

Graphical abstract
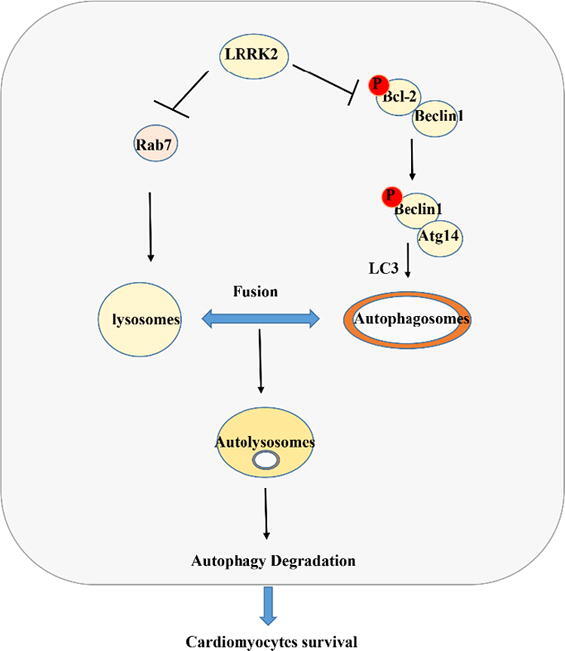
LRRK2 deficiency protected against cardiac remodelling under pressure overload by increasing Bcl-2/beclin1 and Rab7-regulated autophagy levels in the heart.
Keywords: Leucine-rich repeat kinase-2, Cardiac remodelling, Autophagy, Bcl-2, Beclin1, Rab7
Research Highlights
-
•
LRRK2 deficiency protected cardiac remodelling induced by pressure overload
-
•
LRRK2 deficiency preserved cardiac function by regulating both autophagosome formation and degradation.
-
•
LRRK2 regulates autophagosome formation by regulating Bcl2 and Beclin1 interaction.
-
•
LRRK2 regulates autophagosome fusion by interacting with Rab7.
-
•
LRRK2 may become a target of new therapeutic methods for treating heart failure.
Abstract
Introduction
Leucine-rich repetitive kinase-2 (LRRK2) is a Parkinson's disease-related gene that also participates in many inflammatory diseases. However, the functional role of LRRK2 in cardiovascular disease is not clear.
Objective
In this study, we aimed to elucidate the role of LRRK2 in cardiac remodelling under pressure overload.
Methods
Aortic banding surgery was performed to induce cardiac remodelling in a LRRK2 knockout mouse model. A cardiomyocyte remodelling model was established by phenylephrine (PE) stimulation in neonatal rat cardiomyocytes.
Results
LRRK2 was upregulated in remodelled mouse hearts and cardiomyocytes. Cardiac hypertrophy, fibrosis and dysfunction were ameliorated in LRRK2 knockout mice. LRRK2 silencing protected against the PE-induced cardiomyocyte hypertrophic response, while LRRK2 over-expression worsened the PE-induced hypertrophic response in cardiomyocytes. Decreased autophagy was observed in remodelled cardiomyocytes, whereas LRRK2 silencing increased autophagy levels and LRRK2 overexpression reduced autophagy levels. The autophagy inhibitors 3-MA, bafilomycin and chloroquine reversed the protective effects of LRRK2 deficiency. The autophagy activator rapamycin reversed the deleterious effects of LRRK2 overexpression. We found that LRRK2 inhibited Bcl-2 phosphorylation, thus decreasing the phosphorylation of Beclin1. The protective effects of LRRK2 knockout were partly counteracted by Beclin1(+/−) in vivo and Beclin1 silencing in vitro. We also observed an interaction between LRRK2 and Rab7, an autolysosome degradation-associated protein, which caused Rab7 downregulation. Rab7 knockdown almost completely reversed LRRK2 silencing-induced protection of cardiomyocytes
Conclusion
LRRK2 deficiency protected against cardiac remodelling under pressure overload by increasing Bcl-2/Beclin1 and Rab7-regulated autophagy levels in the heart.
Introduction
Heart failure (HF) has developed into one of the leading causes of death in both developed and developing countries [1]. When the heart receives external stimuli, such as hypertension, myocardial infarction, or ischaemic injury, it triggers the process of myocardial remodelling to adapt to changes in myocardial wall tension [2]. Cardiac remodelling depicts the flexibility of the heart to respond to and adapt to these stimuli but ultimately leads to heart failure [2]. The mechanisms involved in cardiac remodelling are complex, with changes in metabolism, redox systems, signalling pathways, endoplasmic reticulum stress, and autophagy in cardiomyocytes [3], as well as changes in other noncardiomyocytes such as fibroblasts, endothelial cells [4] and inflammatory cells [5]. Progress has been made to inhibit and block the process of cardiac remodelling, and clinical drugs such as beta-adrenergic receptor blockers, angiotensin converting enzyme inhibitors (ACEIs), and angiotensin receptor antagonists (ARBs) have been proven to exert anti-remodelling effects [6]. However, despite decades of use of these drugs in patients with heart failure, morbidity and mortality have increased gradually [7]. Thus, a deeper understanding of cardiac remodelling and the identification of new therapeutic methods are urgently needed.
