

Abstract
TDP-43 is a predominantly nuclear DNA/RNA binding protein that is often mislocalized into insoluble cytoplasmic inclusions in post-mortem patient tissue in a variety of neurodegenerative disorders, most notably, Amyotrophic Lateral Sclerosis (ALS), a fatal and progressive neuromuscular disorder. The underlying causes of TDP-43 proteinopathies remain unclear, but recent studies indicate the formation of these protein assemblies is driven by aberrant phase transitions of RNA deficient TDP-43. Technical limitations have prevented our ability to understand how TDP-43 proteinopathy relates to disease pathogenesis. Current animal models of TDP-43 proteinopathy often rely on overexpression of wild-type TDP-43 to non-physiological levels that may initiate neurotoxicity through nuclear gain of function mechanisms, or by the expression of disease-causing mutations found in only a fraction of ALS patients. New technologies allowing for light-responsive control of subcellular protein crowding provide a promising approach to drive intracellular protein aggregation, as we have previously demonstrated in vitro. Here we present a model for the optogenetic induction of TDP-43 aggregation in Drosophila that recapitulates key biochemical features seen in patient pathology, most notably light-inducible persistent insoluble species and progressive motor dysfunction. These data describe a photokinetic in vivo model that could be as a future platform to identify novel genetic and pharmacological modifiers of diseases associated with TDP-43 neuropathology.
Keywords: TDP-43, ALS/FTD, LATE, RNA binding proteins, optoTDP43, Neurodegeneration, optoTDP43
Introduction
Amyotrophic lateral sclerosis (ALS) and Frontotemporal dementia (FTD) are fatal neurodegenerative diseases that lead to a loss of neurons of the spinal cord and motor cortex, or frontal and temporal lobes, respectively. Both are progressive disorders, and no therapies currently exist that meaningfully halt the death of affected neurons. In 2006 it was reported that transactive response element DNA-binding protein 43 kDa (TDP-43) mislocalizes and aggregates in the cytoplasm of affected tissues in ALS/FTD (1,2). It is now estimated that TDP-43 proteinopathy is present in ~97% of sporadic ALS cases and up to 45% of FTD cases (3). Furthermore, TDP-43 proteinopathy, along with amyloid beta deposits and tau tangles, was recently defined as a neuropathological hallmark of a late-onset dementia known as limbic-predominant age-related TDP-43 encephalopathy (LATE) (4). Thus, in vivo models of TDP-43 pathology will allow us to study the consequence of this neuropathology and associated disease mechanisms across a variety of neurodegenerative disorders.
TDP-43 is a tightly regulated and ubiquitously expressed DNA/RNA binding protein. It has two RNA recognition motif (RRM) domains and a C-terminal glycine-rich low complexity, or prion-like, domain (LCD, PrLD) (5). Nuclear TDP-43 functions to regulate alternative splicing of GU-rich intronic RNA sequences and suppresses cryptic exons from inclusion into messenger RNA (6). It also shuttles to the cytoplasm to regulate RNA trafficking (7) and RNA stability both independently (8) and as a part of stress granules (9). In TDP-43 proteinopathies, the protein is often found mislocalized from the nucleus as a diffuse cytoplasmic signal or as detergent-insoluble inclusions that are hyperphosphorylated and ubiquitinated (10). Importantly, the presence of this pathology correlates with regional neurodegeneration observed in affected tissues in ALS/FTD (11). Further implicating TDP-43 proteinopathy in disease pathogenesis, a variety of familial missense mutations causative for ALS and FTD have been identified within the TARDBP gene (12). Many of these disease-causing mutations are crucially located in TDP-43’s LCD/PrLD and have been shown to enhance its propensity to aggregate (13). Some mutations within the TDP-43 LCD/PrLD were recently shown to enhance the aberrant phase transition of the domain leading to the formation of more prolonged, irreversible assemblies upon repeated homotypic interactions (14).
In patients with TDP-43 neuropathology, affected cells can exhibit nuclear clearance of TDP-43 with or without cytoplasmic inclusions, leading to speculation that there are both loss- and gain-of-function mechanisms contributing to neurodegeneration in TDP-43 proteinopathies (15). Consistent with this, cytoplasmic TDP-43 inclusions and cytoplasmic TDP-43 droplets do appear to sequester nuclear TDP-43 indicating the possibility of a feedforward mechanism wherein the mislocalized cytoplasmic TDP-43 promotes loss of nuclear function phenotypes (14,16). Supportive of a loss of TDP-43 function mechanism of toxicity, conditional TARDBP knockout in postnatal mice results in rapid weight loss and death (17). Homozygous embryonic disruption of the TARDBP gene renders mice non-viable, while heterozygous TARDBP loss results in motor dysfunction (18). Furthermore, two recent reports showed that loss of nuclear TDP-43 impairs its ability to block premature polyadenylation sites, resulting in the generation of truncated RNAs for nonfunctional stathmin-2, a microtubule regulatory protein (19,20). These truncated RNAs are present in sporadic ALS patient post mortem motor neurons devoid of nuclear TDP-43 (19).
TDP-43 overexpression has also proven toxic in a variety of in vitro and in vivo models with and without cytoplasmic TDP-43 inclusions (15,21–23). However, it remains unclear whether the motor phenotypes and neurodegeneration observed in these models is causally linked to cytoplasmic TDP-43 aggregation. While many groups observed a strong correlation between the occurrence of TDP-43 pathology and motor neuron degeneration following overexpression (24), there are similar reports of mutant and wild-type TDP-43 overexpression producing ALS/FTD-like phenotypes absent of detectable TDP-43 proteinopathy (25,26). Thus, the precise link between TDP-43 aggregation and toxicity has remained intractable as the tools that we have been limited to largely rely upon manipulation of TDP-43 expression levels.
To address these technical limitations, we developed an optogenetic model of TDP-43 proteinopathy (optoTDP43) through the expression of a chimeric light-responsive and oligomerizing Cryptochrome2 variant (Cry2olig) and human TDP-43 (optoTDP43). These light inducible technologies have been used in vitro to probe biophysical properties of condensates of intrinsically disordered proteins (27) and the role of stress granule resident proteins potentially promoting cytoplasmic protein inclusions (28). We previously showed that blue light is sufficient to induce nuclear clearance and cause neurotoxic cytoplasmic TDP-43 aggregation in human cells (14). Here, we generated an optoTDP43 Drosophila model and show that light-induced aggregation of optoTDP43 recapitulates key biochemical and behavioral features of TDP-43 proteinopathy observed in human patients, namely that nucleation of optoTDP43 aggregates leads to persistent high molecular weight insoluble species and progressive motor dysfunction in vivo.
