

Summary
Measuring cell identity in development, disease, and reprogramming is challenging as cell types and states are in continual transition. Here, we present Capybara, a computational tool to classify discrete cell identity and intermediate ‘hybrid’ cell states, supporting a metric to quantify cell fate transition dynamics. We validate hybrid cells using experimental lineage tracing data to demonstrate the multi-lineage potential of these intermediate cell states. We apply Capybara to diagnose shortcomings in several cell engineering protocols, identifying hybrid states in cardiac reprogramming and off-target identities in motor neuron programming, which we alleviate by adding exogenous signaling factors. Further, we establish a putative in vivo correlate for induced endoderm progenitors, a cell type that has, to date, remained poorly defined. Together, these results showcase the utility of Capybara to dissect cell identity and fate transitions, prioritizing interventions to enhance the efficiency and fidelity of stem cell engineering.
Keywords: Single-cell analysis, cell-type classification, hybrid cells, cell differentiation, cell reprogramming
Graphical Abstract
eTOC Statement
Kong et al. present Capybara, a computational pipeline to classify discrete cell identity and intermediate ‘hybrid’ cell states. They apply Capybara to diagnose shortcomings in several cell engineering protocols, identifying hybrid states in cardiac reprogramming and off-target neural identities in motor neuron programming, leading to improved protocols to increase target cell yield. Further, they demonstrate the utility of Capybara to identify an in vivo correlate for induced endoderm progenitors, a relatively uncharacterized product of direct lineage reprogramming.
Introduction
The accurate quantification of cell identity in the context of stem cell differentiation and reprogramming is crucial to assess and refine cell engineering protocols. Previous methods to assess cell identity using bulk classifiers revealed that directed differentiation of cells from a pluripotent state produces developmentally immature cell types (Cahan et al., 2014). Similarly, direct reprogramming between differentiated states, typically driven by transcription factor (TF) overexpression, yields partially converted cells and off-target identities (Morris et al., 2014). However, precisely characterizing these cell engineering protocols is challenging due to their heterogeneity and the continual transition of cell types and states in dynamic biological systems.
Several computational strategies aim to automate the annotation of cell identity from single-cell data (Abdelaal et al., 2019). For example, Garnett leverages both single-cell RNA-sequencing (scRNA-seq) and single-cell ATAC-sequencing (scATAC-seq) data to classify cell identity (Pliner et al., 2019); ScPred uses scRNA-seq alone to build a prediction model based on a training dataset, estimating the probability of each cell belonging to a cell type category (Alquicira-Hernandez et al., 2019); SingleCellNet is an approach that quantitatively assesses identity using a Random Forest classifier to learn cell type-specific gene pairs from cross-platform and cross-species datasets (Tan and Cahan, 2019). However, many of these current supervised methods require prior biological knowledge to accurately classify cell identity. Furthermore, these approaches deliver discrete cell-type annotation, overlooking hybrid states which represent poorly characterized entities that occupy a space between discrete, fully defined cell identities (MacLean et al., 2018). Hybrid cells, also referred to as intermediate cells, have previously been described in the context of epithelial to mesenchymal transitions (Hong et al., 2015), differentiation of CD4+ T cells (Hong et al., 2012), hematopoiesis (Olsson et al., 2016), and zebrafish development (Farrell et al., 2018). Such mixed identity states can shed light on key transitions or bistable intermediates, yet hybrid cells are challenging to systematically define and capture.
Here, we present Capybara, an unsupervised computational method to quantitatively assess cell identity as a continuous property. In contrast to current approaches to annotate cell identity, we designed Capybara to interrogate the gradual transition of cell identity. To achieve this, we measure the identity scores for each query cell against exhaustive public cell type references using quadratic programming (QP), a method previously used to evaluate the reconfiguration of cell identity during direct lineage reprogramming (Biddy et al., 2018; Treutlein et al., 2016). Unlike existing methods, Capybara uses continuous identity scores to allow multiple identities to be assigned to an individual cell, enabling hybrid cell type identification. We build on this unique feature to develop a ‘transition metric,’ allowing quantification of cell fate transition dynamics.
We benchmark Capybara against a range of existing cell type classifiers, demonstrating its accuracy to annotate discrete cell identity and quantify hybrid cell identity. We validate hybrid cells using experimental lineage tracing of hematopoiesis, in addition to RNA FISH and immunostaining of a hybrid arising during cardiac reprogramming. We also demonstrate the utility of Capybara to diagnose and correct shortcomings in a range of reprogramming methods. Applied to the programming of motor neurons from embryonic stem cells (ESCs) and reprogramming cardiac fibroblasts to cardiomyocytes, our analysis reveals off-target cell identities arising from deficient patterning; additional signaling factors refine motor neuron programming to increase target cell yield over four-fold. Finally, analysis of direct reprogramming from fibroblast to induced endoderm progenitors (iEPs) identifies an in vivo correlate for this relatively uncharacterized reprogrammed cell type, which we validate experimentally. Together, these results showcase the utility of Capybara to dissect cell fate transitions in differentiation and reprogramming, prioritizing strategies to enhance the fidelity of cell engineering. Capybara code and documentation are available via https://github.com/morris-lab/Capybara.
Results
Capybara overview, benchmarking, and validation
To classify cell identity, Capybara assumes that each single-cell transcriptome exists as a combination of fractional identities from an array of candidate cell types. Under this assumption, Quadratic Programming (QP) has previously been used to leverage bulk expression signatures as a reference to classify single cells as a linear combination of different cell types (Biddy et al., 2018; Treutlein et al., 2016). We previously adapted this approach to construct scRNA-seq references of cell identity, supporting high-resolution cell-type classification in intestinal reprogramming (Seiler et al., 2019). Here, we generalize this method via systematic reference construction, using both bulk and annotated single-cell atlas datasets, enabling the unsupervised classification of cell identity in four steps, as follows (Figure 1A; Methods):
Figure 1. Capybara workflow and proof of concept simulation study.
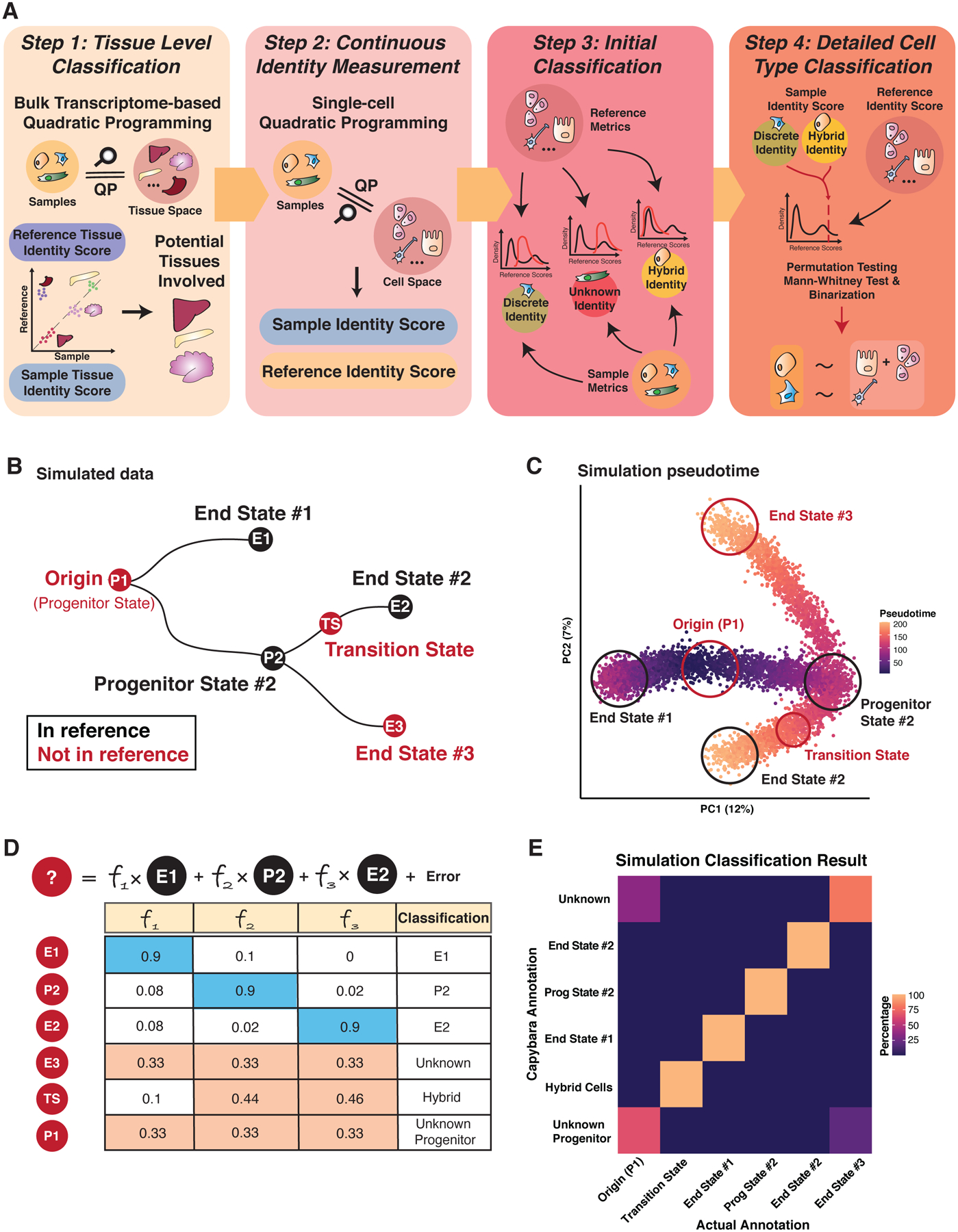
(A) Four steps of the Capybara workflow: 1) Tissue-level classification using bulk expression data to restrict the number of reference cell types in the downstream analysis; 2) Using single-cell atlases, we further identify highly correlated tissues to construct a high-resolution reference. Quadratic programming (QP) provides a continuous measure of cell identity as a linear combination of all cell types within the reference; 3) Initial classification using QP quality metrics to categorize the sample cells into discrete, hybrid, or unknown identities; 4) Detailed cell type classification to map cells to their corresponding cell types using a statistical framework. (B) Simulation study design. Differentiation is simulated from the progenitor state (P1) to two discrete states (E1 and P2). P2 further differentiates into two end states (E2, E3). Red Nodes: Test cells; Black Nodes: Cells included in the reference. (C) Pseudotime presentation of the simulated single-cell dataset, with discrete identities and transition state circled. (D) Expected classification outcomes. (E) Heatmap of percentage agreement between Capybara classifications and simulation ground truth. The transition state receives a hybrid classification; end states are mapped to corresponding discrete identities; unknown cell types are not assigned an identity. See also Figure S1.
First, initial tissue-level classification identifies the most appropriate tissue-specific single-cell reference to use. This step restricts the number of reference cell types included in downstream analysis, reducing excessive noise and dependencies caused by correlation across tissues. This custom single-cell reference is assembled in the second step by subsetting a larger atlas, such as the Mouse Cell Atlas (MCA; (Han et al., 2018)). We overcome gene expression dropout by sampling cells from each defined cell type to create a ‘pseudo-bulk’ reference. Application of QP using this custom reference generates continuous measurements of cell identity as a linear combination of all cell types within the reference. In the third step, we place the assessed cells into one of three identity categories: 1) Discrete, 2) Hybrid, 3) Unknown, using QP quality metrics (Figure S1A; Methods). Finally, we apply a statistical framework to assign a discrete identity to each cell. This step also characterizes the multiple identities harbored by a single cell, representing putative ‘hybrid’ cells. This function distinguishes Capybara from other cell-type classifiers, where cells in transition states are classified as either unknown or are placed within a discrete identity class. This aspect of our workflow enables us to explore the establishment and maintenance of cell identity in complex, continuous biological systems.
We validate the efficacy of Capybara to accurately classify discrete cell identity, using the multiclass area under the receiver operating characteristic (AUROC), together with a recent benchmark algorithm (Abdelaal et al., 2019) and our in-house validation with the Tabula Muris (The Tabula Muris Consortium et al., 2018) (Figure S1B; Methods). When benchmarked against ten other classifiers using five human pancreatic datasets and the Allen Mouse Brain Atlas, Capybara demonstrates a similar and nearly perfect AUROC performance (average = 0.95; rank 5 out of 11; Figure S1C). Further, Capybara classifies the majority of cells as ‘unknown’ when an inappropriate reference is used, suggesting improper choice or insufficient cell-type coverage of the reference. (Figure S1D).
As a further performance validation, we simulate a single-cell dataset comprising distinct differentiation paths to assess if Capybara can: 1) Capture cells with discrete identities; 2) Identify cells that do not correlate with any cell types in the reference; 3) Characterize hybrid cells that are in transition between discrete identities. We use Splatter, a simulation framework based on gamma-Poisson distribution (Methods; Zappia et al., 2017), to simulate distinct differentiation paths from an unknown progenitor state not included in the reference (P1), bifurcating toward two discrete states (E1: End State #1; P2: Progenitor State #2). P2 progenitor cells bifurcate further toward end states #2 and #3 (E2 and E3, respectively; Figure 1B, C), where end state 3 is not included in the reference. Indeed, Capybara accurately classifies cells in the three different identity categories, distinguishing between known discrete identities and cells in transition between them (AUROC = 1; Figure 1B–E). Further, Capybara can distinguish unknown cell types with 100% accuracy and separate unknown progenitor states vs. unknown terminal states, using QP quality metrics (Figure 1D–E; Methods). We benchmark our hybrid cell classification against scMap to illustrate how existing cell type classifiers cannot resolve mixed identity cells (Figure S1E). Furthermore, we show that low-complexity references do not generate artefactual hybrid cell classifications (Figure S1F). Together, our benchmarking and simulation demonstrate the efficacy of our method for cell-type classification of discrete and hybrid cell identities. We next showcase the application of Capybara in a well-characterized continuous differentiation process: hematopoiesis.
Capybara accurately captures cell identity and fate transitions in hematopoiesis
Hematopoiesis represents a cell differentiation process encompassing continuous changes in cell identity to multiple, well-defined terminal states (Orkin and Zon, 2008). We apply Capybara analysis to a published single-cell atlas of early myeloid progenitor differentiation to further test the performance of our method (Paul et al., 2015). Initial tissue-level classification shows a high correspondence of the single-cell data to the bone marrow (Figure 2A: Step 1). From this initial classification, we use the Mouse Cell Atlas (MCA; (Han et al., 2018)) to generate a high-resolution reference. Continuous identity scoring with this reference returns two major cell populations: bone marrow and peripheral blood (Figure 2A: Step 2), consisting of 82.2% discrete cell types, 17.8% hybrid cells, and no unclassified cells (Figure 2A: Step 3; Spreadsheets S1,2). DoubletFinder (McGinnis et al., 2019) and DoubletDecon (DePasquale et al., 2019) analysis labels 7–9% of the hybrid cell population as cell doublets, relative to 4.3–16.9% of the discrete population, ruling out doublets as the source of hybrid signals (Spreadsheet S2).
Figure 2. Capybara classification of hematopoietic cell identity.