A healthy proteostasis network is essential, safeguarding the proper function of the heart [8]. Autophagy is associated with the ubiquitin-proteasomal system (UPS) and the cytoskeleton, maintaining a protein degradation system of cellular protein quality control [9]. Autophagy is a lysosome-mediated degradation pathway that is responsible for removing potentially toxic cytosolic protein aggregates and damaged organelles inside cells [10]. Under physiological conditions, the basal level of autophagy is greatly important in maintaining the normal function of cardiomyocytes. During heart insult, the autophagy level changes in diverse ways [8]. In the ageing heart, the autophagy level is decreased [11]. In ischaemia and infarction hearts, autophagy was found to be activated during oxygen and nutrient deprivation [12], [13]. In diabetic cardiomyopathy, autophagy was found to be adapted in different ways (increased in type 1 diabetes but reduced in type 2 diabetes [14]). In failing hearts, autophagy was found to change in dynamic ways (suppressed in the initial stage, and activated in the end-stage) [15], [16]. These findings suggest the importance of autophagy. Thus, maintaining a healthy autophagy level may be a target for treating HF.
Leucine-rich repeat kinase 2 (LRRK2, a multidomain protein) is a member of the Roco protein family and was first recognized as a Parkinson’s disease (PD)-associated protein [17]. Many studies have also found a functional role of LRRK2 in apoptosis, autophagy, inflammation, proliferation, and synaptogenesis [17], [18], [19], [20]. Six coding changes have been identified with strong evidence for an association with PD pathology [21]. Mutations in the LRRK2 locus are associated with systemic lupus erythematosus [18], Crohn’s disease [22] and inflammatory bowel disease. Recently, LRRK2 was found to be connected with autophagy. LRRK2 suppresses autophagy, which leads to an increased inflammatory response in inflammatory bowel diseases [22]. Wallings R reported that LRRK2 is associated with the a1 subunit of vATPase, leading to a decrease in autophagosome/lysosome fusion in the pathology of PD [19]. LRRK2 mutations impaired depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10 [20]. These data suggest the functional role of LRRK2 in various disease models via regulation of autophagy initiation and degradation. Thus, we hypothesized that LRRK2 participates in the pathology of cardiac remodelling via autophagy regulation. In this study, we used LRRK2 knockout mice and Beclin1 (+/−) mice to elucidate the functional role of LRRK2 in remodelling the heart.
Methods
Animals and animal models
The Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996) and the ARRIVE guidelines and the Animal Care and Use Committee of Zhengzhou University guided our study. LRRK2 KO mice (Jackson Laboratory, 016121) were used in our studies. Beclin1 (+/−) mice (Shanghai Model Organisms, Ensembl ID ENSMUSG00000035086) were crossed with LRRK2 knockout mice to produce LRRK2(−/−)Beclin1(+/−) mice. Aortic banding surgery was performed to establish cardiac remodelling as previously described [23]. In the reversion experiment, mice were divided into the following eight groups: (1) WT-sham (n = 10); (2) LRRK2-sham (n = 10); (3) Beclin1(+/−)-sham (n = 10); (4) LRRK2(−/−)Beclin1(+/−)-sham (n = 10); (5) WT-AB (n = 10); (6) LRRK2-AB (n = 10); (7) Beclin1(+/−)-AB (n = 10); and (8) LRRK2(−/−)Beclin1(+/−)-AB (n = 10). Other methods including adeno associated virus construction and delivery, human heart samples, echocardiographic and haemodynamic analyses, quantitative real-time RT-PCR and western blotting, histological analysis, neonatal rat cardiomyocyte (NRCM) isolation and culture, immunofluorescence and autophagic flux analysis, co-immunoprecipitation assays, and statistical analysis were in the supplementray materials.
Results
Expression of LRRK2 in heart and cardiomyocytes
We first explored the changes in LRRK2 expression in the remodelling of hearts and cardiomyocytes. An increased LRRK2 protein level was observed at 1 week after AB surgery and increased gradually after AB until 8 weeks (Fig. 1a). The phosphorylation status of LRRK2 was also detected, p-LRRK2 (S935) was up-regulated along with LRRK2 post AB ((Fig. 1a). LRRK2 was up-regulated in cardiomyocytes isolated in heart tissue 4 weeks post AB surgery (Fig. 1b). We then detected the expression of LRRK2 in cardiomyocytes in response to various stimuli. As shown in Fig. 1c, the expression level of LRRK2 and p-LRRK2 (S935) were increased sharply in PE-, angiotensin (Ang) II-, and endothelin-1 (ET-1)-stimulated cardiomyocytes. Immunohistochemical staining results showed that LRRK2 was mainly located in cardiomyocytes in mouse hearts (Fig. 1d). In cardiomyocytes, LRRK2 located in cytoplasma as detected by immunofluorescence staining (Fig. 1e). To further confirm the role of LRRK2 in cardiac remodelling and heart failure, human heart sample with heart failure patients undergoing heart transplants as well as control human hearts from normal heart donors was obtained and subjected to protein extraction. As shown in Fig. 1f, the level of LRRK2 was sharply increased in heart sample with heart failure patients as compared with the normal hearts. These results indicate that LRRK2 participates in the pathology of cardiac remodelling. P-Rab10 and Rab10 were detected as it was reported to be the main target of LRRK2. However, the level of both P-Rab10 and Rab10 was not changed in in vivo AB surgery heart (Fig. 1a). We also detected Rab8, but the protein level of Rab8 was hard to be detected by western blot.
Fig. 1.

