

Abstract
Since the discovery of fibroblast growth factor (FGF)-19 over 20 years ago, our understanding of the peptide and its role in human biology has moved forward significantly. A member of a superfamily of paracrine growth factors regulating embryonic development, FGF19 is unique in that it is a dietary-responsive endocrine hormone linked with bile acid homeostasis, glucose and lipid metabolism, energy expenditure, and protein synthesis during the fed to fasted state. FGF19 achieves this through targeting multiple tissues and signaling pathways within those tissues. The diverse functional capabilities of FGF19 is due to the unique structural characteristics of the protein and its receptor binding in various cell types. This review will cover the current literature on the protein FGF19, its target receptors, and the biological pathways they target through unique signaling cascades.
Keywords: Fibroblast growth factor 19, bile acid metabolism, carbohydrate metabolism, lipid metabolism, protein synthesis, farnesoid x receptor
1. Introduction
The fibroblast growth factor (FGF) family is comprised of 22 related proteins. Many members of the FGF family are involved in embryonic development. Thus, the discovery of FGF19 in the human fetal brain in 1999 conformed to the FGF family paradigm of embryonic growth factors (Nishimura, Utsunomiya, Hoshikawa et al., 1999). Further examination of embryonic tissues, mainly in chicken and zebrafish, identified FGF19 in the eye (Kurose, Bito, Adachi et al., 2004) and ear canal (Sanchez-Calderon, Francisco-Morcillo, Martin-Partido et al., 2007). Thus, during organogenesis, FGF19 has clearly defined roles in cellular growth and tissue development. However, in adult tissues, the expression of FGF19 becomes limited to the intestine (Milkiewicz, Klak, Kempinska-Podhorodecka et al., 2016), gall bladder (Zweers, Booij, Komuta et al., 2012), and liver (Milkiewicz et al., 2016,Wunsch, Milkiewicz, Wasik et al., 2015,Hasegawa, Kawai, Bessho et al., 2019,Johansson, Svensson, Almstrom et al., 2020). Unlike the paracrine or autocrine function of other FGF family members, in adult tissues, FGF19 functions as an endocrine factor, marking a unique change in functional activity. The functional purpose of FGF19 in adult tissues did not begin to be deciphered until 2002. FGF19 was identified as both a metabolic regulator (Tomlinson, Fu, John et al., 2002), and as an activator of hepatocellular carcinogenesis (Nicholes, Guillet, Tomlinson et al., 2002). Shortly thereafter, in 2003, FGF19 was found to regulate bile acid metabolism (Holt, Luo, Billin et al., 2003). However, it was not until 2011 that FGF19 was found to promote protein synthesis in the liver (Kir, Beddow, Samuel et al., 2011). A further role of FGF19 function to prevent muscle wasting and increase hypertrophy in skeletal muscle was discovered in 2017 (Benoit, Meugnier, Castelli et al., 2017). The diverse range of function of FGF19 is attributed to its unique structure compared to the other FGFs, and its affinity for binding to its cognate heterodimer surface receptors. The action of FGF19 on these receptors and the downstream pathways that are activated in a tissue-specific manner are still an active area of investigation. This review will focus on our current understanding of FGF19 as an endocrine factor involved in the currently known target pathways for bile acid homeostasis, glucose/lipid/energy metabolism, protein synthesis, and cancer development, as well as the role that FGF19 structure and interaction with its cognate receptors play in regulating these pathways.
2. Structure
There are 18 secreted protein members of the FGF family. The majority of the FGF members function in paracrine signaling and comprise FGF1 subfamily (FGF1 and FGF2), FGF4 subfamily (FGF4, FGF5, and FGF6), FGF7 subfamily (FGF7, FGF10, and FGF22), FGF8 subfamily (FGF8, FGF17, and FGF18), and FGF9 subfamily (FGF9, FGF16, and FGF20). The FGF19 subfamily consisting of FGF19, FGF21, and FGF23 is unique in that its members function as endocrine proteins. The family range in size from 150 to 300 amino acids in length. FGFs all have a conserved 120 amino acid region with highly variable N- and C-terminus regions. Within the conserved region, paracrine subfamily’s structure is formed of 12 anti-parallel β-strands (β1 –β12) (Olsen, Garbi, Zampieri et al., 2003,Zhu, Komiya, Chirino et al., 1991). This conformation forms a β-trefoil fold consisting of three sets of four-stranded β sheets. Within the structure, there are heparin sulfate (HS) binding sequences that help the FGFs bind to their cognate receptors that also contain HS sequences. HS is a large oligosaccharide that is comprised of repeating GlcN(S)6O(S)-IdoA/GlcA(2S) disaccharide units. The endocrine members, FGF19, FGF21, and FGF23 are missing the β11 strand (Goetz, Beenken, Ibrahimi et al., 2007,Harmer, Pellegrini, Chirgadze et al., 2004). The resultant gap leads to the formation of an α-helical structure between strands β10 and β12. This extended loop displaces the HS region in FGF19, blocking the formation of hydrogen bonds at the N-sulfate and 2-O-sulfate groups present in HS (Goetz et al., 2007). Goetz et al. compared the crystal structure of FGF2 bound to HS and FGF19 which showed a four-amino acid region (Lys149, Gln150, Gln152, and Arg157) in FGF19 that could promote FGF19 binding to HS (Goetz, Ohnishi, Kir et al., 2012). Deletion of Lys149, led to complete loss of HS binding by FGF19, but did not alter FGF19’s activation of signaling pathways or suppression of Cyp7a1 and Cyp8b1 gene expression. This decreased interaction with heparin sulfate allows for the FGF19 family of proteins to enter into circulation, rather than be bound by nearby receptors containing HS (Goetz et al., 2007).
3. Receptors
FGF19 enters circulation where it binds to a heterodimer receptor complex comprised of a Fibroblast Growth Factor Receptor (FGFR) and β-Klotho (KLB). The tissue specific expression of both receptors confer target specificity to the action of FGF19. This is unique to the FGF19 receptor subfamily, as the other FGFs bind to homodimer FGFRs in nearby cells where they are expressed (Romero-Fernandez, Borroto-Escuela, Tarakanov et al., 2011).
3.1. Fibroblast Growth Factor Receptors
There are four receptors in the FGFR family, FGFR1, FGFR2, FGFR3, and FGFR4. The FGFRs are relatively homologous with the sequence identity ranging from 53% – 70% (Gong, 2014). The common structural characteristics of the family include an N-terminal signaling sequence, three extracellular immunoglobulin (Ig) domains (IgI, IgII, and IgIII), an acid box region between IgI and IgII, a transmembrane domain, and an intracellular split tyrosine kinase sequence. The IgII and IgIII domains are responsible for ligand binding. Splice variants in the IgIII domain of FGFR1–3 lead to two variants for these receptors, labelled either “b” or “c” (Itoh and Ornitz, 2004). FGFR4 does not have this splice variant in the IgIII domain, so in total there are seven active receptors in the FGFR family: FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3b, FGFR3c, and FGFR4. The FGFRs display wide differential expression in tissues (Hughes, 1997). FGFR1 is expressed in adipose, brain, kidney, lung, heart, and skeletal muscle (Templeton and Hauschka, 1992). FGFR4 is abundantly expressed in liver, lung, gallbladder, and to a lesser extent in the small intestines, colon, pancreas, and adrenal gland (Lin, Wang, Blackmore et al., 2007). FGFR4 is unique to the other FGFRs in that FGF19, but not FGF21 nor FGF23, can bind to it. FGF21 and FGF23 can bind to the receptors FGFR1-FGFR3. FGF19 is able to bind to the “c” variants of FGFR1-FGFR3, but not to the “b” variants. The observed overlap in metabolic effects of FGF19 with FGF21 are due to the shared binding that these receptors have for FGFR1c (Kurosu, Choi, Ogawa et al., 2007,Perry, Lee, Ma et al., 2015,Lan, Morgan, Rahmouni et al., 2017). Whereas, the ability of FGF19 to regulate bile acids in the liver is directly related to FGFR4-FGF19 binding in hepatocytes (Wu, Coulter, Liddle et al., 2011).
3.2. β-Klotho
There are two members of the Klotho family, alpha- or beta-Klotho (KLA or KLB). These single-pass transmembrane proteins have an extracellular domain containing two tandem glycosidase-like domains, also referred to pseudoglcyoside hydrolases, designated KL1 and KL2 (Shiraki-Iida, Aizawa, Matsumura et al., 1998). Given the conformational difference in FGF19 that masks the heparin sulfate binding sequences, it has poor binding affinity to FGFR4. KLB functions to bring FGF19 and FGFRs in close proximity to one another, and therefore increase binding affinity. The C-terminal tail of FGF19 binds to the single pass loop of KLB in two sites (Kuzina, Ung, Mohanty et al., 2019). Site 1 is a multi-turn D-P motif and site 2 is a S-P-S motif. Site 2 binding occurs at the pseudoglycoside hydrolase region in KL2. Modification on the FGF19 C-terminal domain to that of FGF21 causes a loss of binding to KLB (Wu, Lemon, Li et al., 2008). The IgIII domain of FGFRs bind to KLB (Lin et al., 2007,Wu, Ge, Gupte et al., 2007). KLB is only able to interact with the “c” isoforms of FGFR1–3, further limiting the target tissues that FGF19 can exert metabolic effects (Kurosu et al., 2007). For example, FGFR4 and KLB are only predominantly co-expressed in the liver (Lin et al., 2007,Fon Tacer, Bookout, Ding et al., 2010). It should be noted that there is differences in binding affinity for KLB and the FGFRs, with FGFR1c and FGFR4 having the greatest overall binding affinity for KLB (Kurosu et al., 2007). In humans, KLB expression is most abundant in adipose tissue, with moderate expression in liver and pancreas, and low expression in lung, bone, and skeletal muscle (Lin et al., 2007). It is not clear if FGF19 can bind to FGFRs without the presence of KLB in-vivo. In support of KLB dependence for binding of FGF19 to FGFRs, regulation of KLB expression can alter the efficacy of FGF19 signaling. Loss of hepatic KLB increases the bile acid pool and increases expression of genes associated with bile acid production (Katafuchi, Esterhazy, Lemoff et al., 2015). Additionally, KLB deficiency-mediated impairment of FGF19 signaling has been observed in vivo. In obese patients, KLB is repressed by micro-RNA 34a leading to impaired FGF19 response (Fu, Choi, Kim et al., 2012).
4. Rodent Orthologue FGF15
The mouse orthologue Fgf15 was discovered in the embryonic mouse nervous system two years before human FGF19 was identified in brain tissue (McWhirter, Goulding, Weiner et al., 1997). The orthologues only share 51% sequence identity despite sharing similar functions (Nishimura et al., 1999). The expression pattern of FGF15 is well detailed through development. Northern blot analysis detected Fgf15 mRNA appearance at day 11 in mouse embryos (McWhirter et al., 1997). Using in situ hybridization, Fgf15 mRNA expression first appears on day 7.5 – 8 in the neuroectoderm. Fgf15 mRNA displays highly dynamic expression within the central nervous system through gestational day 14. Like FGF19, once organogenesis is complete in the developing embryo, and tissues have reached maturity, the expression pattern of FGF15 shifts, with limited expression in the nervous system and very high expression in the intestine, predominantly in the ileum (Fon Tacer et al., 2010). It is important to note that FGF15 expression does not entirely match that of human FGF19 in mature tissues (Figure 1). Whereas both FGF15 and FGF19 are highly expressed in the ileum, FGF15 is not expressed in the gall bladder, bile ducts (cholangiocytes) or hepatocytes (Fon Tacer et al., 2010,Choi, Moschetta, Bookout et al., 2006). However, the expression pattern of FGFRs and KLB overlap considerably between mouse and human. FGFR1c in the mouse is broadly expressed including the adipose, brain, kidney, lung, heart, and skeletal muscle. FGFR4 is more restricted with high expression in liver, adrenal gland, and kidney, and low expression in intestine, gall bladder, and lung. KLB has the most restricted expression pattern with high abundance in adipose tissue and liver and lesser abundance in the gall bladder, ileum, heart, brain, and skeletal muscle (Fon Tacer et al., 2010,Yang, Jin, Li et al., 2012).
Figure 1.
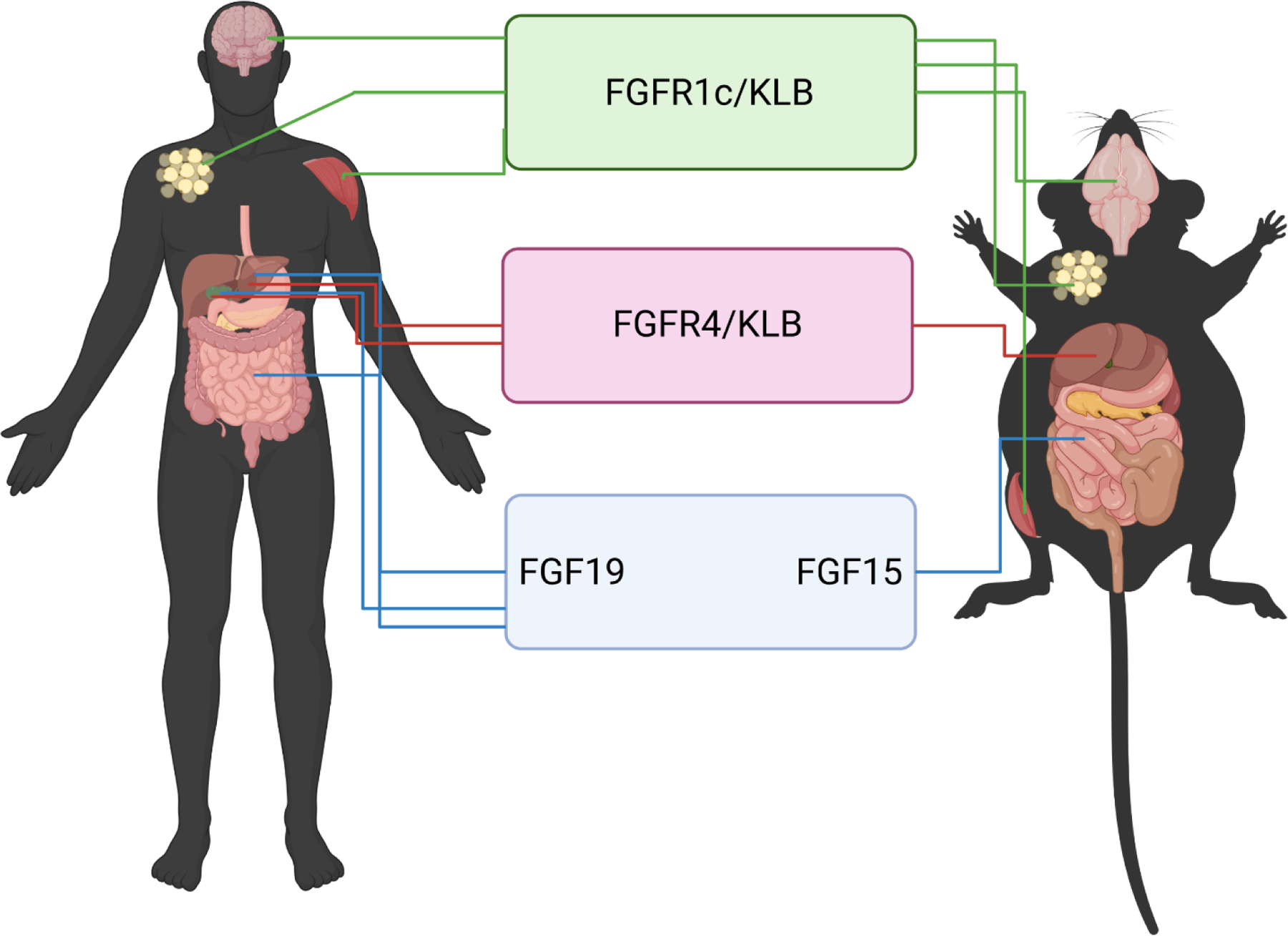
Expression pattern of FGF15/19, FGFR4, FGF1c, and KLB in human and mouse. Fibroblast growth factor (FGF), fibroblast growth factor receptor (FGFR), β-Klotho (KLB)
The differences between FGF15 and FGF19 are not limited to expression pattern, as the binding activity of FGF15 to the cognate receptors FGFR/KLB differ as well. FGF15 can bind with FGFR4 in the liver, in a similar manner as FGF19. Unlike FGF19 that binds to FGFR1c with high affinity, FGF15 binding is weaker (Yang et al., 2012,Zhou, Luo, Chen et al., 2017). Detailed examination of the amino acid sequence of FGF15 identified a single unpaired cysteine (Cys-135) present in the β8-β9 loop region. This unpaired cysteine can lead to dimerization of two FGF15 proteins altering the ability of the peptide to efficiently bind FGFRs (Zhou et al., 2017,Williams, Harper Calderon, E et al., 2021).
The functional relevance of FGF15 and FGF19 binding differences cannot be understated. FGF15 is unable to bind to and activate human FGFR4/KLB, but human FGF19 can bind to and activate mouse FGFR4/KLB and FGFR1c/KLB (Ellis, Naugler, Parini et al., 2013). Further, more recent research on the binding efficiency of FGF19 to FGFR4 uncovered small point mutations that can decrease the binding affinity for FGF19 and FGFR4/KLB reducing the magnitude of downstream signaling pathway activation (Niu, Zhao, Wu et al., 2020). Thus, even small changes in the structure of FGF19 markedly change the response to target receptor. The dimerization of FGF15 likely reduces the diversity of tissue targets compared to FGF19, conferring different overall biological effects. In total, these observations raise important questions about the endogenous function of mouse FGF15 relative to human FGF19.