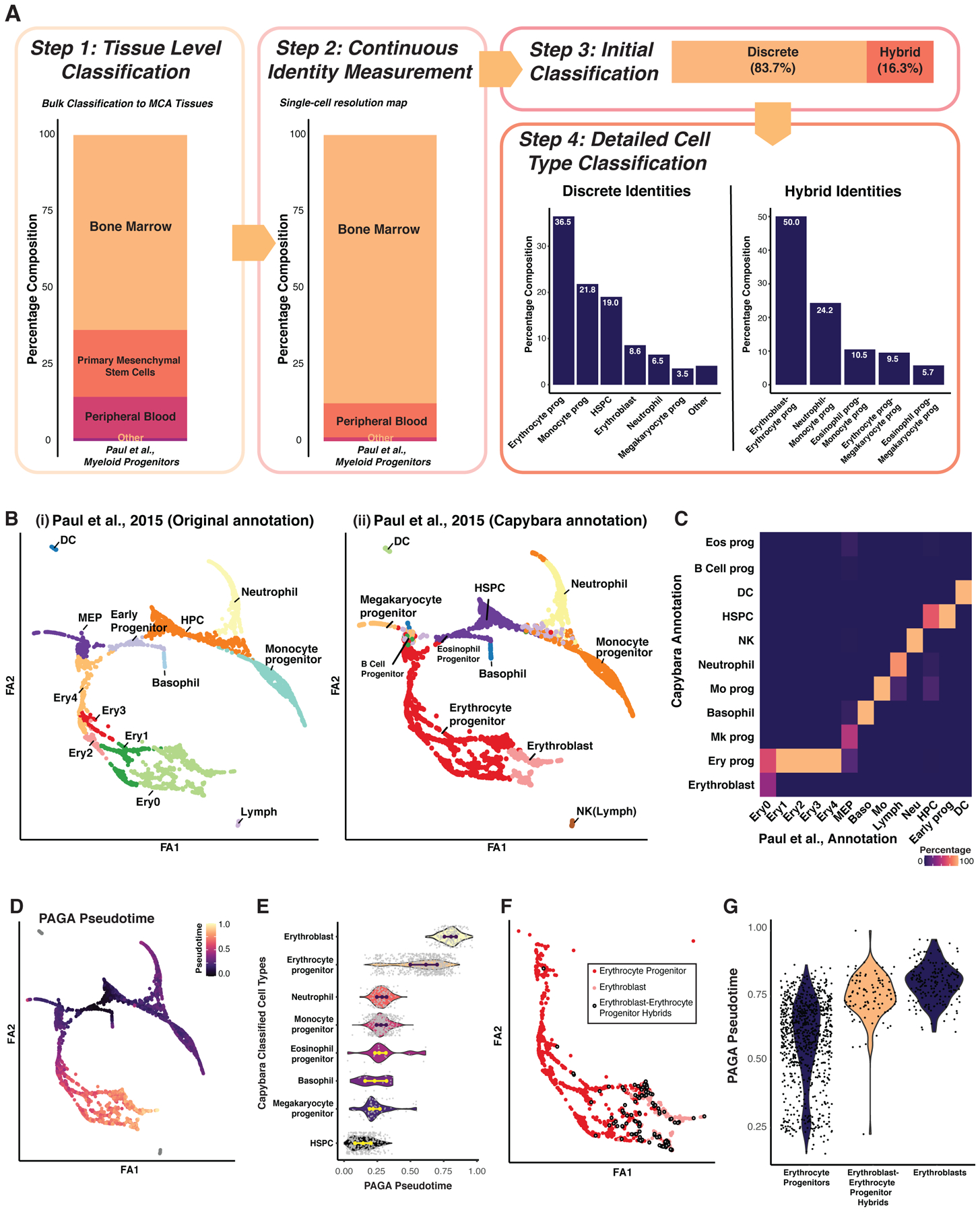
(A) Cell-type classification of an existing myeloid progenitor dataset (n = 2,730 cells; Paul et al., 2015),. ‘Prog’: Progenitor; ‘HSPC’: Hematopoietic Stem and Progenitor Cell. ‘Other’: includes basophils, eosinophil progenitors, B cell progenitors, macrophages, dendritic cells, and NK cells. (B) PAGA embedding. ‘FA’: Force Atlas. (i) Manual annotation of clusters, based on Paul et al., 2015. ‘DC’: Dendritic Cell; ‘MEP’: Megakaryocyte and Erythroid Progenitor; ‘Ery’: Erythroid; ‘Lymph’: Lymphoid; ‘HPC’: Hematopoietic Progenitor Cell. (ii) Capybara annotations. (C) Heatmap comparing manual and Capybara classifications. Color denotes the percentage agreement. (D) Diffusion pseudotime analysis projected onto the PAGA embedding. (E) Pseudotime for each Capybara-classified cell type. (F) Projection of ‘erythrocyte progenitor–erythroblast hybrids,’ along with discrete erythrocyte progenitors and erythroblasts onto the erythroid lineage. (G) Comparison of pseudotime between the hybrid and discrete identities shown in (F). See also Figure S2.
Overall, Capybara cell-type classification identifies the expected myeloid progenitor populations, including erythrocytes, megakaryocytes, hematopoietic stem and progenitor cells (HSPCs), monocytes, and neutrophils (Figure 2A: Step 4). 13 major clusters, resolved using partition-based graph abstraction (PAGA; (Wolf et al., 2019)) and annotated according to Paul et al., agree with Capybara classification (Weighted Cohen’s Kappa = 0.95; Figure 2B, C). Further, each classified population exhibits significant enrichment of established cell-type marker expression (Cd34, Itga2b, Cebpε, Csf1r, and Car2; P < 2.2E-16, Wilcoxon rank-sum test, Figure S2A). In addition, we assess the position of each discrete cell type within pseudotime estimated by PAGA, using modified diffusion pseudotime (Wolf et al., 2019); Capybara-classified HSPCs coincide with early pseudotime, as expected for this relatively undifferentiated cell population (Figure 2D, E).
In addition to discrete cell types, we identify five major hybrid populations, each representing ≥ 0.5% of the overall population: erythroblast–erythrocyte progenitors, megakaryocyte progenitor–erythrocyte progenitors, monocyte progenitor–neutrophils, megakaryocyte progenitor–eosinophil progenitors, and monocyte progenitor–eosinophil progenitors (Figure 2; Step 4). The largest hybrid population constitutes a mixed identity between erythrocyte progenitors and more differentiated erythroblasts, suggesting the hybrids represent a transition state. We leveraged PAGA to evaluate these hybrid cells, assuming hybrids would likely occupy intermediate pseudotime between defined identities. Indeed, erythroblast–erythrocyte progenitor hybrids are located mid-pseudotime, between discrete progenitor and erythroblast states (Figure 2F, G). We observe this trend for all hybrid populations identified (Figure S2B). Further, clusters enriched for hybrid cells are connected, based on PAGA analysis (Figure S2C). Altogether, the application of Capybara to this well-characterized paradigm of cell differentiation accurately identifies major hematopoietic cell populations, in addition to hybrid cell populations.
Lineage tracing reveals the multi-lineage potential of hybrid-classified cells
To further characterize hybrid cells, we leverage single-cell lineage tracing of hematopoiesis (Weinreb et al., 2020). In this prior study, Lin− Sca1+ Kit+ (LSK) HSPCs were isolated and labeled with random heritable barcodes, delivered via lentivirus. The barcoded cells were differentiated in vitro and collected for scRNA-seq at days 2, 4, and 6, yielding 72,946 single-cell transcriptomes. Cells sharing identical barcodes are identified as clonal relatives; thus, early cell state can be directly linked to differentiation outcome, allowing hybrid cell potential to be tested (Figure 3A). For these analyses, we constructed a reference from a small subset (1.7%) of the major day 6 differentiated myeloid populations (Figure S3A, B; Spreadsheet S1; S3). We identify seven major hybrid cell types (Figure 3B). The three largest hybrid populations: monocyte-neutrophil, basophil-mast, and basophil-eosinophil hybrids, contain clones spanning early and late time points. We assessed the cell-type composition of clonal relatives for each hybrid cell population across all time points, revealing significant enrichment of the discrete cell types that constitute each hybrid identity (*: P < 0.05; randomized test; Figure 3C).
Figure 3. Evaluation of hybrid cells using ground-truth lineage tracing.
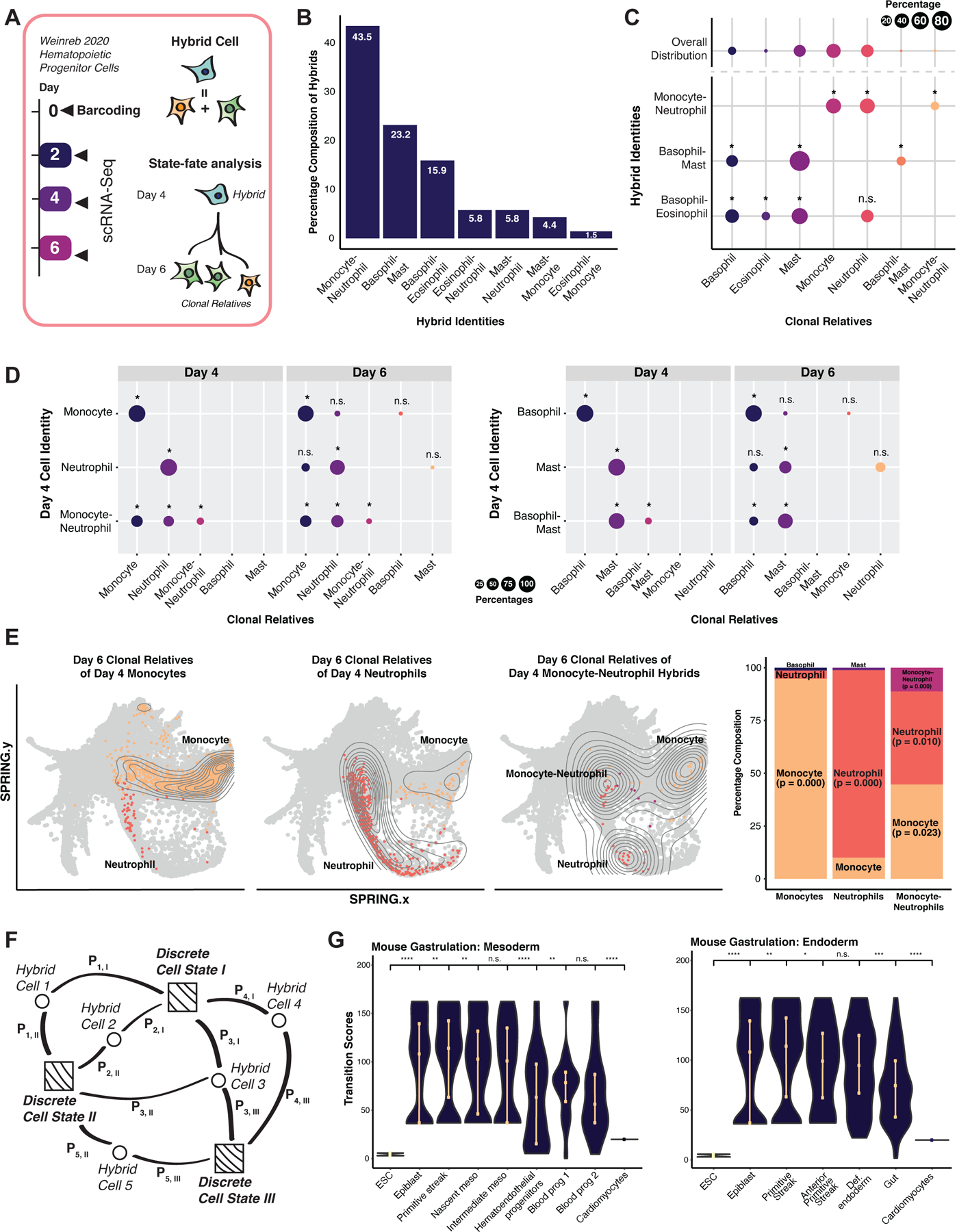
(A) Weinreb et al., 2020 hematopoietic lineage-tracing dataset. Hematopoietic progenitor cells were isolated, barcoded at day 0 and collected for scRNA-seq at day 2. Under myeloid differentiation conditions, cells were collected at days 4 and 6 for scRNA-seq. (B) Major hybrid populations identified by Capybara. (C) Cell-type composition of cells clonally related to major hybrid cell types. Upper row: Cell-type distribution of the overall population. Lower rows: Average cell-type breakdown for all clonal relatives of each major hybrid cell population (*: P <= 0.05, n.s.: P > 0.05, randomization test; 24 +/− 4 cells per clone, 10 clones, 243 cells). (D) State-fate analysis: We identified clones composed of discrete or hybrid identities at day 4 and assessed the cell-type composition of their differentiated clonal relatives at day 6. Top rows: day 6 clonal relatives derived from day 4 lineage-restricted clones. Bottom row: day 6 clonal relatives derived from day 4 clones containing hybrid cells (*: P <= 0.05; randomization test). (E) SPRING projection of cells related to monocyte- and neutrophil-restricted clones and hybrid clones. (F) Capybara’s transition metric. Squares: discrete cells. Circles: hybrid cells. Pi,j: probability of cell i transitioning to cell j. We calculate the transition score of each cell type as the accumulated information received from each cell connection. (G) Transition scores of mouse gastrulation, embryonic stem cells (ESCs), and cardiomyocytes (****: P <= 0.0001, ***: P <= 0.001, **: P <= 0.01, *: P <= 0.05, Wilcoxon test). See also Figure S3.
We next focused on two of the main hybrid populations spanning days 4 and 6 of differentiation: Monocyte-neutrophil and basophil-mast hybrids. We identified clones on day 4 that are composed exclusively of discrete cell identities (i.e., monocytes, neutrophils, basophils, or mast cells only) and found that their day 6 siblings are significantly restricted to their day 4 lineage (P < 0.05, randomized test; Figure 3D). In contrast, day 4 clones containing hybrids generate day 6 populations that are significantly enriched for the discrete cell types that comprise their mixed identity. For example, clones harboring monocyte-neutrophil hybrids on Day 4 generate day 6 populations that are significantly enriched for discrete monocytes, neutrophils, and monocyte-neutrophil hybrids (*: P < 0.05; randomization test; Figure 3D-Left, 3E). Indeed, these monocyte-neutrophil hybrids are transcriptionally similar to a bistable intermediate cell state reported to yield both monocytes and neutrophils (Olsson et al., 2016; Figure S3C–E). Further, day 4 clones harboring basophil-mast hybrid cells generate significant discrete basophil and mast cells populations on day 6 (Figure 3D, right). In summary, experimental lineage tracing data supports the ability of Capybara to capture hybrid cells and that these states are biologically relevant.
A metric to quantify cell fate transition dynamics
Together with previous work, the evidence we present suggests that hybrid cells represent intermediate states (MacLean et al., 2018) – either cells in transition between discrete identities or metastable mixed-lineage-state progenitors. Our unbiased quantification of hybrid cells supports the development of a ‘transition metric,’ where for each discrete cell identity within a population, we measure the strength and frequency of its connection to hybrid states (Figure 3F; Methods). A high transition score represents a high information state where identities converge - a putative cell fate transition.
We first compare transition scores to PAGA-based cell-to-cell connectivity scores. Analyzing myeloid progenitor differentiation (Paul et al., 2015) shows a strong correlation between the total connectivity and transition score (Pearson’s, r = 0.84; Figure S2D). Further, we apply RNA Velocity (La Manno et al., 2018) to identify actively transitioning cell states in cardiomyocyte reprogramming (Stone et al., 2019), observing a strong correlation between transition scores and RNA velocity vectors (Pearson’s, r = 0.77; Figure S3F). Finally, we calculate transition scores for datasets spanning the earliest stages of differentiation to terminally differentiated cardiomyocytes (Klein et al., 2015; Pijuan-Sala et al., 2019; Stone et al., 2019). As development progresses and cells specialize, transition scores progressively and significantly decrease, as expected (Figure 3G). ESCs under maintenance conditions demonstrate low transition scores as they are not actively differentiating. Altogether, our validation of Capybara demonstrates that, in an unbiased manner, we can accurately classify cell identity, hybrid states, and fate transitions. We next apply Capybara to characterize less defined, non-physiological systems, such as cell reprogramming, to diagnose aberrant fate specification and inform protocol refinement strategies.
Characterizing off-target and hybrid cell identity in cardiac lineage reprogramming
We first apply Capybara to assess the direct conversion of fibroblasts to cardiomyocyte-like cells via overexpression of three TFs: Gata4, Mef2c, and Tbx5 (GMT) (Ieda et al., 2010; Qian et al., 2012; Song et al., 2012). We selected a 30,729-cell, two-week time course of cardiac fibroblast to induced cardiomyocyte reprogramming, driven by GMT in the presence of TGFβ and Wnt inhibitors. On day 14, cells expressing the cardiac reporter gene α-Myosin Heavy Chain were sorted (Gulick et al., 1991), and profiled via scRNA-seq (Stone et al., 2019) (Figure 4A).
Figure 4. Capybara analysis of direct cardiac reprogramming.
