

Abstract
The HIV-1 derived gp145 protein is being investigated by research groups as preclinical studies have shown high promise for this protein as a vaccine against HIV. However, one of the main challenges with manufacturing this promising protein has been ascribed to the low yield obtained in mammalian cell cultures. Significant improvements in gp145 production are needed to address this issue to test the gp145 protein as a potentially effective, safe, and affordable HIV vaccine. Here we describe the application of a novel expression technology to create GMP-grade CHO cell lines expressing approximately 50 µg/ml in non-optimized fed-batch culture, which is an order of magnitude higher than that obtained in existing processes. Top producing clones show a high degree of similarity in the glycosylation patterns of the purified protein to the reference standard. Conformational integrity and functionality was demonstrated via high-affinity binding to soluble CD4 using a panel of antibodies including VRC01, F105, Hk20, PG9 and 17b. In summary, we were able to generate CHO cell lines expressing HIV gp145 with significantly higher overall expression yields than currently accessible, and high product quality that could potentially be suitable for future studies assessing the efficacy and safety of gp145-based HIV vaccines.
Keywords: STEP technology, mammalian protein expression, CHO cell line, clone selection, HIV envelope protein, gp145
Introduction
Despite numerous advances in the understanding of human immunodeficiency viruses (HIV) and acquired immunodeficiency syndrome (AIDS), the search for a potent anti-HIV vaccine still continues. The envelope glycoproteins found on the surface of the HIV virus are prime targets for HIV vaccine efforts. Researchers have shown high promise for gp145 as a vaccine candidate against HIV in various preclinical studies [1]. However, further development has remained largely hindered due to low yields of functionally-active recombinant HIV gp145 and, further complicated by the extensive glycosylation pattern that decorates the protein [2].
HIV-1 envelope glycans contribute approximately 50% of its mass represented mainly by N-linked glycans with a small O-linked glycan contribution [2–4]. Multiple hybrid and complex mannose-rich glycans with various levels of sialyation exist and are known to be dependent on the recombinant expression system used for production [2, 3, 5, 6]. Glycosylation of HIV envelope protein remains an important feature related to their production yield, quality, stability and antigenicity [5, 7, 8].
In this article, we report the use of a novel expression technology (referred to as STEP technology) for high-level, stable expression of HIV gp145 with similar glycosylation and in vitro antigenicity and CD-4 binding characteristics as a gp145 reference standard. Lead cell lines generated provided expression levels up to 47.4 µg/ml in non-optimized fed-batch culture which is an order of magnitude higher than the previously described reference standard cell line produced [1].
The STEP technology is comprised of an adaptable, stringent antibiotic (zeocin) selection system and expression enhancing DNA elements. Stringency of selection pressure is introduced by a functionally impaired selection marker (FI-Zeo) thus introducing a minimum threshold for expression of the transgene and selection marker protein in transfected cells. Only cells with sufficiently high expression levels can survive the selection pressure. The gene of interest (GOI) is expressed on the same mRNA as the selection marker which is translationally coupled to the upstream GOI via an IRES, and thus high expression of the selection marker results in high expression levels of the GOI as well. Three STEP vectors with increasing levels of stringency are obtained via insertion of different spacer sequences at the IRES, which enables the selection of the optimal stringency to achieve maximum expression levels for each GOI. Easy to express proteins are typically expressed maximally at the highest stringency, whereas difficult to express proteins are typically expressed maximally at one of the lower stringencies. The expression cassette is flanked by two expression enhancing DNA elements at the 5’ and 3’ sites, which increase expression levels and thereby also facilitates the use of high selection stringency.
In comparison, the reference standard cell line was generated by transfecting CHO-K1 cells with a bicistronic plasmid containing codon-optimized HIV gp145 upstream of an internal ribosomal entry stie (IRES) and the puromycin N-acetyl transferase gene. Stable gp145-producing cells were identified under puromycin selection, and clonal cell lines were established by limiting dilution. The selected clone was adapted for growth in protein-free media, and an RCB was produced [1]. Transient transfections of CHO-S, Expi293F and 293F using PEI were also evaluated and found to yield a maximum expression of 7 µg/ml (data not published), a marginal improvement over the 4 µg/ml expression level of the reference cell line. In both these cases the potential of using gp145 as a potential vaccine would be restricted to Phase 1 studies beyond which the low production yields would be a severe cost-deterrent for vaccine development.
The STEP technology provides a novel expression platform that is the rapid, cost-effective, and provides consistent selection of stable of highly productive, recombinant CHO cell lines for biomanufacturing [9, 10]. Here we report that we have created a stable GMP-grade CHO cell line expressing the gp145 protein with a high degree of similarity in glycosylation pattern when compared to reference standard (purified gp145 produced with non-STEP technology) but with ten times the yield demonstrating the utility of the STEP technology in making difficult to express targets such as HIV gp145 more amenable for vaccine development.
Materials and Methods
Transfection and Pool selection
A vial of suspension CHO cell bank (Batavia Biosciences) was thawed in growth medium (CD DG44 (Catalog #12610–010, Life Technologies, Carlsbad, California USA), 8 mM L-glutamine (Catalog #25030–081, Life Technologies, Carlsbad, California USA), 0.18% Pluronic F-68, (Catalog #24040, Life Technologies, Carlsbad, California USA)) and passaged four times to ensure complete thaw recovery. Cultures were seeded at 0.1 x 106 cells/ml in 150-ml square bottles (Catalog #431430, Corning Inc., China) with 80-ml working volume at each passage until transfection.
Expression vectors were prepared by gene synthesis and subcloning of the gp145 gene [1] into the FI-Zeo1, FI-Zeo2, and FI-Zeo3 STEP vectors (GeneArt, ThermoFisher). Four transfections were performed for each vector type for a total of twelve transfections. Transfections were performed using the Amaxa Nucleofector Kit V from Lonza (VCA-1003) using 5 µg DNA per 2 x 106 cells per sample via electroporation following manufacturer’s instructions. FI-Zeo1 STEP and cells only were used as controls. ITS-X (Insulin-Transferrin-Selenium, Catalog #51500056 Life Technologies, Carlsbad, California USA) was added to the growth medium as a supplement during transfection.
Electroporated cells were transferred to 6-well plates and placed in a humidified incubator set at 37 °C and 5% CO2. After 2h or static incubation, 6-well plates were transferred to a shaking platform set at 90 rpm (2.5 cm orbital diameter) for an additional 2h.
Approximately, 2.5 ml of fresh growth media was added to each pool of transfected cells and they were combined to seed three, T-75 flasks (one T-75 flask per STEP stringency or per 4 transfections). After two days inside the shaker incubator, cells from each T-75 flask were spun down at 100g for 5 minutes at room temperature and resuspended in freshly prepared selection medium [growth medium + 350 µg/ml Zeocin (Catalog #R25005, Life Technologies, Carlsbad, California USA)]. Four new T-25 flasks were created from cells from each parent T-75 flask, seeded at 0.2 x 106 cells/ml in 5-ml culture media. Cell counts and media refreshments were performed periodically, and cells were cryopreserved as soon as cells reached 90% viability. Multiple cell banks were created from each surviving pool. In this case, cell selection lasted for fifteen days and resulted in nine pools all originating from the FI-Zeo1 based expression vector.
