

Abstract
Alternative polyadenylation (APA) generates transcript isoforms that differ in their 3′ UTR content and may therefore be subject to different regulatory fates. Although the existence of APA has been known for decades, quantification of APA isoforms from high-throughput RNA sequencing data has been difficult. To facilitate the study of APA in large datasets, we developed an APA quantification technique called LABRAT (Lightweight Alignment-Based Reckoning of Alternative Three-prime ends). LABRAT leverages modern transcriptome quantification approaches to determine the relative abundances of APA isoforms. In this manuscript we describe how LABRAT produces its calculations, provide a step-by-step protocol for its use, and demonstrate its ability to quantify APA in single-cell RNAseq data.
1. Introduction
The 3′ ends of eukaryotic mRNAs are defined by the processes of cleavage and polyadenylation. In many genes, cleavage and polyadenylation can occur at more than one location, leading to the generation of transcript isoforms that differ in the compositions of their 3′ UTRs. Because the 3′ UTR content of an mRNA can modulate the stability, localization, and translational efficiency of the transcript, APA therefore contributes to the regulation of each of these processes (Cho et al., 2005; Mayr & Bartel, 2009; Sandberg, Neilson, Sarma, Sharp, & Burge, 2008; Taliaferro et al., 2016).
In general, APA events can be classified into two distinct structures. Alternative polyadenylation sites can be located within the same terminal exon, giving rise to a structure sometimes called “Tandem UTRs” (Fig. 1A, left). Conversely, APA sites can be located within different terminal exons, giving rise to an “alternative last exon” (ALE) structure (Fig. 1A, right). Although these two classes of APA have historically been considered separately, recent evidence has demonstrated that they are tightly coregulated, suggesting that they may be regulated by similar mechanisms (Goering et al., 2020; Taliaferro et al., 2016).
Fig. 1.
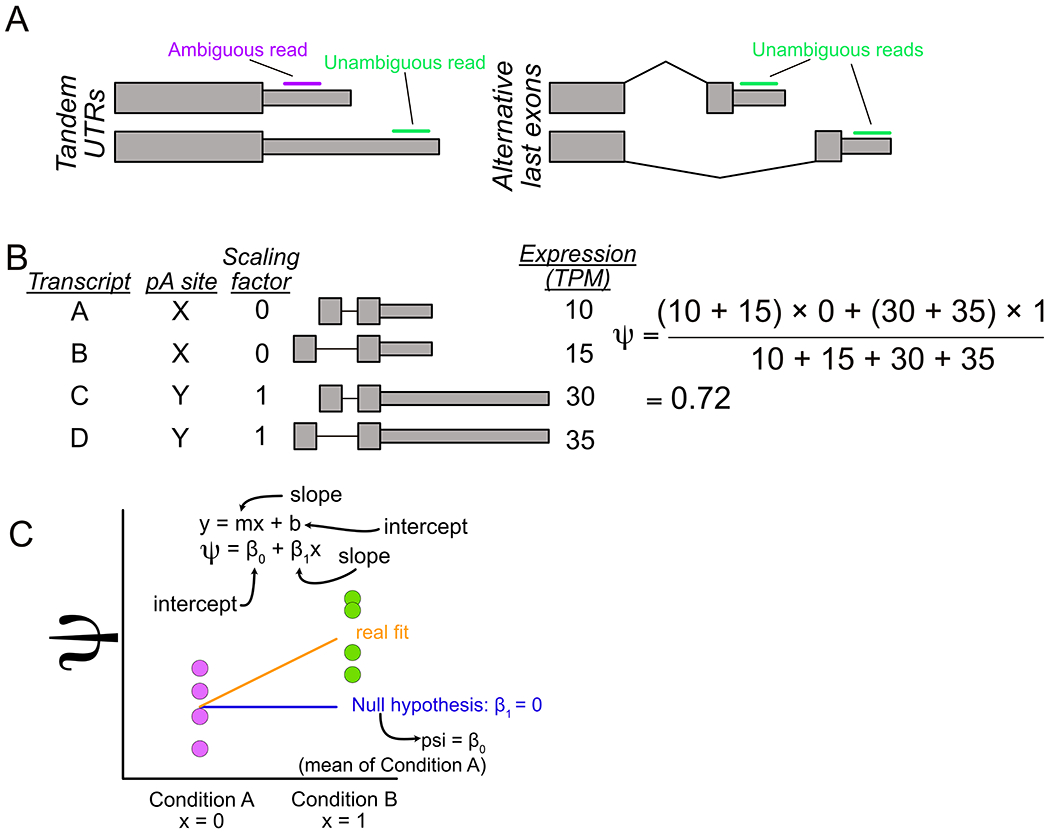
Overview of LABRAT approach. (A) Tandem UTR (left) and alternative last exon (right) gene structures. (B) Visualization of procedure for calculating ψ. (C) Linear model used by LABRAT for identification of genes with significant changes in ψ across conditions.