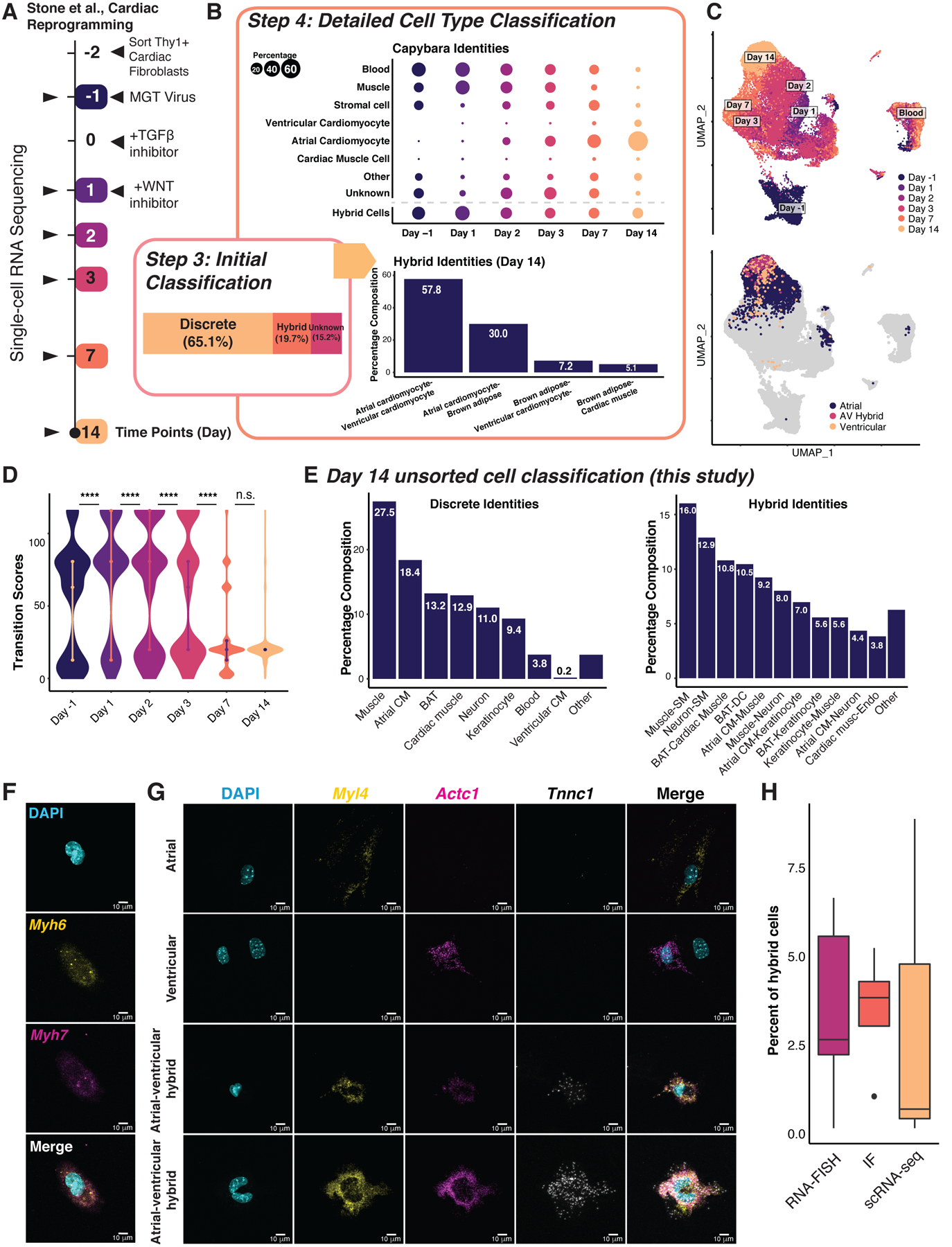
(A) Stone et al., 2019 experimental design. (B) Discrete, hybrid and unknown cell composition. Top: Capybara classified cell type composition over the time course. Dot size is proportional to the discrete population size. Bottom: Hybrid cell identities of the day 14 reprogrammed cells. (C) UMAP plot of the cardiac reprogramming dataset. Top: Collection time points projected onto the UMAP embedding; Bottom: Projection of atrial and ventricular cardiomyocytes, and atrial-ventricular hybrids. (D) Transition scores across the cardiac reprogramming process (****: P <= 0.0001, Wilcoxon test). (E) Detailed cell type and hybrid classification of our unsorted day 14 induced cardiomyocytes (n = 5,107 cells, two independent biological replicates). CM: Cardiomyocyte; BAT: Brown adipose tissue; SM: Smooth muscle; Endo: Endothelium. (F) RNA FISH for Myh6 (atrial) and Myh7 (ventricular) co-expression in a hybrid cell. (G) RNA FISH for Myl4 (atrial) and Actc1, Tnnc1 (ventricular) showing discrete and hybrid cells. Scale bars = 10μm. (H) Hybrid cell percentages measured by RNA FISH, immunofluorescence, and scRNA-seq. See also Figure S4.
Initial tissue-level classification, followed by refinement using the MCA, produces a high-resolution reference containing neonatal heart and skin populations (Figure S4A, B). Two major populations labeled from neonatal skin include macrophages and muscle cells, both mesodermal and resident in the heart (de Soysa et al., 2019). 65.1% of cells in the time course are assigned discrete identities, and 19.7% are assigned hybrid identities (Figure 4B). By reprogramming day 14 (2,320 cells), the majority of cells classify as atrial cardiomyocytes (76%), and ventricular cardiomyocytes (7.7%) (Figure 4B, C; S4C; Spreadsheet S4), confirmed via assessment of region-specific markers (Figure S4B). Non-cardiac cells, such as cardiac fibroblasts, blood, and muscle previously identified by Stone et al., decrease over time (Figure 4B). Hybrid cells in the day 14 sorted population are dominated by an atrial-ventricular (AV) cardiomyocyte intermediate (55.9%; Figure 4B; S4D). Brown adipose also features in discrete and hybrid identities (Figure 4B; S4C, D), which we speculate could be derived from cardiac-resident adipogenic progenitors that function in cardiac repair (Chen et al., 2021; Wu et al., 2010; Yamada et al., 2006). Transition scores significantly increase in the first two days of reprogramming, followed by a progressive decrease (P < 0.0001, Wilcoxon rank-sum Test; Figure 4D), implying an initial period of active fate transitioning, followed by a steady fate commitment. This observation echoes previous findings, where the final reprogramming outcome is determined by within the first 48 hours (Stone et al., 2019).
To gain a more accurate picture of off-target cell types, we generated cardiomyocytes according to the Stone protocol, without α-Myosin Heavy Chain sorting at day 14, yielding 5,107 cells from two independent biological replicates. Integration with the Stone et al. data demonstrates successful recapitulation of the protocol (cosine similarity: 0.71–0.89; Figure S4E). Cell-type classification reveals a similar off-target cell identity profile to the Stone protocol and enrichment of atrial cardiomyocytes (Figure 4E). We confirm the presence of AV hybrids, although at a much lower frequency (<1%; Spreadsheet S1) relative to the Stone protocol, which we attribute to not sorting the cells.
To validate AV hybrids, we performed RNA fluorescence in situ hybridization (FISH) using probes against canonical markers, Myh6 (atrial myosin heavy chain) and Myh7 (ventricular myosin heavy chain) on day 14 reprogrammed cells. We identified hybrid cells co-expressing both markers (Figure 4F; S4F, G). We selected an additional canonical atrial marker, Myl4, along with ventricular markers identified from the scRNA-seq data: Actc1 and Tnnc1 (Figure S4H) and identified further AV hybrids via RNA FISH (Figure 4G). We note that hybrid cells are typically binucleated or possess irregular nuclear morphology. Finally, we performed immunostaining for canonical markers MYL7 (atrial) and MYL2 (ventricular), validating atrial-ventricular hybrid cells at the protein level, in similar proportions to hybrid cells identified by scRNA-seq and FISH (Figure 4H; S4G–J). Together, Capybara can capture critical regionalization dynamics and off-target cell identities, indicating that additional TFs or signal modulation is required to tailor cardiac reprogramming outcomes.
Capybara reveals a dorsal-ventral patterning deficiency in motor neuron programming
Next, we focus on generating spinal motor neurons (MNs) from mouse ESCs. In TF-mediated direct programming (DP), overexpression of Ngn2, Isl1, and Lhx3 (NIL) direct ESCs to spinal MNs, bypassing canonical progenitor states (Mazzoni et al., 2013; Velasco et al., 2017). Alternatively, MNs can be produced by ‘directed differentiation’ (DD), involving sequential treatment with Fibroblast Growth Factors (FGFs), Retinoic Acid (RA), and Sonic hedgehog (SHH) (Wichterle et al., 2002; Wu et al., 2012), designed to recapitulate spinal cord development (Sagner and Briscoe, 2019). These two approaches have been compared via scRNA-seq profiling, confirming the generation of cells resembling MNs (Briggs et al., 2017) (Figure 5A). Here, we primarily focus on the TF-mediated DP protocol, which was reported to generate MNs with higher efficiency (Briggs et al., 2017). To classify cell identity, we use a recent single-cell atlas of mouse embryonic spinal cord, encompassing 118 cell types and states, including non-neuronal cell types surrounding the developing spinal cord (Delile et al., 2019) (Figure 5B). This high-resolution reference, combined with the MCA ESCs, is ideal for our analysis of MN generation, allowing initial tissue-level classification to be bypassed (Figure S5A).
Figure 5. Capybara analysis of spinal motor neuron differentiation and programming.
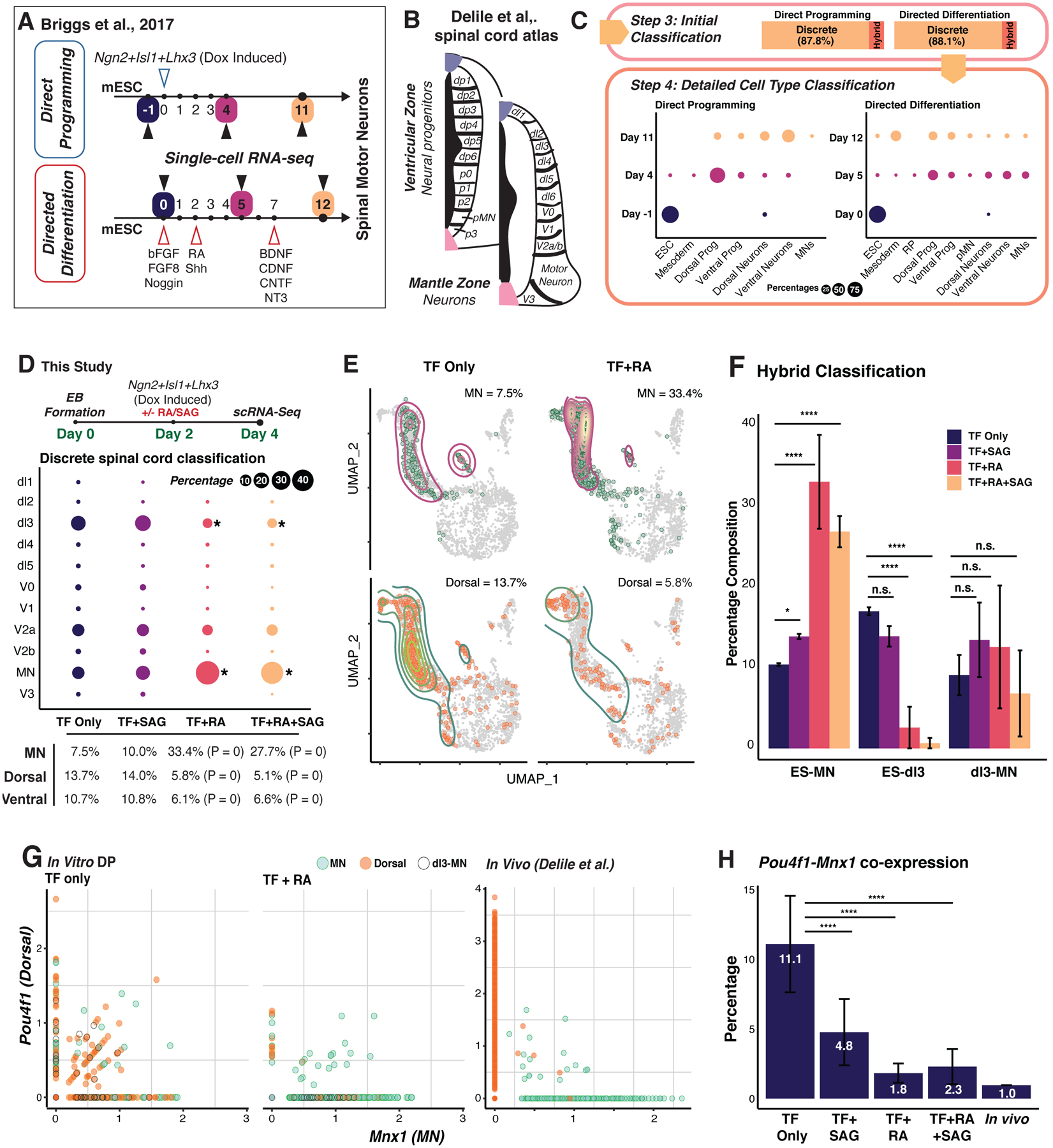
(A) Differentiation vs. direct programming of motor neurons (MNs) from ESCs (Briggs et al., 2017). (B) Spinal cord domains and regions included in the reference atlas (Delile et al., 2019). (C) Cell type composition over the differentiation and programming time courses. Dot size is proportional to the discrete population size. (D) Top: Experimental design in this study. After 48 hr of embryoid body (EB) formation, we induced the original reprogramming cocktail (Ngn2, Isl1, Lhx3: NIL) with retinoic acid (RA) and/or smoothened agonist (SAG). Day 4 cells were collected for scRNA-seq (Cells profiled: TF only: 2,926; TF + SAG: 3,340; TF + RA: 2,828; TF + RA +SAG: 8,042; two independent biological replicates per condition). Bottom: Differentiated spinal cord neuron composition and percentage breakdown of dorsal-ventral populations for each treatment group (*: P <= 0.05, ****: P <= 0.0001, randomization test). (E) UMAP plot of MN and dorsal populations comparing TF-only to TF + RA groups. (F) Major hybrid populations across treatment groups (****: P <= 0.0001, *: P <= 0.05; Two sample Chi-squared test). (G) Expression of the dorsal marker, Pou4f1, and motor neuron marker, Mnx1, comparing this study to the in vivo study (Delile et al., 2019). (H) Quantification of co-expressing cells in across treatment groups and in vivo (****: P <= 0.0001; Two sample Chi-squared test). See also Figure S5.
Capybara assigns discrete identities to 87.8% of cells (n = 4,136/4,704 cells). 12.2% of cells classify as hybrids, and no cells are unclassified (Figure 5C; Spreadsheet S5). Neuronal identity gradually emerges from a dominant ESC classification, with 63.8% of cells classifying as neurons on day 11. However, only 3% of this population classifies as MNs, whereas most cells classify across a range of dorsal-ventral neuronal identities (Figure 5C; S5B). In contrast, MN production in directed differentiation peaks at 13.4% of the early-stage (day 5) population, declining to 3.4% of the overall population (Figure 5C; S5B). Transition scores significantly decrease as TF-mediated programming progresses (P < 2.2E-16; Wilcoxon Test; Figure S5C) with hybrid cell generation peaking at day 4 (Figure S5D, E). Very few hybrid states represent known developmental progressions, particularly in DP compared to DD, suggesting that the mixed identities we observe in this context arise due to aberrant cell fate specification. Together, these observations raise the possibility that dorsal-ventral patterning is incomplete, suggesting that additional patterning signals could enhance MN production.
Retinoic Acid treatment resolves off-target identities to enhance MN generation
Spinal cord regionalization integrates complex spatial and temporal patterning events (Delile et al., 2019), involving different signaling molecules, such as RA and SHH (Lara-Ramírez et al., 2013; Ribes et al., 2009). Hypothesizing that these signals might fine-tune dorsal-ventral patterning to increase MN yield in vitro, we directly programmed ESCs using the originally published protocol (Mazzoni et al., 2013) in the presence and absence of 1 μM all-trans RA and/or 0.5 μM smoothened agonist (SAG - a hedgehog pathway activator) (Figure 5D; Methods). Four days following embryoid body (EB) formation and reprogramming induction, we captured 17,136 cells from two independent biological replicates (cosine similarity = 0.988; Figure S5F, G). 7.5% ± 1.6% of cells classify as MNs with TF induction alone, representing an over three-fold increase on the Briggs protocol, which we speculate is due to the initial EB formation in our protocol. In agreement with the Briggs protocol, we yield 13.7 +/− 1.7% dorsal and 10.3 +/− 1.2% ventral neurons. 35.4% of cells are hybrids (Figure S5H).
We next assessed whether adding RA and/or SAG can increase MN yields by reducing off-target cell generation. Indeed, RA treatment significantly increases MN generation over four-fold, from 7.5 ± 1.6% to 33.4 ± 4.9% (P < 2.2E-16, randomization test), and significantly depletes the off-target dorsal population, mainly dl3, from 13.7 ± 2.0% to 5.8 ± 0.9% (P < 2.2E-16, randomization test) (Figure 5D, E). The off-target ventral (mainly V2a) population is also significantly depleted, from 10.7 ± 1.2% to 6.1 ± 0.5%, upon addition of RA (P < 2.2E-16, randomization test). The addition of SAG alone only slightly enhances MN generation and offers no additional yields when added in combination with RA. Next focusing on the hybrid populations, the ESC-MN population is significantly enriched upon the addition of RA (P < 2.2E-16, randomization test), whereas the ESC-dl3 population is significantly depleted (P < 2.2E-16, randomization test; Figure 5F). Furthermore, upon addition of RA, the number of cells co-expressing the MN marker, Mnx1, and dorsal neuron marker, Pou4f1 is reduced over 6-fold to 1.8 ± 0.7%, in line with the 1% of co-expressing cells observed in vivo (Figure 5G, H; Delile et al., 2019). SAG treatment more than halves the co-expressing population to 4.8% but offers no further reductions when added with RA. Together, these results demonstrate the efficacy of Capybara to diagnose aberrant dorsal-ventral patterning in MN programming, which can be alleviated by the addition of RA to enhance the efficiency and fidelity of MN generation in vitro.
An in vivo correlate for fibroblast to induced endoderm progenitor reprogramming
Finally, we investigate a direct reprogramming process that produces a relatively uncharacterized cell identity with no presently known in vivo correlate. The overexpression of TFs Foxa1 and Hnf4α in mouse embryonic fibroblasts (MEFs) was initially designed to yield hepatocyte-like cells (Sekiya and Suzuki, 2011). However, our previous bulk cell type classification and functional studies revealed that these cells also harbor intestinal potential, in addition to hepatic potential, leading to their designation as progenitor-like ‘induced endoderm progenitors,’ (iEPs) (Guo et al., 2019; Morris et al., 2014). However, an in vivo correlate for these cells has remained elusive.
To better characterize iEP identity, we apply Capybara to our previous 85,010 cell reprogramming time course (Figure 6A) (Biddy et al., 2018). Initial tissue-level classification followed by refinement using the MCA produces a high-resolution reference, mainly consisting of embryonic mesenchyme and several endodermal populations (Figure S6A). Epithelial cells steadily emerge over the time course (5.9% at day 28), with few cells classifying as hepatocytes, agreeing with our previous study (Figure 6B; Spreadsheet S6; Morris et al., 2014). Relative to the above differentiation and reprogramming paradigms, a substantial proportion of cells (35.0%) remain unclassified, suggesting that a key in vivo correlate is missing from the reference.