The role of FGF15 appears to be mainly limited to the functions within the liver achieved through intestinal-derived FGF15 binding to FGFR4/KLB. FGF15 has limited ability, relative to human FGF19, to activate signaling in peripheral tissues through FGFR1c/KLB binding. This paradigm is especially important when using the mouse to model disease states that involve alterations in FGF19 signaling. For example, in human cholestatic liver disease, FGF19 expression is induced in hepatocytes (Milkiewicz et al., 2016,Wunsch et al., 2015,Hasegawa et al., 2019,Johansson et al., 2020), but the functional relevance of hepatic FGF19 expression during cholestasis is unknown. Trying to model this in mice presents a considerable challenge, as even administration of FGF15 agonists to mice does not induce FGF15 expression in the liver (Inagaki, Choi, Moschetta et al., 2005). Similar consideration should be taken to interpreting the effects of activating FGFR1c/KLB in peripheral tissues in mice. As FGF15 does not appear to be regulating peripheral tissues via these pathways, the overlap to human response of FGF19 should be taken with caution. Use of other animal models that more closely model human FGF19 signaling would add more clarity on the role of FGF19 in species where FGF19 endogenously targets FGFR4 and FGFR1c target tissues. Other animal models express FGF19 similar to humans, such as pig (Call, Molina, Stoll et al., 2020,Vonderohe, Guthrie, Stoll et al., 2021,Gavalda-Navarro, Pastor, Mereu et al., 2018) and rabbit (Shang, Guo, Honda et al., 2013). Further research is needed to investigate the function of FGF19 in these species and whether they activate signaling pathways in multiple tissues via FGFR4 or FGFR1c receptors.
5. Transcriptional Regulation of FGF15/19 Expression
5.1. Transcriptional Activators
FGF15/19 is an inducible protein that has multiple sites within its promoter region for activation of its expression. The best-described transcriptional activators of FGF15/19 are members of the nuclear hormone receptor family of transcription factors. The bile acid responsive nuclear hormone receptor, farnesoid x receptor (FXR) is most closely associated with the role of FGF15/19 in regulation of bile acid homeostasis. FXR is highly responsive to primary bile acids and induces FGF19 in a dose-dependent manner when the bile acid chenodeoxycholic acid (CDCA) is administered in both humans and mice (Holt et al., 2003,Li, Pircher, Schulman et al., 2005). Within the FGF19 gene, there are FXR-responsive binding elements (FXRE) in three promoter positions within 2000 kb upstream of the transcription start site (Miyata, Hata, Yamakawa et al., 2012) and within the second intron (Holt et al., 2003). The mouse Fgf15 gene also contains a FXRE in the second intron of the gene, mirroring human FGF19 (Li et al., 2005). In addition to FXR, the xenobiotic sensing nuclear hormone receptor pregnane x receptor (PXR) is also responsive to high concentrations of the toxic secondary bile acid, lithocholic acid (LCA), a derivative of CDCA (Kliewer and Willson, 2002). A PXR binding site was identified in the promoter region of FGF19 within 300 bp of the transcription start site (Wistuba, Gnewuch, Liebisch et al., 2007). The mouse Fgf15 gene does contain a homologous region within the promoter, but mouse and human activation of PXR differ in the downstream regulation of FGF19. FGF19 is potently induced in human intestinal cell culture by LCA or rifampicin treatment (Wistuba et al., 2007) and in piglets administered intravenous rifampicin (Guthrie, Stoll, Chacko et al., 2020); yet in mice, suppression of PXR increases the expression of FGF15 (Zhao, Xu, Shi et al., 2017).
In vitro models of stress have identified non-bile acid related transcriptional regulation of FGF15/19. In response to endoplasmic reticulum stress, the transcription factor activating transcription factor 4 (ATF4) can upregulate FGF19 expression by binding to the amino acid response element (AARE) (Shimizu, Li, Maruyama et al., 2013). In vitro culture of colonic myofibroblasts treated with carbon monoxide show a robust increase in FGF15 expression (Uchiyama, Naito, Takagi et al., 2010). A micro RNA targeted to Fgf15 mRNA, miR-710, was significantly reduced during the treatment, suggesting that FGF15 post-transcriptional regulation is mediated by micro RNAs and can be transiently altered in response to cellular environmental changes.
5.2. Transcriptional Repressors
The transcriptional repression of FGF15/19 occurs through both direct binding and indirect suppression of transcriptional activators. The sterol regulatory element-binding protein 2 (SREBP2) is activated when cholesterol levels are low to upregulate genes in cholesterol biogenesis (Brown and Goldstein, 1997). In the human intestinal cell line LS174T, activation of SREBP2 reduced the expression of FGF19 (Miyata, Hata, Yamazoe et al., 2014). Subsequent analysis confirmed this repression was due to SREBP2 directly interacting with FXR, preventing binding to the FGF19 promoter. This inhibition of FGF19 is somewhat paradoxical in response to low cholesterol levels. As FGF19 decreases the production of bile acids, it spares cholesterol which is the precursor for bile acid synthesis. Therefore suppression of FGF19 by SREBP2 would increase the loss of cholesterol through elevated bile acid metabolism. Given this, whether the mechanism has any physiological relevance in vivo still requires further research.
Other direct mediators of transcriptional repression of the Fgf15 promoter have been identified in mice and not yet confirmed in humans. Mice that have the intestinal deletion of Kruppel-like factor 15 (Klf15−/−), but not liver deletion of Klf15 have decreased bile acid concentrations, which is a key pathway FGF15 targets (Han, Zhang, Jain et al., 2015). Furthermore, in intestinal Klf15−/− mice, the circadian patterning of bile acid synthesis is lost, suggesting that KLF15 is the direct regulator of bile acid cycling. Exploration of the mouse Fgf15 promoter region via chromatin immunoprecipitation assays confirmed that there are at least three promoter regions in the mouse Fgf15 gene that are bound by KLF15. In additional support of the KLF15/FGF15 axis, studies examining the bile acid increasing effects of the type 2 diabetes drug, Teneliglipitn, found KLF15 activated through phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) pathway and downstream suppression of FGF15 (Wang, Wu, Cui et al., 2020). In the intestine, the segmental expression of FGF15 along the intestine is also regulated by direct transcriptional repression. GATA binding protein 4 (GATA4) is highly expressed in the jejunum and has lower expression in the ileum. Mice with ileal knock-in Gata4 have decreased expression of FGF15 and mice with jejunal knock out Gata4 have increased FGF15 (Thompson, Wojta, Pulakanti et al., 2017).
In addition to direct suppression, there is indication that indirect suppression of FGF15 is possible. Treatment of mice with the glucocorticoid receptor (GR) agonist, dexamethasone, reduces the expression of ileal Fgf15 mRNA, along with other FXR target genes (Jia, Zhang, Jia et al., 2019). GR was also capable of upregulating the PXR target gene, Cyp3a11, but treatment with PXR agonist, pregnenolone‑16‑α‑carbonitrile, had no effect of Fgf15 mRNA. The exact mechanism that drives Fgf15 mRNA suppression is not clear. Also, whether this effect is recapitulated on human FGF19 has yet to be established.
6. Functions
6.1. Regulation of Bile Acid Homeostasis
FGF15/19 activation is associated with negative feedback regulation of bile acid synthesis. There are multiple key targets of FGF15/19-mediated regulation in the bile acids synthesis pathway, so a brief overview of the main aspects of bile acid synthesis will be highlighted. Bile acids are amphipathic molecules synthesized by hepatocytes in the liver to facilitate the absorption of dietary lipid. In humans, the primary bile acids are CDCA and cholic acid (CA). The synthesis of these two bile acids occurs via two pathways, the classical (neutral) and alternative (acidic). The neutral pathway is the predominant synthesis pathway in adults and results in the formation of both CA and CDCA (Pullinger, Eng, Salen et al., 2002). The enzyme, cytochrome P450 7A1 (CYP7A1), is the key synthetic enzyme in the neutral pathway for the initiation of bile acid synthesis. CA is formed by an additional modification via CYP8B1. Both CA and CDCA undergo side chain oxidation via the mitochondrial enzyme CYP27A1. Following this, peroxisomal carbon chain cleavage finalizes formation of CA and CDCA. The acidic pathway is initialized by mitochondrial CYP27A1, rather than by CYP7A1. Unlike the neutral pathway, the acidic pathway can be initiated in multiple cell types, including cholangiocytes and macrophages. Several initial oxysterol intermediates are formed via CYP27A1 including 25-hydroxycholesterol and 26-hydroxycholesterol, which are themselves key regulators of cholesterol homeostasis (Cali and Russell, 1991,Li, Pandak, Erickson et al., 2007). The oxysterols formed in the acidic pathway need to be transported to hepatocytes for final processing to form bile acids. However, the acidic pathway can only lead to the synthesis of CDCA. In mice, there is synthesis of CDCA, but rather small amounts of the bile acid in circulation because CDCA is converted to α-muricholic acid (α-MCA) then modified to form β-MCA (Botham and Boyd, 1983).
Originally, α-MCA synthesis was believed to occur through Cyp3a11 (Cuesta de Juan, Monte, Macias et al., 2007), however newer research suggest that Cyp2c70 catalyzes α-MCA formation (Takahashi, Fukami, Masuo et al., 2016). α-MCA is then predominantly converted to β-MCA via C7 epimerization. Other species have a similar modification step following synthesis of CDCA. The pig converts CDCA to γ-muricholic acid, otherwise called hyocholic acid (HCA), via CYP4a21 (Haslewood, 1954,Lundell, Hansson and Wikvall, 2001). β-MCA and HCA are more hydrophilic than CDCA and far less cytotoxic, so species other than humans have an additional mechanism to protect the liver from high bile acid concentrations. After synthesis, bile acids are conjugated with either glycine or taurine in a two-step process of activation by bile acid:CoA synthase enzymes and amidation by bile acid:amino acid transferase enzymes. These bile acids are then transported via the bile salt efflux pump (BSEP) located in the canalicular membrane of hepatocytes to the gall bladder and intestines.
Bile acids that are in the small and large intestine can undergo additional modifications. Multiple species of bacteria, including Lactobacillus (Elkins, Moser and Savage, 2001), Enteroccocus (Wijaya, Hermann, Abriouel et al., 2004), Bifidobacterium (Grill, Schneider, Crociani et al., 1995), and others (Rossocha, Schultz-Heienbrok, von Moeller et al., 2005) can deconjugate bile acids using bile salt hydrolase enzymes. Once deconjugated, a much smaller set of bacteria, some belonging to the genus Clostridium (Wells, Williams, Whitehead et al., 2003), have 7α/β hydroxylase enzymes that convert primary bile acids to secondary bile acids. CDCA is converted to LCA and CA is converted to deoxycholic acid (DCA). The secondary bile acids are much more hydrophobic than primary bile acids and more toxic to the liver (Heuman, 1989). Little LCA is reabsorbed, but the LCA that is gets rapidly re-amidated and sulfonated (Hofmann, 2004). Sulfonated LCA is unable to be reabsorbed once recirculated into the intestines. CDCA, CA, and DCA are reabsorbed in the intestines and recirculated to the liver resulting in approximately 95% of all bile acids recovered in the liver.
The process by which bile acids are synthesized is tightly regulated given the potential for cytotoxicity of high bile acid concentration and the large volume of bile acids that are recirculated back to the liver. Regulation of bile acid synthesis is predominantly focused on the action of CYP7A1. Prior to the identification of FGF19, FXR downregulation of CYP7A1 was thought to be primarily dependent on hepatic FXR-mediated upregulation of the small heterodimeric partner (SHP) (Goodwin, Jones, Price et al., 2000). SHP can bind to transcription factors that activate CYP7A1 expression, including liver receptor homolog 1 (LRH-1, Nr5a2) and hepatocyte nuclear factor 4 alpha (HNF4), and block their promotion of CYP7A1 mRNA transcription (Inoue, Yu, Yim et al., 2006,Lu, Makishima, Repa et al., 2000). However, studies in hepatic Shp−/− mice did not observe large increases in bile acid production suggesting there are SHP-independent mechanisms (Inagaki et al., 2005,Wang, Han, Kim et al., 2003). Early studies examining deletion of Fgf15 in mice observed an impaired ability of mice to suppress CYP7A1 and an elevation in bile acid concentration, pointing to FGF15 as a clear mediator of bile acid regulation (Inagaki et al., 2005). Primary bile acids (predominantly CDCA) in the intestine bind to FXR leading to transcriptional upregulation of FGF15/19. After FGF15/19 is translated, it associates with the protein, DIET1; however, the exact function of DIET1 is not clear. The absence of DIET1 expression results in a decrease in circulating FGF15/19 (Lee, Ong, Vergnes et al., 2018,Vergnes, Lee, Chin et al., 2013).
Once secreted from the intestine, FGF15/19 circulates to the liver to repress bile acid synthesis (Figure 2). The non-receptor tyrosine phosphatase SHP2 complexes with and increases tyrosine phosphorylation of FGFR substrate 2 alpha (FRS2α) to initiate further downstream phosphorylation events (Li, Hsu, Li et al., 2014). In part, SHP2 may also act as a feed forward messenger as it leads to increased tyrosine phosphorylation of FGFR4, which presumably increases FGFR4 signaling activity. The downstream target of FRS2α and SHP2 is the non-receptor tyrosine kinase SRC, which is phosphorylated at amino acid position Tyr278 (Byun, Kim, Ryerson et al., 2018). SRC is responsible for activating the nuclear translocation of FXR via phosphorylation of FXR at Tyr67. FXR translocation allows for activation of bile acid homeostasis genes including hepatic bile export transport genes, BSEP, organic anion solute transporter alpha/beta (OSTα/β), and multidrug resistance protein 2 (MRP2). SRC is also capable of phosphorylating mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 (MAPK/ERK1/2) the canonical pathway associated with CYP7A1 downregulation (Lin et al., 2007,Kurosu et al., 2007,Wu et al., 2007,Song, Li, Owsley et al., 2009). However, SRC knockdown of mouse primary hepatocytes only leads to a marginal decrease in ERK1/2 phosphorylation, so there is likely an additional direct action of FSR2α/SHP2 on ERK1/2 phosphorylation (Byun et al., 2018).
Figure 2.
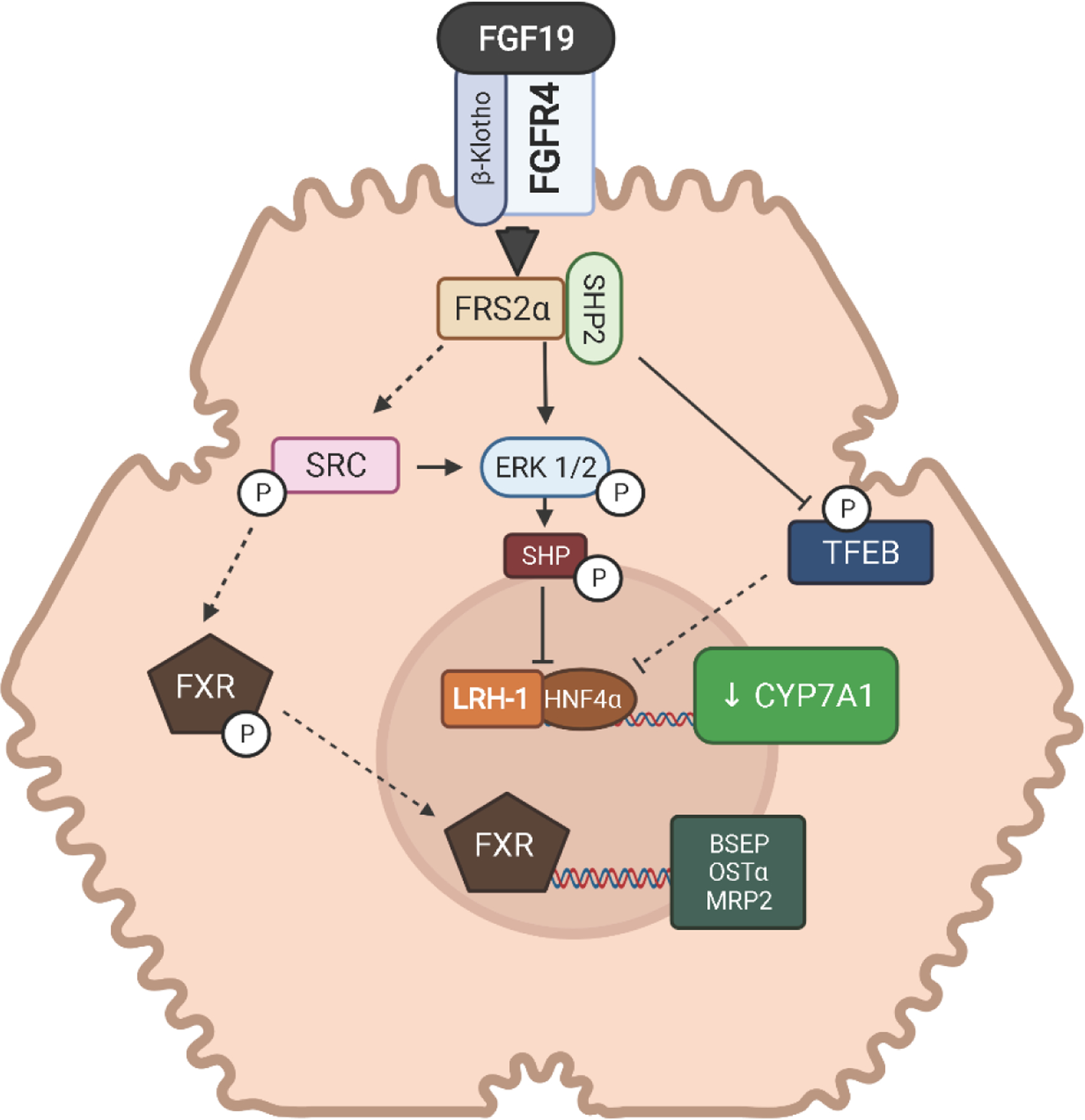
Bile acid signaling of FGF19-FGFR4 in the liver. fibroblast growth factor (FGF), fibroblast growth factor receptor (FGFR), β-Klotho (KLB), farnesoid x receptor (FXR), cytochrome P450 7A1 (CYP7A1), bile salt efflux pump (BSEP), small heterodimeric partner (SHP), liver receptor homolog 1 (LRH-1), hepatocyte nuclear factor 4 alpha (HNF4), FGFR substrate 2 alpha (FRS2α), solute transporter alpha/beta (OSTα/β), multidrug resistance protein 2 (MRP2), extracellular signal-regulated kinase 1/2 (ERK1/2), transcription factor EB (TFEB). Dashed lines represent potential pathways not fully confirmed experimentally.
