) What is its molecular and cellular relationship to other EGIDs and IBD?; and (3) Does EoC show evidence of allergic type 2 immunity? Accordingly, we examined pediatric and adult patients with EoC across multiple sites associated with the Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR)23 and subjected colonic biopsies to genome wide transcriptomic profiling and parallel histological analysis, followed by pathway interrogation, and comparison of the derived findings to other EGIDs and IBD.
Materials and Methods
Study Design and Participants
This study was conducted in CEGIR,23 a national collaborative network of academic centers caring for and researching adults and children with EGIDs. The CEGIR observational study, Outcome Measures in Eosinophilic Gastrointestinal disorders Across the ages (OMEGA), is a longitudinal cohort study investigating the natural history of EoE, EoG, EoD, EGE, and EoC during routine clinical care. Demographic, clinical, endoscopic, and histologic data and GI tissue were prospectively collected starting from 2015; all samples from any CEGIR site that contributed subjects with EoC were used (n = 5 sample-providing institutions) (Supplementary Table S1). The clinical features of subjects were determined during a standard-of-care evaluation using standardized intake forms. All subjects’ clinical data were stored at the Rare Diseases Clinical Research Network (RDCRN) Data Management and Coordinating Center (University of South Florida in Tampa, FL [2015-2019] and Cincinnati Children’s Hospital Medical Center [CCHMC; 2020–2024]).
An EoC diagnosis was made using the combination of: (1) presence of symptoms; symptoms include (but are not limited to) hematochezia, bloody/non-bloody diarrhea, tenesmus, abdominal pain, (2) a history of clinical features indicative of colonic inflammation, such as anemia, peripheral eosinophilia, hemoccult positive stool, EGID, and/or allergic diseases (allergic rhinitis, asthma, food allergy, eczema, or other allergic features suggestive of atopic disease), and (3) colonic mucosal eosinophilia (ascending colon ≥100 eosinophils/high-power field [HPF], descending colon ≥85 eosinophils/HPF, and/or sigmoid colon ≥65 eosinophils/HPF) based on 2X the upper limit of normal for each anatomic site in normal biopsies.24, 25 Inclusion and exclusion criteria are detailed in Supplementary Table S2. In each case, alternative causes of mucosal eosinophilia were ruled out including proctocolitis in infancy; negative tests typically included stool culture for pathogenic bacteria or parasites, viral antibody titers and/or PCR, Celiac and IBD serology.24 For diagnosed EoC cases, EoC disease activity was defined by colonic biopsy eosinophil counts meeting (active EoC) or being lower than the above colonic eosinophilia criteria (inactive EoC). The patients with inactive EoC showed colonic eosinophilia more than the threshold level in the past but less than the threshold level when the biopsy samples were analyzed. Subjects with EoC and concomitant EGID involving other GI segments (esophagus: ≥15 eosinophils/HPF, stomach: ≥30 eosinophils/HPF in 5 HPF) were not excluded.
Non-EoC control subjects (normal [NL], Crohn disease [CD] as an IBD-representative/spectrum disease) from the Cincinnati Center for Eosinophilic Disorders (CCED) EGID database between 2015–2019 included children and adults who had undergone endoscopy, had no history of EoC nor pathologic evidence of EoC surveyed during the index endoscopy, and had colonic biopsies collected for research purposes during the index endoscopy. NL were patients who underwent endoscopic examination due to digestive symptoms but did not show colonic eosinophilia. NL subjects having treatments because of concomitant diseases [e.g., gastroesophageal reflux disease (GERD) and IgE-mediated food allergy] were not excluded. A CD diagnosis was made using previously published guidelines.26 Features include a variable combination of: (1) clinical signs and symptoms including abdominal pain, diarrhea, rectal bleeding, growth delay, and pubertal delay, (2) physical findings including abdominal tenderness, perirectal skin tags, perirectal fistula, and erythema nodosum, (3) endoscopic findings of aphthous, linear or stellate ulcerations, cobble stoning, skip lesions, and strictures in the ileum or colon, (4) histologic findings including ulceration, crypt abscesses, non-caseating granuloma, focal changes within biopsy, and patchy inflammation, and (5) cross-sectional imaging findings including mural thickening, hyperemia, abnormal luminal caliber, altered peristalsis, fibro-fatty proliferation, regional lymphadenopathy, and sinus tracts/fistulae. CD diagnosis and disease activity was based on a combination of the clinical, endoscopic, and histologic characteristics by gastroenterologists and pathologists at CCHMC. The inflammation status (inflamed, non-inflamed) of subjects was defined by assessing histologic features of chronicity and quantifying acute inflammation. A subset of patients with CD who also had a high peak colonic eosinophils/HPF (≥65 eosinophils/HPF) was defined as CD-high colonic eosinophils.
This study was approved by the institutional review boards of the participating institutions via a central institutional review board at CCHMC. An informed consent/assent form was signed by the subjects and/or their legal guardians per institutional guidelines prior to inclusion in the study.
Molecular Evaluation
RNA sequencing was performed using the QuantSeq 3’ mRNA Seq Library Prep Kit FWD for Illumina (Lexogen) as previously described.11 Briefly, total RNA was extracted with the miRNeasy kit (Qiagen, Valencia, Calif) and evaluated with an Agilent Bioanalyzer by the CCHMC Gene Expression Core. Data analyses were performed using DESeq2 in CLC Genomics Workbench software (CLC bio, Waltham, MA, USA) and GeneSpring software ver. 14.9 (Agilent Technologies). Transcripts per kilobase million (TPM) were assessed for statistical significance. Data are available at EGIDExpress (https://egidexpress.research.cchmc.org/data/). Functional enrichment analyses were performed with the ToppGene suite and CluGO.27, 28 Cell type enrichment analysis was performed with xCell.29 EoC score was calculated by summing the normalized expression values of genes dysregulated in the EoC transcriptomes. A real-time reverse-transcription quantitative polymerase chain reaction (RT-qPCR) was performed to determine mucosal expression of genes associated with type 2 inflammation in patients with EGIDs.15, 30 As another relevant disease control, publicly available colonic transcriptome datasets from patients with ulcerative colitis (UC) having active colitis and patient clinical data were comprehensively searched and obtained by the BaseSpace correlation engine (Illumina Inc., San Diego, CA, USA). One dataset having colonic eosinophil counts (GSE109142)31 were also used for EoC score analysis.
Histologic Features
Colonic biopsies were assessed for the peak eosinophil counts and other histologic features of EoC. Hematoxylin and eosin (H&E)–stained biopsy slides from NL, CD, and EoC were reviewed by CEGIR pathologists blinded to the diagnosis. Standardization across centers was performed by a view finder that mimicked a round high-power field and measured 0.27 mm2, an area that is commonly covered at 40X magnification. Histologic features in images of all submitted colon biopsies were as follows: acute crypt abscess, acute cryptitis, acute inflammation, crypt architectural abnormalities, crypt dropout/loss, crypt epithelial injury, crypts partly destroyed by eosinophilic inflammation, eosinophil crypt abscess, eosinophil cryptitis, eosinophils in muscularis mucosa/submucosa, eosinophils in surface epithelium, granulomas, lamina propria eosinophil sheets, lymphocytes in surface epithelium, overall eosinophilic inflammation, pericryptal circumferential eosinophil collars, subcryptal eosinophil aggregates, subcryptal lymphoplasmacytes, and surface epithelial injury. Each feature was scored using a 3-point scale (0 = absent, 1 = mild/moderate, 2 = marked) (Supplementary Table S3).