Figure 6. Capybara analysis of fibroblast to induced Endoderm Progenitor (iEP) Reprogramming.
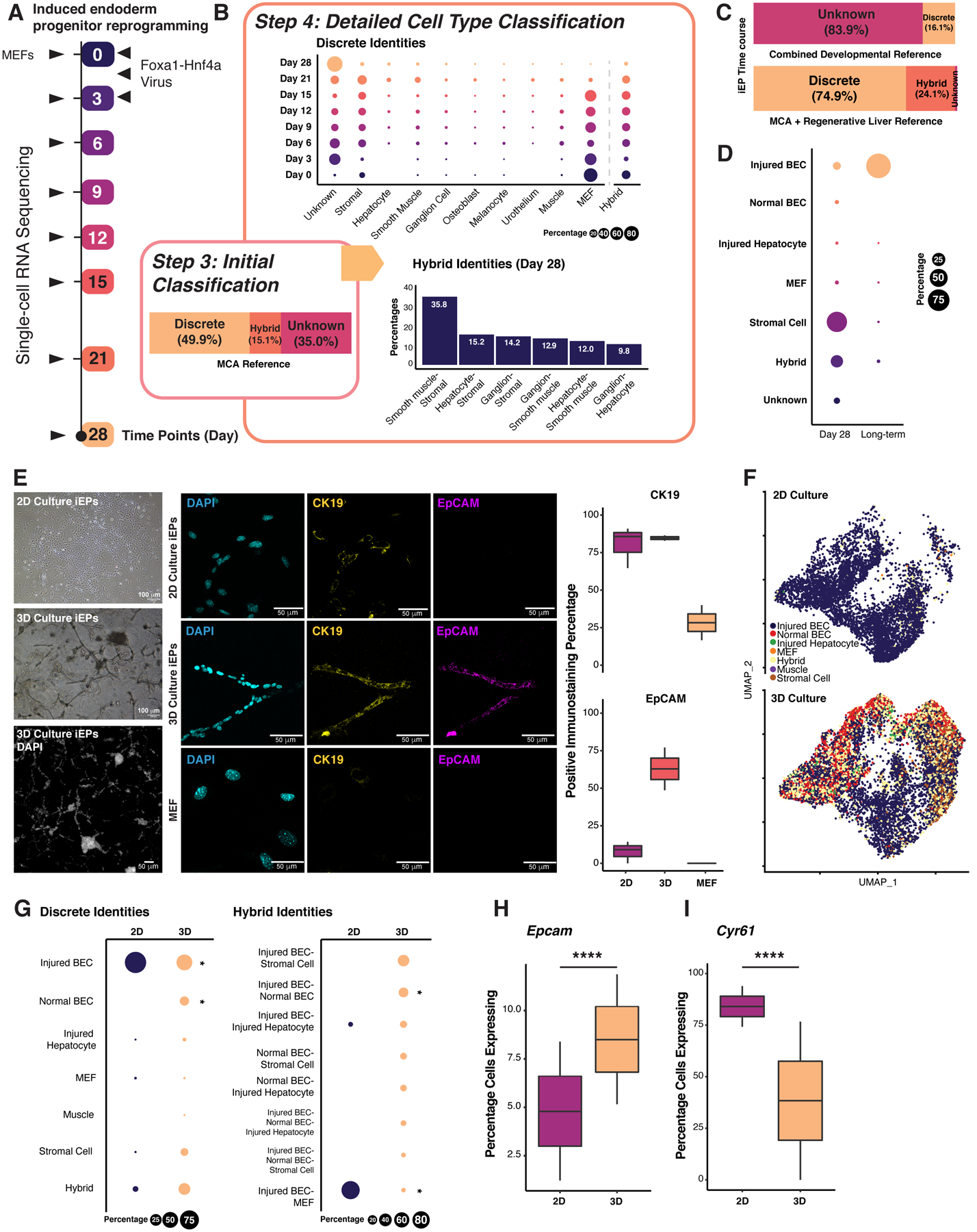
(A) MEF to iEP reprogramming (Biddy et al., 2018). (B) Top: Discrete cell type composition over the time course. Dot size is proportional to the discrete population size. Bottom: Hybrid cell identity proportions of cells after 28 days of reprogramming. (C) Cell composition with a developmental atlas (Han et al., 2020; Nowotschin et al., 2019) or a combined regenerative liver atlas (Han et al., 2018; Pepe-Mooney et al., 2019). (D) Cell type composition of day 28 and long-term cultured iEPs (n = 20,532 and 6,190 cells). (E) Imaging of 2D and 3D-cultured iEPs. Left: Bright-field images and DAPI field of composite z-stack images. Middle: Immunofluorescence images of DAPI, CK19, and EpCAM staining. Right: Quantification of the percentage of positively stained cells. MEFs: negative control (n = two independent biological replicates, two technical replicates each). Scale bars = 100 and 50 μm. (F) UMAP plot of our integrated 2D and 3D single-cell datasets with classified cell types labeled (3D: Two independent biological replicates: n = 9,348 and 4,699 cells). (G) Discrete and hybrid cell type composition of iEPs in 2D and 3D cultures. (*: P <= 0.05, randomization test). Percentage of 2D- and 3D-cultured iEPs expressing Epcam (H), Cyr61 (I) (P <= 0.0001; Two sample Chi-squared test). See also Figure S6.
Hypothesizing that iEPs represent a developmental progenitor, we assembled an embryonic atlas containing endoderm and foregut tissues, spanning E3.5 to E9.5 (Han et al., 2020; Nowotschin et al., 2019). However, iEPs remain unclassified using this reference (Figure 6C; S6B). Alternatively, we consider that iEPs may represent a regenerative cell type, based on their functional repair of liver and colon (Guo et al., 2019; Morris et al., 2014; Sekiya and Suzuki, 2011). Further, evidence supports a role for the Hippo signaling effector Yap1 in iEP generation (Kamimoto et al., 2020) in a process resembling injured liver regeneration (Pepe-Mooney et al., 2019). Thus, we built a high-resolution reference including homeostatic and regenerative liver epithelium, which contains two main regenerative cell types: hepatocytes and biliary epithelial cells (BECs) (Pepe-Mooney et al., 2019). Using this reference, we classify day 28 reprogrammed iEPs, and long-term cultured iEPs (LT-iEPs) that successfully engraft acutely-damaged intestine (Guo et al., 2019; Morris et al., 2014). 8.3 ± 4.7% of day 28 reprogrammed iEPs (n = 20,532 cells) and 95.7 ± 3.5 % of LT-iEPs (n = 6,190 cells, two independent biological replicates) classify as post-injury BECs (Figure 6D; S6B, C). We next sought to experimentally validate the putative identity of iEPs as BECs.
iEPs possess characteristics of biliary epithelial cells in vitro
Under homeostasis, BECs are quiescent and arrange to form tubular, single-epithelial-layered bile ducts in the liver. Upon injury, BECs enter active proliferation and play a key role in regeneration (Kamimoto et al., 2016). BECs isolated from the injured liver can be cultured ex vivo and maintained long-term (Okabe et al., 2009). Thus, we cultured LT-iEPs, harboring the highest proportion of ‘injured BECs,’ in a 3D-gel sandwich culture that promotes tubule formation in vitro (Ogawa et al., 2015), mimicking normal in vivo BEC morphology (Jin et al., 2013; Lewis et al., 2018). We observed branching tubular structures after three days of culture, significantly upregulating established BEC markers, cytokeratin 19 (CK19), and epithelial cell adhesion molecule (EpCAM) by day 5 (Figure 6E; S6D). Moreover, 2D-cultured LT-iEPs express Ck19 but reduced Epcam, recapitulating the reported behavior of injured BECs after expansion in vitro for over 30 days (Okabe et al., 2009).
To further characterize 3D-cultured LT-iEPs, we captured day 5 gel-cultured branching iEPs for scRNA-seq (n = 14,047 cells, two independent biological replicates). Cell-type classification shows the significant emergence of a normal BEC population in 3D-cultured iEPs (14.3 ± 1.7%; P < 2.2E-16, randomization test). This population is absent in 2D culture, which is significantly enriched for post-injury BECs (P < 2.2E-16, randomization test; Figure 6F, G). Additionally, an injured BEC-normal BEC hybrid appears as a unique population under 3D culture conditions (Figure 6G, right). Accompanying the emergence of normal BECs in 3D-culture is a significant expansion of cells expressing Epcam (P < 2.2E-16, randomization test, Figure 6H), in line with the above immunostaining, in addition to a significant reduction in the percentage of cells expressing Cyr61, a marker of injured BECs (P < 2.2E-16; randomization test, Figure 6I)(Pepe-Mooney et al., 2019). Moreover, these BEC-like cells express specific BEC markers, such as Sox9 (Figure S6E, F). Together, the cell type classification and orthogonal validation presented here reveal the previously uncharacterized BEC-like characteristics of iEPs.
Discussion
Here, we have developed and validated Capybara, an unsupervised method to quantitatively assess cell identity and fate transitions. A unique feature of Capybara is the measurement of cell identity as a continuum and its statistical framework to identify hybrid cells. Lineage tracing of hematopoietic differentiation demonstrates the multi-lineage potential of cells classified as monocyte-neutrophil and basophil-mast hybrids. Indeed, the monocyte-neutrophil hybrids we describe here are transcriptionally similar to a reported rare bistable hybrid with the functional potential to generate both monocytic and granulocytic lineages (Olsson et al., 2016). Further, we speculate that basophil-mast hybrids may correspond to a previously described rare basophil-mast progenitor cell (BMCP) which exhibits a hybrid transcriptional profile that primes differentiation toward the mast cell and basophil lineages (Dahlin et al., 2018). Further, we confirm the existence of atrial-ventricular cardiomyocyte hybrids in cardiac reprogramming via RNA FISH and immunostaining, validating the efficacy of Capybara to capture these mixed cell identities.
Hybrid states have been relatively poorly characterized due to their scarcity and transient nature. However, with high-throughput scRNA-seq, more examples of hybrid states are emerging (MacLean et al., 2018), along with computational approaches to characterize them. For example, MuTrans uses multiscale stochastic dynamics to capture transition states from single-cell data (Zhou et al., 2021). Capybara represents a unique method to assess mixed cell identities where deeper profiling of various cell differentiation paradigms may uncover hybrid states representing novel progenitor cell types and transitions. Hybrid cell states have been proposed to fulfill several roles in biological processes: they may serve to control bidirectional transitions between cell types, control fluctuations in cell population size, or create access to new cell identities – which is a crucial component of lineage reprogramming (MacLean et al., 2018). Indeed, we report wide-ranging hybrid states in the reprogramming paradigms we have analyzed here, contrasting with our analysis of hematopoietic hybrids representing rational cell state transitions or reported bistable intermediates. We hypothesize that the high levels of TF overexpression required to convert cell identity yields non-physiological cell states. Alternatively, the diversity of hybrid states may be rooted in the heterogeneity of the starting cell populations. Indeed, characterizing hybrids in this context might provide insight into the origins of successfully reprogramming cells.
The benefits of unsupervised cell-type classification go beyond the characterization of transition states, as we demonstrate via our analysis of diverse cell engineering strategies. For example, we defined regional patterning dynamics in the generation of cardiomyocytes and motor neurons. In cardiomyocyte reprogramming, atrial cardiomyocytes are generated in larger numbers than their ventricular counterparts; an atrial-ventricular hybrid suggests that modification of the protocol could shift this balance. Indeed, inhibition of TGFβ signaling with Wnt activation yields mainly ventricular cardiomyocytes (Wang et al., 2014), whereas both TGFβ and Wnt inhibition generates mostly atrial-like cardiomyocytes. Fine-tuning this balance will be beneficial to increasing yields of atrial or ventricular cardiomyocytes, which are functionally different populations that are both valuable drug-screening targets. In the context of motor neuron programming, we identified a range of off-target dorsal-ventral spinal neuron identities; the addition of retinoic acid to correct this patterning deficiency yielded over 4-fold more motor neurons. Finally, Capybara’s unsupervised cell-type classification identified BECs as a potential in vivo correlate for iEPs, a poorly characterized product of reprogramming. Together, these observations demonstrate the power of Capybara to enable highly quantitative cell type characterization, suggesting new reprogramming strategies.
Limitations of Capybara
It is crucial to note that the performance of Capybara relies on the selection of appropriate reference datasets. We have designed the workflow with this limitation in mind, where initial tissue-level classification identifies the most appropriate tissue-specific single-cell reference to use. Indeed, if an inappropriate reference is used, Capybara will classify cell identity as ‘unknown,’ as we demonstrate in our analysis of iEPs, which subsequently led us to a more suitable reference. A strength of Capybara to note here is that references can be constructed from a minimum of 30 cells, increasing the likelihood that rare cell types can be captured from selected references. As more diverse single-cell datasets become publicly available, we anticipate that this will support a much broader classification of cell identities.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Samantha A. Morris (s.morris@wustl.edu).
Materials Availability:
This study did not generate new unique reagents.
Data and Code Availability:
Single-cell RNA-seq data have been deposited at GEO and are publicly available. Accession numbers are listed in the key resources table. All original code, along with tutorials is available at: https://github.com/morris-lab/Capybara. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Monoclonal Anti-MYL7 Antibody (B-10) | Santa Cruz Biotechnology | RRID:AB_10848272 |
| Rabbit Monoclonal Anti-MYL2 Antibody | Abcam | RRID:AB_10563535 |
| Rat Monoclonal Anti-Mouse CD326 (EpCAM) Antibody | BD Biosciences | RRID:AB_394370 |
| Rabbit Monoclonal Anti-Cytokeratin 19 (CK19) Antibody | Abcam | RRID:AB_2281020 |
| CD90.2 (Thy1.2) Monoclonal Antibody, FITC | Invitrogen | RRID:AB_273503 |
| Alexa Fluor 546 Goat Anti-rabbit IgG | Invitrogen | RRID:AB_2534093 |
| Alexa Fluor 488 Goat Anti-mouse IgG | Invitrogen | RRID:AB_2534088 |
| Alexa Fluor 647 Goat Anti-rat IgG | Invitrogen | RRID:AB_141778 |
| Bacterial and Virus Strains | ||
| Stellar Competent Cells | Takara Bio | Cat #: 636763 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fetal bovine serum (FBS) | Gibco | Cat #: 10082147 |
| Fibroblast Medium-2 | ScienCell Research Laboratories | Cat #: 2331 |
| Matrigel (GFR Membrane Matrix) | Corning | Cat #: CB-40230 |
| β-mercaptoethanol | Life Technologies | Cat #: 21985023 |
| X-tremeGENE9 Transfection Reagent | Sigma Aldrich | Cat #: 6365779001 |
| XAV939 | Cayman | Item #: 13031 |
| SB431542 | Cayman | Item #: 13596 |
| CHIR99021 | BioVision | Cat #: 1677 |
| PD0325901 | Sigma | Cat #: PZ0162 |
| Leukemia Inhibitory Factor | Millipore | Cat #: LIF2050 |
| Retinoic Acid (RA) | ||
| Smoothened Agonist (SAG) | Millipore | Cat #: 566660 |
| Epidermal Growth Factor | Sigma Aldrich | Cat #: E5160 |
| Hepatocyte Growth Factor | Sigma Aldrich | Cat #: H9661 |
| Doxycycline (Dox) | Sigma Aldrich | Cat #: D9891 |
| L-Ascorbic Acid | Sigma Aldrich | Cat #: A8960 |
| Insulin-Transferrin-Selenium-Ethanolamine (ITS-X) | Gibco | Cat #: 51500056 |
| Gentle Cell Dissociation Reagent | STEMCELL Technologies | Cat #: 100–0485 |
| Critical Commercial Assays | ||
| RNAscope Multiplex Fluorescent v2 kit | Advanced Cell Diagnostics | Cat #: 323100 |
| EasySep Mouse FITC Positive Selection Kit II | STEMCELL Technologies | Cat #: 17668 |
| Ampure XP SPRI Beads | Beckman | B23318 |
| Chromium Single Cell 3’ Library and Gel Bead Kit v2 | 10x Genomics | PN-120237 |
| Chromium Single Cell 3’ Chip kit v2 | 10x Genomics | PN-120236 |
| Chromium i7 Multiplex Kit | 10x Genomics | PN-120262 |
| Chromium Next GEM Chip G Single Cell Kit | 10x Genomics | PN-1000127 |
| Library Construction Kit | 10x Genomics | PN-1000196 |
| Chromium Next GEM Single Cell 3’ GEM Kit v3.1 | 10x Genomics | PN-1000130 |
| Chromium Next GEM Single Cell 3’ Gel Bead Kit v3.1 | 10x Genomics | PN-1000129 |
| Dual Index Kit TT Set A | 10x Genomics | PN-1000215 |
| Deposited Data | ||
| scRNA-seq | This paper | GEO: GSE145251 |
| Hematopoiesis Development | Paul et al., 2015 | GEO: GSE72859 |
| Spinal Motor Neuron Differentiation and Programming | Briggs et al., 2017 | GEO: GSE97391 |
| Cardiac Reprogramming | Stone et al., 2019 | GEO: GSE131328 |
| MEF to iEP Reprogramming Time course | Biddy et al., 2018 | GEO: GSE99915 |
| Normal and Post Injury Hepatocytes and BECs | Pepe-Mooney et al., 2019 | GEO: GSE125688 |
| Mouse Gastrulation Atlas | Pijuan-Sala et al., 2019 | GEO: GSE87038 |
| Mouse Cell Atlas | Han et al., 2018 | https://figshare.com/articles/MCA_DGE_Data/5435866 |
| Tabula Muris | Tabula Muris Consortium et al., 2018 | https://figshare.com/projects/Tabula_Muris_Transcriptomic_characterization_of_20_organs_and_tissues_from_Mus_musculus_at_single_cell_resolution/27733 |
| Developing Mouse Spinal Cord Atlas | Delile et al., 2019 | E-MTAB-7320 |
| Experimental Models: Cell Lines | ||
| Mouse Cardiac Fibroblasts (CD1, P0) | ScienCell Research Laboratories | Cat #: M6300 |
| 293T-17 Cells | ATCC | RRID:CVCL_1926 |
| Primary Mouse Embryonic Fibroblast (C57BL/6, E13.5) | ||
| NIL-V5 inducible ESC line | Mazzoni et al., 2013 | |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | The Jackson laboratory | RRID:IMSR_JAX:000664 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Seurat V4 | Satija et al., 2015; Butler et al., 2018; Stuart et al., 2019 | https://satijalab.org/seurat/articles/get_started.html |
| Quadprog | Turlach and Weingessel, 2007 | https://cran.r-project.org/web/packages/quadprog/index.html |
| Cell Ranger v5.0.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| Velocyto | La Manno et al., 2018 | http://velocyto.org/ |
| MASS | Venables and Ripley, 2002 | https://cran.r-project.org/web/packages/MASS/MASS.pdf |
| mixdist | Macdonald and Du, 2018 | https://cran.r-project.org/web/packages/mixdist/mixdist.pdf |
| Splatter | Zappia et al., 2017 | https://github.com/Oshlack/splatter |
| OpenImageR | Mouselimis, 2021 | https://cran.r-project.org/web/packages/OpenImageR/OpenImageR.pdf |
| PAGA | Wolf et al., 2019 | https://github.com/theislab/paga |
| SCANPY | Wolf et al., 2018 | https://scanpy.readthedocs.io/en/stable/ |
| R-4.0.1 | R Core Team, 2021 | https://www.r-project.org/ |
| RStudio | RStudio Team, 2020 | https://www.rstudio.com/ |
| Capybara | This Paper | https://github.com/morris-lab/Capybara |
| Recombinant DNA | ||
| pMx-MGT | Wang et al., 2015 | RRID:Addgene_111810 |
| pGCDNSam-Hnf4α-t2a-Foxa1 | Morris et al., 2014 | |
| pCL-Eco | Novus Biologicals | RRID:Addgene_12371 |
| Other | ||
| RNAscope probe Mm-Tnnc1-C3 | Advanced Cell Diagnostics | Cat #: 511011-C3 |
| Opal 520 Reagent Pack | Akoya | FP1487001KT |
| Opal 570 Reagent Pack | Akoya | FP1488001KT |
| Opal 690 Reagent Pack | Akoya | FP1497001KT |
EXPERIMENTAL MODEL DETAILS
Mouse strain
Mouse Embryonic Fibroblasts (MEFs) were derived from mixed sex E13.5 C57BL/6J embryos (RRID:IMSR_JAX:000664). Timed pregnant C57BL/6 female mice were purchased from the Jackson Laboratory. All procedures were performed according to an IACUC approved protocol at Washington University School of Medicine.