Batch overgrow culture for comparison of transfected cell pools
Transfected pools were compared for growth characteristics and overall productivity in a 30-ml, 7-day batch overgrow culture performed in CD DG44 Medium containing 350 µg/ml Zeocin in 150-ml square bottles at 37 °C, 5% CO2 and 125 rpm shaking. Cultures were monitored for viable and total cell counts using the CASY TT cell counter device (OLS OMNI Life Science, Germany) after seeding, and samples were collected for cell count and ELISA from day 3 onward until the day of harvest (day 7). Culture supernatants at days 5 and 6 were collected after centrifugation and analyzed via ELISA.
Presence of Zeocin in culture medium during protein production is optional. However, selection pressure is maintained for cultures performed during early cell line development. Large-scale production of recombinant proteins does not require the presence of Zeocin.
Subcloning
For clone selection, a suspension containing 5 cells/ml was prepared from a starting culture in its exponential growth phase. Twenty, 96-well plates were seeded at 0.5 cells / well in 100 µl cell suspension and were incubated in a humidified incubator set at 37 °C and 5% CO2. Clones showing >70% confluency / well were expanded to a 12 ml batch cultures in 50 ml mini bioreactors (Catalog#431720, Corning Inc., Mexico) via intermediate stages of 48-well and 6-well plate cultures. The mini bioreactors were incubated for three days inside a humidified shaker incubator set at 37 °C and 5% CO2 and 150 rpm for adequate air-gas exchange. Cultures were constantly monitored for growth characteristics and analyzed for volumetric productivity via ELISA at the end of day 4. After that, 52 out of the initial 80 clones, were selected based on growth characteristics and productivity and were further expanded and evaluated in 60 ml batch cultures.
The top 24 clones from this selection stage were further expanded to 150-ml square bottles with 80 ml working volume, and cell banks were prepared after a 6-day batch culture. Each clone was seeded at a target density of 0.15 x 106 cells/ml. Cultures were monitored daily for VCD and % viability and sampled daily for titer estimation starting at day 4. The top 12 producing clones were evaluated in 7-day batch cultures in a similar fashion.
Fed-batch culture to select top expressing clones
Fed batch cultures were performed using CD DG44 Medium containing 350 µg/ml Zeocin with two different feeding strategies and growth conditions, applied sequentially, to find the three best clones to be used in product quality assessment assays.
80 mL cell cultures were prepared in 250 mL shake flasks at a target seeding density of 0.3 x 106 VC/mL and cultured at 140 rpm/ 5% CO2/ 37 °C. On days 3 and 5, cultures were fed with 5% culture volume using “Efficient Feed A” commercial feed (Catalog # A2502301, Life Technologies, Carlsbad, California USA Laboratories,). The number of viable cells per mL and percent viability was monitored using the CASY TT cell counter device (OLS OMNI Life Science) after seeding, and samples were collected for cell count and ELISA from day 4 onward until the day of harvest (day 7). Culture supernatants were collected after centrifugation and analyzed via ELISA.
Subsequently, 300 mL cell cultures were prepared in a 1L shake flask at a target seeding density of 0.3 x 106 VC/mL and cultured at 140 rpm/ 5% CO2/ 37 °C. Cultures were fed daily starting at day 4 with 4% of CHO CD Efficient Feed™ A supplement (Catalog# A1023401, Life Technologies, Carlsbad, California USA). At day 5, cultures were transferred into a different incubator set at 32 °C and grown there until the day of harvest (day 11). The number of viable cells per mL and percent viability was monitored using the CASY TT cell counter device after seeding and samples were collected for cell count and ELISA from day 4 onwards. Culture supernatants were collected after centrifugation and analyzed via ELISA.
Titer estimation by ELISA
Titers of the gp145 in tissue culture samples were determined using a commercially available HIV-1 gp120 Antigen Capture Assay (Catalog# 5429, ABL Inc., Rockville, Maryland USA) using the manufacturer’s recommended protocol modified to use a purified gp145 reference standard. Briefly, reference standard and test samples were diluted and incubated with disruption buffer on 96-well ELISA plates coated with a mouse monoclonal antibody to HIV-1 gp120. After an hour incubation at 37 °C, plates were washed four times, and incubated for 1 hour at 37 °C with conjugate solution, containing HRP-conjugated human polyclonal antibodies to HIV-1 gp120. Plates were then washed and incubated at ambient temperature with HRP substrate solution for 30 min. Reaction was stopped with stop solution, and the absorbance was read at 450 nm. Concentrations of gp145 in test samples were determined relative to the standard curve.
Purification from cell culture harvest
Clarified harvests from each culture were purified using 2 ml of Galanthus nivalis Lectin (GNL) agarose (Catalog# AL1243–5, Vector Laboratories, Burlingame, California USA) packed in glass columns (Omnifit glass column, 006BCC-10–10-AF, 10 mm/ 100 mm, Kinesis, IL, USA). Each column was equilibrated with 20 mM Tris pH 8.0, 500 mM sodium chloride, 0.1% Triton™ X-100 at 200 cm/h for 7.5 column volumes. Clarified culture media was loaded at 100 cm/h. The column was then washed with 20 mM Tris pH 8.0, 500 mM sodium chloride, 0.1% Triton™ X-100 followed by 50 mM Tris pH 8.0, 150 mM sodium chloride at 200 cm/h for 7 column volumes. Elution was performed using 50 mM Tris pH 8.0, 150 mM sodium chloride, and 0.5 M mannopyranoside at 75 cm/h. Fractions with the highest absorbance were pooled and filtered using 0.8/0.2 µm Acrodisc PF Syringe Filters (Catalog #P4187, Pall Corporation, Baltimore, Maryland USA).
SDS-PAGE
Samples were prepared by diluting in 1x LDS sample loading buffer (Thermofisher) and heating them at 90 °C for 2 min. 5% β-Mercaptoethanol was added to reduced samples. Approximately, 2 µg of protein was loaded per lane of a NuPAGE™ 4–12% Bis-Tris Protein gel (Thermofisher). Gels were run at a constant voltage of 200 V for 50 min or until the dye front reached the bottom of the gel. The gels were then separated from the gel cassette, rinsed briefly with water, and stained with Instant Blue Stain (Catalog #1SB1L, Expedeon, San Diego, California USA) for up to an hour. Gels were imaged directly or after destaining in water until a desired background was achieved using ChemiDoc XRS+ imager (BioRad).
Western blotting
SDS - PAGE gels were run as described above albeit with an approximate load of 0.25 µg per well. The proteins were transferred from the gel to PVDF membrane for antibody staining and detection using the iBlot™ 2 gel transfer device (Thermofisher). Pre-programmed method ‘P0’ was used for the transfer. The method comprises of three stages of increasing voltages, namely a) 20 V for 1 minute, b) 23 V for 4 min and, c) 25 V for 2 min for a total duration of 7 min.
