

Abstract
Idiopathic hypogonadotropic hypogonadism and Kallmann syndrome are rare genetic disorders characterized by isolated gonadotropin-releasing hormone (GnRH) deficiency (IGD) and delayed or absent puberty. Defective GnRH neuron migration during development or secretion of mature GnRH neurons secondary to molecular defects in several key developmental and neuroendocrine pathways are thought to be the primary causes of these disorders. Recent studies have highlighted the importance of semaphorins and their receptors in this system, by showing that these molecules play distinct roles during the development and plasticity of these neurons. Accordingly, mutations in the semaphoring-signaling pathway genes have been found in patients affected by IGD, underlying the importance of semaphorin-mediated signaling pathways in the neuroendocrine axis that control reproduction.
INTRODUCTION
Gonadotropin-releasing hormone (GnRH) is a decapeptide that is produced by a small number of hypothalamic neurons, which originate in the nasal placode during embryonic development and migrate along the terminal nerve (TN) to reach their final position in the hypothalamus. Once there, GnRH neurons send their projections to the median eminence (ME), where GnRH is released into the hypophyseal-portal system to reach the pituitary which responds with the synthesis and release of gonadotropins (LH and FSH). In turn, the gonadotropins stimulate the production of sex steroids in the gonads in both sexes. Defects in the development of GnRH neurons or in GnRH secretion or action lead to isolated gonadotropin-releasing hormone deficiency (IGD), also referred to as idiopathic hypogonadotropic hypogonadism.
Individuals with IGD present with absence of puberty, sexual immaturity, symptoms and signs of hypogonadism, and infertility. Biochemical investigations reveal underlying hypogonadotropic hypogonadism, which is characterized by inappropriately low serum concentrations of the gonadotropins (LH and FSH) in the presence of low circulating concentrations of sex steroids. Infant boys with IGD often exhibit features of neonatal hypogonadism such as microphallus and cryptorchidism (i.e., undescended testes). Adult males with IGD tend to have prepubertal testicular volume (i.e., <4 mL), absence of secondary sexual features (e.g., facial and axillary hair growth, deepening of the voice), decreased muscle mass, diminished libido, erectile dysfunction, and infertility. Adult females typically present with little or no breast development and primary amenorrhea (Shaw et al., 2011). Two broad phenotypic forms of IGD are recognized clinically: when associated with absent sense of smell (anosmia) or diminished sense of smell (hyposmia), IGD is referred as Kallmann Syndrome (KS) and represents 50% of overall IGD cases. The second major form of IGD is referred to as normosmic idiopathic hypogonadotropic hypogonadism (nIHH), wherein IGD presents in the setting of a normal sense of smell. A small subset of IGD patients present with additional nonreproductive features in the setting of multisystemic syndromes such as CHARGE syndrome, Bardet–Biedl syndrome, Waardenburg syndrome, Gordon–Holmes syndrome, Moebius syndrome, septooptic dysplasia, combined pituitary hormone deficiency, adrenal hypoplasia, and congenital obesity (Boehm et al., 2015; Kim, 2015; Stamou and Georgopoulos, 2018). IGD per se can also lead to many comorbidities and long-term effects, such as psychologic and psycho-sexual symptoms (Varimo et al., 2015), osteoporosis (Laitinen et al., 2012), and increased risk of metabolic defects like type II diabetes mellitus (Brand et al., 2014).
Although the precise incidence of IGD is uncertain, the population-based epidemiological studies of Kallmann syndrome show a minimal incidence of 1 in 48,000, with a higher incidence in males (1 in 30,000 vs 1 in 125,000 in females) (Laitinen et al., 2011). Diagnosis of IGD is most commonly suspected in adolescence, when boys/girls present with delayed puberty and physical examination and biochemical testing show characteristic phenotypic features. In boys, prior to the onset of puberty in adolescence, the reproductive cascade is temporarily active in first 6 months of life and this is termed as “mini-puberty.” Defective mini-puberty can result in microphallus and cryptorchidism (undescended testes). Hence, this early neonatal period represents an additional temporal window to obtain an earlier diagnosis of IGD especially if these infants have microphallus and cryptorchidism (Dwyer et al., 2016). An early diagnosis is fundamental for a more effective management and treatment of IGD. Once diagnosed, treatment options consist of administration of sex steroids, gonadotropins, or pulsatile GnRH; therapy is typically chosen based on the goal of treatment: (a) pubertal induction, maintenance of secondary sex features, and restoration of normal sex-steroid levels (testosterone treatment in boys; estrogen and progestin therapy in girls) or (b) induction and maintenance of fertility (gonadotropins or pulsatile GnRH) (Balasubramanian and Crowley, 1993).
As described previously, the dynamic nature of the HPG axis and its myriad and sophisticated regulatory mechanisms make the HPG axis susceptible to dysfunction through disruption of multiple molecular mechanisms. This molecular complexity is reflected in the phenotypic and genetic heterogeneity as well as the low and variable penetrance of genetic mutations observed in IGD. This condition presents as both sporadic and familial cases. In familial pedigrees, multiple modes of inheritance are evident including X chromosome-linked recessive, autosomal recessive and dominant (Boehm et al., 2015). Canonical gene-discovery strategies in affected families if often challenging given the negative effect of IGD on fertility leading to inherent small pedigrees with few affected individuals available for genetic studies. Despite these challenges, over the last three decades, tremendous strides have been made to understand the genetic etiology of IGD. While traditional Mendelian gene-discovery strategies helped to identify several genes initially (described in detail in Stamou et al., 2015), in the last few years, new IGD causative genes have been discovered mainly by applying two complementary approaches: (i) unbiased next-generation genome sequencing of patients along with in vitro and/or in vivo animal models to identify and validate novel candidate genes and (ii) targeted candidate gene studies informed by study of the molecular mechanisms controlling GnRH neuron development or physiology (Sykiotis et al., 2010a,b; Stamou et al., 2015).
