

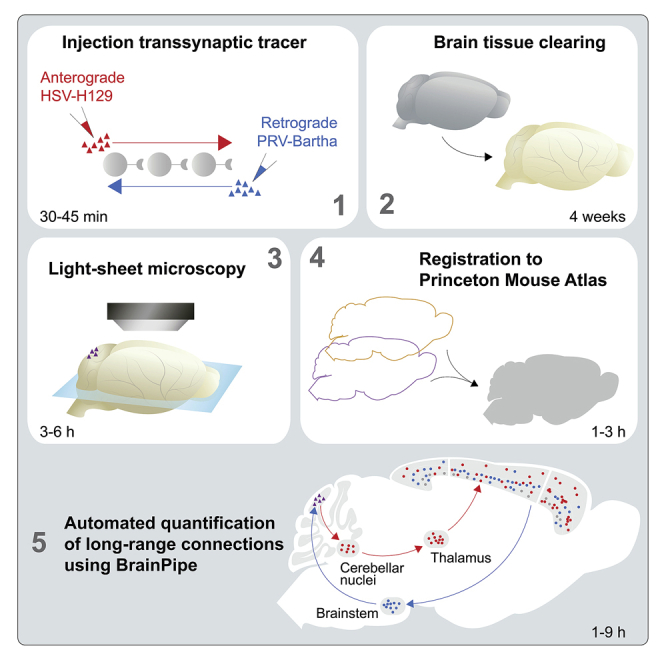
Summary
Transsynaptic viral tracing requires tissue sectioning, manual cell counting, and anatomical assignment, all of which are time intensive. We describe a protocol for BrainPipe, a scalable software for automated anatomical alignment and object counting in light-sheet microscopy volumes. BrainPipe can be generalized to new counting tasks by using a new atlas and training a neural network for object detection. Combining viral tracing, iDISCO+ tissue clearing, and BrainPipe facilitates mapping of cerebellar connectivity to the rest of the murine brain.
For complete details on the use and execution of this protocol, please refer to Pisano et al. (2021).
Subject areas: Bioinformatics, Computer sciences, Microscopy, Neuroscience
Graphical abstract
Highlights
-
•
Using iDISCO+ whole-brain tissue clearing technique for transsynaptic viral tracing
-
•
BrainPipe enables automated registration and analysis of light-sheet microscopy volumes
-
•
Combining viral tracing, iDISCO+, and BrainPipe for high-throughput transsynaptic study
Transsynaptic viral tracing requires tissue sectioning, manual cell counting, and anatomical assignment, all of which are time intensive. We describe a protocol for BrainPipe, a scalable software for automated anatomical alignment and object counting in light-sheet microscopy volumes. BrainPipe can be generalized to new counting tasks by using a new atlas and training a neural network for object detection. Combining viral tracing, iDISCO+ tissue clearing, and BrainPipe facilitates mapping of cerebellar connectivity to the rest of the murine brain.
Before you begin
This protocol comprises 4 primary sections: 1) viral injections, 2) tissue clearing, 3) light-sheet microscopy, and 4) automated computational analysis (volumetric registration to determine anatomical location and cell detection). The viral injections section of the Step-by-step method details describes methodology for injection of transsynaptic viruses that can be used to determine brain-region connectivity. Herpes Simplex Virus (HSV)-H129 is primarily transported anterograde while pseudorabies virus (PRV)-Bartha is transported retrograde. The maximum amount of spread and the number of synapses traveled, depend on the time between injection and sacrifice (hours post-injection, hpi).
Classical histology methods are traditionally used with viral tracing studies, where tissue is extracted, fixed, serially sectioned and imaged using epifluorescence or confocal microscopy. This is time-consuming and error-prone and provides sub-optimal data quality. As an alternative to classic histology, our protocol utilizes iDISCO+ (Renier et al., 2016), a tissue clearing methodology that allows for whole-brain immunofixation followed by steps to extract lipids making the brain transparent allowing for whole-brain imaging without the need for sectioning. To image a cleared brain, light-sheet microscopy is utilized, a non-invasive fluorescent microscopy approach that serially images different planes of tissue. The resulting high-resolution brain images can then be analyzed in an automated fashion using BrainPipe.
BrainPipe consists of several steps, first computationally stitching each tile to generate a brain volume for each acquisition channel. Next, brain volume can be volumetrically aligned to an atlas, known as registration, so that each region can be anatomically categorized. Finally, a convolutional neural network can be used for automated quantification and determination of location for labeled cells. Together BrainPipe allows for whole-brain automated long-range quantifications of transsynaptic tracing studies as done in (Pisano et al., 2021).
Viral injections
Timing: 1–2 h
Viral tracing uses the fact that neurotropic viruses will directionally spread only to synaptically connected neurons. Thus, areas where viral expression is present suggests connectivity with the area initially injected with the virus. The number of synapses traversed by the virus is controlled by the time between injection and sacrifice.
-
1.Prepare glass injection pipettes: using soda lime glass, pull pipettes with a P-97 puller, then bevel them to 30 degrees with an approximate 10 μm tip width (comparable to Liu et al. [2020]).
-
a.If not using a nanoinjector, precise delivery of volume requires calibration of the stereoscope and injection setup.
-
b.Depending on stereoscope setup, distance can be marked by either making millimeter markings with a permanent marker on each pipette or marking on a piece of tape that can be placed onto the pipette.
-
a.
-
2.
Disinfect and prepare stereotaxic surgical setup.
-
3.
Prepare micro-drill burr and dental drill.
-
4.
Turn on surgical body warmer. Set it to 36°C.
-
5.Prepare surgical supplies in surgical area (Figures 1A and 1B):
-
a.Premix 15% mannitol: 0.9% saline solution and fill an injection syringe.
-
b.Premix rimadyl (5 mg/kg carprofen 50 mg/mL, in 0.9% NaCl solution) and fill an injection syringe.
-
c.Fill the vaporizer with isoflurane. Ensure oxygen is connected and that the general anesthesia setup is functional.
-
d.Sterilize surgical tools and cotton swabs according to institutional protocol (we prefer using an autoclave that is capable of dry- or wet-heat sterilization), then place them in the surgical area on a sterile pad.
-
e.Gather ophthalmic ointment.
-
f.Prepare microinjector/microsyringe pump controller per manufacturer’s instruction.
-
g.Prepare surgifoam in case of bleeding by cutting it into small pieces and soaking in 0.9% saline.
-
h.If co-injecting Cholera Toxin B with virus, prepare stock of 0.5% cholera toxin B conjugated to Alexa Fluor 555 in 0.9% saline (CTB-555; Conte et al. [2009]). Take an aliquot and place it on ice (4°C; “wet ice”).
-
i.Thaw −80°C HSV-H129 VC22 viral aliquots by placing them on wet ice.
-
a.
Note: Viral stock is susceptible to titer loss from freeze-thaw cycles. To ensure consistent titers across different injection days we suggest preparing single-day-use 100 microliter aliquots.
Figure 1.

Surgical suite preparation, viral injections and light-sheet imaging
(A and B) Surgical suite preparation before procedures begin.
(C) Injection pipette backfilled and loaded onto injection apparatus.
(D) Posterior interparietal and occipital skull exposed after midline splaying of scalp tissue and removal of posterior neck muscles. Cerebellar vasculature is visible through bone, allowing for anatomical demarcation.
(E) Vermal cerebellum exposed after craniotomy.
(F) Injection of viral solution into the right cerebellar hemisphere.
(G and H) A cleared brain mounted via glue onto a light-sheet imaging cradle.
(I) A cleared brain illuminated by three light-sheets within the imaging chamber of a light-sheet microscope. Scale bars 2.5 mm (D and F), 1 mm (E).
iDISCO+: immunofixation and clearing
Tissue clearing is an alternative methodology to section-based histology. Through a series of permeabilization steps, whole-brain immunostaining, and lipid removal an entire mouse brain can be immunolabeled and subsequently made transparent. This tissue can then be volumetrically imaged using methodology such as light-sheet microscopy.
We recommend preparing stocks of solutions (except for solutions containing antibodies or donkey serum) beforehand. This ensures stocks are at room temperature (20°C–22°C) at use.
-
6.Prepare reagents at benchside (please refer to Data S1- supplementary clearing worksheet for step-by-step instructions and further details):
-
a.1× PBS.
-
b.10% buffered formalin.
-
c.20%, 40%, 60%, 80%, and 100% methanol/H2O stocks (we suggest using double distilled or MilliQ water) in glass bottles. Ensure tight seals to prevent evaporation.
-
d.5% H2O2/methanol stock (1 part 30% H2O2: 5 parts 100% methanol).
-
e.0.2% Triton X-100/1× PBS.
-
f.20% DMSO/0.3 M glycine/0.2% Triton X-100/1× PBS (22.5 mg/mL of glycine).
-
g.PBS + 0.2% Tween-20 + 10 μg/mL heparin (PTwH).
-
a.
Note: 4% Paraformaldehyde in 1× PBS can be used as an alternative to 10% buffered formalin.
Note: For stocks not containing alcohol, we optionally recommend adding 1 g/L of sodium azide to prevent microbial growth. We typically prefer to use these solutions within 2 months of preparation if stored on the lab bench at room temperature (20°C–22°C). Importantly, some antibodies will not work with sodium azide (especially horseradish peroxidase conjugated antibodies), so check with the manufacturer about compatibility prior to use.
Note: We recommend keeping donkey serum frozen at −20°C and only adding donkey serum to solutions on the day reagents are needed. We also recommend keeping primary and secondary antibodies stored per manufacturer recommendations and only added to solutions the day of use.
-
7.The other ingredients in the following solutions can be made beforehand and kept at room temperature (20°C–22°C):
-
a.10% DMSO/6% Donkey Serum/0.2% Triton X-100/1× phosphate-buffered saline (PBS).
-
b.Primary Antibody Solution: 5% DMSO/3% Donkey Serum/PTwH.
-
c.Secondary Antibody Solution: 3% Donkey Serum/PTwH.
-
a.
CRITICAL: Appropriate safety precautions are important as many clearing regents are caustic to skin/eyes. Please work in fume hood when appropriate. Contact your environmental health safety officer for advice before beginning.
Light-sheet microscopy
Light-sheet microscopy uses a plane of light to optically cross-section cleared tissue, with an objective positioned orthogonal to the plane of the light sheet to image fluorescence. The light sheet can be serially stepped through the tissue allowing for rapid volumetric imaging of large chunks of tissue.
-
8.Ensure appropriate excitation and emission filters for imaging of iDISCO+ cleared samples (see problem 4). We suggest the following experimental setup:
-
a.Acquire an autofluorescent channel for registration (volumetric anatomical alignment) purposes using 488 nm excitation and 525 nm emission (FF01-525/39-25, Semrock, Rochester, New York). Cleared tissue has greater autofluorescence at shorter wavelengths, therefore we recommend using the shortest wavelength available for autofluorescence capture.
