

Abstract
Hibiscus hamabo is a semi-mangrove species with strong tolerance to salt and waterlogging stress. However, the molecular basis and mechanisms that underlie this strong adaptability to harsh environments remain poorly understood. Here, we assembled a high-quality, chromosome-level genome of this semi-mangrove plant and analyzed its transcriptome under different stress treatments to reveal regulatory responses and mechanisms. Our analyses suggested that H. hamabo has undergone two recent successive polyploidy events, a whole-genome duplication followed by a whole-genome triplication, resulting in an unusually large gene number (107 309 genes). Comparison of the H. hamabo genome with that of its close relative Hibiscus cannabinus, which has not experienced a recent WGT, indicated that genes associated with high stress resistance have been preferentially preserved in the H. hamabo genome, suggesting an underlying association between polyploidy and stronger stress resistance. Transcriptomic data indicated that genes in the roots and leaves responded differently to stress. In roots, genes that regulate ion channels involved in biosynthetic and metabolic processes responded quickly to adjust the ion concentration and provide metabolic products to protect root cells, whereas no such rapid response was observed from genes in leaves. Using co-expression networks, potential stress resistance genes were identified for use in future functional investigations. The genome sequence, along with several transcriptome datasets, provide insights into genome evolution and the mechanism of salt and waterlogging tolerance in H. hamabo, suggesting the importance of polyploidization for environmental adaptation.
Introduction
Hibiscus hamabo Siebold & Zuccarini, from the Malvaceae family, is a deciduous tree that can reach 5 m in height [1]. H. hamabo, which is endangered, is an appealing ornamental woody plant with obvious seasonal variations; the plant blooms in summer, presenting light green to light lemon-yellow flowers, and its leaves turn red in fall [1]. The plant is native to Asia and mainly distributed in the coastal areas of China, Korea, and Japan, as well as India and some Pacific islands [1, 2]. The distribution of H. hamabo suggests that this species is not adapted to cold zones but prefers the warm regions of coastal areas in the northwestern Pacific. It is known as a semi-mangrove species with long-floating seeds [1]. Plants from the Hibiscus genus show complex ploidy and various chromosomal numbers. Diploids, tetraploids, hexaploids, and octoploids can all be found in the genus, with various chromosomal numbers, ranging from Hibiscus citrinus (2n = 22) [3] to Hibiscus maculatus (2n = 180). H. hamabo is known to contain 92 (2n = 92) chromosomes, but it remains unclear whether it is a diploid species (x = 46) or a diploidized tetraploid species (x = 23) [1]. One phylogenetic study suggested that H. hamabo is related to Hibiscus tiliaceus (2n = 96) and Hibiscus glaber (2n = 82) [4], whereas another study indicated a close relationship between H. hamabo and Hibiscus macrophyllus [5]. However, both studies were based on a very restricted number of markers and limited taxon sampling, and the relationship between H. hamabo and other Hibiscus plants therefore remains ambiguous. H. hamabo may have independently appeared outside the normal range inhabited by H. tiliaceus through the long-distance floating of seeds and/or adaptation to new circumstances [6].
H. hamabo is a semi-mangrove plant that can survive in tidal saline soil and under immersion [7, 8]. How it adapts to such stressful environmental conditions is an intriguing question. Numerous studies have investigated the molecular mechanisms that underlie plant tolerance to salt and waterlogging. Under salt stress, the salt overly sensitive (SOS) pathway, which is conserved in plants, is induced within 2 h to export Na+ out of cells; SOS2 kinase and the SOS1 Na+ antiporter are then induced to increase Na+ efflux [9]. Meanwhile, the SOS pathway coordinates with high-affinity K+ transporter (HKT1) to regulate Na+/K+ homeostasis in plant cells, reduce the accumulation of Na+ in stem tissues, and protect leaves from poisoning caused by Na+ [10]. The vacuolar partitioning of Na+, which is regulated by a Na+/H+ exchanger (NHX), is the main adaption mechanism used by both halophytes and glycophytes to reduce ion toxicity in the cytoplasm [10]. In addition, ABA-dependent and independent SUCROSE nonfermenting1-related PROTEIN KINASE2 genes (SnRKs) in Arabidopsis were found to play key roles in regulation at both the transcriptional and post-transcriptional levels under salt stress, mainly through the targeted regulation and phosphorylation of downstream components [11]. When oxygen is limited because of flooding, anaerobic respiration, which is related to glycolysis and fermentation, is the primary method used by plants to produce energy [12, 13]. Anaerobic proteins (ANPs) such as alcohol dehydrogenase (ADH), pyruvate decarboxylase (PDC), and lactate dehydrogenase (LDH), which play major roles in anaerobic respiration pathways, have been identified as key functional components in many plant species, including Arabidopsis, rice, and maize [12]. Moreover, as the hormone ethylene is critical for coping with waterlogging [13], Group VII ethylene-responsive element binding factors (ERFs) in Arabidopsis are key regulators of genes related to hypoxia and the waterlogging stress response [14].
Recent studies have shown that global warming may result in rising sea levels, which will increase the stresses endured by coastal plants [7]. As a shelterbelt tree species along the coasts, H. hamabo is a good candidate for studying responses and regulatory mechanisms under stress, which are crucial for using this species in the ecological greening of coastal vegetation systems. Some attempts have been made to understand the mechanisms of stress adaptation in H. hamabo, but the absence of genomic data has hindered systematic investigations to date [2, 15]. Here, we report the assembly of the H. hamabo genome at the chromosome level using a combination of PacBio, Hi-C, and Illumina sequencing technology. Using multiple transcriptomic datasets obtained under different treatments and at various time points, we reveal the adaptive mechanisms of H. hamabo to saline and waterlogged environments. This genome should serve as a useful resource, helping the investigation of semi-mangrove plants and promoting the development and utilization of woody coastal plants.
Results
Genome ploidy, sequencing, assembly, and annotation
An investigation of ploidy revealed that H. hamabo is a diploid species with 46 pairs of chromosomes (2n = 2x = 92) (Fig. S2a), and k-mer analysis of Illumina data indicated that its genome size was 1712 Mb. The results of a genome survey indicated that the H. hamabo genome had a low heterozygosity of 0.06% (Fig. S2b, S2c). To obtain a high-quality genome, we adopted an elaborate strategy for genome assembly. We integrated paired-end reads from Illumina (226 G), single-molecule long reads from PacBio (N50 = 7.7 kb, 138 G), and Hi-C sequencing data (161 G) to perform the following steps. First, an initial assembly with a size of 1717 Mb (contig N50 = 1.6 Mb, 4358 contigs) was constructed with the PacBio long reads (80×). Then, Illumina reads (133×) were used to correct the assembled sequences from the previous step. Lastly, we used Hi-C paired-end reads (95×) to produce a final assembly of 1718 Mb (scaffold N50 = 36.3 Mb) in 1915 scaffolds (Table 1, Table S4), similar to the results of the previous assessment. Forty-six of the longest pseudomolecules, accounting for 98.4% (1690 Mb) of the assembly, were selected to correspond to the 46 chromosomes of the haplotype genome of H. hamabo (Fig. 1a, Fig. S5). A genome-wide interaction heatmap shows the grouping and ordering results obtained using the Hi-C data (Fig. S6). Altogether, 52.9% (909 Mb) of the assembly was annotated as repetitive sequences, with long terminal repeat (LTR) elements forming the predominant component (47.4%), including 35.6% Gypsy and 10.3% Copia retro-elements (Fig. 1c, Tables S9, S10). We integrated ab initio, homology-based, and RNA-assisted gene prediction methods to obtain gene models for the H. hamabo genome. We predicted 107 309 protein-coding genes with an average length of 1009 bp and an average of 4.42 exons per gene. RNA sequences obtained using the PacBio and Illumina sequencing platforms from eight different tissues and organs were used to provide transcriptional evidence to support the gene annotations. Of all the protein-coding genes, 90.4% were assigned putative functional annotations, among which 35.7% (38297), 14.2% (15242), and 60.2% (64637) were from the MF (molecular function), CC (cellular component), and BP (biological process) categories, respectively (Table S13).
Table 1.
