

Abstract
Basic and antiviral research on SARS-CoV-2 rely on cellular assays of virus replication in vitro. In addition, accurate detection of virus-infected cells and released virus particles is needed to study virus replication and to profile new candidate antiviral drugs. Here, by flow cytometry, we detect SARS-CoV-2 infection at single cell level and distinguish infected Vero E6 cells from uninfected bystander cells. Furthermore, based on the viral nucleocapsid expression, subpopulations of infected cells that are in an early or late phase of viral replication can be differentiated. Importantly, this flow cytometric technique complements our duplex RT-qPCR detection of viral E and N, and it can be applied to all current SARS-CoV-2 variants of concern, including the highly mutated Omicron variant.
Keywords: : antiviral drug, duplex RT-qPCR, flow cytometry, nucleocapsid, omicron variant, SARS-CoV-2
Method summary
This study describes the characterization of SARS-CoV-2 infected cells using intracellular flow cytometric viral nucleocapsid staining that complements RT-qPCR quantification of viral RNA. The technique makes it possible to distinguish between infected cells in the early (low N) or late phase (high N) of viral replication. It can also be applied to the different variants of concern of SARS-CoV-2, including the Omicron variant.
Since December 2019, COVID-19 has posed a serious threat to global health and has impacted the global economy tremendously. The SARS-CoV-2, a novel Betacoronavirus, was soon identified as the causative agent of COVID-19 [1,2], and quickly spread from the 41 initially reported patients in Hubei province, China to a global pandemic [3]. As of February 2, 2022, there are nearly 377 million confirmed cases of SARS-CoV-2 infection and approximately 5.7 million deaths by COVID-19 worldwide (https://covid19.who.int/).
Despite the great success of the administered vaccines against SARS-CoV-2, the virus can still spread, as evidenced by the current circulation of the highly contagious Omicron variant [4]. This emphasizes the additional need to develop effective antiviral countermeasures. Basic and antiviral research on SARS-CoV-2 rely not only on cellular assays for virus replication but also on the accurate detection of virus-infected cells and released virus particles. The aim of this work is to describe single cell quantification of SARS-CoV-2 infected cells by flow cytometry and to present the complementarity of this technique with the sensitive detection of viral RNA by RT-qPCR. This study is not aiming at comparing both techniques and to determine which technique is the most sensitive one, but at demonstrating the complementarity and the robustness of both techniques as they can detect various variants of SARS-CoV-2, including the (current circulating) highly mutated Omicron variant.
Materials & methods
Cells & virus strains
Cells
African green monkey kidney cells (Vero E6 cells) were obtained from ATCC (CRL-1586) (Manassas, VA, USA) as mycoplasma-free stocks and were grown in Dulbecco's Modified Eagle Medium (DMEM) (Thermo Fisher Scientific, Merelbeke, Belgium) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine (Thermo Fisher Scientific) and 0.075% sodium-bicarbonate (Thermo Fisher Scientific). Cells were maintained at 37°C in a humidified environment with 5% CO2 and were passaged every 3 to 4 days.
Viruses
All virus-related work was conducted in the high-containment biosafety level 3 facilities of the Rega Institute from the Katholieke Universiteit (KU) Leuven (Leuven, Belgium), according to institutional guidelines. SARS-CoV-2 isolates were recovered from nasopharyngeal swabs of RT-qPCR-confirmed human cases obtained from the University Hospital Leuven (Leuven, Belgium). The following SARS-CoV-2 variants of concern were used in this study: Wuhan strain hCoV19/Belgium/GHB-03021/2020 (GISAID accession no. EPI_ISL_407976), 20A.EU2 strain (clinical isolate A1), Alpha strain (UK-501Y.V1_B.1.1.7; EPI_ISL_791333), Beta strain (RSA-501Y.V2_B.1.351; EPI_ISL_896474), Gamma strain (BRA-501Y.V3_P.1; EPI_ISL_1091366), Delta strain (IND-B.1.617.2; EPI_ISL_2425097) and Omicron strain (RSA-B.1.1.529; EPI_ISL_7413964).
SARS-CoV-2 viral stocks were prepared by inoculation of confluent Vero E6 cells in a 150 cm2 culture flask at MOI of 0.02 in viral growth medium (DMEM supplemented with 2% heat-inactivated FBS, 1 mM sodium pyruvate and 1x MEM NEAA). After 1-h incubation at 37°C, viral growth media was added to bring the final volume to 25 ml. Virus-exposed cells were monitored by microscopy for the generation of cytopathic effect (CPE) on consecutive days. When all cells were infected and full CPE was obtained (i.e., at peak of infection), cell supernatant was harvested, cleared by centrifugation and supernatant was aliquoted and stored at 80°C.
To determine replication efficiency, the different SARS-CoV-2 strains were added to Vero E6 cells at an MOI of 0.02, as determined by end-point dilution titration on Vero E6 cells and calculated by the tissue culture infectious dose 50 (TCID50) method of Reed and Muench [5]. The viral genome sequence was verified, and all infections were performed with passage 3 to 5.
Antibodies
The SARS-CoV-2 spike-specific monoclonal rabbit primary antibodies R001 and R007 were obtained from Sino Biological (Wayne, PA, USA; cat. no. 40592-R001 and cat. no. 40150-R007, respectively). The rabbit polyclonal SARS-CoV-2 nucleocapsid-specific antibody was obtained from GeneTex (Hsinchu City, Taiwan; cat. no. GTX135357). The fluorescent-labelled Alexa Fluor 647 (AF647) goat anti-Rabbit IgG monoclonal antibody was from Cell Signaling Technologies (MA, USA; cat. no. 4414).
Wild type SARS-CoV-2 virus infection
One day prior to the experiment, Vero E6 cells were seeded at 20,000 cells/well in 96 well-plates. The test compounds (i.e., the spike-specific antibodies R001 and R007) were diluted in cell infectious media (DMEM + 2% fetal calf serum [FCS]), overlaid on cells and then virus was added to each well (multiplicity of infection [MOI] indicated in the figure legends). Cells were incubated at 37°C under 5% CO2 for the duration of the experiment. The supernatants were collected and stored at -20°C.
