

Abstract
Organic carbon transfer between surface ocean photosynthetic and heterotrophic microbes is a central but poorly understood process in the global carbon cycle. In a model community in which diatom extracellular release of organic molecules sustained growth of a co-cultured bacterium, we determined quantitative changes in the diatom endometabolome and the bacterial uptake transcriptome over two diel cycles. Of the nuclear magnetic resonance (NMR) peaks in the diatom endometabolites, 38% had diel patterns with noon or mid-afternoon maxima; the remaining either increased (36%) or decreased (26%) through time. Of the genes in the bacterial uptake transcriptome, 94% had a diel pattern with a noon maximum; the remaining decreased over time (6%). Eight diatom endometabolites identified with high confidence were matched to the bacterial genes mediating their utilization. Modeling of these coupled inventories with only diffusion-based phytoplankton extracellular release could not reproduce all the patterns. Addition of active release mechanisms for physiological balance and bacterial recognition significantly improved model performance. Estimates of phytoplankton extracellular release range from only a few percent to nearly half of annual net primary production. Improved understanding of the factors that influence metabolite release and consumption by surface ocean microbes will better constrain this globally significant carbon flux.
Subject terms: Biogeochemistry, Metabolomics
Introduction
The transfer of organic carbon from phytoplankton to bacteria via a pool of labile dissolved compounds is a key process in global carbon cycling, involving up to a third of fixed carbon [1, 2]. One important but poorly quantified mechanism for this transfer is the extracellular release of organic compounds by living phytoplankton. Indeed, estimates of the proportion of net primary production (NPP) released extracellularly range from 4 to 47% [3], and of the proportion of heterotrophic bacterial carbon demand supported by extracellular release from 2 to 50% [1]. In part, this largely unconstrained flux can be traced to the difficulties inherent in characterizing labile organic molecules dissolved in surface seawater, such as short turnover times due to rapid bacterial uptake and low concentrations due to efficient scavenging (nmol L−1 to pmol L−1; refs. [4, 5]). Moreover, organic molecules taken up by bacteria become rapidly untraceable because of transformation and respiration inside bacterial cells [2]. Although the dissolved organic carbon (DOC) link between marine phytoplankton and bacteria has long been of interest [6–8], identifying the specific metabolites responsible and measuring their flux is indeed challenging.
Phytoplankton metabolite synthesis exhibits diel cycles in the surface ocean, coordinated with the availability of light energy [9–11]. Similar diel cycles of activity in heterotrophic bacterial communities have also been observed [12–14], often lagging peak phytoplankton activity by a few hours [14, 15]. Gene expression data are providing detailed views of this diel synchronicity between phytoplankton and bacteria in oligotrophic and coastal marine communities. For example, phytoplankton carbon fixation and photosystem gene expression coordinates with bacterial substrate uptake (amino acid and sugar transporters) and citric acid cycle gene expression [16, 17]. Extracellular release of phytoplankton metabolites is the process most likely to underlie these matched patterns.
Intracellular phytoplankton metabolite pools (endometabolites) are the presumptive substrates for the heterotrophic bacteria supported by extracellular release. Yet how faithfully phytoplankton internal concentrations predict exometabolite availability depends on the mechanism of release [3, 18]. In the simplest mechanism, differences in metabolite concentration between phytoplankton cells and ambient seawater drive diffusion [19] (i.e., passive diffusion mechanism), in which case metabolite release to heterotrophic bacteria is largely controlled by intracellular metabolite concentrations of the phytoplankton. Alternatively, active excretion of metabolites to maintain cellular balance can occur by overflow pathways [20], for example to manage redox state [11] or photorespiration (i.e., physiological balance mechanism). Finally, metabolites may be synthesized and excreted in response to associated microbes, potentially to sustain mutualisms or mount defenses [21–23] (i.e., interaction response mechanism).
Here we determined the correspondence between phytoplankton intracellular pools and heterotrophic bacterial substrate availability by examining diel patterns of endometabolites and transcripts, respectively. A model community was established in which marine diatom Thalassiosira pseudonana CCMP1335 [24] was the only source of substrates to bacterium Ruegeria pomeroyi DSS-3 [25]. As diatoms contribute up to 40% of primary production in the surface ocean [26], and R. pomeroyi belongs to a taxon that often dominates diatom bloom communities [27, 28] and responds sensitively to external resources [29–31], this simple community represents a relevant phytoplankton-bacteria association in the surface ocean. Over two day-night cycles, we contemporaneously assayed phytoplankton endometabolite pools by NMR spectroscopy and bacterial metabolite consumption using transcript proxies, and assessed links between the two. Transcript abundance was quantified as the number of mRNA molecules per bacterial cell, enabled by the use of internal mRNA standards; this approach yielded the number of transcripts harbored by a cell for a given gene, matching absolute quantitation in the metabolite data and eliminating ambiguities inherent in proportional expression data [32–34]. The quantitative chemical-biological analytical framework applied to this model system enabled us to assess proposed mechanisms underlying temporal links between microbial autotrophs and heterotrophs via extracellular release of labile metabolites.
Materials and methods
Diel experiment
An axenic strain of marine diatom Thalassiosira pseudonana CCMP1335 was cultured at 18 oC in three replicate 15-L polycarbonate bottles containing 10 L of L1 medium [35] in which NaH13CO3 (Cambridge Isotope Laboratories, CLM-441) was the source of inorganic carbon (labeling efficiency, 78% of C atoms). The light cycle consisted of 16 h light, during which light intensity varied gradually between 0 and 150 µmol photon m−2 s−1 with a maximum intensity at noon, followed by 8 h of dark. Axenic T. pseudonana cultures were pre-incubated for 6 days to achieve cell numbers required for metabolite analysis. The cultures were inoculated with bacterial strain Ruegeria pomeroyi DSS-3 that was grown overnight at 30oC on ½ YTSS liquid medium and washed three times in L1 medium (final concentration, 106 bacterial cells mL−1). Co-cultures (n = 3) were pre-incubated for two days, after which samples were collected every 6 h over the next 48 h for bacterial mRNA sequencing, phytoplankton and bacterial cell counts, and phytoplankton endometabolome analysis.
Bacterial effect experiment
T. pseudonana CCMP1335 and R. pomeroyi DSS-3 co-cultures were prepared as in the diel experiment except that additional treatment was established in which the diatom remained axenic (n = 3 for each). Diatom cells were collected at the 48 h sample time and metabolite concentrations were compared between treatments. Incubation periods, culture media, sample processing protocols, and metabolite analysis schemes were the same as those used in the main experiment.
Light effects experiment
One-week stationary-phase axenic T. pseudonana CCMP1335 grown under similar conditions were sequentially filtered through GF/F filters (Whatman, Maidstone, UK) and 0.2-µm-pore-size PCTE membrane filters (Poretics, Swedesboro, NJ) to remove cells. R. pomeroyi DSS-3 cells prepared as described above were added to the diatom-free filtrate and incubated under light intensities of 150 (100% treatment), 75 (50% treatment), or 0 µmol photon m−2 s−1 (0% treatment), corresponding to light levels at noon, mid-morning and mid-afternoon, and night in the diel experiment (n = 3 for each). Temperature monitoring indicated only a minor temperature increase of 0.5oC in the 100% treatment relative to 50% and 0% treatments. After 4 h, samples were collected for bacterial RNA analysis and cell counts.