Results
optoTDP43 is predominantly expressed in the nucleus but relocates to the cytoplasm and forms inclusions in response to blue light illumination
To study the effects of TDP43 aggregation in vivo, we employed a construct previously validated in vitro to seed physiologically relevant TDP-43 inclusions in response to blue light illumination (14). The optoTDP43 chimeric protein consists of an N-terminal Photolyase-Homologous Region (PHR) of the Cryptochrome 2 protein from Arabidopsis thaliana photoreceptor that reversibly oligomerizes following blue light illumination (29) fused to the full-length human TDP-43 protein and a C-terminal mCherry reporter (Fig 1A, top). We cloned this construct into a pUAST attB expression vector and inserted it in a site-specific manner via pHiC31 integrase into the Drosophila genome to produce a UAS-optoTDP43 fly. We also created UAS-TDP-43-mCh flies using the same method and integrated at the same site (Fig 1A, bottom) to directly compare optoTDP43 with a traditional TDP-43 overexpression model.
Fig. 1. An optogenetic model of TDP-43 leads to light-inducible formation of cytoplasmic aggregates in larval motor neurons.
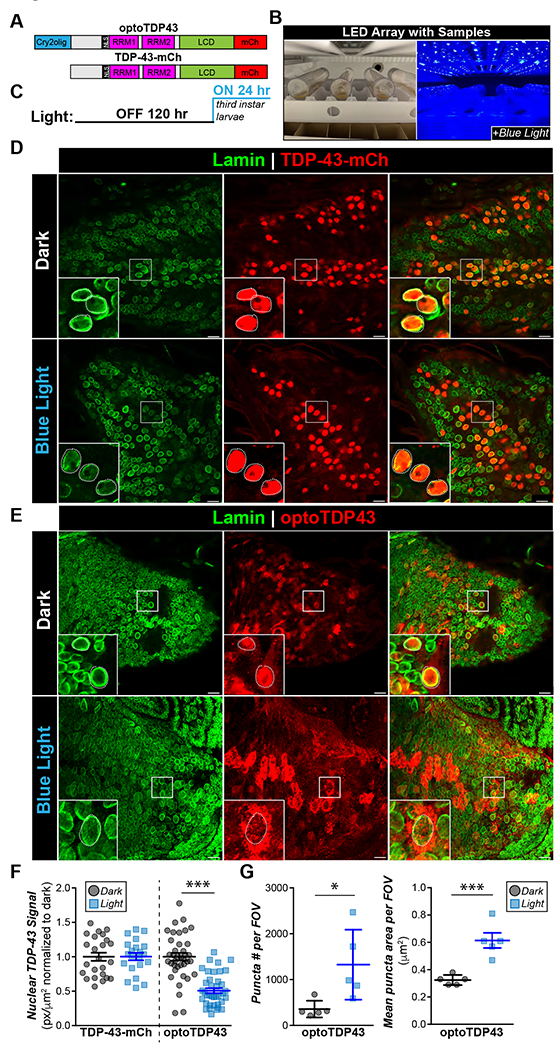
A. Diagram of TDP-43-mCh and optoTDP43 constructs B. Experimental paradigm. Vials were paced sideways 10 cm below a 14-watt 465 nm LED. C. Light paradigm. Parents were crossed in the dark in normal fly vials for 120 hours and then larval early third instar larvae were collected and transferred to new vials and placed under blue light as shown in B. D. Representative images of the ventral nerve cord (VNC) of ok371-GAL4;UAS-TDP-43-mCh larvae motor neurons with Lamin staining to denote the nuclear membrane with and without 24 hours of blue light illumination E. Representative images of the VNC of ok371-GAL4;UAS-optoTDP43 larval motor neurons with and without 24 hours of blue light illumination. F. 24 hours of blue light exposure leads to decreased nuclear TDP-43 signal in optoTDP43 expressing larval motor neurons. Quantification of normalized nuclear TDP-43 staining in larval motor neurons of ok371-GAL4;UAS-TDP-43-mCh and ok371-GAL4;UAS-optoTDP43. n = 25-40 cells across 5 different larvae per light condition ***p<.001. error bars +/− SEM. Data were assumed normal and analyzed by one-tailed student’s t-test. G. Blue light exposure leads to increased number of puncta per field and average puncta size per field in ok371-GAL4;UAS-optoTDP43 motor neurons. N = 5 fields per condition ***p<.001, *p<.05. Error bars +/− S.D. for number of puncta and SEM. for mean puncta size. Data were assumed normal and analyzed by one-tailed student’s t-test. Scale bar = 10 um.
UAS-optoTDP43 or UAS-TDP-43-mCh homozygous females were crossed with ok371-GAL4 males (30) to assess the expression pattern of optoTDP43 and TDP-43-mCh specifically in motor neurons of the larval ventral nerve cord (VNC). Taking advantage of their translucency, third instar larvae were placed in a thin layer of dilute standard cornmeal medium flat under a 14 watt 465 nm blue LED (Fig. 1B) or in darkness for 24 hours prior to immunofluorescent analysis of optoTDP43 localization (Fig. 1C). Nuclear TDP-43 localization was measured in a z-stack of confocal images. Nuclear signal was indicated as a region of interest (ROI) from which we calculated mean signal intensity.
In our traditional overexpression model, TDP-43-mCh protein was overwhelmingly confined to the nucleus regardless of light exposure (Fig. 1D, E), indicating that blue light alone is not sufficient to mislocalize the protein to the cytoplasm in vivo. Interestingly, TDP-43-mCh flies demonstrated 4 times higher protein levels in eye neurons (Fig. S1A, B) and nearly 10 times greater protein levels in larval motor neurons compared to optoTDP43 protein (Fig. S1C, D) despite being integrated at the same site and exhibiting similar RNA expression (Fig. S1E). We failed to observe cytoplasmic inclusions of the TDP-43-mCh protein both in the light and dark conditions regardless of this high expression level in larval motor neurons (Fig. 1D). In contrast, optoTDP43 was largely localized to motor neuron nuclei in animals kept in darkness (Fig. 1E, F), but mislocalized to the cytoplasm (Fig. 1E, F) and formed large cytoplasmic puncta (Fig. 1G) when animals were illuminated for 24 hours.
optoTDP43 aggregates recapitulate key biochemical markers of TDP-43 pathology in ALS/FTD
We next investigated whether light-nucleated cytoplasmic optoTDP43 aggregates observed in larval motor neurons resemble pathologically relevant inclusions found in ALS/FTD patient tissue. As above, third instar optoTDP43 larvae were exposed to 24 hours of blue light or kept in darkness (Fig. 1C). Following these respective light treatments, we immunostained these tissues for two immunohistochemical hallmarks associated with TDP-43 inclusions in ALS/FTD post-mortem tissues: C-terminal (S409/410) phosphorylation and ubiquitination (31). While larvae kept in darkness exhibited predominantly nuclear optoTDP43 localization absent of immunoreactivity, illumination of optoTDP43 larvae for 24 hours led to the formation of cytoplasmic optoTDP43 inclusions that co-stained with phospho-TDP-43 (S409/410) (Fig. 2A) and ubiquitin (Fig. 2B). Interestingly, cytoplasmic optoTDP43 inclusions colocalized with Fmr1 (Fig. 2C), a marker for cytoplasmic RNA granules in Drosophila (32).
Fig. 2. optoTDP43 cytoplasmic aggregates colocalize with key biochemical markers of TDP-43 proteinopathy.