Data from the H. hamabo assembly
| Statistic | H. hamabo |
|---|---|
| Assembly size (bp) | 1718,145,230 |
| Number of scaffolds | 1915 |
| Scaffold N50 size (bp) | 36 349 750 |
| Longest scaffold (bp) | 64 458 935 |
| Number of contigs | 4358 |
| Contig N50 size (bp) | 1 645 376 |
| Longest contig (bp) | 9 364 180 |
| Number of genes | 107 309 |
| BUSCO score | 98.1% |
| GC (%) | 34.90 |
Figure 1.
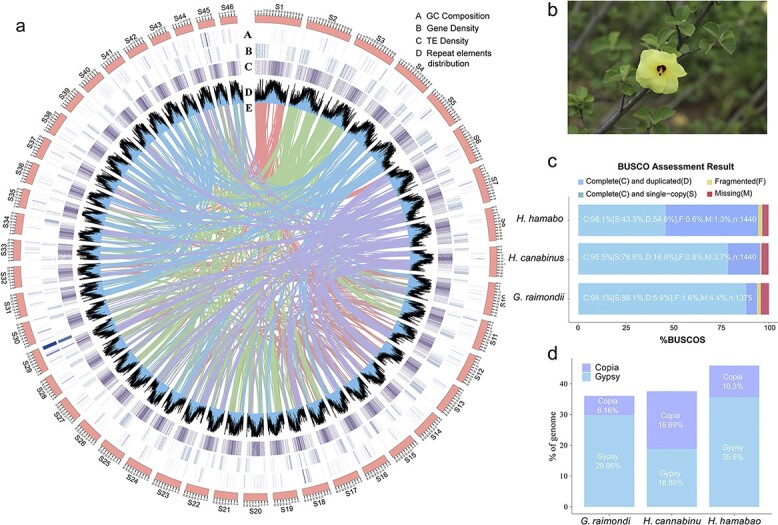
Overview of the Hibiscus hamabo genome (a) Circos plot displaying the general characteristics of the H. hamabo genome. Tracks from outside to inside show GC composition (A), gene density (B), TE density (C), repetitive element distribution (D), and colored bands representing colinear blocks derived from the most recent WGD (whole-genome duplication) event (E). The non-overlapping sliding window for density plots is 500 kb. (b) The flower of H. hamabo. (c) The BUSCO assessment results of the H. hamabo genome compared with those of kenaf (Hibiscus cannabinus) and cotton (G. raimondii). (d) LTR retrotransposons in H. hamabo compared with kenaf and cotton.
To obtain gene models for the H. hamabo genome, we adopted three different strategies for gene prediction, as mentioned above (see Methods). Our annotation captured 98.1% of the Embryophyta BUSCO gene set (odb10, complete and duplicated complete), higher than that in other published Malvaceae genomes, including Hibiscus cannabinus (95.5%) and Gossypium raimondii (97.4%) (Fig. 1b), indicating the high quality of the H. hamabo genome in terms of annotation completeness. Given the high percentage of transposable elements (TEs), we further assessed genome quality by calculating the LTR Assembly Index (LAI) and obtained a final LAI value of 9.89, very close to the “reference” level according to the criteria of Ou et al [16]. We also identified 2743 transfer RNAs, 3936 ribosomal RNAs, 583 small nuclear RNAs (snRNAs), and 439 microRNAs (miRNAs) (Table S14).
Phylogenetic relationships, divergence times, and gene family evolution
Phylogenetic analysis was performed using 385 concatenated low-copy nuclear genes from 14 angiosperm species, including four from the Malvaceae. The phylogenetic inference recovered monophyly of the genus Hibiscus and the family Malvaceae. H. hamabo was resolved as the sister to H. cannabinus, and Hibiscus syriacus was placed as the sister to a lineage composed of H. hamabo and H. cannabinus, suggesting a closer relationship between H. hamabo and H. cannabinus among the three species. Molecular dating based on this topology suggested that Hibiscus originated about 25 million years ago (Mya), and the speciation of H. hamabo was inferred to have occurred approximately 15 Mya with a 95% confidence interval (Fig 2a, Fig S12).
Figure 2.
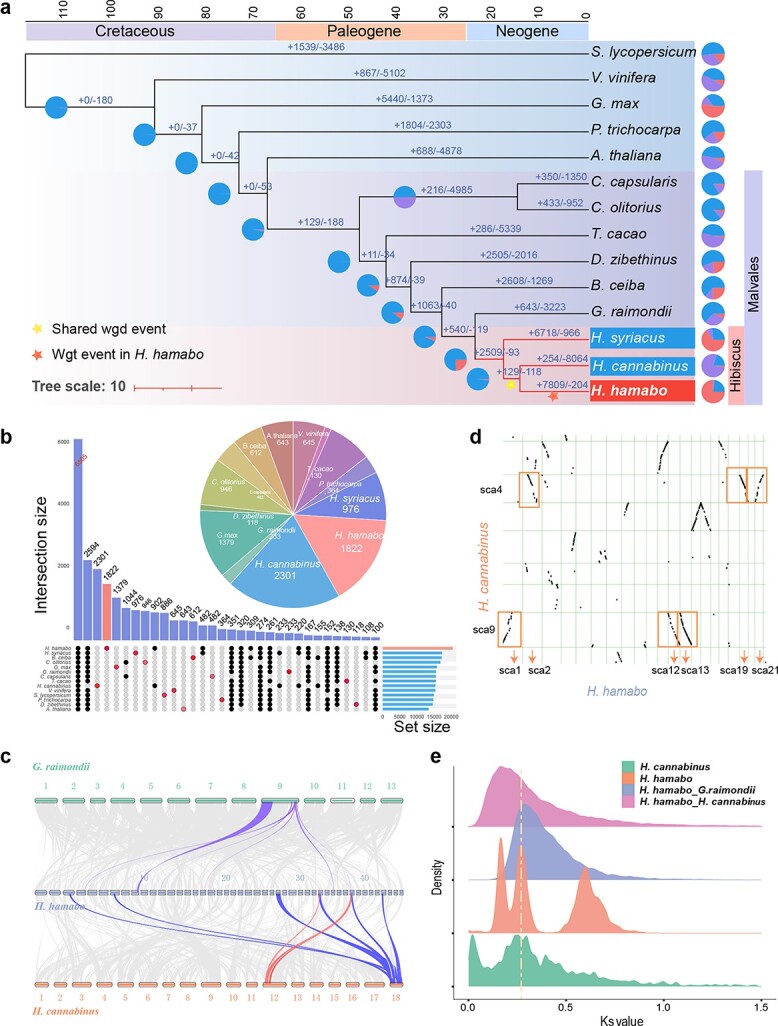
Analysis of evolution and gene families in 14 species (a) Phylogenetic tree of 14 species; the scale at the top represents the divergence time, and the pie charts represent the expansion and contraction of gene families, with red indicating expansion and purple indicating contraction. The yellow and red stars indicate a shared WGD event and a WGT event in H. hamabo, respectively. (b) Upset plots for 14 species; the pie chart shows the number of unique families for each species. (c) Intergenome collinearity plot shows a pattern of 1:6 for G. raimondi and H. hamabo and 2:6 for Hibiscus cannabinus and H. hamabo.(d) Intergenome collinearity dot plot shows a pattern of 1:3 for H. cannabinus and H. hamabo.(e) Distribution of overall synonymous substitution levels (Ks) for paralogs found in syntenic blocks of H. cannabinus and H. hamabo and for orthologs between H. hamabo and H. cannabinus and between H. hamabo and G. raimondii. The yellow dotted line indicates a recent WGD event shared by H. hamabo and H. cannabinus.
The 107 309 protein-coding genes of H. hamabo were classified into 35 408 families, including over 6000 families shared with the other 13 species and 1822 unique families (Fig. 2b). Gene expansion and contraction analysis indicated that H. hamabo has undergone more expansion (7809) than contraction (204) of gene families since its divergence from H. cannabinus (Fig. 2a). Gene ontology (GO) enrichment analysis showed that 2004 significantly expanded (p < 0.05) gene families in H. hamabo were most enriched in the BP terms “cellular response to lipid”, “flower development”, “leaf development”, “plant organ morphogenesis”, “response to water”, “response to auxin” and “regulation of hormone levels” and the MF terms “identical protein binding”, “DNA-binding transcription factor activity”, and “cis-regulatory region sequence-specific DNA binding” (Fig. S11). GO analysis of the 1822 unique gene families showed that they were most enriched in the BP terms “response to endogenous stimulus”, “response to hormone”, “dephosphorylation”, “cell redox homeostasis”, “anatomical structure development”, and “response to auxin” and the MF terms “phosphoprotein phosphatase activity”, “ubiquitin-protein transferase activity”, “protein serine/threonine phosphatase activity”, “protein disulfide oxidoreductase activity” and “protein kinase regulator activity” (Fig. S13). These results suggested an important contribution of expanded and unique genes to functions in environmental adaptability to salt and water, especially with regard to signal transduction.