RT-qPCR detection of SARS-CoV-2
Primers and probes for a duplex RT-qPCR were designed to detect the nucleocapsid (N) and envelope (E) gene of SARS-CoV-2 according to Centers for Disease Control and Prevention (CDC, USA; cat. no. 2019-nCoVEUA-01) and Charité (Berlin, Germany), respectively (Table 1). Those primer sequences were selected to target conserved regions of the viral genome of SARS-CoV-2 specifically and no other human coronaviruses. Primers and probes were obtained from Integrated DNA Technologies (IDT, Leuven, Belgium). To perform a duplex RT-qPCR, probes with matching fluorescent dyes (i.e., FAM and HEX) were designed accordingly. More specifically, for N2 the following probe was used: FAM-ACAATTTGCCCCCAGCGCTTCAG(BHQ1) and for E: HEX-ACACTAGCC(ZEN)ATCCTTACTGCGCTTCG(3IABkFQ). Final concentration of combined primer/probe mix consist of 500 nM forward and reverse primer and 250 nM probe. A stabilized in vitro transcribed universal synthetic single stranded RNA of 880 nucleotides in buffer with known copy number concentration (Joint Research Centre, European Commission, cat. no. EURM-019) was used as a standard to quantitatively measure viral copy numbers.
Table 1. Primer and probe sequences used in duplex RT-qPCR.
| Primer/probe | Sequence (5′–3′) | Reference |
|---|---|---|
| SC2 N Fw | TTACAAACATTGGCCGCAAA | US CDC |
| SC2 N Rev | GCGCGACATTCCGAAGAA | US CDC |
| SC2 N Probe | ACAATTTGCCCCCAGCGCTTCAG | US CDC |
| SC2 E Fw | ACAGGTACGTTAATAGTTAATAGCGT | DE Charité |
| SC2 E Rev | ATATTGCAGCAGTACGCACACA | DE Charité |
| SC2 E Probe | ACACTAGCCATCCTTACTGCGCTTCG | DE Charité |
SC2: SARS-CoV-2.
Total RNA was extracted from cell culture supernatants using QIAamp viral RNA mini kit (Qiagen) following manufacturer's instruction. Viral E and N genes are simultaneously amplified and tested using a multiplex RT-qPCR. All the procedures follow the manufacturer's instructions of the Applied Biosystems TaqMan Fast Virus one-step mastermix (Thermo Fisher Scientific). Briefly, a 20 μl reaction mix was set up containing 5 μl of template, 7 μl of dH2O, 1.5 μl of each combined primer/probe mix (final concentrations of 500 and 250 nM, respectively) and 5 μl of 4X TaqMan Fast Virus 1-step mastermix (Thermo Fisher Scientific). The qPCR plate was sealed and read in the FAM and HEX channels using a QuantStudio™ 5 Real-Time PCR system (Thermo Fisher Scientific) under the following cycling protocol: 50°C for 5 min (reverse transcription), 95°C for 20 s (DNA polymerase activation), followed by 45 cycles of 95°C for 3 s (denaturation) and 55°C for 30 s (annealing and fluorescence collection) followed by an infinite 4°C hold.
Flow cytometry
For the intracellular staining of infected Vero E6 cells, cells were collected at different time points as indicated in the figure legends, and the Fixation/Permeabilization kit from BD Biosciences (NJ, USA) was used (Cat n° 554714). At the time of collection, supernatant was removed, and cells were washed in PBS. Then, trypsin (0.25%) was added and plates were incubated for 3′ at 37°C to detach the cells from the plate, followed by the addition of cold culture medium with 2% FCS. Next, cells were resuspended, transferred to tubes and samples were centrifuged in a cooled centrifuge (4°C) at 500× g for 5′. After removal of the supernatant, cells were fixed and permeabilized by the addition of 250 μl of BD cytofix/cytoperm buffer and incubated at 4°C for 20′. Samples were washed twice with Perm/Wash buffer before the addition of the primary (anti-Nucleocapsid) antibody (0.3 μg per sample). After a 30 min incubation at 4°C, samples were washed twice in BD Perm/Wash buffer, followed by a 30 min incubation at 4°C with the secondary (labeled) antibody, and washed again. Finally, samples were stored in PBS containing 2% formaldehyde (VWR Life Science AMRESCO, PA, USA). Acquisition of all samples was done on a BD FACSCelesta flow cytometer (BD Biosciences) with BD FACSDiva v8.0.1 software. Flow cytometric data were analyzed in FlowJo v10.1 (Tree Star, OR, USA). Subsequent analysis with appropriate cell gating was performed to exclude cell debris and doublet cells, to acquire data on living, single cells only. We opted for not including an extra viability staining as we considered this not to be an added value given that the net effect of virus-induced cytopathic effect would be cell death and cell lysis of infected cells, and thus, depletion of those infected cells from the sample. Also, those lysed cells are mostly removed from the sample by several wash steps during sample collection and an extra viability staining would not rescue these cells.
Results & discussion
Optimization of duplex RTqPCR for SARS-CoV-2
At first, in vitro SARS-CoV-2 basic research in our laboratory was based solely on the CPE readout of virus-infected Vero E6 cells (Figure 1). SARS-CoV-2 infection of a Vero E6 cell monolayer manifests in rounding and detaching of the cells. Administration of the neutralizing antibody R001, that binds to the receptor binding domain (RBD) of the viral spike (S) protein of SARS-CoV-2 [6], completely prevented the development of CPE, demonstrating a strong antiviral effect for R001 due to prevention of virus attachment to the cellular Angiotensin-Converting Enzyme 2 (ACE2) receptor [7] and subsequent virus entry in the cell. In contrast, the antibody R007, that also binds to the S protein but in a non-neutralizing manner, lacked antiviral potency (Figure 1).
Figure 1. Cytopathic effect of SARS-CoV-2 virus on Vero E6 cells.