Primary cell culture
Passage 0 primary cardiac fibroblasts derived from postnatal day 2 CD1 mice (ScienCell, Catalog #M6300; sex not specified) were cultured on gelatin-coated plates in Fibroblast Medium-2 (ScienCell, Cat. #2331). MEFs were cultured on gelatin in DMEM supplemented with 10% FBS, 50 mM β-mercaptoethanol, and penicillin/streptomycin, and reprogrammed before passage 6.
METHOD DETAILS
Capybara Pipeline Overview:
The Capybara pipeline comprises four major steps: 1) Tissue-level classification; 2) High-resolution custom reference generation and continuous identity measurement; 3) Initial classification into discrete, hybrid, or unknown identities; 4) Discrete cell type classification and hybrid identity scoring. Capybara code and documentation are available at: https://github.com/morris-lab/Capybara, along with detailed function descriptions and tutorials.
Basis of Capybara: Quadratic Programming (Setup).
Previous studies have measured continuous changes in cell identity using Quadratic Programming (QP) (Biddy et al., 2018; Treutlein et al., 2016), where The R package QuadProg was used for the calculation of QP scores. In brief, the underlying assumption is that each single-cell transcriptome profile exists as a combination of fractional identities from all possible cell types, described as a linear combination of gene expression profiles from different cell types. This assumption allows us to model cell identity as a multivariate linear regression problem. For ease of biological interpretation, constraints are placed on the coefficients: they are bound between 0 and 1, and the sum of all coefficients does not exceed 1. These constraints limit the use of least squares estimators in this scenario, while QP is an optimization approach that minimizes a quadratic function under the given linear inequalities or equalities.
Let denote the transcriptomic profile of genes g1, g2, …, gn for a query cell, and Xg,t denotes the reference dataset of the same set of genes by cell types t1, t2, …, tm. The goal is then to calculate the identity score vector ft, such that the random error ϵ is minimized, as described below.
In addition to the fractional identity score matrix and the error term, each cell receives a Lagrangian multiplier, gauging how much the solution is pushed toward the constraints. Applying QP offers a quantitative evaluation of cell identity for each cell.
Basis of Capybara: Quadratic Programming (Data processing).
Before QP, using raw count matrices, we first perform log-normalization on both the reference and sample dataset. Let Mg,c be the matrix with each row representing a gene and each column denoting a cell or a cell type.
Let m denote the number of columns, and n denote the number of rows. Then, for each column of the matrix, M*,c,
The normalized matrix is then log-transformed with a base of 2 and pseudo-count of 1. The reference dataset undergoes further scaling to ensure that gene expression levels between datasets are comparable. We calculate the scaling factor as the ratio between of the reference and sample. Further, we filter the gene list of both matrices to include only those genes shared between the reference and sample.
Step 1: Tissue-level classification.
The performance of Capybara hinges on the selection of an appropriate single-cell reference to classify cell identity. Before assessing cell identity at single-cell resolution, we perform a tissue-level classification designed to restrict the number of reference cell types included in downstream analysis, reducing excessive noise and dependencies caused by correlation across tissues in the final single-cell reference. This tissue-level classification is performed using bulk transcriptomics from ARCHS4, an exhaustive resource platform comprising the majority of published RNA-seq datasets (Lachmann et al., 2018). To achieve a relatively comprehensive and clean evaluation, we take a two-step approach: 1) construct a clean bulk RNA-seq reference, and 2) correlation-based tissue classification.
(1). Bulk Reference Construction
ARCHS4, a platform that contains most published RNA-seq and ChIP-seq datasets (Lachmann et al., 2018), was mined for bulk RNA-seq data. ARCHS4 obtained raw datasets from the Gene Expression Omnibus, which were realigned and processed through a uniform pipeline. Using this data bank, we first filtered the available datasets to retain only poly-A and total RNA-seq data from C57BL/6 mice. We then calculated Pearson’s correlations on every sample pair from the same tissue. The top 90 samples with the highest Pearson’s correlation scores for each of 30 tissues comprised the final bulk reference. For tissues with less than 90 samples, we took the entire sample set and randomly sampled with replacement to include 90 total samples. For the selected 90 samples for each tissue, we calculated the average reads per kilobase per million (RPKM) to build the final tissue-level transcriptome profile, containing a total of 30 tissues. We evaluated the quality of this bulk reference by calculating the identity scores of cells from manually annotated single-cell atlases (MCA; (Han et al., 2018) and Tabula Muris; (Tabula Muris Consortium et al., 2018)) based on this reconstructed reference. We randomly selected 90 cells from each tissue of MCA or Tabula Muris and performed QP using the bulk reference, where we observe high scores when mapping the same tissue between single-cell and bulk datasets.
(2). Tissue-Level Classification
A potential concern of using QP to directly classify single cells is the correlation between similar cell types from different tissues. In this scenario, it could be challenging to tease classification results apart if high similarity to the correct cell type drives the high identity score. Thus, we first perform tissue-level classification to restrict the number of reference cell types in the downstream analysis, reducing excessive noise and dependencies caused by correlation across tissues in the final single-cell reference. In general, the three primary inputs of this step include the single-cell reference (e.g., MCA), the sample single-cell dataset, and the constructed bulk reference. Using the tissue reference, we calculate QP scores for the single-cell reference as well as the sample, where we obtain two identity matrices. We then compute the Pearson’s correlations of QP scores between each cell from the single-cell reference and each cell from the sample. We use a threshold at the 90th percentile to binarize the correlation matrix, where a cell-cell pair with a correlation that is greater than the threshold is marked as 1; otherwise, 0. With the binarized matrix, we count the number of cells in each tissue of the reference mapping to the sample. If there is a significant percentage of reference cells of a tissue (over 70%) mapped, we record the tissue label. We then calculate the frequency of each tissue label in the sample. Tissues with a frequency of at least 0.5% sample cells are selected for further analysis at single-cell resolution. Here, it is worth noting that this tissue-level classification removes most irrelevant tissues but still provides a broad range of tissue types, at which point further downstream analysis removes non-relevant cell types (see ‘Cardiomyocyte Reprogramming Analysis,’ below). Additionally, having prior information regarding the tissues involved can be beneficial to narrow down the tissue selection step, as demonstrated by our analysis of hematopoiesis and spinal cord below.
Step 2: Generation of high-resolution custom references and continuous identity measurement.
Having identified the potential tissues present in a sample from the tissue-level classification, we next assemble a custom single-cell reference dataset containing the relevant cell types to classify sample cells. An example of such a reference dataset is the Mouse Cell Atlas (MCA; (Han et al., 2018)), which contains fetal and adult mouse tissues. For each tissue, it offers a detailed cell type breakdown, including the same cell type with different marker genes, offering a high-resolution map of cell-type composition. This reference is assembled based on manual annotation of the specific cell types in the tissue involved. A unique feature of scRNA-seq is dropout - the failure to capture and detect known expressed genes and other technical variation (Lun et al., 2016). Due to the highly sparse nature of scRNA-seq data, an individual cell transcriptome may not provide a complete representation of a cell type. To alleviate the effect of these technical variations, we construct pseudo-bulk references for each cell type of each tissue. We sample 90 cells from each cell type for each tissue. For cell types with more than 90 cells, we calculate Pearson’s correlations between each cell pair. Based on the correlation matrices, we select the most correlated 45 cells to ensure homogeneity and the least correlated 45 cells to capture transcriptional diversity. Cell types with fewer than 90 cells but more than 30 cells are sampled with replacement to achieve a total of 90 cells. Summation of the counts of the selected 90 cells is used to construct the final high-resolution reference, assuming homogeneity in the annotated population of the original single-cell reference. Application of QP using this ‘high-resolution’ reference generates a continuous measurement of cell identity as a linear combination of all cell types within the reference.
Step 3: Initial discrete, hybrid and unknown classification.
As aforementioned, the application of QP generates continuous identity scores, from which we calculate a deviance metric of the scores from the expected score. QP also provides two additional metrics: Error and Lagrangian multiplier. Using these metrics together with the continuous scores, we evaluate the likelihood of a cell to have discrete, hybrid, or unknown identities, compared to the scoring metric of reference cells (Figure S1A). This step can be evaluated in two parts: 1) Deviance, 2) Error, and Lagrangian multiplier.
(1). Deviance
The deviance is calculated via comparison between the identity scores to the expected scores , assuming a cell is equally similar to every cell type in the reference. We consider that cells with unique identities will have major deviations, while those with unknown identities will have minor deviations from the expectation. Let fi,j denote the score of a cell i on cell type j. The deviance is then calculated as follows:
Assuming the reference cells are accurately annotated with discrete identities, we first calculate the total deviance of each reference cell using the identity score matrix of the reference data. We further model the total deviance from the reference cells as a normal distribution, serving as the reference distribution of discrete identity cells. Restricting the hybrid cells to have a maximum of two identities, we establish an ideal distribution for the hybrid cells by shifting the density of discrete identities by 2x standard deviation to the left. Lastly, the unknowns are expected to have an even lower deviation than the hybrid cells. We then calculate the total deviance of each sample cell in the same manner. With the established distributions, we obtain probability scores from the evaluation of each distribution by computing P(X ≥ x). Cells with P(discrete) ≥ 0.01 & P(hybrid) ≥ 0.95 are considered as discrete. Cells with P(discrete) ≤ 0.05 & P(hybrid) ≥ 0.01 & P(unknown) ≥ 0.95 are considered hybrids. Cells with P(hybrid) ≤ 0.01 & P(unknown) ≥ 0 are considered unknowns.
(2). Error & Lagrangian Multiplier
The selection of cells to build the high-resolution custom reference includes both highly correlated and uncorrelated cells in the population of the corresponding cell type. Such a selection scheme provides a multimodal distribution for the error and Lagrangian multiplier metric, serving as background distributions for the extreme cases of matching and unmatching cells. Based on the multimodal density, we build an ideal distribution for the test samples, where the mean is the weighted mean of the mixed normal distribution, and the standard deviation is the weighted standard deviation of the mixed distribution. We consider unknown cells will establish higher error (on the right tail). In contrast, hybrid cells will have comparable error levels but a lower Lagrangian multiplier (on the left tail). In addition, unknowns can be distinguished into unknown progenitors vs. unknown end states by considering the combination of the two distributions. As unknown end states take both higher error and Lagrangian multiplier, unknown progenitors are considered to have a relatively high error but even lower Lagrangian multiplier than the hybrids. Yet, due to the challenges in deconvolving overlapping distributions, we could partially distinguish the two unknown cell types leveraging the combination of the two metrics.
Step 4: Discrete cell type classification and hybrid identity scoring.
While continuous identity scores are informative, discrete cell-type assignment offers a more practical assessment of cell-type composition for a biological system. One approach to call discrete cell types is to apply a threshold to the calculated continuous scores. However, threshold selection and quality of the custom high-resolution reference can bias cell type calling via this approach. To overcome this limitation, we apply QP to score cells in the single-cell reference against the bulk reference. This strategy accounts for reference quality, enabling background matrices to be generated, charting the distributions of possible identity scores for each cell type. We then take a two-step approach to provide discrete and hybrid cell type classification: 1) Empirical p-value calculation via randomized testing; 2) Mann-Whitney-based binarization and classification.
(1). Empirical P-Value Calculation via Randomized Testing
With the constructed single-cell reference, we apply QP to both the sample and reference single-cell datasets to generate continuous measurements of cell identity. Let MR denote the identity score matrix of the reference data with a total of m cell types and 90 · m cells, where fR,i,j denotes the score of reference cell i on cell type j. Let MS denote the identity score matrix of the sample data with a total of m cell types and n cells, where fS,i,j denotes the score of sample cell i on cell type j. We then carry out the following steps to calculate the empirical p-values. (1) For each cell type in MR, we randomly sampled 1000 times and constructed a background density of the identity scores, DR = [fresample,1, …, fresample,1000]. (2) For each score in the identity matrices, we calculate the empirical p-value as follows:
where if (*) is true; otherwise, . (3) Next, we repeat steps (1) and (2) for a total of 50 rounds, recording the empirical p-values matrix for each cell of both the reference and the sample. The result of this step includes two lists of p-value matrices: one for the reference and the other for the sample. For each cell, each column of the p-value matrix denotes a cell type, while each row describes each round of 50.
(2). Binarization and Classification
From randomized testing, we construct two lists of empirical p-value matrices: one for all sample cells, PS, and the other for all reference cells, PR. Using the list for all reference cells and their annotation data, we computed a benchmark empirical p-value for each cell type. Specifically, the annotation data contains cell barcodes and associated annotated cell types. For each cell c and its annotated cell type t0, we identified the corresponding list of empirical p-values, . As a result, we construct a possible range of p-values for each cell type, t, from which we generate the benchmark values. For each cell type t, we eliminate the outlier p-values and select the maximum p-value of the remaining cells as the final benchmark score, Bt = [Bt1,…, Btm]. Outlier p-values are identified based on the definition of outliers in the boxplot (outside of 1.5x the interquartile range above the third quantile or below the first quantile).
Next, we evaluate the sample list with the initial classification results. If the cell is initially considered an unknown, it is skipped for this statistical framework evaluation. The length of the sample list, n, is the number of sample cells. The nth empirical p-value matrix in the list defines empirical p-value for the nth sample cell belonging to reference cell type t under the kth resampling background, where 1 ≤ k ≤ 50. We rank all empirical p-values inside the matrix, from the lowest to the greatest, and break any tie by averaging. The rank-sum for each column t of is then calculated, and the cell type with the lowest rank-sum, t*, is determined to be the putative identity for cell c. We then compare mean to Bt* to assign an identity for cell c. To assign cells harboring hybrid identities, recapitulating those identities, we perform a pairwise Mann–Whitney U test between the t* column and other columns of . For any cell type t’ with rank-sum that is not significantly greater than the rank-sum of t* (significant level=0.05), we consider t’ to be one of multiple identities of query c along with t*. Applying this process to each cell, we generated a binary matrix with 1 = putative identities. Further, we generate a classification table with labeled cell types for each cell barcode.
Transition Scoring.