Diatom endometabolome analysis
Diatom cells were collected by filtering 500 mL of culture onto 2.0-µm-pore-size PCTE membrane filters (Isopore; MilliporeSigma, Burlington, MA) using <10 inHg pressure and stored at −80oC until processing. Cells were removed from filters by sonication in ultra-pure water (MilliporeSigma) and concentrated by freeze-drying [36]. Pellets were mixed with 600 µL of sodium phosphate butter (pH 7.4) with an internal standard of 2,2-dimethyl-2-silapentane-5-sulfonate-d6 (DSS) (1 mmol L−1), vortexed for 5 min, and centrifuged at 20,800 rfc for 10 min, after which supernatants were transferred to 5-mm NMR tubes (Bruker, Billerica, MA). Processing was conducted at 4oC. A blank sample was also included for quality control. Metabolites were analyzed by NMR spectroscopy using a Bruker AVANCE III 800 MHz 5 mm TCI cryoprobe, 800 MHz 1.7 mm TCI cryoprobe, and 600 MHz 5 mm TXI probe. Pulse programs of 1H-13C heteronuclear single quantum correlation (HSQC; Bruker program hsqcetgpprsisp2.2), 1H-13C HSQC-total correlation spectroscopy (HSQC-TOCSY; hsqcdietgpsisp.2), and 1H-13C heteronuclear multiple bond correlation (HMBC; hmbcetgpl2nd) were used. Data were deposited to Metabolomics Workbench (Project ID, PR001019). Data processing was carried out with TopSpin 4.0.3 (Bruker), and peak intensity was extracted using rNMR 1.1.9 [37]. Metabolites were annotated based on chemical shift (HSQC) and coupling information (HSQC-TOCSY and HMBC). The Human Metabolome Database (HMDB) [38] and the Biological Magnetic Resonance Bank (BMRB) [39] were used as reference databases, and additionally the Carbohydrate Structure Database (CSDB) [40] for polysaccharides. Three compounds of interest that are not in these databases were annotated either by obtaining original spectra from chemical standards (DHPS and DMSP; ref. [41]) or based on literature values (homarine; ref. [42]). A confidence level of annotation was assigned to each metabolite [43] (Table S1) where 1 = putative compounds with functional group information; 2 = partially matched to HSQC chemical shift information in the databases or literature; 3 = matched to HSQC chemical shift; 4 = matched to HSQC chemical shift and validated by HSQC-TOCSY or HMBC; 5 = validated by original spectra from chemical standards. Detailed parameter settings are presented in Table S2, with additional information in Metabolomics Workbench. Spectra were standardized to DSS, peak intensities were normalized to cell counts, and data were auto-scaled and presented as Z-scores. Since we did not conduct additional spiking experiments to quantify compounds, we only focus on the difference of the values between samples, and not between compounds. Temporal variations in metabolites were analyzed by extracting peaks using variance-sensitive clustering [44]. The optimal cluster number was selected based on minimum centroid distance and the Xie-Beni index values for the dataset [44]. Background signals originating from labware and solvent were also corrected. Background signals originating from bacteria trapped on the 2.0-µm filters were extremely low in the diatom endometabolite fraction (Supplementary Methods). Periodicity of the temporal patterns for compounds was analyzed using a rhythmicity analysis package RAIN (1.18.0; ref. [45]) in R software (version 3.6.1). Heatmaps were created using the CirHeatmap function (version 1.7) in MATLAB (Mathworks, Natick, MA) [46].
mRNA analysis
For the diel experiment, samples were sequentially filtered through 2.0-µm pore-size PCTE filters (Isopore; MilliporeSigma) to remove diatom cells and 0.2-µm pore-size PES filters (Supor; Pall, New York) to retain bacterial cells. RNA was extracted from the bacterial filters with a phenol-chloroform-isoamyl method [47], which has a better RNA recovery for co-culture samples. Two internal mRNA standards (size, 1,000 nt) were added to each sample before extraction for determining their recovery in the sequence data [48]. For the direct light experiment, samples were filtered through the 0.2-µm pore-size filters to retain bacterial cells. RNA was extracted using the RNeasy Mini Kit (QIAGEN, Hilden, Germany) after cutting filters into pieces under sterile conditions and shaking with 0.5 mL of 0.1-mm zirconia/silica beads (BioSpec Products, Bartlesville, OK) in 1 mL of Denaturation/Lysis Solution (Life Technologies, Carlsbad, CA) for 15 min. For both sample types, filtration was completed within 15 min of collection, filters were flash frozen in liquid nitrogen and then stored at −80oC, DNA was removed (Turbo DNA-free Kit, Ambion, Austin, TX), rRNA was depleted (Ribo-Zero rRNA Removal Kit; Illumina, San Diego, CA), and mRNA was purified (RNA Clean & Concentrator-5; Zymo Research, Irvine, CA). The number of bacteria extracted was ~109 cells.
Sequencing using NextSeq 550 (Illumina) SE50 averaged 19.2 x 106 reads per sample (Table S3). rRNA reads were identified by blast+ (NCBI 2.7.1 and 2.8.1 for the direct light experiment and the diel experiment, respectively) against an rRNA sequence database and removed; rRNA contamination averaged 17.5% of reads. Recovery of the two internal standards was highly consistent (Pearson’s r = 0.96; p < 0.001; n = 26), accounting for 2.2% of mRNA reads per library (Table S3). Remaining reads were mapped to the R. pomeroyi genome and quantified using HTSeq [49]. Differentially expressed genes were identified in pairwise comparisons of sampling times (diel experiment) or light levels (direct light experiment) using MATLAB for absolute analysis and DESeq2 [50] for relative analysis. One of the replicate samples from the initial time point of the experiment was lost; otherwise, n = 3 for all analyses. All other statistical analyses were conducted using MATLAB. Fold-change values and temporal pattern categories for all genes are reported in Table S4.
Cell counts
A 0.5 mL aliquot of culture fixed with glutaraldehyde (final concentration, 1%) was kept at −80oC until analysis. Samples were thawed, stained with SYBR Green I (Thermo Fisher Scientific, Waltham, MA; final concentration, 5 x 10−4 dilution of commercial stock), and injected into a CytoFLEX flow cytometer (Beckman Coulter, Brea, CA). For phytoplankton counts, samples were analyzed without staining. Data were analyzed using CytExpert (Beckman Coulter) and cell density was calculated based on a separate run of a known concentration of bead standards (Beckman Coulter).
Model development
A model was written in R version 3.6.1 with three state variables representing the phytoplankton endometabolome (P), the medium exometabolome (E), and the bacterial endometabolome pools (B). The time evolution of these pools was calculated at 0.1 h intervals. The base model was written to represent only diffusive and fundamental physiological processes, which is largely a passive model, given in the following differential equations:
N is the metabolite biosynthesis rate, derived from light intensity expressed as a cosine function. T is rate at which endometabolites are allocated for biomass and energy generation by phytoplankton cells, calculated as a constant fraction of P from the previous interval. R is release rate of endometabolites from the phytoplankton cell by simple diffusion. U represents bacterial uptake rate from the exometabolome following Michaelis-Menten kinetics. C represents catabolism rate of the metabolite within the bacterial endometabolome, with a constant fraction lost each interval. See Supplemental Methods for information on how variables N, R, T, U, and C were derived.
In addition to the base model, three active terms could be added to the model to represent processes beyond those of simple diffusion and fundamental physiology. The first active term was fixation-irradiance oscillation, o, which represents the process of asymmetric carbon fixation in response to increasing versus decreasing light intensity and impacts the calculation of metabolite production, N. During periods of increasing irradiance (pre-noon), carbon fixation increases proportionally to irradiance, but during periods of decreasing irradiance (post noon), carbon fixation declines more rapidly than irradiance (see Supplemental Methods for details). The second active term was homeostasis, h, representing the increased phytoplankton excretion of metabolite, R, to maintain physiological homeostasis during periods of high light intensity. When h is incorporated into the model, R becomes a function of the rate of metabolite production, N, following the irradiance curve, such that R increases as N increases. The final active term was bacterial response, b, representing a recognition response in the phytoplankton to the presence of bacteria. The b term is multiplicative factor affecting the value of N and representing a change in metabolite production in response to the presence of a co-cultured bacterium.
The model was run for a simulated length of 10 d to replicate experimental conditions. During the first 6 d of ‘axenic growth’ the values B and U were set to zero, followed by ‘inoculation’ with addition of B and U functions for the final 4 d of the modeled experiment. Values for P and U from the final 2 d of model output were used to compare to experimentally measured endometabolome and transcriptome data, respectively.
The model was fit to each matched metabolite/gene pair using an iterative method. A sequence of values was constructed for each of the 6 variables of the base model and 3 active terms (see Supplemental Methods for details), and the model was run for all combinations of each variable and value. For each metabolite/gene pair, two linear correlations (Pearson’s r) were calculated between modeled (P) and experimental diatom endometabolite data, as well as between modeled bacterial metabolite uptake (U) and bacterial transcript expression. Parameter sets that produced significant correlations (p < 0.05; n = 9) between modeled and experimental data for both endometabolite and gene were saved. The r values for each endometabolite/gene pair were averaged and used to compare the overall goodness of fit for each set of model parameters (base, base plus optional active mechanisms in all possible combinations). These r values were adjusted to account for the number of terms beyond the base model according to the formula:
where n = number of samples and k = number of terms. For models with few significant solutions, additional parameter space was explored but did not substantially increase fit.