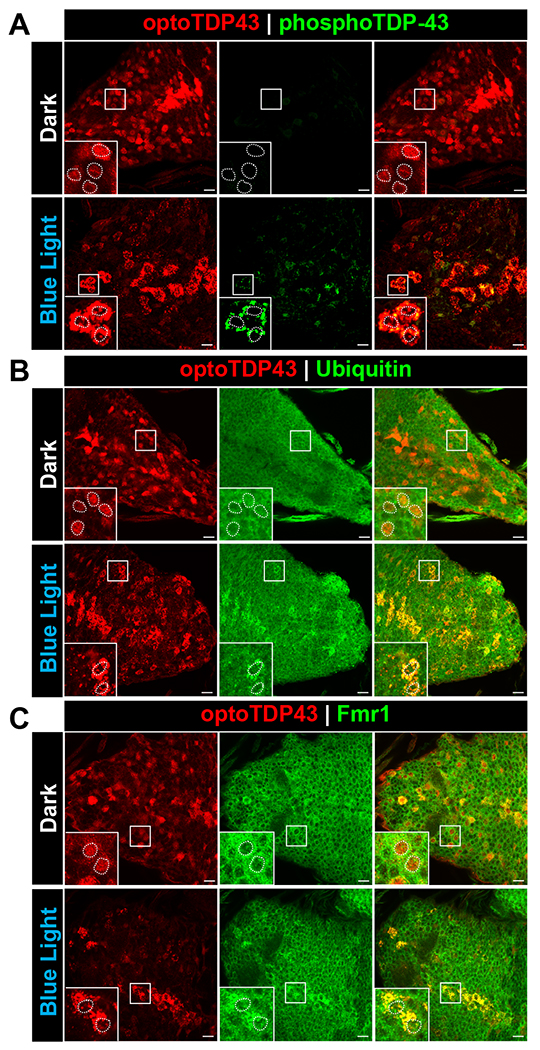
ok371-GAL4;UAS-optoTDP43 larvae were raised at 25 °C and kept under blue light or kept in the dark for 24 hours prior to processing for processing for immunofluorescence. A. Representative images of hyperphosphorylated cytoplasmic optoTDP43 aggregates in ok371-GAL4;UAS-optoTDP43 larvae motor neurons following 24 hours of blue light illumination. These assemblies were not observed in larvae kept in darkness. B. Representative images of ubiquitinated cytoplasmic optoTDP43 aggregates in ok371-GAL4;UAS-optoTDP43 larvae motor neurons following 24 hours of blue light illumination. These assemblies were not observed in larvae kept in darkness. C. Cytoplasmic optoTDP43 colocalize with Fmr1, a marker of Drosophila RNA granules, in ok371-GAL4;UAS-optoTDP43 larvae motor neurons following 24 hours of blue light illumination. n = 5 larvae per staining. Scale bar = 10 um.
optoTDP43 aggregates are detergent insoluble and persist with age
Another feature of pathological TDP-43 inclusions is detergent insolubility. To test if optoTDP43 is detergent insoluble, we expressed optoTDP43 in neurons of the adult Drosophila eye under control of the gmr-GAL4 driver. We used these tissues to provide a longer timespan to test light treatment paradigms while still allowing passage of blue light since there is a limited amount of time to test this in larval motor neurons before pupation. Crosses were maintained in darkness until eclosion and then adult Drosophila were illuminated with blue light or kept in darkness for varying lengths of time prior to sample processing. We observed that increasing the length of blue-light exposure from 48 hours up to 192 hours led to dose-dependent increases in the quantity of detergent-insoluble species (Fig. 3A–B). To test whether these light-induced insoluble species persist following light removal, thus deactivating the oligomerization activity of the Cry2 photoreceptor, adult optoTDP43 flies were next kept under blue-light for 72 hours, then moved to darkness for 72 hours or 144 hours prior to sample processing. Interestingly, insoluble optoTDP43 protein species were present in flies kept in darkness for 72 hours and 144 hours after illumination and the relative insoluble optoTDP43 protein signal increased with time in darkness (Fig. 3C). Furthermore, there was a shift of insoluble species from monomeric bands and cleavage products into higher molecular weight smears following removal of light (Fig. 3C, boxes). This suggests that, once formed, insoluble species are resistant to cellular clearance and promote the formation of additional higher molecular weight optoTDP43 species in the absence of light-induced oligomerization. This shift to an increased insoluble optoTDP43 ratio is not due to increased transgene expression since mCherry levels remains constant throughout the time course (Fig. S2A–B). Notably, expression of optoTDP43 in the absence of blue light results in a mildly degenerative eye phenotype, but less than Drosophila expressing TDP-43-mCh (Fig. S3). This is likely explained by the large relative differences in protein expression levels between TDP-43-mCh and optoTDP43 (Fig. S1A–B). Interestingly, despite the formation of insoluble optoTDP43 species, we did not observe any obvious external eye toxicity after seven days of chronic light exposure (Fig. S4A–B).
Fig. 3. optoTDP43 forms blue light dependent insoluble species that persist following removal of blue light.