The mechanism by which ERK1/2 phosphorylation leads to CYP7A1 suppression is only partially understood. ERK1/2 phosphorylation of SHP at Ser2 may improve protein stability of SHP (Miao, Xiao, Kanamaluru et al., 2009). SHP is post-translationally regulated via ubiquitination at Lys122/Lys123 which leads to its degradation. The ERK-mediated phosphorylation of SHP blocks ubiquitination, increasing the protein abundance of SHP. More recently, the transcription factor EB (TFEB) was identified as a positive regulator of CYP7A1 expression that could be suppressed through ERK1/2 (Wang, Gunewardena, Li et al., 2020). During cholesterol induced lysosomal stress, TFEB translocates to the nucleus and binds to the CYP7A1 promoter upregulating gene expression. In doing so, TFEB reduces cholesterol through enhanced bile acid synthesis. In the liver cell line, HepG2, inhibition of ERK1/2 increases TFEB-targeted upregulation of CYP7A1. Conversely, FGF19-mediated activation of ERK1/2 decreases nuclear translocation of TFEB. Further analysis of TFEB identified a single serine residue, Ser211 that ERK1/2 targets to prevent nuclear translocation of TFEB. (Wang et al., 2020). A second downstream target of the FGF19 signaling pathway in the liver, mammalian target of rapamycin (mTOR), is also capable of phosphorylating TFEB on Ser211 and preventing nuclear translocation (Wang et al., 2020,Wan, Tian, Tan et al., 2016). Despite ERK1/2 being upstream of mTOR in the FGF19 signaling pathway, inhibitor studies strongly indicate that either ERK1/2 directly or mTOR can prevent TFEB translocation, leading to suppression of CYP7A1 expression.
The role of the acidic pathway for bile acid synthesis in adults is typically considered minor. However, infants have high placental transfer and urinary output of metabolites specific to the acidic pathway, suggesting it supplies a greater contribution to their bile acid pool (Tohma, 1996,Tazawa, Yamada, Nakagawa et al., 1984). In adults, there is indication that the acidic pathway has an important role in bile acid synthesis during primary biliary cirrhosis (PBC), wherein CYP7A1 is repressed, but bile acid synthesis is still active (Wunsch et al., 2015). Hepatic FGF19 is significantly increased in PBC patients, and is potentially the source of CYP7A1 repression. However, given continued bile acid synthesis, this would suggest that FGF19 is not able to suppress bile acid synthesis in the acidic pathway in hepatocytes. Since the acidic pathway is not limited to hepatocytes, other cells in the liver, such as cholangiocytes, could supply a source of oxysterols for conversion to bile acids. In cholangiocytes, cholesterol can be converted to oxysterols via the acidic pathway, which can then be transported back to the hepatocyte via the peribiliary plexus to undergo further conversion to bile acids (Xia, Francis, Glaser et al., 2006). In hepatocytes, there does not appear to be a direct regulatory effect of FGF19 on CYP27A1 synthesis of oxysterols in the acidic pathway. However, in cholangiocytes, FGF19 induces a p38-dependent signaling pathway that decreases the expression of CYP27A1 (Jung, York, Wang et al., 2014). This reduction can decrease oxysterol production in cholangiocytes. Whether reduction in the oxysterol production equates to reduced bile acid synthesis from the acidic pathway is not known. It may be that the oxysterols production is locally used to regulate cholesterol metabolism, rather than use for precursors to bile acid synthesis. Yet, this presents and intriguing potential bile acid synthesis mechanism in cholangiocytes that warrants further investigation.
Bile acids upregulate expression of FGF19 in the intestine by binding to the FXR. However, FXR is not the only bile acid responsive receptor in the body. In the intestine and macrophages, G-protein-coupled bile acid receptor (TGR5) is bound by DCA. TGR5 is a key regulator of metabolism that promotes improved glucose control and reduced obesity, thus many metabolic targets of TGR5 overlap with FGF19 (Guo, Chen and Wang, 2016). In the liver, sphingosine 1 phosphate receptor 2 (S1PR2) can activate hepatic Akt and ERK pathways to enhance glucose metabolism, regulate bile acid synthesis, and lipogenesis (Studer, Zhou, Zhao et al., 2012), as well as, enhance liver regeneration (Ikeda, Watanabe, Ishii et al., 2009). Like FXR and TGR5, S1PR2 also binds to bile acids for activation, with taurine conjugated CA being the most potent ligand. Because FGF15/19 can suppress bile acid synthesis, it can alter the production of specific bile acids and indirectly effect metabolism through suppression or generation of bile acids that target TGR5 and S1PR2. In mice, infusion of FGF19 decreases taurine conjugated CA (Wu et al., 2011). This change represents a shift from the neutral pathway to the acidic pathway following suppression on CYP7A1. FGFR4−/− mice conversely have markedly increased concentration of taurine conjugated CA, the ligand for S1PR2. FGF15/19 in this way act as a suppressor of the S1PR2 pathway. Anti-FGF19 antibody treatment in Cynomolgus monkeys causes a large increase in bile acid production, which results in much greater concentration of bile acids in the intestines (Pai, French, Ma et al., 2012). This resultant increased bile acid substrate for bacteria increases the presence of secondary bile acids produced in the gut, including the TGR5 target DCA. Therefore, when FGF19 is active, it may suppress other pathways that contribute to metabolic regulation through indirect methods by reducing the production of bile acid that target them.
FGF15/19 can also impact the flow of bile by mediating gall bladder filling (Choi et al., 2006). Fgf15−/− mice have no bile in their gall bladders after fasting. Administration of either FGF15 or FGF19 causes significant gall bladder filling. In these mice, after cannulation of the common hepatic duct, there was no observed increase in bile flow from the liver, suggesting that filling of the gall bladder is independent of increased outflow of bile from the liver. Whether the target of FGF15/19 is the cholangiocytes or smooth muscle has not been established, nor is the receptor target for gall bladder filling entirely clear. In mice, there is a >10-fold greater expression of FGFR3 compared to FGFR1, FGFR2, and FGFR4. Given this, it has been suggested that FGFR3 may be the primary target for FGF15/19 in the gall bladder. However, in human cholangiocytes, there is abundant expression of FGFR4 (Zweers et al., 2012). In mice, it is likely that FGFR4 is still the primary FGFR involved in gall bladder filling. In part, this would be due to the ability of FGF15 to form dimers, so it is not as potent a ligand for FGFR3c, as FGF19 (Zhou et al., 2017). This is also partially supported in Fgfr4−/− mice, which have gall bladders markedly decreased in size, suggesting that they do not undergo much filling (Yu, Wang, Kan et al., 2000). However, Fgfr4−/− mice can restore gall bladder filling with FGF19 administration (Choi et al., 2006). This finding may suggest that some effects on gall bladder filling are from local binding to FGFR3 in the gall bladder, or activation of sympathetic nervous system signals through FGFR1c, but this has not been established experimentally. In humans, it is also likely that FGFR4 is the main target given the high expression in gall bladder, but may be possible that FGFR3 or other FGFRs have functional activity as well. The action of FGF19 on the gall bladder is mediated, at least in part, through a c-AMP dependent relaxation of smooth muscle within the gall bladder (Choi et al., 2006). It is also capable of preventing cholecystokinin-mediated contraction of gall bladder smooth muscle.
The gall bladder is also a large producer of FGF19 in humans, but mice do not produce any FGF15 in their gall bladders (Zweers et al., 2012,Barrera, Azocar, Molina et al., 2015). The exact purpose of the large quantity of FGF19 in gall bladder is not entirely clear. Though it has been postulated it may have protective effects against bile acid toxicity, there have been no studies to directly support this, or to give insight into how FGF19 could protect from bile acid toxicity. In patients that have undergone cholecystectomy, there is increased synthesis of bile acids and altered diurnal rhythm of FGF19. However, they have no changes in serum bile acid concentration or cholesterol concentration (Barrera et al., 2015). Moreover, in cholecystectomy patients treatment with OCA markedly increased the FGF19 concentration in bile and risk for gallstones (Al-Dury, Wahlstrom, Panzitt et al., 2019).
6.2. Regulation of Glucose and Lipid Metabolism
FGF15/19 displays biphasic fluctuations regulated in part by feeding/fasting patterns of dietary intake. In response to a meal, FGF19 increases transiently for approximately 3 hr and decreases over time during fasting (Lundasen, Galman, Angelin et al., 2006). Thus, the discovery of FGF15/19 as a regulator of metabolism fits with other classic hormones such as insulin and glucagon that also have altered expression patterns in response to dietary intake. Unlike the role of FGF15/19 in regulation of bile acid synthesis, the actions of FGF15/19 on energy expenditure, glucose metabolism, and lipid metabolism are more heterogeneous. Therefore, it is important to delineate the role of FGF15 and FGF19 in the context of metabolism before going into detail on specific activities in various organs. In the liver, FGF15 and FGF19 bind to FGFR4/KLB, and have many shared downstream effects specific to actions in the liver, hence the similar bile acid responses. However, in extrahepatic tissues, the primary targets of FGF15/19 are FGFR1c-3c. Since FGF15 has much lower affinity for FGFR1c-3c compared to FGF19 there is likely a large species divergence in observed metabolic effects, with a much greater effect from FGF19. The literature can be more complicated when examining mouse studies that use endogenous FGF15 response in mice compared to studies that use FGF19. Given this, much of the reviewed literature on metabolism will focus on the role of FGF19 in modulating metabolism, rather than the specific role of FGF15.
6.2.1. Glucose Metabolism
The regulation of glucose in the body is a sophisticated interplay between glucose production (gluconeogenesis), glucose uptake (both peripheral and hepatic), and glucose storage (glycogen synthesis). FGF15/19 exerts regulatory pressure at all three of these key pathways to modify whole body glucose concentrations. As mentioned above, FGF19 likely exerts more global glucose control than FGF15, given extrahepatic targeting, which will be discussed in detail.
Unlike bile acid regulation, which is entirely dependent on FGF15/19 binding to FGFR4/KLB in the liver, control over glucose homeostasis occurs through both hepatic FGF15/19 binding to FGFR4/KLB and extrahepatic tissues activated by FGF19 binding to FGFR1c/KLB (Figure 3). When observing whole body glycemic control, FGF19 is capable of reducing glucose levels in obese models of mice. In high fat diet fed obese mice, transgenic overexpression of FGF19 imparts resistance to post-prandial hyperglycemia, increases sensitivity to insulin, and lowers circulating insulin levels (Tomlinson et al., 2002). Likewise, in ob/ob leptin deficient obese mice, transgenic overexpression of FGF19 improves fasting blood glucose and glucose excursion after glucose tolerance tests (Fu, John, Adams et al., 2004). The injection of FGF19 into ob/ob mice has a similar effect with reduced post-prandial glucose levels and improved glycemic response following glucose tolerance test (Fu et al., 2004,Wu, Ge, Baribault et al., 2013). FGF15 does not confer the same protective effects against hyperglycemia, as does FGF19. In db/db diabetic mice and diet induced obesity mice, adeno-associated virus (AAV) injections of FGF15 failed to lower glucose levels, whereas AAV-FGF19 administration restored blood glucose concentrations to that of healthy control mice (Zhou et al., 2017). This difference highlights that uptake of glucose in peripheral tissues, at least in mice, through FGFR1c/FGF19 is important for whole body glucose uptake. In the liver, the primary mechanism through which FGF15/19 is able to reduce circulating glucose levels and enhance the activity of insulin is through increasing glycogen synthesis and suppressing gluconeogenesis. In fasted mice, administration of FGF19 enhances glycogen synthesis through activation of the ERK1/2 pathway, which targets 90-kDa ribosomal s6 kinase 1 (p90RSK) (Kir et al., 2011). This signaling peptide then interacts with glycogen synthase kinase (GSK) 3α and GSK3β. GSK3α and GSK3β are negative regulators of glycogen synthesis and suppress the action of glycogen synthase (GS). GS is inactive when phosphorylated and there is no synthesis of glycogen. Therefore, through phosphorylation of GSK3α and GSK3β, FGF19 prevents GS phosphorylation and maintains active glycogen synthesis. This function of FGF15/19 acts in parallel with the activity of insulin that is also capable of phosphorylating GSK3α and GSK3β at the same residues (Ser21 and Ser9, respectively) (Sutherland, Leighton and Cohen, 1993). However, insulin acts on these proteins through the Akt pathway.
Figure 3.
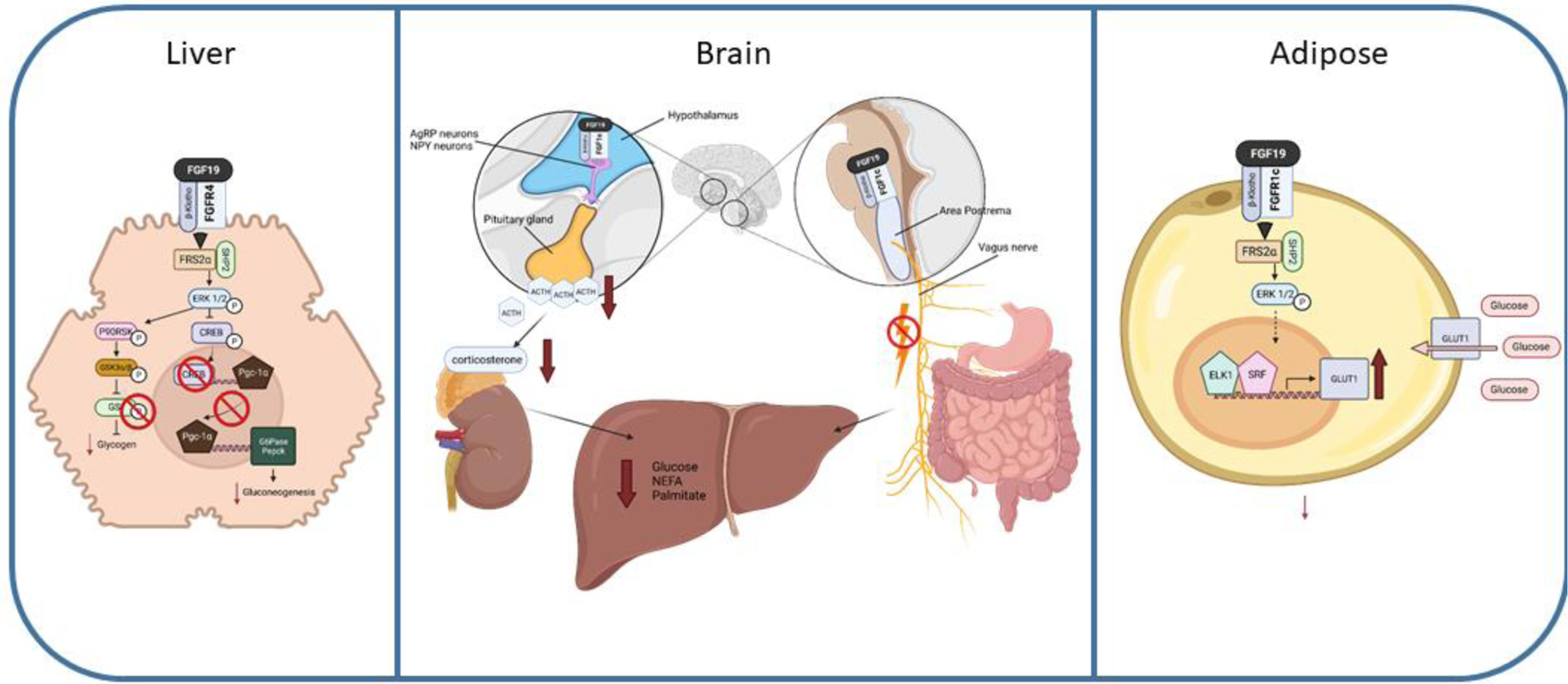
FGF19 regulation of glucose homeostasis in the liver, brain, and adipose tissue. fibroblast growth factor (FGF), fibroblast growth factor receptor (FGFR), β-Klotho (KLB), FGFR substrate 2 alpha (FRS2α), extracellular signal-regulated kinase 1/2 (ERK1/2), 90-kDa ribosomal s6 kinase 1 (p90RSK), glycogen synthase kinase (GSK), glucose-6-phophatase (G6pase), phosphoenoylpyruvate kinase (Pepck), cAMP regulatory element-binding protein (CREB), peroxisome proliferator-activated receptor gamma coactivator (PGC-1α), agouti-related peptide (AgRP), neuropeptide Y (NPY), adrenocorticotropic hormone (ACTH), erythroblast transformation specific transcription factor (ELK1), serum response factor (SRF), glucose transporter (GLUT), non-esterified fatty acid (NEFA). Dashed lines represent potential pathways not fully confirmed experimentally.