Results and discussion
R. pomeroyi was inoculated into axenic T. pseudonana cultures growing under a naturally oscillating light:dark cycle. After a 2-d pre-incubation to allow the bacteria to assimilate labile metabolites that accumulated during the axenic phase, and thus emphasize synchronized production and consumption dynamics during diel cycles, samples were collected every 6 h for the next 48 h at timepoints corresponding to midnight, mid-morning, noon, and mid-afternoon.
Diatom metabolome composition
1H-13C two-dimensional NMR characterization of the diatom endometabolome during the 48 h sampling window revealed 281 major peaks. From these, 31 compounds (accounting for 156 peaks) were identified with high confidence (Table 1; see Table S1 and Fig. S1 for detailed annotation and confidence level information). The number of diatom cells increased ~2-fold over the sampling window, from 0.87 to 1.9 × 105 cells mL−1 (Fig. 1a); metabolite data were normalized to cell number at each sample time.
Table 1.
Diatom endometabolites assigned with high confidence in the diel experiment. For detailed information for compound identification and confidence level, see Table S1 and Fig. S1.
| Compound category/Sub-category | Compound | Function | Group, temporal pattern |
|---|---|---|---|
| Amine | Trimethylamine N-oxide (TMAO) | n.a. | |
| Amino acid | Alanine | Amino acid metabolism | n.a. |
| Arginine | Amino acid metabolism | n.a. | |
| Asparagine | Amino acid metabolism | M-1 (p < 0.001) | |
| Aspartate | Amino acid metabolism | M-4 (p < 0.01) | |
| Glutamine | Amino acid metabolism | M-4 (p < 0.001) | |
| Glutamate | Amino acid metabolism | n.a. | |
| Glycine | Amino acid metabolism | M-1 (p < 0.01) | |
| Lysine | Amino acid metabolism | M-1 (p < 0.001) | |
| Proline | Amino acid metabolism/osmoregulation | M-4 (p < 0.001) | |
| Amino acid/Branched-chain | Valine | Amino acid metabolism | n.a. |
| Isoleucine | Amino acid metabolism | M-1 (p < 0.001) | |
| Leucine | Amino acid metabolism | M-1 (p < 0.001) | |
| Amino acid derivative | Glycine betaine | Osmoregulation | M-1 (p < 0.05) |
| Dimethylglycine | n.a. | ||
| Homarine | Osmoregulation | M-1 (p < 0.05) | |
| Amino alcohol | Ethanolamine | Lipid metabolism | M-1 (p < 0.01) |
| Choline | Choline | Lipid metabolism | n.a. |
| Choline derivative | Phosphorylcholine | Lipid metabolism | M-1 (p < 0.001) |
| Phosphocholine | Glycerophosphocholine | Lipid metabolism | n.a. |
| Glycerol derivative | Glycerol 3-phosphate | Lipid metabolism | M-1 (p < 0.01) |
| Nucleoside | Adenosine | Nucleic acids/ATP constituent | n.a. |
| Guanosine | Nucleic acids/GTP constituent | M-4 | |
| Uridine | M-3 (p < 0.001) | ||
| Organic acid | 3-Hydroxybutyrate | Carbon metabolism | M-2 (p < 0.01) |
| Acetate | Carbon metabolism | M-2 (p < 0.01) | |
| 4-Hydroxyphenylacetate | n.a. | ||
| Sugar/Monosaccharide | Glucose | Carbon/central energy metabolism | M-3 (p < 0.001) |
| Sugar/Polysaccharide | β(1,3)-glucan | Carbon metabolism/storage | M-3 (p < 0.001) |
| Sulfur compound | Dihydroxypropane-sulfonate (DHPS) | Osmoregulation | M-1 (p < 0.001) |
| Dimethylsulfonio-propionate (DMSP) | Osmoregulation | M-1 (p < 0.001) |
Temporal pattern assignments correspond to those in Fig. 1; M-1 = increase, M-2 = decrease, M-3 = diel with a peak at mid-afternoon, and M-4 = diel with a peak at noon. Statistical significance was determined based on linear regression analysis (M-1 and M-2), and RAIN for diel cycles (M-3 and M-4). n.a., not applicable (membership value of >0.5, see text for the detail).
Fig. 1. Diel patterns in microbial cell numbers, phytoplankton metabolites, and bacterial transcripts.

a Cell numbers of co-cultured diatoms and bacteria. b Temporal variations in metabolite peak intensity per diatom cell (left) and transcripts per bacterial cell for genes differentially expressed between noon and night (>2 fold-change and adjusted-p < 0.05, DESeq2) (right). Values were converted to Z-scores and data from each of the three biological replicates are shown. c Temporal patterns identified for metabolites (M-1 through M-4) and gene transcription (G-1 through G-3). Dotted lines indicate 95% confidence intervals. The number of metabolite peaks or genes in each cluster is given in parentheses. Grey shading in panels (a) and (c) indicates night.
To group metabolite peaks that behaved similarly over the diel cycles, cell-normalized abundance data were clustered by variance-sensitive clustering [44] which identified four patterns (Figs. 1b and 1c; Table 1). Group M-1 consisted of 102 peaks (36%) for which a monotonic increase in intensity dominated the 48 h sampling window (Fig. 1c). The twelve compounds annotated with high confidence from this cluster included amino acids (asparagine, glycine, isoleucine, leucine, and lysine), amino acid derivatives (glycine betaine and homarine), an amino alcohol (ethanolamine), a choline derivative (phosphorylcholine), a glycerol derivative (glycerol-3-phosphate), and the sulfur-containing compounds dihydroxypropanesulfonate (DHPS) and dimethylsulfoniopropionate (DMSP) (Table 1, also see Fig. S2 for individual compounds in each cluster). Metabolite group M-2 was characterized by 73 peaks (26%) for which decrease in intensity over time was the dominant pattern, and included two organic acids (3-hydroxybutyrate and acetate). The two other metabolite clusters exhibited diel concentration patterns that peaked in the light and declined in the dark (Table 1, Fig. S2). Group M-3 contained 59 peaks (21%) that reached their maximum intensities at mid-afternoon (R package RAIN, p < 0.001) and included high-confidence annotations of the nucleoside uridine and the carbohydrates glucose and β-1,3-glucan, the latter a subunit of the diatom polysaccharide chrysolaminarin [51]. Group M-4 contained 47 peaks (17%) that reached their maximum intensities at mid-morning or noon (RAIN, p < 0.01) and included high confidence annotations for the amino acids aspartate, glutamine, proline, and guanosine. Overall, four distinct temporal patterns in abundance were observed among components of the T. pseudonana endometabolome during co-growth with a heterotrophic bacterium under a light regime mimicking that of the surface ocean.
Bacterial transcription patterns
We next examined concurrent bacterial transcript inventories indicative of metabolite consumption, normalized to cell counts at the time of sampling (Fig. 1a). The total number of transcripts cell−1 varied significantly over the diel cycle (One-way ANOVA; n = 26, p < 0.01) with ~2.5-fold more mRNAs in the mid-morning and noon cells (95 ± 49 and 114 ± 53 mRNAs cell-1) relative to mid-afternoon and night (42 ± 11 and 58 ± 25 mRNAs cell-1). Correspondingly, the majority of genes had higher transcripts per cell at mid-morning and noon relative to mid-afternoon and night (Fig. S3). This transcript inventory is low compared to exponentially growing Escherichia coli (1,350 mRNAs cell-1; ref. [52]) but comparable to previous measures for marine bacteria in ocean environments [34].
To identify genes that behaved similarly over time, the per cell transcript inventories for each of the 4,278 protein-encoding genes in the R. pomeroyi genome were clustered by variance-sensitive clustering (Fig. 1b, c). Group G-1 consisted of 3,294 genes (76%) exhibiting a diel transcription pattern with a maximum value at noon (Fig. 1c). Group G-2 consisted of 756 genes (17%) exhibiting a diel transcription pattern similar to G-1 but with high values at the first night and mid-morning time points (Fig. 1c). Group G-3 included 271 genes (6%) that did not exhibit a diel pattern, but similar to G-2, the first night and mid-morning values were high (Fig. 1c). We speculate that the higher transcript inventories at initial time points of the G-2 and G-3 clusters reflect incomplete bacterial drawdown of a metabolite accumulating prior to the addition of bacteria.