Given the effect that APA can have on RNA metabolism, the quantification of the relative abundance of APA isoforms for a given gene has been of interest for many years. The advent of high-throughput RNA sequencing opened up the possibility of performing this quantification for many genes at once and of identifying genes whose APA status changes across conditions. A variety of computational tools have been developed for this purpose (Grassi, Mariella, Lembo, Molineris, & Provero, 2016; Ha, Blencowe, & Morris, 2018; Xia et al., 2014).
Early tools (Grassi et al., 2016; Xia et al., 2014) relied on computed alignments between RNAseq reads and a supplied genome or transcriptome. Following this alignment, the abundances of APA isoforms could be calculated by counting the numbers of reads consistent with each isoform. However, these tools suffer from a disadvantage inherent to APA isoforms and tandem UTR structures in particular. In tandem UTR structures, there is often a large amount of shared sequence between the two isoforms. This makes reads that align to this common region less powerful in their ability to discriminate between the isoforms, since they could have arisen from either isoform. This reduces the overall statistical power and accuracy of the analysis.
Alternatively, later approaches, including LABRAT (Goering et al., 2020; Ha et al., 2018), take advantage of modern transcriptome quantification methods like Salmon and Kallisto (Bray, Pimentel, Melsted, & Pachter, 2016; Patro, Duggal, Love, Irizarry, & Kingsford, 2017). These quantification methods can fractionally and probabilistically assign multimapping “ambiguous” reads to individual transcripts, improving the accuracy and power of subsequent quantifications. LABRAT leverages the quantifications produced by Salmon of individual transcripts to compute quantifications of APA site usage. It then compares relative APA site usage across conditions to identify genes whose relative abundance of APA isoforms changes between samples.
2. How LABRAT works
In this section, we will provide an overview of how LABRAT performs its computations. Generally, LABRAT takes a set of transcript-level quantifications produced by Salmon and aggregates them into APA site-level quantifications. These APA site-level quantifications are then used to create an overall value that represents the APA status of the gene called ψ. Values of ψ can range between 0 and 1 with a ψ value of 0 representing exclusive usage of the most upstream (gene-proximal) APA site and a ψ value of 1 representing exclusive usage of the most downstream (gene-distal) APA site. LABRAT then compares ψ values across conditions to identify genes whose APA is differentially regulated between conditions.
2.1. Filtering transcripts
LABRAT takes in a genome annotation in gff format. From this annotation it derives the 3′ ends of transcripts to be quantified. However, it does not consider every transcript. In many annotations, there are dubious transcripts that result from incomplete transcript assemblies, old idiosyncratic ESTs, RNAs that haven’t yet been fully processed, and other error prone sources. Because these may negatively impact the accuracy of APA quantification, LABRAT uses a set of filters to remove these transcripts.
Some of these filters utilize specific transcript tags found in the supplied annotation. These tags may not be found in every annotation, but are always found in Gencode gff annotations. Because Gencode annotations are only offered for human and mouse genomes, this restricts the species compatible to analysis with LABRAT. To ameliorate this limitation, we wrote specific versions of LABRAT that are compatible with Ensembl annotations for rat and Drosophila genomes.
The first filter used ensures that the transcript is protein coding. Although APA may regulate noncoding transcripts including lncRNAs, a large fraction of the undesired, spurious transcripts are not protein coding. To filter these, LABRAT selects transcripts that have the “protein_coding” attribute.
Transcripts whose 3′ end is not well defined have the potential to induce artifacts in APA quantification. These transcripts often arise from degraded or partial transcripts, yet still end up in many genome annotations. To remove these transcripts from the analysis, LABRAT filters out transcripts that contain the attribute “mRNA_end_NF.”