Expression model of LRRK2 in heart and cardiomyocytes, a. Protein level of P-LRRK2 (S935), total LRRK2, P-Rab10, total Rab10 in mouse hearts subjected to AB surgery (n = 6) (*P < 0.05 vs. sham). b Protein level of LRRK2 in cardiomyocytes isolated from heart tissue 4 weeks post AB (n = 6). c. P-LRRK2 (S935), total LRRK2 levels in NRCMs stimulated with AngII, PE, and ET-1 for 24 h (n = 6) (*P < 0.05 vs. CON). d. LRRK2 and α-actin staining in mouse hearts (n = 5). e. LRRK2 staining in PE-stimulated NRCMs (n = 5). f. Protein level of LRRK2 in left ventriculus of heart failure patients and normal heart donors (n = 6).
Cardiac remodelling in LRRK2-deficient mice
Then, we used LRRK2 KO mice to further explore the functional role of LRRK2 in cardiac remodelling under pressure overload. As shown in Fig. 2a, the expression level of LRRK2 almost vanished in KO mouse hearts. LRRK2 KO mice and their WT littermates were subjected to AB surgery. Four weeks after surgery, the hearts were removed. As shown in Fig. 2b, the heart weight/body weight ratio (HW/BW), heart weight, lung weight/body weight ratio (LW/BW), and lung weight/tibia length ratio (LW/TL) were lower in the LRRK2 KO-AB group than in the WT-AB group. HE staining showed a gross decrease in the heart as well as cell surface area in the KO-AB group compared to those of the WT-AB group, while no significant difference was observed in the two sham groups (Fig. 2c). The RT-PCR results revealed reduced transcription levels of hypertrophic markers in the KO-AB group compared to those of the WT-AB group. No significant difference was observed in these two sham groups (Fig. 2b-d). Cardiac fibrosis was also evaluated by PSR staining: perivascular and interstitial fibrosis levels were decreased in the KO-AB group compared to those of the WT-AB group (Fig. 2e). The transcription level of fibrosis markers (Fig. 2f) and the protein level of the matrix metalloproteinases (MMPs) MMP2 and MMP9 were all reduced in KO-AB mouse hearts compared with WT-AB mouse hearts (Fig. 2g). Under physiological conditions, we did not observe a difference in hypertrophy or fibrosis changes in WT and KO mouse hearts.
Fig. 2.

Cardiac remodelling in LRRK2-deficient mice. a. Protein level of LRRK2 in LRRK2 WT and KO mice (n = 6). b. Heart weight/body weight (HW/BW), Heart weight, lung weight/BW (LW/BW), and LW/TL in mice 4 weeks after AB (n = 12 in each group). c. HE staining and cell section area in mouse hearts 4 weeks after AB (n = 6). d. mRNA levels of atrial natriuretic peptide (ANP), B-type Natriuretic Peptide (BNP), myosin heavy chain beta (β-MHC), and α-MHC (n = 6). e. PSR staining and left ventricular collagen volume in mouse hearts 4 weeks after AB (n = 6). f. mRNA levels of collagen I, collagen III, fibronectin and connective tissue growth factor (CTGF) (n = 6).g. Protein levels of MMP2 and MMP9 in mouse hearts (n = 6). *P < 0.05 vs. corresponding sham; # P < 0.05 vs. WT-AB.
The systolic and diastolic wall thickness and volume were evaluated by echocardiography. Four weeks after AB surgery, LV end diastolic dimension (LVEDd) and systolic dimension (LVESd) were increased in WT mice, LV posterior wall thickness at end-diastole (LVPWd) was increased sharply, and LV ejection fraction (LVEF) and fractional shortening (LVFS) were reduced in WT mouse hearts. In KO-AB mice, all these indexes were relatively ameliorated compared with those of WT-AB mice (Fig. S1a, b). A pressure–volume loop was performed to evaluate changes in the haemodynamic index. In WT-AB mice, we observed increased end-diastolic and systolic pressure and decreased maximal rates of pressure development and decay (dp/dt max; dp/dt min) compared with those of WT-sham mice. In contrast, in KO-AB mice, these haemodynamic indexes were also remarkably ameliorated (Fig. S1d, e). Heart rate was not different among the four groups (Fig. S1c). Under physiological conditions, we also did not observe a difference in functional changes in WT or KO mouse hearts.
Cardiac remodelling in mice knockdown of LRRK2
An Adeno associated virus 9 (AAV9)-shLRRK2 delivery system was used to knockdown LRRK2 in mice heart. As shown in Fig. 3a, the LRRK2 protein level was remarkablely dropped after 2, 4, and 6 weeks of AAV9- shLRRK2 injection. Consistent with the results in LRRK2-KO experiments, mice knockdown of LRRK2 revealed reduced HW/BW, HW, lung LW/BW, and LW/TL; dropped cell surface area in heart tissue as well as LV collagen volume (Fig. 3b-d). Mice with knockdown of LRRK2 also showed improved cardiac function as comparing with mice injected with the control AAV9 (shRNA) (Fig. 3e, f).
Fig. 3.

Cardiac remodelling in mice knockdown of LRRK2. a. Protein level of LRRK2 in mice with AAV9-shLRRK2 injection (n = 6). b. Heart weight/body weight (HW/BW), Heart weight, lung weight/BW (LW/BW), and LW/TL in mice 4 weeks after AB (n = 12 in each group). c. HE staining and cell section area in mouse hearts 4 weeks after AB (n = 6). d. PSR staining and left ventricular collagen volume in mouse hearts 4 weeks after AB (n = 6). e. Echocardiography result mice (n = 8). f: Hemodynamic parameters (n = 8). LVEDd: left ventricular end diastolic dimension; LVESd: left ventricular end systolic dimension; LVPWd: LV Postier wall thickness at end-diastole. LVEF: LV ejection fraction; LVFS: LV fractional shorterning. HR: heart rate. EDP/ESP: end-diastolic and systolic pressure. dp/dt max/min: decreased maximal rate of pressure development and decay. *P < 0.05 vs. corresponding sham; # P < 0.05 vs. shRNA-AB.