After transfer, the membrane was blocked using 15 ml of diluent (ABL proprietary formulation) shaking at room temperature for 30–60 min. The membrane was incubated in 15 ml of diluent plus 10 µg mouse anti-gp120 (VRCC 16H3) and 20 µg mouse anti-gp120 (VRCC 18F11), and it was placed on a shaker at room temperature for 60 min. The membrane was washed four times with approximately 25 ml of TBST (Thermofisher, catalog #28360) shaking for 2 min each at room temperature. The membrane was incubated in 1:5000 anti-mouse IgG Alkaline Phosphatase (Catalog #A3562, Millipore-Sigma, St. Louis, Missouri, USA) in 25 ml TBST, pH 8 shaking at room temperature for 60 min. The membrane was washed 4 times in about 25 ml TBST, pH 8 at room temperature shaking for 2 min. The membrane was developed in approximately 15 ml BCIP/NBT (Catalog #50–81-07, KPL pharma, Gaithersburg, Maryland USA) at room temperature shaking until color developed to result in an empirically determined acceptable signal: noise (about 3 min). The membrane was rinsed with water and imaged using the ChemiDoc XRS+ Imager (BioRad).
N-glycan analysis
N-linked oligosaccharides were enzymatically released from the HIV gp145 envelope purified proteins using peptide-N-glycosidase F (Rapid™ PNGase-F, Catalog #P0710S, New England Biolabs, Ipswich, Massachusetts USA). The free oligosaccharides were then derivatized with the fluorophore 2-AB on the reducing terminal N-acetylglucosamine (GlcNAc). Following a cleanup step using GlycoClean™ S Cartridges (Catalog #GKI-4726, Prozyme, Hayward, California USA) to remove protein and excess labeling reagents, the fluorescently labeled oligosaccharides were separated by hydrophilic interaction chromatography (HILIC). Bound oligosaccharides were eluted from the column with an increasing gradient of 0.05% TFA in water and analyzed via HPLC (Waters Inc., Milford, Massachusetts USA). The relative % peak areas of oligosaccharides were calculated in Empower 3 Chromatography software.
Antigenicity ELISA
The antigenicity of purified gp145 to a panel of neutralizing and non-neutralizing antibodies was performed by determining the half maximal effective concentration (EC50) using ELISA. Antibodies evaluated included CD4 binding site antibodies VRC01 (neutralizing) and F105 (non-neutralizing), V1/V2 antibody PG9 (neutralizing) and gp41 hinge region antibody HK20 (neutralizing). To show that the binding of the CD4 inducible neutralizing antibody 17b is enhanced in the presence of CD4, 17b antibody was evaluated in the presence and absence of CD4. For each ELISA, the 96-well ELISA plates were coated with 100 µl antibody diluted to 1 µg/ml (HK20 at 2 µg/ml) in 50 mM sodium bicarbonate, pH 9.6 buffer and incubated at 2–8 °C overnight. The plates were then blocked with 200 µl/well SuperBlock PBS blocking buffer (Thermo Fisher Scientific, catalog #37518) for 1 hour at room temperature. Purified gp145 was serially diluted from 4,000 to 0.98 ng/ml (for F105, gp145 was diluted at 500 to 0.12 ng/ml) in four-fold dilutions. For the 17b + CD4 binding assay, prior to performing the dilution series, samples were diluted to 4,000 ng/ml and combined with a 1:2 molar ratio with soluble CD4 (sCD4) for 1 hour at 37°C to allow for complex formation. Then, 100 µl of diluted samples were added per well, in duplicate. The plates were incubated at 37 °C for 1 hour and washed with 300 µl/well PBS + 0.05% Tween-20 four times.
Next, 100 µl of diluted HRP-conjugated human anti-gp120 polyclonal antibody was added, and the plate were incubated at 37 °C. After an hour of incubation, the plates were washed as before and 100 µl/well K-Blue Aqueous TMB substrate (Neogen Life Sciences, Lexington, Kentucky USA catalog #331177) was added to each well, and the plates were incubated at room temperature for 30 min. The reaction was stopped via addition of 100 µl / well 2N sulfuric acid, and absorbance at 450 nm was measured. The EC50 was determined by plotting the absorbance versus log concentration and performing nonlinear regression analysis with a four-parameter logistic function.
CD4 Affinity
96-well ELISA plates were coated with 100 µl sCD4 diluted to 1 µg/ml in 50 mM sodium bicarbonate, pH 9.6 buffer and set aside at 2–8 °C overnight. The plates were then blocked with 200 µl/well SuperBlock PBS blocking buffer for 1 hour at room temperature. Eight four-fold serial dilutions of purified gp145, 4000 to 0.98 ng/ml, were prepared and 100 µl was added to the plates in duplicate. The plates were incubated at 37 °C for 1h and then washed with 300 µl/well PBS + 0.05% Tween-20 four times. 100 µl/well diluted HRP-conjugated human anti-gp120 polyclonal antibody, was added and the plates were incubated at 37 °C for an additional hour and washed as before. 100 µl/well K-Blue Aqueous peroxidase substrate was added to each well and the plates were incubated at room temperature for 30 min. The reaction was stopped via addition of 100 µl/well 2N sulfuric acid, and absorbance at 450 nm was measured. EC50 was determined by plotting the absorbance versus log concentration and performing nonlinear regression analysis with a four-parameter logistic function.
Results and discussion
STEP vector design and expression analysis
A graphical representation of the DNA elements present in the STEP DNA plasmid is shown in Figure 1A. Briefly, the STEP plasmid is a bicistronic mammalian expression vector that allows the stable, constitutive expression of a selection marker and gene of interest driven by a CMV promoter. The gene of interest (GOI) encoding the target protein (e.g., gp145) is present in the first cistron of the mRNA and its translation is cap dependent. The second cistron is a mutated 14 kDa Sh ble gene product (FI-Zeo) that binds and thereby inactivates the antibiotic Zeocin [11], and it is translated through the internal ribosomal entry site (IRES) [12–16]. As previously reported, the mutation engineered into the Sh ble gene (zeoR) results in an enzyme that is less potent than the wild-type Sh ble gene product in neutralizing Zeocin [17]. To further attenuate Zeocin resistance and drive up recombinant protein expression a small upstream open reading frame (uORF, indicated as spacer in Figure 1) is cloned between the IRES and the ATG start codon of the zeoR gene. Initiation of IRES-mediated translation is described to be less efficient than cap-dependent translation of the upstream cistron [14, 15]. In addition, reinitiation of translation occurs at a downstream open reading frame with reduced translational efficiency, dependent on the size of the uORF [18]. The introduced uORF thus results in impaired translation of the mutated ZeoR gene product [17]. The combination of Zeocin marker mutants and variable sizes of uORFs that variably hamper translation results in an adaptable stringent cell selection system. Since a cell needs to express considerable amounts of ZeoR to survive Zeocin selection that cell per definition makes considerable amounts of the protein of interest, as the gene of interest and ZeoR gene are being expressed on one mRNA [17] Further gains in recombinant protein expression are achieved via the use empirically determined novel enhancer elements [19].
Figure 1.
