

Abstract
Cell-mediated responses to immunological stimuli are often localised in inflammatory sites and involve a number of cell types. These responses can be functionally characterised at the single-cell level on the basis of the types of cytokines expressed either in whole blood or PBMCs. The ability to measure antigen-specific cell responses at the single cell level is an important tool with a wide range of potential applications ranging from studies of disease pathogenesis to the evaluation of vaccines.
A number of experiments were performed in this study in order to establish the optimal conditions for in vitro stimulation of cytokine production by T cells and monocytes in whole blood samples collected from healthy adult Malawian participants and the optimal staining conditions for various cytokine producing cells. Different stimulation methods and conditions, different culture tubes and incubators and different antibody labelling conditions were assessed in order to establish optimal conditions for detecting cytokine-producing cells in whole blood samples.
The use of PMA plus Ionomycin produced highest cytokine-producing T cells whereas LPS was a better stimulant for cytokine producing monocytes. Stimulation of whole blood for 5 h was optimal for cytokine detection in T cells whereas 4 h was optimal for monocytes. BFA was found to be a better Golgi blocker than Monensin and the use of 15 ml Falcon-type polypropylene tubes while stationary resulted in the detection of the highest proportion of cytokine-producing cells. T cells were found to be producers of mainly TNF-α, IFN-γ and IL-2 whereas Monocytes were mainly producing TNF-α and IL-6. Anti-CD3-PerCP (used at a ratio of 1:25), anti-CD14-APC (used at a ratio of 1:50) and anti-cytokine-PE (used at a ratio of 1:12.5) resulted in the best results. The highest cytokine production monocytes were detected when 1 X FACS Lysing solution was used at a volume of 40X that of the whole blood sample compared to the other volumes. These optimal conditions are essential in determination of proportion of cytokine-producing cells using ICS in whole blood.
Keywords: Cytokines, Intracellular
Graphical abstract
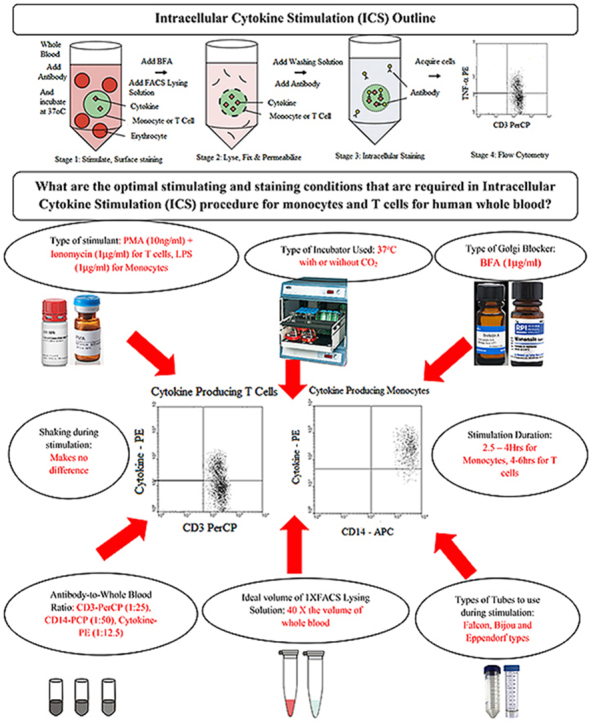
Highlights
-
•
Intracellular Cytokine Staining facilitates measurement of cell responses at single-cell level in immunological studies
-
•
We investigated ideal stimulation and staining conditions for cytokine producing T cells and monocytes in whole blood.
-
•
PMA + Ionomycin was the ideal stimulant for cytokine-producing T cells, LPS was the best for cytokine-producing monocytes.
-
•
Five-hour whole blood stimulation was optimal for cytokine producing T cells whereas four hours was ideal for monocytes.
-
•
BFA was the best Golgi blocker, T cells mainly produced TNF-a, IFN-g and IL-2, Monocytes mainly produced TNF-a and IL-6.
-
•
Anti-CD3-PerCP at a ratio of 1:25, anti-CD14-APC at a ratio of 1:50 and anti-cytokine-PE at a ratio of 1:12.5 were ideal.
-
•
1 X FACS Lysing solution used at a volume of 40X volume of whole blood produced optimal results.
1. Introduction
Cell-mediated responses to immunological stimuli are often localised in inflammatory sites and involve a number of cell types. These responses can be functionally characterised at the single-cell level on the basis of the types of cytokines expressed (Maecker, 2004). The ability to measure antigen-specific T cell responses at the single cell level is an important tool with a wide range of potential applications ranging from studies of disease pathogenesis to the evaluation of vaccines (Gauduin et al., 2004). Several methods have been developed that allow cytokine expression to be measured and these include enzyme-linked immunosorbent assay (ELISA), enzyme-linked immunospot (ELISPOT), limiting dilution assay (LDA), real-time polymerase chain reaction (RT-PCR) and intracellular cytokine staining (ICS) (Godoy-Ramirez et al., 2004; Pala et al., 2000b). Performing flow cytometric analysis on ICS cells allows individual characterisation of large numbers of cells and can fully display the heterogeneity of cell populations.
A great advantage of ICS over the other methods especially ELISA is that multicolour staining can demonstrate exclusive or mutual co-expression of different cytokines in individual cells. This allows the characterisation of T cell subsets on the basis of cytokine production rather than just surface markers (Pala et al., 2000a, 2000b). Thus it is possible to analyse simultaneously CD4+ and CD8+ T cell responses in the same sample and to assess expression of other phenotypic markers on the cells of interest (Gauduin et al., 2004). ICS by flow cytometry not only allows detection of single cell expression of cytokines but facilitates simultaneous detection, quantification, and phenotypic characterisation of antigen-specific T cell subsets in either whole blood or peripheral blood mononuclear cells (PBMCs). Through this method, antigen-specific T cells are identified based on their intracellular accumulation of a cytokine in conjunction with the activation markers such as CD69 following stimulation with specific antigens (Gauduin, 2006; Jung et al., 1993).
ICS involves incubation of whole blood or PBMC with a stimulant while disrupting intracellular Golgi-mediated transport using Brefeldin A (BFA) or Monensin which allows cytokines to accumulate yielding an enhanced cytokine signal that can be detected by flow cytometry. The cells are then permeabilised with a reagent such as FACS Permeabilising solution (FACS Perm) which allows antibodies to detect the intracellular cytokine. This method can detect multiple cytokines per cell and discrete cellular populations that express a particular cytokine. This is crucial especially when studying cytokine response to specific stimuli (Bueno et al., 2001).
Although it is a common practice in most immunological assays to isolate PBMCs either for immediate stimulation, staining and analysis or for cryopreservation, the actual process of PBMCs isolation is not only expensive but labour intensive (Germann et al., 2013) and in some cases, due to ethical reasons, it may not be possible to collect an adequate volume of blood from either a patient or study participant (Peplow et al., 2019; Boahen et al., 2013). In such situations ICS analysis on whole blood samples, which includes lysing and fixations steps, is preferred as it requires relatively less blood.