Polyploidy events in H. hamabo
Intra-genomic synteny analysis of H. hamabo indicated that there were a number of syntenic blocks (Fig. 1A) and a higher density of duplications compared with H. cannabinus [17], suggesting an unusual history of polyploidization in this species. We first calculated the number of synonymous substitutions per synonymous site (Ks) for all the paralogs (paranome) of H. hamabo and found only a single signature peak between 0.15–0.2 (Fig. S15a). After rate adjustment, two peaks at Ks = 0.21 (mode a) and Ks = 0.56 (mode b) emerged, suggesting that two polyploidy events had occurred in H. hamabo. As H. hamabo’s speciation peaks with H. cannabinus and H. syriacus were located at 0.12 and 0.18 (Fig. S15b), it was clear that these two polyploidy events took place in the common ancestor of all Hibiscus plants. To precisely locate the position of the whole-genome duplication (WGD) associated with Ks = 0.56, the paralogs of G. raimondii and the reciprocal best hit (RBH) orthologs were extracted. The Ks distribution showed a WGD peak for G. raimondii at Ks ≈ 0.5 (Fig. S15c), consistent with that of H. hamabo (mode b, Ks ≈ 0.56), and a speciation peak at Ks ≈ 0.3 between H. hamabo and G. raimondii (Fig. S15d), suggesting that there was a shared WGD event between H. hamabo and G. raimondii before speciation. According to these findings, H. hamabo and H. cannabinus should have experienced the same polyploidy events. However, we performed subsequent inter-genomic synteny analyses between the two species using several different methods, and interestingly, all of the results showed a 6:2 or 3:1 pattern between them (Fig. 2c, 2d, Fig. S16), indicating that H. hamabo may have undergone an extra independent whole genome triplication (WGT) event after its split from the common ancestor of H. hamabo and H. cannabinus. With respect to the Ks peak at 0.21, if there had been a species-specific WGT in H. hamabo, the WGT peak would probably be covered by or integrated into this 0.21 peak. To more precisely distinguish the Ks value for the peak of triplicated genes from the peaks of other WGD events, the anchor-pair genes retained in all of the syntenic blocks were extracted. Then, another parameter—the Ks median value of every syntenic block—was used for subsequent analyses [18]. As a result, two very close but independent peaks located at Ks values of 0.1–0.2 and 0.2–0.3 were identified, representing two recent polyploidy events (Fig. 2e, Fig. S17b). Dot plots of the syntenic blocks representing the most recent WGT event showed that the pattern for each block had three syntenic counterparts. Therefore, using H. cannabinus as a reference, we discovered clues to the “hidden” WGT event and successfully distinguished it from another WGD event that occurred not long before. This recent WGT also explains the large number (over 100 000) of protein-coding genes in H. hamabo. In summary, H. hamabo and H. cannabinus shared two polyploidy events before their speciation, the earliest of which was also shared with G. raimondii, and shortly after the divergence of H. hamabo, a WGT event took place in this species (Fig. 2e).
Transcriptomic responses and regulatory networks that are critical to survival in a tidal saline soil and waterlogged environment
To explore the regulatory mechanisms used by H. hamabo to survive under salt and/or waterlogging stress, we sequenced the transcriptomes of its roots and leaves under different treatments: saline soil (S), waterlogging (W), and salt + waterlogging (SW) at 5 min, 9 h, and 3 d after treatments (Table S15). The transcriptomic data showed that the roots and leaves had distinct gene expression profiles, suggesting the presence of different responses and regulatory events in roots and leaves after salt and/or waterlogging treatments (Fig. 3a). To analyze the expression of stress-related genes, all differentially expressed genes (DEGs) in the leaves and roots were classified into 20 clusters according to their expression patterns (Figs. S19, S20).
Figure 3.
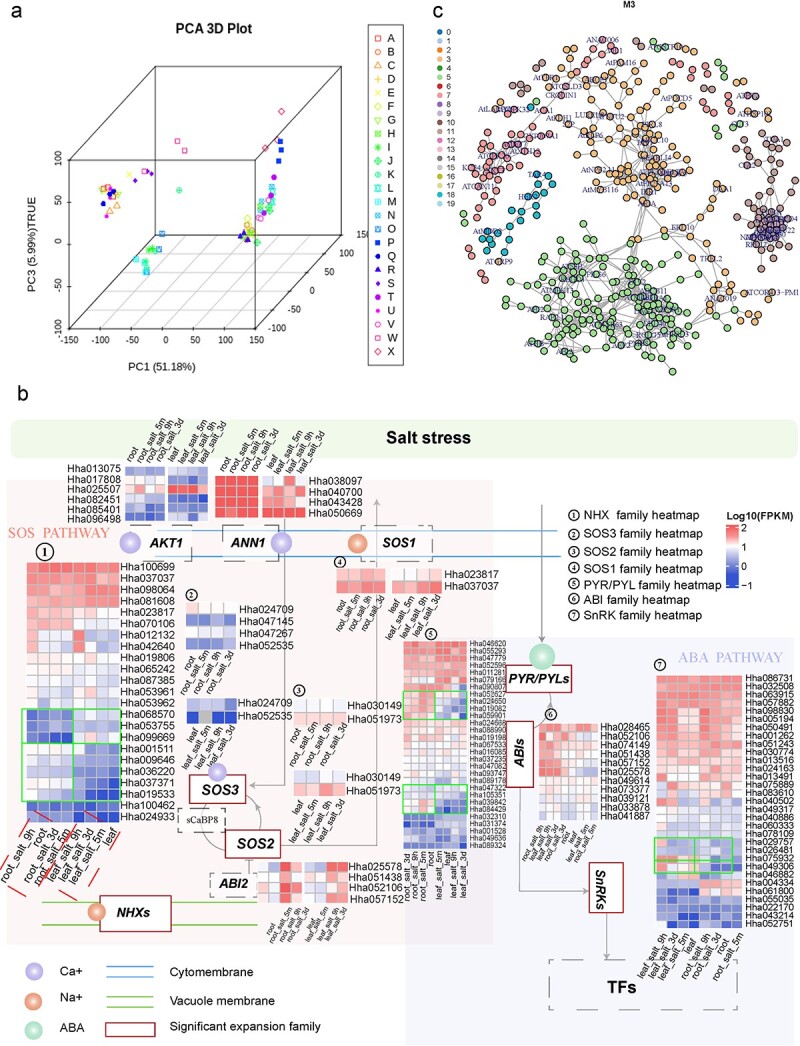
Transcriptome-based analysis of stress-resistance mechanisms in H. hamabo (a) Principal component analysis of transcriptome data from different samples. A–X represent different sample groups. See Table S15 for information about sample groupings. (b) SOS (Salt overly sensitive) and ABA (abscisic acid) pathway responses to salt stress in H. hamabo. ANN1 (annexin1) and AKT1 are a Ca2+ and K+ transporter, respectively. Under salt stress, the SOS pathway is activated by a calcium signal, and SCaBP8 is phosphorylated by SOS2, which may disassociate from AKT1 to promote the concentration of K+. The SOS signaling pathway comprises SOS3, SCaBP8, SOS2, and SOS1 and is important for the regulation of Na+/K+ ion homeostasis. ABI2 (an ABA-responsive gene) negatively regulates the kinase activity of SOS2. NHXs are vacuolar K+/H+ exchangers. In the ABA pathway, the accumulation of ABA activates SnRK2s via the PYR/PYLs-PP2Cs/ABIs-mediated regulatory module. The heatmap shows the differential gene expression patterns in leaves and roots. (c) Correlation networks under salt and waterlogging stress. The numbers 0–19 indicate 20 modules related to salt stress identified from the transcriptomic data.