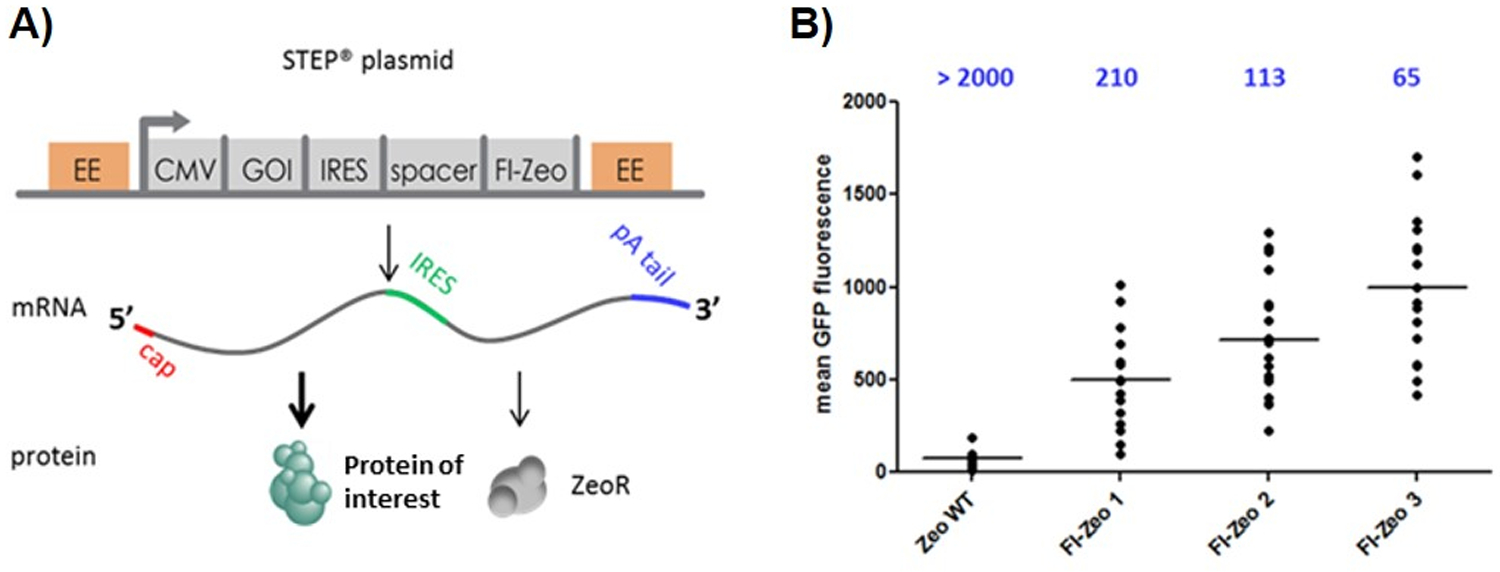
Schematic representation of the STEP technology plasmid (A) and Effects of selection stringency on clone formation and expression level employing GFP as marker protein (B). Plasmids with WT-Zeo and three increasing levels of FI-Zeo-(1, 2, 3) and GFP as GOI were transfected into CHO cells. The number of generated clones was determined (shown on top of the graph) and a small number of clones was subjected to flow cytometry to determine GFP fluorescence. Each dot represents the average GFP fluorescence level from a clone. The horizontal black bar shows the average GFP expression level.
In total, three STEP plasmids, which differ in the mutations carried by the zeoR gene, were generated, resulting in FI-Zeo proteins that differ in their ability to neutralize Zeocin i.e., conferring selection stringency. This three STEP plasmid system forces a cell to maximum expression of the protein of interest. Preliminary studies using Green Fluorescence Protein (GFP) were performed to demonstrate the effect and utility of the three plasmid STEP system. As shown in Figure 1B, mean GFP fluorescence increased in CHO cell clones produced with STEP constructs (FI-Zeo1–2-3) as compared to a construct containing wild type ZeoR protein (Zeo WT). In addition, mean GFP fluorescence further increased using the higher stringency vector FI-Zeo2 as compared to FI-Zeo1. Furthermore, fluorescence was even higher when using the highest stringency vector FI-Zeo3, nearly double that of the FI-Zeo2. Importantly, because of increasing selection stringency, the number of surviving cell clones is reduced while the GFP fluorescence increased, meaning that the number of clones that needs to be screened is lower when compare with other technologies. In this experiment, more than 2,000 surviving cell clones were recovered with the wild-type ZeoR construct, while 210 clones, 113 clones, and 65 clones were obtained with the with FI-Zeo1, FI-Zeo2 and FI-Zeo3, respectively.
The value of this three-plasmid STEP system is that it is very difficult to predict which GOI is optimally expressed at which stringency, for instance if they produce difficult to express or to fold proteins, or proteins that are toxic to the cells. The three-tiered stringency levels ensure that the optimal stringency will be found for any GOI.
Generation of CHO cells expressing gp145 protein
Upon transfection of suspension CHO cells with expression plasmids encoding the gp145 protein, followed by antibiotic selection of transfected cells, nine stable pools expressing gp145 were obtained. These were compared for productivity and population doubling times in a small-scale batch overgrow culture set in duplicate. Figure 2A shows the gp145 expression level at day 5 and population doubling times for all nine CHO pools. Population doubling times (pdt) presented here is an average of pdt calculated at days 3, 5 and 6 with respect to day 0. Pool 8 showed the highest gp145 protein expression level at 16.9 µg/ml and a pdt of 26h. Pools 4 and 9 showed high gp145 expression and favorable doubling time of <24h making them both excellent candidates for further development. As a result, Pool 9 was selected for subcloning.
Figure 2.
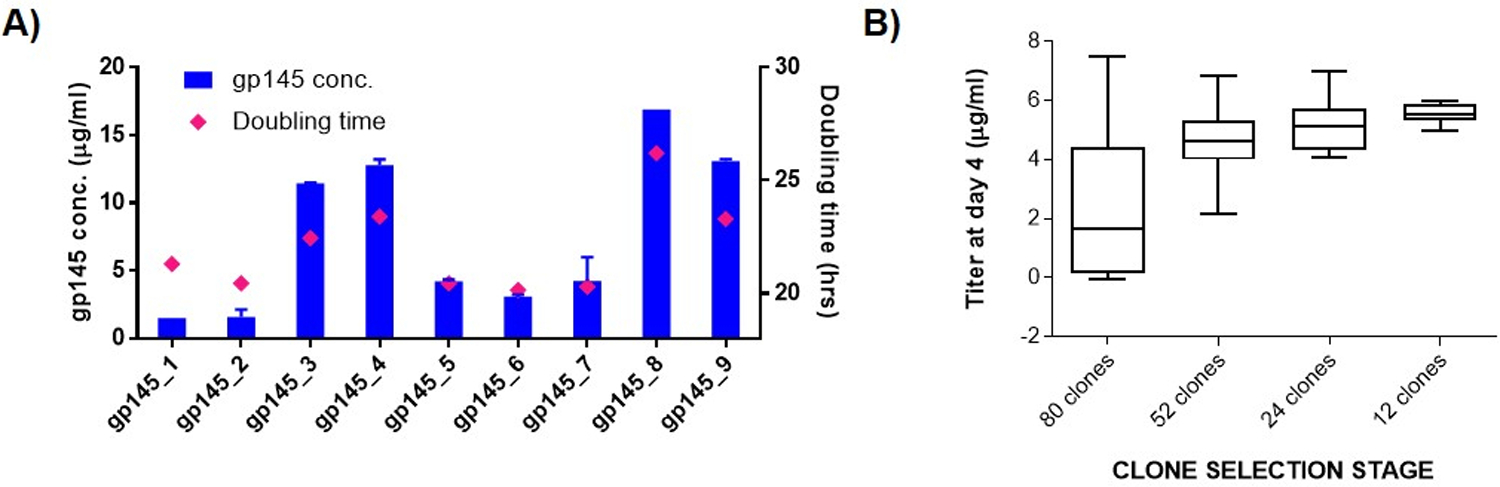
Pool generation and subcloning and clone selection for gp145. (A) Productivity and growth characteristics of STEP™-generated pools for gp145 a 7-day shaking batch overgrow culture. Day 5 titers and average doubling time are depicted as blue bars and magenta diamonds, respectively. (B) Subcloning, starting from pool gp145_9, and clone selection for gp145. The box-and-whiskers plot shows the outcome of the selection steps leading up to selection of the top 12 clones. At each stage, clones with the highest volumetric productivities and better growth characteristics were selected. The box represents the 25th and 75th percentile w.r.t. ELISA titers and whiskers are drawn at the minimum and maximum values of each set.