Expression levels of R. pomeroyi genes annotated with functions in organic carbon influx served as proxies for bacterial substrates made available by phytoplankton extracellular release (372 transporter genes representing 120 transporter systems). In a few cases for which catabolic genes were known but transporter genes were not, we used expression levels of diagnostic catabolic genes as indicators of substrate influx (21 catabolic genes representing 8 catabolic systems) (Table 2). The majority of influx genes exhibited the G-1 diel expression pattern peaking at noon (91%), among them 18 influx systems whose substrates have been experimentally verified [30, 31, 47, 53–55]. Most of these verified G-1 influx systems [15] had large day:night differences in transcript numbers (mean ratio: 33.5 ± 11.7, n = 50 for all genes making up the transport systems), with 13- to 58-fold higher per cell transcript inventories at noon relative to night (adjusted p < 0.05) (Fig. 2). These systems mediate uptake of osmolytes (ectoine and 5-hydroxyectoine), amines (trimethylamine, trimethylamine-N-oxide, and spermidine), organic sulfur compounds (DHPS, isethionate, cysteate, N-acetyltaurine, choline-O-sulfate, and DMSP), and glycolate, xanthine, phosphonate, and ribose. The other three experimentally-verified influx systems with G-1 patterns had modest (mean ratio: 3.7 ± 1.2, n = 9) but nonetheless significant diel expression differences (adjusted p < 0.05) due to high night transcript inventories rather than low noon inventories (Fig. 2); these encode influx of taurine, glucose, and sn-glycerol-3-phosphate. Two experimentally verified influx systems in group G-2 encoded uptake of putrescine and glycine betaine, and one in group G-3 encoded choline uptake. The greater expression levels at noon relative to night were also seen in flagella synthesis genes, implying greater bacterial motility at noon, but not for pathways relating to ATP generation and ribosomal protein synthesis, which had constant numbers of transcripts throughout the diel cycle (Fig. 2). Although expression among replicates followed the same patterns though time, variability within a given time point could be substantial. Potential explanations include physiological offsets among replicate diatom cultures and emergence of bacterial subpopulations within replicates [56].
Table 2.
Bacterial noon/night ratios of transcripts cell−1 for genes indicative of metabolite consumption.
| Compound | Gene locus tag | Gene name | Protein function | Noon/Night transcript ratio | Reference | |
|---|---|---|---|---|---|---|
| Amide | Urea | SPO1707 | urtD | ABC transporter, ATP-binding protein | 43.0 | [25] |
| SPO1708 | urtC | ABC transporter, permease | 37.7 | |||
| SPO1709 | urtB | ABC transporter, permease | 34.6 | |||
| SPO1710 | urtA | ABC transporter, substrate binding | 0.8 (n.s.) | |||
| Amine | TMAO | SPO1548 | tmoX | ABC transporter, periplasmic binding | 38.0 | [53] |
| SPO1550 | tmoV | ABC transporter, permease protein | 41.2 | |||
| SPO1549 | tmoW | ABC transporter, ATP binding | 43.1 | |||
| Amine | TMA | SPO1551 | tmm | TMA monooxygenase | 42.6 | [25] |
| Amino acid derivative | Betaine | SPO3186 | opuD | Glycine-betaine transporter | 1.6 (n.s.) | [25] |
| Amino Acid | Leucine | SPO2793 | ivD | isovaleryl-CoA dehydrogenase | 2.6 | |
| SPO2789 | mccA | methylcrotonyl-CoA carboxylase, alpha subunit | 3.1 | |||
| SPO2790 | mccB | methylcrotonyl-CoA carboxylase, beta subunit | 3.4 | |||
| SPO0390 | glutamate/leucine/phenylalanine/valine dehydrogenase | 1.2 (n.s.) | ||||
| Amino acid derivative | Ectoine/ 5-hydroxyectoine | SPO1146 | uehB | TRAP transporter, small integral membrane protein | 48.4 | [31] |
| SPO1147 | uehA | TRAP transporter, large integral membrane protein | 29.7 | |||
| SPO1145 | uehC | TRAP transporter, periplasmic binding | 36.7 | |||
| Amino Acid | Proline | SPO1031 | hypothetical protein | 21.2 | ||
| SPO2441 | ABC transporter, periplasmic betaine/proline-binding | 16.4 | ||||
| SPOA0231 | ABC transporter, periplasmic substrate-binding | 43.3 | ||||
| Choline | Choline | SPO1087 | betT | Choline transporter | 0.9 (n.s.) | [75] |
| Glycerol derivative | SN-glycerol-3-phosphate | SPO0238 | ugpE | ABC transporter, permease | 4.3 | [25] |
| SPO0239 | ugpA | ABC transporter, permease | 3.3 | |||
| SPO0237 | upgC | ABC transporter, ATP-binding | 3.5 | |||
| SPO0240 | ugpB | ABC transporter, periplasmic substrate-binding protein | 1.3 (n.s.) | |||
| Nuceloside | Xanthine | SPO0654 | xdhA | Xanthine dehydrogenase, A subunit | 33.4 | [76] |
| SPO0653 | xdhB | Xanthine dehydrogenase, B subunit | 33.0 | |||
| SPO0652 | xdhC | Xanthine dehydrogenase accessory factor | 25.8 | |||
| Nucleoside | Uridine | SPO2470 | iunH | inosine-uridine preferring nucleoside hydrolase | 9.9 | |
| Organic Acid | Acetate | SPO1813 | acs | acetyl-coenzyme A synthetase | 0.9 (n.s.) | |
| SPO0325 | phbB | acetoacetyl-CoA reductase | 1.9 | |||
| SPO0326 | phbA | acetyl-CoA acetyltransferase | 1.1 (n.s.) | |||
| Organic acid | Glycolate | SPO3478 | glcD | glycolate oxidase, GlcD subunit | 18.6 | [25] |
| SPO3479 | glcE | glycolate oxidase, GlcE subunit | 19.9 | |||
| SPO3480 | glcF | glycolate oxidase, iron-sulfur subunit | 21.6 | |||
| Phosphonate | Phosphonate | SPO0780 | phnC | ABC transporter, ATP-binding | 32.4 | |
| SPO0781 | phnD | ABC transporter, periplasmic phosphonate-binding | 18.6 | |||
| SPO0782 | phnE-1 | ABC transporter, permease | 38.3 | |||
| SPO0783 | phnE-2 | ABC transporter, permease | 35.6 | |||
| Polyamine | Putrescine | SPO3469 | potF | ABC transporter, periplasmic putrescine-binding | 1.2 | [55] |
| SPO3466 | potI | ABC transporter, permease | 2.5 | |||
| SPO3467 | potH | ABC transporter, permease | 2.6 | |||
| SPO3468 | potG | ABC transporter, ATP-binding | 2.1 | |||
| Polyamine | Spermidine | SPOA0381 | ABC transporter, periplasmic substrate-binding protein | 22.7 | [55] | |
| SPOA0383 | ABC transporter, permease protein | 53.4 | ||||
| SPOA0384 | ABC transporter, permease protein | 42.7 | ||||
| SPOA0382 | ABC transporter, ATP-binding protein | 29.2 | ||||
| Sugar | Ribose | SPOA0253 | ABC transporter, periplasmic substrate-binding | 30.9 | [47] | |
| SPOA0254 | rbsC-1 | ABC transporter, permease | 54.9 | |||
| SPOA0256 | ABC transporter, periplasmic substrate-binding protein | 21.4 | ||||
| SPOA0257 | rbsC-2 | ABC transporter, permease | 50.4 | |||
| SPOA0258 | rbsA | ABC transporter, ATP-binding | 55.7 | |||
| Sugar | Glucose | SPO0861 | xylF | ABC transporter, periplasmic substrate-binding | 2.7 (n.s.) | [47] |
| SPO0862 | xylH | ABC transporter, permease | 6.2 | |||
| SPO0863 | xylG | ABC transporter, ATP-binding | 4.2 | |||
| Sulfur compound | Choline-O-sulfate | SPO1083 | betC | Choline sulfatase | 15.1 | [75] |
| Sulfur compound | Cysteate | SPO2658 | ABC transporter, periplasmic substrate-binding | 22.4 | [41] | |
| SPO2659 | ABC transporter, permease | 38.3 | ||||
| SPO2660 | ABC transporter, permease | 28.0 | ||||
| SPO2661 | ABC transporter, ATP-binding | 32.7 | ||||
| Sulfur compound | Dihydroxypropane-sulfonate (DHPS) | SPO0591 | hpsK | TRAP transporter | 13.0 | [77] |
| SPO0592 | hpsL | TRAP transporter | 32.3 | |||
| SPO0593 | hpsM | TRAP transporter | 24.5 | |||
| Sulfur compound | Dimethylsulfonio-propionate (DMSP) | SPO1913 | dmdA | DMSP demethylase | 15.0 | [41] |
| SPO0453 | dddW | DMSP lyase | 30.7 | |||
| SPO1703 | dddD | DMSP lyase | 41.3 | |||
| SPO2299 | dddP | DMSP lyase | 31.4 | |||
| SPO1596 | dddQ | DMSP lyase | 19.2 | |||
| Sulfur compound | Isethionate | SPO2358 | iseK | TRAP transporter, periplasmic | 30.0 | [41] |
| SPO2357 | iseL | TRAP transporter, small permease | 13.2 | |||
| SPO2356 | iseM | TRAP transporter, DctM | 38.2 | |||
| Sulfur compound | N-acetyltaurine | SPO0660 | naaA | ABC transporter, periplasmic substrate-binding | 54.3 | [41] |
| SPO0661 | naaB | ABC transporter, permease | 32.3 | |||
| SPO0662 | naaB' | ABC transporter, permease | 42.7 | |||
| SPO0663 | naaC | ABC transporter, ATP-binding | 58.2 | |||
| SPO0664 | naaC' | ABC transporter, ATP-binding | 28.2 | |||
| Sulfur compound | Taurine | SPO0674 | tauA | ABC transporter, periplasmic substrate-binding | 1.8 (n.s.) | [41] |
| SPO0675 | tauB | ABC transporter, ATP-binding | 3.7 | |||
| SPO0676 | tauC | ABC transporter, permease | 3.1 |
Bold font indicates the compounds appearing in both the endometabolite and bacterial gene expression datasets.