To date, nearly 30 genes have been demonstrated to cause IGD. However, these known genes only account for ~50% of overall IGD cases and in the remaining cases, the genetic basis remains unknown (Boehm et al., 2015). The causal genes discovered so far can be divided into two groups depending on whether they disrupt the development of GnRH neurons (neurodevelopmental genes) or the action/signaling of GnRH in normally developed GnRH neurons (neuroendocrine genes). The KS phenotype typically results from mutations in neurodevelopmental genes that normally control different steps of migration and development of GnRH neurons. In contrast, nIHH results primarily from mutations affecting neuroendocrine genes that control either GnRH synthesis (GNRH1), action (GNRHR), or its pulse generator (KISS1/KISS1R, TAC3/TACR3, PCSK1, LEP/LEPR) (Balasubramanian and Crowley, 1993; Bianco and Kaiser, 2009; Marino et al., 2014; Boehm et al., 2015; Kim, 2015; Topaloğlu, 2017; Stamou and Georgopoulos, 2018). However, a subset of genes can govern both GnRH development as well as the functional integrity of GnRH neurons and therefore can cause either KS or nIHH. Furthermore, some IGD pedigrees deviate from a simple Mendelian inheritance model wherein affected IGD individuals may harbor deleterious mutations in more than one gene (i.e., digenic/oligogenic inheritance) (Sykiotis et al., 2010a,b; Marino et al., 2014).
Dovetailing the genetic discoveries, detailed clinical studies have shown that IGD individuals, especially those with KS form of IGD, in addition to their reproductive deficits, also display a spectrum of other nonreproductive nonolfactory anomalies including hearing loss, midline facial defects (e.g., cleft lip/palate), renal agenesis, synkinesia (i.e., mirror movements), and cerebellar ataxia (reviewed in Dodé and Hardelin, 2009). Some of these nonreproductive features in IGD individuals often provide specific clues to the underlying genetic etiology and guide clinical genetic testing (e.g., renal agenesis (ANOS1); digital and dental anomalies (FGF-pathway mutations) (Dodé and Hardelin, 2009; Costa-Barbosa et al., 2013). In the same vein, IGD is also recognized as a key phenotypic feature in many multisystemic syndromes (see previously) often caused by single-gene mutations. These observations attest to the multiple pleiotropic roles played by these genes in regulating many different developmental processes across different organ systems during embryogenesis (Boehm et al., 2015; Stamou et al., 2015).
Although KS has been eponymously named after Franz Kallmann who first described the hypogonadal phenotype cosegregating with anosmia in familial KS cases, intriguingly, the link between olfaction and hypogonadism was documented nearly a century earlier by Maestre de San Juan who described an adult male with testes of prepubertal size and absent olfactory bulbs (Maestre de San Juan, 1856). In keeping with this exquisite phenotypic cosegregation, experimental work relating to several KS-associated genes in humans and in other vertebrate models have established the common embryonic origin of olfactory and GnRH neuronal precursors (Dodé and Hardelin, 2009). Several of the known KS genes are now believed to encode an ensemble of molecular guidance cues that direct the olfactory and GnRH axonal pathways to enter the CNS during embryonic development. Recently, mutations in four members (SEMA3A, SEMA3E, SEMA7A, andPLXNA1) of the semaphorin family of axonal guidance cues have emerged as key genes contributing to the pathogenesis of IGD. While SEMA3A and SEMA3E variants are linked exclusively to KS, SEMA7A, and PLXNA1 variants have been linked to both KS and nIHH forms of IGD. Interestingly, mutations in the semaphorin pathway are characterized by two key features: IGD individuals harboring these mutations display variable phenotypic penetrance and these mutations often occur in the context of additional KS/nIHH gene mutations, substantiating the oligogenic basis of IGD. These findings coupled with corroborating findings in several murine models suggest that semaphorin signaling is a critical determinant of vertebrate GnRH neuron development. While mutations in SEMA3E and SEMA7A are extremely rare, SEMA3A and PLXNA1 variants contribute to ~6% of the genetic etiology of IGD. In this chapter, we will focus our attention on the recent works showing the importance of semaphorins and their receptors in the GnRH neuron biology and their involvement in IGD.
THE ROLE OF SEMAPHORINS AND THEIR RECEPTORS/CORECEPTORS IN GnRH NEURONAL ONTOGENY AND MAMMALIAN REPRODUCTION
Semaphorins constitute one of the largest families of guidance cues, highly conserved across species (Yazdani and Terman, 2006). Vertebrate semaphorins are widely expressed in many tissues and their synthesis changes considerably with developmental and pathologic states. The expression of semaphorins has been extensively studied in the developing nervous system, where different members are produced by neuronal as well as by nonneuronal cells. Semaphorins are also expressed in the cardiovascular, endocrine, gastrointestinal, hepatic, immune, musculoskeletal, renal, reproductive, and respiratory systems. Each of the different classes of semaphorins is not defined by a peculiar pattern of expression, rather they are dynamically expressed in specific areas during development and their production often decreases with differentiation and maturation processes. Further, expression changes in adult tissues occur in neuronal and nonneuronal tissues upon injury, during tumorgenesis and in association with other pathologic conditions (Yazdani and Terman, 2006).