-
b.Image injection sites, identified by CTB-555, at 561 nm excitation and 609 nm emission (FF01-609/54-25, Semrock).
-
c.Perform cellular imaging of virally infected cells (anti-HSV) using 640 nm excitation and 680 nm emission (FF01-680/42-25). Cleared tissue has less autofluorescence at longer wavelengths, so it is recommended to use longer wavelength channels for the cell-detection channel.
-
a.
-
9.
For this project we used the LaVision Ultramicroscope II for light-sheet imaging; regardless of the light-sheet microscope being used, it is essential to ensure the microscope is calibrated and that all light paths/sheets are appropriately aligned.
-
10.
Print sample holders (Figures 6C and 6D). See Data S2 in (Pisano et al., 2021) for printing files. We used a Formlabs Form 2 printer with Black resin. Clean printed holders several times with 70% ethanol before use in the microscope.
Note: if using a different printer/resin, ensure resin used is compatible with DBE.
-
11.
Fill DBE to the appropriate level within the microscope imaging chamber.
Figure 6.

Horizontal imaging planes in light-sheet microscopy for cleared mouse brains
(A) The light-sheet microscopy excitation plane (blue) is orthogonal to the detection-path axis from specimen to objective. Figure reproduced from Huisken and Stainier, 2009.
(B) The mouse brain is shortest in the dorsal ventral direction and should be oriented with this direction parallel to the detection axis. The mediolateral dimension is next shortest, and the light sheet should enter from this direction.
(C) Rendering of custom-designed holders to mount mouse brains for whole-brain imaging. The “backstop” (marked by a red asterisk) of the holder provides a surface to glue the caudal and ventral brainstem and prevents glue from obscuring more anterior and ventral surfaces. The ramp (marked by a blue asterisk) provides a positive pitch allowing for complete imaging through the dorsal ventral surface given the working distance of the objective.
(D) Example of cleared mouse brain after mounting on the custom holder.
BrainPipe: light-sheet data registration and cell counting
Light-sheet microscopy produces high-resolution volumetric data on the scale of hundreds of gigabytes. Leveraging these data, BrainPipe first computationally stitches the thousands of images into volumes. Next, it automatically aligns (registers) brain volumes to an atlas for automated determination of anatomical location. Finally, it uses convolutional neural networks to count labeled cells automatically. Through these steps, BrainPipe allows for automated histological analysis of transsynaptic tracing studies.
Install BrainPipe
-
12.
Install Python 3. We recommend Anaconda as a package manager (anaconda.com) and using at least a moderately powered computer. We suggest using a desktop computer with a Linux operating system and with at least 32 gigabytes RAM, 8 core 3.5 GHz processor, 1TB storage and a GPU with 8 GB memory.
-
13.
Download BrainPipe and install it using the following instructions: https://github.com/PrincetonUniversity/BrainPipe.
-
14.
If planning on utilizing a CPU cluster for registration or GPU cluster for convolutional neural network training/inference contact your computing cluster administrator to ensure the appropriate dependencies and packages are available.
Generation of project-specific atlas
-
15.
We recommend starting with a field standard volumetric atlas and annotation set such as the Princeton Mouse Atlas: https://brainmaps.princeton.edu/2020/09/princeton-mouse-brain-atlas-links/. However, often projects require atlases with specific features. We observed better registration performance when registering within a modality (for example, light-sheet to light-sheet) as opposed with across modalities (for example, light-sheet to two-photon). Thus registration might be improved through generating a project-specific atlas, even if it is not technically required. To do so, follow the instructions on https://github.com/PrincetonUniversity/pytlas.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-HSV (1:1,750 for mono-, di-synaptic timepoints, 1:350 for trisynaptic)∗ | Dako | Cat# B011402-2 RRID: AB_2732870 |
| Chicken anti-GFP (1:750 for monosynaptic timepoints, 1:500 for disynaptic)∗ | Aves | Cat# GFP-1020 RRID: AB_10000240 |
| Donkey anti-rabbit AlexaFluor 647 (1:500 for mono-, di-synaptic timepoints, 1:250 for trisynaptic)∗ | Thermo Fisher Scientific | Cat# A31573 RRID: AB_2536183 |
| Donkey anti-chicken AlexaFluor 647 (1:400 for monosynaptic timepoints, 1:300 for disynaptic)∗ | Jackson ImmunoResearch | Cat# 703-606-155 RRID: AB_2340380 |
| Bacterial and virus strains | ||
| HSV-1 strain H129 recombinant VC22 | This paper | H129-VC22 (Princeton Viral Vector Core) |
| HSV-1 strain H129 recombinant 772 | This paper | H129-772 (Princeton Viral Vector Core) |
| PRV Bartha 152 | This paper | PRV-152 (Princeton Viral Vector Core) |
| Chemicals, peptides, and recombinant proteins | ||
| Mannitol | Sigma-Aldrich | M4125 |
| Isoflurane | VetOne | 501017 |
| Puralube, Pharmaderm | Pharmaderm | 37327 |
| Rimadyl (Carprofen) | Zoetis | 141-199 |
| CTB-555 | Sigma-Aldrich | C22843 |
| Ketamine | Vet One | #200-055 |
| Xylazine (Anased) | Akorn | 139-236 |
| 1× PBS | Thermo Fisher Scientific | 14190136 |
| Formalin (10%) | Fisher Scientific | 23-245685 |
| Paraformaldehyde (4%) | Fisher Scientific | AAJ19943 |
| Sodium azide | Sigma-Aldrich | S2002-500G |
| Triton X-100 | Sigma-Aldrich | T8787-50ML |
| Methanol | Carolina Biological Supply | 874195 |
| Hydrogen peroxide | Sigma-Aldrich | H1009-100M |
| Dimethyl sulfoxide (DMSO) | Fisher Scientific | D128-1 |
| Glycine | Sigma-Aldrich | 410225-50G |
| Donkey serum | EMD Millipore | S30-100ml |
| Tween-20 | Sigma-Aldrich | P9416-50ML |
| Heparin | Sigma-Aldrich | H3149-100KU |
| Dichloromethane (DCM) | Sigma-Aldrich | 270997-2L |
| Dibenzyl ether/benzyl ether (DBE) | Sigma-Aldrich | 108014-1KG |
| Super glue - for mounting brains | Loctite | 234796 |
| Krazy glue - for “suturing” after surgical incision | Elmer's | KG48348MR |
| Deposited data | ||
| Princeton Mouse Atlas (PMA) and injection data | Pisano et al., 2021 | https://brainmaps.princeton.edu/2020/09/princeton-mouse-brain-atlas-links/ |
| HSV and PRV injection data aligned to Princeton Mouse Atlas | Pisano et al., 2021 | https://brainmaps.princeton.edu/2021/05/pisano_viral_tracing_injections/ |
| Experimental models: Organisms/strains | ||
| C57BL/6J mice, males and females, age 8–14 weeks | The Jackson Laboratory | 000664 |
| Software and algorithms | ||
| BrainPipe | Github.com/PrincetonUniversity/BrainPipe | zenodo.org/badge/latestdoi/149466560 |
| 3DUNet | Github.com/PrincetonUniversity/3dunet | zenodo.org/badge/latestdoi/157925214 |
| Pyalign | Github.com/PrincetonUniversity/pyalign | zenodo.org/badge/latestdoi/143018489 |
| Pyatlas | Github.com/PrincetonUniversity/pyatlas | zenodo.org/badge/latestdoi/131753598 |
| Python 3+ | (Van Rossum and Drake, 2009) | python.org |
| ImageJ | National Institutes of Health | imagej.net/software/fiji/ |
| Statsmodels 0.9.0 | (Seabold and Perktold, 2010) | statsmodels.org/stable/index.html |
| Blender | Blender | blender.org/ |
| ImSpector microscope controller software V 5.1.293 | LaVision BioTec | imspectordocs.readthedocs.io |
| Numpy 1.14.3 | (Oliphant, 2015) | numpy.org |
| Pandas 0.23.0 | (McKinney et al., 2010) | pandas.pydata.org |
| Matplotlib 2.2.2 | (Hunter, 2007) | matplotlib.org |
| Seaborn 0.9.0 | (Waskom et al., 2014) | seaborn.pydata.org |
| Scikit-Image 0.13.1 | (van der Walt et al., 2014) | scikit-image.org |
| SimpleITK 1.0.0 | (Lowekamp et al., 2013) | simpleITK.org |
| SciPy 1.1.0 | (Virtanen et al., 2020) | scipy.org |
| Scikit-Learn 0.19.1 | (Pedregosa et al., 2011) | scikit-learn.org |
| Other | ||
| Stereotaxic platform | David Kopf Instruments | Kopf Model 1900 |
| Soda lime glass | Kimble | 71900-10 |
| P-97 pipette puller | Sutter Instruments | P-97 |
| Dental drill | Osada | EXL-M40 |
| Burr drill bits | George Tiemann & Co | 160-1293-.24 (.24MM 1 3/4″ LONG 3/32 SHAFT) |
| Surgical body warmer | FHC | DC Temperature Control System |
| Surgifoam | Ethicon | #1972 |
| Black Resin | Formlabs | FLGPBK04 |
| Light-sheet microscope | LaVision BioTec | Ultramicroscope II |
| Emission filters | Semrock | FF01-525/39-25, FF01-609/54-25, FF01-680/42-25 |
| Dental cement | Parkell | S396 (C&B) |
| Kwik-Sil silicone sealant | World Precision Instruments | KWIK-SIL |
| Microinjection syringe pump system | World Precision Instruments | SYS-MICRO4 |
| Opaque magnets | Supermagnetman | D1005A-10 Parylene |
| 50 mL Eppendorf tubes | Fisher Scientific | 22-717-715 |
| 5 mL Eppendorf tubes | Fisher Scientific | 30119401 |
∗Note: Empiric antibody concentrations should be determined for each experiment (see Note at end of subsection viral injections).