Role of LRRK2 in PE-stimulated cardiomyocyte remodelling
To explore the role of LRRK2 in cardiomyocytes, NRCMs were isolated and stimulated with PE for 24 h to establish a cardiomyocyte remodelling model in vitro. Cells were transfected with Ad-LRRK2 to overexpress LRRK2 (Fig. 4a). Increased LRRK2 expression led to an augmented hypertrophic response to PE, as shown by increased cell surface area by α-actin staining and enhanced transcription of hypertrophic markers in the Ad-LRRK2 group compared with that of the control (Ad-NC)-PE group (Fig. 4b, c). Cells were also transfected with LRRK2 siRNA to knock down LRRK2 levels (Fig. 4d). Consistent with the in vivo results, the cell hypertrophic response to PE was lower than that in the control (siRNA)-PE group (Fig. 4e, f). Cardiomyocytes with either enhanced or silenced LRRK2 protein levels did not differ from the normal control under basal conditions (Fig. 4b, d, e, f).
Fig. 4.

Role of LRRK2 in PE-stimulated cardiomyocyte remodelling. a-c: NRCMs were transfected with Ad-LRRK2. a. Protein level of LRRK2 in NRCMs (n = 6). b. α-actin staining and cell surface area in PE-stimulated cells (n = 5). c. mRNA levels of ANP and β-MHC in cells (n = 6). *P < 0.05 vs. corresponding CON; # P < 0.05 vs. Ad-NC-PE.d-f: NRCMs were transfected with LRRK2 siRNA. d. Protein level of LRRK2 in NRCMs (n = 6). e. α-actin staining and cell surface area in PE-stimulated cells (n = 5). f. mRNA levels of ANP and β-MHC in cells (n = 6). *P < 0.05 vs. corresponding CON; # P < 0.05 vs. siRNA-PE.
Effects of LRRK2 on autophagy in cardiomyocytes
Cell autophagy was analysed because LRRK2 was reported to function in autophagy regulation. The autophagy-associated proteins Atg5 and Atg7 were downregulated, while P62 accumulated in PE-stimulated cells (Fig. 5a, b). We observed a robust reduction in Atg5 and Atg7 and a robust increase in P62 accumulation in LRRK2-overexpressing cells under PE stimulation (Fig. 5a, b). Then, cells were transfected with Ad-mRFP-GFP-LC3 (an acidic lysosomal environment quenches acid-sensitive GFP fluorescence, while mRFP is not affected) to evaluate autophagic flux. As shown in Fig. 5c, d, mRFP-stained LC3 (representing the formation of autophagosomes) was decreased in LRRK2-overexpressing cells under both physiological and PE conditions. GFP-LC3 (representing the remaining autophagosomes after degradation) was increased in LRRK2-overexpressing cells under physiological conditions compared to that of the control group but was decreased under PE conditions compared to that of control-PE cells. The degradation rate (indicated by red-green puncta/red puncta) was reduced in LRRK2-overexpressing cells under both physiological and PE conditions. The remaining autophagosome rate (indicated by green puncta/red puncta) was increased in LRRK2-overexpressing cells under both physiological and PE conditions. These data suggest that LRRK2 overexpression affects both the formation and degradation of autophagosomes.
Fig. 5.

Effects of LRRK2 on autophagy in cardiomyocytes. a-d: NRCMs were transfected with Ad-LRRK2. a and b. The protein levels of Atg5, Atg7, and P62 in NRCMs (n = 6). c and d. NRCMs were transfected with Ad-LC3-mCherry-GFP. c: Representative images (n = 5). d: Red puncta number, green puncta number, red-green puncta/red puncta ratio, and green puncta/red puncta ratio. *P < 0.05 vs. corresponding CON; # P < 0.05 vs. Ad-NC-PE.e-h: NRCMs were transfected with LRRK2 siRNA. e and f. Protein levels of Atg5, Atg7, and P62 in NRCMs (n = 6). g and h. NRCMs were transfected with Ad-LC3-mCherry-GFP. g: Representative images (n = 5). h: Red puncta number, green puncta number, red-green puncta/red puncta ratio, and green puncta/red puncta ratio. *P < 0.05 vs. corresponding CON; # P < 0.05 vs. siRNA-PE.
The autophagy level was also detected in LRRK2-silenced cells. Adverse results were observed in LRRK2-silenced cells. As shown in Fig. 5e and f, LRRK2 silencing induced an increase in the autophagy level, with increased Atg5 and Atg7 protein levels and decreased P62 accumulation. In the autophagic flux experiment (Fig. 5g, h), LRRK2-silenced cells had increased red puncta and decreased green puncta under PE stimulation. The degradation rate was enhanced, and the remaining autophagosome rate was reduced in LRRK2-silenced cells under both physiological and PE conditions. These data suggest that LRRK2 silencing helps the formation and degradation of autophagy and maintains an unobstructed autophagic flow.