Subcloning was performed via the limiting dilution method to ensure colony outgrowth from a single cell. Clones were monitored and analyzed by using the CloneSelect™ Imager to assure monoclonality. Pool 9 was expanded and continuously cultured for approximately two weeks to obtain cells in their exponential growth phase for sub cloning. Twenty 96-well plates were seeded at 0.5 cells / well in 100 µl cell suspension. Clones showing >70% confluency per well were carried forward systematically until the number of positive clones was down selected from 80 to 12 top clones, based on growth characteristics and productivity at each stage. Sub-cloning resulted in several clones with acceptable doubling time (less than 24h, data not shown) and an increase in overall volumetric productivity, after 4 days batch culture (5–6 µg/mL), as depicted in Figure 2B.
Top twelve clones originating from the clone expansion and selection scheme were investigated for productivity in a 7-day shaking fed-batch culture. Cultures were sampled for cell counts and titer estimation via ELISA at days 5, 6 and 7. Clones were rank ordered based on their volumetric productivity at day 5. All twelve clones displayed similar titers (Figure 3A). The top three clones in terms of expression, namely 5–3, 6–5, and 13–2, were reevaluated in an 11-day fed-batch culture to study the effect of temperature on overall culture duration and productivity. Figure 3B shows the growth profile and gp145 expression levels for clones 5–3, 6–5, and 13–2. All three clones maintained >90% viability for the entire duration of the fed-batch culture with equivalent peak viable cell densities of ~1.5 x 106 cells/ml. All three clones also showed equivalent gp145 expression levels with an average expression level of 47.4 µg/ml at day 11 (Figure 3C).
Figure 3.
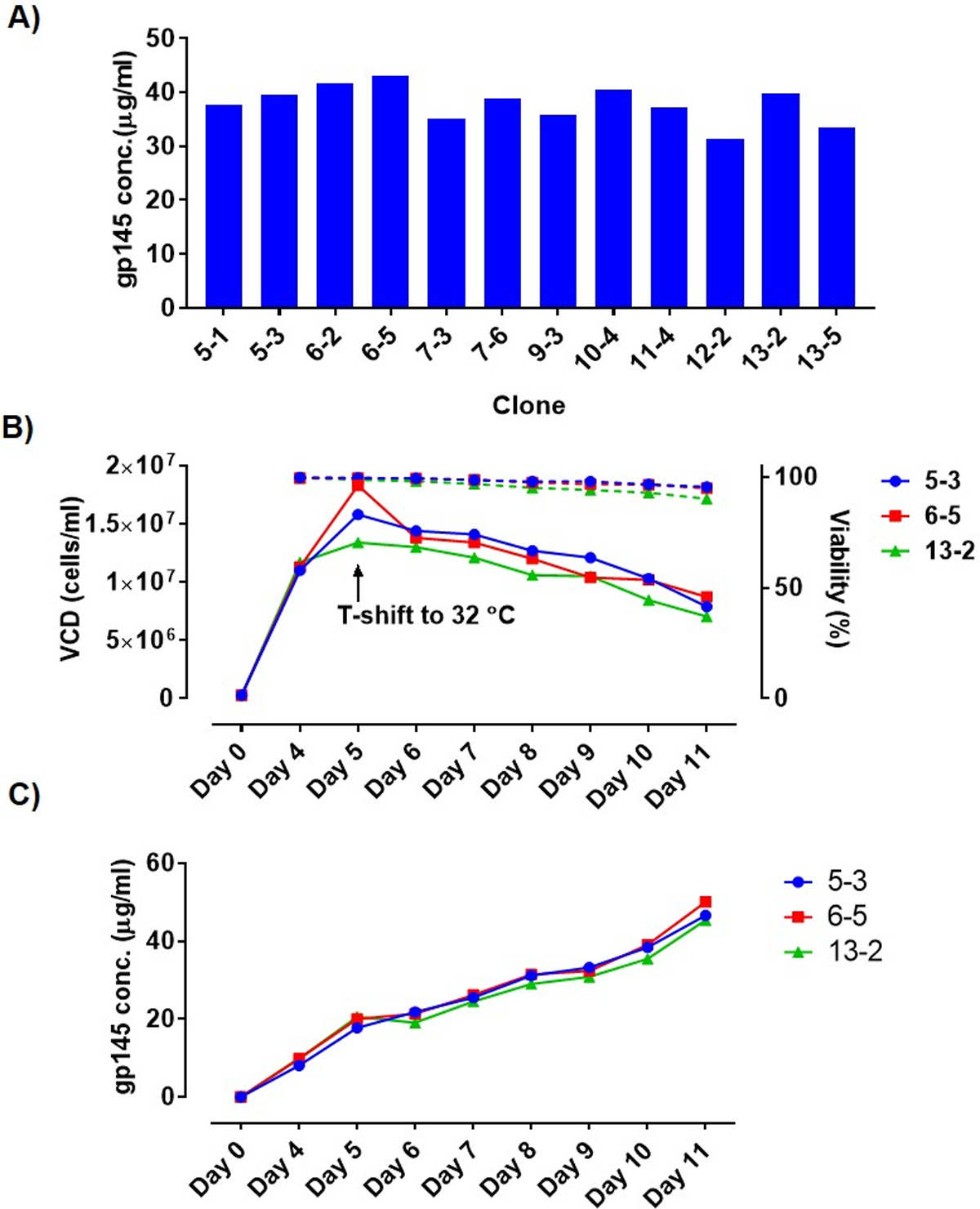
A) gp145 expression for top 12 clones in a 7-day fed-batch culture showing a high level of homogeneity in volumetric productivity at day 7. (B) Growth profile of top 3 gp145-expressing clones from 11-day shaking fed-batch cultures. Viable cell counts and percent viabilities are depicted as solid and dotted lines, respectively. (C) gp145 levels in fed-batch as measured via ELISA showing a steady increase of gp145 for all three clones.
Purification and functional analyses of gp145 envelope protein
To analyze the quality of gp145 protein produced by the three selected cell lines, day 11 harvests from corresponding fed-batch cultures were purified using one step GNL-affinity chromatography. SDS-PAGE and western blotting was used to confirm the identity and purity of the purified protein. An average HIV gp145 envelope protein recovery of 71% (data not shown) was obtained during this one step purification process with 80–90% purity. As shown in Figure 4A, the SDS-PAGE analysis of the affinity-purified gp145 envelope protein from the clones 5–3, 6–5 and 13–2 demonstrated the presence of a protein of the expected size (~150 kDa). Lower molecular protein impurities of 50, 43 and 37 kDa were also observed. However, the relative amount of these impurities with respect to the main band was comparable to that for the reference standard. Subsequent Western blot analyses (Figure 4B) of purified gp145 confirmed the presence of the ~150 kDa gp145 protein band and again minor bands at ~75 and 50 kDa. The latter low molecular weight products were also observed in the reference standard (purified material from a previously generated cell line) albeit at lower intensities. Minor bands at about 50, 80 and >250 kDa are known gp145-related products identified with various monoclonal and polyclonal antisera. The >250 kDa product is presumably an aggregated form that does not fully denature in SDS-PAGE, and the 50 and 80 kDa products are presumably gp145 breakdown products generated during cell culture; the cleavage site is not identified.