Materials and equipment
| Material | Safety considerations |
|---|---|
| HSV H129 and PRV-Bartha | Follow appropriate local Biosafety Level-2 institutional protocols. We suggest personal protective equipment of at least consisting of two layers of nitrile gloves, mask, eye protection, and disposable gown. We prefer to disinfect tools and areas with 70% alcohol or bleach immediately after use followed by sterilization in an autoclave when possible. |
| Paraformaldehyde/Formalin (PFA) | Eye protection, nitrile gloves, work under snorkel or fume hood |
| Methanol | Eye protection, nitrile gloves |
| Hydrogen Peroxide | Eye protection, nitrile gloves |
| Triton X-100 | Eye protection, nitrile gloves |
| Dimethyl sulfoxide (DMSO) | Eye protection, nitrile gloves, work under snorkel or fume hood |
| Donkey Serum | Eye protection, nitrile gloves |
| Antibodies | Eye protection, nitrile gloves |
| Tween-20 | Eye protection, nitrile gloves |
| Dichloromethane (DCM) | Eye protection, double nitrile gloves, work under snorkel or fume hood. Remove gloves immediately if dichloromethane gets on gloves. |
| Dibenzyl ether/benzyl ether (DBE) | Eye protection, nitrile gloves. Dibenzyl ether can be removed from surfaces using 70% ethanol. |
PBS + 0.2% Tween-20 + 10 μg/mL heparin (PTwH)
| Reagent | Final concentration | Amount |
|---|---|---|
| 1× PBS | 99.8% | 998 mL |
| Tween-20 | 0.2% | 2 mL |
| Heparin | 10 μg/mL | 10 mg |
| Total | n/a | 1,000 mL |
Note: Based on experimental needs smaller or larger total volume of this PTwH solution can be made. We (optional) recommend adding 1 g/L of sodium azide to the PTwH solution to prevent microbial growth. We typically prefer to use PTwH within 2 months of making and stored on the lab bench at room temperature (20°C–22°C). Please refer to the table of safety considerations for each reagent used in solution.
20% DMSO/0.3 M glycine/0.2% Triton X-100/1× PBS
| Reagent | Final concentration | Amount |
|---|---|---|
| DMSO | 20% | 50 mL |
| Glycine | 0.3 M (22.5 mg/mL) | 5.625 g |
| Triton X-100 | 0.2% | 0.5 mL |
| 1× PBS | 79.8% | 199.5 mL |
| Total | n/a | 250 mL |
Note: Based on experimental needs smaller or larger total volume of this solution (20% DMSO/0.3M glycine/0.2% Triton X-100/1x PBS) can be made. We (optional) recommend adding 1 g/L of sodium azide to this solution prevent microbial growth. We typically prefer to use within 2 months of making and stored on the lab bench at room temperature (20°C–22°C). Please refer to a table of safety considerations for each reagent used in solution.
10% DMSO/6% Donkey Serum/0.2% Triton X-100/1× PBS
| Reagent | Final concentration | Amount |
|---|---|---|
| DMSO | 10% | 5 mL |
| Donkey Serum | 6% | 3 mL |
| Triton X-100 | 0.2% | 0.1 mL |
| 1× PBS | 83.8% | 41.9 mL |
| Total | n/a | 50 mL |
Note: Based on experimental needs smaller or larger total volume of this solution (10% DMSO/6% Donkey Serum/0.2% Triton X-100/1x PBS) can be made. This solution should be made on the day of use and can be stored at room temperature (20°C–22°C) prior to use. Please refer to the table of safety considerations for each reagent used in solution.
5% DMSO/3% Donkey Serum/PTwH
| Reagent | Final concentration | Amount |
|---|---|---|
| DMSO | 5% | 2.5 mL |
| Donkey Serum | 3% | 1.5 mL |
| PTwH (see recipe above) | 92% | 46 mL |
| Total | n/a | 50 mL |
Note: Based on experimental needs smaller or larger total volume of this solution (5% DMSO/3% Donkey Serum/PTwH) can be made. This solution should be made on the day of use (PTwH used in this solution can be made ahead of time) and can be stored at room temperature (20°C–22°C) prior to use. Please refer to a table of safety considerations for each reagent used in solution.
Step-by-step method details
Viral injections
Viral injections allow for delivery of a viral tracer that is directionally transported from the injection site. Based on duration from injection to animal sacrifice, the approximate number of synapses traversed can be determined (Figure 2). Brain areas expressing virus outside of the injection site suggest transsynaptic connectivity with the area of injection. Transsynaptic tracing methodology is heavily dependent on the tracer used, injection target and model organism. Here we describe murine cerebellar injections of herpes simplex virus-H129 VC22 (HSV-H129; Pisano et al. [2021]). For HSV-H129 or PRV injections, this procedure should be done in a dedicated biosafety level 2 area with appropriate personal protective equipment by trained personnel.
Figure 2.

Application of Kwik-Sil to post-injection craniotomy
(A) Punched-out craniotomy over the vermis cerebellum.
(B) Kwik-Sil two-chamber syringe next to the weigh boat.
(C) Kwik-Sil is put into the weigh boat.
(D) Kwik-Sil being mixed. Kwik-Sil is a binary product, and must be sufficiently mixed to ensure that it will cure.
(E) Application of Kwik-Sil over the craniotomy site. Having a dry skull around the craniotomy helps the product adhere. Take care only to apply the product to the craniotomy site and surrounding skull.
(F) View of craniotomy after application of Kwik-Sil. Scale bars 2.5 mm (A and F).
Please adjust injection protocols according to local Institutional Animal Care and Use Committee as well as Environmental Health and Biological safety requirements. In our case, all experimental procedures were approved by Princeton’s Institutional Animal Care and Use Committee (protocol numbers 1943-19 and 1023-15A1/2).
-
1.
Ensure the surgical area has been cleaned, tools sterilized and all supplies gathered.
-
2.Mix injection solution.
-
a.Mix 18 microliters of viral solution with 2 microliters of 0.5% CTB-555. Ensure proper mixing through gentle pipetting, do not vortex. Place this mixture back on wet ice.
-
a.
Note: We prefer a 9:1 ratio of viral solution to 0.5% CTB-555 as described given the viral solution titers used in our studies. Transsynaptic tracing injection doses (plaque-forming units delivered in a single injection) should just be above the infectivity threshold (Saleeba et al., 2019; Ugolini, 2010). Consequently, this ratio should be adjusted based on viral stock titer and desired injection volume.
-
3.Anesthetize and mount mouse on the stereotaxic apparatus.
-
a.Turn on oxygen flow to the isoflurane induction chamber.
-
b.Place the mouse into the induction chamber and then turn on flow of isoflurane to 5%. Wait for the mouse to become sedated. We find that the mouse will initially breathe rapidly. Breathing normalization is indicative of sufficient dose of anesthetic.
-
c.Adjust flow from induction chamber to stereotax and decrease from 5% to 4% isoflurane.
-
d.Take the mouse out of chamber and mount on stereotax using ear bars and bite bar.
-
e.Slide nose cone over mouse’s nose, then decrease the isoflurane to 2.5%.Note: Adjust level of anesthesia delivered based on mouse breathing rate and level of sedation. Faster breathing may require incremental increases in anesthesia and should be adjusted accordingly throughout the experimental procedure.
-
f.Place ophthalmic ointment on mouse eyes using sterile swab.
-
g.Provide the mouse with a subcutaneous injection of mannitol and rimadyl by pinching the skin between the shoulders and injecting directly underneath, without touching muscle.Note: Mannitol has been shown to increase viral transduction efficiency and viral spread leading to a net increase in expression in neural tissue (Burger et al., 2005; Mastakov et al., 2001).
-
h.Insert rectal temperature probe for real-time temperature readings and adjust mouse warmer accordingly. The mouse body temperature should remain between 33° and 36°C throughout the surgical procedure.
-
a.
-
4.Expose cranium.
-
a.Gently shave the fur from the mouse head. We prefer to use a #10 scalpel blade with warm soapy water.
-
b.Sterilize the scalp using 70% ethanol using a cotton swab.
-
c.Using a scalpel, make a single vertical incision at the midline starting from just posterior to eyes to the occipital bone.
-
d.Splay each layer of tissue open until the skull and posterior neck muscles are exposed. Flush with saline if needed.
-
e.Using tweezers and microscissors remove each layer of neck muscles from their attachment site on the skull.Note: We typically do this in 6 cuts (3 layers for each side). While removing muscle, try to identify and cut around vasculature to prevent unnecessary bleeding. When bleeding occurs, place a saline soaked piece of surgifoam on the site. Then touch a dry swab to the SURGIFOAM sponge to remove additional moisture. Let the sponge remain in place for several minutes to allow for hemocoagulation.
-
f.Then use a cotton swab with 70% ethanol to polish the exposed interparietal and occipital bones. This cleans off any muscle still attached while temporarily making the skull transparent to allow for visualization of cerebellar vasculature (Figure 1D).
-
a.
-
5.Perform craniotomy.
-
a.Determine injection location. For cerebellar injections, injection location is determined through visual identification of vasculature to identify cerebellar lobules.
-
b.We found success with two different approaches listed below.
-
i.Punch-out method: using micro-drill slowly drill a single bore hole through the skull using small circular movements.Note: This method is fast and causes less damage to the skull. However, there is a high risk for overheating the brain tissue deep to the borehole and a risk for drilling into brain tissue. Saline flushes on site and drill tip can help with overheating. A slow deliberate pace can prevent drilling into tissue. Usually CSF can be seen leaking into drill site just before the drill is through the skull.
-
ii.Traditional craniotomy method (Figure 1E): using a micro-drill burr, drill out a circular trench through the skull. This creates a circular piece of skull that can be gently lifted off with #55 forceps.Note: This method has a high success rates, especially when starting. However, it is time-consuming and produces occasional adhesion of dura/brain tissue when removing the inner circular piece of skull. This can be avoided through gently flooding the area with saline to help prevent any suction effects while removing the skull.
-
i.
-
c.Cover exposed brain with saline soaked surgifoam.
-
a.
-
6.Load injection pipette (Figure 1C).
-
a.Using a pipette, pull up 2 microliters of viral solution while working under a fume hood.
-
b.Backfill the glass pipette by slowly dispensing the viral solution into the back of the syringe.
-
c.For safety, wipe the back outside of the pipette with a Kimwipe wetted with 70% ethanol. This ensures that any viral solution that may have gotten on the edge and outside of pipette during loading has been inactivated and removed.
-
d.Now place the injection tubing onto the back of the glass pipette and mount the glass pipette onto the injection apparatus.
-
e.Move the pipette roughly into position and using the stereoscope focus onto the tip of the pipette.
-
f.Ensure that the injection pipette is functional and not clogged. Hold a Kimwipe that has been wetted with 70% ethanol just under the tip of the injection pipette. Slowly apply pressure to the syringe, watching to ensure the cylinder of injection solution moves to the tip of the glass injection pipette. Continue to apply pressure until you see a bead of liquid exit the tip.
-
g.Using the Kimwipe, gently wipe away the bead of the injection solution and discharge into BSL-2 waste.
-
a.
-
7.Inject viral solution (Figure 1F).
-
a.Remove the sponge that was covering the brain. During this time while the brain is exposed, we recommend occasionally wetting the brain surface using saline to prevent it from drying.
-
b.While visualizing the tip under the stereoscope, move the tip of the injection pipette to the brain’s surface. As you gently advance you should see the brain dimpling, confirming you are indeed at the surface of the brain.
-
c.Withdraw the pipette so that dimpling no longer occurs so that it is at the surface again and zero stereotaxic z coordinate.
