

Abstract
A polyvinyl alcohol (PVA) hydrogel loaded with guava leaf extract (GLE) has potential applications as a wound dressing with good antibacterial activity. This study succeeded in fabricating a PVA hydrogel containing GLE using the freeze–thaw (FT) method. By varying the GLE concentration, we can adjust the physical properties of the hydrogel. The addition of GLE results in a decrease in cross-linking during gelation and an increase in the pore size of the hydrogels. The increase of the pore size made the swelling increase and the mechanical strength decrease. The weight loss of the hydrogel also increases because the phosphate buffer saline (PBS) dissolves the GLE. Increasing the GLE concentration caused the Fourier-transform infrared (FTIR) absorbance peaks to widen due to hydrogen bonds formed during the FT process. The crystalline phase was transformed into an amorphous phase in the PVA/GLE hydrogel based on the X-ray diffraction (XRD) spectra. The differential scanning calorimetry (DSC) characterization showed a significant decrease in the hydrogel weight over temperatures of 30–150 °C due to the evaporation of water from the hydrogel matrix. The zone of inhibition of the PVA/GLE hydrogel increased with antibacterial activity against Staphylococcus aureus of 17.93% per gram and 15.79% per gram against Pseudomonas aeruginosa.
A polyvinyl alcohol (PVA) hydrogel loaded with guava leaf extract (GLE) has potential applications as a wound dressing with good antibacterial activity.
Introduction
Guava plants (Psidium guajava) with a high bioactive content are widespread and thriving in Indonesia.1 Guava plants have been widely used to cure fever, diarrhea, diabetes, hypertension, gingivitis, rheumatism, and inflammation. It has been shown that they also accelerate wound healing.2 Guava leaves contain high levels of bioactive molecules, including terpenoids, tannins, steroids, flavonoids, saponins, and alkaloids.3 Flavonoids are natural compounds with a polyphenolic structure consisting of two aromatic rings connected via a heterocyclic pyranic ring.4 One derivative of flavonoids is quercetin (3,3′,4′,5,7-pentahydroxyflavone).5 Quercetin has been shown to contain antioxidants, anti-inflammatory and neuroprotective, anti-cancer, antimicrobial, and anti-allergic compounds.6–9 The quercetin content in guava leaf extract (GLE) is around 2.15–6.78% (w/w).10,11
The high quercetin content in GLE makes the guava plant a good choice for further development and application in medicine and the field of functional food. The current challenges are to find the correct encapsulation method, which is easy, low-cost, and has good performance. The encapsulation method of bioactive materials that is currently being developed is the hydrogel.
Hydrogels are polymers with a three-dimensional cross-linked structure containing hydrophilic groups capable of absorbing water.12 Hydrogels can be made using natural polymers such as alginate, chitosan, gelatin, starch, gellan gum, and cellulose.13–18 In addition, hydrogels manufactured using synthetic polymers such as polyvinyl alcohol (PVA), polyvinyl pyrrolidone (PVP), polyethylene glycol (PEG), and polyacrylic acid (PAA) have the advantage of better mechanical properties.19–22 PVA is the polymer most widely used for hydrogels because it can produce hydrogels with good mechanical properties and is biocompatible.23 PVA-based hydrogel has been successfully applied as a wound dressing, a drug delivery system, and tissue engineering.24–26
The use of hydrogels as wound coverings has been extensively studied since the skin, as the outer covering of the body, is the largest organ that protects internal organs and maintains normal body temperature.27 If there is damage to the skin tissue due to an injury, the function of the skin will be impaired.
Wounds left untreated are vulnerable to infection by bacteria, which ultimately affects the wound healing process.28 According to the World Health Organization (WHO), wound infection causes many deaths every year worldwide. This high mortality rate is caused by poor wound healing abilities and wound covers that cannot kill bacteria in the wound.29 The ideal wound cover must have good mechanical properties (especially flexibility), be non-toxic and hypo-allergenic, absorb wound exudate, prevent bacterial infection, and maintain the necessary moisture around the damage for accelerated wound healing.30
Recently, several researchers have reported the synthesis of PVA hydrogels for wound dressing applications.31–34 PVA has no natural antibacterial activity, so they have combined PVA with active ingredients that have antibacterial activity or have used a combination of natural polymers. Kumar et al. have combined PVA/chitosan with silver nanoparticles to obtain a hydrogel with excellent mechanical properties and promising antibacterial activity.35 However, the scanning electron microscope (SEM) images showed small uniform porosity in this hydrogel due to the physical interaction between the PVA and the chitosan. In their research, Swaroop and Somashekarappa have reported a decrease in the degree of swelling of the PVA hydrogel with the addition of ZnO nanoparticles synthesized by gamma radiation.36 Juby et al. have also previously succeeded in fabricating hydrogels with a mixture of silver nanoparticles, PVA, and gum acacia (polysaccharides) using gamma radiation.37 The addition of acacia gum affected biocompatibility, decreased antibacterial activity against Gram-negative bacteria, and increased the degree of swelling of the hydrogel.
Hydrogel fabrication with gamma radiation has a greater cost and requires more complex equipment than the physical cross-linking method. For the application of wound dressings, a hydrogel fabrication with an easy and inexpensive physical cross-linking method is needed, which also has pores that can absorb wound exudates, good mechanical strength, and antibacterial activity that can inhibit the growth of Gram-positive and Gram-negative bacteria.
We have prepared PVA/guava leaf extract (GLE) hydrogels by varying the GLE concentration using the freeze–thaw (FT) method. The FT method can produce a non-toxic hydrogel because the synthesis process does not require a chemical cross-linking agent.38 During the freezing process, the PVA polymer networks form intermolecular and intramolecular hydrogen bonds, as well as crystallites in which the PVA chains aggregate with ice crystals. The GLE in the PVA polymer network can affect the formation of crystallites, resulting in pores in the hydrogel structure. The presence of pores in the hydrogel structure, the high degree of swelling, and GLE, which has antibacterial activity, make the PVA/GLE hydrogel a good candidate for a wound dressing.
Experimental
Materials
Polyvinyl alcohol (PVA) with molecular weight in the range of 89 000–98 000 Da was purchased from Sigma-Aldrich, Singapore. Dried guava leaves were bought from Babah Kuya's local herbal store, Bandung, Indonesia. Deionized water and technical grade ethanol were purchased from Brataco Chemistry, Indonesia.
Guava leaf extraction
The extraction of guava leaves was carried out by the maceration method as described in our previous study.39 The guava leaf powder (1000 g) was weighed out, then immersed in 10 L of technical grade ethanol. The macerated solution was collected and filtered once every day during this maceration process (three days). The collected macerate was then evaporated using a rotary evaporator to obtain guava leaf extract (GLE) paste. Based on this extraction method, we obtained 165.44 g of GLE paste. Furthermore, this paste was dried using the freeze-drying method at −50 °C for 24 h. Then the frozen paste was placed into a vacuum chamber at a temperature up to 38 °C in the pressure of 8 mBar for 24 h, so that sublimation occurred and the ice in the sample evaporated, producing completely dry GLE.
Preparation of the PVA/GLE hydrogel
The 10 wt% PVA solution was prepared by dissolving 10 g of PVA powder in 90 g of deionized water, then stirring it using a magnetic stirrer at 108 °C for 4 h until a homogeneous solution was obtained. The GLE solution was prepared at a concentration of 10 wt% with deionized water as a solvent, then stirred at 40 °C until it was homogeneous. The precursor solutions were then prepared with the ratios shown in Table 1.
Weight compositions of the hydrogel solutions.
| Sample | PVA/GLE weight ratio | PVA mass (g) | GLE mass (g) |
|---|---|---|---|
| PGL0 | 10 : 0 | 15 | 0 |
| PGL1 | 10 : 1 | 15 | 1.5 |
| PGL2 | 10 : 2 | 15 | 3 |
| PGL3 | 10 : 3 | 15 | 4.5 |
| PGL4 | 10 : 4 | 15 | 6 |
| PGL5 | 10 : 5 | 15 | 7.5 |
The hybrid solutions prepared in this way were then put in the freezer at −25 °C for 20 hours (freeze) then stored at room temperature for four hours (thaw). For each sample, the FT process was repeated six times.
Hydrogel density
As density is a measurement of the mass per unit volume, hydrogel pieces were prepared and weighed and their dimensions were measured. The density (ρ) of hydrogel was determined by using
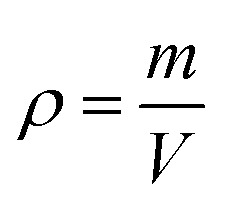 |
1 |
where m corresponds to the mass of the hydrogel (g) and V is the volume of the hydrogel (mL).
Hydrogel morphology
Morphological characterizations of the PVA/GLE hydrogels were performed using a scanning electron microscope (SEM) (JSM-6510LA, JEOL, USA). SEM microphotographs with magnifications of 200× and 1000× were used to observe pore size and cross-linking density of the hydrogels after freeze-drying.
Hydrogel pore size. High porosity is beneficial for the absorption of wound exudate and the transfer of nutrients and oxygen to the cells covered by the wound dressing.40 Then, hydrogel pore size was determined by using ImageJ software (version 64-bit Java 1.8.0_172, NIH, USA).41 Pore sizes of the hydrogels were obtained from SEM images with magnifications of 200×. The number of images analyzed from each hydrogel was 4 images in which each image had a size of 325 μm × 242.5 μm. By calibrating the ImageJ software using SEM images with known sizes, we determined the pore size and then plotted the pore size distribution of the hydrogel.