Figure 4.
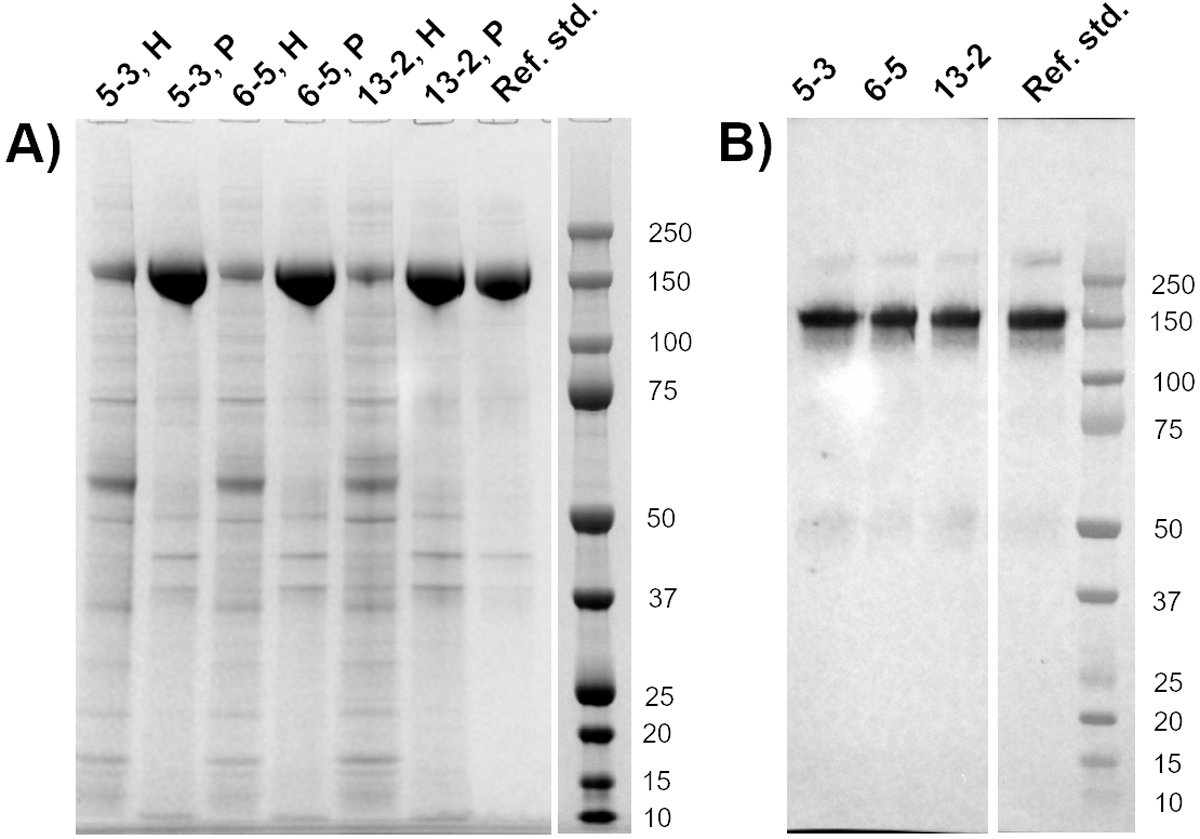
SDS-PAGE and Western Blot gels showing purity and identity of the purified gp145 clones from the top three candidate clones. (A) SDS-PAGE characterization of gp145 harvest (H) and GNL-affinity purified material (P). The gp145 protein appears as a major band of ~150 kDa. Some low-molecular weight impurities were observed. (B) Western blot of affinity purified gp145 for top three clones. Purified gp145 from a previously generated clone was used as a reference standard.
HIV gp145 is a heavily glycosylated protein with glycosylation playing a key role in receptor binding and therefore functionality.
Glycosylation pattern of purified gp145 proteins from all three clones was evaluated using HILIC-based quantitation of enzymatically released 2AB-labeled glycans. Glycan profiles of all three clones show a high degree of similarity with the gp145 reference standard as seen in Figure 5. This shows that the STEP-generated cell lines produce high levels of gp145 while still maintaining correct glycosylation patterns compared to the reference standard. Interestingly, all three CHO clones show matching glycosylation patterns which is a clear indication of the high degree of clonality obtained from the STEP-generated cell pools. Here, it is noteworthy to mention that we have observed in many other programs in progress using STEP technology that cell pools obtained have low heterogeneity, i.e., clones generated from the pools are remarkably similar in both yield and glycosylation patterns (data not shown).
Figure 5.
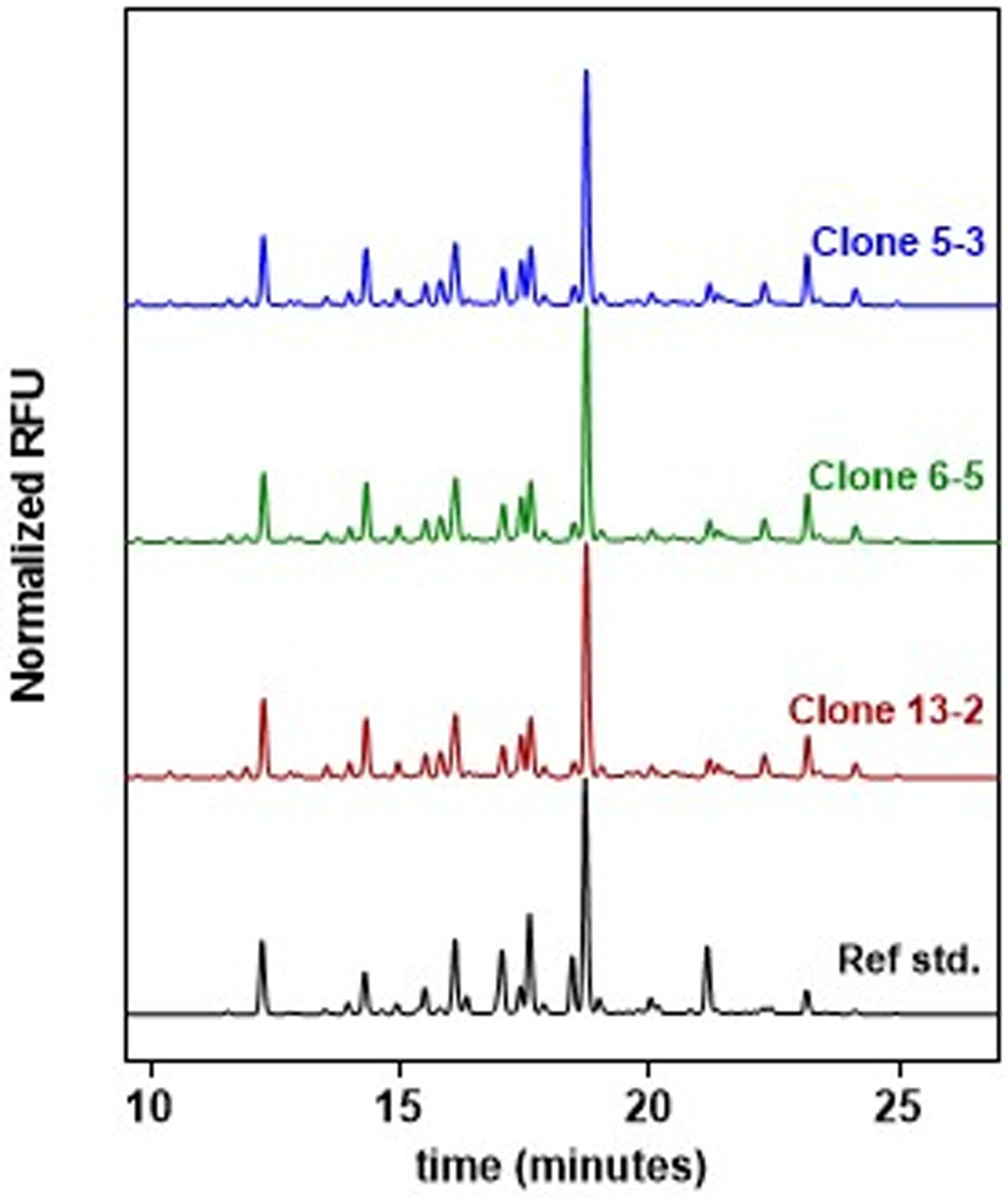
Representation of the dN-linked glycans from gp145 purified from STEP clones, 5–3, 6–5 and 13–2, released by PNGase-F. All clones show a high degree of similarity between them and with the reference standard, being all major peaks of gp145 present in the envelope proteins generated with STEP technology.