n.s., not statistically significant (DeSeq2, adjusted p > 0.05).
Fig. 2. Expression levels of representative R. pomeroyi genes encoding transporters or diagnostic catabolic genes (top) and, for comparison, genes encoding flagella, ATPases, and ribosomal proteins (bottom).

For each panel, the top plot shows noon to night ratios (black circles), and the bottom plot shows average transcripts cell-1 at night, mid-morning, noon, and mid-afternoon. n = 8 or 6; error bars indicate standard deviations. Categories of transcription temporal patterns (G-1, black; G-2, white, G-3, grey) are indicated along the x-axis. Asterisks indicate transporters whose target substrate matches an endometabolite identified with high confidence.
We noticed that R. pomeroyi produced two-fold fewer transcripts for substrate acquisition in the mid-afternoon compared to mid-morning (Fig. 2), despite the fact that illumination was identical. Diel oscillations in the relationship between carbon fixation rate and irradiance have been broadly documented for marine phytoplankton in laboratory and field studies, characterized by pre-noon maxima in photosynthesis rates despite equivalent irradiance levels post-noon [57, 58]. Thus the rapid decrease in expression of most bacterial transporters by mid-afternoon suggests that photosynthetic oscillations may also be manifested in phytoplankton extracellular release.
Control for direct effects of light
Although no light-sensing proteins have been identified in the R. pomeroyi genome, we nonetheless checked whether light could be directly driving changes in gene expression. The bacterium was inoculated into spent medium obtained from axenic T. pseudonana cultures, representing a natural pool of dissolved metabolites, and exposed for 4 h to one of three light levels that matched co-culture irradiance at noon, mid-morning/mid-afternoon, and night in order to assess differential gene expression with light exposure as the only variable. Only 61 genes in the bacterial culture (1.4% of the R. pomeroyi genome) were significantly enriched by one or both light levels compared to the dark treatment (Fig. S4), indicating that the large shifts observed in transporter system expression (Fig. 2) were not directly triggered by light. Nonetheless, the collection of light-enriched genes was not random. Ten genes function in protection against reactive oxygen species (ROS) (Fig. S5, Table S5) which can be formed when light interacts with oxygen or organic compounds [59–61]. Another 16 function in the uptake and metabolism of phosphate (pstSCAB, phoU), and phosphonate (phnDEC, phnIGHLJN) (Fig. S5), and were likely under the control of the similarly enriched phoB regulatory protein [62–64]. Phosphorus acquisition transcript enrichment was surprising, since phosphate concentrations remain non-limiting for many weeks in this model culture system (>10 µmol L−1) [47] and phosphorus availability was identical in all treatments regardless. Higher uptake of phosphorus during morning hours, when photosynthesis rates are low and competition with phytoplankton for phosphorus uptake may be less intense, has been proposed as a niche partitioning strategy for marine bacterioplankton [14, 65]. Based on the concomitant enrichment of phosphorus acquisition genes and ROS-related genes in the absence of light-sensing proteins, we speculate that phosphorus acquisition might be regulated through the cellular detection of ROS dynamics. ROS have known roles in bacterial signaling through redox-sensing transcriptional regulators [66].
Transcript enrichment versus upregulation
Expression of the bacterial influx systems was also calculated as a percent of the transcriptome (Fig. 3), the prevailing analysis approach for RNAseq studies that do not use internal standardization [67]. These calculations identified 57% of influx system genes as having significantly enriched proportions in the noon transcriptome relative to night. In contrast, per cell transcript calculations based on the internal standards identified 94% of influx system genes as having significantly higher transcript inventories at noon relative to night (Fig. 3). These analyses emphasize, on the one hand, the bacterium’s investment in expression of a transporter relative to other cellular functions (percent of transcriptome), and on the other, the absolute number of templates to synthesize the transporter (transcripts per cell). This analysis also revealed that the response of bacterial influx genes is weighted toward higher fold-change compared to the average across all genes for both relative and absolute analyses, implying well-regulated substrate influx. We used per cell bacterial transcript counts and per cell metabolite abundance in the following co-analysis of gene expression and endometabolite data sets.
Fig. 3. Comparison of fold-difference values for absolute versus relative analysis of noon:night ratios of transcripts.
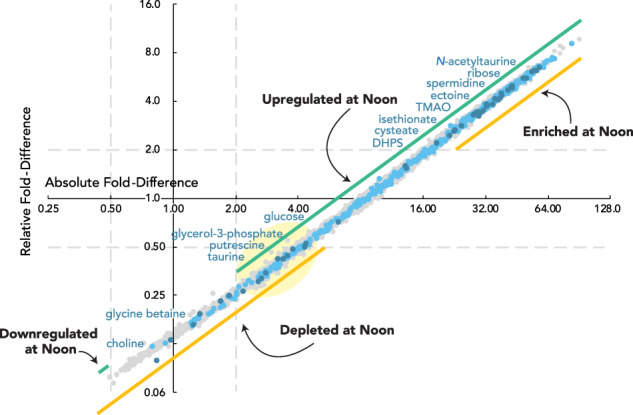
Absolute analysis (x-axis) represents up- or down-regulation of the number of transcripts per bacterial cell. Relative analysis (y-axis) represents enrichment or depletion as a proportion of the transcriptome. Dark blue symbols indicate the transporter genes in Fig. 2; light blue symbols indicate other transporter genes; grey symbols indicate the remaining R. pomeroyi genes. Dashed gray lines mark where fold-difference = 2 on each axis (log2 units). The light yellow oval highlights genes with per cell transcript inventories that are significantly higher at noon yet account for a significantly lower proportion of the cells’ transcriptome.
Coincidence of diatom metabolite accumulation and bacterial transcription
Eight of the bacterial influx systems had target compounds that were also identified with high confidence in the diatom endometabolome, allowing us to compare diel patterns for these matched pairs (Table 2). In all cases, bacterial gene expression followed a diel pattern with peak expression at noon (G-1 or G-2 clusters) (Fig. 4a). The metabolite patterns were more diverse, however. Four metabolites exhibited a continually increasing endometabolome concentration (cluster M-1; leucine, glycerol-3-phosphate, DHPS, and DMSP) (Fig. 4a). One metabolite exhibited a continually decreasing endometabolome concentration (M-2; acetate). Two exhibited diel cycles with mid-afternoon concentration peaks (M-3; glucose and uridine). One exhibited a diel cycle with noon concentration peaks (M-4; proline).
Fig. 4. Modeling diatom endometabolite pools and bacterial transcript inventories.

a Comparison of diel patterns for diatom endometabolite concentration (green symbols) and bacterial transcript inventory for a representative gene encoding uptake or catabolism of the same compound (blue symbols); additional relevant genes are shown in Fig. S6 (mean + standard deviation, n = 3 except for the first night where n = 2). b The base model was optimized with or without optional functions. Each point represents a parameter combination (out of 806,400 tested) for which the model output is significantly correlated with both metabolite and transcript experimental data, and points are colored according to the Pearson’s r values for correlations between experimental data and model simulations, averaged for transcript and metabolite datasets. c The best fit model based on average adjusted r, with individual r values given above the plots for metabolite (green font) and transcript (blue font) inventories. Functions added to the base model to achieve the best fit include two physiological balance mechanisms indicated as +o (irradiance-fixation oscillation) and +h (cellular homeostasis), and a bacterial recognition response indicated as +b. *p < 0.05; **p < 0.01; ***p < 0.001. d Simulation model structure.