Depending on the nature of fixation, unintended changes can occur to the immune cells resulting in epitope modification of certain markers (Maecker et al., 2010; Silva et al., 2020). As a result, there could be reduced binding capacity of antibodies to their specific receptors (Horn et al., 2021). Several researchers (Pinto et al., 2005; Horn et al., 2020; Caraher et al., 2000) have previously investigated on how to negate the effect of lysing and fixation on the accuracy and specificity of ICS methods. Their work mainly concentrated on different lymphocyte subsets such as T, B and NK cells. Despite this, limited work has been done to investigate how different stimulation and staining of whole blood affects the accuracy of the ICS methods on T cells and monocytes.
The objective of this work therefore was to determine the optimal conditions for stimulating cytokine production by T cells and monocytes in vitro using whole blood from Malawian adults and the optimal staining conditions for various cytokines and cell types. Different stimulation methods and conditions, different culture tubes and incubators were assessed to establish optimal conditions. Antibody labelling conditions were assessed using blood stimulated in loosely capped 15 ml Falcon-type tubes in a 5% CO2 at 37 °C, for different durations. T cell stimulations were performed using 50% whole blood/50% Roswell Park Memorial Institute Medium (RPMI)-1640 with Phorbol 12-myristate 13-acetate (PMA) and Ionomycin (IO) and monocyte stimulations were performed using undiluted whole blood with LPS. Whole blood was diluted when stimulated with PMA + IO as per some previous reports that had shown that stimulation of undiluted whole blood resulted in decreased cell viability (Nylander and Kalies, 1999).
2. Materials and methods
2.1. Study participants, ethics clearance and blood samples
Blood samples used in this study were collected from five male Malawian healthy participants aged between 27 and 42 years. Ethical approval for the study was obtained from College of Medicine Research and Ethics Committee (COMREC) and written informed consent was obtained from each participant before taking part in the study. A 10-ml venous blood sample was taken at the time of recruitment for various tests. An aliquot of this sample was collected in a sodium heparin tube for the ICS experiments covered in this paper.
2.2. Data analysis
Each test the results of which are being reported was conducted either in duplicate or in triplicate and data are presented as arithmetic means ± standard deviation. Statistical tests were performed using GraphPad Prism Version 6.01 for Windows (GraphPad Software, San Diego California, USA). The Kruskal-Wallis test was used to compare the means of the cytokine producing monocytes and T cells either when comparing different stimulants, volume of 1 X FACS Lysing solution, type of incubator to use or type of Golgi blocker to use during whole blood stimulation. A P value of <0.05 was considered statistically significant at 95% level of confidence.
2.3. Preparation of various reagents
Phorbol 12-myristate 13-acetate (PMA) was used at 10 ng/mL, Ionomycin, Brefeldin A (BFA), Lipopolysaccharides from Escherichia coli O111:B4 (LPS) and Staphylococcal enterotoxin B from Staphylococcus aureus (SEB) were used at 1 μg/mL, all from Sigma-Aldrich, St Louis, USA. All reagents were prepared according to manufacturer's instructions and aliquots stored at −20 °C (or 4 °C for SEB) until use.
2.4. Cytokine stimulation procedure
Two 1.5 ml polypropylene tubes were appropriately labelled; in one tube 200 μl of heparinised whole blood was mixed with 200 μl of RPMI-1640, and with PMA at a concentration of 10 ng/ml and Ionomycin at a concentration of 1 μg/ml and stimulated at 37 °C for 4hrs. In the second tube, 200 μl of heparinised whole blood was mixed with LPS at a concentration of 1 μg/ml and incubated under the same conditions.
2.5. Labelling (staining) procedure of stimulated whole blood
50 μl of stimulated and unstimulated whole blood samples were labelled in eight FACS tubes at a ratio of 1:25 for anti-CD3-PerCP (Becton Dickinson Pharmingen, Clone G155-78) for the analysis of T cells and at a ratio of 1:50 for anti-CD14-APC (Becton Dickinson, Clone MΦP9) for the analysis of monocytes.
After 15 min incubation, 1X FACS lysis solution (Becton Dickinson) was added to each tube using amount of 40X the blood volume. The tubes were vortexed briefly and incubated again in the dark for 10 min. The tubes were vortexed again and centrifuge at 1600 rpm at 4 °C for 5 min. The supernatant was aspirated and tubes were vortexed before 1 X FACS Perm (Becton Dickinson) was added to each tube using 10 X the blood volume and incubated in the dark for 10 min. PBS +0.5% Bovine Serum Albumin (BSA) (Aldrich) was added to each tube using 30 X the blood volume, vortexed and centrifuged at 1600 rpm at 4 °C for 5 min. The supernatant was aspirated and the tubes vortexed and cells were labelled with PE-conjugated anti-cytokine antibodies at a ratio of 1:12.5.
Each tube was vortexed and incubated for 30 min in the dark at room temperature. PBS +0.5% BSA was added to each tube at 40 X the blood volume, vortexed and centrifuged at 1600 rpm at 4 °C for 5 min. The supernatant was aspirated and the tubes vortexed before the cells were fixed with PBS/1% formaldehyde solution at 4 X the blood volume. Samples were acquired on Flow Cytometer (Two-laser and four parameters Becton Dickinson FacsCalibur) within an hour of being stained and fixed. The data was then analysed using CellQuest based on the gating strategies presented in Fig. 1, Fig. 2 gating for CD3+ (T cells) in R1 gate for lymphocytes (Fig. 1A) and for CD14+ monocytes (Fig. 2A) and in each case an Isotype control was used for setting the gates (Figs. 1B and 2B).
Fig. 1.

Gating Strategy for Cytokine producing T cells: Whole blood samples from healthy controls (n = 5) were stimulated with PMA + ION, labelled with CD3 PerCP, lysed with 2.0 ml of 1 x FACS lysing solution and fixed with BFA before the labelling with Isotype Control (PE) and various cytokine antibodies (PE). The Flow cytometer dot plots illustrate the side scatter plot versus CD3-PerCP (A) with R1 gate for CD3+ lymphocytes (Total T cells), the Isotype Control plot for setting the gates (B), INF-γ producing cells (CD3+IFN-γ+ cells) (C), IL-10 producing T cells (CD3+IL-10+ cells) (D) and TNF-α producing T cells (CD3+TNF-α+ cells) (E).
Fig. 2.

Gating Strategy for Cytokine producing monocytes: Whole blood samples from healthy controls (n = 5) were stimulated with LPS, labelled with CD14 APC, lysed with 2.0 ml of 1 x FACS lysing solution and fixed with BFA before the labelling with Isotype Control (PE) and various cytokine antibodies (PE). Flow cytometer dot plots illustrating the side scatter plot versus CD14 (A) with R1 gate for CD14+ cells (monocytes), the Isotype Control plot for setting the gates (B), IL-6 producing monocytes (CD14+IL-6+ cells) (C) and TNF-α producing monocytes (CD14+TNF-α+ cells) (D).