Hybrid cells label critical transition states in different trajectories. Building on this concept, we measure the strength and frequency of connection to the discrete cell state, which provides a metric that we define as a ‘transition score.’ The calculation of transition scores only involves cells with hybrid identities. In general, using QP, each cell receives fractional identity scores for different cell types in the reference. Interpreting QP as probabilities of the cell transitioning to each discrete cell identity, we use QP scores to measure transition probability.
For a cell marked with multiple identities, we consider a transition between the cell to its terminal cell state as events with the transition probability measured by QP scores Pi,j, where i denotes the cell and j denotes the cell state. Therefore, based on information theory, the information of such transition event can be measured as I(transition) = −log (Pi,j). We further consider how much information the terminal cell state has received, which can be defined as:
Thus, the total amount of information received for cell state j from n connected cells can be computed as:
The measurement appears to be similar to Shannon’s entropy. However, we note that with each cell independently in transition, probabilities from all events do not necessarily add up to 1, distinguishing it from a measure of entropy. Here, to demonstrate this metric, consider an example as demonstrated in Figure 3F, where Cells 1 to 5 harbor multiple identities connecting Cell State I to III. In this example scenario, the transition score for Cell State II can be calculated as:
Using such measurement, we incorporate the frequency and the likelihood of connection such that high information labels a discrete cell state associated with an abundance of dynamic cell transitions.
Benchmarking Capybara.
To assess the efficacy and robustness of Capybara to classify cell identity, we validate each step and demonstrate its basic functionality. In the first step of the Capybara pipeline, tissue-level classification, accuracy is pivotal as it helps reduce noise from other cell types that are not present in the sample. We evaluate the validity of the tissue reference transcriptome based on the identity scores of annotated single-cell atlases (Han et al., 2018; Tabula Muris Consortium et al., 2018). We randomly selected 90 cells from each tissue of MCA and Tabula Muris using the bulk reference, where we observed higher scores mapping of the same tissue between single-cell and bulk.
Next, we assess the classification functionality of Capybara. In this step, we use a benchmarking algorithm that was developed to compare a range of single-cell classification approaches using an array of publicly available datasets (Abdelaal et al., 2019). Briefly, we perform 10-fold cross-validation using various datasets. Here, the predictions from the methods are assessed based on the area under the receiver operating characteristics (AUROC) using the multiclass.roc function in R. Based on five human pancreatic datasets and Allen Mouse Brain Atlas, the performance of Capybara indicates similar accuracy (rank 5) and median F1 score (rank 4.2) with reasonable runtime when benchmarked against ten other classifiers (Figure S1B). In this benchmarking method, 5-fold cross-validation provides a relatively large training set (80%) compared to the test set (20%). A key feature of Capybara is its flexible requirement in terms of training set size. We find that a minimum number of 90 cells sampled from each cell type is required to perform accurate classification. For cell types with fewer than 90 cells, we require a minimum of 30 cells, from which a 90-cell sample will be drawn with replacement from the pool. Using this minimum number of cells, we evaluate our performance using the Tabula Muris mouse cell atlas (Tabula Muris Consortium et al., 2018). Using AUROC scores and accuracy, we benchmark our method against two other classification approaches, scmap (Kiselev et al., 2018) and SingleCellNet (Tan and Cahan, 2019). As a result, we demonstrate the comparable performance of Capybara with excellent performance (AUROC > 0.8).
Generation of simulated data
We use Splatter, an R-based simulation framework based on Gamma-Poisson distribution, to simulate a single-cell dataset comprising distinct differentiation paths (Zappia et al., 2017). We design the cell population to originate from a progenitor state (P1) bifurcating toward two discrete states (E1: End State #1; P2: Progenitor State #2). P2 progenitor cells bifurcate further toward end states #2 and #3 (E2 and E3, respectively; Figure 1B, C). Using this simulated dataset, we assess if Capybara can: 1) Capture cells with unique identities; 2) Identify cells that do not correlate with any cell types in the reference; 3) Characterize transition cells with multiple identities. E1, P2, and E2 cell populations were defined as within 5% variability of the maximum pseudotime at each terminal. We construct a reference using 90 of the most correlated and diverse cells from E1, P2, and E2 cell populations. Cells in E1, P2, and E2 that did not contribute to the reference are used to test the efficacy of accurate classification. The remaining cell populations are not included in the reference to test how Capybara classifies cells with no correlates in the reference.
Capybara Analysis with Previously Published scRNA-seq data
(1). Paul et al. (2015) Mouse Hematopoiesis Analysis
We obtained the raw hematopoiesis count data from GSE72859 (Paul et al., 2015). The data was processed and clustered using SCANPY (Wolf et al., 2018) and PAGA (Wolf et al., 2019). From processing, we included 3,451 genes in the dataset of 2,730 cells. We first perform tissue-level classification with the bulk reference established using ARCHS4, as described in the previous sections. From this, we identified three major relevant tissues: primary mesenchymal stem cells (bone marrow mesenchyme), bone marrow, and bone marrow (c-Kit). Further breakdown of these three major tissues using the MCA (Han et al., 2018) resulted in 49 different cell types. We constructed the high-resolution reference using these 49 cell types. 90 cells were selected from each cell type as described above and saved as the reference single-cell dataset. Followed by preprocessing, we applied QP on the reference and sample single-cell dataset, based on which we further categorized them to discrete, hybrid and unknown, calculated empirical p-values, performed binarization and classification. We projected cells with single identities onto the cluster embedding from PAGA. Cells with hybrid identities were isolated, and we extracted the pseudotime for these cells and their terminal cell identities. We re-assessed these hybrid cells using their scores. If one of the identities scored near zero (score < 10E-3), we considered such identity as inaccurate and discarded it. In this process, we re-evaluated transitioning cells, retaining only those cells with relatively higher shared identity scores. For a hybrid identity to be considered usable, it needs to be represented by more than 0.5% of the sample population. Using this filtering, we alleviate potential transitions due to noise but maintain the more putative transitions. A Wilcoxon test was used to compare if the pseudotime density differs comparing hybrids with their discrete identity parts.
(2). Weinreb et al. (2020) Mouse Hematopoiesis Lineage-Tracing Analysis
We obtained the normalized InDrop single-cell data, annotation, and SPRING embedding for mouse hematopoiesis lineage-tracing dataset from https://github.com/AllonKleinLab/paper-data. In this analysis, we mainly focused on the Lin− Sca+ cKit+ (LSK) population, containing a total of 72,946 cells. We constructed the high-resolution reference using 90 cells in each of the major day 6 differentiated cell types, including basophils, eosinophils, mast cells, monocytes, and neutrophils. Considering the myeloid differentiation culture conditions, we selected these five populations as they represent the continuous expanding populations from day 2 to day 6. Following preprocessing, we generated the continuous identity score measurements for the remaining LSK cells using QP, followed by initial classification, binarization, and classification. Leveraging the lineage information in the dataset, we then identified clones that contained hybrid cells on day 4 to evaluate their siblings on day 4 and progeny on day 6. To compare with the hybrid-containing clones, we also identified day 4 clones that are strictly represented by the discrete compartments of the hybrids. For instance, while assessing monocyte-neutrophil hybrids, we compared the siblings and progeny of day 4 clones strictly represented by monocytes or neutrophils. Enrichment of populations was tested via randomization testing. Briefly, for the clones representing the hybrid and its siblings, we randomly sampled the same number of cells as the clones from the entire population and calculated the proportion of the cell type represented in the sample. We iterated this process 10,000 times to establish a distribution. The likelihood of proportions presented in the hybrid family was evaluated based on this density, providing empirical p-values.
The raw count of the data was obtained by taking the reciprocal of the smallest non-zero gene expression of each cell, following https://github.com/AllonKleinLab/paper-data/issues/7. The data was then processed and clustered using SCANPY (Wolf et al., 2018) and PAGA (Wolf et al., 2019).
(3). Pijuan-Sala et al. (2019) Mouse Gastrulation Transition Score Analysis
We obtained 10x scRNA-seq UMI count data and annotation of mouse gastrulation from GSE87038 (Pijuan-Sala et al., 2019), containing 139,331 cells. The dataset was processed using Seurat (Butler et al., 2018; Satija et al., 2015). We performed classification using all 23 tissues, composed of 361 cell types, in the adult MCA as a reference directly (Han et al., 2018). We constructed the high-resolution reference using these annotated cells. Following preprocessing, we generated continuous identity score measurements for these cells using QP, followed by initial classification, binarization, and classification. We then performed Capybara transition scoring analysis for each sample, analyzing transition score distributions of each annotated cell type from Pijuan-Sala et al.
(4). Stone et al. (2019) Cardiomyocyte Reprogramming Analysis
We obtained the 10x single-cell RNA-sequencing count data from GSE131328 (Stone et al., 2019), containing 30,729 cells. This dataset was processed using Seurat (Butler et al., 2018; Satija et al., 2015) and clustered using UMAP. We used raw data from the filtered cells and genes as input into the Capybara pipeline. We next performed tissue-level classification using ARCHS4 (Lachmann et al., 2018), as described in previous sections, revealing four major tissues, including neonatal skin, neonatal heart, fetal stomach, and fetal lung. Further breakdown of these tissues using MCA (Han et al., 2018) contains 57 cell types. We constructed the high-resolution reference using these annotated cells. Following preprocessing, we generated the continuous identity score measures of these cells using QP, based on which we further performed initial classification, binarization, and classification. We calculated the percentage of each identified cell type in the population. Additionally, we computed the transition scores for the cell states involved in transitions. We performed transition score comparisons using a one-sided Wilcoxon test. We identified region-specific markers from MuscleDB (http://muscledb.org/mouse/mRNA/).
(5). Briggs et al. (2017) In Vitro Spinal Cord Motor Neuron Derivation Analysis
We obtained 10x scRNA-seq UMI count data and annotation of developing mouse spinal cord from https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-7320/ (Delile et al., 2019), including 38,976 cells. We removed unannotated cells and built a high-resolution reference for each cell type at each developmental stage (E9.5 to E13.5), resulting in a total of 118 cell types (19 types for E9.5, 26 for E10.5, 26 for E11.5, 25 for E12.5, 22 for E13.5). We also included embryonic stem cells from the MCA (Han et al., 2018). We obtained the InDrop single-cell dataset for in vitro spinal cord motor neuron derivation from GSE97391 (Briggs et al., 2017). The in vitro datasets were processed and clustered using Seurat (Butler et al., 2018; Satija et al., 2015). From processing, we included 7,860 genes and 7,799 genes in the dataset of 1,984 cells and 2,720 cells in direct programming (DP) and direct differentiation (DD), respectively. We analyzed ESCs from each protocol separately. Following preprocessing, we applied QP using the high-resolution reference on four datasets, including two ESC populations, DP and DD. Based on the identity score matrices, we categorized them into discrete, hybrid, and unknown, calculated the empirical p-value matrices, performed binarization and classification. Cells with discrete identities were separated to calculate the composition in the ventricular zone and mantle zone. The ventricular zone also included the neural crest neurons and mesoderm lineage. Hybrid cells were filtered and refined, as described in the above hematopoiesis section. With the QP scores attached to each identity in the mixed set, we calculated the transition scores for the cell states involved, as described in the transition scoring section. We compared transition scores between different timepoint via a one-sided Wilcoxon test.
(6). Biddy et al. (2018) MEF to Induced Endoderm Progenitor Analysis
We processed scRNA-seq data of induced endoderm progenitor (iEP) reprogramming, as previously described (Biddy et al., 2018). In brief, Scater was used to normalize (McCarthy et al., 2017) the data across time points, and Seurat (Butler et al., 2018; Satija et al., 2015) was used to integrate biological replicates, perform clustering, and visualize cells using t-SNE. We performed tissue-level classification using ARCHS4 (Lachmann et al., 2018), as described in previous sections, highlighting the involvement of 9 potential tissues, containing a total of 73 cell types. Following the construction of a high-resolution reference, we performed preprocessing on the reference and the sample, on which we then applied QP to generate the identity score matrices. Further, we categorized cells into discrete, hybrid, and unknown, calculated the p-value matrices, and performed binarization and classification. We calculated the percent composition of each cell type. Cells with hybrid identities were filtered as described in the above hematopoiesis section, represented by more than 0.5% cells of the population.
We obtained scRNA-seq data of biliary epithelial cells (BECs) and hepatocytes, before and after injury, from GSE125688 (Pepe-Mooney et al., 2019). We built a custom high-resolution reference by incorporating additional tissues from the MCA: fetal liver, MEFs, and embryonic mesenchyme. The long-term iEPs were cultured for 12 months before collection and processing. We had previously used these cells to engraft mouse colon (Guo et al., 2019; Morris et al., 2014). The long-term iEP dataset was processed, filtered, and clustered using Seurat, resulting in 2,008 cells. We also used Seurat to generate module scores of BEC identity, using a panel of markers from (Verhulst et al., 2019). We then constructed the high-resolution reference panel with 20 cell types and performed preprocessing on the reference and single-cell sample. Application of QP using the processed reference and long-term iEP and iEP reprogramming datasets provides us the continuous metric of identity scores, from which we carried out initial classification, binarization, and classification. Gene expression was compared between groups via Wilcoxon test.
10x alignment, digital gene expression matrix generation.
The Cell Ranger v5.0.1 pipeline (https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest) was used to align reads, process, and filter data generated using the 10x Chromium single-cell gene expression platform. Following this step, the default Cell Ranger pipeline was implemented to generate the filtered output data for downstream analysis.
scRNA-seq Data Processing
To process and analyze scRNA-seq data, we used the R package, Seurat V4 (https://satijalab.org/seurat/articles/pbmc3k_tutorial.html). Briefly, each sample was pre-processed based on RNA counts and mitochondrial read percentages and then normalized. The highly variable genes were then identified, followed by scaling and dimensional reduction via PCA. With the selected number of components, graph-based clustering and UMAP plotting were further performed.
(1). Cardiac Reprogramming
When comparing our data with Stone et al., 2019, the data were integrated using canonical correlation analysis and mutual nearest neighbor with the Seurat V4 pipeline (Butler et al., 2018; Stuart et al., 2019). The similarity between the dataset was evaluated based on cosine similarity between the cluster representation in the two datasets.
(2). In Vitro Motor Neuron Programming and iEP Reprogramming
To evaluate reproducibility, datasets for each treatment were integrated across the two biological replicates following the same process described above. The integrated Seurat objects were further integrated to evaluate the effect of different treatment groups.
scRNA-seq Data Capybara Analysis
With the tissues identified from the corresponding publicly available dataset, we started from step 2 of the Capybara pipeline for the single-cell RNA-sequencing data we generated for this study. Using the raw counts, we performed preprocessing on the reference and the sample, on which we then applied QP to generate the identity score matrices. Further, we categorized them into discrete, hybrid, and unknown, calculated the p-value matrices, and performed binarization and classification. We calculated the percent composition of each cell type. Cells with hybrid identities were filtered as described in the above hematopoiesis section, represented by more than 0.5% cells of the population.
Experimental Methods
Reprogramming Virus Production
The retrovirus for cardiac and induced endoderm progenitor (iEP) reprogramming was freshly prepared. 293T cells (RRID:CVCL_1926) were maintained and passaged in fibroblast media (10% FBS, 1x penicillin-streptomycin, 1x β-Mercaptoethanol, in DMEM). 293T cells were seeded at a density of 3 million per 10-cm plate the day before transfection. The following day, the cells were transfected with pMX-MGT (RRID:Addgene_111810) or pGCDN-Sam-Hnf4α-t2a-Foxa1 with 5μg of pCL-Eco (RRID:Addgene_12371), using X-tremeGENE 9 DNA transfection reagent (Sigma, 6365779001) according to the manufacturer’s instructions. Media was replaced with fresh fibroblast media the following day. Retrovirus was harvested the next day by taking the supernatant from the transfected plate and filtered through a 45- μm syringe filter. 500x protamine sulfate was added to the viral media prior to transduction of the mouse cardiac fibroblasts (cardiac reprogramming) or mouse embryonic fibroblasts (iEP reprogramming).
Cardiomyocyte Reprogramming
Direct cardiac reprogramming was performed using primary cardiac fibroblasts derived from a postnatal day 2 CD1 Mouse (ScienCell, Catalog #M6300) following previously published protocols (Ieda et al., 2010; Qian et al., 2013; Stone et al., 2019). Briefly, cardiac fibroblasts (MCFs) were cultured overnight on gelatin-coated plates in Fibroblast Medium-2 (ScienCell, Cat. #2331). MCFs were passaged 1–2 times, cultured for ~5 days for expansion, and prepared for selection of Thy1+ (RRID:AB_273503) cells by MACS. After sorting, MCFs were plated at a density around 100k~200k per 6-cm dish pre-treated overnight with gelatin (day −1). Thy1+ MCFs were transduced with freshly harvested pMX-MGT retrovirus (Wang et al., 2015) (day 0). The viral media was replaced with fresh cardiomyocyte media (10% M199, 10% FBS, 1% NEAA, 1% sodium pyruvate, 1x penicillin-streptomycin, 1x Glutamax, in DMEM) containing 2.6μM SB431542 (Cayman Chemical, Catalog #13031) or DMSO as a vehicle control (day 1). 5μM XAV939 (Cayman Chemical, Catalog #13596) or DMSO was added to the plate without media change (day +2). The media was replaced with fresh cardiomyocyte media two days after the last addition of small molecule (day +4). Media was renewed every 2~3 days. The cells were collected, filtered through a 70μm strainer, resuspended in 1% BSA in PBS, and counted on Day 14 for scRNA-seq (see below).