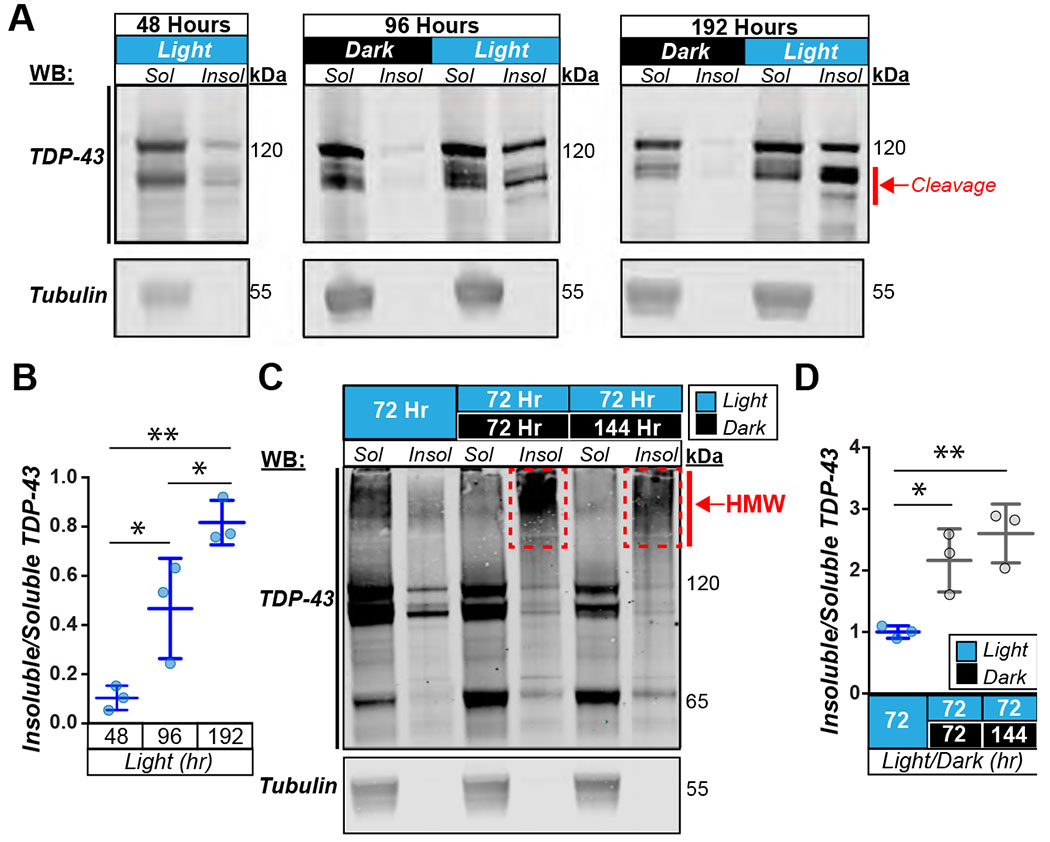
A. Representative blots of soluble/insoluble (S/I) fractionation of gmr-GAL4;UAS-optoTDP43 adult heads staining for TDP-43 and tubulin as loading control with and without light stimulation at 48 hours, 96 hours, and 192 hours of light. B. Quantification of dose dependent formation of insoluble species from representative blots shown in panel A. n=3 blots per condition **p<.01, *p<.05. error bars +/− SD. 1-way ANOVA with test for multiple comparisons between each condition. C. Representative western blots of soluble/insoluble fractionation of gmr-GAL4;UAS-optoTDP43 adult heads staining for TDP-43 after 72 hours of light stimulation, 72 hours of light followed by 72 hours of dark, and 72 hours followed by 144 hours of dark. D. Quantification of relative amounts of insoluble/soluble optoTDP43 after 72 hours of light stimulation and then 72 and 144 hours after removal of blue light. n = 3 blots per condition **p<.01, *p<.05 error bars +/− SD. 1-way ANOVA with Tukey’s test for multiple comparisons.
Nucleation of optoTDP43 inclusions causes larval motor deficits and adult wing phenotypes
We next sought to determine whether blue light-nucleated optoTDP43 inclusion formation in larval motor neurons is sufficient to drive a dysfunctional motor phenotype. We expressed optoTDP43 under control of the motor neuron-specific driver ok371-GAL4 and placed early third instar larvae under blue light for 28 hours prior to assessing their ability to crawl compared to larvae raised in the dark (Fig. 4A). Blue-light exposed optoTDP43 larvae exhibited significantly decreased crawling distance and velocity (Fig. 4B–C) compared to optoTDP43 larvae kept in darkness. This effect was not observed in mCherry expressing larvae (Fig. 4B–C), suggesting a specific effect of optoTDP43 inclusion formation on motor behavior independent of any light-mediated effects. Notably, ~35% of late third instar larvae exposed to blue light into the beginning of pupation for 30 hours exhibited a wrinkled or twisted wing phenotype that was not observed in optoTDP43 flies raised in the dark nor mCherry expressing control flies raised in the dark or with illumination (Fig. 4D–E).
Fig. 4. Blue light induced optoTDP43 aggregation leads to motor dysfunction and a neurotoxic phenotype.
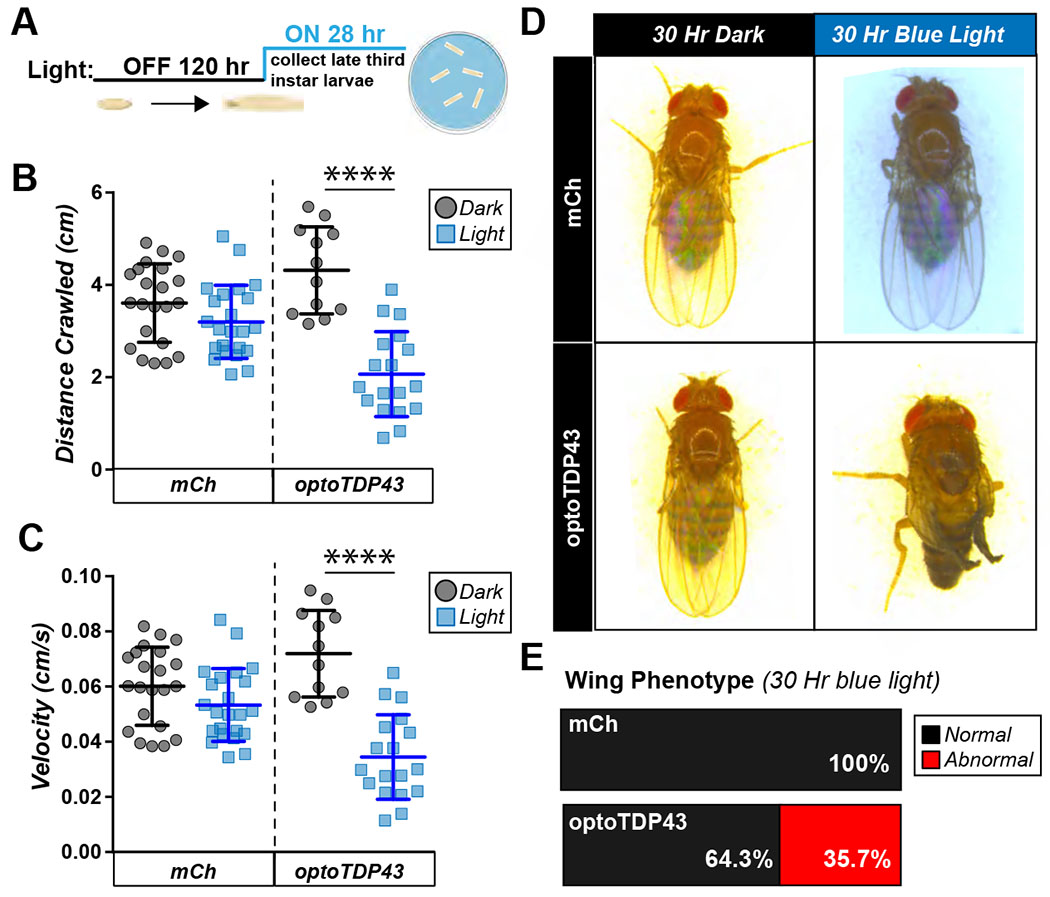
A. Diagram of light protocol for larval crawling assay. B. ok371-GAL4;UAS-optoTDP43 larvae exhibit reduced crawling distance in response to 28 hours of chronic blue light exposure while ok371-GAL4;UAS-mCherry larvae’s crawling is unchanged. N= 20-25 larvae per condition. ****p<.0001, error bars +/− SD. Data were assumed normal and analyzed by a one-way student’s t-test C. ok371-GAL4;UAS-optoTDP43 larvae exhibit reduced crawling velocity in response to 28 hours of chronic blue light exposure while ok371-GAL4;UAS-mCherry larvae’s crawling is unchanged. N= 20-25 larvae per condition. ****p<.0001, error bars +/− SD. Data were assumed normal and analyzed by a one-way student’s t-test. D. Representative images of optoTDP43 flies exhibiting crumpled wing phenotypes in response to blue light exposure. E. Quantification of percentage of flies with abnormal wings, N = 25-30 flies per condition.