Effect of autophagy inhibition/activation on the effect of LRRK2 on cardiomyocytes
We then used three autophagy inhibitors: 3-MA (PI3K inhibitor, to inhibit autophagosome formation), bafilomycin A1 (inhibits the fusion of autophagosomes and lysosomes), and chloroquine (inhibits lysosomal degradation). 3-MA, bafilomycin A1 and chloroquine increased P62 level in NRCMs with LRRK2 knockdown (Fig. 6a). As a result, both bafilomycin A1 and chloroquine completely reversed the protective effect of LRRK2 silencing on cardiomyocyte remodelling, while 3-MA partly counteracted the anti-remodelling effects of LRRK2 silencing, as determined by increased cell surface area and hypertrophic gene transcription levels (Fig. 6b-d). Then, we used the autophagy activator rapamycin (inhibitor of mTOR) and found that rapamycin ameliorated the PE-induced remodelling response in cardiomyocytes. However, rapamycin only partly counteracted the LRRK2 overexpression-induced deleterious effects (Fig. 6e-g). Taken together, autophagosome degradation and formation are equally important in the role of LRRK2 in cardiomyocyte remodelling.
Fig. 6.

Autophagy inhibition/activation and the effect of LRRK2 on cardiomyocytes. a-d: NRCMs were transfected with LRRK2 siRNA and treated with the autophagy inhibitors 3-MA, bafilomycin A1 and chloroquine. a. P62 level in the indicated group (n = 6). b. α-Actin staining (n = 5). c. Cell surface area in PE-stimulated cells (n = 50). d. mRNA levels of ANP and β-MHC in cells (n = 6). *P < 0.05 vs. siRNA; # P < 0.05 vs. PE; εP < 0.05 vs. PE + siLRRK2.e-g: NRCMs were transfected with Ad-LRRK2 and treated with the autophagy activator rapamycin. d. α-Actin staining (n = 5). e. Cell surface area in PE-stimulated cells (n = 50). f. mRNA levels of ANP and β-MHC in cells (n = 6). *P < 0.05 vs. Ad-NC; # P < 0.05 vs. PE; εP < 0.05 vs. PE + Ad-LRRK2.
The autophagy signalling pathway was screened to clarify the mechanism underlying the effect of LRRK2 on autophagy. As shown in Fig. S2a-d, we screened the upstream molecules that regulate autophagy. LC3II level was elevated in both LRRK2-KO mice and in cells with LRRK2 silence (Fig. S2a-d). The upstream proteins AKT, AMPKα, mTOR and ULK1 were not significantly different from those in the control PE group in LRRK2-deficient heart tissue and cells undergoing remodelling. However, Bcl2 was decreased in heart tissue and cells and was not significantly different between the control and LRRK2-deficient groups, but phosphorylated Bcl2 was more abundant in the LRRK2-deficient group than in the control group in both remodelled heart tissue and cells (Fig. S2e, f). Consistently, the extent of decreased Beclin1 was the same between the LRRK2-deficient group and the control group, whereas phosphorylated Beclin1 (T119) was more abundant in the LRRK2-deficient group than in the control group in both remodelled heart tissue and cells (Fig. S2e, f). Thus, Bcl2 and Beclin1 may be targets of LRRK2.
Effect of changes in Beclin1 expression on the functional role of LRRK2 in vitro
Then, we knocked down Beclin1 in cells by using siRNA transfection (Fig. 7a). Beclin1 knockdown accelerated PE-induced remodelling with increased cell surface area and mRNA levels of remodelling markers (Fig. 7b, c). However, Beclin1 knockdown partly counteracted the protective effects of LRRK2 silencing. The remodelling response was reduced in LRRK2-silenced and Beclin 1-knockdown cells compared to that of Beclin1-knockdown cells under PE stimulation (Fig. 7b, c). Beclin1-overexpressing cells were induced by using the adenovirus transfection system (Fig. 7d). Conversely, Beclin1 overexpression protected against the PE-induced remodelling response (Fig. 7e, f). However, Beclin1 overexpression partly counteracted the deleterious effects of LRRK2 overexpression. The remodelling response was still greater in LRRK2- and Beclin 1-overexpressing cells than in Beclin1-overexpressing cells under PE stimulation (Fig. 7e, f). These data suggest that Beclin1 is one of the targets of LRRK2 in the regulation of autophagy.
Fig. 7.

Change in Beclin1 expression and the functional role of LRRK2 in vitro a-c. NRCMs were transfected with Beclin1 siRNA. a. Protein level of Beclin1 (n = 6, *P < 0.05 vs. siRNA). b. α-Actin staining and cell surface area in PE-stimulated LRRK2-silenced cells (n = 5). c. mRNA levels of ANP and β-MHC in cells (n = 6). *P < 0.05 vs. CON; # P < 0.05 vs. PE; εP < 0.05 vs. PE + siLRRK2.d-f: NRCMs were transfected with Ad-Beclin1. d. Protein level of Beclin1 (n = 6, *P < 0.05 vs. Ad-NC). e. α-Actin staining and cell surface area in PE-stimulated LRRK2-overexpressing cells (n = 5). f. mRNA levels of ANP and β-MHC in cells (n = 6). *P < 0.05 vs. CON; # P < 0.05 vs. PE; εP < 0.05 vs. PE + Ad-LRRK2.