Functionally, semaphorins were first discovered as repulsive cues for growing axons and migrating neurons, but several studies have now suggested additional roles for semaphorins including cell adhesion, cell survival, and proliferation.
To date, more than 20 semaphorins have been identified and grouped into eight classes, based on their structural homology and phylogenetic relationship. Class 1 and Class 2 families contain invertebrates semaphorins; Classes 3–7 include vertebrates semaphorins, whereas viral semaphorins are grouped in Class 5 (Bamberg et al., 1999).
The major binding and signaling receptor complexes for semaphorins are neuropilins (NRP1 and NRP2) and plexins (PLXNs), respectively. Neuropilins are transmembrane glycoproteins that mainly act as coreceptors for Class 3 semaphorins in concert with A-type plexins to transduce their signals across a cell membrane, as they possess a very small cytoplasmic domain (Alto and Terman, 2017). Neuropilins are highly conserved through vertebrates, displaying 40% of identical amino acids. Structurally, both NRP1 and NRP2 include an extracellular region, a transmembrane helix, and a short intracellular region composed of a PDZ domain (Parker et al., 2012).
Plexins are the major signaling transducing receptors for semaphorins and comprise a large family of single transmembrane spanning cell surface proteins. Nine different plexins have been identified and divided into four families, according to structure similarity: Plexin A1–4 (PLXNA1–4), Plexin B1–3 (PLXNB1–3), Plexin C1 (PLXNC1), and PlexinD1 (PLXND1) (Tamagnone et al., 1999). PLXNAs usually serve as receptors for Class 3 and Class 6 semaphorins, whereas Class 4 and Class 5 semaphorins preferentially bind to PLXNBs and SEMA7s to PLXNC1. Most of the Class 3 semaphorins interact with NRPs and PLXNs, except SEMA3E which directly binds to PLXND1 (Kruger et al., 2005). Plexins and semaphorins show a high degree of redundancy, thus a single semaphorin can bind to and signal through different plexins according to the tissue Fig. 22.1.
Fig. 22.1.
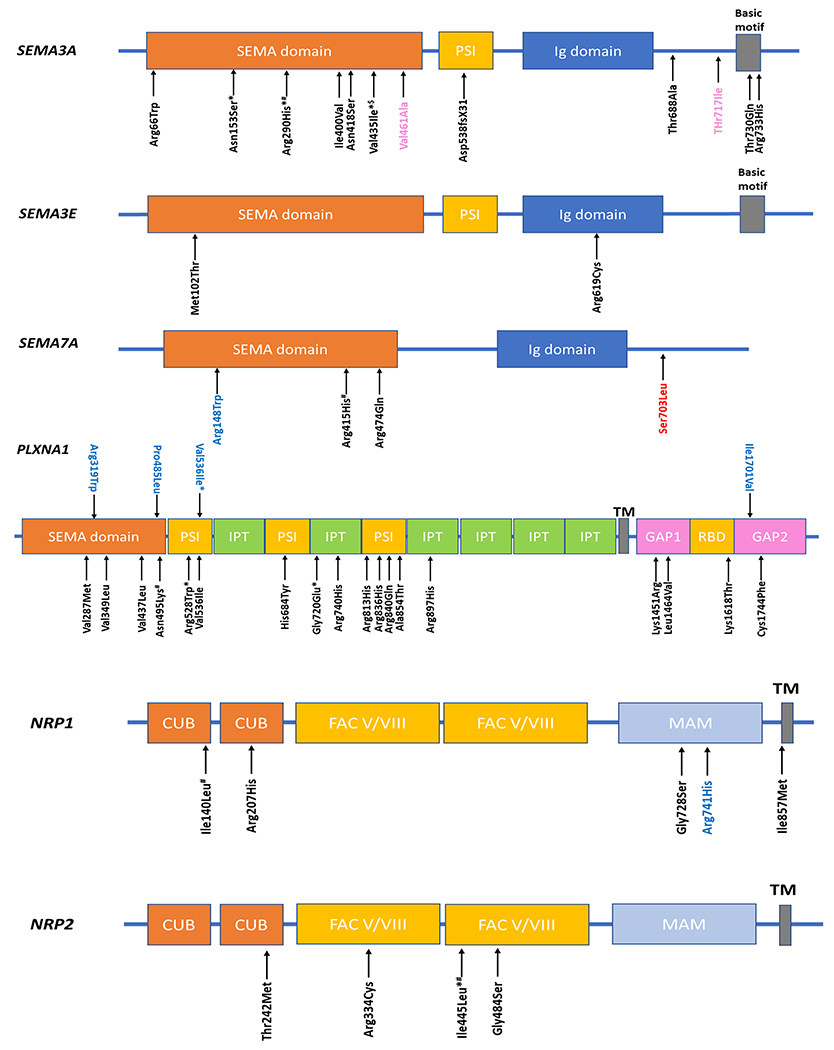
Schematic of SEMA3A, SEMA3E, SEMA7A, PLXNA1, NRP1, and NRP2 proteins and the positions of mutations found in IGD and related phenotypes. Domain abbreviations: CUB, complement binding factors C1r/C1s, Uegf, bone morphogenetic protein 1; FAC V/VIII, coagulation factors V and VIII homology domains; GAP, Ras GTPase-activating protein domain; Ig, Immunoglobulin like; IPT, immunoglobulin-like fold in plexins/transcription factors; MAM, meprin protease-A5 (antigen-receptor tyrosine phosphatase μ and К) domain; PSI, plexin–semaphorin–integrin; RBD, Rho family GTPase-binding domain; SEMA, semaphorin; TM, transmembrane. *—recurrent variants at same position; variants shown in black—KS-associated variants; variants shown in blue—nIHH-associated variants; variants shown in red—CHARGE syndrome-associated variants; variants shown in pink—-congenital-isolated anosmia associated variants; $—variants seen in both KS and isolated anosmia; #—variants seen in IGD but unclear if in KS or nIHH.