Conformational integrity and functionality of the gp145 envelope proteins purified from day-11 fed-batch cultures was demonstrated by high affinity binding to soluble CD4. Hereto, the CD4 binding site of antibodies such as VRC01 (CD4 binding site specific) and F105 (non-neutralizing), Hk20 (gp41 hinge specific), PG9 (quaternary epitope specific) and 17b (CD4-induced epitope specific) were tested in this study. Figure 6A depicts the binding profile of purified gp145 to the 17b antibody in the presence and absence of soluble CD4 (sCD4). Each clone shows a pronounced increase in binding to 17b in the presence of sCD4 thus demonstrating that the gp145 protein undergoes the expected conformational change because of CD4 binding. The binding proved to be specific and occurs in the same range as the reference standard. In Figure 6B the EC50 values for all antibodies tested are shown, and the EC50 values were found to be similar for each clone and in the same range as to the reference standard for the different antibodies tested. Proper display of epitopes and antigenicity are critical attributes of a viable vaccine candidate. Equivalency of the strength and nature of binding of purified gp145 generated using the STEP technology and the reference standard suggest that the epitopes critical to generating a neutralizing humoral response towards HIV-1 gp145 are present in STEP-generated gp145.
Figure 6.
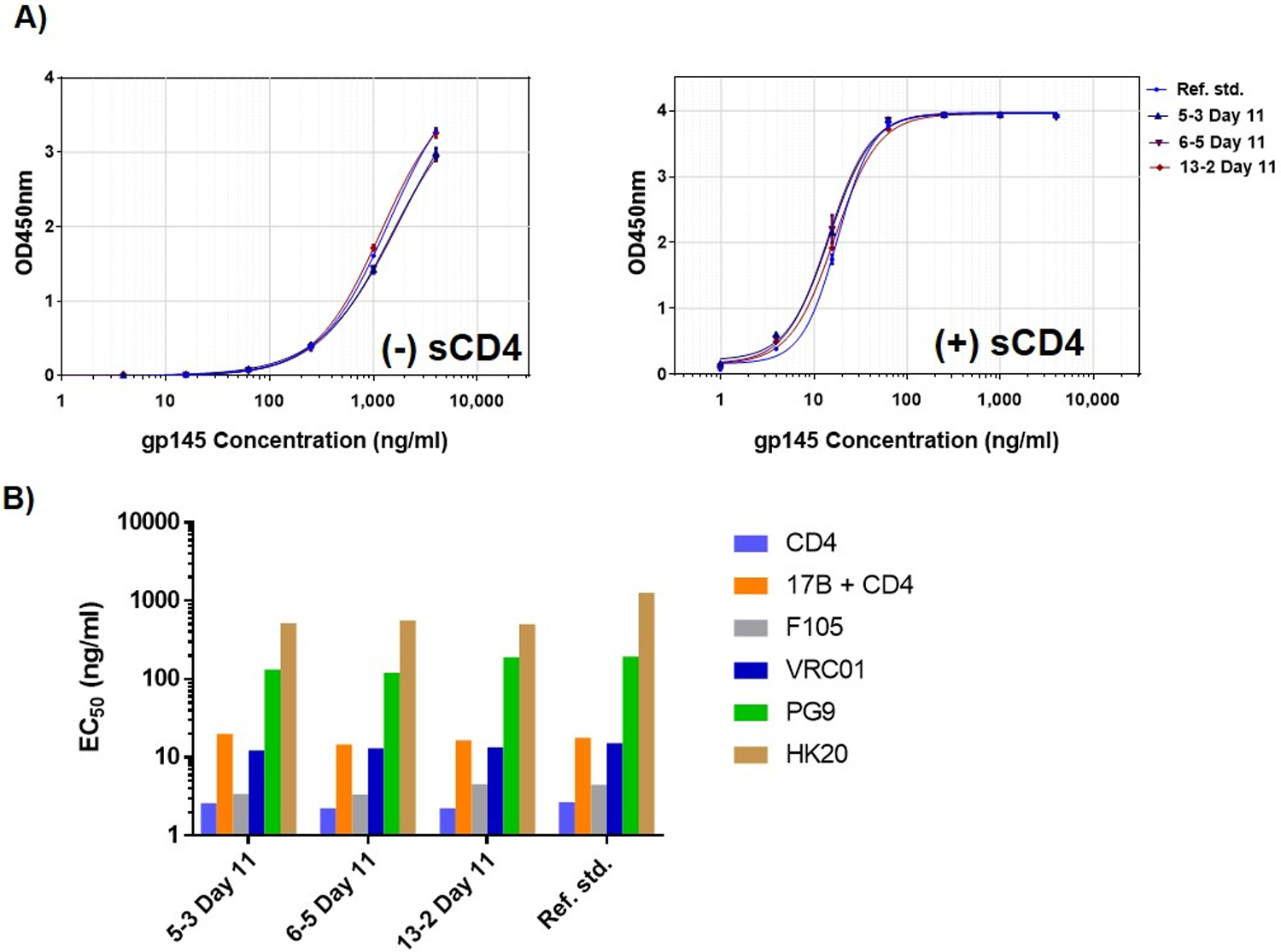
Functionality of purified gp145: In vitro activity. (A) Binding of purified gp145 to 17b in the absence and presence of soluble CD4. The gp145 proteins from the different clones: 5–3 (dark blue), 6–5 (purple), 13–2 (red) were tested and show similar binding to the reference standard (light blue). (B) EC50 values for STEP generated gp145 for clones 5–3, 6–5 and 13–2 against the panel of antibodies. GNL-purified gp145 from a previously developed cell line was used as a reference standard.