Microscopic pictures of infected Vero E6 cells with SARS-CoV-2. Cells were exposed to SARS-CoV-2 (20A.EU2 variant; MOI = 0.015) for 2 h, in the presence or absence of 2.5 μg/ml antibody R001 or R007. Supernatant was then removed, cells were washed and given fresh medium without (= ‘Control’) or with antibody (= ‘R001’ or ‘R007’). Pictures of cytopathic effect (CPE) were taken 3 days postinfection. For R001 a complete protection of virus entry was obtained, whereas R007 had no antiviral effect. Magnification was 4×.
As CPE monitoring and scoring is a fairly subjective evaluation method, we next implemented a highly sensitive RT-qPCR method for the objective quantification of virus infection and virion production. To preserve the detection capacity of our assay for emerging SARS-CoV-2 mutants, we chose the simultaneous detection of two genes in the viral genome, i.e., the gene encoding for the highly abundant nucleoprotein N and that for the envelope protein E (Figure 2A). As proof of principle, we first validated our duplex RT-qPCR assay on a dilution series of a SARS-CoV-2 viral stock. Briefly, 1 over 10 dilutions of the virus stock in culture medium (starting from a 103x dilution) were used for viral RNA extraction and subsequent analysis. The results of the E/N duplex RT-qPCR are shown in Figure 2B, demonstrating a similar accurate detection of virus samples with both genes in a concentration-dependent manner (Figure 2C).
Figure 2. Duplex RT-qPCR detection of SARS-CoV-2 samples.
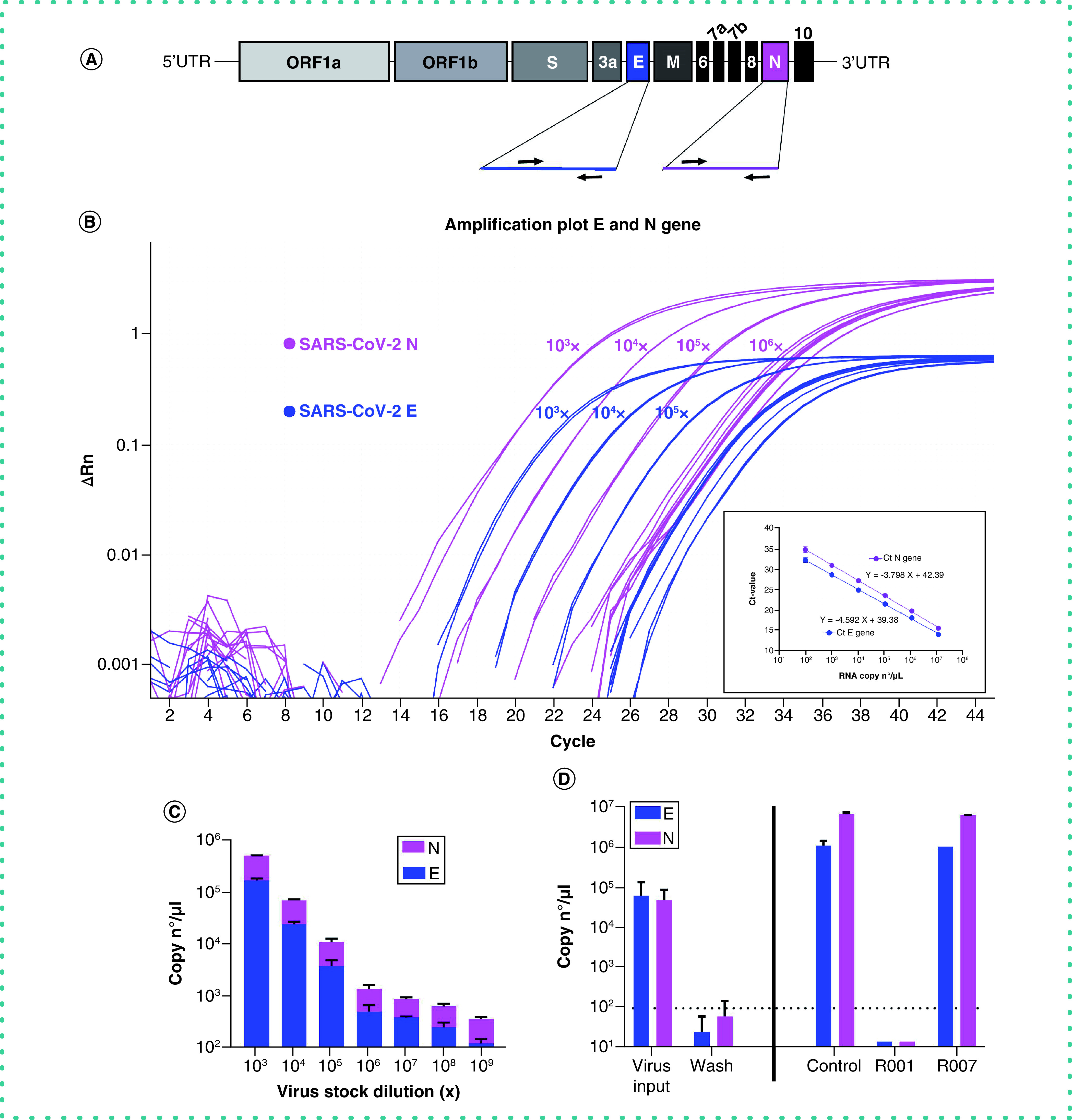
(A) Schematic representation of SARS-CoV-2 genome structure. The full-length RNA genome comprises approximately 30,000 nucleotides and has a replicase complex (comprised of ORF1a and ORF1b) at the 5′UTR. Four genes encode for the structural proteins, i.e., the Spike protein (S), the Envelope protein (E), the Membrane protein (M), and the Nucleocapsid protein (N). The genes for E and N were selected for RT-qPCR detection. (B) Amplification plot of SARS-CoV-2 viral E and N gene. A dilution range (1:10) of a SARS-CoV-2 (20A.EU2 variant) virus stock was measured by duplex RT-qPCR for E and N gene (RNA) copy numbers, starting from a 103x dilution. Graph shows amplification plot for 2 technical replicates of each sample. Insert shows the standard curve for the N and E gene RNA standards. (C) Samples from (B) were analyzed for the quantification of copy numbers of E and N (mean ± standard deviation [SD], n = 2). D) RT-qPCR analysis of the samples from Figure 1. Vero E6 cells were exposed to SARS-CoV-2 (20A.EU2 variant; MOI = 0.015) for 2 h. Supernatant (= virus input) was then removed, and cells were washed in PBS (= wash) and given fresh medium. In the conditions with the antibody-treatment (R001 or R007), 2.5 μg/ml of antibody was administered together with the virus, and administered again after the wash step. At day 3 postinfection, supernatant was collected and a duplex RT-qPCR was performed to quantify the copy numbers of E and N. The dotted line refers to the residual amount (background) of virus that attached aspecifically to the cells. For R001 a complete protection of virus entry was obtained, whereas R007 had no antiviral effect. For each sample, the average (mean ± SD) of two technical replicates is given (n = 2), plotted on a log10 scale.