Maintaining a proper Na+/K+ ratio in the cytoplasm is an important adaptive characteristic of salt-tolerant plants to the presence of excess ions [19]. In H. hamabo roots, SOS2 (Hha031532) and AKT1 (Hha025507) were quickly (5 min) upregulated under S and SW treatments compared with the control (CK), indicating their sensitivity to stimuli and their important roles in short-term responses to the changing Na+/K+ ratio. Based on the clustering results, SOS2 and AKT1 fell into Subclusters 11 and 18 (Figs. S20, S21). The leaf transcriptomes from 5 min after treatment showed no significant changes, indicating a slower response than that seen in the roots. However, when plants were exposed to saline conditions for 9 h and 3 d, H. hamabo NHX2 (Hha100699), encoding a major tonoplast-localized NHX isoform, was induced in leaves to regulate the ion balance. It can influence the Na+/K+ ratio by adjusting the vacuolar absorption of K+, thereby regulating cell turgor and stomatal function [20]. The NHX gene was grouped into Subcluster 14, which was also enriched in genes related to transcription factor activity, suggesting a role for transcription factors in leaves (Figs. S19, S22). Aside from its involvement in calcium signaling, Na+ influx also induces an increase in the production of reactive oxygen species (ROS) [11]. H. hamabo CAT1 (catalase 1, Hha081091 in Subcluster 17), which encodes an antioxidant enzyme, was upregulated in root samples 9 h and 3 d after S and SW treatments, and this is considered to be a reasonable regulatory response to long-term salt stress [21]. In the same subcluster, genes related to proline biosynthetic and metabolic processes, response to gibberellin, and the gibberellin-mediated signaling pathway were also enriched, indicating that solutes and phytohormones also participated in medium- and long-term adjustments under salt stress (Fig. S23). The accumulation of ROS under salt stress subsequently triggers MAP kinase (MAPK) cascades [19], which have been shown to participate in salt-stress signaling responses in Arabidopsis [22, 23] and were also found to respond in H. hamabo roots under S and SW conditions. MPK3 (Hha060729, Hha033359, and Hha018260) and MPK4 (Hha095461) were in Subcluster 18, and MPK6 (Hha011239) was in Subcluster 17. SnRK kinase, a key transcriptional and post-transcriptional regulator under salt stress, has a variety of downstream phosphorylation targets that mediate many processes, such as ion transport, ROS production, and gene transcription [11]. In H. hamabo roots, SnRKs (Hha057882 and Hha063915) were also found in Subcluster 17, together with antioxidant enzymes and MAPK members, demonstrating that ABA may activate ROS signaling and MAPK cascades through SnRKs to regulate the growth of the root system. Another ABA receptor, GCR2 (G-protein-coupled receptor 2, Hha009184) in Subcluster 1, was upregulated in leaf samples treated with S and SW for 9 h and 3d, suggesting that it may be involved in the regulation of stomatal closure and senescence through ABA signal transduction in leaf tissue [24]. Genes related to the metabolism of energy-storing substances and the gibberellin-mediated signaling pathway were also significantly enriched in Subcluster 1 (Fig. S24), suggesting that H. hamabo may also regulate hormone signal transduction and energy metabolism in leaves to defend against stress injury. In summary, genes enriched in root Subclusters 11 and 18 may have regulatory roles in the short-term root salt stress response, whereas Subcluster 17 genes seem to participate more in long-term regulation. The leaves did not appear to respond very quickly to the stress treatments, but genes in Subclusters 1 and 14 may be relevant in the long term.
Waterlogging can reduce gas diffusion, resulting in limited oxygen availability, a condition commonly referred to as hypoxia [25]. To overcome this unfavorable condition, endogenous ethylene accumulates in submerged plant tissues to activate several adaptive signal transduction pathways [25]. In H. hamabo, ADH1 (alcohol dehydrogenase) (Hha098758, Hha081929, Hha098733, Hha005263, Hha005264, and Hha097945) and PDC1 (pyruvate decarboxylase) (Hha038280) were found in Subcluster 9 and were strongly upregulated in W- and SW-treated root samples at all time points to ensure the provision of energy supply under stress (Fig. S25). RAP2.2 (Hha038572, Hha021850, and Hha050179) was also found in Subcluster 9 and was strongly upregulated in W- and SW-treated roots at 5 min to promote the upregulation of ADH and PDC. In Subcluster 9, genes annotated with the terms small molecule metabolic process, single-organism metabolic process, purine-containing compound metabolic process, purine ribonucleotide metabolic process and others were significantly enriched, suggesting that H. hamabo can deploy metabolic systems to clean up damaged nucleotide molecules in response to environmental stress.
Finally, we compared the expression patterns of salt-resistance pathway genes under SW treatment (Fig. 3b). Because of the WGT event, all related genes are expanded in H. hamabo, and some duplicates show different expression patterns in roots and leaves, suggesting further functional divergence among duplicated copies. For example, AKT1 has expanded into six duplicates, with one copy, Hha025507, exhibiting higher expression in leaves than roots, and another, Hha017808, showing the opposite pattern. The NHX family also showed a reciprocal expression pattern, with three members (Hha068570, Hha53755, and Hha099669) more highly expressed in leaves than in roots and five members (Hha001511, Hha009646, Hha036220, Hha037371 and Hha019533) more highly expressed in roots. ANN1 has expanded into four duplicates, almost all of which are extremely highly expressed in roots but less so in leaves. Among six duplicates of AKT1, one gene, Hha025507, shows much higher expression in both roots and leaves than the other five members. Similarly, four members of the NHX family (Hha100699, Hha037037, Hha098064, and Hha081608) show higher levels of expression than the other members. This scenario may reflect the functional redundancy of the recent duplicates.
Because many of the identified genes were involved in different pathways and associated with regulation under stress, it is likely that they form a network to co-regulate responses under stress, especially salt stress. Based on the transcriptomic data, we performed a weighted gene co-expression network analysis (WGCNA) to identify genes potentially involved in the salt-stress response network. Based on their expression patterns, the genes expressed under SW treatment were classified into 20 modules (Fig. S26), among which Modules 3, 5, 7, 11, and 18 contained known functional salt-resistance genes (e.g. AKT1, SOS2, SOS3, and ABI2) (Fig. 3c). Thus, the genes in these modules may play a role in the salt stress regulatory network. The well-known salt-stress genes SOS2 and SOS3 were classified into Modules 11 and 18, respectively, and these two modules contained 125 and 40 genes, respectively (Table S16), providing clues to assist in the investigation of the plant salt-stress regulatory network. Future functional characterization studies should investigate the functions of these candidate genes.
Polyploidy in hibiscus is likely to be associated with stress resistance
As a semi-mangrove plant, H. hamabo resides in a very harsh habitat compared with other Hibiscus plants, and its strong tolerance to stress guarantees its survival in such environments. Because many studies have pointed out that polyploids have stronger resistance to stressful environments [26–29], we wanted to investigate whether there was an association between the specific recent WGT of H. hamabo and its adaptation to stress. The unique gene set, significantly expanded gene set, and WGT gene set were extracted for comparison and analyses. We found that almost all of the significantly expanded genes (over 96.7%) and over a quarter of the unique genes (28.4%) belonged to the WGT gene set, suggesting that the recent WGT event contributed greatly to gene family expansion (Fig. 4a). In total, 1939 “WGT-expansion” gene families were used in a GO enrichment analysis, and these genes were mainly enriched in “response to chemical”, “response to abiotic stimulus”, “response to hormone” and “cellular response” from the “response to stimulus” category and were also significantly enriched in “developmental process”, including “leaf development”, “floral whorl”, “lateral root”, and “xylem and phloem” (Fig. 4b, Fig. S18). The genes that responded to hormones were mainly enriched in GO terms related to ABA, auxin, and ethylene. The ABA pathway is important for plant stress resistance [30], auxin can enhance plant waterlogging tolerance by regulating lateral root initiation and development [11, 31], and the ethylene signal transduction pathway plays a key role in the response to waterlogging [30, 32]. These genes were additionally mapped to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, and they were enriched (p < 0.05) in the “plant hormone signal transduction”, “circadian rhythm-plant” and “ribosome” categories (Fig. 4c). These results suggested that the H. hamabo WGT played a critical role in its adaptability to survival in a high-saline and waterlogged environment. We also annotated and identified SOS and ethylene-response gene families/subfamilies, which regulate salt [9] and waterlogging stress [12, 13, 32, 33], respectively. CIPKs, CBLs, and NHXs, which are supposed to respond to salt stress, were all expanded in H. hamabo compared with other Hibiscus species. Likewise, all ethylene-response gene families were also significantly expanded. Interestingly, H. syriacus, a species with medium salt tolerance, had a pattern of gene family expansion similar to that of H. hamabo (Fig. 4d, 4e). H. syriacus is also likely to have undergone an independent WGD event, implying an underlying mechanistic link between stronger adaptive ability and polyploidy events.