Degree of swelling
The maximum amount of liquid that can be absorbed and held by hydrogel is measured by the degree of swelling. This test uses a phosphate buffer saline (PBS) solution adjusted to the pH of the wound surface (7.4 for chronic wounds). The measurement of the degree of swelling was initiated by drying the hydrogel to a constant weight. Next, the sample was immersed in PBS at 37 °C and then it was weighed 0, 3, 6, 9, 12, 24, and 48 h after immersion. This test aims to investigate the absorption capacity of hydrogels for body fluids at various precursor compositions. The degree of swelling (SD) of hydrogel was obtained from42
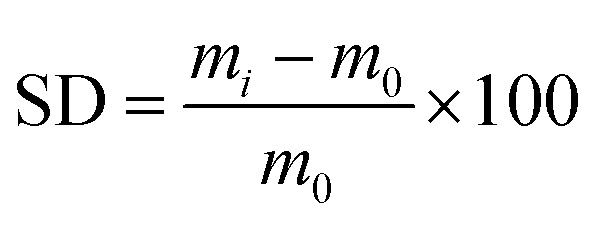 |
2 |
where m0 represents the mass of the dry hydrogel and mi is the mass of the swollen hydrogel at a specific time (gram).
Weight loss
Weight loss expresses the number of gel fractions remaining after the hydrogel was dissolved in PBS. The identification of weight loss was made by drying the hydrogel at a temperature of 50 °C to remove water content. Next, the hydrogel was immersed in a PBS solution with a pH of 7.4 for 48 h at 37 °C. The soaked hydrogel was dried again in the oven to determine the mass of the remaining gel. The amount of insoluble gel indicates the number of cross-links formed in the hydrogel. The weight loss (WL) was calculated using43
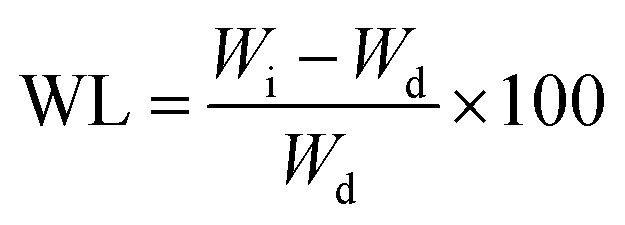 |
3 |
where the weight of the hydrogel after heating is Wi and the weight of the hydrogel after soaking in PBS and drying is Wd.
Fourier transform infrared (FTIR) spectroscopy
The functional group interactions of the hydrogel were characterized using FTIR spectroscopy (IRPrestige21, Shimadzu, Japan). The FTIR spectra of the hydrogel were collected to determine the structural changes that occur in the PVA hydrogel loaded with GLE. The wavenumber range used was 500–4000 cm−1 with the wavenumber resolution of 1.42 cm−1. The transmittance data obtained from various samples were then analyzed to determine any bond structure changes.
X-ray diffraction (XRD) analysis
Crystallinity is one of the factors that significantly affect the mechanical properties of polymers. Hydrogel crystal phase identification was made using an X-ray diffractometer (PW 1700, Philips, USA). The X-ray source used Cu-Kα radiation at 40 kV and 40 mA; 2θ was from 10 to 70°.
Thermal properties
The thermal properties of the hydrogel are used to determine the state of the polymer and evaluate the interactions between the polymer molecules in the hydrogel.44 The resulting differential scanning calorimetry (DSC) curves allow the determination of the melting temperature (Tm) and calculation of the degree of crystallinity (Xc) of hydrogel samples.23 The thermal properties of the PVA powder, GLE powder, and PVA/GLE hydrogels were characterized using a differential scanning calorimetry (DSC) (STA PT1600, Linseis, USA). The measurement was carried out from 30 °C to 600 °C with a temperature increase rate of 10 °C min−1. The degree of crystallinity (Xc) was obtained from 45
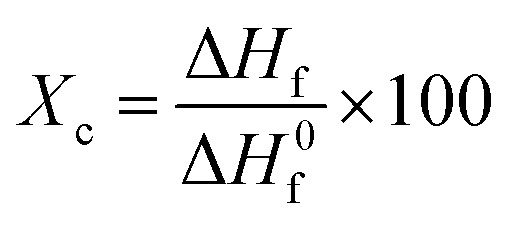 |
4 |
where ΔHf is the melting enthalpy and ΔH0f refers to the melting enthalpy of a fully crystalline polymer (for PVA 138.60 J g−1).46
Mechanical properties
The measurement method often used to determine the elasticity of hydrogels is the compression test.47 The test is carried out by placing the hydrogel between two plates and compressing it. The pressure applied to the hydrogel surface and the change in the hydrogel compression distance are used to obtain a stress–strain curve.48 The compression stress is the ratio of the compressive force to the hydrogel area. In contrast, the compression strain is the ratio of the change in the hydrogel's height when subjected to a compressive force.49 The tool used was a Universal Tensile Machine (SM-200, Sinowon, China). Samples were prepared in blocks with dimensions of 20 mm × 5 mm × 3 mm. The compression modulus was determined by calculating the slope of the linear stress–strain fitting curve.50 In addition, the compressive strength of the hydrogel was calculated using49
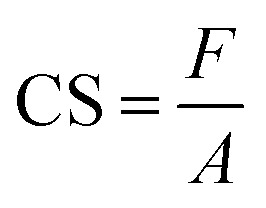 |
5 |
where F represents the compressive force (N) and A is the area of the compressed plane (m2).
Antibacterial study
The PVA/GLE hydrogel antibacterial activity test was performed using the Kirby–Bauer (KB) test and the total plate count (TPC) method against Staphylococcus aureus (ATCC 6538) and Pseudomonas aeruginosa (ATCC 9027). Testing the PVA/GLE hydrogels using this method started with preparing a disc-shaped sample of the hydrogel with a diameter of 20 mm and a thickness of 5 mm. Then the hydrogel was placed over Mueller–Hinton Agar (MHA) solid in a Petri dish. Each hydrogel sample was placed in a separate Petri dish and an inoculum of either S. aureus (1.3 × 106 CFU mL−1) or P. aeruginosa (4.5 × 105 CFU mL−1) was poured on top. The zone of inhibition was measured after 24 h of incubation at 37 °C.
A 10 μL aliquot was taken for each sample and diluted in a physiological sodium chloride solution several times to obtain a calculated bacterial concentration as described in our previous work.51 One mL of diluted bacterial suspension was taken and rubbed evenly onto MHA in a Petri dish, then incubated for 24 h at 37 °C. After incubation, the bacterial colonies from the dilution were counted to determine the amount of bacterial-growth inhibition. The hydrogel antibacterial activity was determined by using51
 |
6 |
The calculation of the number of normal colonies based on the Food & Drug Administration (FDA) standards is 25–250 colonies.52
Statistical analysis
The following experiments, e.g., solution properties, hydrogel density, degree of swelling, weight loss, Young's modulus, and compressive strength were repeated three times. For the antibacterial study, the KB test and the TPC method were repeated three and two times, respectively. Specifically, for the TPC method, the number of samples for each variation in the composition of the PVA/GLE hydrogel was two (n = 2).
The results were expressed in mean ± standard deviation (SD). Statistical differences between groups were analyzed using one-way ANOVA followed by Tukey's HSD (honestly significant difference) post hoc test.53 This statistical test employed the IBM SPSS 20 software (IBM, USA) to determine a significant difference with the confidence level higher than 95% (p < 0.05).54 Values with different superscript letters were significantly different (p < 0.05).
Results and discussion
Solution properties
The PGL0 solution, which is 10 wt% PVA, has distinctive physical characteristics: colourless, odourless, slightly sticky, and thick (Fig. 1a). Noting that the GLE solution is brownish-green and smelly, the addition of the GLE solution into the PVA solution caused the color of the PVA/GLE solution (which are the PGL1-PGL5 solutions) to become diluted.
Fig. 1. (a) Precursor solutions with variation in GLE content, (b) hydrogel PVA/GLE produced by the FT method, (c) solution viscosity, and (d) the density of hydrogel. The values in (c) and (d) are expressed as mean ± SD of 3 samples (n = 3). The * sign indicates a significant difference between groups (p < 0.05).

Moreover, as shown in Fig. 1c, and Table 2, the viscosity, and the density of the PVA/GLE solution decreased because of more solvent contained in the GLE solution. The addition of the solvent made the ratio of the mass of the PVA to the total mass of the solution to decrease. The PGL0 solution had the viscosity and the density of 139.7 ± 1.8u cP and 1.045 ± 0.013u g cm−3, respectively. These values reduced to be 44.0 ± 2.6z cP and 1.028 ± 0.015u g cm−3 for the PGL5 solution. There was a significant difference between groups (p < 0.05) in the solution viscosity due to the addition of GLE but the solution density showed no significant difference (p > 0.05).
Solution properties and hydrogel densitya.
| Sample | Viscosity (cP) | Solution density (g cm−3) | Hydrogel density (g cm−3) |
|---|---|---|---|
| PGL0 | 139.7 ± 1.8u | 1.045 ± 0.013u | 1.32 ± 0.05u |
| PGL1 | 111.4 ± 1.7v | 1.043 ± 0.004u | 1.29 ± 0.02u |
| PGL2 | 94.2 ± 2.3w | 1.041 ± 0.032u | 1.16 ± 0.05v |
| PGL3 | 76.7 ± 2.2x | 1.036 ± 0.030u | 1.15 ± 0.01v |
| PGL4 | 54.2 ± 1.1y | 1.029 ± 0.020u | 1.08 ± 0.01v |
| PGL5 | 44.0 ± 2.6z | 1.028 ± 0.015u | 0.99 ± 0.02w |
The values are expressed as mean ± SD of the 3 samples (n = 3). The use of superscripts on each column represent the statistical significant differences between groups. The groups with the same superscripts within each column indicate that the values are considered not significantly different (p > 0.05).