The aberrant production of glucose through gluconeogenesis is a hallmark characteristic of metabolic syndrome and diabetes (Hatting, Tavares, Sharabi et al., 2018). The ability of FGF15/19 to suppress gluconeogenesis is likely a key component to improve glucose control in mouse models of obesity. In mice, FGF15/19 suppresses the expression of key gluconeogenic genes including glucose-6-phophatase (G6pase) and phosphoenoylpyruvate kinase (Pepck) following 6 hr infusion of either peptide (Potthoff, Boney-Montoya, Choi et al., 2011). Based on promoter binding studies, direct gene regulation occurs through the ability of cAMP regulatory element-binding protein (CREB) being able to bind to the promoter of either gene. In addition, CREB also downregulates expression of a key transcription factor in metabolism, peroxisome proliferator-activated receptor gamma coactivator (PGC-1α), which also displays reduced binding to G6pase and Pepck. As of yet, the mechanism driving the dephosphorylation of CREB in this pathway is not fully known. Some research into CREB target gene silencing by FGF19 suggests that SHP binds to CREB, which can recruit histone methylase LSD1 causing epigenetic silencing of CREB target genes (Byun, Kim, Zhang et al., 2017). However, this mechanism does not entirely align with observed Co-IP of CREB and PGC-1α promoter binding data within G6Pase and Pepck (Potthoff et al., 2011). This suggests two separate mechanisms imparting repression of gluconeogenesis, but more research on these observations is needed.
Independent of the hepatic role of FGF19 on glucose homeostasis, FGF19 binding in the brain confers an additional level of control over serum glucose concentrations through pathways in both the forebrain and hindbrain. In rats with diet induced obesity, injection of FGF19 in the third cerebral ventricles lowers blood glucose without increasing release of insulin (Ryan, Kohli, Gutierrez-Aguilar et al., 2013). A similar glucose lowering effect occurred in ob/ob mice administered intracerebroventricular (ICV) FGF19 injection (Morton, Matsen, Bracy et al., 2013). Unlike mice given FGF19 i.v., ICV administered mice have reduced G6Pase mRNA, but no change to their Pepck mRNA levels and no increase in glycogen synthesis, providing insight into divergent mechanisms within the brain and liver over glucose regulatory control. Detailed examination of the neurons within the arcuate nucleus of the hypothalamus determined that the neuronal targets of FGF19 are not localized in the lateral melanocortin proopiomelanocortin (POMC) neuronal cells. Rather, FGF19 can activate ERK1/2 signaling in the dorsomedial hypothalamic nucleus to suppress agouti-related peptide (AgRP)/ neuropeptide Y (NPY) neuronal activity (Marcelin, Jo, Li et al., 2014). The downstream activity of hypothalamic activation by FGF19 was mapped out in rats with streptozotocin induced type 1 diabetes (T1DM). ICV injection of FGF19 proved effective in decreasing serum glucose concentrations in an insulin-independent mechanism (Perry et al., 2015). The rats had markedly reduced adrenocorticotropic hormone (ACTH). The downstream reduction of circulating corticosterone and subsequent decreased lipolysis and acetyl CoA levels suppresses hepatic glucose production. Research that is more recent identified the dorsal motor nucleus of the vagus (DMV) as expressing both FGFR1 and KLB. Direct targeting of the DMV by 4th ventricle administration of FGF19 reduced serum glucose confirming a hindbrain pathway of FGF19 (Wean and Smith, 2021). It is plausible that FGF19 suppresses central vagal circuitry involved in vago-vagal reflexes, including glucose regulatory circuits within gut-brain-liver axis.
Diabetic mice (db/db) are unable to reduce serum glucose levels when administered FGF15, but effectively do so with FGF19 administration, which is due to FGF19’s ability to bind to FGFR1c in peripheral tissues (Zhou et al., 2017). Adipocytes exclusively express FGFR1 among all FGFRs and express KLB. Administration of FGF19 to adipocytes increases phosphorylation of FRS2α and ERK1/2. Glucose uptake is elevated in adipocytes exposed to FGF19 and knockdown of Klb suppresses glucose uptake (Kurosu et al., 2007,Hansen, Vienberg, Lykkegaard et al., 2018). Further confirmation of FGF19-activiated glucose uptake in adipocytes was observed in DIO mice with adipocyte specific knockout of Klb (Lan et al., 2017). During a euglycemic-hyperinsulemic clamp, glucose infusion rate was significantly higher in wild-type mice after FGF19 infusion compared to KLB−/−. Additionally, whole body glucose uptake increased during FGF19 infusion. However, some data conflicts with the ability of FGF19 to facilitate glucose uptake. A study by Anotonellis et al. did not see any increase in glucose uptake in DIO mice in white adipose tissue in either the absence or presence of insulin. There was a synergistic effect on glucose uptake in brown adipose tissue with FGF19 and insulin administration, however, FGF19 administered alone did not enhance glucose uptake in brown adipose tissue (Antonellis, Droz, Cosgrove et al., 2019). There is not any direct study of FGF19 increasing glucose transporters (GLUT) in adipocytes, but it is highly likely that, like FGF21 which shares the same FGF1c/KLB binding in adipocytes, FGF19 can increase GLUT1 in adipocytes (Ge, Chen, Hui et al., 2011). In pregnant mice, FGF19 can up regulate expression of the glucose transporter, GLUT4, and increase phosphorylation of the insulin signaling molecule, IRS1 and perhaps similar activation in adipocytes is present (Zhao, Wang, Li et al., 2021).
The action of FGF19 to reduce circulating glucose levels is through hepatic, central, and peripheral targeting but there are gaps in our understanding of glucose uptake from FGF19 signaling. Much of the peripheral impact of glucose uptake from FGF19 is focused on adipocytes. It is established that both the brain and muscle are targets of FGF19 and glucose-utilizing tissues. However, the exact contribution they impart on glucose uptake, mediated by FGF19, is unknown. Despite this gap, the ability of FGF19 to reduce serum glucose concentration does present exciting translational possibilities. In obese patients that receive roux-en-y gastric bypass surgery, rapid increases in bile acids and FGF19 are observed postoperatively (Sachdev, Wang, Billington et al., 2016,Gerhard, Styer, Wood et al., 2013,Jansen, van Werven, Aarts et al., 2011). There is some suggestion that this increase in FGF19 directly promotes resolution in type 2 diabetes suppressing gluconeogenesis and improving peripheral glucose uptake (Gerhard et al., 2013,Bozadjieva, Heppner and Seeley, 2018,Pournaras, Glicksman, Vincent et al., 2012). Yet, this finding is not supported in all human studies and is still a topic of debate (Harris, Smith, Mittendorfer et al., 2017,Jorgensen, Dirksen, Bojsen-Moller et al., 2015). However, this presents a unique potential opportunity for research into FGF19 and what role direct administration of the hormone may have in improving glucose control in patients with type 2 diabetes.
6.2.2. Lipid Metabolism
White and brown adipose tissue express FGFR1c and KLB and are therefore potential targets for FGF19 activity (Fon Tacer et al., 2010). FGF19 regulation of adiposity was first observed in the development of the transgenic mice overexpressing the gene. These mice had lower body weight and higher lean mass relative to normal mice (Tomlinson et al., 2002). In various models of obesity in mice, FGF19 also promotes reduced adiposity. In DIO mice fed a diet of high fat, high fructose, and high cholesterol AAV overexpression of reduced accumulation of hepatic lipids and expression of lipogenic enzymes (Zhou, Learned, Rossi et al., 2017). In addition, there were increased un-oxidized cardiolipins in the inner mitochondrial membrane. These cardiolipins are essential for mitochondrial function and efficient energy conversion, therefore providing an additional mechanism to facilitate lipid clearance in these mice (Dudek, 2017). In leptin deficient ob/ob mice fed a high fat diet, transgenic overexpression of FGF19 can reduce body weight and fat pad mass (Fu et al., 2004). However, in ob/ob leptin deficient mice fed standard chow, there were no changes in body weight with exposure to FGF19 (Fu et al., 2004,Wu et al., 2013). Interestingly, despite the chow fed ob/ob mice not losing any weight, they did have increased serum concentrations of triglyceride and cholesterol (Wu et al., 2013). Whereas initially this would appear paradoxical to have a phenotype that can drive a reduction in fat mass, but also cause elevations in serum lipids, it is suggested this is through the dual functions of FGF19. The hepatic role of FGF19 in suppressing bile acid metabolism leads to increased cholesterol and triglycerides, while the metabolic effects on adiposity target utilization of deposited lipid in peripheral tissue. In support of this hypothesis, generation of a C-terminal domain modified FGF19, which leads to loss of FGF1c binding, still increases cholesterol and triglyceride concentration in ob/ob mice, suggesting it is a FGFR4-dependent effect. In humans, it is not clear if the same response will be present. In clinical trials, the use of the FGF19 analog NGM282 reduced triglycerides by up to 47.3 mg/dl compared to baseline, but did see an increase in cholesterol levels. (Harrison, Rinella, Abdelmalek et al., 2018,Rinella, Trotter, Abdelmalek et al., 2019). Also in humans, short term treatment with bile acid sequestrant, cholestyramine, decreased FGF19 and caused a transient increase in triglycerides. However, treatment in subjects for up to one month still observed an increase in FGF19 concentrations, but triglycerides were unchanged (Sjoberg, Straniero, Angelin et al., 2017).
Despite the clear effect that overexpression of FGF19 has on the reduction of hepatic lipids, there is conflicting data on hepatic lipid accumulation from deletion of FGF15. Fgf15−/− mice fed a high fat diet for six months did not differ in the degree of hepatic steatosis or inflammation in the liver. Interestingly, they had a reduction in fibrosis compared to HFD fed controls. The KO mice did have elevated serum triglycerides, alterations in lipogenic gene expression, and altered bile acid homeostasis (Schumacher, Kong, Pan et al., 2017). However, a similarly designed study found slightly different results with Fgf15−/− mice fed high fat diets developing elevated hepatic triglycerides and increased palmitic acid-derived ER stress (Alvarez-Sola, Uriarte, Latasa et al., 2017). Mice that lack the KLB receptor are resistant to diet induced obesity due to an increase in their energy expenditure. They do display altered bile acid composition, suggesting that there are direct changes to FGF19 signaling (Somm, Henry, Bruce et al., 2017). However, there is weak binding that can occur with FGFR4 alone that may contribute to partial signaling of FGF15 (Yang et al., 2012). Though in a similar result, FGFR4 KO mice fed a high fat diet do have lower weight gain than controls and have similar liver triglyceride concentrations. They do, like KLB KO mice, have a higher bile acid pool and larger fecal bile acid output.
The mechanism of how FGF15/19 causes a decrease in lipogenic genes in the liver has only recently started to become clear (Figure 4). Early studies show that peroxisome proliferated associated receptor gamma-2 (PPARγ-2) a key transcriptional regulator of lipogenic genes is suppressed by FGF19 (Alvarez-Sola et al., 2017), along with other associated genes such as fatty acid synthase, sterol regulatory binding protein 1, acyl coA carboxylase and others. A study by Kim et al. recently found that SHP binding regions overlap CpG islands of lipogenic genes (Kim, Seok, Zhang et al., 2020). FGF19 phosphorylation of SHP at these sites allow SHP to recruit dna methyl transferase 3A (DMNT3A) to these regions. DMNT3A increased methylation at the CpG sites represses the expression of the lipogenic genes.
Figure 4.
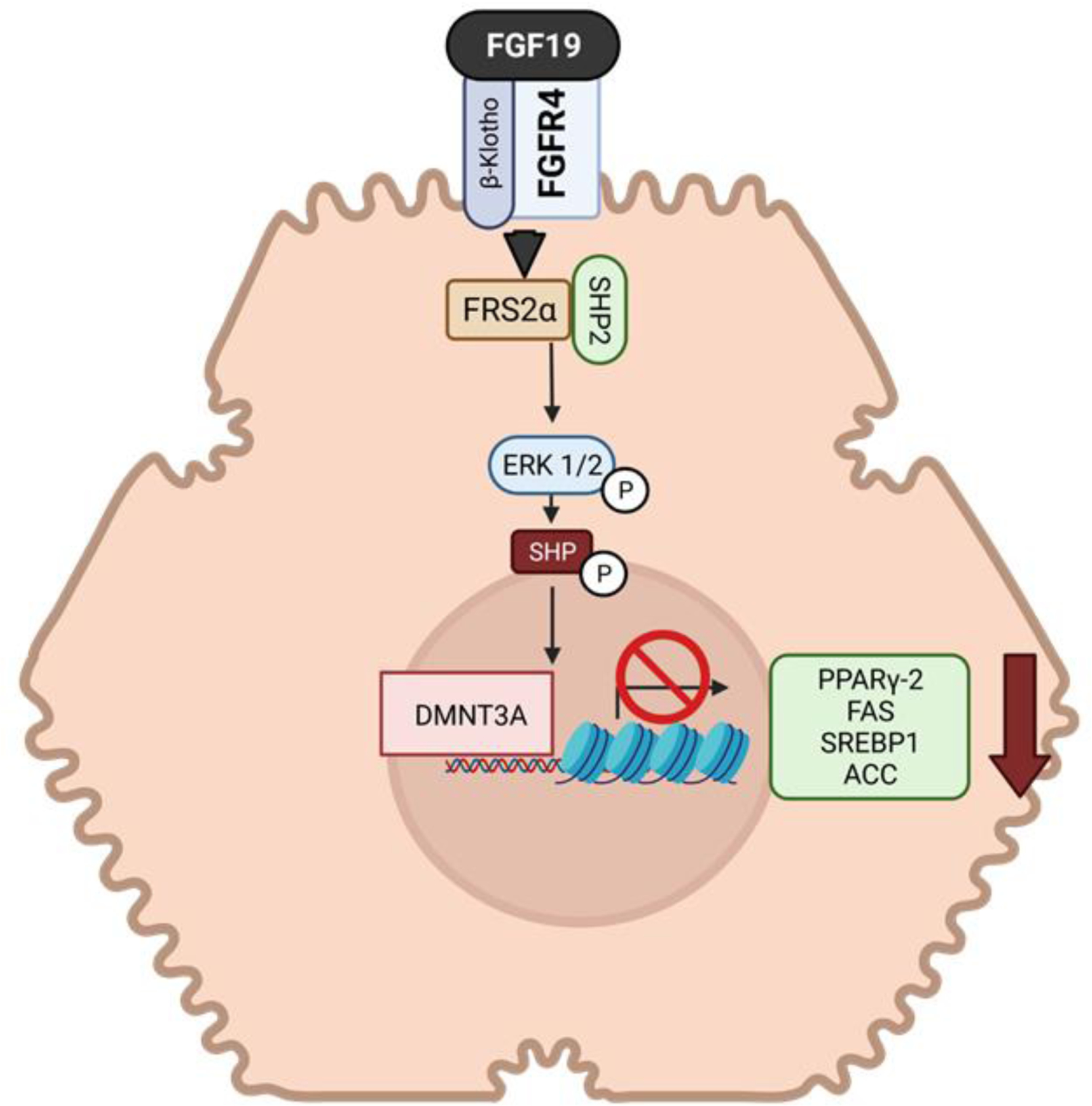
FGF19 regulation of lipid homeostasis in the liver. fibroblast growth factor (FGF), fibroblast growth factor receptor (FGFR), β-Klotho (KLB), small heterodimeric partner (SHP), FGFR substrate 2 alpha (FRS2α), extracellular signal-regulated kinase 1/2 (ERK1/2), peroxisome proliferated associated receptor gamma-2 (PPARγ-2), dna methyl transferase 3A (DMNT3A), sterol regulatory element binding protein 1 (SREBP1), fatty acid synthase (FAS), acyl-coA carboxylase (ACC).
Our current understanding of how adiposity is regulated by FGF19 in the liver is better developed than our current understanding in peripheral tissues. Whereas there is considerable information on the role FGF19 exerts in reduction of adiposity from changes in energy metabolism, which will be discussed in detail in the section below, there is limited data on gene expression and regulation of lipogenic activity of FGF19 in adipocytes. Specifically, research into white adipose tissue, which expresses both FGFR1/KLB, is needed for a better understanding of applicability to human obesity.