Immunostaining of Biopsy
Biopsies were stained as previously described. 13, 32 Ki-67 (a proliferation marker, 790-4286, Roche) or cleaved caspase-3 (an apoptotic marker, ab2302, Abcam) were evaluated for immunohistochemical staining. Ki-67 (MA5-14520; Invitrogen) and phospho-histone H3 (#9706; Cell Signaling Technology) were evaluated for immunofluorescent staining.
Statistical Analysis
Data are presented as n (%) or median (interquartile range [IQR]) unless otherwise stated. Missing data were excluded from all formal statistical analyses. Nonparametric correlation analysis was performed using Spearman’s rank correlation coefficient. For continuous data, statistical significance was determined by the Mann-Whitney U test (nonparametric test, 2 groups) or Kruskal-Wallis test followed by a Dunn multiple-comparison test (nonparametric test, ≥3 groups). Benjamini-Hochberg correction was applied for multiple testing to control the false-discovery rate (FDR). For categorical data, the chi-square test was used. A significant P value was defined as <0.05.
For detailed information, see the Supplementary Material and Methods.
Results
Subject Characteristics
Eighty-seven colonic biopsies (n = 31 EoC [12 active, 19 inactive], 27 CD [16 inflamed, 11 non-inflamed], 29 NL) from 61 subjects (n = 27 EoC, 14 CD, 20 NL) were analyzed, with instances of multiple biopsies (n = 3 EoC, 13 CD, 8 NL subjects) being obtained from different colon sites during a single endoscopy. Demographic and clinical characteristics of the study cohort stratified by group (EoC, CD, NL) are detailed in Table 1 and Supplementary Table S4.
Table 1.
Demographic and clinical characteristics of study subjects.
| NL | CD | EoC | P value | |
|---|---|---|---|---|
| Subjects (n) | 20 | 14 | 27 | |
| Demographic features | ||||
| Age at biopsy (min.–max. years) | 15.1 (4.3–44.9) | 17.3 (11.7–21.6) | 14.0 (7.1–64.4) | .26 |
| Gender (% Male) | 8 (40%) | 7 (50%) | 14 (52%) | .71 |
| Race (% White) | 18 (90%) | 14 (100%) | 25 (93%) | .49 |
| Colonic eosinophil counts | ||||
| Peak (eos/HPF) | 28.5 (23.8–37.5) | 48.5 (31.0–68.3) | 55.5 (23.8–100.8) | .025 |
| Range (min.–max. eos/HPF) | 2–43 | 16–110 | 9–187 | |
| Biopsies (n) | ||||
| Total | 29 | 27 | 31 | - |
| Active or inflamed | - | 16 | 12 | - |
| Normal | 16 | - | - | - |
| Right colon | 10 | 14 | 21 | - |
| Left colon | 19 | 13 | 10 | - |
| History of EGIDs | ||||
| EoE | - | - | 15 (56%) | - |
| EoG | - | - | 5 (19%) | - |
| EoC | - | - | 27(100%) | - |
| History of atopy | ||||
| Atopy (any) | 15 (75%) | 8 (57%) | 15 (56%) | .36 |
| Asthma | 7 (35%) | 2 (14%) | 6 (22%) | .36 |
| Allergic rhinitis | 13 (65%) | 5 (36%) | 11 (41%) | .16 |
| Eczema | 8 (40%) | 3 (21%) | 9 (33%) | .52 |
| Food allergy | 5 (25%) | 0 (0%) | 7 (26%) | .11 |
| Treatment at biopsy | ||||
| Ongoing diet therapy | 9 (45%) | 2 (14%) | 12 (44%) | .12 |
| Proton pump inhibitor | 11 (55%) | 0 (0%) | 9 (33%) | .004 |
| Topical steroids | 3 (15%) | 0 (0%) | 8 (30%) | .059 |
| Systemic steroids | 0 (0%) | 4 (29%) | 2 (7%) | .019 |
| Immune modulator | 0 (0%) | 2 (14%) | 0 (0%) | < .001 |
| Biologies | 0 (0%) | 9 (64%) | 0 (0%) | < .001 |
Data are n (%) or median (interquartile range [IQR]) unless otherwise stated.
CD, Crohn disease; EoC, eosinophilic colitis; EGIDs, eosinophilic gastrointestinal diseases; EoE, eosinophilic esophagitis; EoG, eosinophilic gastritis; eos/HPF, eosinophils per high-power microscopic field; max., maximum; min., minimum; NL, normal.
Ages ranged from 4–64 years, with 43 pediatric (70.5%) and 18 adult (29.5%) subjects. There was a similar proportion of both genders, with 29 male (47.5%) and 32 female (52.5%) subjects. Most subjects self-identified as White (93.4%). Many subjects had a history of atopy (any allergic disease, 62.3%), such as asthma, allergic rhinitis, atopic dermatitis, and food allergy (24.6%, 47.5%, 32.8%, and 19.7%, respectively). Peak colonic eosinophil counts ranged from 2–187 eosinophils/HPF (active EoC 69–187, inactive EoC 9–44, CD 9–110, NL 2–52 eosinophils/HPF).
Focusing on subjects with EoC (n = 27), 15 (56%) had concurrent eosinophilia in the esophagus, 5 (19%) in the stomach, and 1 (0.4%) in both the esophagus and stomach. Whereas demographic features (age at biopsy, gender, race) were similar at baseline among EoC, CD, and NL subjects, EoC subjects had significantly higher peak colonic eosinophil counts (P = .025) and a higher percentage of treatment (proton pump inhibitor therapy at time of biopsy, mainly for concurrent eosinophilia in the esophagus) than CD and NL subjects.
Identification of EoC transcriptome
First, we molecularly profiled EoC employing a stringent diagnostic criteria (more than twice the normal number of mucosal eosinophils in colon).24, 33 To minimize variability and detect meaningful gene dysregulation, we examined the ascending colon, which usually has higher eosinophil counts among colon sites.20 We generated an RNA sequencing data set from colonic tissue of active EoC (n = 6) and NL (n = 8) and compared gene expression. We identified 987 differentially dysregulated genes in active EoC versus NL biopsies (≥ 1.5-fold change, FDR P < .05) (Figure 1A). Unsupervised clustering analysis showed separation between active EoC and NL (Figure 1B). Of these gene signatures (e.g., EoC transcriptome), 577 transcripts were upregulated and 410 were downregulated in active EoC compared to NL (Supplementary Table S5). Notably, despite clinical heterogeneity, there were no substantial molecular differences in several comparisons, such as EoC with coexisting EoE vs. EoC alone, pediatric vs. adult patients, atopic vs. non-atopic, and treated vs. untreated patients (Supplementary Figure S1).
Figure 1. Distinct, conserved pattern of gene expression in active EoC colonic tissue.