Effect of changes in Beclin1 expression on the functional role of LRRK2 in vivo
To confirm the mechanism in vitro, we used Beclin1 (+/−) mice and crossed these mice with LRRK2 KO mice to produce LRRK2(−/−) Beclin1 (+/−) mice. Then, these mice were subjected to AB surgery. Four weeks after surgery, in sham groups, the gross heart, cell section area, heart weight, and lung weight (indicated by HW/BW, HW, LW/BW, LW/TL) in the Beclin1 (+/−) sham group were larger than those in the WT mice but decreased in LRRK2(−/−) Beclin1 (+/−) mice compared with those of the Beclin1 (+/−) sham group. In the AB groups, the gross heart, cell section area, heart weight, lung weight and fibrosis level in the Beclin1 (+/−) AB group were larger than those in the WT mice. These indexes were lower in LRRK2(−/−) Beclin1(+/−) AB mice than in Beclin1(+/−) AB mice but were higher in LRRK2(−/−) AB mice than in LRRK2(+/−) AB mice except fibrosis level (Fig. 8a-d). Cardiac function was also evaluated in these mice. Cardiac function and haemodynamic indexes were not different among the four sham groups but were deteriorated in the Beclin1 (+/−) AB group compared with that of WT mice. Cardiac function in LRRK2(−/−) Beclin1(+/−) AB mice was improved when compared with that of Beclin1(+/−) AB mice but was still worse than that of LRRK2(−/−) AB mice (Fig. 8e-g). These data confirm that LRRK2 regulates autophagy by partly acting on Beclin1.
Fig. 8.

Change in Beclin1 expression and the functional role of LRRK2 in vivo. WT mice, LRRK2−/−mice, Beclin1+/−mice, and LRRK2−/−Beclin1+/−mice were subjected to AB surgery. a and b. HE staining and cell section area in mouse hearts 4 weeks after AB (n = 6). c. HW/BW, Heart weight, LW/BW, and LW/TL in mice 4 weeks after AB (n = 10 in each group). d. PSR staining and left ventricular collagen volume. e and f. Echocardiography results in mice (n = 8). g. Haemodynamic parameters in mice (n = 8). *P < 0.05 vs. corresponding sham; # P < 0.05 vs. WT-AB.
The influence of Rab7 on LRRK2 in cardiomyocytes
Fig. 5, Fig. 6 show that LRRK2 functions in both autophagy formation and degradation. Since Beclin1 regulates autophagosome formation, and we further explored the molecular degradation that is regulated by LRRK2. A previous study reported the functional role of LRRK2 on Rab proteins [20] (participating in the fusion of autophagosomes and lysosomes). We found that LRRK2 interacted with Rab7 in cardiomyocytes (Fig. S3a). Then, we tested Rab7 protein levels in LRRK2-deficient hearts and cells. As expected, Rab7 was increased in LRRK2-deficient hearts and cells compared with those of the control hearts and cells (Fig. S3b, c). However, both P-Rab10 and total Rab10 were not change in LRRK2-deficient cells (Fig. S3c). Then, we used Rab7 siRNA to knockdown Rab7 (Fig. S3d), and as shown in Fig. S3 e and f, the remodelling response in cardiomyocytes was accelerated in Rab7-silenced cells under PE stimulation. Rab7 silencing also completely abolished the LRRK2 silencing-induced protective effects (Fig. S3e, f). These results suggest that the interaction of LRRK2 with Rab7 also participates in the regulation of cardiac remodelling.
To determine whether the effect of LRRK2 is a LRRK2 kinase depend process, we used a selective LRRK2 kinase inhibitor GSK2578215A to treat cardiomyocytes, as shown in Fig. S4a, the P-LRRK2 was down-regulated by GSK2578215A, but the level of LRRK2 was not change by GSK2578215A. PE induced hypertrophic response was inhibited by GSK2578215A (Fig. S4b, c). These data indicated LRRK2 kinase depend process affects cardiac hypertrophy.
Discussion
Short-term cellular stress can maintain the transient balance of cells under stimulation, but long-term stress can lead to disease. Thus, a short break is needed to counteract the activation signal and maintain cell homeostasis [24]. Failure of these braking systems is involved in the pathogenesis of most human diseases. Here, we describe a molecular model of cardiac remodelling in which LRRK2 silencing protects the heart by accelerating autophagy-mediated cellular protein quality control. Furthermore, overexpression of LRRK2 drives the downregulation of autophagy levels and the progression of cardiac remodelling. Although the strong effect of LRRK2 on the heart can be observed under stimulation, knockout of the LRRK2 gene does not change the basic function of the heart. These results suggest that the heart is insensitive to LRRK2 under normal conditions, and LRRK2 does not affect the development of the heart or its normal function.
Cardiomyocytes are terminally differentiated cells and have no proliferative potential, and an appropriate protein quality management system is of great significance in protecting cardiomyocyte function [9]. Autophagy can degrade damaged proteins and organelles and maintain the normal function of cells. In addition, autophagy can recover fatty acids and amino acids and provide energy and nutrition for myocardial cells in starvation conditions [14]. Studies have found that autophagy plays a role in the pathophysiology of HF. In pressure overload-induced cardiac remodelling, autophagy was suppressed in the compensation process and then activated in the decompensated process [15], [16]. Studies have reported that many autophagy activators, such as rapamycin, metformin and 5-aminoimidazole 1 carboxamide ribonucleoside (AICAR), improve cardiac function, reduce cardiac hypertrophy, and delay the progress of HF during pressure overload [25], [26], [27]. In contrast, autophagy suppression was found to be beneficial in the presence of severe pressure overload and in end-stage failing hearts [28]. In our study, we used moderate aortic banding surgery and found that at 4 weeks after surgery, cardiac autophagy was suppressed, and the heart displayed dysfunction, while LRRK2 silencing increased autophagic levels, thus preserving cardiac function. These effects were confirmed by our in vitro experiment showing that autophagy inhibitors reversed the protection of LRRK2 knockdown.