2.6. Type and amount (volume) of different stimulants used
In one tube a mixture of 200 μl whole blood and same volume of RPMI-1640 was mixed with PMA at a concentration of 10 ng/ml and Ionomycin at a concentration of 1 μg/ml for stimulation of T cells. In the second tube 400 μl of whole blood was mixed with LPS at a concentration of 1 μg/ml for the stimulation of monocytes. In the third tube 400 μl of whole blood was mixed with SEB at a concentration of 1 μg/ml and CD28 (Sigma-Aldrich, Clone 15-E8) at a ratio of 1:200 also for the stimulation of monocytes. Each of the three tubes was paired with an unstimulated control tubes to which similar concentrations of the respective stimulants were added. In all six tubes BFA was used as a Golgi body blocker at a concentration of 1 μg/ml. Following the stimulation stage, surface and intracellular labelling for T cells and monocytes was performed as described earlier using anti-CD3-PerCP (at a ratio of 1:25) and anti-CD14-APC (at a ratio of 1:50) and anti-TNF-α (at a ratio of 1:12.5) before the permeabilisation stage.
2.7. Types of cytokines detected in T cells and monocytes
The ability of the study's assay to detect intracellular cytokines in T cells and monocytes in whole blood samples was assessed with particular interest focused on the following cytokines, all PE-conjugated: TNF-α (BD FastImmune, Clone 6401.111), IFN-γ (BD FastImmune, Clone 25723.11), IL-10 (BD Pharmingen, Clone JES3-9D7), IL-6 (BD Pharmingen, Clone MQ2-13A5), IL-4 (BD FastImmune, Clone 3010.211), IL-2 (BD Pharmingen, Clone MQ1-17H12), TGF-β (BD Pharmingen, Clone TW4-2F8), IL-12 p40 (BD Pharmingen, Clone C11.5) and IL-12 p70 (BD Pharmingen, Clone 20C2). Stimulation was performed as described earlier and the antibody labelling was performed using 50 μl of whole blood as already described with anti-CD3-PerCP used at a ratio of 1:25, anti-CD14-APC at a ratio of 1:50 and anti-cytokine-PE at a ratio of 1:12.5. One tube was included with unstimulated blood and labelled with anti-TNF-α−PE at the same ratio to account for any background noise and another tube was included with stimulated blood but labelled with an isotype control at the same ratio.
2.8. Volume of monoclonal antibodies to use for labelling T cells
For this set of experiments 50 μl samples of whole blood stimulated with PMA and Ionomycin (with all stimulations done as previously described) were labelled with anti-TNF-α-PE at four different ratios (1:50, 1:25, 1:12.5 and 1:6.25) using the procedure that has already been explained. Anti-CD69-APC (Becton Dickinson, Clone L78), used at the same four ratios, was included as a positive control for stimulation level. For each ratio, a separate tube was labelled with an isotype control. Two tubes, one for isotype control and another for anti-TNF-α-PE, of unstimulated control samples were included.
2.9. Effect of volume of 1X FACS lysing solution on cytokines detected
Whole blood samples were stimulated using LPS for monocytes and PMA and Ionomycin for T cells at the concentrations and conditions already explained. Stimulation was conducted in 15 ml Falcon tubes which were incubated in a 5% CO2 incubator for 4 h 50 μl of stimulated whole blood from each tube was labelled with anti-CD3-PerCP at a ratio of 1:25 or anti-CD14-APC at a ratio of 1:50 as surface antibody and anti-TNF-α-PE at a ratio of 1:12.5 and the only variable was that at the washing stage after lysis, 1 X FACS lysing solution was added to the lysed mixture using amounts of 20X, 30X or 40X the blood volume. An isotype control was included for each amount of1 X FACS lysis solution used.
2.10. Effect of tube type and contribution of agitation on the amount of cytokine produced
Three types of polypropylene tubes were investigated for stimulations in duplicate: 8 ml Bijou-type tubes, 15 ml Falcon-type tubes and 1.5 ml Eppendorf-type tubes. To each tube a mixture of 200 μl whole blood and RPMI-1640 of the same volume and stimulant were added as previously explained. Tubes were vortexed and incubated at 37 °C for 4hrs. During the incubation, one of the two tubes were placed on a rocker-plate (Bibi-Sterilin, 20 rpm) while the other was left stationary in the incubator. 50 μl of stimulated blood was labelled with anti-CD3-PerCP at a ratio of 1:25 as surface antibody and anti-TNF-α-PE at a ratio of 1:12.5 with an isotype control (also at a ratio of 1:25) included for each tube type used.
2.11. Effect of type of incubator used on the amount of cytokine produced
For this set of experiments four 15 ml Falcon tubes were used. To one set of two tubes 1 ml of whole blood was mixed with LPS at a concentration of 1 μg/ml and in another set of two 15 ml Falcon tubes a mixture of 500 μl whole blood and same volume of RPMI-1640 was mixed with PMA at a concentration of 10 ng/ml and Ionomycin at a concentration of 1 μg/ml. After mixing the contents of the different tubes, each tube was vortexed and their caps left loose to facilitate gas exchange. One tube of each set was placed in a normally aerated incubator (Genlab Ltd, UK, Model PRI/30/TDIG) set at 37 °C and the other tube was placed in an incubator supplied with 5% CO2 at 37 °C (Leec Ltd, UK, Model P50). Stimulated whole blood aliquots from each tube were labelled with anti-CD3-PerCP at a ratio of 1:25 or with anti-CD14-APC at a ratio of 1:50 as surface antibody and anti-TNF-α−PE at a ratio of 1:12.5. An isotype control was included in each case at the same ratios.
2.12. Effect of the type of golgi blocker used on the amount of cytokine detected
In order to compare the effect of the two Golgi blockers, Monensin or BFA, on the proportion of cytokine-producing cells, stimulations were performed in eight 1.5 ml polypropylene tubes. PMA, Ionomycin, LPS and BFA were all prepared and used at concentrations previously mentioned. Monensin was used at a final concentration of 1 μg/ml.
For the T cell experiments, 200 μl whole blood samples were mixed with RPMI-1640, PMA and Ionomycin as previously explained. To the first tube no Golgi blocker (no BFA or Monensin) was added, the second tube only had BFA added at a concentration of 1 μg/ml, the third tube only had Monensin added at a concentration of 1 μg/ml, while the fourth tube had BFA and Monensin added each at final concentration of 500 ng/ml. For the monocyte experiments, four tubes each containing 400 μl whole blood mixed with LPS were used as previously explained. To the first tube no Golgi blocker (no BFA or Monensin) was added, BFA was added to the second tube at a concentration of 1 μg/ml, whereas Monensin added to the third tube at a concentration of 1 μg/ml, and the fourth tube had a mixture of BFA and Monensin added each at final concentration of 500 ng/ml.
All eight tubes were incubated for 4 h at 37 °C with 5% CO2. Surface and intracellular staining for T cells and monocytes were performed as already explained using anti-CD3-PerCP, anti-CD14-APC, anti-TNF-α with an isotype control included.
2.13. Time course experiments for the stimulation of various cytokines
For this set of experiments stimulations were performed in three 15 ml polypropylene tubes. For the detection of cytokine producing T cells a mixture of 500 μl whole blood and same volume of RPMI-1460 was mixed with PMA and Ionomycin as previously explained. For the detection of the cytokine producing monocytes, 1 ml of whole blood was mixed with LPS as already explained. Unstimulated whole blood was used as a control and PMA, Ionomycin, LPS and BFA were all prepared and used at concentrations already provided.