A, Volcano plot (red, upregulated; blue, downregulated) of expression profiles of differentially dysregulated genes between NL and subjects with active EoC (EoC, FDR P < .05, ≥1.5-fold change). B, Clustering analysis based on 987 differentially expressed genes (EoC transcriptome). C, Venn diagram of the number of genes dysregulated in EoC and CD transcriptomes (Supplemental Figure S1). D, Colonic transcriptome data on NL (blue), subjects with inflamed CD (CD, yellow), and subjects with active EoC (red) reduced to 3-dimensional presentation by multidimensional scaling analysis for visual presentation of the expression distance between samples. E, Heat map (red, upregulated; blue, downregulated) and clustering analysis of the expression profiles of the 1,847 genes from the EoC and/or CD transcriptomes with differentially dysregulated expression in active EoC and/or inflamed CD vs. NL (FDR P < .05, ≥1.5-fold change). B and E, each column represents an individual subject or control. CD, Crohn disease; EoC, eosinophilic colitis; NL, normal; FDR, false discovery rate.
Subsequently, we identified a core gene set for subjects with inflamed CD having active colitis to compare with the EoC transcriptome. Using the same approach as for EoC, we identified the 996-gene CD transcriptome (Supplementary Figure S2 and Table S5), which included previously identified genes and pathways associated with inflamed CD (e.g., IBD; C0021390 at DisGeNET)34 (Supplementary Figure S3). Comparing the EoC and IBD [CD, from this study; ulcerative colitis (UC), from 14 published data as summarized in Supplementary Table S6] transcriptomes demonstrated that EoC was distinct (Figure 1C and Supplementary Figure S4), and unsupervised principal component analysis (PCA) and hierarchical clustering analysis demonstrated robust separation of active EoC, inflamed CD, and NL groups (Figure 1D-E).
EoC transcriptome associates with colonic eosinophilia and distinguishes EoC from other EGIDs
The peak colonic eosinophil count from ascending to sigmoid colon significantly correlated with 31% of the EoC transcriptome (Supplementary Table S5), most notably with the expression of eosinophil chemoattractant gene C-C motif chemokine ligand 11 (CCL11, r = 0.78, P < .001) and the eosinophil-specific gene Charcot-Leyden crystal (CLC, r = 0.77, P < .001) (Figure 2A-B). CCL11 and CLC correlated with each other (r = 0.63, P < .001) (Figure 2C). Conversely, the peak colonic eosinophil count correlated with 8% of the CD transcriptome (Supplementary Table S5). The number of genes correlating with the peak colonic eosinophil count significantly differed between the EoC and CD transcriptomes (P < .001) (Figure 2D).
Figure 2. EoC transcriptome associates with colonic eosinophilia and distinguishes EoC from NL and other EGIDs.

A–C, correlation plots for peak colonic eosinophil count and colonic expression of CLC and CCL11, the genes that most correlated with EoC eosinophil count. D, Correlation of peak colonic eosinophil counts with each of the EoC and CD transcriptomes. ****p < .0001, using the chi-square test. E, Venn diagram of the number of genes dysregulated in EGID transcriptomes (EoE, EoG, EoC). F–H, Comparisons of type 2–related gene expression by RT-qPCR in active EGIDs [esophagus (EoE n=82, NL n=50), stomach (EoG n=21, NL n=20), colon (EoC n=12, NL n=16)]. F, eosinophil and mast cells genes; G, eosinophil chemotactic chemokines; H, type 2 cytokines. Data presented as median with interquartile range. Markers represent individual samples. *P < .05, **P < .01, and ****P < .0001, using Mann–Whitney U test. CD, Crohn disease; EoC, eosinophilic colitis; EoE, eosinophilic esophagitis; EoG, eosinophilic gastritis; EGIDs, eosinophilic gastrointestinal diseases; NL, normal; GI, gastrointestinal; HPF, high-power microscopic field; TPM, transcripts per kilobase million.
To determine the relationship between EoC and other EGIDs, we compared the EoC colonic transcriptome with the previously published EoE esophageal and EoG gastric transcriptomes.9, 15 Notably, there was almost no overlap among transcriptomes of these three EGIDs (EoE, EoG, EoC) (9 genes; 1% of EoC transcriptome) (Figure 2E). The common EGID genes, including CLC, which is a hallmark of active eosinophilic inflammation,35 were regulated in similar manners in subjects with EoE, EoG, and EoC (i.e., upregulated in EoE, EoG, and EoC: ALOX5AP, CD9, CLC, CSF2RB, CXCL1, GAPT, MMP12, NCF2, and SOCS1). For upregulated genes, there were some genes in the EoC transcriptome that modestly overlapped with the EoE and EoG transcriptomes whereas downregulated genes in the EoC transcriptome did not overlap with other EGIDs (Supplementary Table S7). Comparing type 2–related gene expression by RT-qPCR (Figure 2F-H) showed that the main chemotactic factor for EoC (lower GI EGID) was likely CCL11, whereas it was CCL26 for both EoE and EoG (upper GI EGIDs) (Figure 2G). Expression of type 2 cytokines (e.g. IL13, IL4, and IL5) were increased in patients with EoE and EoG but not in EoC, although there was substantial heterogeneity (Figure 2H).
Functions and cell types enriched in the EoC transcriptome
To identify EoC-associated molecular pathways, we performed functional annotation enrichment analyses. The highest enrichments were decreased cell cycle functions and increased apoptosis pathways (Figure 3A-B and Supplementary Table S8). Processes downregulated of the EoC transcriptome showed a decrease in cell cycle transcripts (P = 8.6E-7), including proteasome genes. Upregulated processes of the EoC transcriptome were enriched for apoptosis signaling (P = 1.9E-4), including ribosomal genes. There was enrichment in granulocyte activation and degranulation and innate immunity (Supplementary Table S8). Immunohistochemically confirming the pathway analyses showed reduced colonic epithelial and lamina propria cells with positive Ki-67 staining (cell proliferation marker) in biopsy specimens from patients with EoC compared with NL and CD (P < .001) (Figure 3C-D). The number of examined active EoC biopsies was small, but generally Ki-67+ cells appeared reduced in crypts located in areas of dense eosinophilic inflammation. Immunofluorescent staining confirmed reduced Ki-67 staining and also showed decreases in other proliferation markers (phospho-histone H3) (Supplementary Figure S5). In contrast, the number of cleaved caspase-3+ (cell apoptosis marker) cells was significantly increased in EoC and CD versus NL biopsy specimens (P = .02) (Figure 3, C, D).
Figure 3. Functions and cell types enriched in EoC transcriptome.

A–B, Functional annotation enrichment analyses of 410 downregulated (A) and 577 upregulated (B) genes of EoC transcriptome using CluGO overview charts and showing the 5 most significant terms in biological process by ToppGene (full list; Supplementary Table 8). C–D, Decreased cell proliferation and increased apoptosis in patients with EoC. Representative photographs and quantitative evaluation of Ki–67+ (proliferating) and cleaved caspase-3+ (apoptotic) colonic cells from NL, inflamed CD, and active EoC. Ki–67+: left, 4X; right, 20X. Cleaved caspase-3+: left, 10X; right, 20X. *P < .05, versus NL. E–F, Specific increase of gene expression–estimated proportion of cell types in EoC (E) and CD (F). Data presented as mean ± SEM. *P < .05, **P < .01, ***p < .001, ****p < .0001, using Kruskal-Wallis test followed by Dunn multiple-comparison test. aDC, activated dendritic cells; CD, Crohn disease; CD4+ Tem, CD4+ effector memory T cells; EoC, eosinophilic colitis; MPP, multipotent progenitors; NL, normal; FDR, false discovery rate; SEM, standard error of mean.