Figure 4.
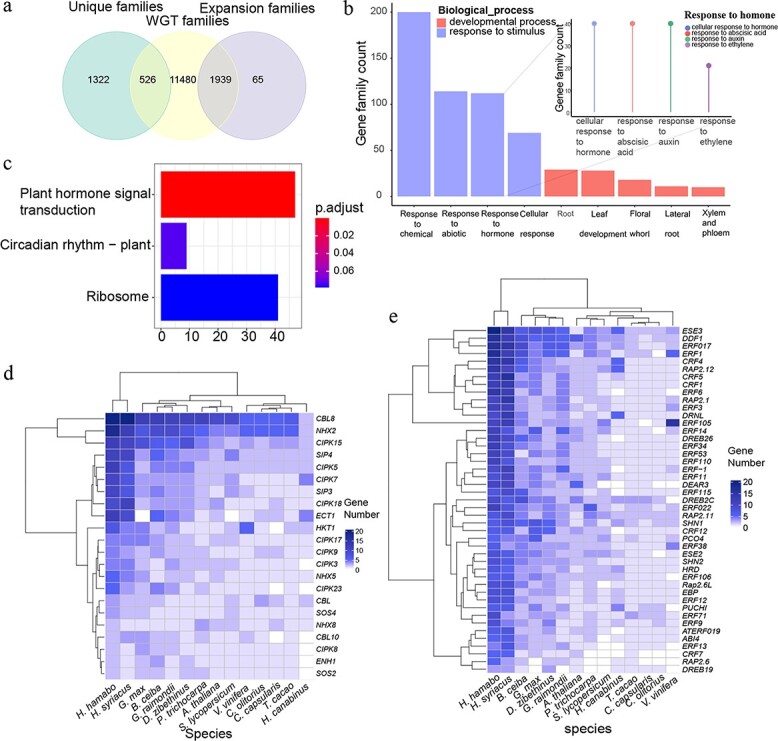
Analysis of “WGT-expansion” gene families (a) Venn diagram showing the relationships among unique families, WGT families, and expanded families in the H. hamabo genome. (b) Summary of the GO enrichment results for the “WGT-expansion” genes; see Fig. S18 for more details. The diagram represents the enriched first-level GO terms “developmental process” and “response to stimulus”, and the lollipop chart shows secondary terms associated with “response to hormone”. (c) KEGG enrichment result for the “WGT-expansion” genes. (d) Expansion of genes involved in the salt stress signal transduction pathway. The different colors represent the copy numbers of each gene. (e) Expansion of genes involved in the response to ethylene. The different colors represent the copy numbers of each gene.
Discussion
The genus Hibiscus comprises over 200 species that are widely distributed in the tropics and subtropics. H. hamabo was originally discovered on the eastern coasts and islands of China and is one of the few species in this genus that can survive in saline and waterlogged habitats. In this study, we sequenced the first semi-mangrove plant genome using a combination of Illumina, PacBio, and Hi-C technology and produced a chromosome-level assembly. The diversity of chromosome numbers in Hibiscus suggests very complicated and distinct evolutionary histories for these species, and polyploidy events are likely to have played important roles in this process. We identified multiple rounds of polyploidy events in three sequenced Hibiscus species, including WGD and WGT, with some events shared by all species and some specific to particular species. Polyploidy events inevitably doubled the chromosome number, whereas post-fragmentation is likely to have exacerbated genome reshuffling, leading to more complex chromosome numbers in this genus. There may also have been hybridization events within and outside the genus, as Hibiscus species do not always form a monophyletic group [6]. Ambiguity in the phylogenetic relationships between Hibiscus and closely related taxa remains and may be associated with frequent polyploidy and possible hybridization events. To achieve a well-resolved phylogeny, broader taxon sampling and genomic markers would be necessary. The identification of a recent WGT helped us to interpret the evolution of the H. hamabo genome and explains the >100 000 protein-coding genes in the species. The identification of this recent WGT also demonstrates the importance of references for comparative analysis. Without H. cannabinus, the evidence for the WGT in H. hamabo would have been difficult to verify. The complex evolutionary history of Hibiscus species subsequently shaped their diverse morphological characteristics. The H. syriacus genome experienced one individual WGD event, and the preferentially retained genes played a critical role in its unique floral morphology [34]. Similarly, a recent WGD took place in the H. cannabinus genome, and the reduplicated gene families are involved in bast fiber formation [17]. Our research revealed one WGD shared with H. cannabinus and another WGT event that occurred after speciation. This high-quality genome will lay the foundation for further research on Hibiscus evolution and the molecular mechanisms of its environmental adaptation.
The WGT also generated more copies of stress-resistance genes, including genes involved in salt and waterlogging resistance that contribute to the survival of this species in a harsh habitat. Polyploids, especially recently formed polyploids, have broad ecological tolerance [29]. WGDs provide abundant genetic resources to enable plants to cope with environmental changes. After a WGD (or WGT), neofunctionalization and subfunctionalization create novel genes, leading to expanded functional diversity and more complex regulatory networks [35]. In H. hamabo, >95% of the significantly expanded gene families belonged to recent WGT-derived genes (Fig. 3A), including multiple stress resistance-related genes, such as SOS pathway and ethylene-related genes (Fig. 4d, 4e). Stress resistance pathways form interrelated regulatory networks. A change in gene number in one pathway inevitably influences other related pathways, and gene dosages must be balanced to enable all of the pathways and networks to work properly [36, 37] (Fig. 3c). For example, salt stress activates the SOS pathway and causes the accumulation of ROS, which subsequently triggers MAPK cascades [19]. These cascades function in ROS homeostasis as central regulators of ROS detoxification and also contribute to ion homeostasis by promoting SOS1 phosphorylation [19].
As a semi-mangrove species with strong resistance to salt, H. hamabo may have many mechanisms of salt tolerance, such as salt excretion, salt rejection, and salt dilution. Furthermore, H. hamabo has a series of structures adapted to the saline environment, such as stellate hairs and crystal cells [38]. The selective absorption of Na+ and K+ and a higher proportion of Na+ accumulation in the roots relative to the whole plant are among the salt exclusion characteristics of H. hamabo [39]. Because it is a salt-dilution halophyte, osmotic regulation and ion compartmentation may also be physiological adaptations of H. hamabo to salt stress [40]. As the transcriptomic data suggest, roots respond very quickly to stress, adjusting ion concentrations in and outside plant cells, as well as inhibiting the production of damaging compounds. In an ion concentration test, the Na+/K+ ratio was significantly higher in the 9-h S group, the 24-h SW group, and the 3-d SW group than in the control group, and it was significantly higher in the root system than in the leaves (Fig. S27), suggesting that H. hamabo may be a kind of K+-preferring and Na+-refusing halophyte. H. hamabo not only is an exclusion-type halophyte that exhibits preferential absorption of K+ over Na+ through the roles of genes like AKT and SOS but also exhibits the mechanism of salt dilution through osmotic regulation and ion zone isolation. Leaves take longer to respond, triggering transcription factors and auxin biosynthesis and regulation. Leaves containing auxin show the greatest capacity for de novo auxin synthesis and commonly show increased resistance to stress; in particular, auxin with stress-induced ROS signals has been reported to correlate plant development with plant responses to environmental changes [41]. With further investigation of the large numbers of genes identified in the H. hamabo genome, the complex mechanisms of salt and waterlogging tolerance in H. hamabo will be resolved, which will also shed light on semi-mangrove plants.