2.2. Truncating transcripts
Transcript models derived from genome annotations necessarily contain all the exons, both constitutive and alternative, that exist within the model. This directly and inflexibly links the inclusion of all exons within that transcript model with cleavage and polyadenylation at the site defined by the model’s 3′ end. However, this may not be biologically meaningful. Although links between alternative exon inclusion and alternative polyadenylation have been observed (Pai et al., 2016), it is not necessarily true that the inclusion or exclusion of an alternative exon upstream in a transcript is always followed by the usage of a given APA site.
To decouple such potentially spurious links, LABRAT truncates each transcript to its final two exons prior to quantification. These transcript terminal fragments are supplied to Salmon for quantification, and the TPM (transcripts per million) values produced are used in downstream steps.
2.3. Calculating expression of APA sites
Salmon quantifies the abundances of individual transcripts. RNA expression at the gene level can be derived from these transcript quantifications by summing expression values across all transcripts within the gene. This strategy is used by the popular RNAseq analysis tool tximport (Soneson, Love, & Robinson, 2015) for usage with differential gene expression analysis tools. LABRAT employs a similar strategy. However, instead of summing expression values across all transcripts that belong to a gene, LABRAT sums expression values across all transcript fragments that belong to a given APA site.
Following quantification, transcript fragments that share 3′ ends are grouped together. By default, transcripts whose 3′ ends are within 25 nt of each other are grouped together, although this parameter is tunable. This allows for some microheterogeneity in APA site location which may arise either for biological reasons or to small inaccuracies in the genome annotation. The expression values (TPMs) of all transcript fragments that belong to a 3′ end are summed, generating an expression quantification for the APA site. Following this process, each gene will be associated with one expression quantification per APA site.
Genes that do not meet a total expression threshold (i.e., the sum across all of the APA sites for the gene) are filtered and removed from further analysis. By default, this threshold is set at 5 TPM, although this parameter is also tunable.
2.4. Calculating ψ values
Following APA site quantification, LABRAT summarizes the APA status of the gene using a metric called ψ. Each gene is therefore assigned one ψ value. A ψ value of 0 represents a gene where the most upstream (gene-proximal) APA site is exclusively used, and a ψ value of 1 represents a gene where the most downstream (gene-distal) APA site is exclusively used.
LABRAT begins by ordering the APA sites for a gene from most upstream to most downstream. For each gene, the number of distinct APA sites, n, is recorded, and each APA site is assigned a value, m, that is equal to its rank order from most upstream to most downstream. LABRAT then calculates a scaling factor for each APA site that is equal to (m − 1)/(n − 1).
The scaled expression value of each APA site is then calculated by multiplying the expression of the APA site by its scaling factor. The scaled expression values are then summed across all APA sites. Unscaled expression values are similarly summed across all APA sites. ψ values are then calculated as the ratio between the scaled and unscaled expression values.
Consider a gene with four transcript fragments that belong to two distinct APA sites (Fig. 1B). Expression values for each APA site are calculated by summing expression values across all transcript fragments that belong to the site. The transcripts that belong to the upstream APA site will have a scaling factor of 0 while the transcripts that belong to the downstream APA site will have a scaling factor of 1. A ψ value can then be calculated by taking the ratio of scaled and unscaled expression values (Fig. 1B).
Importantly, this process scales to genes with more than two APA sites. For example, the scaling factors for a gene with three APA sites would be 0, 0.5 and 1. Each gene, regardless of the number of APA sites it contains, is assigned a single ψ value. For genes with more than two APA sites, ψ therefore gives an overall sense of the relative usage of upstream and downstream APA sites. This approach can be contrasted with the alternative of pairwise comparisons between all possible pairs of APA sites, which, if the gene contains many APA sites, can quickly become unwieldy.
2.5. Comparing ψ values across conditions
After computing ψ values for each gene in each sample, LABRAT compares ψ values across conditions to identify genes whose relative APA usage has changed. This is done using a linear mixed effects model (Fig. 1C). LABRAT fits a model to the ψ values across conditions and then asks if that model is a better fit than a null model that assumes no change in ψ across conditions using a log likelihood test. The raw p value from the log likelihood test is then corrected for multiple hypothesis testing using the Benjamini-Hochberg method (Benjamini & Hochberg, 1995). Importantly, this model can incorporate covariates (e.g., batch, library design).