Semaphorins can also exert their function through other holoreceptor complexes associated to plexins. The ability of semaphorins to bind to different types of receptors makes a wide range of effects possible. Examples of noncanonical semaphorin receptors are cell adhesion molecules (e.g., Nr-CAM and L1-CAM) that associate to neuropilins to transduce Sema3 signals, several tyrosine kinase receptors, including VEGFR2 (vascular endothelial growth factor receptor 2), Met, ErbB2 and off-track (OTK) vascular endothelial growth factor receptors (Vegfr1–2), tyrosine kinase receptors (Met, ErbB2), and integrins (Itgβ1) that specifically bind to Class 7 semaphorins (Alto and Terman, 2017).
SEMA3F, SEMA3A, and their neuropilin and plexin receptors
The observation that several semaphorins are expressed in the olfactory system during development (Pasterkamp et al., 1999; Bashaw, 2007; Rollmann et al., 2007; de Castro, 2009; Matsuda et al., 2010) led researchers to investigate their possible roles also in the development of GnRH neurons given their shared developmental origins in the nasal placode. The involvement of semaphorins and their receptors in the GnRH neuronal system was first suggested by the analysis of mice lacking Nrp2, which is the canonical binding receptor of SEMA3F. Quite strikingly, Nrp2-null mice displayed severe defasciculation of the nasal axons, the scaffolding that the GnRH neurons use to migrate and penetrate the brain. Consequently, these mice are subfertile due to a reduced number of GnRH neurons reaching the hypothalamus. In the same study, both primary and immortalized GnRH neurons (GN11 cells) were found to express not only Nrp2 but also Nrp1 and Plxnal, thus opening the possibility that additional Class 3 semaphorin-mediated signaling pathways are important in this system (Cariboni et al., 2007). In keeping with this notion, in a subsequent study, mice lacking Sema3f or Sema3a, the two canonical ligands for NRP2 and NRP1, respectively, were analyzed (Cariboni et al., 2011). Surprisingly, the analysis of Sema3f-null mice did not show any gross defects in the development of GnRH neurons, implying that another member of the Class 3 semaphorin may be the primary semaphorin, linked to GnRH neurogenesis. Indeed, Sema3a-null mice show arrested migration of GnRH neurons in the nose due to mispatterned projections of the nasal nerves used by GnRH neurons to reach the forebrain. Specifically, these mice show tangles ofperipherin-positive axons accumulating at the level of the nasal-forebrain junction and a nearly complete lack of the terminal nerve in the medial preoptic area (MPOA). This results in a comparable loss of neurons in the forebrain of both embryonic and adult mice, which also display a less innervated ME and a reduced size of the gonads (Cariboni et al., 2011). A similar phenotype is also reported for mice lacking both Nrp1 and Nrp2, suggesting that both coreceptors are needed to mediate SEMA3A-dependent effects on nasal axon patterning and GnRH neuron migration.
Shortly after and in agreement with these findings, two independent groups described mutations in SEMA3A gene (chr7p12.1) in two cohorts of patients affected by KS (Hanchate et al., 2012; Young et al., 2012). In both reports, KS individuals harbored heterozygous variants (frameshift, missense or 11-exon deletion), were predicted to be loss-of-function variants, and a subset of patients harbored heterozygous variants in other KS genes, substantiating an oligogenic inheritance model. In addition to these reports, two studies have reported biallelic SEMA3A mutations in two prepubertal boys resulting in multiple congenital anomalies [postnatal short stature, cardiac defects, skeletal defects) (Hofmann et al., 2013; Baumann et al., 2017). Interestingly, while one of the boys manifested micropenis (Hofmann et al., 2013), the other boy did not have any overt reproductive phenotypes (Baumann et al., 2017). However, his father who harbored a heterozygous deletion was reported to have a delayed growth spurt. More recently, SEMA3A heterozygous variants were also identified in isolated congenital anosmia without any reproductive dysfunction (Alkelai et al., 2017). These observations reiterate the importance of SEMA3A signaling in GnRH neuron ontogeny, olfactory neurogenesis, and its oligogenic contribution to human KS. In addition to these report, dysfunctional SEMA3A signaling has also been linked to other human disease states such as Hirschsprung’s disease (Luzón-Toro et al., 2013) and cardiac arrythmias (Nakano et al., 2013; Boczek et al., 2014). Intriguingly, SEMA3A-associated KS patients do not exhibit gastrointestinal or cardiac phenotypes, and similarly, individuals with SEMA3A-associated Hirschsprung’s disease/cardiac arrythmias do not exhibit KS. These observations demonstrate a pleotropic role for SEMA3A signaling across diverse organ systems and suggest that the other genetic/nongenetic modifiers may influence the phenotypic penetrance and expressivity in individuals with SEMA3A mutations.
A more recent study aimed at exploring the relationship between nasal axon development and the migration of GnRH neurons has highlighted the prominent role of SEMA3A in this context (Taroc et al., 2017). In this working model, SEMA3A, which is highly expressed at the nasal-forebrain junction level, would act on one hand to prevent the OLF and VN nerves from invading the forebrain and on the other to attract both GnRH neurons and the terminal nerve (Taroc et al., 2017). The double role of SEMA3A can be explained by the presence of different sets of receptors and coreceptors on each cell population. According to this model, the TN, as well as GnRH neurons, which express high levels of NRP1, is attracted by SEMA3A, whereas OLF/VN, which express both NRP1 and NRP2, is repelled by SEMA3A, preventing brain invasion.