6.2.3. Energy Metabolism
Like glucose metabolism, energy metabolism is a general term for multiple pathways that balance energy expenditure through changes in activity and metabolic rate, and energy accretion through food intake and macronutrient preference. In humans, obesity can cause a decrease in circulating FGF19 levels (Gallego-Escuredo, Gomez-Ambrosi, Catalan et al., 2015). Whether the decrease in FGF19 is a cause of obesity, or an effect of metabolic changes and elevated inflammation is not entirely clear. However, the animal data strongly supports a direct role of FGF19 in preventing obesity and controlling whole body metabolism. FGF19 overexpression in normal, healthy mice decreases body weight through increased oxygen consumption (VO2), a marker of activity, but does not change respiratory quotient (RQ), an indicator of macronutrient utilization (Tomlinson et al., 2002). These mice are more active, but do not show an altered preference for either fat or carbohydrate intake. They do however increase total food intake, likely as a result of overall increased activity-mediated energy expenditure. In most mouse models of obesity, FGF19 leads to a similar positive effect on body weight. DIO mice administered either AAV-FGF19 or daily iv FGF19 injection lose weight and increase their VO2 uptake, accompanied by higher energy expenditure (Lan et al., 2017,Zhou et al., 2017,Fu et al., 2004). Both transgenic overexpression of FGF19 or daily administration is effective in reducing body weight of ob/ob leptin deficient mice (Fu et al., 2004). Though RQ was not examined in all studies, mice that consumed a high fat diet had a lower RQ after FGF19 administration, suggesting that FGF19 causes a shift to greater fat utilization (Fu et al., 2004). Not all mouse models of obesity respond to FGF19 treatment. In db/db diabetic mice, glucose concentrations were markedly reduced, but there was no change in body weight after 25 weeks for FGF19 treatment (Zhou et al., 2017).
Investigations into the FGF19-mediated decrease in body weight has focused mostly on the role of elevated metabolic rate and activity, based on the findings of initial mouse studies showing high VO2 and energy expenditure. In adipose tissue, energy expenditure can be elevated through an increase in thermogenesis. Brown adipose tissue has high thermogenic potential and in both mice and humans increases energy expenditure and acts as a protective mechanism against cold stress (van der Lans, Hoeks, Brans et al., 2013,van Marken Lichtenbelt, Vanhommerig, Smulders et al., 2009,Cannon and Nedergaard, 2004). Exposure of mice to cold leads to adipose remodeling with white adipose tissue browning as a protective mechanism. A recent study found that AAV-FGF19 administration to mice increased subcutaneous white adipose tissue browning and increased brown adipose tissue thermogenic genes uncoupling protein 1 (Ucp1), iodothyronine deiodinase 2 (Dio2), and Ppargc1a (Moron-Ros, Uriarte, Berasain et al., 2021). Additionally, Fgf15−/− mice were unable to transition white adipose tissue to brown adipose tissue. Interestingly, FGF15/19 have no effect on adipose tissue that is already brown. This result suggests that FGF15/19 are necessary to drive adipocyte remodeling to brown adipose tissue, but do not have an active effect in driving thermogenesis. This result contrasts with other reports on the function of FGF19 in adipocytes. A study be Antonellis et al found there is an increase in uncoupling protein 1 (UCP1) in brown adipose tissue of mice following iv administration of FG19, which increases thermogenesis and weight loss (Antonellis et al., 2019). However, Ucp1−/− mice still lose weight following FGF19 administration. Possibly, following FGF19 administration, weight loss comes from impaired fat absorption due to a decrease in CYP7A1 generated luminal bile acids.
An alternative mechanism that likely plays a much larger role in reducing body weight following FGF19 administration is from central nervous system reduction of metabolism in the hypothalamus. ICV FGF19 injection in mice causes an increase in VO2 consumption (Fu et al., 2004). In both rats and mice on high fat diets, FGF19 ICV administration causes a reduction in food intake and body weight (Ryan et al., 2013,Marcelin et al., 2014). Though these studies give a good indication for the action of FGF19 in the brain, they still do not clarify if there is a strictly tissue-dependent role of FGF19 on weight reduction. The best indication thus far on the tissue specificity for FGF19-meditated weight regulation is from research conducted by Lan et al. using tissue specific knockout of Klb (Lan et al., 2017). Using mice with Klb−/− in the liver, adipose, and brain, the researchers were able to isolate various metabolic roles of FGF19 to each tissue. They observed that Klb−/− liver or adipose mice with diet induced obesity still lost weight following FGF19 administration. Only mice with Klb−/− in the brain failed to lose weight following FGF19 administration. The activation of FGFR1c/KLB in the brain did however increase sympathetic outflow to brown adipose tissue, so at least a partial mechanism of brain-mediated regulation on weight loss by FGF19 takes place in brown adipose.
Given the conflicting data in mouse models, it is difficult to draw conclusions over the mechanism through which FGF19 increases energy expenditure and facilitates weight loss. Though studies that show a UCP1-independent effect combined with research showing the weight loss in intestinal Klb−/− mice, give the best indication that much of the FGF19 weight loss effect is not centralized in the adipocyte in mice. However, the clinical impact of FGF19 on energy expenditure is still unknown. There are no studies that have appropriately looked at changes in VO2 consumption and energy expenditure in humans following FGF19 administration.
6.3. Growth Factor: Protein Synthesis and Tumorigenesis
The primary function of most paracrine FGFs is to facilitate growth and development from the embryonic stage to adulthood. FGF15/19 shares this feature with the rest of the superfamily and can function as a growth factor in both normal cellular responses to nutrient signaling and cellular repair, but also can have aberrant activity leading to the development of neoplasia. In the normal fed state, FGF19 promotes protein synthesis through insulin-independent mechanisms (Figure 5). Mice administered FGF19 i.p. have increased hepatic global protein synthesis rates, increased albumin, and increased liver weight (Kir et al., 2011). The primary pathway for this process starts with FGF15/19 binding to FGFR4/KLB and activating ERK1/2 and then phosphorylation of MAPK interacting protein kinases 1 (Mnk1) at Thr197 and Thr202. Downstream of Mnk1 is phosphorylation of eukaryotic initiation factor 4e (eIF4E) at Ser209. eIF4e is the cap-binding protein that is part of the recruitment complex for ribosomes for the translation of mRNA. Phosphorylation of eIF4E activates it, to enhance protein translation. Parallel to activation of eIF4E, ERK1/2 activation also enhances phosphorylation of residues Ser235 and Ser236 of ribosomal protein S6 (rpS6). Phosphorylation of rpS6 improves global protein synthesis by activating cap-dependent translation (Meyuhas, 2015). The direct upstream mediator of rpS6 is p90RSK, which is the target of ERK1/2 signaling from FGF19. Importantly, this pathway is distinct from insulin, which can also activate rpS6, but signals through p70S6K, rather than p90RSK. Thus, this pathway enhances protein synthesis signaling, but independently of mTOR. More recently, it was suggested that FGF19 can activate S6 through a second independent pathway to create a two-pronged activation of S6 (Wan et al., 2016). Activation occurs through the Ral complex activating mammalian target of rapamycin complex (mTORC)-1 and p70S6K. This finding does somewhat contrast the initial research identifying p90RSK as the main target of FGF19. In the earlier study, treatment of HepG2 liver cells with rapamycin did not prevent FGF19-mediated phosphorylation of rpS6 (Kir et al., 2011). This suggested that the mechanism is entirely mTOR independent. Likely there will need to be more studies to clearly define whether FGF19 activates rpS6 through two redundant pathways, or has a single unique target pathway.
Figure 5.
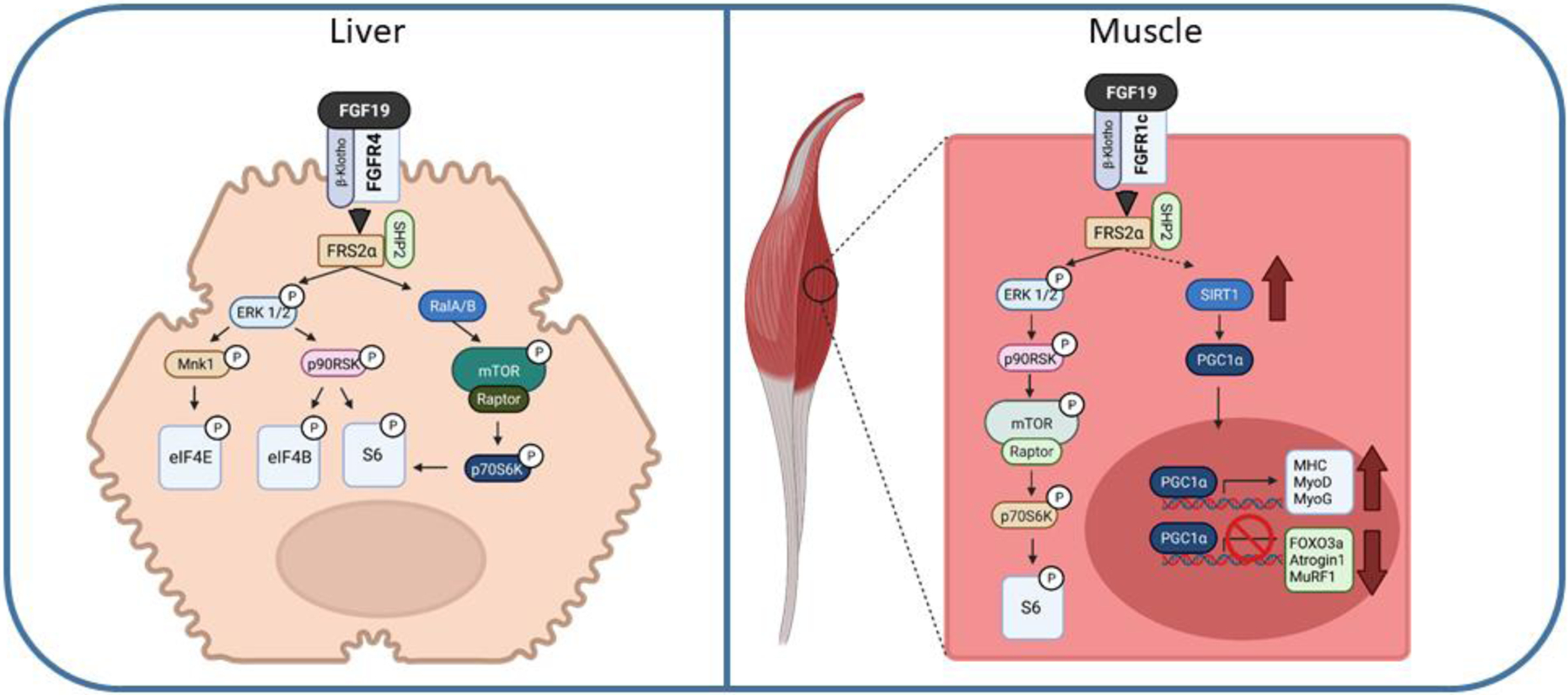
FGF19 signaling in protein synthesis in the liver and skeletal muscle. fibroblast growth factor (FGF), fibroblast growth factor receptor (FGFR), β-Klotho (KLB), FGFR substrate 2 alpha (FRS2α), extracellular signal-regulated kinase 1/2 (ERK1/2), mammalian target of rapamycin (mTOR), regulatory-associated protein of mTOR (Raptor), 90-kDa ribosomal s6 kinase 1 (p90RSK), ribosomal protein S6 kinase beta-1 (p70S6K1), glperoxisome proliferator-activated receptor gamma coactivator (PGC-1α), MAPK interacting protein kinases 1 (Mnk1), eukaryotic initiation factor (eIF), ribosomal protein S6 (S6), sirtuin (SIRT), major histocompatibility complex (MHC), myoblast determination (MyoD), myogenin (MyoG), forkhead/winged helix box gene, group (FOXO), E3 ubiquitin-protein ligase (MuRF). Dashed lines represent potential pathways not fully confirmed experimentally.
FGF19 stimulates protein synthesis in other tissues besides the liver, where in skeletal muscle, FGF19 has anabolic effects. FGF19 can increase muscle fiber size and prevent against skeletal muscle atrophy (Benoit et al., 2017). Both in vivo and in vitro, FGF19-mediated protein synthesis signals through activation of the mTOR – S6K1 pathway. Recent research has also shown that obesity induced muscle atrophy can be reduced with FGF19 administration via activation of the AMPK/SIRT-1/PGC-alpha pathway (Guo, Li, Tian et al., 2021).
In addition to skeletal muscle and hepatic protein synthesis, FGF15/19 is important in liver growth and regeneration. Fgf15−/− or Fgfr4−/− in mice impairs their capacity to regenerate liver following partial hepatectomy (Kong, Huang, Zhu et al., 2014,Uriarte, Fernandez-Barrena, Monte et al., 2013,Padrissa-Altes, Bachofner, Bogorad et al., 2015). In Fgf15−/− mice, there is a reduction in ERK signaling and an impairment to induce the key regulators of cytokine transcription signal transducer and activator of transcription 3 (STAT3) and nuclear-localized nuclear factor kappa B (NF-κB) (Kong et al., 2014). Cytokine response is important for priming the liver to activate regeneration and the loss of this feature by FGF15 may impair the early signaling necessary for priming liver regeneration. In a recent human clinical study examining the role of FGF19 during early phase liver regeneration, there was no change in FGF19 expression post-hepatectomy, but there was a large increase in bile acid production, suggesting low FGF19 activity (Koelfat, van Mierlo, Lodewick et al., 2021). The authors concluded that FGF19 might only play a minor role in human liver regeneration and that bile acids may be the more relevant molecules regulating early phase events. Thus, more detailed molecular analysis of FGF19 during liver regeneration would be needed to for definitive conclusions.
Even if not specific to a function in liver regeneration, it is clear FGF19 does play an important role in hepatic growth. Ectopic overexpression of FGF19 in skeletal muscle of mice causes hepatic tumor growth (Nicholes et al., 2002). Hepatic tumorigenesis also occurs in mice administered an AAV-FGF19 vector (Zhou et al., 2017); however, in the same study mice administered AAV-FGF15 did not develop tumors. Thus, identifying FGF19 tumorigenic effect as orthologue-specific. The tumorigenic effect is not isolated to FGF19 treatment in mice, as FGF19 expression in patients correlates with tumor progression and incidence of hepatocellular carcinoma (Miura, Mitsuhashi, Shimizu et al., 2012). FGF19-mediated tumorigenesis can be blocked as both FGFR4−/− mice and mice treated with an antibody targeting FGFR4, do not develop hepatic tumors when FGF19 is overexpressed (French, Lin, Wang et al., 2012). This result is particularly interesting as it suggests that FGF15 and FGF19 binding to FGFR4 differs enough to alter the magnitude of downstream signaling cascades and result in divergent biological effects.
FGF19 functions in an autocrine feed forward loop in hepatocellular carcinoma (HCC). FGF19 activates signaling pathways that lead to the progression of HCC and then tumor cells upregulate expression of FGF19 to facilitate continued growth (Kang, Haq, Sung et al., 2019,Sawey, Chanrion, Cai et al., 2011,Latasa, Salis, Urtasun et al., 2012,Ahn, Jang, Shim et al., 2014). The same autocrine feed forward function is also active in gall bladder cancer, another site of both FGF19 expression and FGFR4 receptors (Chen, Liu, Liu et al., 2021). FGF19 and FGFR4 target extra hepatic cancers as well including colon cancer (Pai, Dunlap, Qing et al., 2008,Heinzle, Gsur, Hunjadi et al., 2012), lung cancer (Li, Li, Han et al., 2020), head and neck squamous cell carcinoma (Gao, Lang, Zhao et al., 2019) and potentially breast (Tiong, Tan, Choo et al., 2016), and ovarian cancers (Zaid, Yeung, Thompson et al., 2013). It is out of the scope of this review to cover all the pathways and mechanisms FGF19 activates to promote all the types of associated cancer. FGF19/FGFR4 activity in HCC functions through multiple pathways to enhance tumorigenesis and metastasis (Goetz and Mohammadi, 2013). Activation of the Raf-Ras-MAPK pathway generates a mitogenic cell response. Cell motility is enhanced through phospholipase Cγ activation of protein kinase C. Suppression of apoptosis occurs through PI3K-AKT pathways. FGF19 can also enhance the metastatic potential for HCC through induction of epithelial–mesenchymal transition (EMT) via activation of the GSK3-cantenin pathway (Zhao, Lv, Liang et al., 2016). Additionally by activation of the STAT3 pathway through JAK-STAT signaling, FGF19 can enhance tumor growth (Zhou, Wang, Phung et al., 2014).
7. Therapeutics
With a functional understanding of the biological pathways FGF19 targets and the structure/function relationship of FGF19, there is great interest in use of the FGF19 pathway to treat diseases associated with bile acid homeostasis, metabolism and cancers. The two predominant approaches to pharmaceutically increase circulating FGF19 is either directly by hormone administration or indirectly through activation of the FXR pathway. There are several drug compounds that function as FXR agonists to activate the FGF19 pathway through FXR and are either FDA approved (e.g. Obeticholic acid) or in clinical trials (e.g. Tropifexor). However, given that these drugs have a greater array of biological effects through FXR than to just increase FGF19, they are outside the scope of this review.
Native human FGF19 is not an ideal candidate for activation and treatment of diseases related to cholestasis and metabolism given its mitogenic properties. Various analogues are in development to mimic the effects of FGF19 on bile suppression and glucose/lipid metabolism that lack the mitogenic effect. One such peptide is NGM282 (Aldafermin), which has three amino acid substitutions (A30S, G31S, and H33L) and five-amino acid deletions in the N-terminal region of FGF19 (Zhou et al., 2014,Luo, Ko, Elliott et al., 2014). Early mouse studies showed that NGM282 could suppress bile acid synthesis and reduce hepatic lipotoxicity associated with obesity in mice (Zhou et al., 2017,Zhou, Learned, Rossi et al., 2016). Currently in human trials, NGM282 can reduce bile acid synthesis is patients with primary sclerosing cholangitis and reduce markers of fibrosis, but does not reduce serum markers of liver injury, such as alkaline phosphatase (Hirschfield, Chazouilleres, Drenth et al., 2019,Sanyal, Ling, Beuers et al., 2021). In clinical trials for obesity-associated non-alcoholic steatohepatitis, NGM282 treatment can reduce hepatic lipid content and improve markers of fibrosis, without any indication of tumorigenesis (Harrison et al., 2018,Rinella et al., 2019,Sanyal et al., 2021,Harrison, Neff, Guy et al., 2021). Other than NGM282, there are additional variants that have modified single point mutations in FGF19 binding domains to FGFR4 (Y115A), HS (K149A), and KLB (D198A) display decreasing mitogenic signaling, respectively, while still reducing bile acids synthesis (Niu et al., 2020). Interestingly, these single point mutations are enough to impair formation of a quaternary structure of two FGFR4/KLB dimers, limiting the signaling potential of FGF19.