2.6. RNAseq library design
Although the majority of publicly available RNAseq data is derived from RNAseq libraries that cover the whole transcript, these are perhaps not the best library design for quantifying APA. Newer approaches that specifically profile the 3′ ends of reads (Zheng, Liu, & Tian, 2016) offer high sensitivity and accuracy for APA quantification. To deal with these designs, LABRAT includes the –librarytype parameter. If this value is set to “RNAseq” then the quantification of ψ values proceeds as described in Sections 2.1–2.5. However, if this value is set to “3pseq,” then some minor deviations are employed.
First, instead of quantifying the last two exons of every transcript, the last 300 nt of every transcript are used. This value is used because the insert sizes in many 3′ end sequencing libraries are 100–300 nt long. Second, the transcript quantification of these libraries uses Salmon’s count output instead of the length-normalized TPM value. This is because length normalization of 3′ end data is not necessary nor desirable.
3. Quantifying alternative polyadenylation with LABRAT
3.1. Installing LABRAT
Although this depends on the size of the transcriptome being analyzed, LABRAT generally uses between 5 and 20Gb of memory and takes between 30min and 4h to complete. Multithreading is supported during the quantification of APA isoform abundance with salmon (Patro et al., 2017). See the README at https://github.com/TaliaferroLab/LABRAT for detailed installation instructions, requirements and detailed documentation. The above GitHub repository contains a file (labratenv.yaml) that contains the prerequisites necessary to run LABRAT. The code in Fig. 2 uses this file and the conda package manager (available at https://docs.conda.io/projects/conda/en/latest/user-guide/install/) to install everything needed for running LABRAT.
Fig. 2.

Example code to install python environments compatible with LABRAT.
3.2. Generating transcript ends for quantification
LABRAT is run in three steps. The first filters transcripts in a genome annotation for those with high confidence 3′ ends. The second quantifies the abundances of those ends. The third calculates the relative usage of APA sites in each gene as ψ values and identifies genes whose ψ value changes significantly across conditions.
LABRAT creates and utilizes a database constructed from a gff genome annotation file to relate transcripts and genes. To create this database, LABRAT requires these genome annotations in uncompressed gff3 format. It is recommended to use annotation files from GENCODE (https://www.gencodegenes.org) as they contain specific flags used by LABRAT. Once created, this database will be stored in the same directory as the gff file from which it was made. Database generation only needs to be performed once but can take a few hours. If interrupted, the partial database created should be deleted before attempting again. Once this database is created, LABRAT uses it to make a fasta file of transcript ends to be quantified.
The relevant options for the creation of this fasta file are as follows:
–mode: This argument defines which of the three steps in APA quantification LABRAT will be performing. For the generation of the transcript end fasta file, it should be set to “makeTFfasta.”
–gff: This is the path to the gff annotation to be used.
–genomefasta: This is the path to the sequence of the genome in fasta format.
–lasttwoexons: This flag tells LABRAT if the fasta file it creates should contain the entire sequence of a transcript, or just it’s 3′ end. If included, the 3′ end is generated. If omitted, the entire sequence is generated. Including this flag can lead to higher accuracy of APA quantifications. This is because it removes the contribution of upstream alternative exons, which are rigidly associated with specific 3′ ends in the annotation, to the quantification of APA.
–librarytype: The allowed values of this parameter are “RNAseq” and “3pseq.” These correspond to the library design strategies used in generating the data (see Section 2.6).
The example in Fig. 3 will generate a fasta file containing the last two exons of filtered transcripts from a human genome annotation file in preparation for quantification with RNAseq data. This fasta file will be named TFseqs.fa.
Fig. 3.
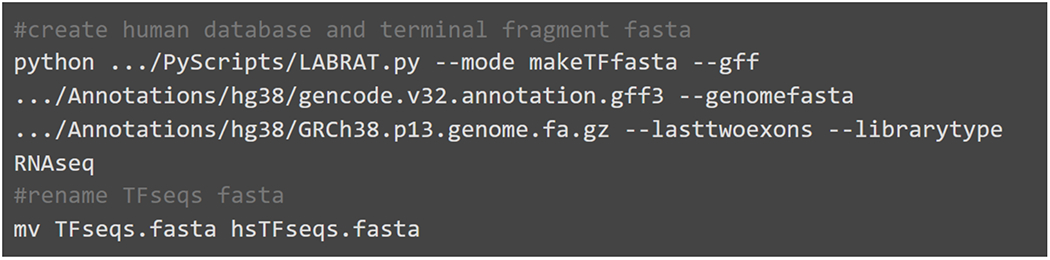
Example code for creating both the terminal fragment fasta file and a genome annotation database. Gencode’s genome annotation gff file and genome fasta are required inputs. TFseqs.fasta is created in the current directory while the database file is generated in the same directory as the gff.