Hydrogel density
The freeze–thaw (FT) method changed the sample from a solution to a gel, as shown in Fig. 1b. The FT method works through the formation of solvent molecule crystallites, which generates cross-linking and hydrogen bonding with water to form a hydrogel.
The measurement results depicted in Fig. 1d and listed in Table 2 showed a decrease in the hydrogel density due to changes in the internal structure of the hydrogel with the addition of the GLE.
The freezing stage results in the formation of hexagonal networks of hydrogen bonds with the water molecules present in the hydrogel network.55 The one-way ANOVA test showed that there was a significant difference between groups (p < 0.05) in the hydrogel density. The PGL0 and PGL1 hydrogels significantly had the highest density compared to other groups. For the PGL2, PGL3, and PGL4 hydrogels, they have densities ranging from 1.08 to 1.16 g cm−3. There was no significant difference between these groups (p > 0.05).
The PGL5 hydrogel (Fig. 1b and c) is more tenuous than the PGL0 hydrogel. Our initial assumption was that pores are formed, which will increase in size with GLE concentration. Furthermore, we investigated this assumption using an SEM.
Hydrogel morphology
The morphological observations of the PVA/GLE freeze-dried hydrogels were intended to evaluate the hydrogel porosity distribution under dry conditions.56 As shown in Fig. 2a, the pore size greatly affects the feasibility of the hydrogels for applications in the biomedical field. The PGL0 hydrogel (pure PVA hydrogel) shows a porous hydrogel morphology, which correlates with existing studies.57,58
Fig. 2. (a) Morphologies of PVA/GLE hydrogels with different GLE concentrations (PGL0, PGL1, PGL2, PGL3, PGL4, and PGL5), (b) pore size distributions of PVA/GLE hydrogels based on non-linear curve fitting on OriginPro 8.5.1 SR2 software with the Gaussian as the peak function.

In the PGL1, PGL2, PGL3, PGL4, and PGL5 hydrogels, when the GLE concentration was increased, it was seen that the pore sizes of the hydrogels had increased. Thangprasert et al. who investigated the effect of the addition of PVA concentrate on the resulting gelatin/PVA hydrogel morphology, concluded that a decrease in PVA concentration would cause an increase in the pore size.26 The presence of PVA as a cross-linking agent decreases when GLE is added. Thus, in the present study, the sample PGL5 had the largest water content. When the hydrogel is freeze-dried, a sublimation process (ice crystal evaporation) occurs in the hydrogel and leaves a large pore.
Hydrogel pore size
The pore characteristics of hydrogels, such as pore structure, pore size, and porosity, can affect properties such as the absorbance capacity of water, swelling rate, mechanical strength, separation efficiency, and degree of sensitivity.59 The optimal pore diameter for neovascularization (blood vessel) applications is 5 μm; for fibroblast growth, it is 5–15 μm; for skin regeneration of adult mammals, 20–125 μm; and for bone regeneration, 100–350 μm.40
The pore size distribution of each hydrogel given in Fig. 2b was non-linearly fitted to the Gaussian function using OriginPro 8.5.1 SR2 software (version 8.5.1 SR2, OriginLab Corporation, Northampton, MA, USA) to determine its average pore diameter. The results of pore size measurements in detail to see their distribution can be seen in Table S1†. In principle, the hydrogel pore size can be controlled by varying the freezing temperature in the freeze-drying process, resulting in hydrogel pores with a diameter of 1–250 μm.60 In this study, however, the freeze-drying parameters used were kept constant. Therefore, the hydrogel pore size was only influenced by the variation of the precursor composition.
The average pore diameter of the PGL0 (pure PVA) hydrogel was 20.17 ± 5.62a μm. On the other hand, Ceylan et al. synthesized PVA hydrogels using PVA with a molecular weight of 89 000–98 000 Da (in which the molecular weight was identical to that in the present study) using the FT method (freeze temperature of −16 °C for 20 h, thawing at room temperature for 3 h, and one FT cycle).58 They obtained the average pore diameter of 3.69 μm, which is smaller than that of the PGL0. The reason is due to the differences in the freeze temperature and the number of FT cycles, resulting in different degrees of cross-linking and size of the ice crystals formed during the synthesis process.
The average pore diameters of the PGL0 and PGL1 hydrogels had no significant difference (p > 0.05). The statistical test is given in Table S2† also did not show a significant difference in the average pore diameter between PGL2 and PGL3, as well as between PGL4 and PGL5 (p > 0.05). Significant differences were obtained between the PGL0 and PGL1 hydrogels against the PGL2 and PGL3 hydrogels, and between the PGL4 and PGL5 hydrogels. Moreover, the average pore diameter increased with increasing GLE concentration for the PGL2, PGL3, PGL4, and PGL5 hydrogels. This addition caused a decrease in the PVA polymer content so that the degree of cross-linking decreased. The decrease in the degree of cross-linking resulted in a reduction of hydrogel density (Fig. 1d and Table 2) and an increase in water content. The high-water content of the PGL5 hydrogel will leave large diameter pores (63.35 ± 23.96c μm) when it is freeze dried.
Degree of swelling
The degree of swelling indicates the ability of the hydrogel to absorb water. It was found that the degree of swelling of the PVA/GLE hydrogels increased with increasing the GLE concentration, as demonstrated in Fig. 3.
Fig. 3. Degree of swelling of PVA/GLE hydrogels in PBS solutions. Each point represents the average of the results from three different samples (n = 3). The * sign indicates a significant difference between groups (p < 0.05).

All variations of the samples showed a significant increase from 0 to 12 hours. The expansion continued (but the increase was less significant) until the 24th hour, then reached a swollen equilibrium state.61 The PGL0 hydrogel showed the lowest degree of swelling, around 135 ± 5a% because it had the smallest pore size, and thus its ability to absorb PBS solution was also smaller than other hydrogels. This finding was confirmed by the previous study of Croitoru et al.62
The PGL1, PGL2, and PGL3 hydrogels had degrees of swelling that increased with the addition of GLE, namely 151 ± 7a,b%, 155 ± 5a,b%, and 165.3 ± 6.5b%, respectively. The increase that occurred was not significant because the pore sizes of the PGL1, PGL2, and PGL3 hydrogels did not differ significantly (p > 0.05). The PGL4 hydrogel shows a degree of swelling around 189.3 ± 8.5c% because it has an average pore diameter larger than those of the PGL0, PGL1, PGL2, and PGL3 hydrogels but smaller than that of the PGL5 hydrogel. The PGL5 hydrogel has the largest average pore diameter among the hydrogels. This allows the absorption of large amounts of PBS solution, up to 207 ± 12c%.
Weight loss
The reduction in hydrogel weight expresses the number of parts of the hydrogel dissolved in PBS while measuring the degree of swelling, as shown in Fig. 4. Statistical tests on weight loss showed that there was a significant difference between groups (p < 0.05). The PGL0 hydrogel had a sturdier structure due to their higher cross-linked content. When immersed in PBS, the PGL0 hydrogel absorbed less liquid PBS than other hydrogels. The higher amount of cross-linking content causes the PGL0 hydrogel tend to retain its hydrogel structure. The weight loss of PGL0 was the smallest at 2.47 ± 0.46a%.
Fig. 4. Weight loss of PVA/GLE hydrogels after immersion in PBS for 48 hours. The values are expressed as mean ± SD of the 3 samples (n = 3). The * sign indicates a significant difference between groups (p < 0.05).

The increase in GLE content in the PGL1, PGL2, PGL3, PGL4, and PGL5 hydrogels made the cross-linking content and the ability to maintain the hydrogel structure to decrease. The hydrogel dissolution process is characterized by a change in the PBS solution's colour from bright green to a more concentrated green. The PGL5 hydrogel showed the largest weight loss of 25.88 ± 0.48e%, with a decreased sample size compared to the initial size before the process of measuring the degree of swelling.
Fourier transform infrared (FTIR) spectroscopy
The FTIR spectra of PVA powder (Fig. 5) showed absorbance peaks at 3200–3600 cm−1 (O–H stretch), 2932 cm−1 (C–H stretch), 1449 cm−1 (C–H bend), and 1094 cm−1 (C–O–C stretch). These results are consistent with previous study performed by Khorasani et al.56 The 1094 cm−1 peak indicates a crystalline phase of PVA.63
Fig. 5. FTIR spectra of the PVA powder, the GLE powder, and the PVA/GLE hydrogels with varied GLE contents.

The GLE has a broad absorbance peak due to the phenolic hydroxyl group (O–H stretch) at 3200–3600 cm−1. The peaks at 2929 cm−1 shows the C–H stretch, at 2075 cm−1 due to the C–O stretch, and at 1616 cm−1 related to an aromatic ring of the carbonyl group (C O). The peak at 1451 cm−1 confirms the C–H bending of the methyl groups', while the peak at 1051 cm−1 indicates the C–O–C stretch. These observations were also previously confirmed by Jeyasundari et al. and Rehan et al.64,65
The PGL0 hydrogel showed absorbance peaks similar to those of PVA powder. The difference is in the width of the peak of the hydroxyl group (stretching O–H). Sirousazar et al. indicated that the widening peaks of the hydroxyl groups of the PVA hydrogels could be caused by the crystallization of the PVA polymer chain and the formation of hydrogen bonds during the freeze–thaw process.66 This also applies to the stretching of the C O on the aromatic rings derived from the active ingredient of GLE. The addition of GLE causes the absorbance peak at the 1616 cm−1 to widen and the wavenumber to shift.