Next, we applied our duplex RT-qPCR analysis on the virus production of SARS-CoV-2 infected Vero E6 cells that were treated with virus-neutralizing antibodies. Briefly, virus yield analysis (i.e., new virions released by the infected cells in the culture medium) was performed at 3 days postinfection (p.i.). Thus, supernatant of Vero E6 cells infected with SARS-CoV-2 was collected and subjected to RNA extraction and RT-qPCR. To assess if there was successful replication of the virus, we compared the samples with the virus input, i.e., the diluted virus stock solution to which the cells were exposed to for 2 h. Also, we collected the wash sample (after the removal of virus input) that represents the amount of residual virus particles that were attached to the cells ‘aspecifically’ without productive viral entry in the cell, to determine the lower limit of detection of our virus yield assay. As shown in Figure 2D, Vero E6 cells were successfully infected with SARS-CoV-2 with productive virus release in the supernatant (‘Control’), as evidenced by the high copy numbers of viral RNA that exceeded the amount of virus input. Treatment with antibody R001 completely abolished virus infection whereas R007 did not exert any antiviral activity, thus, confirming the CPE analysis of Figure 1. Thus, as comparable data were obtained for both SARS-CoV-2 genes, we concluded that we successfully developed a E/N gene duplex RT-qPCR.
To examine the robustness of our assay, we next determined the virus yield in Vero E6 cells infected with the different SARS-CoV-2 variants of concern. As summarized in Figure 3, our duplex RT-qPCR assay simultaneously detected the viral genes E and N from all tested variants, also, from the current circulating Omicron variant. This also demonstrates that the many reported mutations in the different variants seem not to affect the primer binding sites of our assay.
Figure 3. Duplex RT-qPCR analysis of virus yield from different SARS-CoV-2 variants of concern.

Vero E6 cells were exposed to a fixed virus stock dilution (10×) of different variants of concern of SARS-CoV-2 (that correlates with the following MOI: Wuhan = 3.4; 20A.EU2 = 7.4; Alpha = 1.6; Beta = 15; Delta = 0.016; Omicron = 1.6). After a 2 h incubation, supernatant (with virus) was then removed, and cells were washed in PBS and given fresh medium. At day 3 postinfection, supernatant (that contained newly produced virus particles) was collected and a duplex RT-qPCR was performed to quantify the copy numbers of the E gene (left panel) and the N gene (right panel). For each condition, two biological replicates were included, and for the RT-qPCR quantification two technical replicates of each sample were analyzed to calculate the average value. Bars represent mean ± standard deviation (SD) (n = 2).
Intracellular flow cytometric nucleoprotein staining of SARS-CoV-2 infected cells
Complementary to the RT-qPCR analysis described earlier, we implemented an assay to analyze virus infection at a single cell level, which effectively determines what cells from a heterogeneous cell population are susceptible to SARS-CoV-2. We opted for flow cytometry and selected an antibody that is raised against the viral nucleoprotein [8], a conserved viral protein that is abundantly expressed in virus-infected cells and stabilizes and protects the viral RNA [9]. A big advantage of flow cytometry is that by analysis of the forward (FSC) and side (SSC) scatter, important information can be retrieved regarding the morphology of the cells, allowing for exclusion of cell debris originating from (virus-infected) dead cells from healthy, intact cells.
Briefly, Vero E6 cells were infected with SARS-CoV-2, collected at selected time points p.i., fixed and permeabilized. Cells were then stained with a primary antibody against N, followed by incubation with a secondary antibody that is fluorescently labeled, and analyzed by flow cytometry. A kinetic study determined the optimal time point of cell collection, given that the cells need to be infected sufficiently, but not yet at that level of full blown CPE where the lytic effect of the virus destroys the infected cell population. As summarized in Figure 4A, viral N protein could be detected in Vero E6 cells at 24 h p.i. (30% infected cells), but longer incubation of the cells resulted in nearly a complete infected cell population (87%). Interestingly, N detection by flow cytometry was not restricted to the SARS-CoV-2 Wuhan-related variant 20A.EU2, but also a 40-h incubation of Vero E6 cells with the Gamma (Brazilian) variant returned a clear detection of viral N protein, showing an infection rate of 75% (Figure 4B).
Figure 4. Kinetics of viral nucleoprotein expression in SARS-CoV-2 infected Vero E6 cells.