Further evaluating the relative composition of immune cell subsets, epithelia, and other stromal cell types in EoC, we applied a computational gene expression deconvolution approach using xCell.29 Of the 64 cell types represented by gene expression, several immune cells were specifically increased in EoC and CD. Active EoC had increased gene expression associated with eosinophils, basophils, CD4+ effector memory T cells, and multipotent progenitors, whereas inflamed CD had increased gene expression associated with monocytes, plasma cells, neutrophils, activated dendritic cells, and megakaryocytes (Figure 3E-F).
Colonic histologic features and transcript association with disease
All subjects with active EoC showed marked, though uneven, colonic eosinophilic inflammation (Figure 4A) even within the same biopsy specimen. In subjects with EoC, the ascending colon had higher peak eosinophil counts than the left colon (mean 96.3 eosinophils/HPF vs. 43.7 eosinophils/HPF, respectively; P = .007), consistent with the normally higher counts in the right than left colon, whereas histologic features other than eosinophil count were similar regardless of disease activity (active vs. inactive EoC). Notably, tissue eosinophilia with no additional crypt architectural abnormalities was the most common finding (87%) in EoC colonic biopsies.
Figure 4. Colonic histologic features and associations with colonic transcripts.

A, Hematoxylin and eosin–stained colon biopsy specimen of a representative subject with EoC (200X magnification). Eosinophils densely populate crypts (arrow) and pericryptal circumferential collars (arrowhead). B, Histologic feature clustering in colon biopsies with features arranged to ensure that members of the same cluster are adjacent in the correlation plot and in the same order as in the cluster members. Color map shows correlations among histologic features; darker red shades indicate stronger positive correlations. C, Comparison of histologic features among NL, inflamed CD, and active EoC. Data are mean ± SEM. *P < .05, **P < .01, and ****p < .0001, using Kruskal-Wallis test followed by Dunn multiple-comparison test. D, Spearman r correlations of eosinophilic histologic features with cell proliferation/apoptosis in the epithelium. *P < .05. E, Hierarchic relationships between histologic features on the basis of EoC transcriptome gene expression profile correlations, showing a Spearman r–based heat diagram for gene-level correlations. Darker red shades indicate stronger positive correlations, whereas darker blue shades indicate stronger negative correlations. EoC, eosinophilic colitis; CD, Crohn disease; NL, normal; SEM, standard error of mean.
Assessing the relationships among the EoC colonic histologic features, we generated a correlation plot with clustering arrangement (Figure 4B). Consistent with features commonly reported by pathologists examining such biopsies, there were strong correlations for inflammatory and structural changes in crypts; the most significant was between crypt epithelial injury and crypt dropout/loss (r = 0.80). Also, there were significant correlations within eosinophilic features, the most significant being between pericryptal circumferential eosinophil collars and lamina propria eosinophil sheets (r = 0.52). However, possibly due to the low occurrence, features related to eosinophilic and acute crypt abscess had low correlations with other features. Some features were included to distinguish EoC from IBD, including CD, and were not expected to correlate with EoC-related features; for example, sarcoid-like granulomas are a characteristic finding in CD but not EoC and therefore are not expected to correlate with EoC histopathology.
Notably, some colonic histologic features specifically associated with diseases, reflecting the intent of the histopathologic examination to distinguish among various colonic diseases. Among the histologic features, 5 features showed differences among the active EoC, inflamed CD, and NL. As expected, overall eosinophilic inflammation, pericryptal circumferential eosinophil collars, and eosinophilic cryptitis were significantly increased in active EoC compared with inflamed CD or NL (Figure 4C, upper), whereas acute cryptitis and acute inflammation were significantly increased in inflamed CD compared with active EoC or NL (Figure 4C, lower).
To understand the potential link between eosinophil-associated histologic features and the identified EoC-related functional pathways, we assessed correlations between eosinophilic histologic features and cell proliferation (Ki-67) and apoptosis (cleaved caspase-3) in the epithelium. Pericryptal circumferential eosinophil collars were negatively correlated with cell proliferation (r = −0.45, P < .05) and positively correlated with apoptosis (r = 0.48, P < .05) (Figure 4D), suggesting that epithelial-eosinophil cross-talk occurs in areas of eosinophilic collar formation.
Further dissecting the molecular basis for colonic histopathology in EoC, we evaluated associations between the EoC transcriptome and histologic features using Spearman r at the gene level (Figure 4E). We observed that histologic features commonly observed in EoC biopsies, such as overall eosinophilic inflammation, pericryptal circumferential eosinophil collars, and lamina propria eosinophil sheets, highly correlated with the EoC transcriptome and clustered together. Genes associated with each major histologic feature (r > 0.3, P < .05) showed enrichment in several biological processes: overall eosinophilic inflammation, purine ribonucleotide biosynthetic process (P = 2.85E-07); pericryptal circumferential eosinophil collars, mitochondrion organization (P = 3.24E-07); and lamina propria eosinophil sheets, protein targeting to endoplasmic reticulum (P = 8.63E-06) (Supplementary Table S9). Overall, we found that EoC had unique pathogenic gene sets and histologic manifestations, suggesting clinical utility of these features because of correlation with pathogenic gene sets.
EoC transcriptome as a function of differential diagnosis and disease activity
Generating quantitative values to reflect molecular changes, we developed an EoC score by summing the normalized expression values of the dysregulated EoC transcriptome genes (987) (Figure 5A) to distinguish active EoC from other conditions and quantify EoC severity.
Figure 5. EoC transcriptome as a function of disease activity and differential diagnosis.

A, Schematic summary of EoC score generation based on dimensionality reduction of the EoC transcriptome to distinguish active EoC vs. NL and quantify EoC disease severity. B, Discovery and replication of the EoC score with independent patients and from different colon sites (discovery: ascending, replication: descending/sigmoid colon). Peak colonic eosinophil count (left) and EoC score (right) are shown. Data are mean ± SEM. ***P < .001, using Mann–Whitney U test. C, EoC score as a function of disease activity in EoC. Peak colonic eosinophil count (left) and the EoC score (right) are shown. Data are mean ± SEM. **P < .01, ***p < .001, and ****p < .0001, using Kruskal-Wallis test followed by Dunn multiple-comparison test. D, Unsupervised principal component analysis of the EoC transcriptome showed complete separation of active EoC from inactive EoC and controls, whereas controls and inactive EoC overlapped. E, Comparison between active EoC and the challenge cases of IBD (CD and UC) with high colonic eosinophil count (High eos). Peak colonic eosinophil count (left) and the EoC-IBD differential score (right) are shown. The dashed line indicates 65 eosinophils/HPF. Data are mean ± SEM. NS, not significant, ***P < .001, and ****P < .0001, using Mann–Whitney U test. F, A receiver operating characteristic curve analysis showing utility of the EoC-IBD differential score to differentiate active EoC from IBD (CD and UC) (High eos). AUC, area under the curve; EoC, eosinophilic colitis; IBD, inflammatory bowel disease; CD, Crohn disease; UC, ulcerative colitis; NL, normal; HPF, high-power microscopic field; SEM, standard error of mean.