X-ray diffraction (XRD) analysis
The X-ray diffractograms obtained are presented in Fig. 6. The PVA powder showed a diffraction pattern with a sharp peak at an angle of 2θ = 19.8° (d = 4.58 Å) related to the crystal plane orientation (101). Another diffraction peak is at 2θ = 22.6° (d = 3.93 Å) related to the crystal plane orientation of (200). This diffraction pattern reveals that the PVA powder is crystalline, resulting from strong intermolecular interactions between the PVA chains due to the intermolecular hydrogen bonds.67 The GLE powder only has one diffraction peak at 2θ = 15.1° (d = 5.3 Å). In the range of 2θ = 19°–50°, the diffraction pattern was widened and had a greater slope (halo). This indicates that the GLE powder is semicrystalline and tends to become amorphous.68
Fig. 6. XRD spectra of the PVA powder, the GLE powder, and the PVA/GLE hydrogels. The inset image shows that the diffraction peaks on the hydrogel widened with the addition of GLE concentrations for PGL0, PGL1, PGL2, PGL3, PGL4, and PGL5.

The PGL0, PGL1, PGL2, PGL3, PGL4, and PGL5 hydrogels showed a wide peak shift (halo) at an angular range of 2θ = 26°–29°, and there was a significant decrease in intensity as the GLE concentration was increased.
Specifically, the location of the halo peaks formed for each hydrogel is as follows: PGL0 at 2θ = 26.9°; PGL1 at 2θ = 27°; PGL2 at 2θ = 26.7°; PGL3 at 2θ = 28.9°; PGL4 at 2θ = 29.1°; and PGL5 at 2θ = 29.1°. The shift of the hydrogel peak towards the right, away from the PVA powder peaks at 2θ = 19.8° and 22.6°, occurred due to increased water content and the decreased concentration of PVA. Thus the degree of crystallinity is also reduced.69,70
Thermal properties
The hydrogel decomposition due to thermal effects is demonstrated by the thermogravimetry-derivative thermogravimetry (TG-DTG) curves in Fig. 7a. The TG-DTG curves shows information about the decomposition phase, the maximum temperature of the DTG, and the residual weight percentage during the heating process at 600 °C. The weight losses in the temperature range of 30–150 °C, for PVA powder and GLE powder, were 6.21% and 7.73%, respectively. The highest percentage of residual weight at 600 °C was the GLE powder at 29.70%, followed by the PVA powder at 3.75%. The two precursors were dry and had less water content than the hydrogels. The GLE powder had the most significant percentage of residual weight at 600 °C because its thermal stability is achieved at higher temperatures, around 980 °C.71
Fig. 7. (a)Thermal decomposition of the PVA powder, the GLE powder, and the PVA/GLE hydrogel at 30–600 °C, (b) DTA curves of the PVA and GLE powders, (c) DTA curves of the PVA/GLE hydrogel.

The decomposition process of the hydrogel is divided into the evaporation phase of the water content and the decomposition of the solid hydrogel phase.72 Evaporation of the water content in the hydrogel network starts at a 30 °C and continues to 150 °C. The evaporation process resulted in a weight loss of more than 80% for all hydrogels. The decomposition phase of the hydrogels that have lost their liquid components started at 155 °C and continued to 400 °C. The weight loss in this phase ranged from 9% to 11%, related to the degradation of the polyene structure of the PVA chain.73 Consequently, the hydrogel with the highest PVA concentration, which is the PGL0 hydrogel, had a greater shrinkage, 11.30%, than the other hydrogels. The PVA decomposition process continued at temperatures from 400 °C to 600 °C. There is a break in the PVA polymer main chain, resulting in carbon residues at this stage.73,74 The phase change in the PVA/GLE hydrogels can be observed in the differential thermal analysis (DTA) curves in Fig. 7b and c.
The DTA curves of the PVA powder and the GLE powder did not show endothermic peaks at temperatures below 150 °C, because these two precursors were dry, so there was no water evaporation process, unlike in the hydrogels. These DTA characterization results are corroborated by the previous TG-DTG results, which showed a minimum weight percentage reduction. The DTA curves of the hydrogels shows broad endothermic peaks at heating temperatures below 150 °C. The width of the endothermic curve indicates a significant fusion of the sample. The melting in question is the evaporation of the water content of the hydrogel that has been confirmed in the TG-DTG curve. The DTA characterization can also be used to determine the melting temperature (Tm) of the sample, as shown in Table 3. We can determine the heat of fusion from the melting temperature value by calculating the area under the curve.75
Melting temperature, the heat of fusion, and the degree of crystallinity of PVA powder, GLE powder, and PVA/GLE hydrogela.
| Sample | T m (°C) | Δ m (J g−1) during PVA melting | Degree of crystallinity (%) |
|---|---|---|---|
| PVA | 222; 253; 439 | 44.80 ± 1.28t | 32.31 ± 0.08t |
| GLE | 192; 325 | — | — |
| PGL0 | 221; 250; 423 | 10.81 ± 0.85u | 7.80 ± 0.03u |
| PGL1 | 218; 414 | 7.41 ± 0.56v | 5.34 ± 0.02v |
| PGL2 | 214; 340 | 5.17 ± 0.64w | 3.73 ± 0.04w |
| PGL3 | 213; 337 | 4.36 ± 0.38w | 3.14 ± 0.03x |
| PGL4 | 211; 336 | 3.52 ± 0.35w,x | 2.53 ± 0.05y |
| PGL5 | 209; 334 | 1.78 ± 0.42x | 1.28 ± 0.03z |
The value of heat of fusion, and degree of crystallinity was expressed as mean ± SD of 3 samples (n = 3). The use of superscripts on each column represent the statistical significant differences between groups. The groups with the same superscripts within each column indicate that the values are considered not significantly different (p > 0.05).
The PVA powder starts to melt at 222 °C, while the GLE powder starts to melt at 192 °C. The melting points of the hydrogels from PGL0 to PGL5 decreased to lower temperatures (222 °C to 209 °C and 439 °C to 334 °C) due to the reduction in PVA concentration. Similar results were shown in the previous studies by S. Gupta et al.76 Calculated area under the heat of fusion curve and degree of crystallinity are presented in Table 3.
The heat of fusion and degree of crystallinity of PVA had a significant difference (p < 0.05). The PVA powder has the largest heat of fusion of 44.80 ± 1.28t J g−1. The high value of the heat of fusion of the PVA powder caused the degree of crystallinity to increase (32.31 ± 0.08t%). The hydrogels showed a decrease in the heat of fusion and degree of crystallinity as they ranged from the PGL0 to PGL5 hydrogel. However, for the heat of fusion, there was no significant difference between groups of the PGL2, PGL3, and PGL4 hydrogels (p > 0.05). The reduced PVA content and increased GLE concentration in the PVA/GLE hydrogels were the leading causes of this downward trend. The PGL5 hydrogel had the lowest heat of fusion and the lowest degree of crystallinity at 1.78 ± 0.42x J g−1 and 1.28 ± 0.03z%, respectively.
Mechanical properties
The compression stress–strain curve in this study is depicted in Fig. 8. Wang et al. have presented the stress–strain curve of a PVA/carbon dots (CDs) composite hydrogel, which has a significant increase in stress values at strains of 30–85%.77
Fig. 8. Stress–strain curves of the PVA/GLE hydrogels.

The stress–strain curves obtained in this study show similar characteristics, increasing stress values at the percentage strain of 50–95% for all samples. The curve of the PGL0 hydrogel has a sharper gradient than the other hydrogels. This result has also been confirmed in the previous study of Jayaramudu et al. when observing the effect of variations in the concentration of cellulose nanocrystals (CNCs) on the mechanical properties of PVA/CNC hydrogels.78 The sharper gradient of the PGL0 hydrogel indicates a larger compression modulus. We observed the compression modulus values at the three strain levels given in Fig. 9.
Fig. 9. The compressive modulus of the PVA/GLE hydrogels at strain levels of 0–49%, 50–69%, and 70–95%.

A compression strain level of 0–49% indicates a tiny increase in the value of the compressive stress (the slope of the curve is very small), with the compression modulus ranges from 0.00224–0.00714 kPa. The best scenario is that the compressive force acting on the hydrogel is evenly distributed over each part of the hydrogel. The strain level of 50–69% shows an increase in the compression modulus because the stress–strain curve has a sharper gradient than for the 0–49% strain. At this stage, the compressive force exerted on the sample is not evenly distributed in each part of the hydrogel sample because there is a reduction in the area (the sample begins to crumble). As a result, the compressive stress value will experience a greater increase than the previous level and produce a sharper stress–strain curve. Compression modulus values at the 50–69% strain level ranged from 0.25–0.62 kPa.
The strain level of 70–95% is accompanied by a very significant increase in compression stress. A probable cause is the hydrogel sample experiencing massive destruction when the greater compressive force is used. The destruction of the sample results in a smaller contact area, resulting in an uneven distribution of the compressive force. Therefore, the value of the compression stress, which is inversely proportional to the compressed area, has increased. The modulus of compression based on a curve gradient showed values ranging from 2.64–7.76 kPa. According to Li et al. the compression modulus is highly dependent on the resulting strain ratio with an increase of up to 40 times the initial value.49 In this study, we found that the increase in the modulus of elasticity reached 1000 times the initial value at a strain ratio of 0–95% and 15 times at 50–95% strain ratio. The variation in the composition of the PVA/GLE hydrogels affects the compressive modulus at each strain level, as given in Table 4.