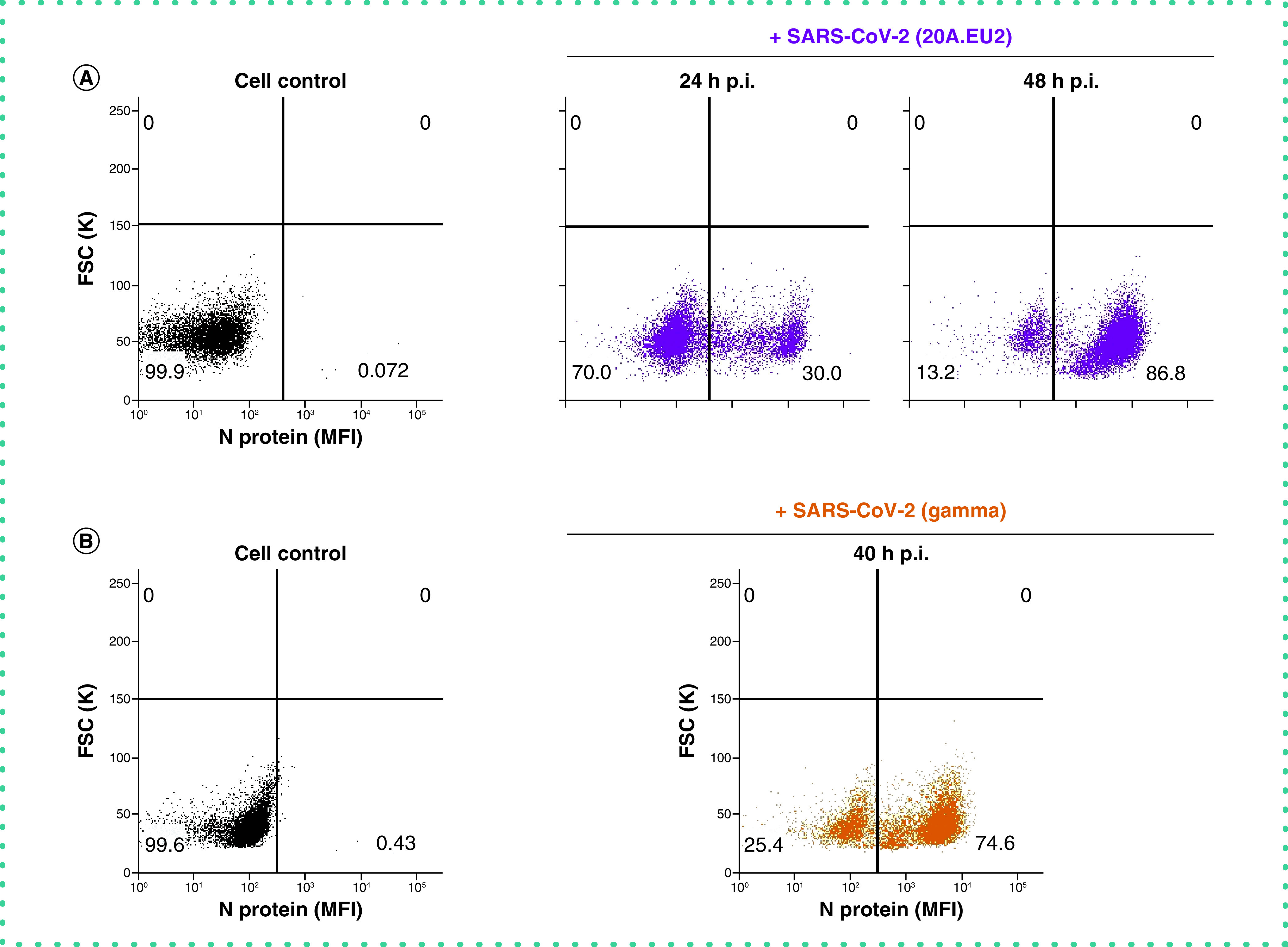
(A) Vero E6 cells were exposed to SARS-CoV-2 (20A.EU2 variant; MOI = 1.5) for 2 h, washed and incubated with culture medium. At the indicated time points, cells were collected, fixed, permeabilized and stained with an antibody that binds to the viral nucleoprotein N. (B) Same as in (A) but for the infection with the Gamma variant (MOI = 2). Dot plots show N expression in noninfected (lower left quadrant) and infected (lower right quadrant) Vero E6 cells from a representative experiment with multiple individual samples. Single cell analysis was done on numerous independent cells, and the individual plots shown in the figure are based on the analysis of 8,000 – 10,000 cells by flow cytometry, resulting in a very accurate analysis. The numbers in each quadrant refer to the distribution of the cells (i.e., percentage of total cell population). The dot plot in black (left panel) represents the non-infected cell control.
FSC: Forward scatter; MFI: Mean fluorescence intensity; p.i.: Postinfection.
Next, we assessed the robustness of this assay for the detection of different variants of concern of SARS-CoV-2 (Figure 5). For 6 different variants, including the recent circulating Omicron strain, a dilution range of virus stock was administered to Vero E6 cells. Next, 48 h p.i. cells were stained for viral N protein. As summarized in Figure 5, Vero E6 cells are highly permissive for all tested variants of concern of SARS-CoV-2, with infection efficiencies up to 68%. For most variants with a concentrated virus stock (i.e., Wuhan, 20A.EU2, Beta and Omicron), within each variant dilution of virus input was related to higher infection rates. This discrepancy can be explained by the gradual increase in CPE with higher virus input which results in relatively more cell lysis and detached (infected) cells that are removed from the sample during wash steps. In most cases, the first dilution of the virus stock contains an excessive amount of virus that has a destructive impact on the cells. The longer survival of the infected cells in the condition of low virus input (right column of Figure 5) will be reflected by a higher relative number of infected cells as compared to the non-infected ones. Also, in this condition multiple rounds of virus infection can take place, resulting in a gradual increase in the absolute number of infected cells. For the Alpha variant, a 100x dilution seems to give the highest number of infected cells, whereas a more diluted virus input resulted in less infected cells. Of note, the virus stock of the Delta variant contained the least infectious particles, as evidenced by a concentration-dependent reduction in N positive cells. In fact, a 1,000x dilution of this stock returned an infection rate of less than 2%. Importantly, our intracellular flow cytometry assay consistently detected all different variants of SARS-CoV-2, demonstrating the broad spectrum usage of this technique.
Figure 5. Viral N staining of Vero E6 cells infected with different variants of SARS-CoV-2.