3.3. Quantification of transcript fragments
After generation of the transcript fragments, their relative abundance in the supplied high-throughput sequencing data will be calculated. This step relies on the transcriptome quantification tool Salmon (Patro et al., 2017).
This step should be run in an empty directory. Compressed or uncompressed fastq or fasta read files that contain either single or paired end reads can be used. The code in Fig. 4 outputs a directory for each sample containing salmon quantification files.
Fig. 4.
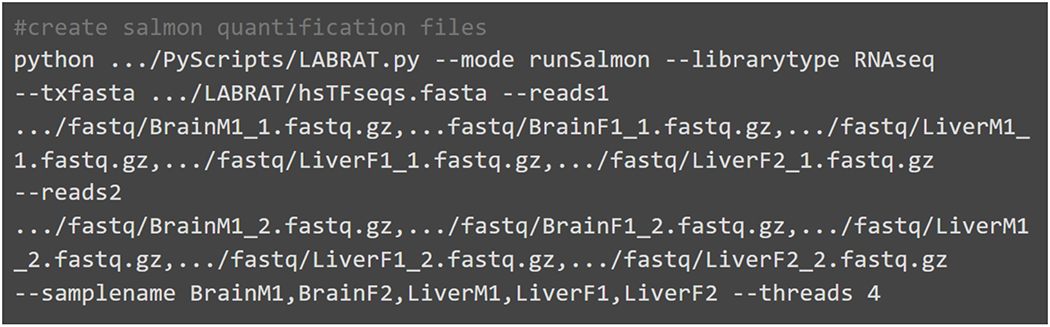
Example code for running LABRAT’s runSalmon function. RNAseq forward reads (reads1), reverse reads (reads2) and sample names are required inputs. Three prime end sequencing reads can also be used however the librarytype option should reflect the type of library provided. This code must be run in an empty directory as it outputs quantifications in new salmon directories for each sample.
The relevant options for the quantification of these transcript fragments are as follows:
–mode: This argument defines which of the three steps in APA quantification LABRAT will be performing. For the quantification of transcript abundances, it should be set to “runSalmon.”
–txfasta: This is the path to the fasta file of transcript fragments to be quantified that was generated in Section 3.2.
–reads1: A comma separated list of files containing the forward sequencing reads.
–reads2: A comma separated list of files containing the reverse sequencing reads. This can be omitted if single end data is being used. Importantly, the order of this list must be consistent with the order of the samples in – reads1.
–samplename: A comma separated list of names for the samples being quantified. Importantly, the order of this list must be consistent with the order of the samples in –reads1.
3.4. Calculating ψ values
Transcript abundances are then used to calculate ψ values for every gene with at least two alternative polyadenylation sites. Gene-level ψ values are then compared across conditions to identify genes whose ψ value has significantly changed.
This requires knowledge of which samples belong to which conditions. This is supplied to LABRAT using a tab-delimited file. Minimally, this file contains two columns with the headers “sample” and “condition.” Optionally additional columns can be added that specify covariates. It is required that the header for any covariate column contain the string “covariate.” Sample names must match those given to LABRAT during the Salmon quantification, and the condition column must contain exactly two factors. An example of this file in tabular form is shown in Table 1.
Table 1.
Example of sampconds text file in tabulated format.
| Sample | Condition | Covariate1 |
|---|---|---|
| BrainM1 | Brain | M |
| BrainF1 | Brain | F |
| LiverM1 | Liver | M |
| LiverF1 | Liver | F |
| LiverF2 | Liver | F |
Sample and condition columns are required with additional “covariate” containing columns being optional. This file defines the conditions that psi values are calculated across.
Following quantification, ψ values for all genes in all conditions as well as raw and Benjamini-Hochberg corrected p-values are reported in a file named “LABRAT.psis.pval.” Differences in mean ψ values across the conditions are also reported. Additionally the APA structure for each gene is reported. The possible values for this structure are “TUTR” (tandem UTR), “ALE” (alternative last exon), or if both structures are present within the gene, “mixed.” A second file called “numberofposfactors.txt” is also generated. This file contains information of how each transcript was assigned to an APA site. The code in Fig. 5 uses LABRAT to quantify ψ values in human brain and liver samples.