Compressive moduli and compressive strengths of PVA/GLE hydrogelsa.
| Sample | Compressive modulus at specified strain (kPa) | Compressive strength at 95% strain level (kPa) | ||
|---|---|---|---|---|
| 0–49% | 50–69% | 70–95% | ||
| PGL0 | 0.0071 ± 0.0021u | 0.52 ± 0.02u,v | 7.76 ± 0.11u | 208.47 ± 12.16u |
| PGL1 | 0.0043 ± 0.0006v | 0.55 ± 0.02u,v | 5.98 ± 0.16v | 184.65 ± 26.33u |
| PGL2 | 0.0038 ± 0.0007v | 0.62 ± 0.02u | 4.65 ± 0.02w | 162.25 ± 35.90u,v |
| PGL3 | 0.0022 ± 0.0004v | 0.45 ± 0.01v | 3.96 ± 0.04x | 129.97 ± 3.38v,w |
| PGL4 | 0.0026 ± 0.0006v | 0.32 ± 0.06w | 3.64 ± 0.01y | 114.50 ± 5.17v,w |
| PGL5 | 0.0022 ± 0.0003v | 0.25 ± 0.06w | 2.64 ± 0.08z | 108.15 ± 0.73w |
The value of compression modulus, and compressive strength were expressed as mean ± SD of 3 samples (n = 3). The use of superscripts on each column represent the statistical significant differences between groups. The groups with the same superscripts within each column indicate that the values are considered not significantly different (p > 0.05).
There was no significant difference between groups of the PGL1, PGL2, PGL3, PGL4, and PGL5 hydrogels (p > 0.05) for the compression modulus at 0–50% strain ratio. However, the PGL0 hydrogel showed a significant difference with other groups for the same strain ratio (p < 0.05). The PGL0 hydrogel had a compression modulus of 0.52 kPa, which increased to 0.62 kPa in the PGL2 hydrogel, then gradually decreased to 0.25 kPa for the PGL5 hydrogel at a strain level of 50–69%. Similar results have been identified in previous studies by Wang et al. when they observed the effect of variations in the concentration of CDs on the compression modulus of PVA/CDs composite hydrogel.77 When the variation of CDs had increased from 0% to 3%, the modulus of hydrogel compression increased from 0.4 kPa to 0.8 kPa, then began to decrease to 0.1 kPa when the CDs concentration was increased to 6%. At the strain level of 70–95%, an anomaly in the compression modulus is not observed as in the previous level. In this strain ratio, statistical tests showed a significant difference between groups for all hydrogels (p < 0.05). The compression modulus of the PGL0 hydrogel, which is 7.76 ± 0.11u kPa, decreases to 2.64 ± 0.08z kPa for the PGL5 hydrogel. Jayaramudu et al. concluded that there was a decrease in the compression modulus of the PVA hydrogel with variations in the concentration of CNC.78 They had investigated a pure PVA sample which showed a compression modulus value of 82 kPa and decreased with increased concentration of CNC (7%) to 7 kPa.
Human tissues, including skin, have Young's modulus values ranging from 1 to 100 kPa.79,80 Based on the measurement results in this study, all samples at the 70–95% strain level met the application criteria as a wound dressing. They had a compression modulus value consistent with the range of Young's moduli of body tissues. Compressive strength is the ability to withstand induced loads to reduce size.81 The stress–strain curve in Fig. 7a shows that all hydrogel samples have a curve resembling a “J,” which indicates a high compressive strength in the hydrogel.82
According to Stauffer and Peppast, the compressive strength of the PVA hydrogel based on three freeze–thaw cycles, 10 hours of freezing, and one hour thawing, was 3.5 kPa.83 The PGL0 hydrogel did not show a significant difference with the PGL1 and PGL2 hydrogels (p > 0.05) but did a significant difference to the other hydrogels (p < 0.05). In this study, we obtained a compressive strength of 208.47 ± 12.16u kPa for the PGL0 hydrogel. This high compressive strength is influenced by the number of FT cycles: six cycles with a combination of FT time durations (20 hours to 4 hours). The addition of the number of cycles and a longer freeze duration can increase the cross-linked density resulting in a PVA hydrogel with better mechanical properties.84
The addition of GLE concentrate in the PGL1 to PGL5 hydrogels caused a very significant decrease in compressive strength values. However, the PGL3, PGL4, and PGL5 hydrogels had no significant differences between groups (p > 0.05).
The presence of GLE macromolecules and a reduction in the PVA concentration causes the cross-linked density to decrease.85 This has previously been proven using SEM imaging, which shows an increase in pore size, and DTA characterization, which demonstrates a decrease in the degree of crystallinity of the hydrogel when the GLE concentration is increased.
Antibacterial study
The antibacterial properties of hydrogels are expressed by the zone of inhibition and the percentage of activity per mass unit.86 An inhibition zone is created around the hydrogel during the incubation process due to the hydrogel sample's antibacterial activity, as presented in Fig. 10a. There was a significant difference between groups in the zone of inhibition both against S. aureus and P. aeruginosa (p < 0.05). Both S. aureus and P. aeruginosa were resistant to the PGL0 hydrogel, as evidenced by the absence of a clear zone around the sample. Based on previous research, PVA does not have antibacterial activity, so it cannot inhibit bacterial growth.87
Fig. 10. (a) Zone of inhibition and (b) antibacterial activity of PVA/GLE hydrogels after 24 hours of incubation against S. aureus and P. aeruginosa.

The PGL1 hydrogel, which contains GLE, produces an inhibition zone of 14.93 ± 0.05a mm for S. aureus and 13.94 ± 0.04a mm for P. aeruginosa. Biswas et al. previously reported that the ethanol extract of guava leaves was able to form an inhibition zone of 11 mm against S. aureus.88 Meanwhile, Hoque et al. obtained an inhibition zone for guava leaves extracted with ethanol of 15.3 mm against S. aureus and an inhibition zone for aqueous extract of guava leaves of 12 mm against P. aeruginosa.89
The ability of GLE to inhibit bacteria is influenced by the presence of polyphenol and flavonoid groups. These polyphenol and flavonoid groups can interact with bacterial cell membranes through hydrophobic–hydrophobic interactions, thereby increasing cell membrane leakage and damaging the structure of bacterial cells.90 The inhibition zone formed against both types of bacteria was enlarged when the GLE concentration increased. Based on the classification of inhibition zone characteristics by Ouchari et al. the PGL1, PGL2, and PGL3 hydrogels (against P. aeruginosa) have “strong” inhibition zones (between 10–20 mm), whereas the PGL3, PGL4, and PGL5 hydrogels (against S. aureus) have “very strong” zones of inhibition (greater than 20 mm).91 The inhibition zone diameter against S. aureus is greater than P. aeruginosa because of the difference in cell wall thickness of the two bacteria.56 Furthermore, to determine the hydrogel antibacterial activity, we conducted tests using the total plate count method. The bacterial suspension incubated for 24 hours at 37 °C will form a bacterial colony on MHA media, as shown in Fig. 10b. The best antibacterial activity can be seen in the number of bacterial colonies that are the least formed on the Petri dishes compared to the number of control colonies.51 The number of P. aeruginosa bacteria was greater because of its smaller size with cell wall thicknesses ranging from 1.5–10 nm, while S. aureus had bacterial cell wall sizes ranging from 20–80 nm.92 The calculation of antibacterial activity with eqn (6) is shown in Table 5.
Zone of inhibition and antibacterial activity of PVA/GLE hydrogels after 24 hours incubation against S. aureus and P. aeruginosa.
| Sample | Zone of inhibition (mm) | Weight (mg) | Log bacterial colonies after 24 h incubation (CFU mL−1) | Antibacterial activity of hydrogel (% per gram) | ||||
|---|---|---|---|---|---|---|---|---|
| S. aureus | P. aeruginosa | S. aureus | P. aeruginosa | S. aureus | P. aeruginosa | S. aureus | P. aeruginos a | |
| PGL0 | 0 | 0 | 20.9 | 28.6 | 14.15 | 14.22 | 0.33 ± 0.02a | 0.19 ± 0.01a |
| PGL1 | 14.93 ± 0.05a | 13.94 ± 0.04a | 23.9 | 29.1 | 10.88 | 13.04 | 9.85 ± 0.82b | 3.01 ± 0.04b |
| PGL2 | 16.46 ± 0.10b | 15.42 ± 0.13b | 23.2 | 30.2 | 10.16 | 11.38 | 12.32 ± 0.65c | 6.73 ± 0.15c |
| PGL3 | 20.31 ± 0.04c | 17.52 ± 0.06c | 22.8 | 28.5 | 9.07 | 8.28 | 15.92 ± 0.46d | 14.73 ± 0.47d |
| PGL4 | 23.84 ± 0.06d | 23.32 ± 0.08d | 22.3 | 29.4 | 8.92 | 7.98 | 16.70 ± 0.55d,e | 15.01 ± 0.63d,e |
| PGL5 | 34.55 ± 0.12e | 26.90 ± 0.15e | 21.2 | 29.0 | 8.80 | 7.74 | 17.93 ± 0.78e | 15.79 ± 0.29e |
| Control | — | — | — | — | 14.25 | 14.30 | — | — |
Antibacterial activity against S. aureus showed significant differences between groups for the PGL0, PGL1, PGL2, and PGL3 hydrogels (p < 0.05). However, between the PGL 3 and PGL 4 hydrogels, and between the PGL4 and PGL5 hydrogels, there were no significant differences (p < 0.05). The PGL0 hydrogel had antibacterial activity of 0.33 ± 0.02a% per gram and 0.19 ± 0.01a% per gram against S. aureus and P. aeruginosa, respectively. The number of bacteria formed in PGL0 is almost as greater as the number in the control, indicating that PGL0 has a weak ability to fight bacteria. The PGL1 hydrogel gave the reduction in the number of bacterial colonies formed of 23.10% against S. aureus and 8.29% against P. aeruginosa compared to the PGL0. The presence of GLE inhibited the number of bacterial colonies formed, reaching 9.85 ± 0.82b% per gram against S. aureus and 3.01 ± 0.04b% per gram against P. aeruginosa. It appears that GLE is more effective in inhibiting the growth of S. aureus colonies than P. aeruginosa colonies. P. aeruginosa has an effective permeability barrier as a Gram-negative bacteria, consisting of a thin lipopolysaccharide exterior membrane called peptidoglycan, limiting the penetration of plant extrusions.88 Peptidoglycan is a type of polysaccharide responsible for maintaining the integrity of the bacterial cell structure.92 The additional GLE in the PGL2, PGL3, PGL4, and PGL5 hydrogels resulted in a reduction in the number of bacterial colonies. The smaller number of bacterial colonies formed indicates that the antibacterial activity of the PVA/GLE hydrogels is increasing. The PGL5 hydrogel contains the most GLE, allowing high hydrophobic interactions with bacterial cell membranes and damaging these cell structures for S. aureus and P. aeruginosa. The antibacterial activity of the PGL5 hydrogel was 17.93 ± 0.78e% per gram against S. aureus and 15.79 ± 0.29e% per gram against P. aeruginosa.