The tubes were incubated for 4 h for LPS stimulation and for 8 h for PMA + Ionomycin. All stimulations were performed at 37 °C, 5% CO2. At the following time intervals; 15min, 30min, 1hr, 1.5hrs, 2hrs, 2.5hrs, 3hrs, 3.5hrs, 4hrs, 6hrs and 8hrs for T cells and 15min, 30min, 1hr, 1.5hrs, 2hrs, 2.5hrs, 3hrs, 3.5hrs and 4hrs for monocytes, a 50 μl aliquot was taken from each tube and each aliquot was labelled with either anti-CD3-PerCP or anti-CD14-APC added as surface antibodies as previously explained before permeabilisation and anti-Cytokine (TNF-α, IFN-γ, IL-2, IL-6) added after the permeabilisation stage as mentioned previously. An isotype control was included.
3. Results
3.1. Type and amount of different stimulants used
In ICS procedure, unlike where purified forms of specific antigens are used, various reagents can be used to stimulate the intracellular cytokine production. In this study we found that using the combination of PMA and Ionomycin as a stimulant resulted in a significantly (p < 0.0001) higher proportion of TNF-α producing T cells (48.86%) compared to the combination of SEB and CD28 (6.36%) or LPS (1.74%) (Fig. 3A and Fig. S1).Stimulation with LPS resulted in the highest (p < 0.0001) percentage of TNF-α- producing monocytes (84.29%) compared to the combination of SEB and CD28 (4.70%) and PMA and Ionomycin (2.98%) (Fig. 3A, Fig. S1). Once it was established that the best stimulants were LPS for monocytes and the combination of PMA and Ionomycin for T cells, the next set of experiments was to determine which other cytokines are produced by these cell types when subjected to this nature of stimulation.
Fig. 3.

Means (±standard deviation) of the percentage of TNF-α producing T cells and monocytes detected in whole blood samples (n = 5). Fig. 3A shows the proportion of TNF-α producing monocytes after stimulation with PMA + Ionomycin (PO), SEB + CD28 (SEB) or LPS. Fig. 3B shows the percentage of TNF-α, IFN-γ and IL-6 producing T cells and monocytes in whole blood samples following stimulation with PMA and Ionomycin (for CD3+Cytokine + subsets) and with LPS (for CD14+cytokine + subsets). Fig. 3C is for IL-4, IL-2 and TGF-b whereas Fig. 3D shows the IL-12p40 and IL-12p70 producing T cells and Monocytes. 50 μl of stimulated and unstimulated whole blood samples were labelled in eight FACS tubes at a ratio of 1:25 for anti-CD3-PerCP for the analysis of T cells and at a ratio of 1:50 for anti-CD14-APC. After permeabilisation, the following cytokines, all PE-conjugated: TNF-α (BD FastImmune, Clone 6401.111), IFN-γ (BD FastImmune, Clone 25723.11), IL-10 (BD Pharmingen, Clone JES3-9D7), IL-6 (BD Pharmingen, Clone MQ2-13A5), IL-4 (BD FastImmune, Clone 3010.211), IL-2 (BD Pharmingen, Clone MQ1-17H12), TGF-β (BD Pharmingen, Clone TW4-2F8), IL-12 p40 (BD Pharmingen, Clone C11.5) and IL-12 p70 (BD Pharmingen, Clone 20C2) were added anti-cytokine-PE at a ratio of 1:12.5 before flow cytometric analysis.
3.2. Type of cytokines detected in T cells and monocytes
Although both monocytes, representing the innate arm of the immune system, and T cells, which are part of the adaptive immunity, are capable of producing cytokines, the type and quantity of cytokines they are capable of producing vary. In this study we found that TNF-α was one cytokine that was produced in higher levels by both T cells and monocytes (Fig. 3B and Fig. S1) either as whole blood or when PBMCs were used (Fig. S5). For monocytes, two cytokines, TNF-α and IL-6 were detected in substantially higher levels compared to the other cytokines (Fig. 3B) with TNF-α and IL-6 producing monocytes detected in significantly (p < 0.0001) higher proportions than T cells producing the same cytokines (Fig. 3B) . In contrast, higher percentages of TNF-α, IFN-γ, IL-2 and a bit of IL-4 producing T cells were detected compared to the percentage T cells producing the other cytokines such as IL-6 with the proportion of IFN-γ (Fig. 3B) and IL-2 (Fig. 3C) producing T cells being significantly (p < 0.0001) higher than that of IFN-γ and IL-2 producing monocytes. Surprisingly very low levels of IL-10, TGF-β, IL-12 p40 and IL-12 p70 T cells and monocytes were detected although the proportion of IL-12p70 producing monocytes were significantly (p = 0.008) higher than that of IL-12p70 producing T cells (Fig. 3D). Having established that the main cytokines that could be detected were TNF-α, IFN-γ, IL-2 and IL-6, the next set of experiments was the antibody titration. This was aimed at determining what ratio of stimulated whole blood to monoclonal antibodies would result in the detection of the highest percentage of cytokine (with TNF-α being used as a standard cytokine in most subsequent experiments)-producing T cells.
3.3. The volume of monoclonal antibodies to use for labelling T cells
Although suppliers of various monoclonal antibodies will normally provide some guidance on the volume of each antibody that should be used for optimal results, conducting titration experiments for each antibody helps to determine the ideal volume to use under the ensuing conditions. In this study we found that the lowest percentage of TNF-α producing T cells (18.64%) was detected when the anti-TNF-α monoclonal antibody was used at a ratio of 1:50 (Fig. 4A). However, there was no difference in the percentage of TNF-α−producing T cells when TNF-α was used at the other three ratios of 1:25 (23.80%), 1:12.5 (25.25%) and 1:6.25 (24.79%). The percentage of activated T cells expressed as CD3+CD69+ T cells did not differ at all four ratios (1:50, 1:25, 1:12.5 and 1:6.25) of the antibodies used. Once we established that there was no difference in using the antibodies at any of these four ratios, the next parameter to investigate was the volume of 1 X FACS lysing solution to be used especially for the detection of cytokine-producing monocytes.
Fig. 4.

Means (±standard deviation) of the percentage of TNF-α producing T cells and monocytes detected in whole blood samples (n = 5) that had been subjected to different stimulants. Fig. 4A shows the percentage of TNF-α producing T cells (stimulated with PMA + Ionomycin) and TNF-α producing monocytes (stimulated with LPS) when the volume of antibodies used to label the stimulated samples was varied. Fig. 4B shows the effect of the volume of the 1 X FACS Lysing solution (20X or 30X or 40X the volume of whole blood used) on the proportion of TNF-α producing T cells and monocytes. Fig. 4C shows the effect of performing the stimulations in different containers and conditions on the proportion of TNF-α producing T cells and whereas Fig. 4D shows the same effect on monocytes. The following were the container types and conditions used: Bijou tube on a rocker (BR). Stationary Bijou tube (BS), 15 mL Falcon tube on a rocker (FR), Stationary 15 mL Falcon tube (FS), Eppendorf tube on a rocker (ER) Stationary Eppendorf tube (ES). Fig. 4E shows the effect of provision of CO2 in the incubator during stimulation on the proportion of TNF-α producing T cells or monocytes. Fig. 4F shows the effect of using either BFA or Monensin (MNSIN) or a combination of the two Golgi blockers (B + M) on the proportion of TNF-α producing T cells and monocytes. For all plots the Y-axis is the percentage of cytokine-producing cells. The concentrations and volumes of various reagents and antibodies are similar to those provided in the legend for Fig. 3.