Fig. 5.
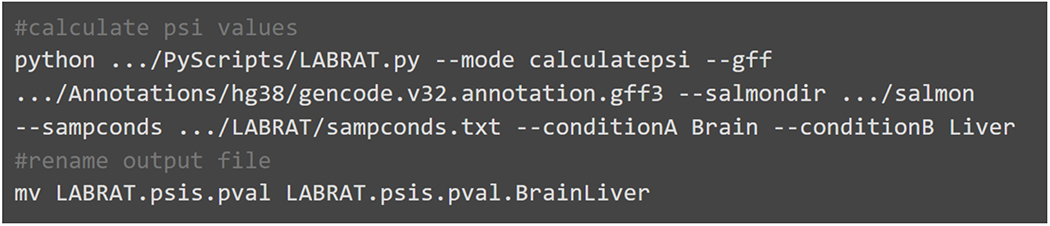
Example code for running LABRAT’s calculatepsi function. Gencode’s genome annotation gff, the directories produced by runSalmon, a tab-delimited sampconds text file and defined conditions are required inputs. This code produces several output files within the current directory.
The relevant options for the quantification of the ψ values are as follows:
–mode: This argument defines which of the three steps in APA quantification LABRAT will be performing. For the calculation of ψ values, it should be set to “calculatepsi.”
–gff: This is the path to the gff annotation to be used. It should be the same annotation used in earlier steps.
–salmondir: A directory containing salmon quantification subdirectories with one for each sample. The names of these subdirectories are the sample names supplied during the runSalmon step.
–sampconds: A tab delimited text file relating samples, conditions, and optionally, covariates. The names of the samples should match those found on the subdirectories in the salmondir directory.
–conditionA and –conditionB: In order to define a difference in ψ across conditions (Δψ), the direction of comparison must be defined. Δψ for each gene is defined as the mean ψ value in condition B minus the mean ψ value in condition A. Both conditionA and conditionB must be found in the condition column of the sampconds file.
3.5. Directory architecture
LABRAT expects and will create a defined directory structure in regard to the Salmon quantifications. An example of this directory structure is shown in Fig. 6.
Fig. 6.
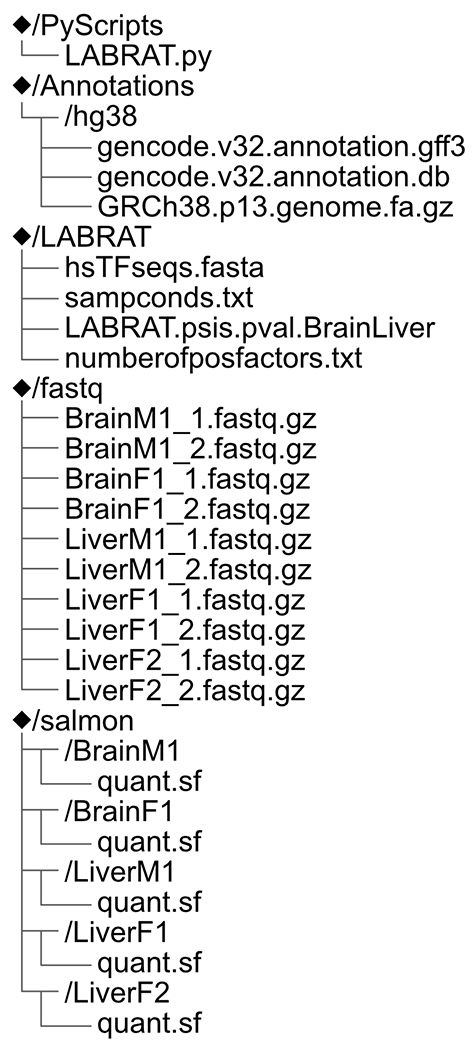
Schematic of resulting directories after completing this LABRAT quickstart guide. While not explicitly required, similar directory organization for LABRAT projects is best practice.