Practically, hydrogels that have antibacterial activity are needed for wound dressing applications, bone implant, dental infections, osteomyelitis, gastrointestinal infections, catheter infections, and prosthesis implant infections.86 The practical applications mentioned above require a hydrogel with good antibacterial activity, in order to reduce the possibility of bacterial penetration into our bodies. Even though the bacteria had already entered, with the high ability to damage the bacterial membrane, the bacteria could not grow more because their growth had been inhibited by the antibacterial hydrogel which is very important to treat infections effectively.93
The PGL0 hydrogel has the highest PVA composition than the other hydrogels in the present study. The high PVA content allows the formation of more cross-links in the polymer chain during the gelation process. When there are more cross-links, the space around these cross-links will be smaller, this space is known as the mesh size, also known as the pore size of the hydrogel.94 With decreasing PVA composition sequentially from the PGL1 to PGL5 hydrogel, the number of cross-links formed will also decrease and cause the pore size to increase. This pore size will further affect the antibacterial activity of the hydrogel. The porous morphology of the hydrogel allows the controlled release of antibacterial extract from the hydrogel matrix with different release profiles depending on the structure and pore size. Zou et al. stated that the hydrogel which has a large pore size and interconnected pores has excellent and long-term antibacterial activity against both S. aureus and E. coli bacteria.95
The large and interconnected pores can facilitate the rate of antibacterial release so that the antibacterial activity of the hydrogel is more effective. Antibacterial measurements using both the KB test and TPC method showed that the highest antibacterial activity was possessed by the hydrogels with the largest pore sizes (the PGL4 and PGL5 hydrogels). Furthermore, the hydrogels exhibiting homogeneous and interconnected pore sizes allow good nutrient exchange and cell migration for biomedical applications injected into the human body.96
The application of PVA in the biomedical field has been approved by the FDA due to its good biocompatibility and non-toxicity.97 The PVA hydrogel has a high-water content, similar to the human tissue, and thus provide excellent biocompatibility.98 The cytotoxicity test of PVA hydrogel was performed using the L929 mouse fibroblast cell culture. With a maximum PVA concentration at 10%, the IC80 value has been obtained and the cell viability will be reduced by 20% compared to the initial number of cultured cells. Therefore, PVA hydrogel can be applied as an extracellular matrix (ECM).99
PVA hydrogels can also be loaded with antibacterial components such as silver nanoparticles (AgNPs), chitosan, or gelatin.37,100 The MTT test conducted by Rodríguez-Rodríguez et al. on HT29-MTX-E12 cells (a human colorectal adenocarcinoma) on PVA/chitosan/gelatin hydrogel showed that the cell viability percentage only decreased after incubation for 3 and 7 days but still had a value above 80%. Hence, it proved that the PVA/chitosan/gelatin hydrogel has a good biocompatibility and thus can be applied as a tissue engineering material.100
Regarding GLE, the biocompatibility has been demonstrated in many previous studies with the cytotoxicity tests included.101–103 The cell viabilities of CCD-45 SK cells (human healthy skin fibroblasts) due to the presence of aqueous GLE and ethanolic GLE reached 63% and 93%, respectively. The percentage cell viability of HepG2 (human hepatocellular carcinoma cells) showed more promising results with a value of 50% or even lower. Based on the results, the GLE can thus be applied as a cytotoxic agent in medical treatment.103 It was also found that the IC50 values in GLE ranged from 200 μg mL−1 to 250 μg mL−1 against Kasumi-1 leukaemia cells.101,104 Thus, the cytotoxicity activity of GLE is very significant to inhibit the growth of these cancer cells. Therefore, the PVA hydrogels loaded with GLE could have good biocompatibility and cytotoxic activity.
Conclusions
We have successfully synthesized PVA/GLE hydrogels using the freeze–thaw method. Increasing the GLE concentration causes the pore size, degree of swelling, and weight loss to increase. FTIR analysis showed functional groups from the PVA powder and the GLE powder in the PVA/GLE hydrogel samples. According to XRD analysis, the increase in GLE content caused the crystalline phase to become amorphous, and the degree of crystallinity decreased, based on the DSC analysis. The DTA interpretation showed a reduction in the melting temperature due to a decrease in PVA concentration. The decreasing concentration of PVA caused the density of the hydrogel cross-linking of the PVA and GLE to decrease so that the compressive modulus and compressive strength are lower. However, with increasing GLE, the inhibition zone against S. aureus and P. aeruginosa was more significant, and the antibacterial activity was higher.
Author contributions
W. X. W. worked on conceptualization, methodology, data curation and original drafting. H. R. L. oversees conceptualization, methodology, and data curation. D. E. and T. S. provide resources and carry out supervision. F. A. N. is responsible for resources, writing-reviewing and editing, and supervision. K. K. is responsible for resources, writing-review and editing, supervision, and funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This research was financially supported by the Institute for Research and Community Service, ITB Research Grant in the fiscal years of 2020–2021 and Ristek-BRIN PTUPT Research Grant in the fiscal years of 2019–2020. We gratefully acknowledge the Indonesian Endowment Fund for Education (LPDP) for the provision of the master scholarships of W. X. W.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04092h
References
- Susanto S. Melati M. Aziz S. A. Agrivita. 2019;41:48–54. [Google Scholar]
- Gutiérrez R. M. P. Mitchell S. Solis R. V. J. Ethnopharmacol. 2008;117:1–27. doi: 10.1016/j.jep.2008.01.025. [DOI] [PubMed] [Google Scholar]
- Raj A. Menon V. Sharma N. Vegetos. 2020;33:750–758. doi: 10.1007/s42535-020-00151-4. [DOI] [Google Scholar]
- Ghorbani A. Rashidi R. Shafiee-Nick R. Biomed. Pharmacother. 2019;111:947–957. doi: 10.1016/j.biopha.2018.12.127. [DOI] [PubMed] [Google Scholar]
- Wang Y. Tao B. Wan Y. Sun Y. Wang L. Sun J. Li C. Biomed. Pharmacother. 2020;128:110372. doi: 10.1016/j.biopha.2020.110372. [DOI] [PubMed] [Google Scholar]
- Caddeo C. Gabriele M. Fernàndez-Busquets X. Valenti D. Fadda A. M. Pucci L. Manconi M. Int. J. Pharm. 2019;565:64–69. doi: 10.1016/j.ijpharm.2019.05.007. [DOI] [PubMed] [Google Scholar]
- Tang S. M. Deng X. T. Zhou J. Li Q. P. Ge X. X. Miao L. Biomed. Pharmacother. 2020;121:109604. doi: 10.1016/j.biopha.2019.109604. [DOI] [PubMed] [Google Scholar]
- Güran M. Şanlıtürk G. Kerküklü N. R. Altundağ E. M. Süha Yalçın A. Eur. J. Pharmacol. 2019;859:172486. doi: 10.1016/j.ejphar.2019.172486. [DOI] [PubMed] [Google Scholar]