3.4. Effects of volume of 1X FACS lysing solution on the amount of cytokine detected
A red blood cells (RBCs) lysing solution, such as FACS lysing solution (BD Biosciences) or ammonium chloride solution, is required when staining whole blood unlike where PBMCs are used. In addition to lysing RBCs, FACS lysing solution also fixes cells. We therefore conducted this set of experiments with the aim of assessing the effect of the volume of 1 X FACS lysing solution on the proportion of cytokine-producing monocytes and T cells.
We found that for monocytes, as the volume of 1 X FACS lysing solution was increased from 20X to 40X the blood volume, the percentage of TNF-α producing monocytes significantly increased (p < 0.0001) from 36.72% to 91.13% (Fig. 4B) and Fig. S2) and the percentage of TNF-α producing monocytes at 40X the volume of whole blood was also significantly (p < 0.0001) higher than at 30X volume (49.42%) compared to 91.13%). However the percentage of TNF-α producing T cells was not affected by the volume of 1 X FACS lysing solution since this remained within the range of 52.54 to 50.24% (Fig. 4B). We then investigated if the tubes used for stimulation and shaking the tubes during the stimulation stage had any effect on the stimulation process.
3.5. Effect of tube type and agitation on the amount of cytokine detected
We conducted this series of experiments firstly to determine if the type of tubes used during stimulation affects the proportion of cytokine-producing cells. Tubes used for this purpose can either be made from polypropylene or polyvinyl chloride (PVC) or polystyrene. Secondly we wanted to investigate if continuous agitation of the incubation tubes during the stimulation stage could have an effect on the proportion of cytokine-producing cells.
In addressing the first part, we found that the type of tubes in which the whole blood samples were stimulated had negligible effect on the percentage of TNF-α producing T cells (Fig. 4C) and monocytes (Fig. 4D). Falcon tubes, either stationary (59.49%) or agitated (52.97%) resulted in higher proportion of TNF-a producing T cells than either Bijou or Eppendorf tubes (Fig. 4C). In contrast, continuous shaking of the tubes seemed to be ideal since we found that the highest percentage of TNF-α producing monocytes (89.72%) was detected in the samples stimulated in 15 ml Falcon tubes which were placed on a rocker during the whole stimulation process whereas the lowest percentage of TNF-α producing monocytes (75.76%) was obtained when stimulation was done in stationary Eppendorf polypropylene tubes with the difference between the use of shaken and stationary Eppendorf tubes being significant (p < 0.01).
Considering that the conditions under which the stimulation is done are fundamental we then explored if the type of incubator used during stimulation had any effect on the proportion of cytokine producing cells.
3.6. Effect of type of incubator used on the amount of cytokine detected
The stimulation stage of cells in ICS can either be done in a standard incubator set at a particular temperature (usually 37 °C) or in an incubator continuously supplied with 5% CO2 or merely in a water bath set at the same temperature. We therefore conducted these experiments to determine if the type of incubator used would affect the proportion of cytokine-producing cells. The observed that the percentages of TNF-α producing T cells (54.06%) and monocytes (86.58%) obtained when the stimulation was performed in an incubator supplied with 5% CO2 were similar (p = 0.2667 for T cells and p = 0.3656 for monocytes) to those observed when stimulation was performed in a normally aerated incubator without CO2 (55.29% for TNF-α producing T cells and 85.86% for TNF-α producing monocytes) (Fig. 4E). The next set of experiments was aimed at determining which Golgi blocker was ideal for intracellular cytokine detection between BFA and Monensin.
3.7. Effect of the type of golgi blocker on the amount of cytokine detected
Cytokines are produced in vitro when cells are activated by the use of an appropriate stimulant. In order to accumulate the cytokines within the cells as they are produced, protein secretion needs to be blocked by the addition of reagents that inhibit Golgi apparatus/endoplasmic reticulum function. The most widely used secretion blocking reagents are Monensin and BFA. These two reagents differ slightly in their mode of action with Monensin acting as an inhibitor of trans-Golgi function, whereas BFA inhibits protein transport between the endoplasmic reticulum (ER) and the Golgi [9]. We conducted this series of experiments in order to determine which of these two Golgi blockers would result in detection of more cytokine-producing monocytes and T cells.
We found that using a combination of BFA and Monensin produced similar levels of TNF-α producing T cells (59.36%) as when BFA was used on its own (52.36%) (Fig 4F, Fig. S3 and Fig. S5). Using Monensin on its own resulted in the detection of significantly (p < 0.0001) lower percentage of TNF-α producing T cells (30.73%) compared to when either BFA on its own or in combination with Monensin was used. The highest percentage of TNF-α producing monocytes (89.46%) was detected when BFA was used on its own and the lowest percentage (60.17%), just as was the case with TNF-α producing T cells, was observed when Monensin was used on its own (Fig. 4F, Fig. S3 and Fig. S5) and the difference was also statistically significant (p < 0.0001). Next we wanted to determine what the optimal stimulation period for the detection of different cytokine-producing cells.
3.8. Outcome of the time course experiments
One crucial variable that has a fundamental bearing on the amount of cytokines produced during in vitro stimulations is the incubation duration from the time a stimulant is added. Ideally, ICS procedure aims to detect cytokine-producing cells at the optimal stage of cytokine production and before the cytokine levels start to decline. We found that for monocytes, after an hour of stimulation 53% were already producing TNF-α and only 15% were producing IL-6 (Fig. 5), indicating that TNF-α is produced more rapidly by monocytes compared with IL-6. The peak proportion of cytokine-producing monocytes (about 85% for both TNF-α and IL-6) was detected after the samples had been incubated for 3 h and this was maintained at the 4-h stimulation stage. However the proportion of TNF-α producing monocytes was already above 80% after 2 h of stimulation whereas the proportion of IL-6 producing monocytes only reached the ≥80% mark after 3-h of stimulation.
Fig. 5.

Percentage of TNF-α and IL-6 producing monocytes at different stages of stimulation. Whole blood samples from healthy controls (n = 5) were stimulated with LPS for a duration of 4 h. At different time points (15 min, 30 min, 1 h, 1.5 h, 2 h, 2.5 h, 3 h, 3.5 h and 4 h) an aliquot of the stimulated blood was collected, labelled with CD14 APC, lysed with 2.0 ml of 1 x FACS lysing solution and fixed with BFA before the labelling with Isotype Control (PE) and either IL-6 PE or TNF-α PE and analysis by Flow Cytometry.