The EoC score was increased in patients with active EoC compared to non-EoC (P < .001) (Figure 5B, discovery). This finding was replicated in an independent patient cohort, with EoC and non- EoC, regardless of the colon sites (descending and sigmoid colon, P < .001) (Figure 5B, replication).
Exploring the potential reversibility of the EoC transcriptome according to disease activity, we compared the EoC score among active EoC, inactive EoC, and NL. Similar to the peak colonic eosinophil count (P < .001) (Figure 5C, left), the EoC score was specifically increased in patients with active EoC compared to non-EoC patients and patients with inactive EoC (P < .001) (Figure 5C, right; 5D).
We created a score with the use of a more limited number of genes by using different cut-offs (e.g., 5-fold change, 3-fold change). Although these gene-subset EoC scores (5-fold change or 3-fold change, respectively) showed similar results (Supplementary Figure S6), the EoC score based on the entire EoC transcriptome (987 genes) showed a better correlation with peak colonic eosinophil counts (Spearman r = 0.63, P < .0001).
Finally, assessing the potential utility of the EoC transcriptome for definitive diagnosis, we utilized our dataset (EoC and CD) and one dataset (UC) having colonic eosinophil counts (GSE109142)31 to generate a modified EoC score, the EoC-IBD differential score, for differential diagnosis against clinically challenging cases. Genes for the modified EoC score were selected from the EoC transcriptome on the basis of the following considerations: dysregulation between EoC and IBD defined by P values and fold changes and bidirectional changes of gene expression. On the basis of the EoC-IBD differential score derived from 17 genes (Supplementary Table S10), we compared active EoC and a subset of inflamed IBD (CD and UC) having high colonic eosinophil levels (clinically challenging cases). Although there was no difference in the peak colonic eosinophil count between active EoC and inflamed IBD with high colonic eosinophilia (P = .211) (Figure 5E, left), the EoC-IBD differential score separated these groups (P < .0001) (Figure 5E, right). A receiver operating characteristic curve analysis demonstrated excellent diagnostic merit for the EoC-IBD differential score (P = .0001, AUC = 1.00) (Figure 5F).
DISCUSSION
Herein, we report fundamental information about the molecular and histologic features of EoC. First, we defined the EoC transcriptome, a core gene set conserved across colon sites in patients with EoC. Second, we demonstrated that the EoC transcriptome is associated with tissue eosinophil levels and disease activity, is markedly distinct from upper GI EGID transcriptomes. Although we cannot fully rule out type 2 immunity, there was no evidence of strong type 2 allergic inflammation in EoC when compared with the type 2 signature seen in EoE and EoG. Third, robust EoC gene expression revealed functional pathways in EoC pathogenesis, including molecular evidence for reduced cell proliferation and increased apoptosis, which were substantiated in biopsies by Ki-67 and cleaved caspase-3 staining. Reduced cell proliferation was unexpected and suggests that distinct cellular mechanisms might be locally operational in EoC. Fourth, based on cell deconvolution, we identified the involvement of eosinophils, basophils, CD4+ effector memory T cells, and multipotent progenitors in EoC. Fifth, we linked the magnitude of molecular changes to histologic changes. Strong correlations with the EoC transcriptome were observed in pericryptal circumferential eosinophil collars, providing a better understanding of histologic features of clinical biopsies. Finally, we showed that the EoC score, based on the EoC transcriptome, readily assessed disease activity and distinguished EoC from the clinically challenging cases of IBD with high eosinophilia. This collective evidence establishes that EoC is a discrete disease entity involving pathways distinct from those of upper EGIDs and IBD.
We identified CLC as the gene most highly induced in EoC. Given this gene’s specificity to eosinophils and basophils and that eosinophilic inflammation is a hallmark of EoC,16, 35 this finding substantiates the data’s integrity. Indeed, colonic CLC expression levels strongly correlated with eosinophilia-quantified disease severity. CLC protein (i.e., galectin 10) is an eosinophil specific granule protein that is secreted by activated eosinophils and promotes type 2 immune activity. Antibodies directed against key epitopes of the CLC crystallization interface have been shown to dissolve preexisting CLCs in mucus from patients with asthma and were effective in controlling disease in a humanized mouse model.36 As CLCs can be found in EoC stool,37-39 these antibodies may be beneficial for relieving EoC tissue inflammation.
Though CLC and other eosinophil products likely promote proinflammatory changes in EoC, we observed that eosinophil regulation may differ in patients with EoC compared to other EGIDs. Notably, CCL11 (eotaxin-1), but not CCL24 (eotaxin-2) nor CCL26 (eotaxin-3), was highly upregulated in tissue from patients with EoC compared with control tissue and exhibited a significant, positive correlation with colonic CLC expression. This finding is consistent with an essential role for CCL11 (eotaxin-1) in regulating eosinophil-associated GI pathology, from the small intestine to the colon, in a mouse model and humans.40, 41 CCL11 (eotaxin-1) upregulation is also observed in IBD,41, 42 suggesting similar colonic eosinophil regulation. Why CCL11 (eotaxin-1) is specifically overexpressed compared with CCL24 (eotaxin-2) and CCL26 (eotaxin-3) deserves further study and may relate to the lack of strong type 2 cytokines, such as IL-13, which drives CCL26 expression in EoE. Several possibilities could account for these findings, such as differences in tissue composition (e.g., resident cell types) or distinct disease mechanisms (e.g., differential cell recruitment or altered gene expression programs of resident cells). The dissimilarities in differentially regulated transcripts, especially CCL26 (eotaxin-3) in patients with upper EGIDs (EoE, EoG) and CCL11 (eotaxin-1) in those with lower EGID (EoC) might arise, at least partially, from the distinct structural cells and immunocytes present in those tissues. The apparent weaker type 2 immune response deserves further analysis given the small sample size and the heterogeneity observed.