Materials and methods
Sample preparation and sequencing
The H. hamabo tissue was originally obtained from the Institute of Botany, Jiangsu Province, and the Chinese Academy of Sciences. We obtained high-quality genomic DNA from H. hamabo leaves using a modified CTAB method [42]. Several tools and techniques were used to examine DNA quantity and quality: 0.8% agarose gel electrophoresis, a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) with the Qubit dsDNA HS Assay Kit, and a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). We extracted total RNA using the TRIzol reagent (Invitrogen, CA, USA) and assessed its purity and integrity on a NanoDrop 2000 spectrophotometer and a Bioanalyzer 2100 system (Agilent Technologies, CA, USA). We assessed RNA density using 1.5% agarose gel electrophoresis. The Sequel Binding Kit 2.1, Sequel Sequence Kit 2.1, and Sequel SMRT Cell 1 M V2 were used for sequencing, and data were analyzed using SMRT Link 5.1 software. Initial filtering was performed based on the read quality values of the original data, and the effective data output after filtering was counted.
Transcriptome sequencing and analysis
The transcriptome analysis was performed with seedlings grown from seeds of the H. hamabo individual that was used for genome sequencing. Four treatments were applied to seedlings with 8–10 true leaves: (i) Control (CK), the seedlings were irrigated with 1/4 Hoagland’s nutrient solution; (ii) Salt stress (S), the NaCl concentration of the irrigation solution was set to 3.5 wt% (simulated seawater); (iii) Waterlogging (W), a 1–2 cm layer of nutrient solution was maintained above the soil surface; and ( ) A combination of S and W (SW). Leaves and roots were collected 5 min, 9 h, and 3 d after treatment, and RNA samples were isolated for Illumina sequencing using the paired-end 150-bp sequencing mode. All N bases, low-quality reads, and adapter sequences were removed to acquire the final clean reads. HISAT2 and Bowtie 2 (v2.2.2) were used to align clean reads to the H. hamabo reference genome with default parameters. FPKM (fragments per kb per million reads) was used for the normalization of gene and isoform expression values. All differentially expressed genes (DEGs) were identified using the R package edgeR with the criteria of false discovery rate < 0.05 and fold change ≥2. Several other R packages were also used to analyze gene expression patterns, including NbClust for DEG clustering and ggplot2 for visualization.
) A combination of S and W (SW). Leaves and roots were collected 5 min, 9 h, and 3 d after treatment, and RNA samples were isolated for Illumina sequencing using the paired-end 150-bp sequencing mode. All N bases, low-quality reads, and adapter sequences were removed to acquire the final clean reads. HISAT2 and Bowtie 2 (v2.2.2) were used to align clean reads to the H. hamabo reference genome with default parameters. FPKM (fragments per kb per million reads) was used for the normalization of gene and isoform expression values. All differentially expressed genes (DEGs) were identified using the R package edgeR with the criteria of false discovery rate < 0.05 and fold change ≥2. Several other R packages were also used to analyze gene expression patterns, including NbClust for DEG clustering and ggplot2 for visualization.
For WGCNA, we used the R package WGCNA (v1.66) to construct co-expression networks. Genes with FPKM values less than 1 were filtered out. The parameters of blockwiseModules () were set to soft power = 16, minModulesSize = 30, mergeCutHeight = 0.25, and default parameters were used for other settings. The function pickSoftThreshold () in WGCNA was used to choose the soft power. Candidate hub genes were identified based on a thresholding of 0.6. Cytoscape was used to visualize the co-expression network.
Assembling third-generation long reads
After removal of adaptor sequences from sequences generated by the PacBio Sequel platform, we obtained 17 800 118 subreads. The longest 150x subreads were chosen for subsequent genome assembly. First, the H. hamabo genome was assembled using FALCON (v0.2.2) (https://github.com/PacificBiosciences/FALCON/tree/v0.2.2) with default parameters. The Arrow pipeline in the SMRT Link 4 toolkit was used to polish the primary assembly. Finally, short reads from Illumina sequencing were used to further correct any remaining errors using Pilon v1.22 [43]. To evaluate the integrity of the assembly and the uniformity of the sequencing coverage, CLR (Continuous Long Reads) subreads of H. hamabo were selected and matched back to the assembled genome using the comparison tool minimap2 [44] with default parameters. The comparison rate of reads, the extent of genome coverage, and the depth distribution were determined to evaluate the integrity of the assembly and the uniformity of the sequencing coverage.
Genome quality evaluation
To assess assembly completeness, we first mapped the CLRs to the final assembly with the default parameters of minimap2 (v2.5) [44]. We then evaluated the assembly completeness using BUSCO v3.0.2 [45]. We also mapped the Illumina paired-end short reads to the genome to confirm the high quality of the contig assembly based on a high ratio of alignment and a high distribution peak of the insertion length distribution. BWA 0.7.17 software was used to align the Illumina short reads to the reference genome to evaluate its accuracy at the single-base level [46]. Homozygous SNP loci were identified with the GATK 4.0.8.1 [47] package. We also assessed genome quality by calculating the LAI using the LTR_Finder [48] and LTR_retriever [49] packages.
Genome annotation
Repetitive sequences were identified in the H. hamabo genome using two combined methods: homologous annotation and de novo prediction. For homology-based analysis, the RepeatMasker (open-4.0.9) [50] package was used to identify known TEs from the Repbase TE library [51]. For de novo prediction, the comprehensive pipeline RepeatModeler (http://www.repeatmasker.org/RepeatModeler/) was used to construct a H. hamabo genome repeat library, and the subprograms RECON (v1.08) [52] and RepeatScout (v1.0.5) [53] were used to identify decentralized repeat sequences in the genome. LTR_Finder (v1.0.7) [48] was used to extract all LTR sequences. Tandem repeats were extracted with the TRF [54] package. Finally, RepeatMaker was used to merge the results from the two methods to identify the repetitive content.
A combination of homology-based, RNA-seq-guided and ab initio methods were used to predict protein-coding genes. Repetitive sequences were masked with RepeatMasker, and ab initio gene prediction was performed with the Augustus (v3.3.1) package [55–57] and GeneScan software [58]. The previous RNA-seq dataset was chosen as a source for model training. For homology-based prediction, we used Exonerate to align query sequences to the genome and predict coding genes. For RNA-seq-guided gene prediction, the TopHat (v2.1.1) package [59] was used to assemble all transcripts, and Cufflinks (v2.2.1) [60] was used for gene structure identification. We used Maker (v3.00) [61] to integrate the results of all gene-prediction methods.
The National Center for Biotechnology Information (NCBI) non-redundant (NR) database, the TrEMBL [62], InterPro [63], and Swiss-Prot [62] protein databases, and the KEGG database [64] were used to predict gene functions based on the best matched alignments using BLASTP [65, 66] with an E-value threshold of 1e−5. The InterPro protein database was used for protein domain annotation with PfamScan [67] and InterProScan v5.35–74.0 [68]. Motifs and domains within gene models were identified with PFAM databases [69], and Blast2GO [70] was used to map GO [71] terms to each gene.
Analysis of gene families
The longest transcripts of each protein-coding gene in H. hamabo and other carefully selected species (Arabidopsis thaliana [72], Corchorus olitorius [73], Gossypium raimondii [74], H. syriacus [34], Theobroma cacao [75], Bombax ceiba [76], Durio zibethinus [77], H. cannabinus [17], Populus trichocarpa [78], Vitis vinifera [79], Corchorus capsularis [80], Glycine max [81] and Solanum lycopersicum [82]) were used to cluster the gene families. All longest transcripts were extracted using a python script from the OrthoFinder repository at Github (https://github.com/davidemms/OrthoFinder). BLASTP [65] was used to search for all possible homologs within all species with an E-value threshold of 1e−5. OrthoFinder v14 [83] was used to classify genes into gene families with default parameters. To exclude false positive effects, genes with amino acid sequences shorter than 50 amino acids were filtered out. To assign functions to genes related to stress resistance, orthologous genes were identified from H. hamabo and A. thaliana using OrthoFinder, and functional annotations were obtained from the TAIR website. All stress resistance-related subfamilies were used to construct a phylogenetic tree, and all orthologs were identified.
Phylogenetic analysis
To determine the evolutionary relationships among H. hamabo and other angiosperm species, we constructed a phylogenetic tree using protein-coding sequences of 586 low-copy orthologous genes. The translated amino acids of the genes were aligned using MUSCLE v3.8.31 [84] and then concatenated using a python script. We then used RAxML v8.2.11 [85] to reconstruct a consensus tree using the maximum likelihood (ML) algorithm with the rapid bootstrap method.