In addition to the role of SEMA3A signaling during GnRH neurons development, further studies have demonstrated that SEMA3A may also exerts a crucial role in adulthood by remodeling GnRH fibers that project to the ME during the estrus cycle (Giacobini et al., 2014). Specifically, brain endothelial cells release SEMA3A, which promotes GnRH neuron axonal sprouting through NRP1, expressed by GnRH neuron axon terminals. In addition, the amount of released SEMA3A increases dramatically during proestrus, when GnRH is released in massive surges to sustain LH surge (Giacobini et al., 2014). Thus it is likely that, given the extensive expression in the territories relevant to GnRH neurons development and reproductive function as well as the important roles of this molecule during GnRH neuron development and plasticity, Sema3a should be considered a master gene in this system and it is not surprising that mutations are found in ~6% KS patients.
Various combinations of A-type plexins may also contribute to the distinct responses to SEMA3A signaling in this system. Indeed, NRPs are not able to transduce a signaling cascade, but need to be coupled to a member of the PLXNA family to signal intracellularly. The role of the plexins in human GnRH neuron biology became evident when heterozygous missense mutations in the PLXNA1 gene were identified in patients with KS and nIHH, with an oligogenic pattern of inheritance. Complementary studies in PLXNA1-deficient mice showed that GnRH neuron and the olfactory system are only mildly affected (Marcos et al., 2017) raising the possibility that PLXNA1 may act in partial redundancy with another A-type Plexin. In keeping with this hypothesis, it has been recently shown that only the combined loss of PLXNA1 and PLXNA3 phenocopied the full spectrum of nasal axon and GnRH neuron defects of Sema3a knockout mice, suggesting that both PLXNA1 and PLXNA3 are required for the proper nasal axon targeting and GnRH neuron migration. These findings also suggest that together with PLXNA1, the human orthologue of PLXNA3 should be also investigated as a candidate gene for IGD (Oleari et al., 2019). Similar to SEMA3A mutations, which demonstrate pleiotropy in terms of their associated human phenotypes, heterozygous PLXNA1 mutations, in addition to IGD, have also been linked to epilepsy (Oliver et al., 2016; Park et al., 2017) and schizophrenia (Fromer et al., 2014).
In addition to SEMA3A and PLXNA1 gene mutations, NRP1 and of NRP2 missense variants have also been identified in the heterozygous state in some patients with KS, in the absence of mutations in nine KS-linked genes (Marcos et al., 2017). Notably, one of these patients carried monoallelic mutations of PLXNA1 and NRP1, both of which, however, were inherited from his clinically unaffected mother, therefore indicating that the two mutations are not sufficient to produce the disease phenotype. A more recent study also reported the presence of one IGD patient carrying NRP1 mutation in heterozygosity with PLXNA1 (Kotan et al., 2019), suggesting that neuropilin pathway mutations also contribute to the oligogenic basis of IGD.
SEMA3E
SEMA3E is a molecule involved in many developmental processes, including nervous and cardiovascular system formation. It exerts its function through either PLXND1 or VEGFR2, hence it acts independently by neuropilins, differently from all the other Class 3 semaphorins (Oh and Gu, 2013). In the GnRH neuronal system, both PLXND1 and SEMA3E are strongly expressed in the nasal compartment and in the MPOA, during development. Interestingly, PLXND1 is dispensable for the correct vascular patterning and axonal pathfinding in the embryonic nose, but essential to maintain GnRH neuronal number once in the MPOA. Indeed, GnRH neurons start to express detectable levels of PLXND1 only after entering in the brain, where they are likely to respond to the SEMA3E source. Accordingly, mice lacking Sema3e or Plxnd1 have a reduced number of GnRH neurons in the brain. In this context, SEMA3E acts as a prosurvival signal for GnRH neurons in the hypothalamus. This was confirmed by in vitro studies which showed that SEMA3E acts as a potent prosurvival factor for GT1–7 cells, an immortalized model of hypothalamic GnRH neurons (Mellon et al., 1990). Importantly, sporadic mutations in the SEMA3E gene (chr7q21.11) have previously been linked with CHARGE syndrome (Lalani et al., 2004), a complex syndrome whose phenotypic spectrum also includes syndromic IGD, and few years later in two brothers affected by KS (Cariboni et al., 2015). Interestingly, in the same study, the SEMA3E mutation was accompanied by the presence of a heterozygous mutation in the CHD7 gene and compound heterozygous mice for Chd7 and the SEMA3E receptor Plxnd1 displayed a more pronounced reduction in the GnRH neuron innervation of the ME compared to their single heterozygous littermates. Altogether these findings suggest that CHD7 and SEMA3E signaling interact genetically to control the GnRH neuron system.