Pathway analysis of the EoC transcriptome identified a robust reduction in cell cycle pathways, which was substantiated by a decreased number of proliferating (Ki-67) cells in EoC colonic biopsies. In contrast, upper GI EGIDs (EoE and EoG) feature expansion of the basal epithelium and increased cell proliferation.13, 16 A series of downregulated genes, including NADPH oxidase 1 (NOX1), stratifin (SFN), and several 26S proteasome (PSMC1, 3, 6, PSMD4, 7), may relate to the decreased cell proliferation, as decreased NOX1 expression is known to produce a significant decline in reactive oxygen species (ROS) production and cell cycle arrest.43 Interestingly, NOX2-deficient mice have interstitial pneumonitis with eosinophilic crystals and granulomas.44 Inhibiting SFN expression increases apoptosis and cell cycle arrest.45 In addition, the 26S proteasome is known as the end point of the ubiquitin-proteasome pathway that is chiefly required for cell cycle progression. The observed enrichment of decreased expression in 26S proteasome–associated genes might relate to decreased proliferation in patients with EoC. Notably, several cases of colitis were reported after taking bortezomib, an inhibitor of the 26S proteasome.46, 47 Functional pathway analysis of the EoC transcriptome also showed evidence of increased apoptosis in agreement with an increased number of apoptotic (cleaved caspase-3) cells in EoC colonic biopsies. Positive regulation of apoptosis could slow down epithelial turnover and proliferation in colonic tissue, leading to impaired intestinal barrier function and facilitating inflammatory processes. Relatedly, in colon biopsy specimens from infants with allergic/eosinophilic colitis, high numbers of apoptotic epithelial cells were identified by apoptotic cell–specific histochemical assay.48 Interestingly, previous microRNA analysis of patients with EoC also suggested this phenomenon.49 Furthermore, the dominance of caspase 3, in contrast to caspase 8 and the ripoptosome, further contrasts the tissue-specific responses related to EoC and EoE pathogenesis.50 Our collective data suggest distinct molecular and cellular mechanisms are locally operational in patients with EoC.
Although specific clinicopathologic consensus diagnostic criteria have not been established for EoC, our study highlights colonic histologic changes as having utility in EoC diagnosis. Applying a stringent threshold values for EoC diagnosis,24 our data suggest that additional pathologic changes, including the presence of eosinophil sheets, cryptitis or crypt abscesses, and muscular involvement, are also present and may facilitate the diagnosis of EoC. Furthermore, the lack of acute inflammation and cryptitis (features of IBD) should raise suspicion for EoC. These data support the importance of a systematic survey of histologic features other than eosinophil counts in colonic biopsies from GI diseases. Further research is needed to establish the appropriate histologic criteria and guidelines for EoC that will assist pathologists to differentiate between normal findings and disease and differential diagnoses. Similarly, efforts to raise awareness of the importance of quantifying GI tract eosinophilia and reporting associated histologic findings are needed.
EoC had a distinct molecular profile and correlating histologic features. Of the EoC histologic features, eosinophilic features were highly associated with the EoC transcriptome, with the strongest association being pericryptal circumferential eosinophil collars. As expected, not all histologic features showed strong associations with the EoC transcriptome, possibly due to the low occurrence in patients with EoC of some histologic features that were anticipated to be prominent in CD but not EoC, namely, acute inflammatory cells, surface erosion/ulceration, and lamina propria fibroplasia.51 Indeed, some colonic histologic features, including pericryptal circumferential eosinophil collars, were specifically associated with the EoC-associated functions (decreased cell proliferation, increased apoptosis). The imbalance of cell proliferation and cell death, normally maintained in cellular homeostasis, and its correlation with unique histologic features associated with EoC suggests epithelial-eosinophil cross-talk particularly at the interface of eosinophilic collars. The eosinophilic features best reflected the molecular signature changes in EoC, warranting close attention to them when interpreting disease diagnosis and activity.
Despite promising results, our study has limitations. First, the small sample size of EoC (n = 27) due to the rarity of the disease limits the impact of results. Also, heterogeneity in EoC (e.g., comorbid EoC-EoE vs. EoC alone) might affect the results, although it might be practical as reflecting the real-world manifestations. The study definition for EoC and its activity was applied for balanced feasibility and accuracy, warranting future analyses with further accurate evaluations (e.g., controlled comorbidity, validated symptom assessment, and standardized endoscopic/histomolecular follow-up). Second, our findings included patients with active EoC and CD who had mixed treatment status (Table 1) and patients who had treatment-refractory disease, which might influence the results. However, patients still exhibited signs of active disease clinically, histologically, and molecularly. Therefore, the treatments were not effective in eradicating the disease, and key molecular pathways involved in pathogenesis were likely still active, at least partially. Third, though we utilized unbiased, highly sensitive, genome-wide transcriptome approaches to identify key gene signatures, the analyses were performed on whole biopsies, composed of a mixture of cellular components, rather than single cells. We performed computational deconvolution of cell subset proportions to address this limitation; however, future studies using single-cell preparations will be important for further cellular subset characterizations. Finally, the data were limited by the cross-sectional approach, highlighting the importance of additional replication, particularly in prospective and longitudinal studies.
In conclusion, we established EoC as a unique GI disease by identifying a conserved colonic transcriptome that associates with colonic eosinophilia, is markedly distinct from that of other GI diseases, and is uniquely associated with distinct histologic features, especially pericryptal circumferential eosinophil collars. Mechanistically, we uncovered that EoC is not related to strong type 2 immunity but rather apoptosis and reduced epithelial cell proliferation. Our data propel more mechanistic studies that will lead to new insights regarding EoC pathogenesis and to future molecular-based prevention and therapies.
Supplementary Material
What You Need to Know.
BACKGROUND AND CONTEXT:
Eosinophilic colitis (EoC) is a poorly understood disease process. Classification of EoC as part of a spectrum of eosinophilic gastrointestinal disorders (EGID) or inflammatory bowel disease (IBD) has not been determined.
NEW FINDINGS:
We identified a conserved colonic transcriptome in EoC patients, which was proportional to the degree of colonic eosinophilia, markedly distinct from other gastrointestinal diseases, and uniquely associated with mechanistic processes distinct from other EGID.
LIMITATIONS:
Although this study deeply examined the largest number of samples from EoC to date, the cohort size is still limited in size and scope.
IMPACT:
We establish EoC as a disease markedly distinct from other EGID and IBD, with a disease mechanism that does not involve allergic inflammation, thereby providing a foundation for understanding the disease and improving diagnosis and treatment.
Acknowledgments
The authors would like to thank all patients who participated in the study. The authors are also grateful to their colleagues and clinical support staff for procuring biopsies, blood samples, and clinical data.
Grant Support
This study was supported by NIH grant K99/R00 AI158660 (to T.S.) and CEGIR (U54 AI117804), which is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS), and is co-funded by National Institute of Allergy and Infectious Diseases (NIAID), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NCATS and, the Intramural Research Program of the NIH. CEGIR is also supported by patient advocacy groups including the American Partnership for Eosinophilic Disorders (APFED), Campaign Urging Research for Eosinophilic Disease (CURED), and Eosinophilic Family Coalition (EFC). As a member of the RDCRN, CEGIR is also supported by its Data Management and Coordinating Center (DMCC) (U2CTR002818). Funding support for the DMCC is provided by the National Center for Advancing Translational Sciences (NCATS) and the National Institute of Neurological Disorders and Stroke (NINDS). This project was supported in part by NIH P30 DK078392 (Gene Expression Core, Pathology Research Core, and Confocal Imaging Core) of the Digestive Diseases Research Core Center in Cincinnati.