8. Conclusions
The field of FGF19 research is still rapidly expanding. Our understanding of the signaling events that mediate FGF19 action and the various target tissues and signaling pathways of FGF19 are still being delineated. There is still a limited literature defining the physiological and metabolic function of FGF19 during early development after birth or at later stages in life, such as aging. There are still gaps in our knowledge of signaling pathways and the exact role of the brain, adipose, and liver for regulation of whole body metabolism and glucose regulation. One issue of particular concern is how well studies in mice administered FGF19 translate to the human response of FGF19 given the species difference in receptor expression of FGFR1c-FGFR3c. This limitation can be overcome with use of more translational preclinical models, like the pig, that naturally express FGF19 and have receptors that more closely match human FGFRs and KLB.
Financial disclosure:
This work was supported in part by federal funds from the USDA, Agricultural Research Service under Cooperative Agreement Number 3092-51000-060-01, and National Institutes of Health NIDDK grants R01-DK094616 (D. Burrin), T32-DK007664 (C. Vonderohe), and K01-DK129408 (G. Guthrie).
Abbreviations:
- FGF
fibroblast growth factor
- HS
heparin sulfate
- FGFR
fibroblast growth factor receptor
- KLB
β-Klotho
- Ig
immunoglobulin
- KLA
α-Klotho
- FXR
farnesoid x receptor
- CDCA
chenodeoxycholic acid
- FXRE
FXR-responsive binding elements
- PXR
pregnane x receptor
- LCA
lithocholic acid
- ATF4
activating transcription factor 4
- AARE
amino acid response element
- SREBP2
sterol regulatory element-binding protein 2
- Klf15
Kruppel-like factor 15
- PI3K
phosphoinositide 3-kinase
- Akt
protein kinase B
- GATA4
GATA binding protein 4
- GR
glucocorticoid receptor
- CA
cholic acid
- CYP7A1
cytochrome P450 7A1
- α-MCA
α-muricholic acid
- HCA
hyocholic acid
- BSEP
bile salt efflux pump
- DCA
deoxycholic acid
- SHP
small heterodimeric partner
- LRH-1, Nr5a2
liver receptor homolog 1
- HNF4
hepatocyte nuclear factor 4 alpha
- FRS2α
FGFR substrate 2 alpha
- OSTα/β
solute transporter alpha/beta
- MRP2
multidrug resistance protein 2
- MAPK/ERK1/2
mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2
- TFEB
transcription factor EB
- mTOR
mammalian target of rapamycin
- PBC
primary biliary cirrhosis
- TGR5
G-protein-coupled bile acid receptor
- S1PR2
sphingosine 1 phosphate receptor 2
- AAV
adeno-associated virus
- p90RSK
90-kDa ribosomal s6 kinase 1
- S6K1
ribosomal protein S6 kinase beta-1
- GSK
glycogen synthase kinase
- G6pase
glucose-6-phophatase
- Pepck
phosphoenoylpyruvate kinase
- CREB
cAMP regulatory element-binding protein
- PGC-1α
peroxisome proliferator-activated receptor gamma coactivator
- ICV
intracerebroventricular
- POMC
proopiomelanocortin
- AgRP
agouti-related peptide
- NPY
neuropeptide Y
- T1DM
type 1 diabetes
- ACTH
adrenocorticotropic hormone
- DMV
dorsal motor nucleus of the vagus
- DIO
diet induced obesity
- PPARγ-2
peroxisome proliferated associated receptor gamma-2
- DMNT3A
dna methyl transferase 3A
- RQ
respiratory quotient
- UCP1
uncoupling protein 1
- Mnk1
MAPK interacting protein kinases 1
- eIF4E
eukaryotic initiation factor 4e
- rpS6
ribosomal protein S6
- mTORC
mammalian target of rapamycin complex
- STAT3
signal transducer and activator of transcription 3
- NF-κB
nuclear factor kappa B
- HCC
hepatocellular carcinoma
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
All figure artwork created with Biorender.com
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- [1].Nishimura T, Utsunomiya Y, Hoshikawa M, Ohuchi H and Itoh N, 1999. Structure and expression of a novel human FGF, FGF-19, expressed in the fetal brain, Biochim Biophys Acta 1444, 148–51. [DOI] [PubMed] [Google Scholar]
- [2].Kurose H, Bito T, Adachi T, Shimizu M, Noji S and Ohuchi H, 2004. Expression of Fibroblast growth factor 19 (Fgf19) during chicken embryogenesis and eye development, compared with Fgf15 expression in the mouse, Gene Expr Patterns 4, 687–93. [DOI] [PubMed] [Google Scholar]
- [3].Sanchez-Calderon H, Francisco-Morcillo J, Martin-Partido G and Hidalgo-Sanchez M, 2007. Fgf19 expression patterns in the developing chick inner ear, Gene Expr Patterns 7, 30–8. [DOI] [PubMed] [Google Scholar]
- [4].Milkiewicz M, Klak M, Kempinska-Podhorodecka A, Wiechowska-Kozlowska A, Urasinska E, Blatkiewicz M, Wunsch E, Elias E and Milkiewicz P, 2016. Impaired Hepatic Adaptation to Chronic Cholestasis induced by Primary Sclerosing Cholangitis, Sci Rep 6, 39573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zweers SJ, Booij KA, Komuta M, Roskams T, Gouma DJ, Jansen PL and Schaap FG, 2012. The human gallbladder secretes fibroblast growth factor 19 into bile: towards defining the role of fibroblast growth factor 19 in the enterobiliary tract, Hepatology 55, 575–83. [DOI] [PubMed] [Google Scholar]
- [6].Wunsch E, Milkiewicz M, Wasik U, Trottier J, Kempinska-Podhorodecka A, Elias E, Barbier O and Milkiewicz P, 2015. Expression of hepatic Fibroblast Growth Factor 19 is enhanced in Primary Biliary Cirrhosis and correlates with severity of the disease, Sci Rep 5, 13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hasegawa Y, Kawai M, Bessho K, Yasuda K, Ueno T, Satomura Y, Konishi A, Kimura T, Ikeda K, Tachibana M, Miyoshi Y, Michigami T, Kondou H and Ozono K, 2019. CYP7A1 expression in hepatocytes is retained with upregulated fibroblast growth factor 19 in pediatric biliary atresia, Hepatol Res 49, 314–323. [DOI] [PubMed] [Google Scholar]
- [8].Johansson H, Svensson JF, Almstrom M, Van Hul N, Rudling M, Angelin B, Nowak G, Fischler B and Ellis E, 2020. Regulation of bile acid metabolism in biliary atresia: reduction of FGF19 by Kasai portoenterostomy and possible relation to early outcome, J Intern Med 287, 534–545. [DOI] [PubMed] [Google Scholar]
- [9].Tomlinson E, Fu L, John L, Hultgren B, Huang X, Renz M, Stephan JP, Tsai SP, Powell-Braxton L, French D and Stewart TA, 2002. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity, Endocrinology 143, 1741–7. [DOI] [PubMed] [Google Scholar]
- [10].Nicholes K, Guillet S, Tomlinson E, Hillan K, Wright B, Frantz GD, Pham TA, Dillard-Telm L, Tsai SP, Stephan JP, Stinson J, Stewart T and French DM, 2002. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice, Am J Pathol 160, 2295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, Goodwin B and Jones SA, 2003. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis, Genes Dev 17, 1581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino-Powell K, Xu HE, Shulman GI, Kliewer SA and Mangelsdorf DJ, 2011. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis, Science 331, 1621–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Benoit B, Meugnier E, Castelli M, Chanon S, Vieille-Marchiset A, Durand C, Bendridi N, Pesenti S, Monternier PA, Durieux AC, Freyssenet D, Rieusset J, Lefai E, Vidal H and Ruzzin J, 2017. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice, Nat Med 23, 990–996. [DOI] [PubMed] [Google Scholar]
- [14].Olsen SK, Garbi M, Zampieri N, Eliseenkova AV, Ornitz DM, Goldfarb M and Mohammadi M, 2003. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs, J Biol Chem 278, 34226–36. [DOI] [PubMed] [Google Scholar]
- [15].Zhu X, Komiya H, Chirino A, Faham S, Fox GM, Arakawa T, Hsu BT and Rees DC, 1991. Three-dimensional structures of acidic and basic fibroblast growth factors, Science 251, 90–3. [DOI] [PubMed] [Google Scholar]
- [16].Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, Xu C, Neubert TA, Zhang F, Linhardt RJ, Yu X, White KE, Inagaki T, Kliewer SA, Yamamoto M, Kurosu H, Ogawa Y, Kuro-o M, Lanske B, Razzaque MS and Mohammadi M, 2007. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members, Mol Cell Biol 27, 3417–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Harmer NJ, Pellegrini L, Chirgadze D, Fernandez-Recio J and Blundell TL, 2004. The crystal structure of fibroblast growth factor (FGF) 19 reveals novel features of the FGF family and offers a structural basis for its unusual receptor affinity, Biochemistry 43, 629–40. [DOI] [PubMed] [Google Scholar]
- [18].Goetz R, Ohnishi M, Kir S, Kurosu H, Wang L, Pastor J, Ma J, Gai W, Kuro-o M, Razzaque MS and Mohammadi M, 2012. Conversion of a paracrine fibroblast growth factor into an endocrine fibroblast growth factor, J Biol Chem 287, 29134–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Romero-Fernandez W, Borroto-Escuela DO, Tarakanov AO, Mudo G, Narvaez M, Perez-Alea M, Agnati LF, Ciruela F, Belluardo N and Fuxe K, 2011. Agonist-induced formation of FGFR1 homodimers and signaling differ among members of the FGF family, Biochem Biophys Res Commun 409, 764–8. [DOI] [PubMed] [Google Scholar]
- [20].Gong SG, 2014. Isoforms of receptors of fibroblast growth factors, J Cell Physiol 229, 1887–95. [DOI] [PubMed] [Google Scholar]
- [21].Itoh N and Ornitz DM, 2004. Evolution of the Fgf and Fgfr gene families, Trends Genet 20, 563–9. [DOI] [PubMed] [Google Scholar]
- [22].Hughes SE, 1997. Differential expression of the fibroblast growth factor receptor (FGFR) multigene family in normal human adult tissues, J Histochem Cytochem 45, 1005–19. [DOI] [PubMed] [Google Scholar]
- [23].Templeton TJ and Hauschka SD, 1992. FGF-mediated aspects of skeletal muscle growth and differentiation are controlled by a high affinity receptor, FGFR1, Dev Biol 154, 169–81. [DOI] [PubMed] [Google Scholar]
- [24].Lin BC, Wang M, Blackmore C and Desnoyers LR, 2007. Liver-specific activities of FGF19 require Klotho beta, J Biol Chem 282, 27277–27284. [DOI] [PubMed] [Google Scholar]
- [25].Kurosu H, Choi M, Ogawa Y, Dickson AS, Goetz R, Eliseenkova AV, Mohammadi M, Rosenblatt KP, Kliewer SA and Kuro OM, 2007. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21, J Biol Chem 282, 26687–26695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Perry RJ, Lee S, Ma L, Zhang D, Schlessinger J and Shulman GI, 2015. FGF1 and FGF19 reverse diabetes by suppression of the hypothalamic-pituitary-adrenal axis, Nat Commun 6, 6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lan T, Morgan DA, Rahmouni K, Sonoda J, Fu X, Burgess SC, Holland WL, Kliewer SA and Mangelsdorf DJ, 2017. FGF19, FGF21, and an FGFR1/beta-Klotho-Activating Antibody Act on the Nervous System to Regulate Body Weight and Glycemia, Cell Metab 26, 709–718 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wu AL, Coulter S, Liddle C, Wong A, Eastham-Anderson J, French DM, Peterson AS and Sonoda J, 2011. FGF19 regulates cell proliferation, glucose and bile acid metabolism via FGFR4-dependent and independent pathways, PLoS One 6, e17868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shiraki-Iida T, Aizawa H, Matsumura Y, Sekine S, Iida A, Anazawa H, Nagai R, Kuro-o M and Nabeshima Y, 1998. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein, FEBS Lett 424, 6–10. [DOI] [PubMed] [Google Scholar]
- [30].Kuzina ES, Ung PM, Mohanty J, Tome F, Choi J, Pardon E, Steyaert J, Lax I, Schlessinger A, Schlessinger J and Lee S, 2019. Structures of ligand-occupied beta-Klotho complexes reveal a molecular mechanism underlying endocrine FGF specificity and activity, Proc Natl Acad Sci U S A 116, 7819–7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wu X, Lemon B, Li X, Gupte J, Weiszmann J, Stevens J, Hawkins N, Shen W, Lindberg R, Chen JL, Tian H and Li Y, 2008. C-terminal tail of FGF19 determines its specificity toward Klotho co-receptors, J Biol Chem 283, 33304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wu X, Ge H, Gupte J, Weiszmann J, Shimamoto G, Stevens J, Hawkins N, Lemon B, Shen W, Xu J, Veniant MM, Li YS, Lindberg R, Chen JL, Tian H and Li Y, 2007. Co-receptor requirements for fibroblast growth factor-19 signaling, J Biol Chem 282, 29069–72. [DOI] [PubMed] [Google Scholar]
- [33].Fon Tacer K, Bookout AL, Ding X, Kurosu H, John GB, Wang L, Goetz R, Mohammadi M, Kuro-o M, Mangelsdorf DJ and Kliewer SA, 2010. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse, Mol Endocrinol 24, 2050–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Katafuchi T, Esterhazy D, Lemoff A, Ding X, Sondhi V, Kliewer SA, Mirzaei H and Mangelsdorf DJ, 2015. Detection of FGF15 in plasma by stable isotope standards and capture by anti-peptide antibodies and targeted mass spectrometry, Cell Metab 21, 898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fu T, Choi SE, Kim DH, Seok S, Suino-Powell KM, Xu HE and Kemper JK, 2012. Aberrantly elevated microRNA-34a in obesity attenuates hepatic responses to FGF19 by targeting a membrane coreceptor beta-Klotho, Proc Natl Acad Sci U S A 109, 16137–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].McWhirter JR, Goulding M, Weiner JA, Chun J and Murre C, 1997. A novel fibroblast growth factor gene expressed in the developing nervous system is a downstream target of the chimeric homeodomain oncoprotein E2A-Pbx1, Development 124, 3221–32. [DOI] [PubMed] [Google Scholar]
- [37].Choi M, Moschetta A, Bookout AL, Peng L, Umetani M, Holmstrom SR, Suino-Powell K, Xu HE, Richardson JA, Gerard RD, Mangelsdorf DJ and Kliewer SA, 2006. Identification of a hormonal basis for gallbladder filling, Nat Med 12, 1253–5. [DOI] [PubMed] [Google Scholar]
- [38].Yang C, Jin C, Li X, Wang F, McKeehan WL and Luo Y, 2012. Differential specificity of endocrine FGF19 and FGF21 to FGFR1 and FGFR4 in complex with KLB, PLoS One 7, e33870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhou M, Luo J, Chen M, Yang H, Learned RM, DePaoli AM, Tian H and Ling L, 2017. Mouse species-specific control of hepatocarcinogenesis and metabolism by FGF19/FGF15, J Hepatol 66, 1182–1192. [DOI] [PubMed] [Google Scholar]
- [40].Williams CM, Harper Calderon J, E, H., Jimenez Y, Barringer K, Carbonaro M, Molina-Portela MDP, Thurston G, Li Z and Daly C, 2021. Monomeric/dimeric forms of Fgf15/FGF19 show differential activity in hepatocyte proliferation and metabolic function, FASEB J 35, e21286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ellis EC, Naugler WE, Parini P, Mork LM, Jorns C, Zemack H, Sandblom AL, Bjorkhem I, Ericzon BG, Wilson EM, Strom SC and Grompe M, 2013. Mice with chimeric livers are an improved model for human lipoprotein metabolism, PLoS One 8, e78550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Niu J, Zhao J, Wu J, Qiao G, Gu J, Zhou C, Li Q, Ying L, Wang D, Lin H, Li X, Mohammadi M and Huang Z, 2020. Curtailing FGF19’s mitogenicity by suppressing its receptor dimerization ability, Proc Natl Acad Sci U S A 117, 29025–29034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ and Kliewer SA, 2005. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis, Cell Metab 2, 217–25. [DOI] [PubMed] [Google Scholar]
- [44].Call L, Molina T, Stoll B, Guthrie G, Chacko S, Plat J, Robinson J, Lin S, Vonderohe C, Mohammad M, Kunichoff D, Cruz S, Lau P, Premkumar M, Nielsen J, Fang Z, Olutoye O, Thymann T, Britton R, Sangild P and Burrin D, 2020. Parenteral lipids shape gut bile acid pools and microbiota profiles in the prevention of cholestasis in preterm pigs, J Lipid Res 61, 1038–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Vonderohe C, Guthrie G, Stoll B, Chacko S, Dawson H and Burrin DG, 2021. Tissue-specific Mechanisms of Bile Acid Homeostasis and Activation of FXR-FGF19 Signaling in Preterm and Term Neonatal Pigs, Am J Physiol Gastrointest Liver Physiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gavalda-Navarro A, Pastor JJ, Mereu A, Villarroya F and Ipharraguerre IR, 2018. Developmental regulation of the intestinal FGF19 system in domestic pigs, Am J Physiol Gastrointest Liver Physiol 314, G647–G654. [DOI] [PubMed] [Google Scholar]