In conclusion, a stable gp145-protein expressing CHO cell-line was developed using our novel expression plasmid technology. Starting from an initial expression level of 4–7 µg/ml in batch culture, a fed-batch process was developed, yielding gp145 expressed at the 50 µg/ml level. This is an order of magnitude higher than the 4 µg/ml achieved by the previously described cell line produced using a non-STEP bicistronic expression system [1]. Moreover, the STEP vectors deliver a high-quality protein with native-like glycosylation patterns. The novel expression technology allowed for the rapid generation of CHO pools with low heterogeneity. During clonal selection, 80 clones were screened and top 12 producers selected based on growth characteristics and titers. The top 3 selected clones proved similar in titer and provided a gp145 protein product with similar molecular weight, glycan profile, functionality, and activity as the reference standard developed using non-STEP methodology. The flexible stringency selection system of STEP vectors ensures that our expression platform achieves optimal protein expression levels in CHO cells for any GOI without additional need for gene amplification or multiple rounds of subcloning and provides stable clones with low heterogeneity in about a 12 week timeframe.
Acknowledgements
The authors thank Deborah Deane, Nandini Sane, Cyndy Rhodes and Aeliya Jafri for coordinating and managing the project, and Dr. Remco Spanjaard for insightful discussions during writing of the manuscript. We also thank Mihir Shukla, Rizwan Manzer, Mohamed Shaker, Jeff Caron, and Andy Wasserman from Batavia for their contributions in executing the experiments described in this paper. The authors acknowledge Dr. Barton Haynes (Duke University) for the provision of the antibodies VRCC 16H3 and 20 VRCC 18F11 used in this study as well as ABL and the Military HIV Research Program (MHRP) for providing the gp145 gene sequence.
Funding
This work was funded, in part, with federal funds from the Division of AIDS (DAIDS), National Institute of Allergy and Infectious Diseases (NIAID), NIH, Department of Health and Human Services, under contract HHSN2722011000021I task order HHSN27200022 that was awarded to ABL. Additional support was provided by Batavia Biosciences. The views expressed here are our private opinions and are not considered official views of the U.S. government or an expression of U.S. government endorsement.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
All authors have no competing interests to declare. Declarations of interest: none.
References
- [1].Wieczorek L, Krebs SJ, Kalyanaraman V, Whitney S, Tovanabutra S, Moscoso CG, et al. Comparable Antigenicity and Immunogenicity of Oligomeric Forms of a Novel, Acute HIV-1 Subtype C gp145 Envelope for Use in Preclinical and Clinical Vaccine Research. J Virol 2015;89:7478–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Go EP, Irungu J, Zhang Y, Dalpathado DS, Liao HX, Sutherland LL, et al. Glycosylation site-specific analysis of HIV envelope proteins (JR-FL and CON-S) reveals major differences in glycosylation site occupancy, glycoform profiles, and antigenic epitopes’ accessibility. J Proteome Res 2008;7:1660–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Irungu J, Go EP, Zhang Y, Dalpathado DS, Liao HX, Haynes BF, et al. Comparison of HPLC/ESI-FTICR MS versus MALDI-TOF/TOF MS for glycopeptide analysis of a highly glycosylated HIV envelope glycoprotein. J Am Soc Mass Spectrom 2008;19:1209–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhu X, Borchers C, Bienstock RJ, Tomer KB. Mass spectrometric characterization of the glycosylation pattern of HIV-gp120 expressed in CHO cells. Biochemistry 2000;39:11194–204. [DOI] [PubMed] [Google Scholar]
- [5].Raska M, Takahashi K, Czernekova L, Zachova K, Hall S, Moldoveanu Z, et al. Glycosylation patterns of HIV-1 gp120 depend on the type of expressing cells and affect antibody recognition. J Biol Chem 2010;285:20860–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gonzalez-Feliciano JA, Akamine P, Capo-Velez CM, Delgado-Velez M, Dussupt V, Krebs SJ, et al. A recombinant gp145 Env glycoprotein from HIV-1 expressed in two different cell lines: Effects on glycosylation and antigenicity. PLoS One 2020;15:e0231679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].McCaffrey RA, Saunders C, Hensel M, Stamatatos L. N-linked glycosylation of the V3 loop and the immunologically silent face of gp120 protects human immunodeficiency virus type 1 SF162 from neutralization by anti-gp120 and anti-gp41 antibodies. J Virol 2004;78:3279–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pollakis G, Kang S, Kliphuis A, Chalaby MI, Goudsmit J, Paxton WA. N-linked glycosylation of the HIV type-1 gp120 envelope glycoprotein as a major determinant of CCR5 and CXCR4 coreceptor utilization. J Biol Chem 2001;276:13433–41. [DOI] [PubMed] [Google Scholar]
- [9].Ghaderi D, Zhang M, Hurtado-Ziola N, Varki A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev 2012;28:147–75. [DOI] [PubMed] [Google Scholar]
- [10].Kim JY, Kim YG, Lee GM. CHO cells in biotechnology for production of recombinant proteins: current state and further potential. Appl Microbiol Biotechnol 2012;93:917–30. [DOI] [PubMed] [Google Scholar]
- [11].Drocourt D, Calmels T, Reynes JP, Baron M, Tiraby G. Cassettes of the Streptoalloteichus hindustanus ble gene for transformation of lower and higher eukaryotes to phleomycin resistance. Nucleic Acids Res 1990;18:4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gurtu V, Yan G, Zhang G. IRES bicistronic expression vectors for efficient creation of stable mammalian cell lines. Biochem Biophys Res Commun 1996;229:295–8. [DOI] [PubMed] [Google Scholar]
- [13].Ho SC, Bardor M, Li B, Lee JJ, Song Z, Tong YW, et al. Comparison of internal ribosome entry site (IRES) and Furin-2A (F2A) for monoclonal antibody expression level and quality in CHO cells. PLoS One 2013;8:e63247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Houdebine LM, Attal J. Internal ribosome entry sites (IRESs): reality and use. Transgenic Res 1999;8:157–77. [DOI] [PubMed] [Google Scholar]
- [15].Kaufman RJ, Davies MV, Wasley LC, Michnick D. Improved vectors for stable expression of foreign genes in mammalian cells by use of the untranslated leader sequence from EMC virus. Nucleic Acids Res 1991;19:4485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ho SC, Bardor M, Feng H, Mariati, Tong YW, Song Z, et al. IRES-mediated Tricistronic vectors for enhancing generation of high monoclonal antibody expressing CHO cell lines. J Biotechnol 2012;157:130–9. [DOI] [PubMed] [Google Scholar]
- [17].Van Blokland HJ, Hoeksema F, Siep M, Otte AP, Verhees JA. Methods to create a stringent selection system for mammalian cell lines. Cytotechnology 2011;63:371–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kozak M Constraints on reinitiation of translation in mammals. Nucleic Acids Res 2001;29:5226–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hoeksema F, van Blokland R, Siep M, Hamer K, Siersma T, den Blaauwen J, et al. The use of a stringent selection system allows the identification of DNA elements that augment gene expression. Mol Biotechnol 2011;48:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]