- Ding H. Liang X. Wang Q. Wang M. Li Z. Sun G. Carbohydr. Polym. 2020;248:116797. doi: 10.1016/j.carbpol.2020.116797. [DOI] [PubMed] [Google Scholar]
- Batubara I. Suparto H. Wulandari N. S. IOP Conf. Ser. Earth Environ. Sci. 2017;58:012060. doi: 10.1088/1755-1315/58/1/012060. [DOI] [Google Scholar]
- Hirudkar J. R. Parmar K. M. Prasad R. S. Sinha S. K. Jogi M. S. Itankar P. R. Prasad S. K. Microb. Pathog. 2020;138:103807. doi: 10.1016/j.micpath.2019.103807. [DOI] [PubMed] [Google Scholar]
- Cao J. Wang Y. He C. Kang Y. Zhou J. Carbohydr. Polym. 2020;242:116420. doi: 10.1016/j.carbpol.2020.116420. [DOI] [PubMed] [Google Scholar]
- Homaeigohar S. Tsai T. Y. Young T. H. Yang H. J. Ji Y. R. Carbohydr. Polym. 2019;224:115112. doi: 10.1016/j.carbpol.2019.115112. [DOI] [PubMed] [Google Scholar]
- Pawar V. Dhanka M. Srivastava R. Colloids Surf., B. 2019;173:776–787. doi: 10.1016/j.colsurfb.2018.10.034. [DOI] [PubMed] [Google Scholar]
- Lai D. Li E. Yan Y. Liu Y. Zhong J. Lv D. Ke Y. Chen H. Guo T. Org. Electron. 2019;75:105409. doi: 10.1016/j.orgel.2019.105409. [DOI] [Google Scholar]
- Koev T. T. Muñoz-García J. C. Iuga D. Khimyak Y. Z. Warren F. J. Carbohydr. Polym. 2020;249:116834. doi: 10.1016/j.carbpol.2020.116834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M. Giri T. K. J. Drug Deliv. Sci. Technol. 2020;56:101586. doi: 10.1016/j.jddst.2020.101586. [DOI] [Google Scholar]
- Palantöken S. Bethke K. Zivanovic V. Kalinka G. Kneipp J. Rademann K. J. Appl. Polym. Sci. 2020;137:23–25. doi: 10.1002/app.48380. [DOI] [Google Scholar]
- Nafo W. Al-Mayah A. Int. J. Non Lin. Mech. 2020;124:103515. doi: 10.1016/j.ijnonlinmec.2020.103515. [DOI] [Google Scholar]
- Kuźmińska A. Butruk-Raszeja B. A. Stefanowska A. Ciach T. Colloids Surf., B. 2020;192:4–9. doi: 10.1016/j.colsurfb.2020.111066. [DOI] [PubMed] [Google Scholar]
- Brown A. He H. Trumper E. Valdez J. Hammond P. Griffith L. G. Biomaterials. 2020;243:119921. doi: 10.1016/j.biomaterials.2020.119921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C. Tang S. Meng Q. Colloids Surf., B. 2019;181:734–739. doi: 10.1016/j.colsurfb.2019.06.019. [DOI] [PubMed] [Google Scholar]
- Bersanetti P. A. Escobar V. H. Nogueira R. F. Ortega F. dos S. Schor P. Morandim-Giannetti A. de A. Eur. Polym. J. 2019;112:610–618. doi: 10.1016/j.eurpolymj.2018.10.024. [DOI] [Google Scholar]
- Fang H. Wang J. Li L. Xu L. Wu Y. Wang Y. Fei X. Tian J. Li Y. Chem. Eng. J. 2019;365:153–164. doi: 10.1016/j.cej.2019.02.030. [DOI] [Google Scholar]
- Muchová M. Münster L. Capáková Z. Mikulcová V. Kuřitka I. Vícha J. Mater. Sci. Eng. C. 2020;116:111242. doi: 10.1016/j.msec.2020.111242. [DOI] [PubMed] [Google Scholar]
- Thangprasert A. Tansakul C. Thuaksubun N. Meesane J. Mater. Des. 2019;183:108113. doi: 10.1016/j.matdes.2019.108113. [DOI] [Google Scholar]
- Yadav N., Parveen S., Chakravarty S. and Banerjee M., Skin anatomy and morphology, 2020 [Google Scholar]
- Moura L. I. F. Dias A. M. A. Carvalho E. de Sousa H. C. Acta Biomater. 2013;9:7093–7114. doi: 10.1016/j.actbio.2013.03.033. [DOI] [PubMed] [Google Scholar]
- Tao G. Wang Y. Cai R. Chang H. Song K. Zuo H. Zhao P. Mater. Sci. Eng. C. 2019;101:341–351. doi: 10.1016/j.msec.2019.03.111. [DOI] [PubMed] [Google Scholar]
- Boateng J. Catanzano O. J. Pharm. Sci. 2015;104:3653–3680. doi: 10.1002/jps.24610. [DOI] [PubMed] [Google Scholar]
- He F. Jiao H. Tian Y. Zhao L. Liao X. Fan Z. Liu B. J. Biomater. Sci. Polym. Ed. 2018;29:325–343. doi: 10.1080/09205063.2017.1417002. [DOI] [PubMed] [Google Scholar]
- Wang J. Zhang C. Yang Y. Fan A. Chi R. Shi J. Zhang X. Appl. Surf. Sci. 2019;494:708–720. doi: 10.1016/j.apsusc.2019.07.224. [DOI] [Google Scholar]
- Gao T. Jiang M. Liu X. You G. Wang W. Sun Z. Ma A. Chen J. Polymers. 2019;11:11010171. doi: 10.3390/polym11010171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamloo A. Aghababaie Z. Afjoul H. Jami M. Bidgoli M. R. Vossoughi M. Ramazani A. Kamyabhesari K. Int. J. Pharm. 2020;592:120068. doi: 10.1016/j.ijpharm.2020.120068. [DOI] [PubMed] [Google Scholar]
- Kumar A. Behl T. Chadha S. Int. J. Biol. Macromol. 2020;149:1262–1274. doi: 10.1016/j.ijbiomac.2020.02.048. [DOI] [PubMed] [Google Scholar]
- Swaroop K. Somashekarappa H. M. Mater. Today: Proc. 2018;5:21314–21321. [Google Scholar]
- Juby K. A. Dwivedi C. Kumar M. Kota S. Misra H. S. Bajaj P. N. Carbohydr. Polym. 2012;89:906–913. doi: 10.1016/j.carbpol.2012.04.033. [DOI] [PubMed] [Google Scholar]
- Nagakawa Y. Kato M. Suye S. I. Fujita S. RSC Adv. 2020;10:38045–38054. doi: 10.1039/D0RA07322A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezeki Y. A. Hapidin D. A. Rachmawati H. Munir M. M. Khairurrijal K. Adv. Powder Technol. 2020;31:1811–1824. doi: 10.1016/j.apt.2020.02.016. [DOI] [Google Scholar]
- Annabi N. Nichol J. W. Zhong X. Ji C. Koshy S. Khademhosseini A. Dehghani F. Tissue Eng., Part B. 2010;16:371–383. doi: 10.1089/ten.teb.2009.0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. A. Rasband W. S. Eliceiri K. W. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi S. Wang P. Hu S. Li S. Pang J. Zhou Z. Sun G. Huang L. Cheng X. Xing S. Chen X. Carbohydr. Polym. 2019;224:115176. doi: 10.1016/j.carbpol.2019.115176. [DOI] [PubMed] [Google Scholar]
- Sittiwong J. Niamlang S. Paradee N. Sirivat A. AAPS PharmSciTech. 2012;13:1407–1415. doi: 10.1208/s12249-012-9869-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y. Bian J. Peng F. Zhang X. M. Sun R. C. Carbohydr. Polym. 2014;101:272–280. doi: 10.1016/j.carbpol.2013.08.085. [DOI] [PubMed] [Google Scholar]
- Butylina S. Geng S. Oksman K. Eur. Polym. J. 2016;81:386–396. doi: 10.1016/j.eurpolymj.2016.06.028. [DOI] [Google Scholar]
- de Lima G. G. Ferreira B. D. Matos M. Pereira B. L. Nugent M. J. D. Hansel F. A. Magalhães W. L. E. Carbohydr. Polym. 2020;245:116612. doi: 10.1016/j.carbpol.2020.116612. [DOI] [PubMed] [Google Scholar]
- Buckley C. T. Thorpe S. D. O'Brien F. J. Robinson A. J. Kelly D. J. J. Mech. Behav. Biomed. Mater. 2009;2:512–521. doi: 10.1016/j.jmbbm.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Xing J. Luo Y. Zhan J. Kang Z. Int. J. Solids Struct. 2018;144–145:301–312. doi: 10.1016/j.ijsolstr.2018.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. Wang D. Yang W. Song Y. RSC Adv. 2016;6:20166–20172. doi: 10.1039/C6RA02166B. [DOI] [Google Scholar]
- Lee D., Zhang H. and Ryu S., Cellul. Superabsorbent Hydrogels, 2019, pp. 1–21 [Google Scholar]
- Edikresnha D. Suciati T. Munir M. M. Khairurrijal K. RSC Adv. 2019;9:26351–26363. doi: 10.1039/C9RA04072B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David A. Davidson C. E. J. Microbiol. Methods. 2014;107:214–221. doi: 10.1016/j.mimet.2014.08.023. [DOI] [PubMed] [Google Scholar]
- Bahadoran M. Shamloo A. Nokoorani Y. D. Sci. Rep. 2020;10:7–9. doi: 10.1038/s41598-019-56411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S. P. Lo K. Y. Tseng T. N. Liu J. M. Shih T. Y. Cheng K. C. Cell. Polym. 2019;38:15–30. doi: 10.1177/0262489319839211. [DOI] [Google Scholar]
- Long J. Etxeberria A. E. Kornelsen C. V Nand A. Ray S. Bunt C. R. Seyfoddin A. ACS Appl. Bio Mater. 2019;2:2766–2779. doi: 10.1021/acsabm.9b00190. [DOI] [PubMed] [Google Scholar]
- Khorasani M. T. Joorabloo A. Adeli H. Mansoori-Moghadam Z. Moghaddam A. Carbohydr. Polym. 2019;207:542–554. doi: 10.1016/j.carbpol.2018.12.021. [DOI] [PubMed] [Google Scholar]