Immunostaining for day 14 Reprogrammed Cells in Cardiac Reprogramming
Mouse cardiac fibroblasts were generated as described above. On day 13 of reprogramming, the cells were transferred to 4-Chamber Culture Slides (Falcon). On the next day, the cells were rinsed with 1x DPBS and fixed in 4% paraformaldehyde for 20 minutes at room temperature. The samples were then washed with 1x DPBS three times, permeabilized, and blocked with blocking buffer (0.2% TritonX-100 and 3% FBS in DPBS) for one hour. The primary antibodies, MYL2 (RRID:AB_10563535) and MYL7 (RRID:AB_10848272), were diluted 1:250 (MYL2 and MYL7) in blocking buffer. The blocking buffer was then removed from the sample, and the primary antibodies were added. The samples were incubated with the primary antibody at 4°C overnight (12hr). The samples were then washed for 5 minutes three times. The secondary antibodies, Alexa Fluor 546 Goat Anti-rabbit IgG (RRID:AB_2534093) and Alexa Fluor 488 Goat Anti-mouse (RRID:AB_2534088), were diluted 1:1000 in blocking buffer. Secondary antibodies were added and incubated at 4°C overnight (12hr). The samples were washed again for 5 minutes, three times. 100 μl of 300 nM DAPI (Invitrogen) was added to each slide chamber and incubated at room temperature for 1 minute. The samples were washed for 5 minutes three times. In the last wash, we aspirate all the DPBS and remove the chamber from the slides. A coverslip was then applied with ProLong Gold Antifade Mountant (Invitrogen). The slides were imaged using an Olympus FV1200 Confocal Microscope with 10x, 20x, and 40x water objectives. The number of positive cells was counted in each channel using ImageJ with “Analyze Particles” function. The total number of cells was determined based on the DAPI counts.
RNA Fluorescent in Situ Hybridization
On day 13 of mouse cardiac reprogramming (above), the cells were transferred to 4-Chamber Culture Slides (Falcon). The next day, cells were rinsed with 1x DPBS and fixed with 10% Neutral Buffered Formalin for 30 minutes at room temperature. RNAscope Multiplex Fluorescent v2 kit (Advanced Cell Diagnostics) was used to perform RNA-FISH to probe Myh6, Myh7, Myl4, Actc1, and Tnnc1 mRNA, following the protocol for cultured adherent cell samples. Briefly, the slides were treated with hydrogen peroxide and RNAscope protease III (Advanced Cell Diagnostics). Then, the slides were incubated to hybridize with the specified probes using the RNAscope HybEZ II Oven (Advanced Cell Diagnostics). Probes were then amplified, and the HRP signal was developed using Opal dyes (Akoya, Opal 520: FP1487001KT; Opal 570: FP1488001KT; Opal 690: FP1497001KT). The dyes were reconstituted following the manufacturer’s instruction in DMSO and diluted 1 to 2000 in TSA Buffer for signal development. Finally, DAPI (Advanced Cell Diagnostics) staining was applied to the slide, and a coverslip was then applied with ProLong Gold Antifade Mountant (Invitrogen). The slides were imaged using an Olympus FV1200 Confocal Microscope with 40x and 60x water objectives. Images were then analyzed using computational quantification: RNA-FISH images were first processed through ImageJ to ensure the same maximum intensity across images. Through ImageJ, individual cells were segmented into smaller regions. All three channels of each selection were stored for further processing with a custom R script to quantify intensity at single-cell resolution. Individual cells were read in as individual matrices, where averaged green and red intensity were calculated and compared.
Motor Neuron Programming from mouse ESCs
The NIL (Ngn2-Isl1-Lhx3)-V5 inducible ESC line was previously described (Mazzoni et al., 2013). All the inducible ESC lines were grown in 2-inhibitors medium (Advanced DMEM/F12:Neurobasal (1:1) Medium (Gibco), supplemented with 2.5% ESC-grade fetal bovine serum (vol/vol, Corning), N2 (Gibco), B27 (Gibco), 2mM L-glutamine (Gibco), 0.1 mM β-mercaptoethanol (Gibco), 1000 U/ml leukemia inhibitory factor (Millipore), 3μM CHIR (BioVision) and 1 μM PD0325901 (Sigma). To obtain Embryoid bodies (EBs) ESC were trypsinized (Gibco) and 3 × 105 cells were plated in each 100 mm dish in AK medium (Advanced DMEM/F12:Neurobasal (1:1) Medium, 10% Knockout SR (vol/vol) (Gibco), Pen/Strep (Gibco), 2mM L-glutamine and 0.1 mM 2-mercaptoethanol) (day −2). After 48 hr, EBs were passed 1:2, and the inducible cassette was induced by adding 3 μg/ml of Doxycycline (Sigma) and/or 1 μM all-trans retinoic acid and/or 0.5 μM smoothened agonist (SAG) (Millipore, 566660). Differentiating EBs were washed three times with PBS, dissociated with Trypsin, and pipetted into single-cell suspensions. After 48 hr, cells were preserved in methanol (Alles et al., 2017) before processing for single-cell profiling (below).
Long-term iEP culture
Mouse Embryonic Fibroblasts were derived from the C57BL/6J strain (RRID:IMSR_JAX:000664). All animal procedures were based on animal care guidelines approved by the Institutional Animal Care and Use Committee. Mouse embryonic fibroblasts were reprogrammed to iHeps/iEPs, as in Sekiya and Suzuki (2011). Briefly, fibroblasts were prepared from E13.5 embryos and serially transduced with polyethylene glycol concentrated Hnf4α-t2a-Foxa1, followed by culture on gelatin for two weeks in hepato-medium (DMEM:F-12, supplemented with 10% FBS, 1 mg/ml insulin (Sigma-Aldrich), dexamethasone (Sigma-Aldrich), 10 mM nicotinamide (Sigma-Aldrich), 2 mM L-glutamine, 50 mM β-mercaptoethanol (Life Technologies), and penicillin/streptomycin, containing 20 ng/ml hepatocyte growth factor (Sigma-Aldrich), and 20 ng/ml epidermal growth factor (Sigma-Aldrich)), after which the emerging iEPs were cultured on collagen and passaged twice per week for three months.
Matrigel Sandwich Culture of Long-term iEPs
We adapted the culturing method from Ogawa et al., 2015 and Okabe et al., 2009. Briefly, 70% Matrigel in DMEM was added as a bottom layer to the plate, 96-well glass-bottom plate (20 μl) or glass-bottom 35mm μ-Dish (100 μl; iBidi) or 6-well plate (100 μl). The bottom layer was allowed to solidify at 37°C for 30 minutes. Long-term iEPs were dissociated using 0.05% Trypsin-EDTA (diluted from 0.25%; Gibco, Cat #: 25200056). The cells were resuspended in pre-chilled OVM-medium (William’s E medium, supplemented with 10% FBS, dexamethasone, 10 mM nicotinamide, 2 mM L-glutamine, 0.2 mM ascorbic acid, 20 mM HEPES, 1% penicillin/streptomycin, 1% sodium pyruvate, 0.15% of 7.5% sodium bicarbonate, 14 mM glucose, containing 1x ITS-X (Gibco), 20 ng/ml hepatocyte growth factor (Sigma-Aldrich), and 20 ng/ml epidermal growth factor (Sigma-Aldrich)). The top layer was prepared with 40% Matrigel, with 1.2mg/ml Collagen Type I (Gibco, stock of 3mg/ml), mixed with 20k cells for each well of 96-well plate, or 80k for each well of a 6-well plate. After 30 minutes, the top layer was added to the plate and allowed to solidify and set in the incubator for 45 minutes. After the top layer solidified, pre-warmed OVM medium was added. The medium was changed every other day. After five days of gel culture, the cells were imaged and processed for single-cell RNA-sequencing.
iEP Preparation from Matrigel Culture for Single-Cell Profiling
Cells in 3D gel-culture were dissociated using a combination of Type I Collagenase (Gibco; 100 μl of 500 mg/ml Type I Collagenase in 1 ml of OVM) and 1ml of Gentle cell dissociation reagent (STEMCELL Technologies). Briefly, the medium was carefully pipetted off, and 1 ml of enzyme mix was added to each well of the 6-well plate. The plate was incubated at 37°C for 10 minutes. The partially dissociated gel was collected into a 15-ml Falcon tube, further mixed on a rocker for 15 minutes at room temperature. The cells were then pelleted at 300xg for 5 minutes and washed with 1ml HBSS. The solution was passed through a 27-gauge needle using a 3 ml syringe. The cells were counted, centrifuged, resuspended in 0.04% BSA in 1x DPBS, and passed through a 70 μm cell strainer before loading onto the 10x Chromium Single Cell Chip.
Immunofluorescence Staining of Branching iEPs
iEPs were 3D-cultured in a 96-well glass-bottom plate or glass-bottom 35mm μ-Dish for imaging. For immunostaining, cells were washed with 1x DPBS three times and fixed overnight in 4% paraformaldehyde at 4°C. The fixed sample was then washed twice with 1x DPBS for 15 minutes, permeabilized, and blocked with blocking buffer (0.2% TritonX-100 and 3% FBS in DPBS) for 10 minutes at room temperature. The primary antibodies, EpCAM (RRID:AB_394370) and CK19 (RRID:AB_2281020), were diluted at 1:100 (EpCAM) and 1:200 (CK19) in the blocking buffer. The samples were incubated with the primary antibodies at 4°C overnight (12hr). The sample was then washed for 15 minutes three times. Secondary antibodies Alexa Fluor 546 Goat Anti-rabbit (RRID:AB_2534093), and Alexa Fluor 647 Goat Anti-rat IgG (RRID:AB_141778), were diluted 1:500 (Alexa Fluor 546 and Alexa Fluor 647) in the blocking buffer. 50 μl of 300 nM DAPI (Invitrogen) was added with the secondary antibodies and incubated at 4°C overnight (12hr). The samples were washed again for 15 minutes three times. Cells in 96-wells were imaged as a 3D z-stack using a Zeiss LSM 880 Confocal with Airyscan, with a 40x air objective. Samples in the 35-mm dish were transferred to a slide, covered with a coverslip with ProLong Gold Antifade Mountant (Invitrogen), and imaged using an Olympus FV1200 Confocal Microscope with 40x water objective. Representative images were chosen.
Single-cell profiling
For single-cell library preparation on the 10x Genomics platform, we used: the Chromium Single Cell 3′ Library & Gel Bead Kit v2 (PN-120237), Chromium Single Cell 3′ Chip kit v2 (PN-120236), and Chromium i7 Multiplex Kit (PN-120262), according to the manufacturer’s instructions in the Chromium Single Cell 3′ Reagents Kits V2 User Guide. Prior to cell capture, methanol-fixed cells were placed on ice, then spun at 3000rpm for 5 minutes at 4°C, followed by resuspension and rehydration in PBS, according to Alles et al., 2017. 17,000 cells were loaded per lane of the chip, aiming to capture 10,000 single-cell transcriptomes. The resulting cDNA libraries were quantified on an Agilent Tapestation and sequenced on an Illumina HiSeq 2500. For analysis of cardiomyocyte reprogramming, The Chromium Single Cell 3’ (v2) Reagent Kits (PN-120237, PN-120236, PN-120262) were used to prepare single-cell RNA-seq libraries, according to manufacturer’s guidelines. Libraries were pooled and sequenced on an Illumina NextSeq 550. For motor neuron programming, prior to loading the 10x chip, methanol-fixed cells were counted, spun, resuspended in 1% BSA in PBS, and counted again, according to 10x Genomics methanol fixation protocol. The Chromium Single Cell 3’ (v2) Reagent Kits (PN-120237, PN-120236, PN-120262) were used to prepare single-cell RNA-seq libraries, according to manufacturer’s guidelines. Libraries were pooled and sequenced on an Illumina NextSeq 550.
QUANTIFICATION AND STATISTICAL ANALYSIS
See METHODS DETAILS; Capybara Pipeline Overview for software details and statistical approach. Group sizes and statistical tests are indicated in the text. In all figures, error bars indicate standard deviations. Sample sizes and numbers of replicates are indicated in the figure legends.
Supplementary Material
Supplemental Spreadsheet 1. Other cell-type compositions for all datasets analyzed in this study. Related to Figures 2–6; S2–6.
Supplemental Spreadsheet 2. Detailed cell type classification for Paul et al., data; Related to Figures 2 and S2.
Supplemental Spreadsheet 3. Detailed cell type classification for Weinreb et al., data; Related to Figures 3 and S3.
Supplemental Spreadsheet 4. Detailed cell type classification for Stone et al., data and the cardiac reprogramming in this study; Related to Figures 4 and S4.
Supplemental Spreadsheet 5. Detailed cell type classification for the motor neuron programming in this study; Related to Figures 5 and S5.
Supplemental Spreadsheet 6. Detailed cell type classification for Biddy et al., data the iEP reprogramming in this study; Related to Figures 6 and S6.
Highlights.
Capybara uses reference atlases to measure the identity of single cells
Capybara captures hybrid cell states, supporting a fate transition metric
Defining off-target cell identity yields improved reprogramming protocols
In vivo correlates for poorly defined reprogramming products are identified
Acknowledgments
We thank members of the Morris laboratory for their helpful discussions; Barbara Treutlein for sharing the original QP code; Dennis Oakley of the Washington University Center for Cellular Imaging (WUCCI) supported by Washington University School of Medicine, The Children’s Discovery Institute (CDI-CORE-2015-505 and CDI-CORE-2019-813) and the Foundation for Barnes-Jewish Hospital (3770 and 4642); Lee Grimes and Nathan Salomonis for helpful discussion on hybrid cell identity. Thank you also to Colin. This work was funded by National Institute of General Medical Sciences R01 GM126112, and Silicon Valley Community Foundation, Chan Zuckerberg Initiative Grant HCA2-A-1708-02799, both to S.A.M.; S.A.M. is supported by an Allen Distinguished Investigator Award (through the Paul G. Allen Frontiers Group), a Vallee Scholar Award, a Sloan Research Fellowship, and a New York Stem Cell Foundation Robertson Investigator Award; W.K. is supported by a Douglas Covey Fellowship; E.M.H. is supported by NIH/NHLBI T32 HL007317-44
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
S.A.M. is on the advisory board of Cell Stem Cell.