4. Quantification of APA in single cell RNAseq data
LABRAT was originally designed to quantify APA from bulk RNAseq data. However, the growing plethora of available single cell RNAseq datasets presented an opportunity to look at the heterogeneity of APA regulation with cell populations. To take advantage of this, a companion script was written, LABRATsc (LABRAT single cell). LABRATsc uses the same approach that LABRAT does by using available tools to quantify transcript abundances, grouping transcripts that share polyadenylation sites together, and reporting APA status using ψ values that range between 0 and 1. In this section, we briefly detail how LABRATsc deals with single cell RNAseq data, special considerations to keep in mind, and present an example workflow and outputs.
4.1. How LABRATsc quantifies APA in single cell RNAseq data
LABRAT relies on Salmon to quantify transcripts from bulk RNAseq data. LABRATsc relies on an analogous tool for single cell RNAseq quantification, alevin (Srivastava, Malik, Smith, Sudbery, & Patro, 2019). While LABRAT compares ψ values between two conditions (e.g., treatment and control), LABRATsc compares ψ values between predefined groups or clusters of cells. These clusters can be defined using standard approaches such as tSNE and UMAP. Following quantification with alevin, transcripts are filtered to keep only those with confidently defined 3′ ends exactly as they are in LABRAT-based quantification. Quantification then proceeds upon one of two paths as indicated by the –mode parameter.
If the –mode parameter is set to “cellbycell” then a ψ value is calculated for every gene in every cell. In practice, the coverage for most genes in most cells is extremely low or nonexistent. Genes that do not pass a read coverage threshold (indicated by the –readcount in a given cell) have ψ values of NA in that cell. For each gene, ψ values are then compared across cell clusters using the ψ value in each individual cells as an independent observation.
If the –mode parameter is set to “subsampleClusters,” then read counts for each transcript are first summed across all of the cells within a cluster. This has the advantage of raising the number of reads associated with each gene, but single cell resolution is lost. Statistical tests to identify genes with regulated APA across cell clusters are performed by creating a distribution of ψ values for each gene in each cluster through bootstrapping resampling.
4.2. Important considerations
Droplet-based single-cell RNA-sequencing libraries (10 × Genomics, Drop-seq, etc.) are particularly well suited to APA analysis as their construction is virtually identical to that of bulk PAS-seq libraries that capture 3′ ends directly using anchored oligo-d(T) primers (“TVN” priming) (Yao & Shi, 2014). However, several important caveats must be considered when using LABRATsc with these libraries.
First, low read depth, relative to bulk RNA-seq, and the so-called drop out effect limit the reliable detection of APA events in individual cells to relatively highly expressed (and robustly captured) genes. These inherent limitations make the selection of appropriate minimum read thresholds (set using the –readcountfilter argument) critical to identifying robust APA events with LABRATsc. We suggest thresholds of ≥100 counts per gene for cluster-level ψ value calculation (–mode subsampleClusters) and ≥5 counts for per-cell ψ value calculation (–mode cellbycell) as reasonable starting points, but these thresholds—particularly in the per-cell case—vary considerably between experiments due to differences in sequencing depth, per-cell RNA content, and cell type-specific gene expression patterns, among other factors. It is important to note that the accuracy of per-cell ψ value calculations with low read thresholds will be less accurate for genes with large numbers of 3′ end isoforms, but this concern is mitigated somewhat by the fact that 94% of the human GENCODE genes considered by LABRATsc have five or fewer isoforms.
Second, while these libraries putatively capture 3′ ends of mRNAs, they also contain substantial internal priming artifacts derived from TVN priming on genomically encoded poly(A) tracts. This is not a rare event—we have observed that internal priming accounts for up to 40% of molecules captured in 10 × Genomics 3′ libraries, and RNA velocity methods rely on this internal priming in introns to quantify pre-mRNAs (La Manno et al., 2018). If these internal priming events occur in close proximity to bonafide polyadenylation sites, they may skew raw ψ values substantially by “double counting” some fragments. However, while this may impact the accuracy of raw ψ values for some genes, the relative ψ values between cells are unaffected as the rate of internal priming is largely consistent across cells and we are thus still able to reliable identify APA events.
4.3. Generating input matrices for LABRATsc with alevin
Prior to running LABRATsc, cell-by-isoform count matrices must be produced. This step relies on the single-cell transcriptome quantification tool alevin, which is distributed with Salmon. For general information about alevin, please see https://salmon.readthedocs.io/en/latest/alevin.html and Srivastava et al. (2019).
The following arguments must be passed to alevin for use with LABRATsc, ideally in this order:
–l: Library type. For most single-cell libraries, this will be “ISR.”