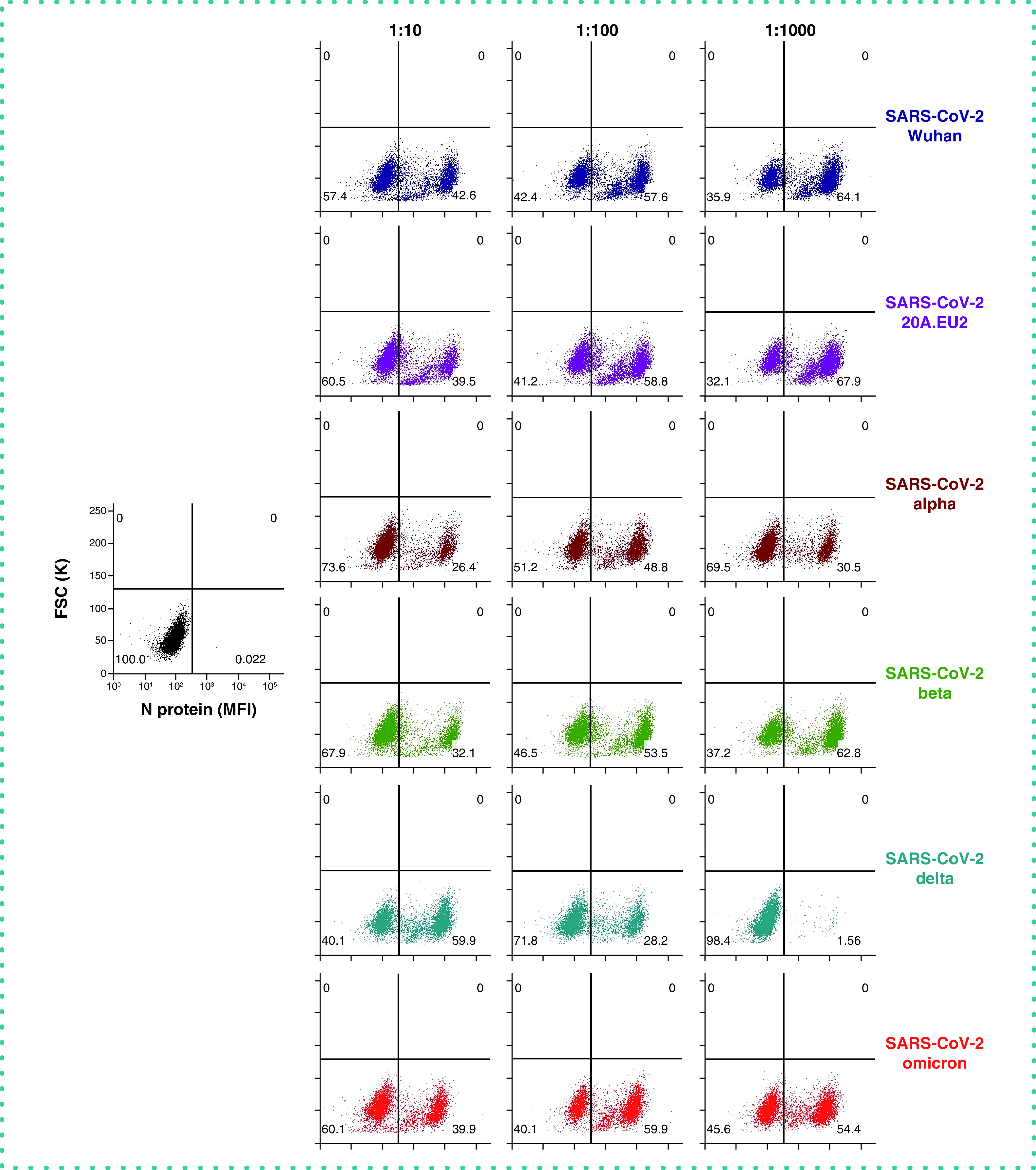
Vero E6 cells were exposed to a virus stock dilution (10x, 100x or 1000x) of different variants of concern of SARS-CoV-2 (the 10x dilution correlates with the following MOI: Wuhan = 3.4; 20A.EU2 = 7.4; Alpha = 1.6; Beta = 15; Delta = 0.016; Omicron = 1.6). At day 2 post-infection, cells were collected, fixed, permeabilized and stained with an antibody that binds to the viral N protein. Dot plots show N expression in noninfected (lower left quadrant) and infected (lower right quadrant) cells from a representative experiment with multiple individual samples. Single cell analysis was done on numerous independent cells, and the individual plots shown in the figure are based on the analysis of 8,000 – 10,000 Vero E6 cells by flow cytometry. The numbers in each quadrant refer to the distribution of the cells (i.e., percentage of total cell population). The dot plot in black (left panel) represents the non-infected cell control.
FSC: Forward scatter; MFI: Mean fluorescence intensity.
Finally, we analyzed the flow cytometric data in more detail. As shown in Figure 6A, infection of Vero E6 cells with low virus input of SARS-CoV-2 for 2 days resulted in a large population of infected cells. Moreover, two distinct cell populations of infected cells could be distinguished depending on the level of N protein expression. We hypothesized that the cells with low N protein levels are the newly infected cells in an early stage of the replication cycle. More specifically, those cells are infected cells that have taken up the virus but have not started the synthesis of new viral proteins yet. As the N protein is abundantly present in each virus particle to coat the viral genome [9], cells that contain virus particles should return detectable amounts of N protein. On the other hand, cells that are active replicating virus have started the biosynthesis of N protein and will produce N protein in high(er) amounts [10]. As the analysis of the flow cytometric data also includes a doublet discrimination gating step, the cells with high level N expression are not simple pairs of Vero E6 cells with double amount of viral N protein. To prove our hypothesis, and to investigate the nature of the two distinct N-positive cell populations, we performed an experiment in which we exposed Vero E6 cells to a high virus input (MOI of 28) for a short time (4 h). This should allow enough time for the virus to enter the cells, but without starting the replication process (which is estimated at 6 to 8 h post infection). As a control, we included the spike-neutralizing antibody R001 as a virus entry inhibitor. Several staining controls were performed (data not shown) to verify that the R001 rabbit antibody is not binding to the cells in a non-specific way that could give a false-positive staining with the fluorescent-labelled goat anti-rabbit IgG monoclonal antibody to detect the anti-nucleoprotein rabbit antibody. As depicted in Figure 6B, already after 4 h p.i., a substantial amount (19%) of virus-infected cells could be visualized (red square area on the dot plot), which was completely prevented by R001 treatment. The full protection with R001 antibody (i.e., identical dot plot as compared to the non-infected control) also proofs that there is no enhanced non-specific background staining with the anti-spike antibody. The substantial amount of virus-infected cells suggests that within 4 h virus has attached to the cells and/or has entered the cells. As expected, within this short time frame, only low levels of N staining were obtained in the N-positive cells. Of course, as Vero E6 cells are known for their efficient endocytic viral uptake mechanisms [11,12], multiple viral particles can be taken up by a single cell. This can also explain the variation in N expression of the N low subpopulation. Longer incubation of the cells (24 h) resulted in the propagation of high N expressing cells (blue square area), thus, active virus-replicating cells. During this 24 h time window, new virions will be released from the infected cells and can enter uninfected bystander cells, generating a new population of low N expressing cells. Thus, our flow cytometric analysis of SARS-CoV-2 infected cells can also indicate at a single cell level the status in the virus replication cycle. This information can be very helpful to profile new antiviral compounds as virus entry versus replication inhibitors.