We next tested if light induced oligomerization of Cry2 produced motor dysfunction regardless of the presence of TDP-43. Despite not observing any obvious changes in aggregation via IF following 24 hours of blue light stimulation (Fig. S5A) and no chronic light induced external eye toxicity (Fig. S5B) following seven days of chronic blue light stimulation, third instar larvae expressing Cry2-mCh in motor neurons and exposed to 28 hours of blue light exhibited decreased crawling ability compared to Cry2-mCh larvae kept in the dark (Fig. S5C). This suggests that widespread light-induced Cry2-mediated protein oligomerization in general may confer acute cytotoxicity to motor neurons in vivo.
optoTDP43 aggregation initiates progressive motor dysfunction in adult flies
To address whether induction of optoTDP43 inclusions affects adult motor function, we placed late third instar optoTDP43 and mCherry larvae under blue-light for 30 hours and allowed them to initiate pupation under blue light before being removed from light as pupae (Fig. 5A). Once eclosed, we assayed adult flies’ ability to climb using the RING assay (33). OptoTDP43 flies that eclosed following late larval/early pupal light exposure exhibited significantly decreased climbing ability compared to those kept in darkness, while no effect of light exposure alone was observed in mCherry-expressing flies, demonstrating that climbing dysfunction was not an artifact of our light treatment (Fig. 5B). Interestingly, Cry2-mCherry expressing adult flies exhibited climbing deficits following blue light exposure, in keeping with the observation in larvae that blue-light induced Cry2-mediated protein oligomerization confers acute motor neuron toxicity in vivo.
Fig. 5. optoTDP43 motor deficits persist with age.
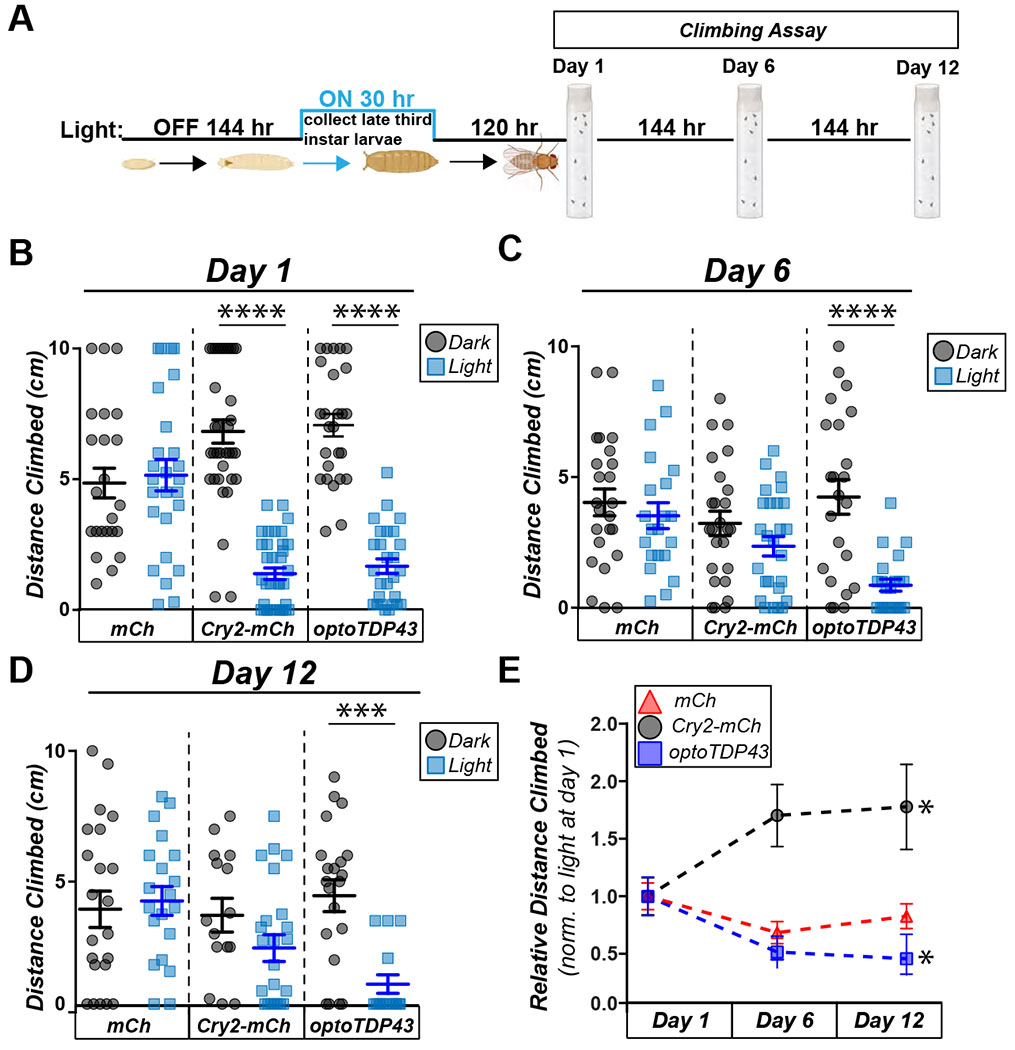
A. Diagram of light protocol for the climbing assay. B. ok371-GAL4;UAS-Cry2-mCherry and ok371-GAL4;UAS-optoTDP43 adults exhibit reduced climbing ability in response 30 hours of chronic blue light exposure while ok371-GAL4;UAS-mCherry adults do not. N =20-30 flies in triplicate per condition. ****p<.0001, error bars +/− SEM. Data was assumed normal and analyzed by a one-way ANOVA with post hoc Tukey’s multiple comparisons within each genotype. C. ok371-GAL4;UAS-Cry2-mCherry flies lose the difference in climbing ability between light and dark exposed flies, while light exposed ok371-GAL4;UAS-optoTDP43 show continued motor deficits compared to those kept in the dark. N =20-30 flies in triplicate per condition. ****p<.0001, error bars +/− SEM. Data were assumed normal and analyzed by a one-way ANOVA with Tukey’s multiple comparisons within each genotype. D. At 12 days post eclosion, ok371-GAL4;UAS-optoTDP43 show continued motor deficits compared to those kept in the dark, whereas both ok371-GAL4;UAS-mCherry and ok371-GAL4;UAS-Cry2-mCherry show no difference. N =20-30 flies in triplicate per condition. ****p<.0001, error bars +/− SEM. Data were assumed normal and analyzed by a one-way ANOVA with Tukey’s multiple comparisons within each genotype. E. Blue light exposed ok371-GAL4;UAS-Cry2-mCherry flies show improved climbing with age. n = 25-30 flies per condition. Blue light exposed ok371-GAL4;UAS-optoTDP43 flies show worsened climbing with age. N = 20-30 flies per condition. *p<.05. error bars +/− S.E.M. Data were assumed normal and analyzed by one-way ANOVA and then day 12 was compared to day 1 with post hoc with Sidak’s post hoc multiple comparisons test.