-
d.Quickly lower injection pipette to 200 microns perpendicular to the brain surface. We find that rapid movement of the needle helps pierce the brain.Note: If there is significant brain dimpling, this usually means the injection pipette was not advanced rapidly enough. This can be overcome by moving the pipette back and forth slowly to advance the pipette through the dimpled surface.
-
e.Move the stereoscope so the meniscus is visualized. Deliver the desired volume by measuring the distance traversed by the meniscus.Note: We have found that 80–240 nanoliters provides reproducible infections for our viral titers. We prefer to deliver volume no faster than 100 nL/min.
-
f.Do not withdraw the pipette right after injection. Wait at least 1 min to allow the injection solution to spread locally into tissue. During this time, move the stereoscope to focus on the injection site.Note: Because our injection solution was pink in color, we were usually able to visualize it in local tissue, further confirming a successful injection.
-
g.Slowly withdraw the injection pipette and dispose of it in an appropriate sharps container.Note: For injections targeting deeper regions stereotactically, after piercing the brain surface we recommend slow deliberate descent and eventual ascent of the injection needle. For injections of higher volumes, we recommend keeping the injection pipette in target location for several minutes after injection solution delivery.
-
a.
-
8.Cover injection site.
-
a.Gently wet the brain surface with saline.
-
b.Apply Kwik-Sil to the brain surface using an applicator after mixing Kwik-Sil using disposable tips or separately on sterile surface (Figure 2). Ensure Kwik-Sil covering touches the bone around the entire craniotomy to form a protective barrier over the brain.
-
a.
Note: Allowing the exposed bony surface around the craniotomy to dry slightly at this step has helped keep the Kwik-Sil elastomer plug in place. We prefer to use a sterile swab to gently dry the surrounding cranium for this reason. It is also important to wait for Kwik-Sil to dry before proceeding to the next step, this can be assessed using the wooden back end of a swab to gently touch the Kwik-Sil and ensure it is tacky/rubbery.
Optional: Once dry, dental cement can be added on top of Kwik-Sil plug and bone. Since injections were never extended past 80 h post injection, we never had problems with Kwik-Sil plugs falling off.
-
9.Close scalp.
-
a.Using tweezers, pull together two halves of the scalp.Note: Skin can sometimes adhere to the underlying tissue, if this is the case, gently flush saline onto tissue.
-
b.Moving in a rostral to caudal fashion, pinch scalp together and slowly apply glue to the incision.Note: The goal is to seal the skin. Take care to not allow glue to penetrate through to the cranium which could lead to adhesion of scalp to cranium. Alternatively, sutures can be used for this step. Stop anesthesia, release ear bars, pull back nose cone and remove the mouse from the sterotax. Turn off all oxygen and isoflurane flow.
-
a.
-
10.Place mouse into new cage.
-
a.After glue has dried, remove ear bars and place the mouse into a singly-housed cage (or as protocol requires) with water and food.
-
a.
Optional: Have a warming light placed in part of the cage to ensure mouse body temperature remains appropriate during recovery.
Optional: If protocol allows, a small injection of normal saline (for example 10 mL/kg subcutaneous injection) can be given for post-surgical fluid resuscitation.
-
11.
Clean up the surgical area, disinfect and dispose of BSL-2 material appropriately.
Note: After viral injection protocol has been standardized, we strongly recommend performing an empirical time series to benchmark viral spread characteristics, identifying ideal timepoints for targeted number of synapses spread (Figure 3; see problems 1 and 5). For discussion regarding time point determination of transsynaptic viral tracers, see Pisano et al. (2021): Methods, In Vivo Virus injections, Transsynaptic time point determination and Discussion, Study limitations.
Figure 3.

Modified neuroinvasive viruses can be used to determine neuron connectivity
(A) Directionality of spread of transsynaptic viral tracers. Schematic of anterograde spread of herpes simplex virus 1 H129 (HSV-H129), and retrograde spread of the Bartha strain of pseudorabies virus (PRV-Bartha) and the N2c strain of rabies which is glycoprotein deleted (Rb N2c ΔG). H129 and PRV-Bartha spread continuously across a chain of synaptically connected neurons, while spread of Rb N2c ΔG requires the trans-complementation of the missing glycoprotein in each cell. Adeno-associated virus (AAV) usually does not spread transsynaptically and is shown for comparison.
(B) Various viral expression levels across a network of connected cells, including local interneurons. Viral expression levels vary continuously along synaptically-connected neurons. The continuous distribution requires consideration of the amount of fluorescence that constitutes a positively infected cell.
iDISCO+: immunofixation and clearing
Although many clearing techniques have been developed, we found that the iDISCO+ methodology (Renier et al., 2016) generates reproducible high signal to noise, which can be difficult to attain with transsynaptic viral tracing relatively to other viral-driven techniques such as AAV (Figure 3). It is first important to identify methanol-compatible antibodies (Figure 4; see https://idisco.info/idisco-protocol/ for antibody validation protocol) In our experience the most challenging feature of clearing optimization is determining appropriate antibody concentration for each experiment (see problems 4, 5, and 6). We recommend initially clearing several samples, each with different antibody concentrations. Despite clearing taking several weeks, it is not labor intensive and can be efficient because many samples can be cleared in parallel. To further increase efficiency, we also recommend preparing stocks of solutions (except for primary and secondary antibody solutions) beforehand. This also allows all stocks to be at room temperature (20°C–22°C) before use (see problem 3 for consequences of rapid temperature changes during clearing). For stocks not containing alcohol we optionally recommend adding 1 g/L of sodium azide to prevent microbial growth. Appropriate safety precautions are important as many clearing regents are caustic to skin/eyes.
Note: Please see accompanying worksheet sheet for clearing. To prevent errors and facilitate communication amongst lab members, we recommend printing one worksheet for each batch of cleared brains and keeping it in a lab clearing binder for members to document when each step was performed.
Note: Concentrations and timing of incubation steps have been optimized for mouse cerebellar injections of HSV-H129 and PRV at timepoints used in (Pisano et al., 2021). We strongly recommend optimizing parameters related to antibody concentration as well as primary and secondary antibody incubation times before full experimental cohorts. Larger chunks of tissue (e.g., rat brain) will require longer duration for most steps in this protocol.
-
12.Fix tissue (please refer to Data S1- supplementary clearing worksheet for step-by-step instructions and further details).
-
a.After tissue extraction, fix tissue for at least 4 h in 4% PFA/PBS.
-
a.
Note: We prefer transcardial perfusion before tissue extraction, comparable to Gage et al. (2012). It is important that this step should be performed per local Institutional Animal Care and Use Committee protocol to ensure humane treatment.
Note: We have had best results using transcardial perfusions with initial PBS flushes with 10 μg/mL heparin. This helps prevent spurious signals in cleared tissue secondary to unremoved blood in vasculature.
Note: It is important to be careful not to damage the brain tissue from sharp skull edges or any tools used during the extraction process.
Note: We recommend fixing tissue overnight on a rocking table, using a 50 mL Eppendorf tube.
-
13.Dehydrate tissue.
-
a.Transfer brains to 5 mL Eppendorf tubes and wash with 1× PBS for 30 min, three times rocking at room temperature (R@RT; 20°C–22°C).Note: We prefer printing labels, rather than using a marker to write on the tube, as the many wash steps over the 4-week course tends to remove any markings, increasing risk for sample misidentification.
-
b.For 1 h R@RT, serially dehydrate brains by placing in increasing concentrations of methanol (20, 40, 60, 80, 100, 100% methanol).
 Pause point: Samples can be stored long term in 100% methanol at 4°C (wrapped in foil, without rocking). Timing works the best to start hydrogen peroxide permeabilization (step 13c) on a Tuesday evening.
Pause point: Samples can be stored long term in 100% methanol at 4°C (wrapped in foil, without rocking). Timing works the best to start hydrogen peroxide permeabilization (step 13c) on a Tuesday evening. -
c.Permeabilize brains in 5% H2O2/methanol overnight R@RT.
-
a.
-
14.Rehydrate brains.
-
a.For 1 h R@RT, serially rehydrate brains by placing in decreasing concentrations of methanol (100%, 80%, 60%, 40%, 20% methanol).
-
b.Wash in 1× PBS for 1 h R@RT.
-
c.Wash in 0.2% Triton X-100/PBS for 1 h R@RT, two times.
-
d.Wash in 20% DMSO/0.3 M glycine/0.2% Triton X-100/PBS at rocking at 37°C for 2 days.
-
a.
-
15.Block brains to reduce non-specific antibody binding.
-
a.Remove samples from the 37°C incubator and allow samples to reach room temperature (20°C–22°C; approximately 1.5 h). Problem 3 depicts consequences of rapid temperature changes during clearing.Note: We have found rapid changes in temperature of tissue leads to tissue tears. An alternative approach to these steps is to store the 10%DMSO/6% donkey serum/0.2% Triton X-100/PBS in a 37°C incubator. Then quickly change solutions into this pre-warmed solution, placing them back into the incubator at 37°C.
-
b.Block in 10%DMSO/6% donkey serum/0.2% Triton X-100/PBS for 2–3 days rocking at 37°C for 2 days.
-
a.
-
16.Primary immunostaining.
-
a.Remove samples from the 37°C incubator and allow samples to reach room temperature (20°C–22°C; approximately 1.5 h).
-
b.While samples are adjusting to room temperature (20°C–22°C), mix primary antibody solution:
-
i.Spin down antibody at 4°C for 10 min at 17–18 g (∼14,000 rpm). We find this helps remove antibody aggregates that can lead to false positive signal in superficial brain tissue (problem 6).
-
ii.Add antibodies in appropriate concentration (See Table S3 in Pisano et al., 2021) to 5% DMSO/3% donkey serum/PTwH.
-
i.
-
c.Wash in PBS + 0.2% Tween-20 + 10 μg/mL heparin (PTwH) R@RT for 1 h, two times.
-
d.Immunostain in primary antibody solution rocking at 37°C for 7 days.
-
a.
Note: Ideal primary and secondary antibody concentrations depend on experimental parameters including the properties of the virus and primary and secondary antibodies, injection location and size, and hours post-injection. Too much antibody can lead to nonspecific staining, and too little antibody will produce insufficient signal to detect neurons using convolutional neural networks. Optimize antibody concentrations for the specific experimental paradigm before running a full experimental cohort.
-
17.Primary wash day.
-
a.Remove samples from the 37°C incubator and allow samples to reach room temperature (20°C–22°C; approximately 1.5 h).
-
b.Wash in PTwH R@RT for 10 min, then 15 min, then 30 min, then 1 h, then 2 h. Then wash once more in PTwH and leave until the next day.
-
a.
Note: These are the minimum number of washes. Adding several extra washes can help decrease false positive signals.
-
18.Secondary immunostaining.