SEMA7A
SEMA7A is the only glycosylphosphatidylinositol (GPI)-anchored semaphorin and it can act either as a membrane-bound or secreted protein, after cleavage (Xu et al., 1998). SEMA7A binds with equal affinity to PLXNC1, reducing the integrin-mediated cell adhesion, and to β-integrin (ITGβ1), promoting integrin clustering and therefore cell attachment (Suzuki et al., 2007). Since SEMA7A is expressed in the nasal pit and in the olfactory bulb (Pasterkamp et al., 2007), a role for SEMA7A in the GnRH neuronal system was proposed. Indeed, SEMA7A was found to promote GnRH neuron migration in the nasal compartment, where it is highly expressed. Specifically, SEMA7A is abundant on OLF/VN nerves during development while the two SEMA7A receptors are differentially expressed in GnRH neurons: in the nasal mesenchyme, GnRH neurons express ITGβ1 and migrate along the SEMA7A-expressing axonal scaffold, instead once crossing the nasal-forebrain junction, they begin to express PLXNCC1 to stop migration. Consistently Sema7a-null mice display defective GnRH migration, a less innervated ME, and dysfunctional gonads (Messina et al., 2011). Like SEMA3A, SEMA7A exerts an additional function on the ME plasticity. Specifically, SEMA7A is expressed by tanycytes, specialized glial cells lining the third ventricle; here, it induces retraction of GnRH neuron terminals and promotes the expansion of tanycytes end feet by acting through PLXNC1 and ITGβ1, respectively (Parkash et al., 2015). In agreement with the essential role exerted by SEMA7A in the development and functionality of GnRH neurons in mice, mutations in SEMA7A (chr15q22.3-q23) have been found in both KS and nIHH (Känsakoski et al., 2014).
SEMA4D
SEMA4D is a membrane-bound semaphorin that is proteolytically cleaved to bind PLXNB1 in combination with MET tyrosine kinase receptor (Conrotto et al., 2005). These interactions are thought to mediate growth cone collapse of developing axons or to induce chemotaxis of epithelial and endothelial cells. Interestingly, the SEMA4D receptors PLXNB1 and PLXNB2 are highly expressed in the developing olfactory structures (Perälä et al., 2005. Consistently, SEMA4D regulates GnRH neuron migration through the PLXNB1-MET receptor complex (Giacobini et al., 2008). Specifically, PLXNB1 colocalizes with NCAM-positive nasal axons in the nasal mesenchyme only at early stages of development (E12.5 but not E14.5 nor E17.5). Consistent with a temporary effect only at early stages of GnRH neuron migration, Plxnbl-null mice showed a reduced number of GnRH neurons reaching the forebrain with accumulation in the olfactory bulb region. Decreased GnRH neurons in the adult brains also resulted in decreased GnRH fibers in the ME. Yet, Sema4d knockout mice do not have a reproductive phenotype, with normal migration of GnRH neurons, suggesting compensation by different semaphorins and no human mutations in this pathway have been documented so far.
CONCLUSIONS
Semaphorin-signaling pathways play pivotal roles during neural development and network assembly as well as in the unique migratory journey of GnRH neurons from the nose to the hypothalamus. In this context, semaphorins have been shown to control both axon guidance and neuronal migration during GnRH neuron development as well as neuronal plasticity during GnRH neuron secretion in adulthood. These fundamental biological processes underlie the genetic basis of reproductive disorders such as IGD, which is largely caused by mutations in genes that affect either the development or the function of GnRH neurons. Accordingly, the recent identification of mutations in semaphorin and their receptor genes in patients with IGD strongly support the importance of semaphorins in this process.
Given the several roles of semaphorins in axon guidance, neuronal migration, and neuronal plasticity and the unique development and physiology of GnRH neurons, it is not surprising if additional members of the semaphorin-plexin signaling pathways will be found to be implicated. Further, given the important dual role of semaphorin signaling in regulating neuronal and vascular patterning, future studies will be needed to prove their reciprocal influence in the GnRH system.
References
- Alkelai A et al. (2017). Next-generation sequencing of patients with congenital anosmia. Eur J Hum Genet. Springer US, 25 (12): 1377–1387. 10.1038/s41431-017-0014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alto LT, Terman JR (2017). Semaphorins and their signaling mechanisms. Methods Mol Biol 1493 (2): 1–25. 10.1007/978-1-4939-6448-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian R, Crowley WF (1993). Isolated gonadotropin-releasing hormone (GnRH) deficiency, GeneReviews® Available at http://www.ncbi.nlm.nih.gov/pubmed/20301509.
- Bamberg JA et al. (1999). Unified nomenclature for the semaphorins/collapsins. Semaphorin nomenclature committee. Cell 97 (5): 551–552. 10.1016/S0092-8674(00)80766-7. [DOI] [PubMed] [Google Scholar]
- Bashaw GJ (2007). Semaphorin directs axon traffic in the fly olfactory system. Neuron 53 (2): 157–159. 10.1016/j.neuron.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Baumann M et al. (2017). A recognizable type of syndromic short stature with arthrogryposis caused by bi-allelic SEMA3A loss-of-function variants. Clin Genet 92 (1): 86–90. 10.1111/cge.12967. [DOI] [PubMed] [Google Scholar]
- Bianco SDC, Kaiser UB (2009). The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol 5 (10): 569–576. 10.1038/nrendo.2009.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boczek NJ et al. (2014). Characterization of SEMA3A-encoded semaphorin as a naturally occurring Kv4.3 protein inhibitor and its contribution to Brugada syndrome. Circ Res 115 (4): 460–469. 10.1161/aRCRESAHA.115.303657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm U et al. (2015). Expert consensus document: European consensus statement on congenital hypogonadotropic hypogonadism—pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. Nature Publishing Group, 11 (9): 547–564. 10.1038/nrendo.2015.112. [DOI] [PubMed] [Google Scholar]