OptoTDP43 flies exposed to light exhibited progressive climbing dysfunction (Fig. 5C–E) with age, whereas Cry2-mCherry flies exposed to light paradoxically recovered some climbing function with age (Fig. 5C–E, Supplementary videos 1–6). Due to the observation that light-induced insoluble optoTDP43 increases with age (Fig. 3), we hypothesized that the different patterns in toxicity may be related to optoTDP43’s propensity to form persistent insoluble species. To test if Cry2-mCherry formed light-dependent insoluble species, we expressed Cry2-mCherry in adult eye neurons, illuminated the animals for 72 hours, and removed from the light for 72 hours and 144 hours before assessing presence of soluble and insoluble Cry2-mCherry. Cry2-mCherry flies did not form insoluble species in response to blue light nor show an age-related accumulation of further insoluble species (Fig. S5D) following the removal of light as observed with optoTDP43 protein in vivo (Fig. 3). We propose that optoTDP43’s ability to form these persistent insoluble species allows it to exert continued toxicity while the toxicity of Cry2-mCherry is more transient, likely due to its inability to form stable toxic species following the initial light induced oligomerization.
Discussion
The present study characterizes a light-responsive Drosophila model of TDP-43 proteinopathy. Using blue light, we can nucleate optoTDP43 protein into inclusions that recapitulate key features of ALS/FTD neuropathology. This includes nuclear clearance, hyperphosphorylation, ubiquitination, detergent insolubility, and N-terminal cleavage. The resulting insoluble optoTDP43 assemblies form higher molecular weight species that persist for days following the removal of light. Furthermore, the formation of cytoplasmic optoTDP43 inclusions are sufficient to drive motor dysfunction in larval and adult Drosophila.
TDP-43 Drosophila models are frequently employed to study ALS/FTD (34). Traditional overexpression of both human TDP-43 (23,35,36) and Drosophila TBPH (37,38) produces neurodegenerative phenotypes. Genetic deletion or RNAi knockdown of TBPH induces motor dysfunction and neuronal dendritic branching deficits in Drosophila (39,40). Through these various manipulations of TDP-43 and TBPH expression levels, it is clear that there are both gain of function and loss of function routes toward toxicity, and it can be difficult to determine how these toxic processes relate to sporadic disease.
It remains unclear if and how TDP-43 proteinopathy contributes to disease pathogenesis in ALS/FTD, and whether the formation of cytoplasmic protein inclusions is causative, epiphenomenal, or protective in relation to neurodegeneration. Familial mutations in the TARDBP gene are causative for ALS/FTD (41,42) and often promote its aggregation both in vivo and in vitro suggesting that TDP-43 aggregation is intrinsically linked to disease (13). Our data demonstrate that light-nucleated optoTDP43 mislocalization/aggregation produces a gain of function toxicity in larval and adult Drosophila motor neurons. These data are supported by a recent study utilizing a similar light inducible Cry2-based TDP-43 fusion construct (opTDP-43) in zebrafish spinal neurons (43). Using a Tg[SAIGFF213A] x Tg[UAS:opTDP-43z] x Tg[mnr2b-hs:EGFP-TDP-43z] triple transgenic fish lacking the zebrafish TDP-43 ortholog and expressing both eGFP-TDP43 and an opTDP-43 that similarly mislocalizes to the cytoplasm in vivo, they demonstrate motor neuron axon growth defects and myofiber denervation that precedes trapping of nuclear non-optogenetic TDP-43 with blue light illumination (43). This raises the possibility that the optoTDP43 Drosophila system is uniquely reliant on a gain of function cytoplasmic TDP-43 toxicity since it is unclear if TBPH is recruited to optoTDP43 inclusions. A previous study where E. coli derived TDP-43 protein aggregates were transfected into human neuroblastoma (SH-SY5Y) cells showed that cytoplasmic TDP-43 protein aggregates were sufficient to produce ubiquitinated/hyperphosphorylated TDP-43 aggregates that exerted neurotoxic effects all without a significant loss of endogenous nuclear-localized TDP-43 (44). Notably, the light-induced optoTDP43 inclusions shown here colocalize with Fmr1, a marker of RNA transport granules and frequently used as a stress granule marker in Drosophila (45), suggesting optoTDP43 protein recruitment to these structures may confer toxicity, or that optoTDP43 assemblies recruit RNA binding proteins (RBPs) leading to their loss of function.
Unexpectedly, we found that light treatment in Drosophila larvae expressing Cry2-mCherry produced an acute motor phenotype despite showing no significant changes in Cry2-mCherry localization when compared to animals kept in darkness. However, while both adult fly lines initially exhibited motor defects, only the optoTDP43 flies’ motor function worsened with age, whereas the Cry2-mCherry expressing flies recovered. Given our observation that, once nucleated, optoTDP43’s insoluble species persist with age whereas Cry2-mCherry does not, the presence of these persistent insoluble TDP-43 species is likely driving the progressive motor deficits following the removal of light (Fig. 5, Supplemental Videos 1–6). Why this aggregation doesn’t lead to external eye toxicity is unclear, however a recent study demonstrated that trapping overexpressed TBPH in insoluble aggregates via simultaneous overexpression of an aggregation prone artificial construct based on TDP-43’s Q/N-rich C-terminal tail is protective against TBPH overexpression toxicity in the fly eye (38). This suggests possible intrinsic differences between eye neurons and motor neurons in Drosophila in response to TBPH/TDP-43 protein aggregation.
Drosophila model systems have uncovered profound insights into the mechanisms of neurodegenerative disease (46). Overexpression models have provided important insight into TDP-43 mediated toxicity, but are limited by their reliance on expression-induced toxicity. The optoTDP43 model described here allows us to further study the role of TDP-43 in neurodegeneration in vivo by dissecting aggregation-induced toxicity from expression-induced toxicity. These data suggest that light nucleated optoTDP43 cytoplasmic insoluble aggregates are sufficient to drive progressive motor dysfunction. Future studies will explore novel genetic and pharmacological modifiers of TDP-43 mislocalization and aggregation in vivo.
Materials and Methods
Cloning
Previously described constructs (14) were cloned into pUAST attB expression vectors using Gibson assembly method and inserted into chromosome 3 of the Drosophila genome by Bestgene Inc. via the PhiC31 integrase transgenesis system.
Drosophila Stocks
All Drosophila stocks were kept at 18/25 C in light-dark controlled incubators. All experiments involving blue light-sensitive crosses were raised at 25/29 C in vials wrapped in foil and sorted in the dark under red light.