There are four steps in a full autophagic process: first, the formation of a double membrane structure; second, engulfment of damaged organelles and aberrant proteins; third, fusion with a lysosome to form the autolysosome; and fourth, degradation of the cargo by lysosomal hydrolases [8]. In our autophagic flux study, we observed that LRRK2 regulates not only the formation but also the degradation of autophagosomes. These results were confirmed by autophagy inhibition and activation experiments. Autophagy inhibitors completely reversed the protective effects of LRRK2 silencing, but autophagy activation only partly reversed the deleterious effects of LRRK2 overexpression. There are several signalling pathways that regulate autophagy [8]. In nutrient-rich conditions, mTOR is activated and thereby inhibits autophagosome formation through inactivation of ULK1 [29]. Under nutrient-poor conditions, AMPKα is activated. Through inhibition of mTOR and direct activation of ULK1, AMPKα positively regulates autophagy [30]. In our study, we found that the activation of AMPKα was decreased and AKT (another upstream activator of mTOR) was increased in both remodelled heart tissue and cardiomyocytes. Activation of mTOR was increased in heart tissue and cardiomyocytes. We also found decreased ULK1 expression in heart tissue and cardiomyocytes. LRRK2 did not affect the levels of these proteins.
Beclin1 is a central protein in autophagosome formation and forms a complex with vacuolar protein sorting 34 (VPS34) and VPS15 to initiate the formation of the phagophore [31]. Under normal conditions, the BH3 domain of Beclin1 interacts with Bcl2/Bcl-XL to stabilize Beclin1 dimers, which effectively suppress autophagosome biogenesis [32]. Once stimulated, the upstream molecule mediates phosphorylation of Bcl2, which induces the dissociation of the Beclin1-Bcl2 complex, facilitating heterodimerization of Beclin1 with Atg14 and activation of autophagy [33]. In our study, we observed decreased phosphorylation of Bcl2 in remodelled heart tissue and cardiomyocytes, as well as decreased phosphorylation (T119, activation) of Beclin1. However, LRRK2 increased the phosphorylation of Bcl2 and Beclin1 without influencing the total Bcl2 and Beclin1 levels. These results suggest that LRRK2 regulates autophagy initiation by suppressing the interaction of Bcl2 and Beclin. These effects were confirmed by using hemizygous deletion of Beclin1. As hemizygous deletion in Beclin1 in mice revealed a deteriorating remodelling process and severe cardiac dysfunction, LRRK2 deficiency could partly alleviate these unfavourable phenotypes induced by hemizygous deletion of Beclin1 under pressure overload.
Fig. 5, Fig. 6 show that we confirmed that LRRK2 affects autophagosome formation and degradation, but the mechanism by which LRRK2 regulates autophagosome degradation was unclear. Studies have reported that LRRK2 is associated with Rab10, which regulates fusion in mitophagy [20]. Rabs play important roles in the fusion of autophagosomes and lysosomes to form autolysosomes [34]. Thus, we screened those Rabs. As a result, we observed an interaction between LRRK2 and Rab7. Knockdown of Rab7 completely reversed the anti-remodelling effect of LRRK2 silencing. This suggests that LRRK2 affects autophagosome degradation by targeting Rab7. In addition, inhibiting autophagy could partly reverse the anti-remodelling effect of LRRK2 deficiency, but inhibiting autophagosome degradation almost completely abolished the protection exerted by LRRK2 deficiency.
In summary, LRRK2 deficiency protected cardiac remodelling induced by pressure overload and preserved cardiac function by regulating both autophagosome formation and degradation. By functioning in the Bcl2 and Beclin1 interaction, LRRK2 regulates autophagosome formation, and by interacting with Rab7, LRRK2 regulates autophagosome fusion. Thus, LRRK2 may become a target of new therapeutic methods for treating HF.
Availability of data and material
The original data will be available when requiring to the corresponding author.
Funding
This study was supported by the National Natural Science Foundation of China (Grant nos. 82070233), the Scientific and Technological Project of Henan Province (202102310364).
Compliance with Ethics Requirements
All Institutional and National Guidelines for the care and use of animals (fisheries) were followed.
The Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and the ARRIVE guidelines and the Animal Care and Use Committee of Zhengzhou University guided our study.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2021.07.004.
Contributor Information
Yuan Liu, Email: fccliuy2@zzu.edu.cn.