We asked whether the three mechanisms proposed for phytoplankton extracellular release (passive diffusion, physiological balance, and interaction response) could explain the paired patterns in endometabolites (the presumed source) and bacterial transcription (the proxy for extracellular release). A simulation model was established with passive diffusion as the default release mechanism by phytoplankton, governed solely by a metabolite concentration gradient between the inside and outside of cells. Beyond the base model, additional parameters permitted two active physiological balance release mechanisms, fixation-irradiance oscillation and cell homeostasis (for example due to photorespiration or redox imbalance), and one active bacterial interaction mechanism (for bacterial influence over metabolite release rate) (Fig. 4d), and these were tested for improved representation of the experimental data over that of the base model. The model assumed that R. pomeroyi transporter systems were regulated by the availability of their substrate, which has been supported in previous studies [30, 31, 53, 55, 68], and that substrates are taken up by the bacteria according to Michaelis-Menten kinetics.
Models in which passive diffusion was the sole mechanism of extracellular release best reproduced the observed concentration pattern for acetate, which steadily decreased through time in the diatom endometabolome (M-2 pattern); and for proline, which had a diel pattern with a peak at noon (M-4 pattern). Adding additional parameters to the base model did not significantly increase fit for these compounds (adjusted r, p > 0.5; Table S6). Glucose and uridine, which had diel peaks in concentration in the afternoon (M-3 pattern), were best represented in models that included one or both physiological balance mechanisms (fixation-irradiance oscillation and cell homeostasis). Finally, leucine, glycerol-3-phosphate, DMSP, and DHPS, the compounds exhibiting monotonically increasing endometabolite concentrations (M-1 pattern), were best reproduced by the model when the bacterial interaction mechanism was invoked, and may represent shifts to higher steady state concentrations. In contrast to metabolite concentrations, the experimental gene expression data for the 8 compounds all showed a diel peak at noon. While most best-fit models reproduced this transcription pattern, they failed to replicate the noon peaks in expression for acetate, glucose, and leucine (Fig. 4b, c; Fig. S6; Table S6).
The inability of diffusion alone to reproduce the experimental data for the majority of metabolites suggests that passive movement of molecules through cell membranes is insufficient to explain metabolite release [69]. Physiological balance mechanisms represented by the photosynthetic oscillations and cell homeostasis parameters are consistent with evidence that light intensity affects photorespiration rates [3, 70] as well as the biosynthetic balance between metabolite classes (e.g., amino acids versus storage carbohydrates; ref. [71]).
The role of bacterial recognition is more controversial, since the underlying processes by which recognition could be achieved are not yet resolved, nor is it clear how widespread or important such interactions may be in natural microbial communities. Chemicals released from bacteria or bacterial alteration of environmental conditions, such as nutrient pools or ROS, might serve as signals [22]. In previous research, cultured T. pseudonana exhibited transcriptional changes consistent with a recognition response acting via leucine-rich repeat (LRR) proteins following addition of R. pomeroyi [22]. Similarly, marine diatom Pseudo-nitzchia multiseries released tryptophan in response to bacterium Sulfitobacter sp. S11 [21] and the bacterium converted tryptophan to the plant hormone indole-3-acetic acid. The model prediction of a role for R. pomeroyi in triggering concentration increases of specific T. pseudonana endometabolites was tested experimentally by comparing co-cultures to axenic cultures. Three of the four M1 compounds (leucine, glycerol-3-phosphate, and DHPS) had significantly higher concentrations in the diatom endometabolome when incubated with bacteria relative to axenic incubations (Fig. S7). DMSP was the exception, with no significant difference in the T. pseudonana endometabolome in the presence or absence of bacteria. DMSP carries out a number of functions in phytoplankton cells, including roles as an osmolyte [72], predator deterrent [73], and ROS scavenger [74], and therefore its regulation in the diatom endometabolome may be more complex than what the model structure could represent (Table S6). Considering these eight focal compounds, computational matching of endometabolite and transcript abundance patterns revealed dynamics consistent with active diatom exudation of labile compounds.
Conclusions
The quantitative importance of marine phytoplankton-bacteria carbon flux has motivated inquiries into the factors that regulate phytoplankton extracellular release, such as light, temperature, and nutrient limitation [71]. Here we examined diatom internal metabolite accumulation patterns to ask how well they predict external release patterns. We found that blueprints for endometabolite accumulation are diverse (at least four daily patterns), that diatom release strategies vary by molecule, and that active mechanisms involving both diatom physiological balance and bacterial interactions may play key roles. The composition of metabolite release by phytoplankton determines rates and efficiencies of carbon processing by surface ocean bacteria, while the magnitude of metabolite release determines the allocation of recent photosynthate into dissolved versus particulate organic carbon reservoirs, with the former having a lower likelihood of participating in ocean sequestration.
Supplementary information
Acknowledgements
C. Smith and S. Sharma provided sequencing and bioinformatic assistance, J. Gluhska and C. Panagos offered expertise on NMR analysis, M. Landa and B. Nowinski provided valuable comments on experimental design, F. Ferrer-González and J. Schreier assisted with sampling, and the University of Georgia Genomics and Bioinformatics Core (GGBC) provided sequencing services.
Author contributions
MU and MAM conceived of the study, MU and MO collected the data, MU, MO, ASE, and MAM analyzed data, WS developed the model, and MU and MAM wrote the paper with input from all authors.
Funding
This work was supported by NSF (IOS-1656311 and OCE-2019589 to MAM and ASE), The Simons Foundation (grant 542391 to MAM) within the Principles of Microbial Ecosystems (PriME) Collaborative, The Gordon and Betty Moore Foundation (5503 to MAM and ASE), JSPS (Research Fellowship for Young Scientists and Grant-in-Aid for JSPS Fellows to MU), and the Swedish Research Council (2018-06571 to MO). This is the NSF Center for Chemical Currencies of a Microbial Planet (C-CoMP) publication #002.
Data availability
Data that support the findings of this study have been deposited in NCBI SRA with BioProject accession number PRJNA649292 (sequencing data), and Metabolomics Workbench with Project ID PR001019, dx.doi.org/10.21228/M80408 (metabolome data).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41396-021-01172-w.
References
- 1.Baines SB, Pace ML. The production of dissolved organic matter by phytoplankton and its importance to bacteria: patterns across marine and freshwater systems. Limnol Oceanogr. 1991;36:1078–90. doi: 10.4319/lo.1991.36.6.1078. [DOI] [Google Scholar]
- 2.Williams PJLeB. Heterotrophic bacteria and the dynamics of dissolved organic material. In: Kirchman DL (ed). Microbial Ecology of the Oceans, 1st edn. New York: Wiley-Liss; 2000. p. 153–200.
- 3.Thornton DCO. Dissolved organic matter (DOM) release by phytoplankton in the contemporary and future ocean. Eur J Phycol. 2014;49:20–46. doi: 10.1080/09670262.2013.875596. [DOI] [Google Scholar]
- 4.Nagata T. Organic matter-bacteria interactions in seawater. In: Kirchman DL, editor. Microbial Ecology of the Oceans. Hoboken: John Wiley and Sons, Inc; 2008. pp. 207–41. [Google Scholar]
- 5.Kujawinski EB. The impact of microbial metabolism on marine dissolved organic matter. Ann Rev Mar Sci. 2011;3:567–99. doi: 10.1146/annurev-marine-120308-081003. [DOI] [PubMed] [Google Scholar]
- 6.Azam F, Fenchel T, Field JG, Gray JS, Meyerreil LA, Thingstad F. The ecological role of water-column microbes in the sea. Mar Ecol Prog Ser. 1983;10:257–63. doi: 10.3354/meps010257. [DOI] [Google Scholar]
- 7.Cole JJ, Findlay S, Pace ML. Bacterial production in fresh and saltwater ecosystems - a cross-system overview. Mar Ecol Prog Ser. 1988;43:1–10. doi: 10.3354/meps043001. [DOI] [Google Scholar]
- 8.Moran MA, Kujawinski EB, Stubbins A, Fatland R, Aluwihare LI, Buchan A, et al. Deciphering ocean carbon in a changing world. Proc Nat Acad Sci. 2016;113:3143–51. doi: 10.1073/pnas.1514645113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Becker KW, Collins JR, Durham BP, Groussman RD, White AE, Fredricks HF, et al. Daily changes in phytoplankton lipidomes reveal mechanisms of energy storage in the open ocean. Nat Comm. 2018;9:5179. doi: 10.1038/s41467-018-07346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boysen AK, Carlson LT, Durham BP, Groussman RD, Aylward FO, Ribalet F, et al. Diel oscillations of particulate metabolites reflect synchronized microbial activity in the North Pacific Subtropical Gyre. bioRxiv. 2020: 2020.05.09.086173.