Larval Preparations, IHC and quantification
Third instar larvae were dissected, fixed and stained as previously described (47). Briefly, larvae or adult were dissected in iced-cold phosphate buffered saline (PBS) (Lonza, #17–516F). Dissected larvae were fixed in 4% formaldehyde, washed 3× in PBS, then washed 3× in 0.1% PBST (0.1% Triton X-100 in PBS) and incubated overnight with primary antibodies (mouse anti-lamin, DSHB, 1:200, mouse anti-mCherry 1:100 rabbit anti-TDP-43, Proteintech 10782-2-AP, 1:200; mouse anti-mono and poly ubiquitinylated conjugates, Enzo Life Sciences BML-PW8810, 1:100; rat anti-phospho Ser409/410 TDP-43, EMD Millipore clone 1D3, 1:50) in 0.1% PBST. Then, Larvae were washed 3× in 0.1% PBST (0.1% Triton X-100 in PBS) and incubated for 2 hours in secondary antibodies (anti-rabbit Alexa Flour 568, Invitrogen, # 651727, 1:100; anti-mouse Alexa Flour 647, Invitrogen, # 28181, 1:100; anti-rabbit Alexa Flour 405, Life Technologies, # 157554, 1:100; anti-rabbit Alexa Flour 647, Life Technologies, # 1660844, 1:100; anti-rat Alexa Fluor 488, Invitrogen, #A-11006), and mounted using Fluoroshield (SIGMA, #F6182). Slides were visualized using a Nikon A1 laser-scanning confocal microscope system utilizing 60X oil immersion objectives (CFI Plan Apo Lambda 60X Oil, Nikon).
Adult and larval light induced aggregation protocol
255 ml of water was added to a 50 mL bottle of standard cornmeal medium, boiled in a microwave, and then 2 mL (for larval light exposure) or 13 mL (for adult exposure) of medium was laid down along the length of a standard Drosophila vial. These tubes were then placed sideways 10 cm below a 14 W blue, 465 nm LED (HQRP 12″ light system, Amazon). Larva and adults in the “dark” condition had their vials wrapped in aluminum foil and kept under the same blue light for the amount of time required for each experiment. For multi day light exposure of adult eyes, flies were moved to a new vial every two days.
Soluble/insoluble fractionation and western blotting
12 heads were cut off with a razor blade and immediately stored at −80 C or crushed on dry ice and allowed to incubate in RIPA buffer (0.5% NP40, 10 mM Tris HCl pH 7.8, 10 mM EDTA, 150 mM NaCl, 1X Pierce protease inhibitor cocktail, 2.5 mM Sodium Vanadate). Crushed heads were sonicated for 2 cycles of 3 x 15 s pulses with 5 s of rest between pulses. Samples were then centrifuged at 21,000 x g for 20 min at 4 °C to pellet the insoluble material. Supernatant was removed as the soluble fraction. The remaining pellet was washed with 1 mL of washing buffer (50 mM Tris HCl pH 7.4 150 mM NaCl), vortexed and centrifuged for 5 minutes and supernatant was removed. 48 microliters of 5% SDS resolubilization buffer (50mM Tris HCl pH 6.8, 5% SDS, 10% glycerol) was added to the pellet and was re-sonicated for 2 cycles of three 15 s pulses with 5 s of rest between pulse. Tubes were then vortexed and briefly spun down <1 sec. Supernatant was removed as the insoluble fraction. Lysates were then sonicated and centrifuged to remove debris. Supernatants were boiled at 95 °C in Laemmli Buffer (Boston Bioproducts, #BP-111R) for 5 min and loaded onto a 4–12% NuPage Bis-Tris gel (Novex/Life Technologies). Proteins were transferred using the iBlot2 (Life Technologies, #13120134) onto nitrocellulose (iBlot 2 NC regular Stacks, Invitrogen, #IB23001). Western blots were blocked with milk solution (BLOT-QuickBlocker reagent, EMB Millipore, #WB57–175GM) and incubated in primary antibody (rabbit anti-TDP-43, Proteintech 10782-2-AP,1:1500; mouse anti-mCherry, Novus Biologicals 1C81, 1:1000; mouse anti-tubulin, SIGMA Life Science, 1:10 000) at 4 °C overnight. Blots were washed and incubated in secondary antibody (anti-mouse IRDye 680D, LICOR, 1:10 000; anti-rabbit, DYLight 800, Pierce, 1:10 000, anti-rat, DYLight 800, Pierce, 1:10 000) for 1 h prior to imaging on Licor imager (Odyssey CLx). All western blots were run in triplicate using biological replicates. Protein levels were quantified in Licor Image studio and statistical analysis were performed in GraphPad Prism 6.
External eye imaging
All external eye images were taken at the dates indicated immediately after removal of the fly head at the dates indicated in the respective figures with a Leica M205C light microscope. Fly eye degeneration was quantified as previously described (48).
Adult climbing/wing phenotype assay
Vials of third instar larvae were either placed wrapped in foil or placed under blue light for 30 hours at 25 °C. After 30 hours, these vials were taken out from under the blue light and wrapped in foil and allowed to finish pupating while not exposed to any light. Then, 0-1 day old flies were anesthetized placed into vials, and allowed to acclimatize for 15 min in the new vials, under normal light. Racks of vials containing the flies were knocked three times on the base of the bench and a video camera was used to record the flies climbing up the wall of the vials. The distance climbed in 3 s was quantified and the mean for each group was calculated and analyzed using GraphPad Prism 6 software. The proportion of flies showing non-wildtype wing phenotypes were counted and that proportion was quantified. All genotypes were run in triplicate using biological replicates. Flies were then aged in the dark and reassessed at days 6 and 12 post eclosion. and that proportion was quantified. All genotypes were run in triplicate.
Larval Crawling Assay
Early third instar larvae were collected and placed under blue light or wrapped in foil and placed under blue light for 28 hours at 25 °C. They were then collected, on a blue agar plate, allowed to acclimate there for 1 minute and then recorded crawling for 30 s. The distance crawled over that minute was quantified using Kinovea software.
Statistical Analyses
All statistical analysis was done using GraphPad Prism 6 software using either Wilcoxon sum rank test, student’s t-test or one-way ANOVAs with Tukey’s or Dunnet’s multiple comparisons test as deemed appropriate. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health R01NS105756 to CJD, R21AG064940 and R21NS100055 to CJD and UBP, R01NS081303, R21NS111768, and R21NS101661 to UBP. the LiveLikeLou Center for ALS Research at the University of Pittsburgh Brain Institute and the Pittsburgh Foundation to CJD, the Robert Packard Center for ALS at Johns Hopkins to CJD and UBP, the Muscular Dystrophy Association and the ALS Association to UBP. AMG was supported by the Milton Safenowitz Postdoctoral Fellowship from the ALS Association and a generous grant from Barbara McCormick. JRM was supported by a predoctoral fellowship from the University of Pittsburgh Center for Protein Conformational Diseases. We would like to thank members of the Donnelly and Pandey lab for help with troubleshooting experiments and comments on the manuscript.