- [47].Shang Q, Guo GL, Honda A, Saumoy M, Salen G and Xu G, 2013. FGF15/19 protein levels in the portal blood do not reflect changes in the ileal FGF15/19 or hepatic CYP7A1 mRNA levels, J Lipid Res 54, 2606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li J, Pircher PC, Schulman IG and Westin SK, 2005. Regulation of complement C3 expression by the bile acid receptor FXR, J Biol Chem 280, 7427–34. [DOI] [PubMed] [Google Scholar]
- [49].Miyata M, Hata T, Yamakawa H, Kagawa T, Yoshinari K and Yamazoe Y, 2012. Involvement of multiple elements in FXR-mediated transcriptional activation of FGF19, J Steroid Biochem Mol Biol 132, 41–7. [DOI] [PubMed] [Google Scholar]
- [50].Kliewer SA and Willson TM, 2002. Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor, J Lipid Res 43, 359–64. [PubMed] [Google Scholar]
- [51].Wistuba W, Gnewuch C, Liebisch G, Schmitz G and Langmann T, 2007. Lithocholic acid induction of the FGF19 promoter in intestinal cells is mediated by PXR, World J Gastroenterol 13, 4230–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Guthrie G, Stoll B, Chacko S, Lauridsen C, Plat J and Burrin D, 2020. Rifampicin, not vitamin E, suppresses parenteral nutrition-associated liver disease development through the pregnane X receptor pathway in piglets, Am J Physiol Gastrointest Liver Physiol 318, G41–G52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhao LY, Xu JY, Shi Z, Englert NA and Zhang SY, 2017. Pregnane X receptor (PXR) deficiency improves high fat diet-induced obesity via induction of fibroblast growth factor 15 (FGF15) expression, Biochem Pharmacol 142, 194–203. [DOI] [PubMed] [Google Scholar]
- [54].Shimizu M, Li J, Maruyama R, Inoue J and Sato R, 2013. FGF19 (fibroblast growth factor 19) as a novel target gene for activating transcription factor 4 in response to endoplasmic reticulum stress, Biochem J 450, 221–9. [DOI] [PubMed] [Google Scholar]
- [55].Uchiyama K, Naito Y, Takagi T, Mizushima K, Hayashi N, Harusato A, Hirata I, Omatsu T, Handa O, Ishikawa T, Yagi N, Kokura S and Yoshikawa T, 2010. Carbon monoxide enhance colonic epithelial restitution via FGF15 derived from colonic myofibroblasts, Biochem Biophys Res Commun 391, 1122–6. [DOI] [PubMed] [Google Scholar]
- [56].Brown MS and Goldstein JL, 1997. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor, Cell 89, 331–40. [DOI] [PubMed] [Google Scholar]
- [57].Miyata M, Hata T, Yamazoe Y and Yoshinari K, 2014. SREBP-2 negatively regulates FXR-dependent transcription of FGF19 in human intestinal cells, Biochem Biophys Res Commun 443, 477–82. [DOI] [PubMed] [Google Scholar]
- [58].Han S, Zhang R, Jain R, Shi H, Zhang L, Zhou G, Sangwung P, Tugal D, Atkins GB, Prosdocimo DA, Lu Y, Han X, Tso P, Liao X, Epstein JA and Jain MK, 2015. Circadian control of bile acid synthesis by a KLF15-Fgf15 axis, Nat Commun 6, 7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang G, Wu B, Cui Y, Zhang B, Jiang C and Wang H, 2020. Teneligliptin Promotes Bile Acid Synthesis and Attenuates Lipid Accumulation in Obese Mice by Targeting the KLF15-Fgf15 Pathway, Chem Res Toxicol 33, 2164–2171. [DOI] [PubMed] [Google Scholar]
- [60].Thompson CA, Wojta K, Pulakanti K, Rao S, Dawson P and Battle MA, 2017. GATA4 Is Sufficient to Establish Jejunal Versus Ileal Identity in the Small Intestine, Cell Mol Gastroenterol Hepatol 3, 422–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jia K, Zhang D, Jia Q and Zhang QY, 2019. Regulation of Fgf15 expression in the intestine by glucocorticoid receptor, Mol Med Rep 19, 2953–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK, Verhagen A, Rivera CR, Mulvihill SJ, Malloy MJ and Kane JP, 2002. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype, J Clin Invest 110, 109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Cali JJ and Russell DW, 1991. Characterization of human sterol 27-hydroxylase. A mitochondrial cytochrome P-450 that catalyzes multiple oxidation reaction in bile acid biosynthesis, J Biol Chem 266, 7774–8. [PubMed] [Google Scholar]
- [64].Li X, Pandak WM, Erickson SK, Ma Y, Yin L, Hylemon P and Ren S, 2007. Biosynthesis of the regulatory oxysterol, 5-cholesten-3beta,25-diol 3-sulfate, in hepatocytes, J Lipid Res 48, 2587–96. [DOI] [PubMed] [Google Scholar]
- [65].Botham KM and Boyd GS, 1983. The metabolism of chenodeoxycholic acid to betamuricholic acid in rat liver, Eur J Biochem 134, 191–6. [DOI] [PubMed] [Google Scholar]
- [66].Cuesta de Juan S, Monte MJ, Macias RI, Wauthier V, Calderon PB and Marin JJ, 2007. Ontogenic development-associated changes in the expression of genes involved in rat bile acid homeostasis, J Lipid Res 48, 1362–70. [DOI] [PubMed] [Google Scholar]
- [67].Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, Wolf CR, Henderson CJ and Gonzalez FJ, 2016. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans, J Lipid Res 57, 2130–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Haslewood GA, 1954. “Hyocholic acid”, a trihydroxy bile acid from pig bile, Biochem J 56, xxxviii. [PubMed] [Google Scholar]
- [69].Lundell K, Hansson R and Wikvall K, 2001. Cloning and expression of a pig liver taurochenodeoxycholic acid 6alpha-hydroxylase (CYP4A21): a novel member of the CYP4A subfamily, J Biol Chem 276, 9606–12. [DOI] [PubMed] [Google Scholar]
- [70].Elkins CA, Moser SA and Savage DC, 2001. Genes encoding bile salt hydrolases and conjugated bile salt transporters in Lactobacillus johnsonii 100–100 and other Lactobacillus species, Microbiology (Reading) 147, 3403–12. [DOI] [PubMed] [Google Scholar]
- [71].Wijaya A, Hermann A, Abriouel H, Specht I, Yousif NM, Holzapfel WH and Franz CM, 2004. Cloning of the bile salt hydrolase (bsh) gene from Enterococcus faecium FAIR-E 345 and chromosomal location of bsh genes in food enterococci, J Food Prot 67, 2772–8. [DOI] [PubMed] [Google Scholar]
- [72].Grill J, Schneider F, Crociani J and Ballongue J, 1995. Purification and Characterization of Conjugated Bile Salt Hydrolase from Bifidobacterium longum BB536, Appl Environ Microbiol 61, 2577–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Rossocha M, Schultz-Heienbrok R, von Moeller H, Coleman JP and Saenger W, 2005. Conjugated bile acid hydrolase is a tetrameric N-terminal thiol hydrolase with specific recognition of its cholyl but not of its tauryl product, Biochemistry 44, 5739–48. [DOI] [PubMed] [Google Scholar]
- [74].Wells JE, Williams KB, Whitehead TR, Heuman DM and Hylemon PB, 2003. Development and application of a polymerase chain reaction assay for the detection and enumeration of bile acid 7alpha-dehydroxylating bacteria in human feces, Clin Chim Acta 331, 127–34. [DOI] [PubMed] [Google Scholar]
- [75].Heuman DM, 1989. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions, J Lipid Res 30, 719–30. [PubMed] [Google Scholar]
- [76].Hofmann AF, 2004. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity, Drug Metab Rev 36, 703–22. [DOI] [PubMed] [Google Scholar]
- [77].Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, Maloney PR, Willson TM and Kliewer SA, 2000. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis, Mol Cell 6, 517–26. [DOI] [PubMed] [Google Scholar]
- [78].Inoue Y, Yu AM, Yim SH, Ma X, Krausz KW, Inoue J, Xiang CC, Brownstein MJ, Eggertsen G, Bjorkhem I and Gonzalez FJ, 2006. Regulation of bile acid biosynthesis by hepatocyte nuclear factor 4alpha, J Lipid Res 47, 215–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J and Mangelsdorf DJ, 2000. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors, Mol Cell 6, 507–15. [DOI] [PubMed] [Google Scholar]
- [80].Wang L, Han Y, Kim CS, Lee YK and Moore DD, 2003. Resistance of SHP-null mice to bile acid-induced liver damage, J Biol Chem 278, 44475–81. [DOI] [PubMed] [Google Scholar]
- [81].Lee JM, Ong JR, Vergnes L, de Aguiar Vallim TQ, Nolan J, Cantor RM, Walters JRF and Reue K, 2018. Diet1, bile acid diarrhea, and FGF15/19: mouse model and human genetic variants, J Lipid Res 59, 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Vergnes L, Lee JM, Chin RG, Auwerx J and Reue K, 2013. Diet1 functions in the FGF15/19 enterohepatic signaling axis to modulate bile acid and lipid levels, Cell Metab 17, 916–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Li S, Hsu DD, Li B, Luo X, Alderson N, Qiao L, Ma L, Zhu HH, He Z, Suino-Powell K, Ji K, Li J, Shao J, Xu HE, Li T and Feng GS, 2014. Cytoplasmic tyrosine phosphatase Shp2 coordinates hepatic regulation of bile acid and FGF15/19 signaling to repress bile acid synthesis, Cell Metab 20, 320–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Byun S, Kim DH, Ryerson D, Kim YC, Sun H, Kong B, Yau P, Guo G, Xu HE, Kemper B and Kemper JK, 2018. Postprandial FGF19-induced phosphorylation by Src is critical for FXR function in bile acid homeostasis, Nat Commun 9, 2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Song KH, Li T, Owsley E, Strom S and Chiang JY, 2009. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression, Hepatology 49, 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Miao J, Xiao Z, Kanamaluru D, Min G, Yau PM, Veenstra TD, Ellis E, Strom S, Suino-Powell K, Xu HE and Kemper JK, 2009. Bile acid signaling pathways increase stability of Small Heterodimer Partner (SHP) by inhibiting ubiquitin-proteasomal degradation, Genes Dev 23, 986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wang Y, Gunewardena S, Li F, Matye DJ, Chen C, Chao X, Jung T, Zhang Y, Czerwinski M, Ni HM, Ding WX and Li T, 2020. An FGF15/19-TFEB regulatory loop controls hepatic cholesterol and bile acid homeostasis, Nat Commun 11, 3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wan ZY, Tian JS, Tan HW, Chow AL, Sim AY, Ban KH and Long YC, 2016. Mechanistic target of rapamycin complex 1 is an essential mediator of metabolic and mitogenic effects of fibroblast growth factor 19 in hepatoma cells, Hepatology 64, 1289–301. [DOI] [PubMed] [Google Scholar]
- [89].Tohma M, 1996. [Determination of fetal bile acids and related steroidal compounds and their profile in neonatal biological fluids], Yakugaku Zasshi 116, 753–75. [DOI] [PubMed] [Google Scholar]
- [90].Tazawa Y, Yamada M, Nakagawa M, Konno T and Tada K, 1984. Direct measurement of urinary bile acids of infants by high-performance liquid chromatography connected with an enzyme immobilized column, Tohoku J Exp Med 143, 361–71. [DOI] [PubMed] [Google Scholar]
- [91].Xia X, Francis H, Glaser S, Alpini G and LeSage G, 2006. Bile acid interactions with cholangiocytes, World J Gastroenterol 12, 3553–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Jung D, York JP, Wang L, Yang C, Zhang A, Francis HL, Webb P, McKeehan WL, Alpini G, Lesage GD, Moore DD and Xia X, 2014. FXR-induced secretion of FGF15/19 inhibits CYP27 expression in cholangiocytes through p38 kinase pathway, Pflugers Arch 466, 1011–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Guo C, Chen WD and Wang YD, 2016. TGR5, Not Only a Metabolic Regulator, Front Physiol 7, 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Studer E, Zhou X, Zhao R, Wang Y, Takabe K, Nagahashi M, Pandak WM, Dent P, Spiegel S, Shi R, Xu W, Liu X, Bohdan P, Zhang L, Zhou H and Hylemon PB, 2012. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes, Hepatology 55, 267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Ikeda H, Watanabe N, Ishii I, Shimosawa T, Kume Y, Tomiya T, Inoue Y, Nishikawa T, Ohtomo N, Tanoue Y, Iitsuka S, Fujita R, Omata M, Chun J and Yatomi Y, 2009. Sphingosine 1-phosphate regulates regeneration and fibrosis after liver injury via sphingosine 1phosphate receptor 2, J Lipid Res 50, 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Pai R, French D, Ma N, Hotzel K, Plise E, Salphati L, Setchell KD, Ware J, Lauriault V, Schutt L, Hartley D and Dambach D, 2012. Antibody-mediated inhibition of fibroblast growth factor 19 results in increased bile acids synthesis and ileal malabsorption of bile acids in cynomolgus monkeys, Toxicol Sci 126, 446–56. [DOI] [PubMed] [Google Scholar]
- [97].Yu C, Wang F, Kan M, Jin C, Jones RB, Weinstein M, Deng CX and McKeehan WL, 2000. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4, J Biol Chem 275, 15482–9. [DOI] [PubMed] [Google Scholar]
- [98].Barrera F, Azocar L, Molina H, Schalper KA, Ocares M, Liberona J, Villarroel L, Pimentel F, Perez-Ayuso RM, Nervi F, Groen AK and Miquel JF, 2015. Effect of cholecystectomy on bile acid synthesis and circulating levels of fibroblast growth factor 19, Ann Hepatol 14, 710–21. [PubMed] [Google Scholar]
- [99].Al-Dury S, Wahlstrom A, Panzitt K, Thorell A, Stahlman M, Trauner M, Fickert P, Backhed F, Fandriks L, Wagner M and Marschall HU, 2019. Obeticholic acid may increase the risk of gallstone formation in susceptible patients, J Hepatol 71, 986–991. [DOI] [PubMed] [Google Scholar]
- [100].Lundasen T, Galman C, Angelin B and Rudling M, 2006. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man, J Intern Med 260, 530–6. [DOI] [PubMed] [Google Scholar]
- [101].Fu L, John LM, Adams SH, Yu XX, Tomlinson E, Renz M, Williams PM, Soriano R, Corpuz R, Moffat B, Vandlen R, Simmons L, Foster J, Stephan JP, Tsai SP and Stewart TA, 2004. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes, Endocrinology 145, 2594–603. [DOI] [PubMed] [Google Scholar]
- [102].Wu X, Ge H, Baribault H, Gupte J, Weiszmann J, Lemon B, Gardner J, Fordstrom P, Tang J, Zhou M, Wang M and Li Y, 2013. Dual actions of fibroblast growth factor 19 on lipid metabolism, J Lipid Res 54, 325–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Sutherland C, Leighton IA and Cohen P, 1993. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling, Biochem J 296 (Pt 1), 15–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Hatting M, Tavares CDJ, Sharabi K, Rines AK and Puigserver P, 2018. Insulin regulation of gluconeogenesis, Ann N Y Acad Sci 1411, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Potthoff MJ, Boney-Montoya J, Choi M, He T, Sunny NE, Satapati S, Suino-Powell K, Xu HE, Gerard RD, Finck BN, Burgess SC, Mangelsdorf DJ and Kliewer SA, 2011. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1alpha pathway, Cell Metab 13, 729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Byun S, Kim YC, Zhang Y, Kong B, Guo G, Sadoshima J, Ma J, Kemper B and Kemper JK, 2017. A postprandial FGF19-SHP-LSD1 regulatory axis mediates epigenetic repression of hepatic autophagy, EMBO J 36, 1755–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ryan KK, Kohli R, Gutierrez-Aguilar R, Gaitonde SG, Woods SC and Seeley RJ, 2013. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats, Endocrinology 154, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Morton GJ, Matsen ME, Bracy DP, Meek TH, Nguyen HT, Stefanovski D, Bergman RN, Wasserman DH and Schwartz MW, 2013. FGF19 action in the brain induces insulin-independent glucose lowering, J Clin Invest 123, 4799–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Marcelin G, Jo YH, Li X, Schwartz GJ, Zhang Y, Dun NJ, Lyu RM, Blouet C, Chang JK and Chua S Jr., 2014. Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism, Mol Metab 3, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Wean JB and Smith BN, 2021. FGF19 in the Hindbrain Lowers Blood Glucose and Alters Excitability of Vagal Motor Neurons in Hyperglycemic Mice, Endocrinology 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Hansen AMK, Vienberg SG, Lykkegaard K, Zhao X, Tingqing G, Han D, Zhang X, Thogersen H, Sass-Orum K, Tagmose T, Raun K and Andersen B, 2018. Differential receptor selectivity of the FGF15/FGF19 orthologues determines distinct metabolic activities in db/db mice, Biochem J 475, 2985–2996. [DOI] [PubMed] [Google Scholar]