For the T cells, production of TNF-α was observed to occur at a faster rate compared to that of IL-2 and that of IFN-γ with over 25% of T cells already producing TNF-α after 1 h of stimulation while IL-2 only reached this percentage after 2 h and 45 min of stimulation (Fig. 6). Forty-five percent was the highest proportion of T cells producing TNF-α and this was observed after 4 h of stimulation after which it started to decline. The highest percentage of IL-2-producing T cells (about 48%) was observed after 6 h of stimulation (Fig. 6) after which it started to decrease. The highest percentage of IFN-γ-producing T cells was 28% and this was observed after 4 h of stimulation (Fig. 6) and the levels begun to decrease after the 4-h mark.
Fig. 6.

Percentage of T cells producing IL-2, IFN-γ and TNF-α at different time points following stimulation with PMA and Ionomycin. Whole blood samples from healthy controls (n = 5) were stimulated with PMA + ION. At different time points (from 15 min to 8 h) an aliquot of the stimulated blood was collected and labelled with CD3-PerCP, lysed with 2.0 ml of 1 x FACS lysing solution and fixed with BFA before the labelling with Isotype Control PE and IL-2 PE, INF-γ PE and TNF-α PE. The samples were then analysed by flow cytometry.
4. Discussion
The combination of PMA and Ionomycin (PMA + IO) has been used as an activating stimulus to induce cytokine expression in several studies with good results as was the case in this study. Since the number of antigen-specific cytokine responding T cells is usually low and therefore can be difficult to detect, the PMA + IO combination is often used since it can potentially activate all T cells independent of their antigen-specific receptors (Godoy-Ramirez et al., 2004). One of the drawbacks of PMA + IO is that it induces a marked decrease in the levels of CD4 expression and as such the proportion of cytokine producing CD4+ T cells can be detected to be artificially lower than normal due to the effect of the PMA + IO (Godoy-Ramirez et al., 2004).
This study also showed that when stimulating whole blood with PMA-IO, only TNF-α, IFN-γ and IL-2 producing T cells could be detected in substantial amounts compared with background numbers. In addition, only TNF-α and IL-6 producing monocytes were observed in substantial amounts in blood samples stimulated with LPS. Some researchers investigated the influence of the degree of dilution of whole blood and the incubation period on whole blood and PBMCs cultured with various stimulants. The investigators assessed the expression of the cytokines IFN-γ, TNF-α, IL-2, IL-4, IL-10, and IL-13 and found that the level of cells producing the cytokines other than IFN-γ were generally much lower, and those cells producing IL-4 and IL-13 were difficult to distinguish from background levels of unstimulated cultures (Godoy-Ramirez et al., 2004).
In one study, high levels of TNF-α, IFN-γ, IL-2 and TGF-β producing CD4+ and CD8+ T cells, IL1-α, IL-6 and IL-8 producing monocytes were observed but only minimal amounts producing IL-10 (Hodge et al., 2005). Other studies have also reported equally high levels of IL-12 and TNF-α producing monocytes in LPS stimulated cultures (Bueno et al., 2001) although our study only detected very low levels of IL-12 p40 and IL-12 p70 producing monocytes. These variations in the type of cytokines observed in different studies suggest that the detection of each cytokine requires different stimulation and labelling conditions.
Although most, if not all, monoclonal antibodies come with instructions that include the volume of the antibody to be used per a specified volume of whole blood or PBMCs, it is always good practice that before one embarks on a major project, one performs a proper and systematic dilution exercise of the antibodies to determine the appropriate and ideal ratio of antibody to whole blood to use and also the optimal volume of other reagents such as lysing solutions that need to be used. The observation that the antibody volumes used did not have any major effect on the proportion of TNF-a producing T cells was used as a basis for using the appropriate blood-to-antibody ratio in subsequent experiments and studies.
Although the FACS Lysing solution is mainly used for lysing the RBCs in the whole blood cells, it was surprising to observe that the volume of this solution had a major bearing on the proportion of cytokine-producing monocytes. This study established that the highest percentage of TNF-α producing monocytes was obtained when 1 X FACS Lysing solution was used at a volume 40 times that of the whole blood sample. However, the amount of FACS Lysing solution did not affect the percentage of TNF-α producing T cells. The BD recommendation is to use 2 ml of the 1 X FACS Lysing solution and 500 μl of 1 X FACS permeabilising solution when 50 μl of activated cells are used (Becton Dickinson, 1999). In this study using 1 X FACS lysing solution in any lower ratio than 50 μl whole blood to 2 ml of 1 X FACS lysing solution resulted in lower percentages of TNF-α and IL-6 producing monocytes. Since lower volumes of 1 X FACS lysing solution had sufficed for conventional immunophenotyping with surface marker labelling, it is possible that this high volume of 1 X FACS lysing solution is necessary to fix intracellular cytokines within monocytes thereby preventing them diffusing out of these cells following permeabilisation.
The results of the study also showed that the type of tubes used during stimulation does not make much difference in the proportion of cytokine-producing cells. This might be because all three were made from polypropylene, but of different sizes and from different suppliers. Very few investigators have studied this aspect of cytokine stimulation. Various groups have used polyvinyl chloride (PVC) tubes Hodge et al. (2005), capped polystyrene round bottom tubes (Godoy-Ramirez et al., 2004; Sewell et al., 1997), and Falcon-type 2063 non-stick polypropylene round-bottom tubes (Nomura et al., 2000; Baran et al., 2001; Gauduin, 2006). None of these groups have investigated the effect of different tubes on cytokine stimulation although cells, especially monocytes, have been observed to adhere to the sides of polystyrene tubes (Hodge et al., 2005).
The findings of this study also showed that shaking the tubes on a rocker during stimulation did not enhance cytokine production. Some investigators (Chaka et al., 1997) explored the influence of shaking on the kinetics of TNF-α release induced by Cryptococcus neoformans. They found that shaking resulted in a very rapid release of TNF-α and IFN-γ which was then followed by a fast decrease in the levels of these cytokines for both C. neoformans-stimulated and LPS-stimulated PBMCs. The reduction in levels of the cytokines was attributed to them being broken down to fragments that could not be detected. Chaka et al. (1997) also found that when tubes were left stationary the kinetics of TNF-α release appeared to be protracted, with detectable levels of TNF-α observed after 3 h of stimulation and levels still increasing even after 18 h of stimulation.
The study also found that using an incubator supplied with 5% CO2 during stimulation does not result in the release of higher percentage of cytokines compared with a non-CO2 incubator. In an attempt to determine the device that generated the most consistent values across two time points from the same donor, some researchers investigated the effect of performing stimulations in a Dubnoff water bath set at 37 °C, a humidified 5% CO2 incubator and a dry heat incubator on the frequencies of TNF-α production (Ray et al., 2006). The investigators found that incubation in a water bath resulted in higher TNF-α production compared to when humidified CO2 incubator or the dry heat incubator were used (Ray et al., 2006).