Conflicts of Interest
M.E.R. is a consultant for Pulm One, Spoon Guru, ClostraBio, Serpin Pharm, Allakos, Celgene, Astra Zeneca, Adare/Ellodi Pharma, GlaxoSmith Kline, Guidepoint and Revolo Biotherapeutics and has an equity interest in the first five listed and royalties from reslizumab (Teva Pharmaceuticals), PEESSv2 (Mapi Research Trust) and UpToDate. M.E.R. is an inventor of patents owned by Cincinnati Children’s Hospital Medical Center. M.H.C. is a consultant for Allakos, AstraZeneca, Calypso, Esocap, GlaxoSmithKline, Receptos/Celgene, Regeneron, and Shire a Takeda company and has received research funding from AstraZeneca, Shire a Takeda company, Receptos/Celgene, and Regeneron. V.A.M. is a consultant for Shire and has received research funding from Shire. E.S.D. is a consultant for Abbott, Abbvie, Adare/Ellodi, Aimmune, Allakos, Amgen, Arena, AstraZeneca, Avir, Biorasi, Calypso, Celgene/Receptos/BMS, Celldex, Eli Lilly, EsoCap, GSK, Gossamer Bio, Landos, Morphic, Parexel/Calyx, Regeneron, Robarts/Alimentiv, Salix, Sanofi, and Shire/Takeda; has received research funding from Adare/Ellodi, Allakos, AstraZeneca, GSK, Meritage, Miraca, Nutricia, Celgene/Receptos/BMS, Regeneron, and Shire/Takeda; and has received educational grants from Allakos, Banner, and Holoclara. N.G. is a consultant for Allakos, Astra-Zeneca, Sanofi-Regeneron, Abbvie, and Nutricia. I.H. is a consultant for Regeneron, Receptos, Shire, Allakos, and Adare and has received research funding from Regeneron, Receptos, Shire, and Adare. G.T.F. is a consultant for Sanofi and a co-founder of EnteroTrack. S.K.G. is a consultant for Abbott, Allakos, Adare Pharmaceuticals, DBV Technologies, Gossamer Bio, QOL Medical, Medscape, Receptos/Celgene and UpToDate, and has received research support from Shire, a Takeda company. M.C. is a consultant for Regeneron, Allakos, Adare, Shire/Takeda, AstraZeneca, Sanofi and Bristol Myers Squibb; and has received research funding from Regeneron, Allakos, Shire, AstraZeneca and Danone. C.M.D. has received research grant support from the National Institutes of Health/National Institute of Allergy and Infectious Diseases (UM2 AI130836, U01 AI126614, R01 AI135197, U54 AI117804), DBV Technologies, Aimmune Therapeutics, Nutricia North America, Regeneron Pharmaceuticals, and the Scurlock Foundation and is a consultant for Moonlight Therapeutics. J.B.W is a consultant for medical advisory for Regeneron, Sanofi, and Allakos, and has received clinical trial funding from Regeneron and Allakos. S.S.A. is a consultant for Aimmune, AstraZeneca, and Medscape and is a co-inventor of oral viscous budesonide, patented by UCSD and licensed by Shire/Takeda and has received research grant support from the National Institutes of Health. G.W.F. has received research support from Lucid, Allakos, Regeneron, Takeda/Shire, and Adare/Ellodi and is a consultant for Adare/Ellodi, Allakos, Bristol Myers Squibb, Lucid and Takeda/Shire. All other authors declare that they have no competing interests.
List of Abbreviations
- APFED
American Partnership for Eosinophilic Disorders
- CCED
Cincinnati Center for Eosinophilic Disorders
- CCHMC
Cincinnati Children’s Hospital Medical Center
- CCL
C-C Motif Chemokine Ligand
- CD
Crohn's disease
- CEGIR
Consortium of Eosinophilic Gastrointestinal Disease Researchers
- CLC
Charcot-Leyden crystal
- CURED
Campaign Urging Research for Eosinophilic Disease
- DMCC
Data Management and Coordinating Center
- EFC
Eosinophilic Family Coalition
- EoC
eosinophilic colitis
- EoG
eosinophilic gastritis
- EGID
eosinophilic gastrointestinal diseases
- EoE
eosinophilic esophagitis
- FDR
false-discovery rate
- GI
gastrointestinal
- H&E
hematoxylin and eosin
- HPF
high-power microscopic field
- IBD
inflammatory bowel disease
- IQR
interquartile range
- NCATS
National Center for Advancing Translational Sciences
- NIAID
National Institute of Allergy and Infectious Diseases
- NIDDK
National Institute of Diabetes and Digestive and Kidney Diseases
- NL
normal
- OMEGA
Outcomes Measures in Eosinophilic Gastrointestinal disorders Across the ages
- ORDR
Office of Rare Diseases Research
- PCA
principal component analysis
- RDCRN
Rare Diseases Clinical Research Network
- TPM
transcripts per kilobase million
- UC
ulcerative colitis
Footnotes
Writing Assistance
Shawna Hottinger provided editorial assistance as a medical writer funded by Cincinnati Children’s Hospital Medical Center.
The list of participants is provided in this article’s Supplementary Material
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol 2004;113:11–28. [DOI] [PubMed] [Google Scholar]
- 2.Jensen ET, Martin CF, Kappelman MD, et al. Prevalence of Eosinophilic Gastritis, Gastroenteritis, and Colitis: Estimates From a National Administrative Database. J Pediatr Gastroenterol Nutr 2016;62:36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mansoor E, Saleh MA, Cooper GS. Prevalence of Eosinophilic Gastroenteritis and Colitis in a Population-Based Study, From 2012 to 2017. Clin Gastroenterol Hepatol 2017;15:1733–1741. [DOI] [PubMed] [Google Scholar]
- 4.DiTommaso LA, Rosenberg CE, Eby MD, et al. Prevalence of eosinophilic colitis and the diagnoses associated with colonic eosinophilia. J Allergy Clin Immunol 2019;143:1928–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jensen ET, Aceves SS, Bonis PA, et al. High Patient Disease Burden in a Cross-sectional, Multicenter Contact Registry Study of Eosinophilic Gastrointestinal Diseases. J Pediatr Gastroenterol Nutr 2020;71:524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner KO, Sinkre RA, Neumann WL, et al. Primary Colonic Eosinophilia and Eosinophilic Colitis in Adults. Am J Surg Pathol 2017;41:225–233. [DOI] [PubMed] [Google Scholar]
- 7.Mark J, Fernando SD, Masterson JC, et al. Clinical Implications of Pediatric Colonic Eosinophilia. J Pediatr Gastroenterol Nutr 2018;66:760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanchard C, Wang N, Stringer KF, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 2006;116:536–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherrill JD, Kiran KC, Blanchard C, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun 2014;15:361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shoda T, Matsuda A, Nomura I, et al. Eosinophilic esophagitis versus proton pump inhibitor-responsive esophageal eosinophilia: Transcriptome analysis. J Allergy Clin Immunol 2017;139:2010–2013. [DOI] [PubMed] [Google Scholar]
- 11.Shoda T, Wen T, Caldwell JM, et al. Loss of Endothelial TSPAN12 Promotes Fibrostenotic Eosinophilic Esophagitis via Endothelial Cell-Fibroblast Crosstalk. Gastroenterology 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shoda T, Kaufman KM, Wen T, et al. Desmoplakin and periplakin genetically and functionally contribute to eosinophilic esophagitis. Nat Commun 2021;12:6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caldwell JM, Collins MH, Stucke EM, et al. Histologic eosinophilic gastritis is a systemic disorder associated with blood and extragastric eosinophilia, TH2 immunity, and a unique gastric transcriptome. J Allergy Clin Immunol 2014;134:1114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato M, Shoda T, Shimizu H, et al. Gene Expression Patterns in Distinct Endoscopic Findings for Eosinophilic Gastritis in Children. J Allergy Clin Immunol Pract 2017;5:1639–1649. [DOI] [PubMed] [Google Scholar]