- 11.Durham BP, Boysen AK, Carlson LT, Groussman RD, Heal KR, Cain KR, et al. Sulfonate-based networks between eukaryotic phytoplankton and heterotrophic bacteria in the surface ocean. Nat Microbiol. 2019;4:1706–15. doi: 10.1038/s41564-019-0507-5. [DOI] [PubMed] [Google Scholar]
- 12.Burney CM, Davis PG, Johnson KM, Sieburth JM. Diel relationships of microbial trophic groups and in situ dissolved carbohydrate dynamics in the Caribbean Sea. Mar Biol. 1982;67:311–22. doi: 10.1007/BF00397672. [DOI] [Google Scholar]
- 13.Gasol JM, Doval MD, Pinhassi J, Calderon-Paz JI, Guixa-Boixareu N, Vaque D, et al. Diel variations in bacterial heterotrophic activity and growth in the northwestern Mediterranean Sea. Mar Ecol Prog Ser. 1998;164:107–24. doi: 10.3354/meps164107. [DOI] [Google Scholar]
- 14.Kuipers B, van Noort GJ, Vosjan J, Herndl GJ. Diel periodicity of bacterioplankton in the euphotic zone of the subtropical Atlantic Ocean. Mar Ecol Prog Ser. 2000;201:13–25. doi: 10.3354/meps201013. [DOI] [Google Scholar]
- 15.Ottesen EA, Young CR, Gifford SM, Eppley JM, Marin R, Schuster SC, et al. Multispecies diel transcriptional oscillations in open ocean heterotrophic bacterial assemblages. Science. 2014;345:207–12. doi: 10.1126/science.1252476. [DOI] [PubMed] [Google Scholar]
- 16.Aylward FO, Eppley JM, Smith JM, Chavez FP, Scholin CA, DeLong EF. Microbial community transcriptional networks are conserved in three domains at ocean basin scales. Proc Nat Acad Sci. 2015;112:5443–8. doi: 10.1073/pnas.1502883112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frischkorn KR, Haley ST, Dyhrman ST. Coordinated gene expression between Trichodesmium and its microbiome over day–night cycles in the North Pacific Subtropical Gyre. ISME J. 2018;12:997–1007. doi: 10.1038/s41396-017-0041-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seymour JR, Amin SA, Raina JB, Stocker R. Zooming in on the phycosphere: the ecological interface for phytoplankton-bacteria relationships. Nat Microbiol. 2017;2:17065. doi: 10.1038/nmicrobiol.2017.65. [DOI] [PubMed] [Google Scholar]
- 19.Bjornsen PK. Phytoplankton exudation of organic-matter - why do healthy cells do it. Limnol Oceanogr. 1988;33:151–4. doi: 10.4319/lo.1988.33.1.0151. [DOI] [Google Scholar]
- 20.Fogg GE. The ecological significance of extracellular products of phytoplankton photosynthesis. Bot Mar. 1983;26:3–14. doi: 10.1515/botm.1983.26.1.3. [DOI] [Google Scholar]
- 21.Amin SA, Hmelo LR, van Tol HM, Durham BP, Carlson LT, Heal KR, et al. Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature. 2015;522:98–101. doi: 10.1038/nature14488. [DOI] [PubMed] [Google Scholar]
- 22.Durham BP, Dearth SP, Sharma S, Amin SA, Smith CB, Campagna SR, et al. Recognition cascade and metabolite transfer in a marine bacteria‐phytoplankton model system. Environ Microbiol. 2017;19:3500–13. doi: 10.1111/1462-2920.13834. [DOI] [PubMed] [Google Scholar]
- 23.Guerrini F, Mazzotti A, Boni L, Pistocchi R. Bacterial-algal interactions in polysaccharide production. Aquat Micro Ecol. 1998;15:247–53. doi: 10.3354/ame015247. [DOI] [Google Scholar]
- 24.Armbrust EV, Berges JA, Bowler C, Green BR, Martinez D, Putnam NH, et al. The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science. 2004;306:79–86. doi: 10.1126/science.1101156. [DOI] [PubMed] [Google Scholar]
- 25.Moran MA, Buchan A, Gonzalez JM, Heidelberg JF, Whitman WB, Kiene RP, et al. Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature. 2004;432:910–3. doi: 10.1038/nature03170. [DOI] [PubMed] [Google Scholar]
- 26.Uitz J, Claustre H, Gentili B, Stramski D. Phytoplankton class-specific primary production in the world’s oceans: Seasonal and interannual variability from satellite observations. Global Biogeochem Cycles. 2010;24.
- 27.Buchan A, LeCleir GR, Gulvik CA, Gonzalez JM. Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat Rev Microbiol. 2014;12:686–98. doi: 10.1038/nrmicro3326. [DOI] [PubMed] [Google Scholar]
- 28.Luo HW, Moran MA. Evolutionary ecology of the marine Roseobacter clade. Microbiol Mol Biol Rev. 2014;78:573–87. doi: 10.1128/MMBR.00020-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nowinski B, Moran MA. Niche dimensions of a marine bacterium are identified using invasion studies in coastal seawater. Nat Microbiol. 2021;6:524. doi: 10.1038/s41564-020-00851-2. [DOI] [PubMed] [Google Scholar]
- 30.Denger K, Lehmann S, Cook AM. Molecular genetics and biochemistry of N-acetyltaurine degradation by Cupriavidus necator H16. Microbiology. 2011;157:2983–91. doi: 10.1099/mic.0.048462-0. [DOI] [PubMed] [Google Scholar]
- 31.Schulz A, Stoveken N, Binzen IM, Hoffmann T, Heider J, Bremer E. Feeding on compatible solutes: a substrate-induced pathway for uptake and catabolism of ectoines and its genetic control by EnuR. Environ Microbiol. 2017;19:926–46. doi: 10.1111/1462-2920.13414. [DOI] [PubMed] [Google Scholar]
- 32.Crossette E, Gumm J, Langenfeld K, Raskin L, Duhaime M, Wigginton K. Metagenomic quantification of genes with internal standards. mBio. 2021;12:e03173-20. doi: 10.1128/mBio.03173-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gifford SM, Becker JW, Sosa OA, Repeta DJ, DeLong EF. Quantitative transcriptomics reveals the growth-and nutrient-dependent response of a streamlined marine methylotroph to methanol and naturally occurring dissolved organic matter. mBio. 2016;7:e01279-16. doi: 10.1128/mBio.01279-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moran MA, Satinsky B, Gifford SM, Luo HW, Rivers A, Chan LK, et al. Sizing up metatranscriptomics. ISME J. 2013;7:237–43. doi: 10.1038/ismej.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guillard RRL, Hargraves PE. Stichochrysis immobilis is a diatom, not a chyrsophyte. Phycologia. 1993;32:234–6. doi: 10.2216/i0031-8884-32-3-234.1. [DOI] [Google Scholar]
- 36.Uchimiya M, Tsuboi Y, Ito K, Date Y, Kikuchi J. Bacterial substrate transformation tracked by stable-isotope-guided NMR metabolomics: application in a natural aquatic microbial community. Metabolites. 2017;7:52. doi: 10.3390/metabo7040052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis IA, Schommer SC, Markley JL. rNMR: open source software for identifying and quantifying metabolites in NMR spectra. Mag Res Chem. 2009;47:S123–S6. doi: 10.1002/mrc.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu YF, et al. HMDB 3.0-the human metabolome database in 2013. Nuc Acids Res. 2013;41:D801–D7. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, et al. BioMagResBank Nuc Acids Res. 2008;36:D402–D8. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toukach PV, Egorova KS. Carbohydrate structure database merged from bacterial, archaeal, plant and fungal parts. Nuclic Acids Res. 2016;44:D1229–D36. doi: 10.1093/nar/gkv840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Landa M, Burns AS, Durham BP, Esson K, Nowinski B, Sharma S, et al. Sulfur metabolites that facilitate oceanic phytoplankton-bacteria carbon flux. ISME J. 2019;13:2536–50. doi: 10.1038/s41396-019-0455-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boroujerdi AFB, Lee PA, DiTullio GR, Janech MG, Vied SB, Bearden DW. Identification of isethionic acid and other small molecule metabolites of Fragilariopsis cylindrus with nuclear magnetic resonance. Anal Bioanal Chem. 2012;404:777–84. doi: 10.1007/s00216-012-6169-2. [DOI] [PubMed] [Google Scholar]
- 43.Walejko JM, Chelliah A, Keller-Wood M, Gregg A, Edison AS. Global metabolomics of the placenta reveals distinct metabolic profiles between maternal and fetal placental tissues following delivery in non-labored women. Metabolites. 2018;8:10. doi: 10.3390/metabo8010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwämmle V, Jensen ON. VSClust: feature-based variance-sensitive clustering of omics data. Bioinformatics. 2018;34:2965–72. doi: 10.1093/bioinformatics/bty224. [DOI] [PubMed] [Google Scholar]
- 45.Thaben PF, Westermark PO. Detecting rhythms in time series with RAIN. J Biol Rhythms. 2014;29:391–400. doi: 10.1177/0748730414553029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Welsh J (2020). CirHeatmap. Available from: https://github.com/joadwe/cirheatmap.