The finding that the use of BFA results in higher detection of TNF-α compared to Monensin is consistent with the findings of other studies (Nylander et al., 1999; Schuerwegh et al., 2001; Vicetti Miguel et al., 2012). In one study BFA, and not Monensin, was found to be capable of completely blocking extracellular CD69 expression after in vitro stimulation with PMA-IO (Nylander et al., 1999). Other investigators also found that BFA was a more potent, effective, and less toxic inhibitor of cytokine secretion than Monensin (Schuerwegh et al., 2001).
Working with human peripheral blood, some investigators showed that BFA was superior to a combination of BFA and Monensin as a secretion-blocking agent (Bueno et al., 2001). Their results showed BFA being associated with a higher percentage of cytokine-positive cells and greater amounts of detectable cytokines per cell compared to BFA in combination with Monensin. Monensin is an inhibitor of trans-Golgi function, whereas BFA inhibits protein transport between the endoplasmic reticulum (ER) and the Golgi (Nylander et al., 1999). BFA mode of action seems to make it a better Golgi blocker compared to Monensin.
One variable that has a greater bearing on the amount of cytokines produced in vitro is the incubation period after the addition of stimulus. Some studies (Suni et al., 1998; Scheibenbogen et al., 2005; Nomura et al., 2000) have shown that a-six hour incubation with the addition of BFA for the last 4 h provided substantial levels of cytokine expression. Results of this study show that 4 h was the optimal stimulation time for production of TNF-α and IL-6 in monocytes (using LPS as stimulant) and TNF-α and IL-2 in T cells (using PMA + IO as stimulant). However, this study found that 6 h was the optimal stimulation time for the production of IFN-γ in T cells.
One study found that more diluted cells could be stimulated for a longer period without the integrity of the cells being affected. Activation with PMA + IO resulted in an increased frequency of CD4+IFN-γ+ and CD8+IFN-γ+ over time in cultures with whole blood diluted 1/5 or 1/10 in contrast to blood cultured at lower dilutions like 1/1 or ½ with the highest frequency observed in samples cultured for 72 h Godoy-Ramirez et al. (2004). We found that the main problem with 72-h, or longer, stimulations is that cell integrity was affected and this makes gating for lymphocytes or T cells during data analysis very difficult.
This study had several limitations the main one of which is the sample size. The study only recruited five healthy adults who consented to provide a blood sample for various analyses. Despite that limitation, all experiments were still conducted in triplicates. Secondly, although the use of 96 wells is now commonly used for stimulation of blood samples for ICS in order to optimize the use of small volumes of blood, we were unable to include this means of stimulation in our study. Thirdly, the use of a combination of PMA and Ionomycin could potentially induce the production of a much broader spectrum of cytokines by T cells than what could normally be observed under in vivo stimulation of T cells by antigens via antigen presenting cells (APCs) in real life (Pala et al., 2000b).
Although in this study some stimulation procedures were done using crude malarial schizont lysate, the levels of cytokine-producing cells were far lower than what was detected when PMA + IO and LPS were used. Ideally it would have been more informative if the proportions of cells reported after PM + IO and LPS also included proportions of cytokine producing cells resulting from stimulation with antigens obtained from known microbes. Lastly although negligible proportions of IL-10, IL-4 and TGF-β producing cells were observed in this study, appropriately conducted and longer time course experiments might be worth considering in order to establish the optimal stimulation duration for whole blood samples at which significant levels of any of these cytokines can be detected.
5. Conclusion
The results of these sets of experiments could be useful for other researchers as they optimize stimulating and staining procedures for intracellular cytokine staining work just like our group did (Mandala et al., 2016). They provide a starting point for the optimal stimulation conditions for T cells and monocytes and on which cytokines to study as well as the optimal labelling conditions to use.
Grant information
This was funded by a grant from the Bill and Melinda Gates Foundation (BMGF) and an institution grant from the Wellcome Trust to MLW.
Authors’ contribution
WLM and HL conceived the various hypotheses and designed the experiments. WLM, HL and VH performed the experiments. WLM, AM and VH were involved in data analysis. WLM, HL, VH, MS and AM wrote, reviewed and edited the manuscript. All authors read and approved the final version of the manuscript.
Author summary
Intracellular Cytokine Staining (ICS) enables us to measure antigen-specific cell responses at the single cell level and this allows us to study disease pathogenesis and even evaluate vaccine efficacy.
We performed a number of experiments with the aim of establishing the ideal conditions for stimulating in vitro cytokine production by lymphocytes and monocytes in whole blood samples and the optimal staining conditions for various cytokines and cell types.
We found that the use of PMA plus Ionomycin produced highest cytokine-producing T cells whereas LPS was a better stimulant for cytokine producing monocytes. Stimulation of whole blood for 5 h was optimal for cytokine detection in T cells whereas 4 h was optimal for monocytes. BFA was found to be a better Golgi blocker than Monensin and that the use of 15 ml Falcon-type polypropylene tubes while stationary resulted in the detection of the highest proportion of cytokine-producing cells. T cells were found to be producers of mainly TNF-α, IFN-γ and IL-2 whereas Monocytes were mainly producing TNF-α and IL-6. Anti-CD3-PerCP (used at a ratio of 1:25) and anti-CD14-APC (used at a ratio of 1:50) and anti-cytokine-PE (used at a ratio of 1:12.5) resulted in the best results. The highest cytokine production monocytes were detected when FACS Lysing solution was used at 40 times the volume of whole blood compared to the other volumes.