Figure 6. Viral N staining distinguishes two subpopulations of SARS-CoV-2 infected Vero E6 cells.
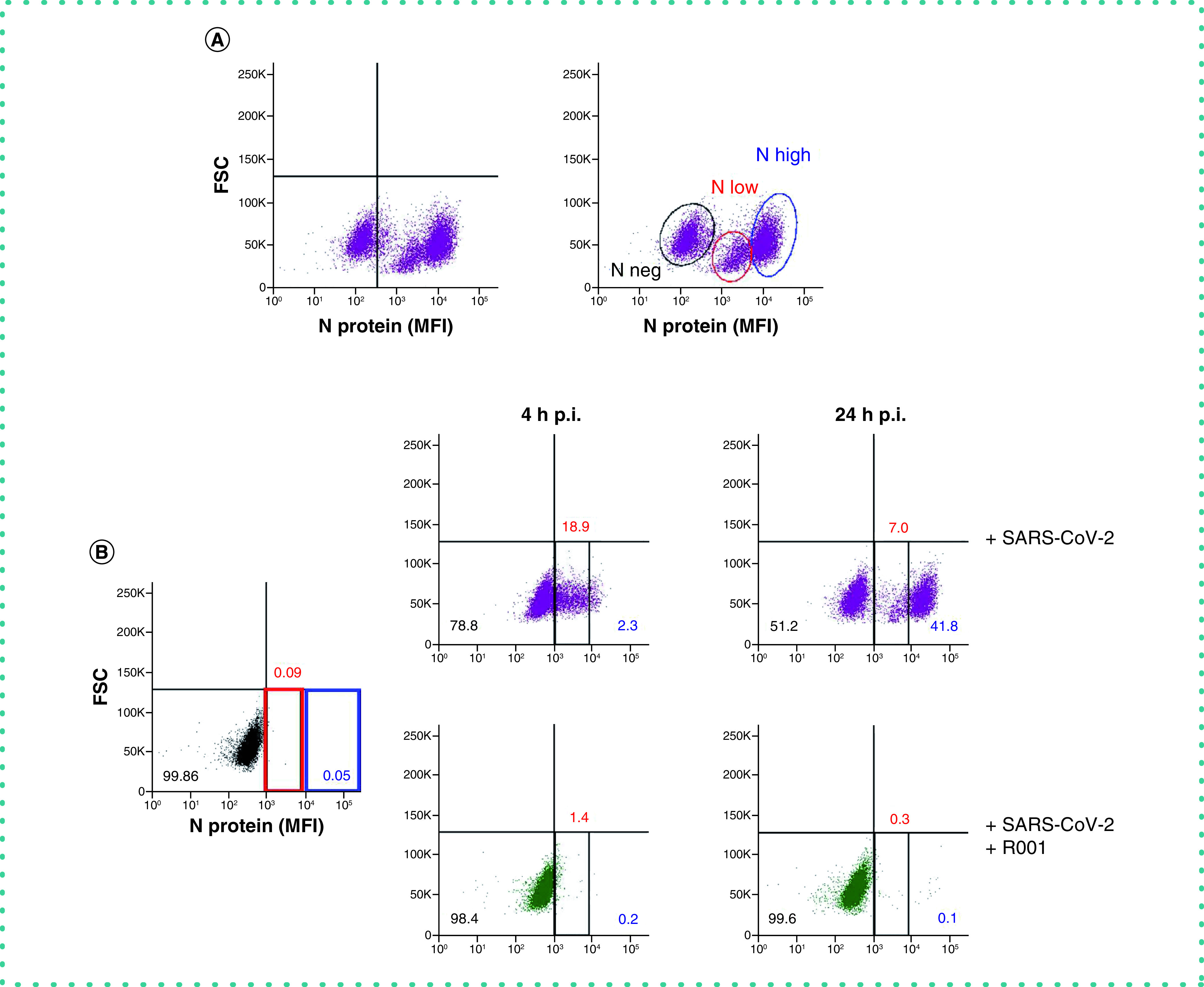
(A) Vero E6 cells were exposed to a low virus input of SARS-CoV-2 (20A.EU2 variant; MOI = 0.28). At day 2 p.i., cells were collected, fixed, permeabilized and stained with an antibody that binds to the viral nucleoprotein N. Dot plots show N expression in noninfected (lower left quadrant) and infected (lower right quadrant) cells from a representative experiment with multiple individual samples. Single cell analysis was done on numerous independent cells, and the individual plots shown in the figure are based on the analysis of 8,000 – 10,000 Vero E6 cells by flow cytometry. The graph shows two distinct subpopulations of infected cells, i.e., ‘N low’ (marked by a red line) and ‘N high’ (marked by a blue line). (B) Vero E6 cells were exposed to a high virus input of SARS-CoV-2 (20A.EU2 variant 1:10; MOI = 28), without (top panels) or with 10 μg/ml of spike-neutralizing antibody R001 (bottom panels). At 4 h p.i., half of the samples were washed to remove virus input and were administered fresh medium (without or with R001) and incubated for another 20 h followed by N staining. The other half of the cells were collected at 4 h p.i., fixed, permeabilized and stained with an antibody that binds to the viral N protein. Dot plots show N expression in infected cells from a representative experiment with multiple individual samples. Single cell analysis was done on numerous independent cells, and the individual plots shown in the figure are based on the analysis of 8,000 – 10,000 Vero E6 cells by flow cytometry. The two distinct subpopulations of infected cells, i.e., ‘N low’ (marked in red) and ‘N high’ (marked in blue) are indicated. The numbers refer to the distribution of the cells (i.e., percentage of total cell population). The dot plot in black (left panel) represents the noninfected cell control. To determine non-specific background of the spike-neutralizing antibody R001, in the same experiment non-infected controls were also incubated with R001 and stained with the fluorescently labeled secondary anti-rabbit antibody. As those dot-plot panels were identical to the non-infected control sample (without R001), they are not presented in the figure for clarity of the figure.