- Brand JS et al. (2014). Testosterone, sex hormone-binding globulin and the metabolic syndrome in men: an individual participant data meta-analysis of observational studies. PLoS One 9 (7): e100409. 10.1371/journal.pone.0100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariboni A et al. (2007). Neuropilins and their ligands are important in the migration of gonadotropin-releasing hormone neurons. J Neurosci 27 (9): 2387–2395. 10.1523/JNEUROSai.5075-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariboni A et al. (2011). Defective gonadotropin-releasing hormone neuron migration in mice lacking SEMA3A signalling through NRP1 and NRP2: implications for the aetiology of hypogonadotropic hypogonadism. Hum Mol Genet 20 (2): 336–344. 10.1093/hmg/ddq468. [DOI] [PubMed] [Google Scholar]
- Cariboni A et al. (2015). Dysfunctional SEMA3E signaling underlies gonadotropin-releasing hormone neuron deficiency in Kallmann syndrome. J Clin Invest 125 (6): 2413–2428. 10.1172/JCI78448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrotto P et al. (2005). Sema4D induces angiogenesis through met recruitment by Plexin B1. Blood 105 (11): 4321–4329. 10.1182/blood-2004-07-2885. [DOI] [PubMed] [Google Scholar]
- Costa-Barbosa FA et al. (2013). Prioritizing genetic testing in patients with Kallmann syndrome using clinical phenotypes. J Clin Endocrinol Metab 98 (5): E943–E953. 10.1210/jc.2012-4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro F (2009). Wiring olfaction: the cellular and molecular mechanisms that guide the development of synaptic connections from the nose to the cortex. Front Neurosci 3 (DEC): 52. 10.3389/neuro.22.004.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodé C, Hardelin J-P (2009). Kallmann syndrome. Eur J Hum Genet 17 (2): 139–146. 10.1038/ejhg.2008.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer AA, Jayasena CN, Quinton R (2016). Congenital hypogonadotropic hypogonadism: implications of absent minipuberty. Minerva Endocrinol 41 (2): 188–195 Available at http://www.ncbi.nlm.nih.gov/pubmed/27213784. [PubMed] [Google Scholar]
- Fromer M et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506 (7487): 179–184. 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacobini P et al. (2008). Semaphorin 4D regulates gonadotropin hormone-releasing hormone-1 neuronal migration through PlexinB1-Met complex. J Cell Biol 183 (3): 555–566. 10.1083/jcb.200806160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacobini P et al. (2014). Brain endothelial cells control fertility through ovarian-steroid-dependent release of semaphorin 3A. PLoS Biol 12 (3): e1001808. 10.1371/journal.pbio.1001808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanchate NK et al. (2012). SEMA3A, a gene involved in axonal pathfinding, is mutated in patients with Kallmann syndrome. PLoS Genet 8 (8): e1002896. 10.1371/journal.pgen.1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K et al. (2013). Biallelic SEMA3A defects cause a novel type of syndromic short stature. Am J Med Genet A 161A (11): 2880–2889. 10.1002/ajmg.a.36250. [DOI] [PubMed] [Google Scholar]
- Känsäkoski J et al. (2014). Mutation screening of SEMA3A and SEMA7A in patients with congenital hypogonadotropic hypogonadism. Pediatr Res 75 (5): 641–644. 10.1038/pr.2014.23. [DOI] [PubMed] [Google Scholar]
- Kim SH (2015). Congenital hypogonadotropic hypogonadism and Kallmann syndrome: past, present, and Future. Endocrinol Metab 30 (4): 456–466. 10.3803/EnM.2015.30.4.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotan LD et al. (2019). Prevalence and associated phenotypes of PLXNA1 variants in normosmic and anosmic idiopathic hypogonadotropic hypogonadism. Clin Genet 95 (2): 320–324. 10.1111/cge.13482. [DOI] [PubMed] [Google Scholar]
- Kruger RP, Aurandt J, Guan K-L (2005). Semaphorins command cells to move. Nat Rev Mol Cell Biol 6 (10): 789–800. 10.1038/nrm1740. [DOI] [PubMed] [Google Scholar]
- Laitinen E-M et al. (2011). Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland. Orphanet J Rare Dis. BioMed Central Ltd, 6 (1): 41. 10.1186/1750-1172-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitinen E-M et al. (2012). Bone mineral density, body composition and bone turnover in patients with congenital hypogonadotropic hypogonadism. Int J Androl 35 (4): 534–540. 10.1111/j.1365-2605.2011.01237.x. [DOI] [PubMed] [Google Scholar]
- Lalani SR et al. (2004). SEMA3E mutation in a patient with CHARGE syndrome. J Med Genet 41 (7): e94. 10.1136/jmg.2003.017640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzón-Toro B et al. (2013). Mutational spectrum of semaphorin 3A and semaphorin 3D genes in Spanish Hirschsprung patients. PLoS One 8 (1): e54800. 10.1371/journal.pone.0054800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestre de San Juan A (1856). Falta total de los nervios olfactorios con anosmía en un individuo en quien existia una atrofía congénita de los testículos y miembro viril. Siglo Medico 131: 211. [Google Scholar]
- Marcos S et al. (2017). Defective signaling through plexin-A1 compromises the development of the peripheral olfactory system and neuroendocrine reproductive axis in mice. Hum Mol Genet 26 (11): 2006–2017. 10.1093/hmg/ddx080. [DOI] [PubMed] [Google Scholar]
- Marino M et al. (2014). Central hypogonadotropic hypogonadism: genetic complexity of a complex disease. Int J Endocrinol. Hindawi Publishing Corporation, 2014: 649154. 10.1155/2014/649154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda I et al. (2010). Development of the somatosensory cortex, the cerebellum, and the main olfactory system in Semaphorin 3F knockout mice. Neurosci Res. Elsevier Ireland ltd and Japan neuroscience Society, 66 (3): 321–329. 10.1016/j.neures.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Mellon PL et al. (1990). Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 5 (1): 1–10. Available at: papers://c32175d7-d89d-4b27-af31-837cef862ad3/Paper/p417. [DOI] [PubMed] [Google Scholar]