- 15.Shoda T, Wen T, Caldwell JM, et al. Molecular, endoscopic, histologic, and circulating biomarker-based diagnosis of eosinophilic gastritis: Multi-site study. J Allergy Clin Immunol 2020;145:255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins MH, Capocelli K, Yang GY. Eosinophilic Gastrointestinal Disorders Pathology. Front Med (Lausanne) 2017;4:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Shea KM, Aceves SS, Dellon ES, et al. Pathophysiology of Eosinophilic Esophagitis. Gastroenterology 2018;154:333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunn JLM, Shoda T, Caldwell JM, et al. Esophageal type 2 cytokine expression heterogeneity in eosinophilic esophagitis in a multisite cohort. J Allergy Clin Immunol 2020;145:1629–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyles JL, Martin LJ, Shoda T, et al. Very early onset eosinophilic esophagitis is common, responds to standard therapy, and demonstrates enrichment for CAPN14 genetic variants. J Allergy Clin Immunol 2021;147:244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeBrosse CW, Case JW, Putnam PE, et al. Quantity and distribution of eosinophils in the gastrointestinal tract of children. Pediatr Dev Pathol 2006;9:210–8. [DOI] [PubMed] [Google Scholar]
- 21.Sonnenberg A, Turner KO, Singhal A, et al. Prevalence and concordant occurrence of esophageal, gastric, duodenal, and colonic eosinophilia. Dis Esophagus 2020;33. [DOI] [PubMed] [Google Scholar]
- 22.Pesek RD, Rothenberg ME. Eosinophilic gastrointestinal disease below the belt. J Allergy Clin Immunol 2020;145:87–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta SK, Falk GW, Aceves SS, et al. Consortium of Eosinophilic Gastrointestinal Disease Researchers: Advancing the Field of Eosinophilic GI Disorders Through Collaboration. Gastroenterology 2019;156:838–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collins MH. Histopathologic features of eosinophilic esophagitis and eosinophilic gastrointestinal diseases. Gastroenterol Clin North Am 2014;43:257–68. [DOI] [PubMed] [Google Scholar]
- 25.Pesek RD, Greuter T, Lopez-Nunez O, et al. Clinicopathologic Correlations in Eosinophilic Gastrointestinal Disorders. J Allergy Clin Immunol Pract 2021;9:3258–3266. [DOI] [PubMed] [Google Scholar]
- 26.Bousvaros A, Antonioli DA, Colletti RB, et al. Differentiating ulcerative colitis from Crohn disease in children and young adults: report of a working group of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the Crohn's and Colitis Foundation of America. J Pediatr Gastroenterol Nutr 2007;44:653–74. [DOI] [PubMed] [Google Scholar]
- 27.Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009;25:1091–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Bardes EE, Aronow BJ, et al. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009;37:W305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wen T, Stucke EM, Grotjan TM, et al. Molecular diagnosis of eosinophilic esophagitis by gene expression profiling. Gastroenterology 2013;145:1289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haberman Y, Karns R, Dexheimer PJ, et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun 2019;10:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Travers J, Rochman M, Caldwell JM, et al. IL-33 is induced in undifferentiated, non-dividing esophageal epithelial cells in eosinophilic esophagitis. Sci Rep 2017;7:17563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker MM, Potter MD, Talley NJ. Eosinophilic colitis and colonic eosinophilia. Curr Opin Gastroenterol 2019;35:42–50. [DOI] [PubMed] [Google Scholar]
- 34.Piñero J, Ramírez-Anguita JM, Saüch-Pitarch J, et al. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res 2020;48:D845–d855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klion AD, Ackerman SJ, Bochner BS. Contributions of Eosinophils to Human Health and Disease. Annu Rev Pathol 2020;15:179–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Persson EK, Verstraete K, Heyndrickx I, et al. Protein crystallization promotes type 2 immunity and is reversible by antibody treatment. Science 2019;364. [DOI] [PubMed] [Google Scholar]
- 37.Cello JP. Eosinophilic gastroenteritis--a complex disease entity. Am J Med 1979;67:1097–104. [DOI] [PubMed] [Google Scholar]
- 38.Moore D, Lichtman S, Lentz J, et al. Eosinophilic gastroenteritis presenting in an adolescent with isolated colonic involvement. Gut 1986;27:1219–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uppal V, Kreiger P, Kutsch E. Eosinophilic Gastroenteritis and Colitis: a Comprehensive Review. Clin Rev Allergy Immunol 2016;50:175–88. [DOI] [PubMed] [Google Scholar]
- 40.Hogan SP, Mishra A, Brandt EB, et al. A critical role for eotaxin in experimental oral antigen-induced eosinophilic gastrointestinal allergy. Proc Natl Acad Sci U S A 2000;97:6681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahrens R, Waddell A, Seidu L, et al. Intestinal macrophage/epithelial cell-derived CCL11/eotaxin-1 mediates eosinophil recruitment and function in pediatric ulcerative colitis. J Immunol 2008;181:7390–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coburn LA, Horst SN, Chaturvedi R, et al. High-throughput multi-analyte Luminex profiling implicates eotaxin-1 in ulcerative colitis. PLoS One 2013;8:e82300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Juhasz A, Markel S, Gaur S, et al. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J Biol Chem 2017;292:7866–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trocme C, Deffert C, Cachat J, et al. Macrophage-specific NOX2 contributes to the development of lung emphysema through modulation of SIRT1/MMP-9 pathways. J Pathol 2015;235:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shiba-Ishii A, Noguchi M. Aberrant stratifin overexpression is regulated by tumor-associated CpG demethylation in lung adenocarcinoma. Am J Pathol 2012;180:1653–62. [DOI] [PubMed] [Google Scholar]
- 46.Moon SJ, Min CK, Lee DG, et al. Pseudomembranous colitis following bortezomib therapy in a myeloma patient. Acta Haematol 2007;117:211–4. [DOI] [PubMed] [Google Scholar]
- 47.Nogales Rincón O, Huerta Madrigal A, Merino Rodriguez B, et al. Rectal bleeding and diarrhea caused by bortezomib-induced colitis. Gastroenterol Hepatol 2010;33:753–4. [DOI] [PubMed] [Google Scholar]
- 48.Kumagai H, Masuda T, Maisawa S, et al. Apoptotic epithelial cells in biopsy specimens from infants with streaked rectal bleeding. J Pediatr Gastroenterol Nutr 2001;32:428–33. [DOI] [PubMed] [Google Scholar]
- 49.Kiss Z, Béres NJ, Sziksz E, et al. Specific MicroRNA Pattern in Colon Tissue of Young Children with Eosinophilic Colitis. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brusilovsky M, Rochman M, Rochman Y, et al. Environmental allergens trigger type 2 inflammation through ripoptosome activation. Nat Immunol 2021;22:1316–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li J, Mao R, Kurada S, et al. Pathogenesis of fibrostenosing Crohn's disease. Transl Res 2019;209:39–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.