-
a.Mix secondary antibody solution:
-
i.Spin down antibodies at 4°C for 10 min at 17–18 g (∼14K RPM) before using.
-
ii.Add secondary antibody to 3% donkey serum/PTwH.
-
i.
-
b.Immunostain samples in secondary antibody solution rocking at 37°C for 7 days.
-
a.
Note: Secondary antibody is light-sensitive and should be covered in foil or light-protected to prevent degradation of signal.
-
19.Secondary wash day.
-
a.Remove samples from the 37°C incubator and allow samples to reach room temperature (20°C–22°C; approximately 1.5 h).
-
b.Wash in PTwH R@RT for 10 min, then 15 min, then 30 min, then 1 h, then 2 h. Then wash once more in PTwH and leave until the next day.
-
a.
Note: This is the minimum number of washes. Adding several extra washes can help decrease false positive signal.
-
20.Dehydration and clearing.
-
a.Serially dehydrate brains by placing in increasing concentrations of methanol (20%, 40%, 60%, 80%, 100%, 100% methanol). Each dehydration step should be at least one hour and samples should R@RT.
-
b.Place tissue in 67% dichloromethane (DCM)/33% methanol R@RT.Note: when working with DCM it is important to wear two layers of nitrile gloves and work under a fume hood or snorkel.
-
c.Place into 100% DCM R@RT for 15 min, two times.
-
d.Place into 100% dibenzyl ether (DBE) for 1 h, without rocking, but initially invert tube several times.
-
e.Place into the final solution of 100% DBE and be sure to protect brains from light.Note: DBE can be cleaned off the outside of the tube with 70% ethanol.Note: Both primary and secondary antibodies must be compatible with iDISCO+ (they must not be sensitive to methanol fixation). We suggest using the validation method described here for new untested antibodies: https://idisco.info/idisco-protocol/. Figure 4 shows an example of methanol testing of anti-HSV-1 antibodies (see problem 4).CRITICAL: Appropriate safety precautions are important as many clearing regents are caustic to skin/eyes. Please work in fume hood when appropriate.CRITICAL: Dichloromethane (DCM) is toxic and challenging to work with. It must be used in a fume hood or area with appropriate ventilation. For this reason, we suggest preparing the day of use. Certain plastics are not resistant to DCM, thus alway test different plastic containers before their use.
-
a.
Figure 4.

Methanol resistance of the Dako anti-HSV-1 antibody
(A) Red-green overlays of tissue stained using different anti-HSV-1 antibodies. Red: anti-HSV-1 immunofluorescence. Green: H129-driven green fluorescent protein (GFP) expression. Dako antiHSV-1 antibody worked well with methanol-fixed tissues, while V2 antibody was less reactive and V1 was almost completely inactive.
(B) Antibody detection of viral proteins greatly increased the identification of virus-infected neurons. Scale bars: 250 μm (A) and 20 μm (B).
Figure 5.

Clearning a mouse brain using iDISCO+
(A) Fixed mouse brain before clearing.
(B) Mouse brain after clearing but without refractive-matching dibenzyl ether.
(C) Mouse brain after clearing and in dibenzyl ether.
(D) Grayscale image of cleared mouse brain in dibenzyl ether on top of a Ramón y Cajal drawing to illustrate transparency. The red arrows indicate brain edges. Scale bars 0.5 cm.
Light-sheet microscopy
Light-sheet microscopy takes advantage of refractive index matching of immersion media with cleared tissue to decrease light scattering. iDISCO+ utilizes dibenzyl ether (DBE) for imaging (Figures 5B and 5C contrasts brain with and without index matching). DBE can dissolve certain plastics. It is important that all microscope hardware that is exposed to solvent be DBE-compatible. Appropriate safety precautions are again important. For this project we used the LaVision Ultramicroscope II in conjunction with custom printed holders that enable gluing of the ventral brainstem for sample mounting. Regardless of light-sheet microscope used, it is essential to ensure the microscope is calibrated and that all light paths/sheets are appropriately aligned per microscope manufacturer recommendations.
Typically, light-sheet microscopy is limited by the working distance of the objective. Because of this, it should be placed along the shortest axis of the brain (dorsal-ventral) to define the detection axis (Figures 6A and 6B). Optimal illumination by the light sheet is achieved when the light-sheet plane enters from the medial-lateral direction, the next smallest dimension. For imaging at cellular resolution, we have had success with a 4× objective providing 1.63 μm/pixel resolution (1× magnification, 4× objective, 0.28 NA, 5.6–6.0 mm working distance, 3.5 mm × 4.1 mm field of view, LVMI-FLuor 4×, LaVision BioTec; Figure 7). With this objective, we find that 3 × 3 tiling (with typically 10% overlap) per horizontal plane (see problem 7). Separate left- and right-sided illumination images were taken every 7.5 micrometers step size using an excitation-sheet with a numerical aperture of 0.008. Light diffracts as the light-sheet passes through tissue causing tissue further away (in X) from the light-sheet source to be less in-focus. Thus, ipsilateral regions will be better illuminated by an ipsilateral light-sheet than a contralateral light-sheet. Towards this goal, BrainPipe automatically utilizes a computational stitching approach (Bria and Iannello, 2012) to separately stitch together the same z-plane acquired during left-sided illumination (Figure 7A) and during right sided-illumination. To utilize the fact that ipsilateral areas will be more in-focus from the ipsilateral sheet, the two stitched images are blended together, using midline sigmoidal-blending of the two planes (Figure 7D). This approach helps to reduce light-diffraction imaging artifacts.
-
21.Mount the sample (Figures 1G and 1H).
-
a.Clean a 3D-printed mounting puck and clean with ethanol to remove dust and/or residual glue from any prior imaging. Make sure that the mounting puck is dry.
-
b.Remove a cleared brain from DBE vial and place onto paper towel ventral-side up.
-
c.Use a Kimwipe to gently dry the ventral surface of the brain.Optional: Occasionally remaining pieces of dura can be seen on the brain surface. Gently sweep a Kimwipe across these areas to remove unwanted dura mater. Alternatively, use tweezers to gently pull off any remaining dura.
-
d.Put a small drop of glue on the mounting puck where the dorsal brainstem will be placed.Note: We find this orientation and gluing scheme is optional for whole-brain imaging excluding the most caudal brainstem. Depending on project goals orientation and gluing location might change. Gluing can cause light scattering, so use the smallest amount possible that prevents brain movement.
-
e.Using either DBE-compatible tweezers or gloved fingers, gently place the brain into location on the mounting puck. Wait at least 90 s for glue to dry.Note: We find less glue is typically better and occasionally smearing the glue before placing the brain hastens the drying process and prevents excess glue from attaching to brain decreasing imaging artifacts.Note: Keep in mind there is a balance between allowing the glue to dry for sufficient timing and increased risk of generating bubbles in ventricles (Figure 8A and 8B).
-
f.Gently turn the brain upside down to ensure glue is dry. Place it into imaging cradle.
-
g.Place the imaging cradle with brain into the microscope imaging chamber.
-
a.
-
22.Turn on microscope, initialize software and begin acquisition.
-
a.Turn on applicable lasers, camera, stage controller and then initialize software.
- b.
-
a.
Note: it is important to ensure that when setting the inferior (ventral) limit of the scan that the objective does not contact the superior (dorsal) brain surface.
Note: Imaging time can be minimized by imaging one set of tiles at a specific x-y location at all levels of z and again in the second channel, then moving to a different x-y location for the next set of tiles (Figure 7A).
Note: While initializing scans, inspect ventricles for air bubbles (Figures 8A and 8B). If air bubbles are present, we recommend placing the brain back into DBE vial, inverting several times and attempting to reimage it several days later, after the air bubble has had a chance to escape the ventricle.
-
23.Removing brain from mounting puck.
-
a.Using DBE-compatible tweezers or spatula, apply downward pressure to the puck and gently scrape towards the brainstem to separate the brain from the puck.Note: We typically find only a small amount of residual glue remains on the brainstem that can either be removed by gently passing Kimwipes across the brainstem surface or using DBE-compatible tweezers to lightly pull off glue.
-
b.Place the brain back into DBE and protect the brain from light.
-
c.We have had success reimaging brains several times over the course of several years.
-
a.
Figure 7.

Light-sheet imaging parameters for cellular imaging across the entire mouse brain
(A) Scan configuration for the fastest acquisition. Typically, a 3 × 3 tile scan, using 10% overlap, covers a large enough field of view to encompass the mouse brain. Empirically the left/right illumination, z, channel, x, y acquisition order was found to be the fastest.
(B) The tradeoff between focus depth and waist curvature of the light-sheet. LaVision’s approach to adjusting the sheet width is by effectively adjusting the excitation numerical aperture (NA). A higher NA gives a thinner horizontal focus but with steeper curvature; this gives better center focus, but focus decays rapidly with increasing distance from the center, increasing risk for photobleaching of tissue.
(C) Computational stitching between tiles reduces seam artifacts. The red arrows point to seams in images without (top) and with (bottom) computational stitching.
(D) Sigmoidal blending between stitched left- and right- sided illumination images to produce the final image. Scale bars 250 μm.
Figure 8.

Light-sheet images of mouse brains with and without imaging artifacts
(A and B) Two examples of autofluorescent images where air bubbles in ventricles caused imaging artifacts. Air bubble formation has been found to be related to the amount of time exposed to air while mounting the brain. Red arrows point to air bubbles. Images taken using a 1.3× objective with 5 μm/pixel resolution. (A) Brain with a small air bubble, causing minimal imaging artifacts. (B) Brain with large ventricular air bubbles causing significant shadow artifacts between two lateral ventricles as well centrally in the third ventricle.
(C and D) Example corresponding pair of autofluorescent (C) and H129 signal (D) Images after the complete stitching process. Images acquired using 4× objective with 1.63 μm/pixel resolution. Full-resolution images are 6050 × 7160 pixels (XY). 1 mm scale bars.
Analysis: registration and cell-counting
1–3 weeks to generate training annotations and train CNN.
BrainPipe is an automated transsynaptic tracing and labeling analysis pipeline (Figure 9). Generally BrainPipe workflow consists of three steps: registration to a common atlas, cell detection and injection site recovery. Registration requires three things: 1) atlas volume, 2) corresponding annotation volume and file, and 3) elastix registration parameter files. BrainPipe includes elastix registration parameter files that work well with the Princeton Mouse Atlas (Pisano et al., 2021). BrainPipe is compatible with other atlases, only possibly requiring adjustment of some of the elastix parameter files (see problem 8).
Figure 9.

Methodological comparison of traditional anatomical assignment with automated anatomical assignment
(A and B) Traditional manual-based tissue-section registration and subregional anatomical assignment example. Modified from Pomeranz et al. (2017). (A) Gross cytoarchitecture and tissue-section shape is used for traditional visual registration. Human-user visually defined coronal section atlas correspondence of fluorescent tissue after PRV injection. (B) Cytoarchitecture can be used for traditional anatomical assignment. After section registration, subregional anatomical assignment is hand-drawn.