- 47.Landa M, Burns AS, Roth SJ, Moran MA. Bacterial transcriptome remodeling during sequential co-culture with a marine dinoflagellate and diatom. ISME J. 2017;11:2677–90. doi: 10.1038/ismej.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Satinsky BM, Gifford SM, Crump BC, Moran MA Use of internal standards for quantitative metatranscriptome and metagenome analysis. In: DeLong EF (ed). Methods in Enzymology. 2013. 531: p. 237-50. [DOI] [PubMed]
- 49.Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Becker S, Tebben J, Coffinet S, Wiltshire K, Iversen MH, Harder T, et al. Laminarin is a major molecule in the marine carbon cycle. Proc Nat Acad Sci. 2020;117:6599–607. doi: 10.1073/pnas.1917001117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neidhardt F, Ingraham J, Schaechter S Physiology of the bacterial cell: a molecular approach. Massachusetts: Sinauer Associates Inc.; 1990.
- 53.Lidbury I, Murrell JC, Chen Y. Trimethylamine N-oxide metabolism by abundant marine heterotrophic bacteria. Proc Nat Acad Sci. 2014;111:2710–5. doi: 10.1073/pnas.1317834111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mayer J, Huhn T, Habeck M, Denger K, Hollemeyer K, Cook AM. 2,3-Dihydroxypropane-1-sulfonate degraded by Cupriavidus pinatubonensis JMP134: purification of dihydroxypropanesulfonate 3-dehydrogenase. Microbiology. 2010;156:1556–64. doi: 10.1099/mic.0.037580-0. [DOI] [PubMed] [Google Scholar]
- 55.Mou XZ, Sun SL, Rayapati P, Moran MA. Genes for transport and metabolism of spermidine in Ruegeria pomeroyi DSS-3 and other marine bacteria. Aquat Micro Ecol. 2010;58:311–21. doi: 10.3354/ame01367. [DOI] [Google Scholar]
- 56.Biller SJ, Coe A, Roggensack SE, Chisholm SW Heterotroph interactions alter Prochlorococcus transcriptome dynamics during extended periods of darkness. mSystems. 2018; 3 10.1128/mSystems.00040-18. [DOI] [PMC free article] [PubMed]
- 57.Harding L, Meeson B, Prézelin B, Sweeney B. Diel periodicity of photosynthesis in marine phytoplankton. Mar Biol. 1981;61:95–105. doi: 10.1007/BF00386649. [DOI] [Google Scholar]
- 58.Harding L, Prezelin B, Sweeney B, Cox J. Diel oscillations of the photosynthesis-irradiance (PI) relationship in natural assemblages of phytoplankton. Mar Biol. 1982;67:167–78. doi: 10.1007/BF00401282. [DOI] [Google Scholar]
- 59.Blough NV, Zepp RG Reactive oxygen species in natural waters. Active oxygen in chemistry. Dordrecht: Springer; 1995. p. 280–333.
- 60.Zafiriou OC, Joussot-Dubien J, Zepp RG, Zika RG. Photochemistry of natural waters. Environ Sci Technol. 1984;18:358A–71A. doi: 10.1021/es00130a711. [DOI] [Google Scholar]
- 61.Ziegelhoffer EC, Donohue TJ. Bacterial responses to photo-oxidative stress. Nat Rev Microbiol. 2009;7:856–63. doi: 10.1038/nrmicro2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lubin EA, Henry JT, Fiebig A, Crosson S, Laub MT. Identification of the PhoB regulon and role of PhoU in the phosphate starvation response of Caulobacter crescentus. J Bacteriol. 2016;198:187–200. doi: 10.1128/JB.00658-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang C, Huang TW, Wen SY, Chang CY, Tsai SF, Wu WF, et al. Genome-wide PhoB binding and gene expression profiles reveal the hierarchical gene regulatory network of phosphate starvation in Escherichia coli. Plos One. 2012;7:e47314. doi: 10.1371/journal.pone.0047314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hsieh YJ, Wanner BL. Global regulation by the seven-component Pi signaling system. Curr Opin Microbiol. 2010;13:198–203. doi: 10.1016/j.mib.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muratore D, Boysen AK, Harke MJ, Becker KW, Casey JR, Coesel SN, et al. Community-scale synchronization and temporal partitioning of gene expression, metabolism, and lipid biosynthesis in oligotrophic ocean surface waters. bioRxiv. 2020: 2020.05.15.098020.
- 66.Giedroc DP. Hydrogen peroxide sensing in Bacillus subtilis: it is all about the (metallo)regulator. Mol Microbiol. 2009;73:1–4. doi: 10.1111/j.1365-2958.2009.06752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131:281–5. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- 68.Weinitschke S, Sharma PI, Stingl U, Cook AM, Smits TH. Gene clusters involved in isethionate degradation by terrestrial and marine bacteria. Appl Environ Microbiol. 2010;76:618–21. doi: 10.1128/AEM.01818-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hellebust JA. Excretion of some organic compounds by marine phytoplankton 1. Limnol Oceanogr. 1965;10:192–206. doi: 10.4319/lo.1965.10.2.0192. [DOI] [Google Scholar]
- 71.Behrenfeld MJ, Halsey KH, Milligan AJ. Evolved physiological responses of phytoplankton to their integrated growth environment. Philos Trans R Soc B: Biol Sci. 2008;363:2687–703. doi: 10.1098/rstb.2008.0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kiene RP, Linn LJ, Bruton JA. New and important roles for DMSP in marine microbial communities. J Sea Res. 2000;43:209–24. doi: 10.1016/S1385-1101(00)00023-X. [DOI] [Google Scholar]
- 73.Fredrickson KA, Strom SL. The algal osmolyte DMSP as a microzooplankton grazing deterrent in laboratory and field studies. J Plankton Res. 2009;31:135–52. doi: 10.1093/plankt/fbn112. [DOI] [Google Scholar]
- 74.Sunda W, Kieber DJ, Kiene RP, Huntsman S. An antioxidant function for DMSP and DMS in marine algae. Nature. 2002;418:317–20. doi: 10.1038/nature00851. [DOI] [PubMed] [Google Scholar]
- 75.Lidbury I, Kimberley G, Scanlan DJ, Murrell JC, Chen Y. Comparative genomics and mutagenesis analyses of choline metabolism in the marine Roseobacter clade. Environ Microbiol. 2015;17:5048–62. doi: 10.1111/1462-2920.12943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cunliffe M. Purine catabolic pathway revealed by transcriptomics in the model marine bacterium Ruegeria pomeroyi DSS-3. FEMS Microbiol Ecol. 2016;92:fiv150. doi: 10.1093/femsec/fiv150. [DOI] [PubMed] [Google Scholar]
- 77.Durham BP, Sharma S, Luo HW, Smith CB, Amin SA, Bender SJ, et al. Cryptic carbon and sulfur cycling between surface ocean plankton. Proc Nat Acad Sci. 2015;112:453–7. doi: 10.1073/pnas.1413137112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data that support the findings of this study have been deposited in NCBI SRA with BioProject accession number PRJNA649292 (sequencing data), and Metabolomics Workbench with Project ID PR001019, dx.doi.org/10.21228/M80408 (metabolome data).