- Cascone M. G. Lazzeri L. Sparvoli E. Scatena M. Serino L. P. Danti S. ACS Appl. Bio Mater. 2004;15:1309–1313. doi: 10.1007/s10856-004-5739-z. [DOI] [PubMed] [Google Scholar]
- Ceylan S., Göktürk D. and Bölgen N., Biomed. Mater. Eng., 2016, 27, pp. 327–340 [DOI] [PubMed] [Google Scholar]
- Xing L. Li Z. Zhang Q. Zhang Y. Liu P. Zhang K. RSC Adv. 2018;8:2622–2631. doi: 10.1039/C7RA10904K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X. Shamshina J. L. Berton P. Gurau G. Rogers R. D. Green Chem. 2015;18:53–75. doi: 10.1039/C5GC02396C. [DOI] [Google Scholar]
- Münster L. Capáková Z. Fišera M. Kuřitka I. Vícha J. Carbohydr. Polym. 2019;218:333–342. doi: 10.1016/j.carbpol.2019.04.091. [DOI] [PubMed] [Google Scholar]
- Croitoru C. Pop M. A. Bedo T. Cosnita M. Roata I. C. Hulka I. Polymers. 2020;12:560. doi: 10.3390/polym12030560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou K. Dong X. Qin C. Ji X. He J. Mater. Sci. Eng. C. 2017;77:1017–1026. doi: 10.1016/j.msec.2017.03.287. [DOI] [PubMed] [Google Scholar]
- Jeyasundari J. Praba P. S. Jacob Y. B. A. Vasantha V. S. Shanmugaiah V. Chem. Sci. Rev. Lett. 2017;6:1244–1252. [Google Scholar]
- Rehan M. Ahmed-Farid O. A. Ibrahim S. R. Hassan A. A. Abdelrazek A. M. Khafaga N. I. M. Khattab T. A. ACS Sustainable Chem. Eng. 2019;7:18612–18623. doi: 10.1021/acssuschemeng.9b04952. [DOI] [Google Scholar]
- Sirousazar M. Khodamoradi P. Mater. Today Commun. 2020;22:100719. doi: 10.1016/j.mtcomm.2019.100719. [DOI] [Google Scholar]
- Liu T. Jiao C. Peng X. Chen Y. N. Chen Y. He C. Liu R. Wang H. J. Mater. Chem. B. 2018;6:8105–8114. doi: 10.1039/C8TB02556H. [DOI] [PubMed] [Google Scholar]
- Osorio C. Carriazo J. G. Barbosa H. Quim. Nova. 2011;34:636–640. doi: 10.1590/S0100-40422011000400016. [DOI] [Google Scholar]
- Pingan H. Mengjun J. Yanyan Z. Ling H. RSC Adv. 2017;7:2450–2459. doi: 10.1039/C6RA25579E. [DOI] [Google Scholar]
- Ricciardi R. Auriemma F. Gaillet C. De Rosa C. Lauprêtre F. Macromolecules. 2004;37:9510–9516. doi: 10.1021/ma048418v. [DOI] [Google Scholar]
- Athmaselvi K. A. Kumar C. Balasubramanian M. Roy I. J. Food Process Eng. 2014;2014:1–10. doi: 10.1155/2014/524705. [DOI] [Google Scholar]
- Reguieg F. Ricci L. Bouyacoub N. Belbachir M. Bertoldo M. Polym. Bull. 2020;77:929–948. doi: 10.1007/s00289-019-02782-3. [DOI] [Google Scholar]
- Sunaryono S. Taufiq A. Mufti N. Hidayat N. Rugmai S. Soontaranon S. Putra E. G. R. Darminto D. IOP Conf. Ser. Mater. Sci. Eng. 2017;202:012041. doi: 10.1088/1757-899X/202/1/012041. [DOI] [Google Scholar]
- Abasian M. Hooshangi V. Moghadam P. N. Iran. Polym. J. 2017;26:313–322. doi: 10.1007/s13726-017-0521-5. [DOI] [Google Scholar]
- Hassan C. M. Peppas N. A. Adv. Polym. 2000;153:37–65. [Google Scholar]
- Gupta S. Webster T. J. Sinha A. ACS Appl. Bio Mater. 2011;22:1763–1772. [Google Scholar]
- Wang Y. Xue Y. Wang J. Zhu Y. Zhu Y. Zhang X. Liao J. Li X. Wu X. Qin Y. X. Chen W. Polymers. 2019;11:11071112. [Google Scholar]
- Jayaramudu T. Ko H. U. Kim H. C. Kim J. W. Muthoka R. M. Kim J. Materials. 2018;11:1–11. doi: 10.3390/ma11091615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X. Gu X. Wang Y. Chen X. Ling J. Yang Y. RSC Adv. 2020;10:17280–17287. doi: 10.1039/D0RA02017F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. Yuk H. Parada G. A. Wu Y. Liu X. Nabzdyk C. S. Youcef-Toumi K. Zang J. Zhao X. Adv. Mater. 2019;31:1–9. doi: 10.1002/adma.201807101. [DOI] [PubMed] [Google Scholar]
- Hadrys D. Węgrzyn T. Piwnik J. Wszołek L. Węgrzyn D. Arch. Metall. Mater. 2016;61:123–126. [Google Scholar]
- Guan Y. Qi X. M. Zhang B. Chen G. G. Peng F. Sun R. C. Bioresources. 2015;10:1378–1393. [Google Scholar]
- Stauffer S. R. and Peppast N. A., 1992, 33, 3932–3936.
- Kim T. H. An D. B. Oh S. H. Kang M. K. Song H. H. Lee J. H. Biomaterials. 2015;40:51–60. doi: 10.1016/j.biomaterials.2014.11.017. [DOI] [PubMed] [Google Scholar]
- Figueroa-Pizano M. D. Vélaz I. Peñas F. J. Zavala-Rivera P. Rosas-Durazo A. J. Maldonado-Arce A. D. Martínez-Barbosa M. E. Carbohydr. Polym. 2018;195:476–485. doi: 10.1016/j.carbpol.2018.05.004. [DOI] [PubMed] [Google Scholar]
- Li S. Dong S. Xu W. Tu S. Yan L. Zhao C. Ding J. Chen X. Adv. Sci. 2018;5:1700527. doi: 10.1002/advs.201700527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal-Ballen A. Lopez-Garcia J. Merchan-Merchan M. A. Lehocky M. Molecules. 2018;23:3109. doi: 10.3390/molecules23123109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas B. Rogers K. McLaughlin F. Daniels D. Yadav A. Internet J. Microbiol. 2013:746165. doi: 10.1155/2013/746165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahfuzul Hoque M. D. Bari M. L. Inatsu Y. Juneja V. K. Kawamoto S. Foodborne Pathog. Dis. 2007;4:481–488. doi: 10.1089/fpd.2007.0040. [DOI] [PubMed] [Google Scholar]
- Olatunde O. O. Benjakul S. Vongkamjan K. J. Food Biochem. 2018;42:1–11. doi: 10.1111/jfbc.12600. [DOI] [Google Scholar]
- Ouchari L. Boukeskasse A. Bouizgarne B. Ouhdouch Y. Biol. Open. 2018:035410. doi: 10.1242/bio.035410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai-Prochnow A. Clauson M. Hong J. Murphy A. B. Sci. Rep. 2016;6:1–11. doi: 10.1038/s41598-016-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. Han Q. Chen B. Zheng Y. Zhang K. Li Q. Wang J. Int. J. Nanomed. 2018;13:2217–2263. doi: 10.2147/IJN.S154748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera D. Medini M. Seethamraju D. Falkowski R. White K. Olabisi R. M. J. Microencapsul. 2018;35:475–481. doi: 10.1080/02652048.2018.1526341. [DOI] [PubMed] [Google Scholar]
- Zou S. Wei Z. Hu Y. Deng Y. Tong Z. Wang C. Polym. Chem. 2014;5:4227–4234. doi: 10.1039/C4PY00436A. [DOI] [Google Scholar]
- Pierau L. Versace D.-L. Materials. 2021;14:787. doi: 10.3390/ma14040787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerroni B. Cicconi R. Oddo L. Scimeca M. Bonfiglio R. Bernardini R. Palmieri G. Domenici F. Bonanno E. Mattei M. others Heliyon. 2018;4:e00770. doi: 10.1016/j.heliyon.2018.e00770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Mooney D. J. Nat. Rev. Mater. 2016;1:1–18. doi: 10.1038/natrevmats.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankari L. Fernandes B. L. Rebelatto C. L. K. Brofman P. R. S. Int. J. Polym. Mater. Polym. Biomater. 2020;69:653–658. doi: 10.1080/00914037.2019.1596915. [DOI] [Google Scholar]
- Rodríguez-Rodríguez R. Velasquillo-Martínez C. Knauth P. López Z. Moreno-Valtierra M. Bravo-Madrigal J. Jiménez-Palomar I. Luna-Bárcenas G. Espinosa-Andrews H. García-Carvajal Z. Y. J. Biomed. Mater. Res., Part A. 2020;108:81–93. doi: 10.1002/jbm.a.36794. [DOI] [PubMed] [Google Scholar]
- Levy A. S. Carley S. K. Trop. J. Pharm. Res. 2012;11:201–207. [Google Scholar]
- Lee W. C. Mahmud R. Noordin R. Pillai Piaru S. Perumal S. Ismail S. J. Essent. Oil-Bear. Plants. 2013;16:32–38. doi: 10.1080/0972060X.2013.764196. [DOI] [Google Scholar]
- Kemegne G. A. Bettache N. Nyegue M. A. Etoa F.-X. Menut C. Res. J. Pharm., Biol. Chem. Sci. 2020;8:58–64. [Google Scholar]
- Chen K.-C. Hsieh C.-L. Peng C.-C. Hsieh-Li H.-M. Chiang H.-S. Huang K.-D. Peng R. Y. Nutr. Canc. 2007;58:93–106. doi: 10.1080/01635580701308240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.