- [112].Antonellis PJ, Droz BA, Cosgrove R, O’Farrell LS, Coskun T, Perfield JW 2nd, Bauer S, Wade M, Chouinard TE, Brozinick JT, Adams AC and Samms RJ, 2019. The anti-obesity effect of FGF19 does not require UCP1-dependent thermogenesis, Mol Metab 30, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Ge X, Chen C, Hui X, Wang Y, Lam KS and Xu A, 2011. Fibroblast growth factor 21 induces glucose transporter-1 expression through activation of the serum response factor/Ets-like protein-1 in adipocytes, J Biol Chem 286, 34533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Zhao S, Wang D, Li Z, Xu S, Chen H, Ding W, Yang J, Zhao W, Sun B and Wang Z, 2021. FGF15/FGF19 alleviates insulin resistance and upregulates placental IRS1/GLUT expression in pregnant mice fed a high-fat diet, Placenta 112, 81–88. [DOI] [PubMed] [Google Scholar]
- [115].Sachdev S, Wang Q, Billington C, Connett J, Ahmed L, Inabnet W, Chua S, Ikramuddin S and Korner J, 2016. FGF 19 and Bile Acids Increase Following Roux-en-Y Gastric Bypass but Not After Medical Management in Patients with Type 2 Diabetes, Obes Surg 26, 957–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Gerhard GS, Styer AM, Wood GC, Roesch SL, Petrick AT, Gabrielsen J, Strodel WE, Still CD and Argyropoulos G, 2013. A role for fibroblast growth factor 19 and bile acids in diabetes remission after Roux-en-Y gastric bypass, Diabetes Care. 36, 1859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Jansen PL, van Werven J, Aarts E, Berends F, Janssen I, Stoker J and Schaap FG, 2011. Alterations of hormonally active fibroblast growth factors after Roux-en-Y gastric bypass surgery, Dig Dis 29, 48–51. [DOI] [PubMed] [Google Scholar]
- [118].Bozadjieva N, Heppner KM and Seeley RJ, 2018. Targeting FXR and FGF19 to Treat Metabolic Diseases-Lessons Learned From Bariatric Surgery, Diabetes 67, 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Pournaras DJ, Glicksman C, Vincent RP, Kuganolipava S, Alaghband-Zadeh J, Mahon D, Bekker JH, Ghatei MA, Bloom SR, Walters JR, Welbourn R and le Roux CW, 2012. The role of bile after Roux-en-Y gastric bypass in promoting weight loss and improving glycaemic control, Endocrinology 153, 3613–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Harris LLS, Smith GI, Mittendorfer B, Eagon JC, Okunade AL, Patterson BW and Klein S, 2017. Roux-en-Y Gastric Bypass Surgery Has Unique Effects on Postprandial FGF21 but Not FGF19 Secretion, J Clin Endocrinol Metab 102, 3858–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Jorgensen NB, Dirksen C, Bojsen-Moller KN, Kristiansen VB, Wulff BS, Rainteau D, Humbert L, Rehfeld JF, Holst JJ, Madsbad S and Clausen TR, 2015. Improvements in glucose metabolism early after gastric bypass surgery are not explained by increases in total bile acids and fibroblast growth factor 19 concentrations, J Clin Endocrinol Metab 100, E396–406. [DOI] [PubMed] [Google Scholar]
- [122].Zhou M, Learned RM, Rossi SJ, DePaoli AM, Tian H and Ling L, 2017. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice, Hepatol Commun 1, 1024–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Dudek J, 2017. Role of Cardiolipin in Mitochondrial Signaling Pathways, Front Cell Dev Biol 5, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Harrison SA, Rinella ME, Abdelmalek MF, Trotter JF, Paredes AH, Arnold HL, Kugelmas M, Bashir MR, Jaros MJ, Ling L, Rossi SJ, DePaoli AM and Loomba R, 2018. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial, Lancet 391, 1174–1185. [DOI] [PubMed] [Google Scholar]
- [125].Rinella ME, Trotter JF, Abdelmalek MF, Paredes AH, Connelly MA, Jaros MJ, Ling L, Rossi SJ, DePaoli AM and Harrison SA, 2019. Rosuvastatin improves the FGF19 analogue NGM282-associated lipid changes in patients with non-alcoholic steatohepatitis, J Hepatol 70, 735–744. [DOI] [PubMed] [Google Scholar]
- [126].Sjoberg BG, Straniero S, Angelin B and Rudling M, 2017. Cholestyramine treatment of healthy humans rapidly induces transient hypertriglyceridemia when treatment is initiated, Am J Physiol Endocrinol Metab 313, E167–E174. [DOI] [PubMed] [Google Scholar]
- [127].Schumacher JD, Kong B, Pan Y, Zhan L, Sun R, Aa J, Rizzolo D, Richardson JR, Chen A, Goedken M, Aleksunes LM, Laskin DL and Guo GL, 2017. The effect of fibroblast growth factor 15 deficiency on the development of high fat diet induced non-alcoholic steatohepatitis, Toxicol Appl Pharmacol 330, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Alvarez-Sola G, Uriarte I, Latasa MU, Fernandez-Barrena MG, Urtasun R, Elizalde M, Barcena-Varela M, Jimenez M, Chang HC, Barbero R, Catalan V, Rodriguez A, Fruhbeck G, Gallego-Escuredo JM, Gavalda-Navarro A, Villarroya F, Rodriguez-Ortigosa CM, Corrales FJ, Prieto J, Berraondo P, Berasain C and Avila MA, 2017. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: development of an FGF19-based chimeric molecule to promote fatty liver regeneration, Gut 66, 1818–1828. [DOI] [PubMed] [Google Scholar]
- [129].Somm E, Henry H, Bruce SJ, Aeby S, Rosikiewicz M, Sykiotis GP, Asrih M, Jornayvaz FR, Denechaud PD, Albrecht U, Mohammadi M, Dwyer A, Acierno JS Jr., Schoonjans K, Fajas L, Greub G and Pitteloud N, 2017. beta-Klotho deficiency protects against obesity through a crosstalk between liver, microbiota, and brown adipose tissue, JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Kim YC, Seok S, Zhang Y, Ma J, Kong B, Guo G, Kemper B and Kemper JK, 2020. Intestinal FGF15/19 physiologically repress hepatic lipogenesis in the late fed-state by activating SHP and DNMT3A, Nat Commun 11, 5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Gallego-Escuredo JM, Gomez-Ambrosi J, Catalan V, Domingo P, Giralt M, Fruhbeck G and Villarroya F, 2015. Opposite alterations in FGF21 and FGF19 levels and disturbed expression of the receptor machinery for endocrine FGFs in obese patients, Int J Obes (Lond) 39, 121–9. [DOI] [PubMed] [Google Scholar]
- [132].van der Lans AA, Hoeks J, Brans B, Vijgen GH, Visser MG, Vosselman MJ, Hansen J, Jorgensen JA, Wu J, Mottaghy FM, Schrauwen P and van Marken Lichtenbelt WD, 2013. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis, J Clin Invest. 123, 3395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P and Teule GJ, 2009. Cold-activated brown adipose tissue in healthy men, N Engl J Med. 360, 1500–8. [DOI] [PubMed] [Google Scholar]
- [134].Cannon B and Nedergaard J, 2004. Brown adipose tissue: function and physiological significance, Physiol Rev 84, 277–359. [DOI] [PubMed] [Google Scholar]
- [135].Moron-Ros S, Uriarte I, Berasain C, Avila MA, Sabater-Masdeu M, Moreno-Navarrete JM, Fernandez-Real JM, Giralt M, Villarroya F and Gavalda-Navarro A, 2021. FGF15/19 is required for adipose tissue plasticity in response to thermogenic adaptations, Mol Metab 43, 101113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Meyuhas O, 2015. Ribosomal Protein S6 Phosphorylation: Four Decades of Research, Int Rev Cell Mol Biol 320, 41–73. [DOI] [PubMed] [Google Scholar]
- [137].Guo A, Li K, Tian HC, Fan Z, Chen QN, Yang YF, Yu J, Wu YX and Xiao Q, 2021. FGF19 protects skeletal muscle against obesity-induced muscle atrophy, metabolic derangement and abnormal irisin levels via the AMPK/SIRT-1/PGC-alpha pathway, J Cell Mol Med 25, 3585–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Kong B, Huang J, Zhu Y, Li G, Williams J, Shen S, Aleksunes LM, Richardson JR, Apte U, Rudnick DA and Guo GL, 2014. Fibroblast growth factor 15 deficiency impairs liver regeneration in mice, Am J Physiol Gastrointest Liver Physiol 306, G893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Uriarte I, Fernandez-Barrena MG, Monte MJ, Latasa MU, Chang HC, Carotti S, Vespasiani-Gentilucci U, Morini S, Vicente E, Concepcion AR, Medina JF, Marin JJ, Berasain C, Prieto J and Avila MA, 2013. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice, Gut 62, 899–910. [DOI] [PubMed] [Google Scholar]
- [140].Padrissa-Altes S, Bachofner M, Bogorad RL, Pohlmeier L, Rossolini T, Bohm F, Liebisch G, Hellerbrand C, Koteliansky V, Speicher T and Werner S, 2015. Control of hepatocyte proliferation and survival by Fgf receptors is essential for liver regeneration in mice, Gut 64, 1444–53. [DOI] [PubMed] [Google Scholar]
- [141].Koelfat KVK, van Mierlo KMC, Lodewick TM, Bloemen JG, van der Kroft G, Amygdalos I, Neumann UP, Dejong CHC, Jansen PLM, Olde Damink SWM and Schaap FG, 2021. Bile Salt and FGF19 Signaling in the Early Phase of Human Liver Regeneration, Hepatol Commun 5, 1400–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Miura S, Mitsuhashi N, Shimizu H, Kimura F, Yoshidome H, Otsuka M, Kato A, Shida T, Okamura D and Miyazaki M, 2012. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma, BMC Cancer 12, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].French DM, Lin BC, Wang M, Adams C, Shek T, Hotzel K, Bolon B, Ferrando R, Blackmore C, Schroeder K, Rodriguez LA, Hristopoulos M, Venook R, Ashkenazi A and Desnoyers LR, 2012. Targeting FGFR4 inhibits hepatocellular carcinoma in preclinical mouse models, PLoS One 7, e36713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Kang HJ, Haq F, Sung CO, Choi J, Hong SM, Eo SH, Jeong HJ, Shin J, Shim JH, Lee HC, An J, Kim MJ, Kim KP, Ahn SM and Yu E, 2019. Characterization of Hepatocellular Carcinoma Patients with FGF19 Amplification Assessed by Fluorescence in situ Hybridization: A Large Cohort Study, Liver Cancer 8, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Sawey ET, Chanrion M, Cai C, Wu G, Zhang J, Zender L, Zhao A, Busuttil RW, Yee H, Stein L, French DM, Finn RS, Lowe SW and Powers S, 2011. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening, Cancer Cell 19, 347–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Latasa MU, Salis F, Urtasun R, Garcia-Irigoyen O, Elizalde M, Uriarte I, Santamaria M, Feo F, Pascale RM, Prieto J, Berasain C and Avila MA, 2012. Regulation of amphiregulin gene expression by beta-catenin signaling in human hepatocellular carcinoma cells: a novel crosstalk between FGF19 and the EGFR system, PLoS One 7, e52711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Ahn SM, Jang SJ, Shim JH, Kim D, Hong SM, Sung CO, Baek D, Haq F, Ansari AA, Lee SY, Chun SM, Choi S, Choi HJ, Kim J, Kim S, Hwang S, Lee YJ, Lee JE, Jung WR, Jang HY, Yang E, Sung WK, Lee NP, Mao M, Lee C, Zucman-Rossi J, Yu E, Lee HC and Kong G, 2014. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification, Hepatology 60, 1972–82. [DOI] [PubMed] [Google Scholar]
- [148].Chen T, Liu H, Liu Z, Li K, Qin R, Wang Y, Liu J, Li Z, Gao Q, Pan C, Yang F, Zhao W, Zhang Z and Xu Y, 2021. FGF19 and FGFR4 promotes the progression of gallbladder carcinoma in an autocrine pathway dependent on GPBAR1-cAMP-EGR1 axis, Oncogene 40, 4941–4953. [DOI] [PubMed] [Google Scholar]
- [149].Pai R, Dunlap D, Qing J, Mohtashemi I, Hotzel K and French DM, 2008. Inhibition of fibroblast growth factor 19 reduces tumor growth by modulating beta-catenin signaling, Cancer Res 68, 5086–95. [DOI] [PubMed] [Google Scholar]
- [150].Heinzle C, Gsur A, Hunjadi M, Erdem Z, Gauglhofer C, Stattner S, Karner J, Klimpfinger M, Wrba F, Reti A, Hegedus B, Baierl A, Grasl-Kraupp B, Holzmann K, Grusch M, Berger W and Marian B, 2012. Differential effects of polymorphic alleles of FGF receptor 4 on colon cancer growth and metastasis, Cancer Res 72, 5767–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Li F, Li Z, Han Q, Cheng Y, Ji W, Yang Y, Lu S and Xia W, 2020. Enhanced autocrine FGF19/FGFR4 signaling drives the progression of lung squamous cell carcinoma, which responds to mTOR inhibitor AZD2104, Oncogene 39, 3507–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Gao L, Lang L, Zhao X, Shay C, Shull AY and Teng Y, 2019. FGF19 amplification reveals an oncogenic dependency upon autocrine FGF19/FGFR4 signaling in head and neck squamous cell carcinoma, Oncogene 38, 2394–2404. [DOI] [PubMed] [Google Scholar]
- [153].Tiong KH, Tan BS, Choo HL, Chung FF, Hii LW, Tan SH, Khor NT, Wong SF, See SJ, Tan YF, Rosli R, Cheong SK and Leong CO, 2016. Fibroblast growth factor receptor 4 (FGFR4) and fibroblast growth factor 19 (FGF19) autocrine enhance breast cancer cells survival, Oncotarget 7, 57633–57650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Zaid TM, Yeung TL, Thompson MS, Leung CS, Harding T, Co NN, Schmandt RS, Kwan SY, Rodriguez-Aguay C, Lopez-Berestein G, Sood AK, Wong KK, Birrer MJ and Mok SC, 2013. Identification of FGFR4 as a potential therapeutic target for advanced-stage, high-grade serous ovarian cancer, Clin Cancer Res 19, 809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Goetz R and Mohammadi M, 2013. Exploring mechanisms of FGF signalling through the lens of structural biology, Nat Rev Mol Cell Biol 14, 166–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Zhao H, Lv F, Liang G, Huang X, Wu G, Zhang W, Yu L, Shi L and Teng Y, 2016. FGF19 promotes epithelial-mesenchymal transition in hepatocellular carcinoma cells by modulating the GSK3beta/beta- catenin signaling cascade via FGFR4 activation, Oncotarget 7, 13575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Zhou M, Wang X, Phung V, Lindhout DA, Mondal K, Hsu JY, Yang H, Humphrey M, Ding X, Arora T, Learned RM, DePaoli AM, Tian H and Ling L, 2014. Separating Tumorigenicity from Bile Acid Regulatory Activity for Endocrine Hormone FGF19, Cancer Res 74, 3306–16. [DOI] [PubMed] [Google Scholar]
- [158].Luo J, Ko B, Elliott M, Zhou M, Lindhout DA, Phung V, To C, Learned RM, Tian H, DePaoli AM and Ling L, 2014. A nontumorigenic variant of FGF19 treats cholestatic liver diseases, Sci Transl Med 6, 247ra100. [DOI] [PubMed] [Google Scholar]
- [159].Zhou M, Learned RM, Rossi SJ, DePaoli AM, Tian H and Ling L, 2016. Engineered fibroblast growth factor 19 reduces liver injury and resolves sclerosing cholangitis in Mdr2-deficient mice, Hepatology 63, 914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Hirschfield GM, Chazouilleres O, Drenth JP, Thorburn D, Harrison SA, Landis CS, Mayo MJ, Muir AJ, Trotter JF, Leeming DJ, Karsdal MA, Jaros MJ, Ling L, Kim KH, Rossi SJ, Somaratne RM, DePaoli AM and Beuers U, 2019. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: A multicenter, randomized, double-blind, placebo-controlled phase II trial, J Hepatol 70, 483–493. [DOI] [PubMed] [Google Scholar]
- [161].Sanyal AJ, Ling L, Beuers U, DePaoli AM, Lieu HD, Harrison SA and Hirschfield GM, 2021. Potent suppression of hydrophobic bile acids by aldafermin, an FGF19 analogue, across metabolic and cholestatic liver diseases, JHEP Rep 3, 100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Harrison SA, Neff G, Guy CD, Bashir MR, Paredes AH, Frias JP, Younes Z, Trotter JF, Gunn NT, Moussa SE, Kohli A, Nelson K, Gottwald M, Chang WCG, Yan AZ, DePaoli AM, Ling L and Lieu HD, 2021. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients With Nonalcoholic Steatohepatitis, Gastroenterology 160, 219–231 e1. [DOI] [PubMed] [Google Scholar]