The MCMCTree program in the PAML (version 4.9 h) package was used for divergence time estimation, and the ultrametric phylogenetic tree with divergence times as branch lengths and the previously identified gene families were used for the analysis of gene family expansion and contraction using the CAFÉ package [86]. A random birth and death model was used to determine how gene families changed with time. All p-values of gene families were calculated, and a threshold of p < = 0.05 was used as the criterion for identification of rapidly evolving gene families.
Analysis of WGD events
WGD software [87] was used for Ks-based paralog age distributions. All potential paralogs were detected with all-vs-all protein sequence blast using BLASTP with an E-value threshold of 10−10, and the MCL package was used for gene family construction. Mafft [88] was used to align each family. Gene families (with n members) for which n × (n − 1)/2 > `max pairwise` were removed, and a phylogenetic tree was built for each family using FastTree [89]. Ks values for each pair resulting from pairwise comparisons were obtained using a maximum likelihood algorithm in the CODEML program of the PAML v.4.4c package [90], and the weighted Ks values were used to construct the Ks distribution. Finally, we performed mixture modeling for all possible WGD inferences using the BGMM method. The ksrates package (https://github.com/VIB-PSB/ksrates) was used for rate adjustment, and the JCVI [91] and minimap packages [44] were used for syntenic visualization. The WGDI package [18] was used for collinearity anchor pair identification and analysis. First, all syntenic blocks were identified using the improved collinearity pipeline in WGDI with “p-value = 0.05”. Then the Ks value for each anchor gene pair located in syntenic blocks was calculated using the Ks pipeline in WGDI. A Ks dotplot of all anchor pairs was obtained using the blocks pipeline in WGDI, and the kspeaks pipeline in WGDI was used for distribution analysis of the Ks median value for each syntenic block. Finally, all of the above results from the Ks distributions were summarized in a single picture using the ggplot2 package.
Acknowledgments
This work was supported by the Six Talent Peaks Project of Jiangsu Province (NY-042), the Open Fund of the Jiangsu Key Laboratory for the Research and Utilization of Plant Resources (JSPKLB201928), and the Talent Training Funds of the Institute of Botany, Jiangsu Province and Chinese Academy of Sciences.
Author contributions
Conceived and designed the experiments: GC. Performed the experiments: WZ, ZF, and NL. Analyzed the data: WZ, XJ, HS, ZF, YR, CD, JJ, SA, YV, and GC. Contributed reagents/materials/analysis tools: HJ, LZ, YC, and YY. Wrote the paper: WZ, XJ, HS, and GC. All authors read and approved the final manuscript.
Data availability
The data supporting the findings of this work are available within the paper and its Supporting Information files. The data sets generated and analyzed during this study are available from the corresponding author upon request. All the whole-genome raw data generated during this study have been deposited in the SRA database under BioProject number PRJNA759075. Transcriptome clean data have been deposited in the SRA database under BioProject number PRJNA759717. The final chromosome-scale genome assembly and annotation data have been deposited in the Figshare database (https://doi.org/10.6084/m9.figshare.19142558.v1).
Conflict of interest statement
The authors declare no competing financial interests.
Supplementary data
Supplementary data is available at Horticulture Research online.
Supplementary Material
References
- 1. Sakhanokho HF, Islam-Faridi N, Babiker EMet al. Determination of nuclear DNA content, ploidy, and FISH location of ribosomal DNA in hibiscus hamabo. Sci Hortic. 2020;264:109167. [Google Scholar]
- 2. Li J, Liao J, Guan Met al. Salt tolerance of hibiscus hamabo seedlings: a candidate halophyte for reclamation areas. Acta Physiol Plant. 2012;34:1747–55. [Google Scholar]
- 3. Fryxell PA, Stelly DM. Documented chromosome numbers 1993:2. Additional chromosome counts in the Malvaceae. SIDA Contributions to Botany. 1993;15:639–47. [Google Scholar]
- 4. Takayama K, Ohi-Toma T, Kudoh Het al. Origin and diversification of Hibiscus glaber, species endemic to the oceanic Bonin Islands, revealed by chloroplast DNA polymorphism: origin and diversification of Hibiscus glaber. Mol Ecol. 2005;14:1059–71. [DOI] [PubMed] [Google Scholar]
- 5. Tanmoy AM, Alam MM, Moosa Met al. Corchorus L. and hibiscus L.: molecular phylogeny helps to understand their relative evolution and dispersal routes. Bioresearch Communication. 2015;1. [Google Scholar]
- 6. Takayama K, Kajita T, Murata Jet al. Phylogeography and genetic structure of Hibiscus tiliaceus- speciation of a pantropical plant with sea-drifted seeds: Phylogeography of Hibiscus tiliaceus. Mol Ecol. 2006;15:2871–81. [DOI] [PubMed] [Google Scholar]
- 7. Wu T, Gu S, Zhou Het al. Photosynthetic and physiological responses of native and exotic tidal woody seedlings to simulated tidal immersion. Estuar Coast Shelf Sci. 2013;135:280–4. [Google Scholar]
- 8. Wang Z, Xu X, Ni Let al. Efficient virus-induced gene silencing in hibiscus hamabo Sieb. Et Zucc. Using tobacco rattle virus. PeerJ. 2019;7:e7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin H, Yang Y, Quan Ret al. Phosphorylation of SOS3-LIKE CALCIUM BINDING PROTEIN8 by SOS2 protein kinase stabilizes their protein complex and regulates salt tolerance in Arabidopsis. Plant Cell. 2009;21:1607–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang Y, Guo Y. Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytol. 2018;217:523–39. [DOI] [PubMed] [Google Scholar]
- 11. Van Zelm E, Zhang Y, Testerink C. Salt tolerance mechanisms of plants. Annu Rev Plant Biol. 2020;71:403–33. [DOI] [PubMed] [Google Scholar]
- 12. Yin D, Sun D, Han Zet al. PhERF2, an ethylene-responsive element binding factor, plays an essential role in waterlogging tolerance of petunia. Hortic Res. 2019;6:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou W, Chen F, Meng Yet al. Plant waterlogging/flooding stress responses: from seed germination to maturation. Plant Physiol Biochem. 2020;148:228–36. [DOI] [PubMed] [Google Scholar]
- 14. Zhao N, Li C, Yan Yet al. Comparative transcriptome analysis of waterlogging-sensitive and waterlogging- tolerant Chrysanthemum morifolium cultivars under waterlogging stress and reoxygenation conditions. Int J Mol Sci. 2018;19:1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yuan Y, Brunel C, Kleunen Met al. Salinity-induced changes in the rhizosphere microbiome improve salt tolerance of hibiscus hamabo. Plant Soil. 2019;443:525–37. [Google Scholar]
- 16. Ou S, Chen J, Jiang N. Assessing genome assembly quality using the LTR assembly index (LAI). Nucleic Acids Res. 2018;46:e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang L, Xu Y, Zhang Xet al. The genome of kenaf (Hibiscus cannabinus L.) provides insights into bast fibre and leaf shape biogenesis. Plant Biotechnol J. 2018;18:1796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun P, Jiao B, Yang Yet al. WGDI: a user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Biorxiv. 2021. [DOI] [PubMed] [Google Scholar]
- 19. Yang Y, Guo Y. Unraveling salt stress signaling in plants: salt stress signaling. J Integr Plant Biol. 2018;60:796–804. [DOI] [PubMed] [Google Scholar]
- 20. Barragán V, Leidi EO, Andrés Zet al. Ion exchangers NHX1 and NHX2 mediate active potassium uptake into vacuoles to regulate cell turgor and stomatal function in Arabidopsis. Plant Cell. 2012;24:1127–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumar, V., Khare, T., Sharma, M.et al. ROS-Induced signaling and gene expression in crops under salinity stress. in reactive oxygen species and antioxidant systems in plants: Role and regulation under abiotic stress. Springer Singapore 159–184 (2017).