References
- Abdelaal T, Michielsen L, Cats D, Hoogduin D, Mei H, Reinders MJT, and Mahfouz A (2019). A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biology 20, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alles J, Karaiskos N, Praktiknjo SD, Grosswendt S, Wahle P, Ruffault P-L, Ayoub S, Schreyer L, Boltengagen A, Birchmeier C, et al. (2017). Cell fixation and preservation for droplet-based single-cell transcriptomics. BMC Biology 15, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alquicira-Hernandez J, Sathe A, Ji HP, Nguyen Q, and Powell JE (2019). scPred: accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biology 20, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddy BA, Kong W, Kamimoto K, Guo C, Waye SE, Sun T, and Morris SA (2018). Single-cell mapping of lineage and identity in direct reprogramming. Nature 564, 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JA, Li VC, Lee S, Woolf CJ, Klein A, and Kirschner MW (2017). Mouse embryonic stem cells can differentiate via multiple paths to the same state. ELife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahan P, Li H, Morris SA, Lummertz da Rocha E, Daley GQ, and Collins JJ (2014). CellNet: Network Biology Applied to Stem Cell Engineering. Cell 158, 903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, Meng T, Gao PJ, and Ruan CC (2021). The Role of Brown Adipose Tissue Dysfunction in the Development of Cardiovascular Disease. Frontiers in Endocrinology 12, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin JS, Hamey FK, Pijuan-Sala B, Shepherd M, Lau WWY, Nestorowa S, Weinreb C, Wolock S, Hannah R, Diamanti E, et al. (2018). A single-cell hematopoietic landscape resolves 8 lineage trajectories and defects in Kit mutant mice. Blood 131, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delile J, Rayon T, Melchionda M, Edwards A, Briscoe J, and Sagner A (2019). Single cell transcriptomics reveals spatial and temporal dynamics of gene expression in the developing mouse spinal cord. Development (Cambridge, England) 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePasquale EAK, Schnell DJ, van Camp PJ, Valiente-Alandí Í, Blaxall BC, Grimes HL, Singh H, and Salomonis N (2019). DoubletDecon: Deconvoluting Doublets from Single-Cell RNA-Sequencing Data. Cell Reports 29, 1718–1727.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell JA, Wang Y, Riesenfeld SJ, Shekhar K, Regev A, and Schier AF (2018). Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick J, Subramaniam A, Neumann J, and Robbins J (1991). Isolation and characterization of the mouse cardiac myosin heavy chain genes. The Journal of Biological Chemistry 266, 9180–9185. [PubMed] [Google Scholar]
- Guo C, Kong W, Kamimoto K, Rivera-Gonzalez GC, Yang X, Kirita Y, and Morris SA (2019). CellTag Indexing: genetic barcode-based sample multiplexing for single-cell genomics. Genome Biology 20, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Chaturvedi P, Kishimoto K, Koike H, Nasr T, Iwasawa K, Giesbrecht K, Witcher PC, Eicher A, Haines L, et al. (2020). Single cell transcriptomics identifies a signaling network coordinating endoderm and mesoderm diversification during foregut organogenesis. Nature Communications 2020 11:1 11, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, et al. (2018). Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091–1107.e17. [DOI] [PubMed] [Google Scholar]
- Hong T, Xing J, Li L, and Tyson JJ (2012). A simple theoretical framework for understanding heterogeneous differentiation of CD4 + T cells. BMC Systems Biology 6, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong T, Watanabe K, Ta CH, Villarreal-Ponce A, Nie Q, and Dai X (2015). An Ovol2-Zeb1 Mutual Inhibitory Circuit Governs Bidirectional and Multi-step Transition between Epithelial and Mesenchymal States. PLOS Computational Biology 11, e1004569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, and Srivastava D (2010). Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Ji S, and Sun A (2013). Efficient generation of biliary epithelial cells from rabbit intrahepatic bile duct by Y-27632 and Matrigel. In Vitro Cellular and Developmental Biology - Animal 49, 433–439. [DOI] [PubMed] [Google Scholar]
- Kamimoto K, Kaneko K, Kok CYY, Okada H, Miyajima A, and Itoh T (2016). Heterogeneity and stochastic growth regulation of biliary epithelial cells dictate dynamic epithelial tissue remodeling. ELife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimoto K, Hoffmann C, and Morris S (2020). CellOracle: Dissecting cell identity via network inference and gene function prediction. [DOI] [PMC free article] [PubMed]
- Kiselev VY, Yiu A, and Hemberg M (2018). scmap: projection of single-cell RNA-seq data across data sets. Nature Methods 15, 359–362. [DOI] [PubMed] [Google Scholar]
- Klein AMM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DAA, and Kirschner MWW (2015). Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A, Torre D, Keenan AB, Jagodnik KM, Lee HJ, Wang L, Silverstein MC, and Ma’ayan A (2018). Massive mining of publicly available RNA-seq data from human and mouse. Nature Communications 9, 1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Ramírez R, Zieger E, and Schubert M (2013). Retinoic acid signaling in spinal cord development. The International Journal of Biochemistry & Cell Biology 45, 1302–1313. [DOI] [PubMed] [Google Scholar]
- Lewis PL, Su J, Yan M, Meng F, Glaser SS, Alpini GD, Green RM, Sosa-Pineda B, and Shah RN (2018). Complex bile duct network formation within liver decellularized extracellular matrix hydrogels. Scientific Reports 2018 8:1 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Bach K, and Marioni JC (2016). Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biology 17, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean AL, Hong T, and Nie Q (2018). Exploring intermediate cell states through the lens of single cells. Current Opinion in Systems Biology 9, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- la Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni EO, Mahony S, Closser M, Morrison CA, Nedelec S, Williams DJ, An D, Gifford DK, and Wichterle H (2013). Synergistic binding of transcription factors to cell-specific enhancers programs motor neuron identity. Nature Neuroscience 16, 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Campbell KR, Lun ATL, and Wills QF (2017). Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 347, btw777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Systems 8, 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Cahan P, Li H, Zhao AM, San Roman AK, Shivdasani RA, Collins JJ, and Daley GQ (2014). Dissecting Engineered Cell Types and Enhancing Cell Fate Conversion via CellNet. Cell 158, 889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S, Setty M, Kuo Y-Y, Liu V, Garg V, Sharma R, Simon CS, Saiz N, Gardner R, Boutet SC, et al. (2019). The emergent landscape of the mouse gut endoderm at single-cell resolution. Nature 2019 569:7756 569, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M, Ogawa S, Bear CE, Ahmadi S, Chin S, Li B, Grompe M, Keller G, Kamath BM, and Ghanekar A (2015). Directed differentiation of cholangiocytes from human pluripotent stem cells. Nature Biotechnology 2015 33:8 33, 853–861. [DOI] [PubMed] [Google Scholar]
- Okabe M, Tsukahara Y, Tanaka M, Suzuki K, Saito S, Kamiya Y, Tsujimura T, Makamura K, and Miyajima A (2009). Potential hepatic stem cells reside in EpCAM+ cells of normal and injured mouse liver. Development 136, 1951–1960. [DOI] [PubMed] [Google Scholar]
- Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, and Grimes HL (2016). Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 2016 537:7622 537, 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH, and Zon LI (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, Winter D, Lara-Astiaso D, Gury M, Weiner A, et al. (2015). Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 163, 1663–1677. [DOI] [PubMed] [Google Scholar]
- Pepe-Mooney BJ, Dill MT, Alemany A, Ordovas-Montanes J, Matsushita Y, Rao A, Sen A, Miyazaki M, Anakk S, Dawson PA, et al. (2019). Single-Cell Analysis of the Liver Epithelium Reveals Dynamic Heterogeneity and an Essential Role for YAP in Homeostasis and Regeneration. Cell Stem Cell 25, 23–38.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijuan-Sala B, Griffiths JA, Guibentif C, Hiscock TW, Jawaid W, Calero-Nieto FJ, Mulas C, Ibarra-Soria X, Tyser R.C. v., Ho DLL, et al. (2019). A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliner HA, Shendure J, and Trapnell C (2019). Supervised classification enables rapid annotation of cell atlases. Nature Methods 16, 983–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu J, and Srivastava D (2012). In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485, 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribes V, le Roux I, Rhinn M, Schuhbaur B, and Dollé P (2009). Early mouse caudal development relies on crosstalk between retinoic acid,Shh and Fgf signalling pathways. Development 136, 665–676. [DOI] [PubMed] [Google Scholar]
- Sagner A, and Briscoe J (2019). Establishing neuronal diversity in the spinal cord: a time and a place. Development (Cambridge, England) 146. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nature Biotechnology 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler KM, Waye SE, Kong W, Kamimoto K, Bajinting A, Goo WH, Onufer EJ, Courtney C, Guo J, Warner BW, et al. (2019). Single-Cell Analysis Reveals Regional Reprogramming During Adaptation to Massive Small Bowel Resection in Mice. Cellular and Molecular Gastroenterology and Hepatology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya S, and Suzuki A (2011). Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 475, 390–393. [DOI] [PubMed] [Google Scholar]
- Song K, Nam Y-J, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, et al. (2012). Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Soysa TY, Ranade SS, Okawa S, Ravichandran S, Huang Y, Salunga HT, Schricker A, Del Sol A, Gifford CA, and Srivastava D (2019). Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 572, 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone NR, Gifford CA, Thomas R, Pratt KJB, Samse-Knapp K, Mohamed TMA, Radzinsky EM, Schricker A, Ye L, Yu P, et al. (2019). Context-Specific Transcription Factor Functions Regulate Epigenomic and Transcriptional Dynamics during Cardiac Reprogramming. Cell Stem Cell 25, 87–102.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabula Muris Consortium, Overall coordination, Logistical coordination, Organ collection and processing, Library preparation and sequencing, Computational data analysis, Cell type annotation, Writing group, Supplemental text writing group, and Principal investigators (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, and Cahan P (2019). SingleCellNet: A Computational Tool to Classify Single Cell RNA-Seq Data Across Platforms and Across Species. Cell Systems 9, 207–213.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein B, Lee QY, Camp JG, Mall M, Koh W, Shariati SAM, Sim S, Neff NF, Skotheim JM, Wernig M, et al. (2016). Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco S, Ibrahim MM, Kakumanu A, Garipler G, Aydin B, Al-Sayegh MA, Hirsekorn A, Abdul-Rahman F, Satija R, Ohler U, et al. (2017). A Multi-step Transcriptional and Chromatin State Cascade Underlies Motor Neuron Programming from Embryonic Stem Cells. Cell Stem Cell 20, 205–217.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhulst S, Roskams T, Sancho-Bru P, and van Grunsven LA (2019). Meta-Analysis of Human and Mouse Biliary Epithelial Cell Gene Profiles. Cells 2019, Vol. 8, Page 1117 8, 1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Cao N, Spencer CI, Nie B, Ma T, Xu T, Zhang Y, Wang X, Srivastava D, and Ding S (2014). Small Molecules Enable Cardiac Reprogramming of Mouse Fibroblasts with a Single Factor, Oct4. Cell Reports 6, 951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu Z, Yin C, Asfour H, Chen O, Li Y, Bursac N, Liu J, and Qian L (2015). Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circulation Research 116, 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb C, Rodriguez-Fraticelli A, Camargo FD, and Klein AM (2020). Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, and Jessell TM (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397. [DOI] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biology 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Göttgens B, Rajewsky N, Simon L, and Theis FJ (2019). PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biology 20, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-Y, Whye D, Mason RW, and Wang W (2012). Efficient Differentiation of Mouse Embryonic Stem Cells into Motor Neurons. Journal of Visualized Experiments e3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Liu Z, Wang H, Zhang Y, Zhou J, Lin Q, Wang Y, Duan C, and Wang C (2010). Efficient isolation of cardiac stem cells from brown adipose. Journal of Biomedicine and Biotechnology 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Wang X. di, Yokoyama SI, Fukuda N, and Takakura N (2006). Cardiac progenitor cells in brown adipose tissue repaired damaged myocardium. Biochemical and Biophysical Research Communications 342, 662–670. [DOI] [PubMed] [Google Scholar]
- Zhou P, Wang S, Li T, and Nie Q (2021). Dissecting transition cells from single-cell transcriptome data through multiscale stochastic dynamics. Nature Communications 2021 12:1 12, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Spreadsheet 1. Other cell-type compositions for all datasets analyzed in this study. Related to Figures 2–6; S2–6.
Supplemental Spreadsheet 2. Detailed cell type classification for Paul et al., data; Related to Figures 2 and S2.
Supplemental Spreadsheet 3. Detailed cell type classification for Weinreb et al., data; Related to Figures 3 and S3.
Supplemental Spreadsheet 4. Detailed cell type classification for Stone et al., data and the cardiac reprogramming in this study; Related to Figures 4 and S4.
Supplemental Spreadsheet 5. Detailed cell type classification for the motor neuron programming in this study; Related to Figures 5 and S5.
Supplemental Spreadsheet 6. Detailed cell type classification for Biddy et al., data the iEP reprogramming in this study; Related to Figures 6 and S6.
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available. Accession numbers are listed in the key resources table. All original code, along with tutorials is available at: https://github.com/morris-lab/Capybara. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Monoclonal Anti-MYL7 Antibody (B-10) | Santa Cruz Biotechnology | RRID:AB_10848272 |
| Rabbit Monoclonal Anti-MYL2 Antibody | Abcam | RRID:AB_10563535 |
| Rat Monoclonal Anti-Mouse CD326 (EpCAM) Antibody | BD Biosciences | RRID:AB_394370 |
| Rabbit Monoclonal Anti-Cytokeratin 19 (CK19) Antibody | Abcam | RRID:AB_2281020 |
| CD90.2 (Thy1.2) Monoclonal Antibody, FITC | Invitrogen | RRID:AB_273503 |
| Alexa Fluor 546 Goat Anti-rabbit IgG | Invitrogen | RRID:AB_2534093 |
| Alexa Fluor 488 Goat Anti-mouse IgG | Invitrogen | RRID:AB_2534088 |
| Alexa Fluor 647 Goat Anti-rat IgG | Invitrogen | RRID:AB_141778 |
| Bacterial and Virus Strains | ||
| Stellar Competent Cells | Takara Bio | Cat #: 636763 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fetal bovine serum (FBS) | Gibco | Cat #: 10082147 |
| Fibroblast Medium-2 | ScienCell Research Laboratories | Cat #: 2331 |
| Matrigel (GFR Membrane Matrix) | Corning | Cat #: CB-40230 |
| β-mercaptoethanol | Life Technologies | Cat #: 21985023 |
| X-tremeGENE9 Transfection Reagent | Sigma Aldrich | Cat #: 6365779001 |
| XAV939 | Cayman | Item #: 13031 |
| SB431542 | Cayman | Item #: 13596 |
| CHIR99021 | BioVision | Cat #: 1677 |
| PD0325901 | Sigma | Cat #: PZ0162 |
| Leukemia Inhibitory Factor | Millipore | Cat #: LIF2050 |
| Retinoic Acid (RA) | ||
| Smoothened Agonist (SAG) | Millipore | Cat #: 566660 |
| Epidermal Growth Factor | Sigma Aldrich | Cat #: E5160 |
| Hepatocyte Growth Factor | Sigma Aldrich | Cat #: H9661 |
| Doxycycline (Dox) | Sigma Aldrich | Cat #: D9891 |
| L-Ascorbic Acid | Sigma Aldrich | Cat #: A8960 |
| Insulin-Transferrin-Selenium-Ethanolamine (ITS-X) | Gibco | Cat #: 51500056 |
| Gentle Cell Dissociation Reagent | STEMCELL Technologies | Cat #: 100–0485 |
| Critical Commercial Assays | ||
| RNAscope Multiplex Fluorescent v2 kit | Advanced Cell Diagnostics | Cat #: 323100 |
| EasySep Mouse FITC Positive Selection Kit II | STEMCELL Technologies | Cat #: 17668 |
| Ampure XP SPRI Beads | Beckman | B23318 |
| Chromium Single Cell 3’ Library and Gel Bead Kit v2 | 10x Genomics | PN-120237 |
| Chromium Single Cell 3’ Chip kit v2 | 10x Genomics | PN-120236 |
| Chromium i7 Multiplex Kit | 10x Genomics | PN-120262 |
| Chromium Next GEM Chip G Single Cell Kit | 10x Genomics | PN-1000127 |
| Library Construction Kit | 10x Genomics | PN-1000196 |
| Chromium Next GEM Single Cell 3’ GEM Kit v3.1 | 10x Genomics | PN-1000130 |
| Chromium Next GEM Single Cell 3’ Gel Bead Kit v3.1 | 10x Genomics | PN-1000129 |
| Dual Index Kit TT Set A | 10x Genomics | PN-1000215 |
| Deposited Data | ||
| scRNA-seq | This paper | GEO: GSE145251 |
| Hematopoiesis Development | Paul et al., 2015 | GEO: GSE72859 |
| Spinal Motor Neuron Differentiation and Programming | Briggs et al., 2017 | GEO: GSE97391 |
| Cardiac Reprogramming | Stone et al., 2019 | GEO: GSE131328 |
| MEF to iEP Reprogramming Time course | Biddy et al., 2018 | GEO: GSE99915 |
| Normal and Post Injury Hepatocytes and BECs | Pepe-Mooney et al., 2019 | GEO: GSE125688 |
| Mouse Gastrulation Atlas | Pijuan-Sala et al., 2019 | GEO: GSE87038 |
| Mouse Cell Atlas | Han et al., 2018 | https://figshare.com/articles/MCA_DGE_Data/5435866 |
| Tabula Muris | Tabula Muris Consortium et al., 2018 | https://figshare.com/projects/Tabula_Muris_Transcriptomic_characterization_of_20_organs_and_tissues_from_Mus_musculus_at_single_cell_resolution/27733 |
| Developing Mouse Spinal Cord Atlas | Delile et al., 2019 | E-MTAB-7320 |
| Experimental Models: Cell Lines | ||
| Mouse Cardiac Fibroblasts (CD1, P0) | ScienCell Research Laboratories | Cat #: M6300 |
| 293T-17 Cells | ATCC | RRID:CVCL_1926 |
| Primary Mouse Embryonic Fibroblast (C57BL/6, E13.5) | ||
| NIL-V5 inducible ESC line | Mazzoni et al., 2013 | |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | The Jackson laboratory | RRID:IMSR_JAX:000664 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Seurat V4 | Satija et al., 2015; Butler et al., 2018; Stuart et al., 2019 | https://satijalab.org/seurat/articles/get_started.html |
| Quadprog | Turlach and Weingessel, 2007 | https://cran.r-project.org/web/packages/quadprog/index.html |
| Cell Ranger v5.0.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| Velocyto | La Manno et al., 2018 | http://velocyto.org/ |
| MASS | Venables and Ripley, 2002 | https://cran.r-project.org/web/packages/MASS/MASS.pdf |
| mixdist | Macdonald and Du, 2018 | https://cran.r-project.org/web/packages/mixdist/mixdist.pdf |
| Splatter | Zappia et al., 2017 | https://github.com/Oshlack/splatter |
| OpenImageR | Mouselimis, 2021 | https://cran.r-project.org/web/packages/OpenImageR/OpenImageR.pdf |
| PAGA | Wolf et al., 2019 | https://github.com/theislab/paga |
| SCANPY | Wolf et al., 2018 | https://scanpy.readthedocs.io/en/stable/ |
| R-4.0.1 | R Core Team, 2021 | https://www.r-project.org/ |
| RStudio | RStudio Team, 2020 | https://www.rstudio.com/ |
| Capybara | This Paper | https://github.com/morris-lab/Capybara |
| Recombinant DNA | ||
| pMx-MGT | Wang et al., 2015 | RRID:Addgene_111810 |
| pGCDNSam-Hnf4α-t2a-Foxa1 | Morris et al., 2014 | |
| pCL-Eco | Novus Biologicals | RRID:Addgene_12371 |
| Other | ||
| RNAscope probe Mm-Tnnc1-C3 | Advanced Cell Diagnostics | Cat #: 511011-C3 |
| Opal 520 Reagent Pack | Akoya | FP1487001KT |
| Opal 570 Reagent Pack | Akoya | FP1488001KT |
| Opal 690 Reagent Pack | Akoya | FP1497001KT |