(C and D) Automated registration and subregional assignment. The blue star illustrates how a point (cell center) can be transformed into different space for eventual anatomical assignment. (C) Volumetric registration transforms light-sheet volumes into atlas space. Automated mapping between the pre-transformed volume and the atlas is established. Deformation fields illustrate two-dimensional plane deformation. (D) Anatomical assignment is automated after volumetric registration to atlas space. The pixel value in annotation space of voxel-coordinates is an identity lookup table and can be used to determine region identity. Scale bars are 500 μm (A), 250 μm (B), 1 mm (C and D). Abbreviations: DRD, dorsomedial raphe nucleus; DRL, dorsolateral raphe nucleus, DRV, ventromedial raphe nucleus.
To automate cell detection, BrainPipe utilizes a three-dimensional convolutional neural network (CNN). We found that CNNs for our task required about 3,600 human-annotated cell centers (see Figure 1E and Table 2 of Pisano et al. [2021]). Cell centers from the reconstructed volumes were obtained by thresholding that was established during training evaluation (see Figure 1F of Pisano et al. [2021] and Methods, Automated detection of virally labeled cells, evaluation of CNN). For maximum detection accuracy, cell detection is performed on unregistered full-sized volumes. Then detected cells are transformed to atlas space. The final output consisted of a comma-separated-value file that included the xyz coordinates as well as measures of perimeter, sphericity, and number of voxels in the z-dimension for each detected cell in the brain volume.
Note: As a starting point we pre-trained CNNs for both H129 and PRV detection are available at https://lightsheetatlas.pni.princeton.edu/public/brainpipe_demo_datasets/CNN_pretrained_models.tar.gz. Although we found high accuracy both in registration and cell-detection, BrainPipe’s performance is highly dependent on the input data (see problems 8 and 9; problem 9 includes a discussion on CNN’s use in cell detection). Before starting CNN-training, we strongly suggest ensuring previous steps in this protocol (viral injection, antibody clearing and light-sheet imaging) optimization be performed. Pre-trained networks are often a good point to start for troubleshooting or transfer learning but better likely performance can be obtained by training CNNs to each experimenters’ datasets.
Note: Before analysis, it is important to manually curate the dataset. For our study, we screened each brain (1) clearing quality (ensuring each brain is sufficiently transparent; see problem 2), (2) significant tissue deformation from extraction process (Figures 10C and 10D, see problem 8), (3) viral spread from injection site (see problem 1), (4) antibody penetration (see problems 4 and 5), (5) blending artifacts related to microscope misalignment, (6) injection site within target location, (7) successful registration (see problem 8), and (8) convolutional neural network (CNN) overlay of detected cells with brain volume in signal channel (see problem 9).
-
24.Download and install BrainPipe: github.com/PrincetonUniversity/BrainPipe.
-
a.Instructions for installation can be found under README.md.
-
a.
-
25.
To work through basic use cases of BrainPipe, we recommend going through the EXAMPLES.md file.
-
26.
Demonstration datasets to ensure proper usage of BrainPipe can be found at: https://lightsheetatlas.pni.princeton.edu/public/brainpipe_demo_datasets/.
-
27.
For a deeper dive into the BrainPipe pipeline, please see IMPORTANT_FILES.md for details.
-
28.
When starting to use your own data, BrainPipe expects certain formatting of images. Please see FILE-FORMATTING.md for details.
-
29.
Finally the folder tutorials has several Jupyter notebooks to analyze injection sites, generating a CNN (3D U-Net) training set, scaling the CNN, and mapping regions of interest (ROIs) to an atlas.
-
30.We recommend starting with the BrainPipe information and tutorials listed on Github; however, please see below for some further details to Train and Evaluate a CNN (see problem 9).
-
a.Generate annotated training volumes by randomly selecting volumes of at least 200 × 200 × 50 pixels (XYZ) from full-sized cell channel volumes (BrainPipe.tools.conv_net.utils.training).Note: To ensure training data is representative of the animal variability across the whole-brain, training volumes should be selective from different anatomical regions in different brains with various amounts of labeling.Note: We recommend having several users annotate similar training volumes - this allows for a human-human F1 score which can be used to compare with a CNN-human F1 score. If these two scores are approximately the same, then it indicates that the CNN approaches human-human performance.
-
b.Annotations were recorded by marking cell centers using ImageJ and imported into Python (BrainPipe.tools.conv_net.utils.training.make_training_inputs).
-
c.Generate labeled volumes using Otsu’s thresholding method (skimage.filters.threshold_otsu, Scikit-Image; 30 × 30 × 8 voxels, XYZ) around each center to label somas.
-
d.Using annotated volumes, trained CNN (BrainPipe.tools.conv_net.pytorchutils.run_exp). Select the CNN window, stride, and training-validation-testing split in the run_exp.py module based on project needs and computational power available.Params["in_spec"] = dict(input=(1,20,192,192))Params["output_spec"] =collections.OrderedDict(cleft=(1,20,192,192))Params["width"] = [32, 40, 80]params["patchsz"] = (20,192,192)Note: We used 192 × 192 × 20 CNN window size with 0.75 strides. Our training dataset was split into a 70% training, 20% validation, and 10% testing subset.Note: We suggest stopping training when the loss value, a measure of CNN learning, stabilizes (in our case around 295,000 training iterations). This helps prevent overfitting, i.e., the possibility that the neural network learns particular training examples rather than learning the category.
-
e.To determine CNN performance on H129 data, calculate an F1 score to compare CNN output with ground truth annotations by quantifying true positives (TP), false negatives (FN), and false positives (FP) (BrainPipe.tools.conv_net.training.sweep).
-
i.Connected-component analysis first (scipy.ndimage.label) groups islands of nonzero voxels to identify each island as a putative cell.
-
ii.Next calculate pairwise Euclidean distances (scipy.spatial.distance.euclidean) between CNN-predicted cell centers and human-annotated ground truth centers.
-
iii.Using bipartite matching, serially pair closest predicted and ground truth centers and remove each from unpaired pools. Unmatched predicted or ground truth centers should be considered as FPs or FNs, respectively. In our case prediction-ground truth center pairs with a Euclidean distance greater than 30 voxels (∼49 μm) were likely inaccurate and not paired.Note: Training the CNN is an interactive process. It is important to not just look at learning statistics but to visualize what the CNN has learned, as well as false negatives and false positives. This way the training dataset can be enriched to facilitate generalized learning applicable to project needs.
-
i.
-
a.
Figure 10.

Examples of poorly cleared and torn tissue
(A) A well-cleared brain in dibenzyl ether (DBE).
(B) A poorly cleared tissue in DBE. Note the darker amber color.
(C and D) Two horizontal z-planes showing examples of torn tissue resulting from rapid temperature shifts during the clearing process (red arrows) and tears that occur during brain-from-skull extraction process (yellow arrows). (D) There are also two large tears in tissue, one in the cerebellum and the on the right side of the brain. This often occurs when the brain gets damaged during the tissue extraction process. Scale bars: 2 mm (A and B), 1 mm (C and D).
Expected outcomes
Viral injections
In our experience delivery of 104 PFUs of HSV-H129 should provide consistently successful infections leading to transsynaptic spread of virus from the injection site. Because iDISCO+ clearing can be time-consuming, a way to confirm successful delivery of a virus is to traditionally section brain tissue using a vibratome. Then tissue can be immunostained over several days and quickly imaged on epifluorescent or confocal microscope (Figure 4 shows epifluorescent-imaged tissue). Also, we typically find that around 60 h post-injection, mice start to show visible signs of viral infection, a signal of successful delivery of sufficient viral quantities.
Clearing and light-sheet imaging
There are typically two features of successful iDISCO+ based clearing. The first is successful tissue clearing, which can only be observed during the final steps, when tissue is transitioned from DCM to DBE. This clearing quality will also be noticeable during light-sheet imaging by seeing a low background level of autofluorescence (particularly in the far-red channels). The second feature of successful iDISCO+ clearing is level of antibody staining (and specificity) which can only be identified using microscopy.
Typically, we observed at 24 h post injection (hpi) one synapse spread from the injection site. Around 50 hpi two synapses, and around 80 hpi three synapses. As mentioned previously, determination of viral spread should be done empirically in the particular neural circuit being studied. HSV-H129 using anti-HSV immunostaining typically causes somatic labeling with very proximal processes also being labeled. PRV-152 using anti-GFP immunostaining tends to highlight proximal processes more so than H129 labeling (Figures 11 and 12). Inspecting cell morphology using either method of labeling should reveal specificity of labeling (non-specific labeling: smaller punctuated labeling particularly at brain edges, suggests too high primary and/or secondary antibody concentrations).
Figure 11.

Whole brain horizontal sections of PRV and HSV stained tissue
(A–C) Posterior-vermal output paths reach the frontal cortex and striatum. Example viral labeling after a vermal injection. (A) Autofluorescent horizontal plane for anatomical reference. (B) Deep frontal neocortical layers receive input from the cerebellum. (C) The striatum receives cerebellar input in dense clusters. Projections are 300 μm across the dorsal-ventral axis. Scale bars: 1 mm (A), 250 μm (B and C). (D–I) Input and output of the lateral cerebellar cortex is bilateral at the neocortical level.
(D) Horizontal maximum projection of 188 μm from a mouse brain 78 h post PRV injection.
(E and F) The left (E) and right (F) neocortex sends input to the right cerebellar hemisphere.
(G) Horizontal maximum projection of 375 μm from a mouse brain 83 h post H129 injection.
(H and I) The left (H) and right (I) neocortex receive input from the right cerebellar hemisphere. Red arrows highlight injection sites. Scale bars: 1 mm (D and G), 150 μm (E, F, H, and I).
Figure 12.

Cellular resolution light-sheet microscopy of HSV-H129
(A–C) Cerebellar labeling just peripheral to HSV-H129 injection sites with clearly visible Purkinje cells.
(D–F) Neocortical labeling after cerebellar injection of HSV-H129 showing clusters of cortical neurons that receive trisynaptic Purkinje cell input. Pyramidal neurons with apical dendrites are visible. Maximum intensity projection images of 100 μm (A–D) and 300 μm (E and F). Images were acquired with a SmartSPIM microscope with 0.20 sheet NA, 1.87 μm/pixel resolution and a step size of 2 μm. Scale bars: 500 μm (A and B), 250 μm (C and D), 100 μm (E and F).
BrainPipe
With successful brain registration, the BrainPipe-generated elastix subfolder should have post-registered volumes. These volumes can be easily overlaid with the atlas using a program such as ImageJ. From there it is possible to manually inspect the registered volume to determine if registration is sufficient.