Yintao Zhao, Email: fcczhaoyt@zzu.edu.cn.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Nichols M., et al. Cardiovascular disease in Europe 2014: epidemiological update. Eur Heart J. 2014;35(42):2950–2959. doi: 10.1093/eurheartj/ehu299. [DOI] [PubMed] [Google Scholar]
- 2.Mann D.L., Bogaev R., Buckberg G.D. Cardiac remodelling and myocardial recovery: lost in translation? Eur J Heart Fail. 2010;12(8):789–796. doi: 10.1093/eurjhf/hfq113. [DOI] [PubMed] [Google Scholar]
- 3.Haque ZK, W.D. How cardiomyocytes sense pathophysiological stresses for cardiac remodeling. Cell Mol Life Sci 2017;74(6): 983–1000. [DOI] [PMC free article] [PubMed]
- 4.Zouein F.A., Booz G.W., Altara R. STAT3 and endothelial cell-cardiomyocyte dialog in cardiac remodeling. Front Cardiovasc Med. 2019;6:50. doi: 10.3389/fcvm.2019.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, H Z, Li H. Insights into innate immune signalling in controlling cardiac remodelling. Cardiovasc Res 2017;113(13): 1538–50. [DOI] [PubMed]
- 6.Papadimitriou L., et al. The limitations of symptom-based heart failure management. Card Fail Rev. 2019;5(2):74–77. doi: 10.15420/cfr.2019.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith J.G. Molecular epidemiology of heart failure: translational challenges and opportunities. JACC Basic Transl Sci. 2017;2(6):757–769. doi: 10.1016/j.jacbts.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Z D, Wiersma M, Brundel BJJM. Role of autophagy in proteostasis: friend and foe in cardiac diseases. Cells 2018;7(12): E279. [DOI] [PMC free article] [PubMed]
- 9.Arrieta A., Blackwood E.A., Glembotski C.C. ER protein quality control and the unfolded protein response in the heart. Curr Top Microbiol Immunol. 2018;414:193–213. doi: 10.1007/82_2017_54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saha S., et al. Autophagy in health and disease: a comprehensive review. Biomed Pharmacother. 2018;104:485–495. doi: 10.1016/j.biopha.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Taneike M., et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6(5):600–606. doi: 10.4161/auto.6.5.11947. [DOI] [PubMed] [Google Scholar]
- 12.Matsui Y., et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 13.Yan L., et al. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102(39):13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanamori H., et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy. 2015;11(7):1146–1160. doi: 10.1080/15548627.2015.1051295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakai A., et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13(5):619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 16.Takemura G., et al. Autophagic degeneration and death of cardiomyocytes in heart failure. Autophagy. 2006;2(3):212–214. doi: 10.4161/auto.2608. [DOI] [PubMed] [Google Scholar]
- 17.Price A., et al. The LRRK2 signalling system. Cell Tissue Res. 2018;373(1):39–50. doi: 10.1007/s00441-017-2759-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang M, Y C, Cai J, Liu S, Liu XN, Chen Y, et al. LRRK2 is involved in the pathogenesis of system lupus erythematosus through promoting pathogenic antibody production. J Transl Med 2019; 17(1): 37. [DOI] [PMC free article] [PubMed]
- 19.Wallings R., Connor-Robson N., Wade-Martins R. LRRK2 interacts with the vacuolar-type H+-ATPase pump a1 subunit to regulate lysosomal function. Hum Mol Genet. 2019 doi: 10.1093/hmg/ddz088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wauters F, C T, Imberechts D, Martin S, Koentjoro B, Sue C, Vangheluwe P, et al. LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy, 2019; 16(2): 203–22. [DOI] [PMC free article] [PubMed]
- 21.Mamais A., et al. Analysis of macroautophagy related proteins in G2019S LRRK2 Parkinson's disease brains with Lewy body pathology. Brain Res. 2018;1701:75–84. doi: 10.1016/j.brainres.2018.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takagawa T., et al. An increase in LRRK2 suppresses autophagy and enhances Dectin-1-induced immunity in a mouse model of colitis. Sci Transl Med. 2018;10(444) doi: 10.1126/scitranslmed.aan8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y., et al. Toll-like receptor 5 deficiency attenuates interstitial cardiac fibrosis and dysfunction induced by pressure overload by inhibiting inflammation and the endothelial-mesenchymal transition. Biochim Biophys Acta. 2015;1852(11):2456–2466. doi: 10.1016/j.bbadis.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 24.Deng K.Q., et al. Targeting transmembrane BAX inhibitor motif containing 1 alleviates pathological cardiac hypertrophy. Circulation. 2018;137(14):1486–1504. doi: 10.1161/CIRCULATIONAHA.117.031659. [DOI] [PubMed] [Google Scholar]
- 25.McMullen J.R., et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109(24):3050–3055. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- 26.Zhang C.X., et al. Metformin attenuates ventricular hypertrophy by activating the AMP-activated protein kinase-endothelial nitric oxide synthase pathway in rats. Clin Exp Pharmacol Physiol. 2011;38(1):55–62. doi: 10.1111/j.1440-1681.2010.05461.x. [DOI] [PubMed] [Google Scholar]
- 27.Li H.L., et al. Long-term activation of adenosine monophosphate-activated protein kinase attenuates pressure-overload-induced cardiac hypertrophy. J Cell Biochem. 2007;100(5):1086–1099. doi: 10.1002/jcb.21197. [DOI] [PubMed] [Google Scholar]
- 28.Pasquier B. Autophagy inhibitors. Cell Mol Life Sci. 2016;73(5):985–1001. doi: 10.1007/s00018-015-2104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alers S., et al. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y., Chen Y. AMPK and autophagy. Adv Exp Med Biol. 2019;1206:85–108. doi: 10.1007/978-981-15-0602-4_4. [DOI] [PubMed] [Google Scholar]
- 31.Pattingre S., et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Menon MB, D S. Beclin 1 phosphorylation - at the center of autophagy regulation. Front Cell Dev Biol 2018;6(137). [DOI] [PMC free article] [PubMed]
- 33.Song X., et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc(-) activity. Curr Biol. 2018;28(15):2388–2399 e5. doi: 10.1016/j.cub.2018.05.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamb C.A., Longatti A., Tooze S.A. Rabs and GAPs in starvation-induced autophagy. Small GTPases. 2016;7(4):265–269. doi: 10.1080/21541248.2016.1220779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original data will be available when requiring to the corresponding author.