References:
- 1.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006. Oct 6;314(5796):130–133. [DOI] [PubMed] [Google Scholar]
- 2.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006. Dec 22;351(3):602–611. [DOI] [PubMed] [Google Scholar]
- 3.Buratti E Functional Significance of TDP-43 Mutations in Disease. Adv Genet. 2015. Aug 7;91:1–53. [DOI] [PubMed] [Google Scholar]
- 4.Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019. Jun 1;142(6):1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012. Jun 26;1462:61–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ling JP, Pletnikova O, Troncoso JC, Wong PC. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015. Aug 7;349(6248):650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014. Feb 5;81(3):536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Volkening K, Leystra-Lantz C, Yang W, Jaffee H, Strong MJ. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 2009. Dec 11;1305:168–182. [DOI] [PubMed] [Google Scholar]
- 9.Liu-Yesucevitz L, Bilgutay A, Zhang Y-J, Vanderweyde T, Citro A, Mehta T, et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS ONE. 2010. Oct 11;5(10):e13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saberi S, Stauffer JE, Schulte DJ, Ravits J. Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol Clin. 2015. Nov;33(4):855–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baloh RH. TDP-43: the relationship between protein aggregation and neurodegeneration in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. FEBS J. 2011. Oct;278(19):3539–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moreno F, Rabinovici GD, Karydas A, Miller Z, Hsu SC, Legati A, et al. A novel mutation P112H in the TARDBP gene associated with frontotemporal lobar degeneration without motor neuron disease and abundant neuritic amyloid plaques. Acta Neuropathol Commun. 2015. Apr 3;3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009. Jul 24;284(30):20329–20339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis-Marks MR, Needham PG, et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron. 2019. Apr 17;102(2):321–338.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Giorgio F, Maduro C, Fisher EMC, Acevedo-Arozena A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis Model Mech. 2019. Jan 2;12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasset-Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, et al. Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron. 2019. Apr 17;102(2):339–357.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiang P-M, Ling J, Jeong YH, Price DL, Aja SM, Wong PC. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci U S A. 2010. Sep 14;107(37):16320–16324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kraemer BC, Schuck T, Wheeler JM, Robinson LC, Trojanowski JQ, Lee VMY, et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010. Apr;119(4):409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019. Jan 14;22(2):180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019. Jan 14;22(2):167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miguel L, Frébourg T, Campion D, Lecourtois M. Both cytoplasmic and nuclear accumulations of the protein are neurotoxic in Drosophila models of TDP-43 proteinopathies. Neurobiol Dis. 2011. Feb;41(2):398–406. [DOI] [PubMed] [Google Scholar]
- 22.Langellotti S, Romano V, Romano G, Klima R, Feiguin F, Cragnaz L, et al. A novel Drosophila model of TDP-43 proteinopathies: N-terminal sequences combined with the Q/N domain induce protein functional loss and locomotion defects. Dis Model Mech. 2016. Jun 1;9(6):659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Ray P, Rao EJ, Shi C, Guo W, Chen X, et al. A Drosophila model for TDP-43 proteinopathy. Proc Natl Acad Sci U S A. 2010. Feb 16;107(7):3169–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y-F, Gendron TF, Zhang Y-J, Lin W-L, D’Alton S, Sheng H, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010. Aug 11;30(32):10851–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnold ES, Ling S-C, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A. 2013. Feb 19;110(8):E736–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 2010. Jan 13;30(2):639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin Y, Berry J, Pannucci N, Haataja MP, Toettcher JE, Brangwynne CP. Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell. 2017. Jan 12;168(1-2):159–171.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang P, Fan B, Yang P, Temirov J, Messing J, Kim HJ, et al. Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology. elife. 2019. Mar 20;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taslimi A, Vrana JD, Chen D, Borinskaya S, Mayer BJ, Kennedy MJ, et al. An optimized optogenetic clustering tool for probing protein interaction and function. Nat Commun. 2014. Sep 18;5:4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993. Jun;118(2):401–415. [DOI] [PubMed] [Google Scholar]
- 31.Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008. Jul;64(1):60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008. Mar;33(3):141–150. [DOI] [PubMed] [Google Scholar]
- 33.Nichols CD, Becnel J, Pandey UB. Methods to assay Drosophila behavior. J Vis Exp. 2012. Mar 7;(61). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casci I, Pandey UB. A fruitful endeavor: modeling ALS in the fruit fly. Brain Res. 2015. May 14;1607:47–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berson A, Sartoris A, Nativio R, Van Deerlin V, Toledo JB, Porta S, et al. TDP-43 Promotes Neurodegeneration by Impairing Chromatin Remodeling. Curr Biol. 2017. Dec 4;27(23):3579–3590.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choksi DK, Roy B, Chatterjee S, Yusuff T, Bakhoum MF, Sengupta U, et al. TDP-43 Phosphorylation by casein kinase Iε promotes oligomerization and enhances toxicity in vivo. Hum Mol Genet. 2014. Feb 15;23(4):1025–1035. [DOI] [PubMed] [Google Scholar]
- 37.Diaper DC, Adachi Y, Sutcliffe B, Humphrey DM, Elliott CJH, Stepto A, et al. Loss and gain of Drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum Mol Genet. 2013. Apr 15;22(8):1539–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cragnaz L, Klima R, Skoko N, Budini M, Feiguin F, Baralle FE. Aggregate formation prevents dTDP-43 neurotoxicity in the Drosophila melanogaster eye. Neurobiol Dis. 2014. Nov;71:74–80. [DOI] [PubMed] [Google Scholar]
- 39.Feiguin F, Godena VK, Romano G, D’Ambrogio A, Klima R, Baralle FE. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009. May 19;583(10):1586–1592. [DOI] [PubMed] [Google Scholar]
- 40.Lu Y, Ferris J, Gao F-B. Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Mol Brain. 2009. Sep 25;2:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen H-J, Topp SD, Hui HS, Zacco E, Katarya M, McLoughlin C, et al. RRM adjacent TARDBP mutations disrupt RNA binding and enhance TDP-43 proteinopathy. Brain. 2019. Dec 1;142(12):3753–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014. Jan;17(1):17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asakawa K, Handa H, Kawakami K. Optogenetic modulation of TDP-43 oligomerization accelerates ALS-related pathologies in the spinal motor neurons. Nat Commun. 2020. Feb 21;11(1):1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Capitini C, Conti S, Perni M, Guidi F, Cascella R, De Poli A, et al. TDP-43 inclusion bodies formed in bacteria are structurally amorphous, non-amyloid and inherently toxic to neuroblastoma cells. PLoS ONE. 2014. Jan 30;9(1):e86720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farny NG, Kedersha NL, Silver PA. Metazoan stress granule assembly is mediated by P-eIF2alpha-dependent and -independent mechanisms. RNA. 2009. Oct;15(10):1814–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGurk L, Berson A, Bonini NM. Drosophila as an in vivo model for human neurodegenerative disease. Genetics. 2015. Oct;201(2):377–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson EN, Gochenaur L, Singh A, Grant R, Patel K, Watkins S, et al. Traumatic injury induces stress granule formation and enhances motor dysfunctions in ALS/FTD models. Hum Mol Genet. 2018. Apr 15;27(8):1366–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007. Jun 14;447(7146):859–863. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.