With successful CNN-based cell detection, an output will consist of a csv file of post-transformed xyz coordinates of detected cells in the brain volume. While initially optimizing the CNN, we recommend overlaying these cell detected centers onto post-registered cell-channel volume. This ensures that both the CNN is accurately identifying cell centers as well as appropriate transformation of coordinates from signal to atlas space. Alternatively, pre-transformed cell centers can be overlaid onto the pretranformed brain volumes.
Limitations
Tissue clearing is an efficient way to immunostain large chunks of tissue without the need to section. This preserves tissue, allowing for volumetric imaging of cleared chunks of tissue. Limitations of clearing are primarily related to three things: the size of the tissue to be cleared, size capacity of the lightsheet microscope (Figures 6A and 6B) and antibody compatibility. We have found that mouse brains can be cleared and successfully imaged as a single unit. However, for larger chunks of tissue such as non-human primate tissue, the tissue must be cut into several smaller units. Diffusion of antibodies and removal of lipids is diffusion limited. Thus, with larger tissue the ability to stain and clear deeper portions decreases. Also with larger tissue, the light-sheet microscope must be able to accommodate the larger chunks of tissue. This is the case both for XY, via the stage motors’ translation distance as well as the working distance of the objective. Regarding antibody compatibility, methanol testing must be done to find antibodies that are resistant to methanol (Figure 4).
Drawbacks of light-sheet microscopy. Whole-brain anatomical tracing and tissue clearing with light-sheet microscopy can be initially challenging to deploy within a lab for primarily two reasons. First access to a compatible light-sheet microscope is needed. Although not orders of magnitude more expensive than confocal microscopes, light-sheet microscopes can be more expensive. The light-sheet microscope must also be capable of imaging the brain or tissue being studied. Some commercial turn-key microscopes are already capable of imaging most/all of a mouse brain; however, with large species such as the rat, this might require a custom microscope or at the very least custom configurations to the imaging chamber, microscope objective, and x, y and z motors to accommodate the larger tissue. Second, for laboratories not already experienced with light-sheet microscopy, it requires dedicated person-hours to learn the microscopy technique and to learn to handle the data that is produced. Epifluorescent and confocal microscope data analysis typically are done using visual-guided analyses (i.e., human labeling/quantification) while light-sheet microscopy data, being significantly larger in size, realistically requires analysis through programming techniques (i.e., automated anatomical registration and cell quantification discussed here). However, once access to a compatible microscope is achieved and experience gained on data handling, the data quality and richness is far superior to that of classical section-based microscopy.
Considerations for when to use light-sheet microscopy. For a project that is focused on a specific, single circuit of interest and where microscopy is only needed to verify appropriate expression or recording location, then perhaps the initial undertaking required for tissue clearing and analysis of light-sheet data is too great for the gains it provides. However, if a lab already has familiarity, then even a project without anatomical focus greatly benefits from this process as anatomical volumetric reconstruction and registration allows for better inspection of the data. It is hard to understate the benefit that is gained from being able to serially step through serial images of an entire cleared brain. Light-sheet microscopy provides quantification across the entire brain. This allows for unbiased analysis as opposed to previous classical studies. These classical studies, because they relied on manual analysis, could not realistically quantify every brain region. These studies likely missed meaningful relationships as a result. Given automated analysis and the high-throughput nature of our pipeline, another major advantage is the ability to have larger sample sizes for experimental cohorts which enables studies to have enough power significance detection.
Troubleshooting
Problem 1
Poor viral spread from injection site.
When looking at imaging results from either a cleared brain (step 22) or traditionally sectioned and epifluorescent/confocal microscope imaged brain, there is little viral labeling outside of the injection site.
Potential solution
Co-injection with CTB, not only is useful for marking the precise injection site, but it also serves to ensure that viral injection solution was delivered into the brain.
If CTB labeling is not present: If there is no CTB labeling at site of injection, then no viral injection solution was delivered. This means the pipette tip was likely clogged (step 6f). Next surgery, ensure that the tip is not clogged right before piercing the brain. If this problem occurs often, then we recommend beveling the glass pipette tips to have a large pore to decrease likelihood of clogging.
If CTB labeling is present: this signals that viral injection solution was successfully delivered. This suggests that the viral tracer did not infect successfully. Ensure viral aliquots have not gone through multiple freeze thaw cycles (we suggest making single-day use 100 microliter aliquots). Ensure a sufficiently large injection (step 2a). We find that at least 104 PFUs are needed for reproducible infection for HSV-H129 (this will vary based on virus, mouse strain and injection location). Try increasing the volume of delivery to ensure a greater number of PFUs is delivered. If there is still no success with significant increase in PFUs delivered, a bad batch of viral stock should be considered. Try a new stock.
Problem 2
Poor clearing of tissue.
When looking at tissue in index matching media like DBE (step 20e), the tissue appears too dark amber in color and cloudy (Figures 10A and 10B).
Potential solution
Because tissue clearing is a long process with many steps and reagents, tissue clearing can fail for many reasons and can be a difficult problem to troubleshoot. We recommend taking an already fixed brain and performing the last clearing protocol day (step 20) on that brain. Although this brain will not be immunostained, within one day a test of clearing can be performed. This will ensure that reagents and the most important clearing steps (final alcohol dehydration as well as DCM and DBE washes) are working.
We have noticed that brains from older animals (especially mice around 1 year or older) have poorer clearing (they will be a darker amber color, Figures 10A and 10B).
If clearing is successful but suboptimal, wait extra days up to a week before imaging the brain, as tissue continues to clear over time (with minimal loss of antibody signal). If there is an inhomogeneity of clearing with outer, more superficial, tissue being “clearer” than deeper tissue this means that incubation times for wash steps were cut too short. We suggest then adding 15–30 min to each wash step, particularly on the final clearing day (step 20).
Note: Although our protocol was optimized for mouse brains, we have successfully cleared larger chunks of neural tissue (rat, nonhuman primate). Diffusion time is proportional to the square of the distance diffused. Thus, in general for large tissue, we suggest substantial increases in the duration of all steps (washes and antibody incubation).
Problem 3
Cleared brains have torn tissue.
When visualizing tissue (step 22), there are tears at the edges or deep within tissue with edges that look like cartoon drawn clouds (Figures 10C and 10D).
Potential solution
We believe that most of these tears come from tissue that has changed temperature too quickly (see subsection IDISCO+: immunofixation and clearing and steps 14d–19). This results in inhomogeneous tissue shrinking/expansion causing tears. It is important to ensure that samples can be allowed to gradually warm/cool, and when new fluid is added to samples, that these fluids be the same temperature as the previous fluid. With each wash step, ensure temperature matching of washes.
In addition if there are tears or divots noted to be at the edge of tissue (yellow arrows, Figures 10C and 10D) this is likely related to the extraction process (step 12a). During the process of removing the skull from the recently perfused and fixed brain, it is very easy to accidentally damage the edge of the brain while using extraction tools. While breaking away the skull, often there can be sharp skull edges that can protrude into the brain further damaging it. It is important that while removing the skull during brain extraction that great care is taken to not damage the tissue, as this can certainly affect the registration and cell detection process.
Problem 4
No labeling, very weak labeling in brain.
When visualizing the brain using light-sheet microscopy (step 22), there is almost no/very little labeling in the brain.
Potential solution
Three common causes when starting clearing are:
-
•
Inappropriate primary-to-secondary antibody compatibility. Secondary antibody chosen does not target the primary antibody species.
-
•
The excitation/emission filter set is not optimized (step 8) for the chosen secondary antibody fluorophores.
-
•
Primary and/or secondary antibodies are not compatible with iDISCO+. If antibodies are sensitive to methanol fixation, binding during the primary and secondary immunostaining will not occur. It is important to iDISCO+ validate any new antibodies. The method is described here for new untested antibodies: https://idisco.info/idisco-protocol/ (Figure 4 depicts an example of methanol testing).
Problem 5
Too little immunostaining within deep structures.
When visualizing the brain on a light-sheet microscope (step 22), there is visible labeling in outer structures such as neocortex, but deeper structures such as thalamus have little or no labeling.
Potential solution
We have identified 3 reasons for this:
-
•
The brain does not actually have labeling in deeper structures. Given injection location and number of synapses the virus was allowed to spread (see Note at end of subsection viral injections), there is actually no connectivity of deeper structures with injected structure. For example, we observed essentially no thalamic labeling in our disynaptic PRV cerebellar cortical injections. This agrees with known anatomy (see Pisano et al. [2021] for discussion) that there are essentially no anterograde disynaptic projections from thalamus to cerebellar cortex. If deep structure labeling is expected, given the experimental injection paradigm, then we suggest serially sectioning a brain, classically immunostaining it and confirming presence of deep structure labeling. If this is the case then it suggests an issue with clearing, see next two points below.
-
•
Incubation time of clearing was too short (see second Note of subsection iDISCO+: immunofixation and clearing of the Step-by-step method details). If deep structure labeling is expected but not observed. It could suggest that the incubation time of primary and secondary antibody solutions was too short. Meaning antibodies through diffusion did not have enough time to reach deep structures. A solution is to increase the primary and secondary antibody times, for example from 7 days to 10 days.
-
•
Similar to the previous point, if deep structure labeling is expected but not observed, it could be related to having insufficient antibody concentration. That is, there is so much antigen of interest in the brain that an inadequate number of antibodies diffuse to deep structures since more superficial structures uptake almost all available antibodies. A solution in this case is increasing the antibody concentration in solution (as opposed to a large solution volume of the same antibody concentration).
To save time and the number of iterations needed for clearing optimization, in the correct situations we suggest following bullet points 2 and the 3 in parallel, that is increasing the incubation time and antibody concentration for the following clearing batch.
Problem 6
Too much staining/non-specific staining especially in superficial structures.
When imaging a brain using light-sheet microscopy (step 22), small nonspecific punctate labeling is observed throughout the brain. The labeling can sometimes appear denser in superficial regions, but generally is homogeneously present throughout much of the brain without anatomical specificity. This nonspecific labeling does not have cellular morphology.
Potential solution
Often we find this nonspecific labeling pattern occurs when the primary and/or secondary antibody solution is too high in concentration (see second Note of subsection iDISCO+: immunofixation and clearing). If this is the case we recommend decreasing antibody concentrations (usually we will decrease both primary and secondary solutions at the same time). It is important to empirically determine appropriate concentrations during initial clearing optimization before clearing batches of brains to be used for analysis.
Other, less often, causes for this nonspecific staining could be inadequate blocking (step 15). Increasing the length of time for this might modestly decrease nonspecific staining. One other cause could be bad antibody stock containing “clumped” antibodies. Spinning down antibodies (step 16bi) before mixing into the primary antibody solution used in clearing helps prevent this issue.
Problem 7
Poor stitching of tiled images.
Brain tissue at edges of XY tiles appears to be misaligned (step 25). Figure 7C shows an example of how poor stitching might look. Figures 8C and 8D shows appropriate stitching in XY.