FSC: Forward scatter; MFI: Mean fluorescence intensity; p.i.: Postinfection.
Conclusion & future perspective
In the context of basic research on viruses, a repertory of cell-based assays and sensitive techniques for virus detection is requested. In response to the COVID-19 pandemic, major efforts have been made to quickly adapt or develop techniques for the detection of SARS-CoV-2, either its viral genome or the viral proteins [13–18]. In this study, we successfully optimized a SARS-CoV-2 duplex RT-qPCR for the detection of viral RNA copies by the simultaneous amplification of the nucleocapsid and envelope genes. This sensitive technique efficiently detected viral E and N from all tested variants, also, from the current circulating Omicron variant. This demonstrates that the many reported mutations in the different variants seem not to affect the primer/probe binding sites of our PCR. For the quantification of virus replication and virion production, samples for RT-qPCR are mostly based on cell culture supernatant from infected cells or cell lysate, thus originating from thousands of cells, either infected or uninfected. However, with our flow cytometric technique, based on intracellular viral nucleocapsid staining, we succeeded to analyze virus infection at the single cell level, which is highly valuable to determine what cells from a heterogeneous cell population are susceptible to SARS-CoV-2. Furthermore, this technique makes it possible to distinguish between subpopulations of infected cells that are in an early (low N) or late phase (high N) of viral replication. This can be an added value in the profiling of new candidate antiviral drugs, to discriminate between entry inhibitors and viral polymerase inhibitors that block viral replication. Importantly, this flow cytometric technique complements RT-qPCR detection and can be applied to all current SARS-CoV-2 variants of concern, including the highly mutated Omicron variant. Furthermore, with the expansion of RNA flow cytometry, in which target-specific probe sets hybridize to the target RNA transcripts inside cells, combination of protein and RNA detection at a single cell level will become feasible. We believe that this method will be a valuable supplement, not only for the ongoing research on COVID-19, but also in future research on virology in general.
Author contributions
K Vermeire, E Vanhulle and J Stroobants designed the research. E Vanhulle, A Camps, B Provinciael and J Stroobants performed the research. E Vanhulle and K Vermeire analyzed the data. E Vanhulle and K Vermeire wrote the manuscript. P Maes contributed new reagents/analytic tools and critically reviewed the manuscript. All of the authors discussed the results and commented on the manuscript.
Acknowledgments
The authors thank G Schoofs for technical support to the flow cytometric experiments.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Chan JF-W, Yuan S, Kok K-H et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. The Lancet 395(10223), 514–523 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F, Zhao S, Yu B et al. A new coronavirus associated with human respiratory disease in China. Nature 579(7798), 265–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen L, Liu W, Zhang Q et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg. Microbes Infect. 9(1), 313–319 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Planas D, Saunders N, Maes P et al. Considerable escape of SARS-CoV-2 omicron to antibody neutralization. Nature 602(7898), 671–675 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 27(3), 493–497 (1938). [Google Scholar]
- 6.Wrapp D, Wang N, Corbett KS et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367(6483), 1260–1263 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann M, Kleine-Weber H, Schroeder S et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2), 271–280 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Key publication showing that ACE2 is the main receptor for virus entry of SARS-CoV-2.
- 8.Clausen TM, Sandoval DR, Spliid CB et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell 183(4), 1043–1057 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai Z, Cao Y, Liu W, Li J. The SARS-CoV-2 nucleocapsid protein and its role in viral structure, biological functions, and a potential target for drug or vaccine mitigation. Viruses 13(6), e13061115 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romano M, Ruggiero A, Squeglia F, Maga G, Berisio R. A structural view of SARS-CoV-2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells 9(5), e9051267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mercer J, Helenius A. Virus entry by macropinocytosis. Nat. Cell Biol. 11(5), 510–520 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Murgolo N, Therien AG, Howell B et al. SARS-CoV-2 tropism, entry, replication, and propagation: considerations for drug discovery and development. PLoS Pathog. 17(2), e1009225 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bloemen M, Rector A, Swinnen J et al. Fast detection of SARS-CoV-2 variants including omicron using one-step RT-PCR and Sanger sequencing. J. Virol. Methods 304, e114512 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Domenico M, De Rosa A, Boccellino M. Detection of SARS-COV-2 proteins using an ELISA test. Diagnostics (Basel) 11(4), e11040698 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eftekhari A, Alipour M, Chodari L et al. A comprehensive review of detection methods for SARS-CoV-2. Microorganisms 9(2), e9020232 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• A comprehensive overview of the different techniques to detect SARS-CoV-2
- 16.Liu J, Babka AM, Kearney BJ, Radoshitzky SR, Kuhn JH, Zeng X. Molecular detection of SARS-CoV-2 in formalin-fixed, paraffin-embedded specimens. JCI Insight 5(12), e139042 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Safiabadi Tali SH, LeBlanc JJ, Sadiq Z et al. Tools and techniques for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)/COVID-19 detection. Clin. Microbiol. Rev. 34(3), e00228–20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Massoth LR, Desai N, Szabolcs A et al. Comparison of RNA in situ hybridization and immunohistochemistry techniques for the detection and localization of SARS-CoV-2 in human tissues. Am. J. Surg. Pathol. 45(1), 14–24 (2021). [DOI] [PubMed] [Google Scholar]