CRediT authorship contribution statement
Wilson Mandala: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. Visopo Harawa: Conceptualization, Data curation, Investigation, Writing – original draft, Writing – review & editing. Alinane Munyenyembe: Data curation, Formal analysis, Investigation, Software, Writing – original draft, Writing – review & editing. Monica Soko: Data curation, Investigation, Software, Writing – original draft, Writing – review & editing. Herbert Longwe: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
The Authors would like to acknowledge the following people for their contribution to this work in one way or another; Esther Gondwe, Calman MacLennan, and Chisomo Msefula. Special thanks go to the study participants.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.crimmu.2021.10.002.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- Baran J., Kowalczyk D., Ozog M., Zembala M. Three-color flow cytometry detection of intracellular cytokines in peripheral blood mononuclear cells: comparative analysis of phorbol myristate acetate-ionomycin and phytohemagglutinin stimulation. Clin. Diagn. Lab. Immunol. 2001;8:303–313. doi: 10.1128/CDLI.8.2.303-313.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boahen O., Owusu-Agyei S., Febir L.G., Tawiah C., Tawiah T., Afari S., et al. Community perception and beliefs about blood draw for clinical research in Ghana. Trans. R. Soc. Trop. Med. Hyg. 2013;107(4):261–265. doi: 10.1093/trstmh/trt012. [DOI] [PubMed] [Google Scholar]
- Bueno C., Almeida J., Alguero M.C., Sanchez M.L., Vaquero J.M., Laso F.J., San Miguel J.F., Escribano L., Orfao A. Flow cytometric analysis of cytokine production by normal human peripheral blood dendritic cells and monocytes: comparative analysis of different stimuli, secretion-blocking agents and incubation periods. Cytometry. 2001;46:33–40. [PubMed] [Google Scholar]
- Caraher E.M., Parenteau M., Gruber H., Scott F.W. Flow cytometric analysis of intracellular IFN-gamma, IL-4 and IL-10 in CD3(+)4(+) T-cells from rat spleen. J Immunol Methods. 20. 2000;244(1–2):29–40. doi: 10.1016/s0022-1759(00)00249-0. PMID: 11033016. [DOI] [PubMed] [Google Scholar]
- Chaka W., Verheul A.F., Hoepelman A.I. Influence of different conditions on kinetics of tumor necrosis factor alpha release by peripheral blood mononuclear cells after stimulation with Cryptococcus neoformans: a possible explanation for different results. Clin. Diagn. Lab. Immunol. 1997;4:792–794. doi: 10.1128/cdli.4.6.792-794.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson Becton. 1999. Intracellular Cytokine Staining for Flow Cytometric Analysis. [Google Scholar]
- Gauduin M.C. Intracellular cytokine staining for the characterization and quantitation of antigen-specific T lymphocyte responses. Methods. 2006;38:263–273. doi: 10.1016/j.ymeth.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Gauduin M.C., Kaur A., Ahmad S., Yilma T., Lifson J.D., Johnson R.P. Optimization of intracellular cytokine staining for the quantitation of antigen-specific CD4+ T cell responses in rhesus macaques. J. Immunol. Methods. 2004;288:61–79. doi: 10.1016/j.jim.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Germann A., Oh Y.J., Schmidt T., Schon U., Zimmermann H., von Briesen H. Temperature fluctuations during deep temperature cryopreservation reduce PBMC recovery, viability and T-cell function. Cryobiology. 2013;67(2):193–200. doi: 10.1016/j.cryobiol.2013.06.012. [DOI] [PubMed] [Google Scholar]
- Godoy-Ramirez K., Franck K., Mahdavifar S., Andersson L., Gaines H. Optimum culture conditions for specific and nonspecific activation of whole blood and PBMC for intracellular cytokine assessment by flow cytometry. J. Immunol. Methods. 2004;292:1–15. doi: 10.1016/j.jim.2004.04.028. [DOI] [PubMed] [Google Scholar]
- Hodge G., Markus C., Nairn J., Hodge S. Effect of blood storage conditions on leucocyte intracellular cytokine production. Cytokine. 2005;32:7–11. doi: 10.1016/j.cyto.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Jung T., Schauer U., Heusser C., Neumann C., Rieger C. Detection of intracellular cytokines by flow cytometry. J. Immunol. Methods. 1993;159:197–207. doi: 10.1016/0022-1759(93)90158-4. [DOI] [PubMed] [Google Scholar]
- Maecker H.T. Cytokine flow cytometry. Methods Mol. Biol. 2004;263:95–108. doi: 10.1385/1-59259-773-4:095. [DOI] [PubMed] [Google Scholar]
- Maecker H.T., McCoy J.P., Jr., Amos M., Elliott J., Gaigalas A., Wang L., et al. A model for harmonizing flow cytometry in clinical trials. Nat. Immunol. 2010;11(11):975–978. doi: 10.1038/ni1110-975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandala W.L., Msefula C.L., Gondwe E.N., Drayson M.T., Molyneux M.E., MacLennan C.A. Monocyte activation and cytokine production in Malawian children presenting with P. falciparum malaria. Parasite Immunol. 2016;38(5):317–325. doi: 10.1111/pim.12319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura L.E., Walker J.M., Maecker H.T. Optimization of whole blood antigen-specific cytokine assays for CD4+ T cells. Cytometry 2000. 2000;40:60–68. doi: 10.1002/(sici)1097-0320(20000501)40:1<60::aid-cyto8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Nylander S., Kalies I. Brefeldin A, but not monensin, completely blocks CD69 expression on mouse lymphocytes: efficacy of inhibitors of protein secretion in protocols for intracellular cytokine staining by flow cytometry. J. Immunol. Methods. 1999;224:69–76. doi: 10.1016/s0022-1759(99)00010-1. [DOI] [PubMed] [Google Scholar]
- Pala P., Verhoef A., Lamb J.R., Openshaw P.J. Single cell analysis of cytokine expression kinetics by human CD4+ T-cell clones during activation or tolerance induction. Immunology. 2000;100:209–216. doi: 10.1046/j.1365-2567.2000.00036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pala P., Hussell T., Openshaw P.J. Flow cytometric measurement of intracellular cytokines. J. Immunol. Methods. 2000;21(1–2):107–124. doi: 10.1016/s0022-1759(00)00230-1. 243, PMID: 10986410. [DOI] [PubMed] [Google Scholar]
- Peplow C., Assfalg R., Beyerlein A., Hasford J., Bonifacio E., Ziegler A.G. Blood draws up to 3% of blood volume in clinical trials are safe in children. Acta Paediatr. 2019;108(5):940–944. doi: 10.1111/apa.14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray C.A., Dumaual C., Willey M., Fill J., O'Brien P.J., Gourley I., Devanarayan V., Konrad R.J. Optimization of analytical and pre-analytical variables associated with an ex vivo cytokine secretion assay. J. Pharmaceut. Biomed. Anal. 2006;41:189–195. doi: 10.1016/j.jpba.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Scheibenbogen C., Letsch A., Asemissen A.M., Schmittal A., Thiel E., Keilholz U. In: Analyzing T Cell Responses. Nagorsen D., Marincola F., editors. Springer; Dordrecht: 2005. Intracellular cytokine staining cytokine flow cytometry for characterization of tumor specific T cell response. [DOI] [Google Scholar]
- Schuerwegh A.J., Stevens W.J., Bridts C.H., De Clerck L.S. Evaluation of monensin and brefeldin A for flow cytometric determination of interleukin-1 beta, interleukin-6, and tumor necrosis factor-alpha in monocytes. Cytometry. 2001. 2001;46:172–176. doi: 10.1002/cyto.1102. [DOI] [PubMed] [Google Scholar]
- Sewell W.A., North M.E., Webster A.D., Farrant J. Determination of intracellular cytokines by flow-cytometry following whole-blood culture. J. Immunol. Methods. 1997;209:67–74. doi: 10.1016/s0022-1759(97)00150-6. [DOI] [PubMed] [Google Scholar]
- Silva M.H., Lepzien R., Ols S., Dahlberg B., Grunewald J., Loré K., et al. Stabilization of blood for long-term storage can affect antibody-based recognition of cell surface markers. J. Immunol. Methods. 2020;481–482:112792. doi: 10.1016/j.jim.2020.112792. [DOI] [PubMed] [Google Scholar]
- Suni M.A., Picker L.J., Maino V.C. Detection of antigen-specific T cell cytokine expression in whole blood by flow cytometry. J. Immunol. Methods. 1998;212:89–98. doi: 10.1016/s0022-1759(98)00004-0. [DOI] [PubMed] [Google Scholar]
- Vicetti Miguel R.D., Maryak S.A., Cherpes T.L. Brefeldin A, but not monensin, enables flow cytometric detection of interleukin-4 within peripheral T cells responding to ex vivo stimulation with Chlamydia trachomatis. J. Immunol. Methods. 2012;384(1–2):191–195. doi: 10.1016/j.jim.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.