Potential solution
Reasons include:
-
•
Percent overlap performed using microscope controller software does not match percent overlap input into BrainPipe (Figures 7A, 7C, and 7D). Brainpipe, using terastitcher, starts with an expected calculated overlap inputted by the user. Terasticher will then make smaller adjustments to optimize stitching; however, with enough discrepancy between actual and inputted overlap percentage, terastitcher will fail.
-
•
Light-sheets on a microscope are misaligned (see subsection light-sheet microscopy of Before you begin), not parallel with each other or parallel with the objective focal plane. This leads to mismatch of excitation tissue at edges when stitching left and right edges of adjacent tiles (Figure 7D).
Problem 8
Poor registration.
Post-registered brain volume appears to be misaligned with the atlas (step 25). This is most apparent when overlaying the post-registered brain with the atlas volume. These findings suggest improper registration.
Potential solution
First confirm there is indeed poor registration, by overlaying the post-registered brain volume with the atlas volume. We suggest using a program like ImageJ/FIJI to do a two-color overlay (ImageJ>Colors>Merge Channels…) to do this overlay. If there is poor registration, next ensure that the orientation of the atlas matches that of the volume being registered (registration often fails when attempting to register a horizontally oriented brain to a sagittal atlas). Orientation can be adjusted using BrainPipe using the “finalorientation” input in the run_tracing.py file.
If misorientation issues have been addressed and registration is still poor, then likely the registration parameter files in need to adjusted. These will be called in the “parameterfolder” input in the run_tracing.py file.
We notice that brains with very poor clearing (Troubleshooting problem 2) or with significant damage (step 12), as may occur during the extraction process with indentations or large tissue tears, cannot be overcome using elastix nonlinear deformation. Typically we exclude these brains during our quality control screening process.
Problem 9
Poor cell detection when using CNN.
The CNN does not appear to be accurately detecting objects of interest (step 30). When visualizing CNN output, there appears to be a high number of false positives and or false negative labels.
Potential solution
CNN’s have only more recently been used for cell detection. Prior to this, approaches such as ClearMap, have utilized cell detectors that typically consist of a combination of filters (intensity, morphology, gradients, volumes, sphericity, etc). These “out of the box” cell detectors have in the past been successfully used for objects with high signal-to-noise (SNR) and simple morphologies (e.g., c-Fos staining which has a spherical relatively high SNR labeling). However, their performance tends to be outmatched by CNN, especially when either there is ample training data and/or there is greater variability in object morphology and intensity. We successfully applied CNN’s for virally-labeled neuron detection. Another benefit of CNN’s is that once sufficient labeling for a training dataset is completed, the automated training process is all that is needed to optimize detector performance. This decreases the need for user-based parameter tuning of most other non-CNN-based cell detectors.
Increasing CNN performance requires both increasing the number of training examples in a dataset as well as ensuring that the training dataset includes the variability within and between datasets. Neurons in different brain regions will differ in size, morphology, labeling intensity, labeling location (perhaps more somatic and more axonal/dendritic), and proximity of labeling (clusters of adjacent labeled neurons versus sparse distributed labeled neurons). Also, different areas of the brain will have different baseline levels of background autofluorescence and tissue texture (i.e., the striatum looks very different than the thalamus, even in cleared tissue). Finally, brains from different clearing batches will differ in baseline autofluorescence. The training dataset should include enough examples that cover the differences noted.
It is important to visualize each step (just like tiling and registration), rather than to only rely on performance statistics like accuracy or F1 scores (these are of course important!). By visualizing what the CNN is wrongly marking (false positive) or wrongly missing (false negative), one can specifically enrich their training dataset to address performance deficits identified.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: Samuel S.-H. Wang, (sswang@princeton.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact without restriction. Transsyanptic tracing viruses, including HSV-H129 VC22, are available through the Princeton Neuroscience Institute Viral Core Facility: https://vnl.princeton.edu/.
Acknowledgments
We thank Nicolas Renier for help with tissue-clearing optimization; Stephan Thiberge for microscopy help; Esteban Engel for viral use guidance; Shruthi Deivasigamani, Joseph Gotto, Joyce Lee, Laura Lynch, Caroline Jung, Sanjeev Janarthanan, Dafina Pacuku, Federico Uquillas, and Thaddeus Weigel for technical assistance; and Pavel Osten for project advice. This work was supported by NIH R01 NS045193, R01 MH115750, and U19 NS104648 (S.W.), F31 NS089303 (T.P.), Netherlands Organization for Scientific Research Veni ZonMW, 91618112 (H.-J.B), and Erasmus MC Fellowship 106958 (H.-J.B).
Author contributions
T.P. and S.W. conceived and designed studies. T.P. performed virus injections and prepared tissue. T.P., J.V., S.G., and C.J. did tissue extraction and clearing. T.P. and K.S. optimized tissue clearing. Z.D. and T.P. imaged tissue and ran the computational data analysis pipeline for light-sheet data. T.P. and H.-J.B. performed data analysis and prepared figures. T.P. designed image analysis algorithms. A.H. built visualizations, created tutorials, and manages BrainPipe on Github. T.P. and S.W. wrote the initial draft of the manuscript, which was edited by all authors.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101289.
Contributor Information
Thomas J. Pisano, Email: tpisano@alumni.princeton.edu.
Samuel S.-H. Wang, Email: sswang@princeton.edu.
Supplemental information
Data and code availability
BrainPipe is available at https://github.com/PrincetonUniversity/BrainPipe. The Princeton Mouse Atlas data have been deposited at https://brainmaps.princeton.edu/2020/09/princeton-mouse-brain-atlas-links/ and are publicly available as of the date of publication. Aligned viral tracing injection data have been deposited at https://brainmaps.princeton.edu/2021/05/pisano_viral_tracing_injections/ and are publicly available as of the date of publication. Unprocessed data reported in this paper will be shared by the lead contact upon reasonable request. All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Bria A., Iannello G. TeraStitcher - a tool for fast automatic 3D-stitching of teravoxel-sized microscopy images. BMC Bioinform. 2012;13:316. doi: 10.1186/1471-2105-13-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger C., Nguyen F.N., Deng J., Mandel R.J. Systemic mannitol-induced hyperosmolality amplifies rAAV2-mediated striatal transduction to a greater extent than local co-infusion. Mol. Ther. 2005;11:327–331. doi: 10.1016/j.ymthe.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Conte W.L., Kamishina H., Reep R.L. Multiple neuroanatomical tract-tracing using fluorescent Alexa Fluor conjugates of cholera toxin subunit B in rats. Nat. Protoc. 2009;4:1157–1166. doi: 10.1038/nprot.2009.93. [DOI] [PubMed] [Google Scholar]
- Gage G.J., Kipke D.R., Shain W. Whole animal perfusion fixation for rodents. J. Vis. Exp. 2012;65:3564. doi: 10.3791/3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisken J., Stainier D.Y.R. Selective plane illumination microscopy techniques in developmental biology. Development. 2009;136:1963–1975. doi: 10.1242/dev.022426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter J.D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 2007;9:90–95. [Google Scholar]
- Liu L., Chen S., Li N., Svoboda K. ZappyLab, Inc.; 2020. Virus Injection v1. [Google Scholar]
- Lowekamp B.C., Chen D.T., Ibáñez L., Blezek D. The design of SimpleITK. Front. Neuroinform. 2013;7:45. doi: 10.3389/fninf.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastakov M.Y., Baer K., Xu R., Fitzsimons H., During M.J. Combined injection of rAAV with mannitol enhances gene expression in the rat brain. Mol. Ther. 2001;3:225–232. doi: 10.1006/mthe.2001.0246. [DOI] [PubMed] [Google Scholar]
- McKinney W. Proceedings of the 9th Python in Science Conference. SciPy; 2010. Data structures for statistical computing in python; pp. 51–56. [Google Scholar]
- Oliphant T.E. Continuum Press; 2015. Guide to NumPy. [Google Scholar]
- Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- Pisano T.J., Dhanerawala Z.M., Kislin M., Bakshinskaya D., Engel E.A., Hansen E.J., Hoag A.T., Lee J., de Oude N.L., Venkataraju K.U., et al. Homologous organization of cerebellar pathways to sensory, motor, and associative forebrain. Cell Rep. 2021;36:109721. doi: 10.1016/j.celrep.2021.109721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomeranz L.E., Ekstrand M.I., Latcha K.N., Smith G.A., Enquist L.W., Friedman J.M. Gene expression profiling with cre-conditional pseudorabies virus reveals a subset of midbrain neurons that participate in reward circuitry. J. Neurosci. 2017;37:4128–4144. doi: 10.1523/JNEUROSCI.3193-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renier N., Adams E.L., Kirst C., Wu Z., Azevedo R., Kohl J., Autry A.E., Kadiri L., Umadevi Venkataraju K., Zhou Y., et al. Mapping of brain activity by automated volume analysis of immediate early genes. Cell. 2016;165:1789–1802. doi: 10.1016/j.cell.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleeba C., Dempsey B., Le S., Goodchild A., McMullan S. A student’s guide to neural circuit tracing. Front. Neurosci. 2019;13:897. doi: 10.3389/fnins.2019.00897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabold S., Perktold J. Proceedings of the 9th Python in Science Conference. Scipy; 2010. Statsmodels: econometric and statistical modeling with python; p. 61. [Google Scholar]
- Ugolini G. Advances in viral transneuronal tracing. J. Neurosci. Methods. 2010;194:2–20. doi: 10.1016/j.jneumeth.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Van Rossum G., Drake F.L. CreateSpace Independent Publishing Platform; 2009. Python 3 Reference Manual: (Python Documentation Manual Part 2) [Google Scholar]
- Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J., et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 2020;17:261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt S., Schönberger J.L., Nunez-Iglesias J., Boulogne F., Warner J.D., Yager N., Gouillart E., Yu T., scikit-image contributors scikit-image: image processing in Python. PeerJ. 2014;2:e453. doi: 10.7717/peerj.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskom M., Botvinnik O., Hobson P., Cole J.B., Halchenko Y., Hoyer S., Miles A., Augspurger T., Yarkoni T., Megies T., et al. Zenodo; 2014. Seaborn: v0.5.0.https://zenodo.org/record/12710#.YlXMUijMK44 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
BrainPipe is available at https://github.com/PrincetonUniversity/BrainPipe. The Princeton Mouse Atlas data have been deposited at https://brainmaps.princeton.edu/2020/09/princeton-mouse-brain-atlas-links/ and are publicly available as of the date of publication. Aligned viral tracing injection data have been deposited at https://brainmaps.princeton.edu/2021/05/pisano_viral_tracing_injections/ and are publicly available as of the date of publication. Unprocessed data reported in this paper will be shared by the lead contact upon reasonable request. All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.