- 22. Teige M, Scheikl E, Eulgem Tet al. The MKK2 pathway mediates cold and salt stress signaling in Arabidopsis. Mol Cell. 2004;15:141–52. [DOI] [PubMed] [Google Scholar]
- 23. Xu J, Li Y, Wang Yet al. Activation of MAPK kinase 9 induces ethylene and camalexin biosynthesis and enhances sensitivity to salt stress in Arabidopsis. J Biol Chem. 2008;283:26996–7006. [DOI] [PubMed] [Google Scholar]
- 24. Raghavendra AS, Gonugunta VK, Christmann Aet al. ABA perception and signalling. Trends Plant Sci. 2010;15:395–401. [DOI] [PubMed] [Google Scholar]
- 25. Phukan UJ, Mishra S, Shukla RK. Waterlogging and submergence stress: affects and acclimation. Crit Rev Biotechnol. 2016;36:956–66. [DOI] [PubMed] [Google Scholar]
- 26. Van de Peer Y, Maere S, Meyer A. The evolutionary significance of ancient genome duplications. Nat Rev Genet. 2009;10:725–32. [DOI] [PubMed] [Google Scholar]
- 27. Van de Peer Y, Mizrachi E, Marchal K. The evolutionary significance of polyploidy. Nat Rev Genet. 2017;18:411–24. [DOI] [PubMed] [Google Scholar]
- 28. Zhang L, Li X, Ma Bet al. The tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance. Mol Plant. 2017;10:1224–37. [DOI] [PubMed] [Google Scholar]
- 29. Van de Peer Y, Ashman T-L, Soltis PSet al. Polyploidy: an evolutionary and ecological force in stressful times. Plant Cell. 2021;33:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao C, Zhang H, Song Cet al. Mechanisms of plant responses and adaptation to soil salinity. The Innovation. 2020;1:100017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vidoz ML, Mignolli F, Aispuru HTet al. Rapid formation of adventitious roots and partial ethylene sensitivity result in faster adaptation to flooding in the aerial roots (aer) mutant of tomato. Sci Hortic. 2016;201:130–9. [Google Scholar]
- 32. Wang S, Huang J, Wang Xet al. PagERF16 of populus promotes lateral root proliferation and sensitizes to salt stress. Front Plant Sci. 2021;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hinz M, Wilson IW, Yang Jet al. Arabidopsis RAP2.2: an ethylene response transcription factor that is important for hypoxia survival. Plant Physiol. 2010;153:757–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim Y-M, Kim S, Koo Net al. Genome analysis of Hibiscus syriacus provides insights of polyploidization and indeterminate flowering in woody plants. DNA Res. 2017;24:71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vanneste K, Baele G, Maere Set al. Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the cretaceous–Paleogene boundary. Genome Res. 2014;24:1334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Birchler JA, Veitia RA. Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc Natl Acad Sci. 2012;109:14746–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Conant GC, Birchler JA, Pires JC. Dosage, duplication, and diploidization: clarifying the interplay of multiple models for duplicate gene evolution over time. Curr Opin Plant Biol. 2014;19:91–8. [DOI] [PubMed] [Google Scholar]
- 38. Shang H. Studies on Mechanism of Salt Ions Transportation and Distribution in Hibiscus Hamabo Sieb.Et Zucc. Shandong Normal University; 2010. [Google Scholar]
- 39. Bo P. Effects of NaCl Stress on Seeds Germination and Seedlings Physiological Characteristics of Hibiscus Hamabo Sieb.Et Zucc. Shandong Normal University; 2008. [Google Scholar]
- 40. Bo P, Sun X, Sun Tet al. Antioxidative system and osmotic regulation in seedlings of annual hibiscus hamabo sieb.Et Zucc under salt stress. Acta Botanica Boreali. 2008;28:0113–8. [Google Scholar]
- 41. Bielach A, Hrtyan M, Tognetti V. Plants under stress: involvement of auxin and cytokinin. Int J Mol Sci. 2017;18:1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sahu SK, Thangaraj M, Kathiresan K. DNA extraction protocol for plants with high levels of secondary metabolites and polysaccharides without using liquid nitrogen and phenol. ISRN Molecular Biology. 2012;2012:205049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walker BJ, Abeel T, Shea Tet al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Simão FA, Waterhouse RM, Ioannidis Pet al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2. [DOI] [PubMed] [Google Scholar]
- 46. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:13033997 [q-bio]. 2013. [Google Scholar]
- 47. McKenna A, Hanna M, Banks Eet al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ou S, Jiang N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018;176:1410–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tarailo-Graovac M, Chen N. Using repeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009;25. [DOI] [PubMed] [Google Scholar]
- 51. Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–20. [DOI] [PubMed] [Google Scholar]
- 52. Bao Z. Automated De novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002;12:1269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:i351–8. [DOI] [PubMed] [Google Scholar]
- 54. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stanke M, Steinkamp R, Waack Set al. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32:W309–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stanke M, Morgenstern B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33:W465–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stanke M, Keller O, Gunduz Iet al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:W435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. [DOI] [PubMed] [Google Scholar]
- 59. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Trapnell C, Williams BA, Pertea Get al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cantarel BL, Korf I, Robb SMCet al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2007;18:188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Boeckmann B. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mitchell A, Chang H-Y, Daugherty Let al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015;43:D213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kanehisa M, Goto S, Sato Yet al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Altschul S. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Camacho C, Coulouris G, Avagyan Vet al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mistry J, Bateman A, Finn RD. Predicting active site residue annotations in the Pfam database. BMC Bioinformatics. 2007;8:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jones P, Binns D, Chang H-Yet al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Punta M, Coggill PC, Eberhardt RYet al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Conesa A, Götz S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genom. 2008;2008:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ashburner M, Ball CA, Blake JAet al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Michael TP, Jupe F, Bemm Fet al. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat Commun. 2018;9:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sarkar D, Mahato AK, Satya Pet al. The draft genome of Corchorus olitorius cv. JRO-524 (Navin). Genomics Data. 2017;12:151–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Udall JA, Long E, Hanson Cet al. De novo genome sequence assemblies of Gossypium raimondii and Gossypium turneri. G3 Genes|Genomes|Genetics. 2019;9:3079–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Argout X, Salse J, Aury J-Met al. The genome of Theobroma cacao. Nat Genet. 2011;43:101–8. [DOI] [PubMed] [Google Scholar]
- 76. Gao Y, Wang H, Liu Cet al. De novo genome assembly of the red silk cotton tree (Bombax ceiba). GigaScience. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Teh BT, Lim K, Yong CHet al. The draft genome of tropical fruit durian (Durio zibethinus). Nat Genet. 2017;49:1633–41. [DOI] [PubMed] [Google Scholar]
- 78. Hofmeister BT, Denkena J, Colomé-Tatché Met al. A genome assembly and the somatic genetic and epigenetic mutation rate in a wild long-lived perennial Populus trichocarpa. Genome Biol. 2020;21:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Badouin H, Velt A, Gindraud Fet al. The wild grape genome sequence provides insights into the transition from dioecy to hermaphroditism during grape domestication. Genome Biol. 2020;21:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Islam MS, Saito JA, Emdad EMet al. Comparative genomics of two jute species and insight into fibre biogenesis. Nat Plants. 2017;3:1–7. [DOI] [PubMed] [Google Scholar]
- 81. Shen Y, Liu J, Geng Het al. De novo assembly of a Chinese soybean genome. Sci China Life Sci. 2018;61:871–84. [DOI] [PubMed] [Google Scholar]
- 82. Takei H, Shirasawa K, Kuwabara Ket al. De novo genome assembly of two tomato ancestors, Solanum pimpinellifolium and Solanum lycopersicum var. cerasiforme, by long-read sequencing. DNA Res. 2021;28:dsaa029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Han MV, Thomas GWC, Lugo-Martinez Jet al. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol. 2013;30:1987–97. [DOI] [PubMed] [Google Scholar]
- 87. Zwaenepoel A, Van de Peer Y. Wgd—simple command line tools for the analysis of ancient whole-genome duplications. Bioinformatics. 2019;35:2153–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rozewicki J, Li S, Amada KMet al. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 2019;47:W5–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Bioinformatics. 1997;13:555–6. [DOI] [PubMed] [Google Scholar]
- 91. Tang H, Bowers JE, Wang Xet al. Synteny and collinearity in plant genomes. Science. 2008;320:486–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this work are available within the paper and its Supporting Information files. The data sets generated and analyzed during this study are available from the corresponding author upon request. All the whole-genome raw data generated during this study have been deposited in the SRA database under BioProject number PRJNA759075. Transcriptome clean data have been deposited in the SRA database under BioProject number PRJNA759717. The final chromosome-scale genome assembly and annotation data have been deposited in the Figshare database (https://doi.org/10.6084/m9.figshare.19142558.v1).