- Messina A et al. (2011). Dysregulation of Semaphorin7A/β1-integrin signaling leads to defective GnRH-1 cell migration, abnormal gonadal development and altered fertility. Hum Mol Genet 20 (24): 4759–4774. 10.1093/hmg/ddr403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano Y et al. (2013). A nonsynonymous polymorphism in semaphorin 3A as a risk factor for human unexplained cardiac arrest with documented ventricular fibrillation. PLoS Genet 9 (4): e1003364. 10.1371/journal.pgen.1003364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh W-J, Gu C (2013). The role and mechanism-of-action of Sema3E and Plexin-D1 in vascular and neural development. Semin Cell Dev Biol 24 (3): 156–162. 10.1016/j.semcdb.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleari R et al. (2019). PLXNA1 and PLXNA3 cooperate to pattern the nasal axons that guide gonadotropin-releasing hormone neurons. Development 146 (21). 10.1242/dev.176461. [DOI] [PubMed] [Google Scholar]
- Oliver KL et al. (2016). In silico prioritization based on coexpression can aid epileptic encephalopathy gene discovery. Neurol Genet 2 (1): e51. 10.1212/NXG.0000000000000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K et al. (2017). PLXNA1 developmental encephalopathy with syndromic features: a case report and review of the literature. Am J Med Genet A 173 (7): 1951–1954. 10.1002/ajmg.a.38236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkash J et al. (2015). Semaphorin7A regulates neuroglial plasticity in the adult hypothalamic median eminence. Nat Commun 6: 6385. 10.1038/ncomms7385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MW et al. (2012). Function of members of the neuropilin family as essential pleiotropic cell surface receptors. Biochemistry 51 (47): 9437–9446. 10.1021/bi3012143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasterkamp RJ, Ruitenberg MJ, Verhaagen J (1999). Semaphorins and their receptors in olfactory axon guidance. Cell Mol Biol (Noisy-le-Grand) 45 (6): 763–779 Available at: http://www.ncbi.nlm.nih.gov/pubmed/10541474. [PubMed] [Google Scholar]
- Pasterkamp RJ et al. (2007). Expression patterns of semaphorin7A and plexinC1 during rat neural development suggest roles in axon guidance and neuronal migration. BMC Dev Biol 7: 98. 10.1186/1471-213X-7-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perälä NM, Immonen T, Sariola H (2005). The expression of plexins during mouse embryogenesis. Gene Expr Patterns 5 (3): 355–362. 10.1016/j.modgep.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Rollmann SM et al. (2007). The early developmental gene Semaphorin 5c contributes to olfactory behavior in adult drosophila. Genetics 176 (2): 947–956. 10.1534/genetics.106.069781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw ND et al. (2011). Expanding the phenotype and genotype of female GnRH deficiency. J Clin Endocrinol Metab 96 (3): E566–E576. 10.1210/jc.2010-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamou MI, Georgopoulos NA (2018). Kallmann syndrome: phenotype and genotype of hypogonadotropic hypogonadism. Metab Clin Exp 86: 124–134. 10.1016/j.metabol.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamou MI, Cox KH, Crowley WF (2015). Discovering genes essential to the hypothalamic regulation of human reproduction using a human disease model: adjusting to life in the “-Omics” era. Endocr Rev 36 (6): 603–621. 10.1210/er.2015-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K et al. (2007). Semaphorin 7A initiates T-cell-mediated inflammatory responses through alpha1beta1 integrin. Nature 446 (7136): 680–684. 10.1038/nature05652. [DOI] [PubMed] [Google Scholar]
- Sykiotis GP et al. (2010a). Deciphering genetic disease in the genomic era: the model of GnRH deficiency. Sci Transl Med 2 (32): 32rv2. 10.1126/scitranslmed.3000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykiotis GP et al. (2010b). Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci U. S. A 107 (34): 15140–15144. 10.1073/pnas.1009622107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagnone L et al. (1999). Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99 (1): 71–80. 10.1016/S0092-8674(00)80063-X. [DOI] [PubMed] [Google Scholar]
- Taroc EZM et al. (2017). The terminal nerve plays a prominent role in GnRH-1 neuronal migration independent from proper olfactory and vomeronasal connections to the olfactory bulbs. Biol Open 6 (10): 1552–1568. 10.1242/bio.029074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topaloğlu AK (2017). Update on the genetics of idiopathic hypogonadotropic hypogonadism. J Clin Res Pediatr Endocrinol 9 (Suppl. 2): 113–122. 10.4274/jcrpe.2017.S010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varimo T et al. (2015). Health-related quality of life in male patients with congenital hypogonadotropic hypogonadism. Clin Endocrinol (Oxf) 83 (1): 141–143. 10.1111/cen.12701. [DOI] [PubMed] [Google Scholar]
- Xu X et al. (1998). Human semaphorin K1 is glycosyl-phosphatidylinositol-linked and defines a new subfamily of viral-related semaphorins. J Biol Chem 273 (35): 22428–22434. 10.1074/jbc.273.35.22428. [DOI] [PubMed] [Google Scholar]
- Yazdani U, Terman JR (2006). The semaphorins. Genome Biol 7 (3): 211. 10.1186/gb-2006-7-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J et al. (2012). SEMA3A deletion in a family with Kallmann syndrome validates the role of semaphorin 3A in human puberty and olfactory system development. Hum Reprod 27 (5): 1460–1465. 10.1093/humrep/des022. [DOI] [PubMed] [Google Scholar]