–1: A list of files containing the forward sequencing reads (also supports shell expansion of wildcards).
–2: A list of files containing the forward sequencing reads (also supports shell expansion of wildcards) in the same order as −1.
–dropseq/–chromium/–chromiumV3: One of these arguments must be provided depending on the sequencing platform used.
-i: A salmon index, generated with LABRAT using the –librarytype 3pseq argument (as in Section 3.2).
-p: Number of threads used by alevin (default is all available threads).
-o: Output path for count matrix and metadata.
–tgMap: A transcript-to-gene map file, which consists of each transcript ID in the salmon index listed twice per-line, separated by a tab.
–fldMean 250: Expected mean fragment length (250 for consistency with LABRAT’s execution of salmon).
–fldSD 20: Expected standard deviation of mean fragment length (20 for consistency with LABRAT’s execution of salmon).
–validateMappings: Enables selective alignment of the sequencing reads.
–whitelist: A whitelist of cell barcodes from a previous analysis to restrict quantitation to previously identified valid barcodes [Optional].
The example in Fig. 7 will generate a count matrix in the folder “sample1” from a hypothetical 10 × Genomics 3′ v3 single-cell RNA-seq library (lists of FASTQs indicated with placeholders [read1 FASTQs] and [read2 FASTQs]) against a pre-build salmon reference (hsTFseqs3pseq.fasta) with a transcript-to-gene map (hsTFseqs3pseq.fasta.tgMap) and optional whitelist of cell barcodes (sample1_barcodes.txt).
Fig. 7.
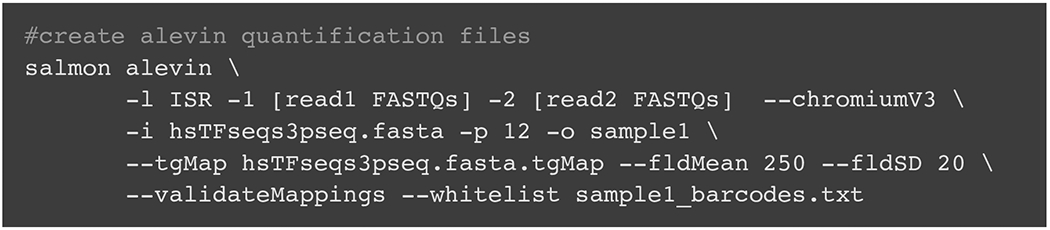
Example code showing the use of alevin to generate input matrices for LABRATsc.
4.4. Calculating ψ values with LABRATsc
Transcript counts from one or more single-cell libraries are next used to calculate ψ values for every gene with at least two alternative polyadenylation sites. As when running LABRAT on bulk RNA-seq, gene-level ψ values are then compared across conditions to identify genes with significant ψ value changes. As described in Section 4.1, LABRATsc provides two different approaches for ψ calculation and significance testing: per-cell or using subsampled clusters.
The relevant options for the quantification of the ψ values with LABRATsc are as follows:
–mode: cellbycell or subsampleClusters, as described in Section 4.1.
–gff:This is the path to the gff annotation to be used. It should be the same annotation used to generate the salmon index provided to alevin.
–alevindir: A directory containing alevin quantification subdirectories with one for each sample. The names of these subdirectories will be appended to the cell names in each sample matrix to form a “sample_barcode” cell id for each cell.
–conditions: A tab delimited text file with column names “sample” and “condition.” The first column contains cell ids and the second column contains cell condition or cluster. The cell ids in the sample column must follow the “sample_barcode” structure described above. Note that unlike LABRAT, LABRATsc does not currently support covariates.
–readcountfilter: Minimum read count necessary for calculation of ψ values. Genes that do not pass this filter will have reported ψ values of NA. For “cellbycell” mode, this is the number of reads mapping to a gene in that single cell, while in “subsampleClusters” mode, this is the summed number of reads mapping to a gene across all cells in a predefined cluster.
–conditionA and –conditionB: In order to define a difference in ψ across conditions (Δψ), the direction of comparison must be defined. Δψ for each gene is defined as the mean ψ value in condition B minus the mean ψ value in condition A. Both conditionA and conditionB must be found in the condition column of the sampconds file.
The code in Fig. 8 uses LABRATsc to quantify ψ values using both available modes in example data provided in the LABRAT github repo (paths are relative to the root directory of the repository).
Fig. 8.
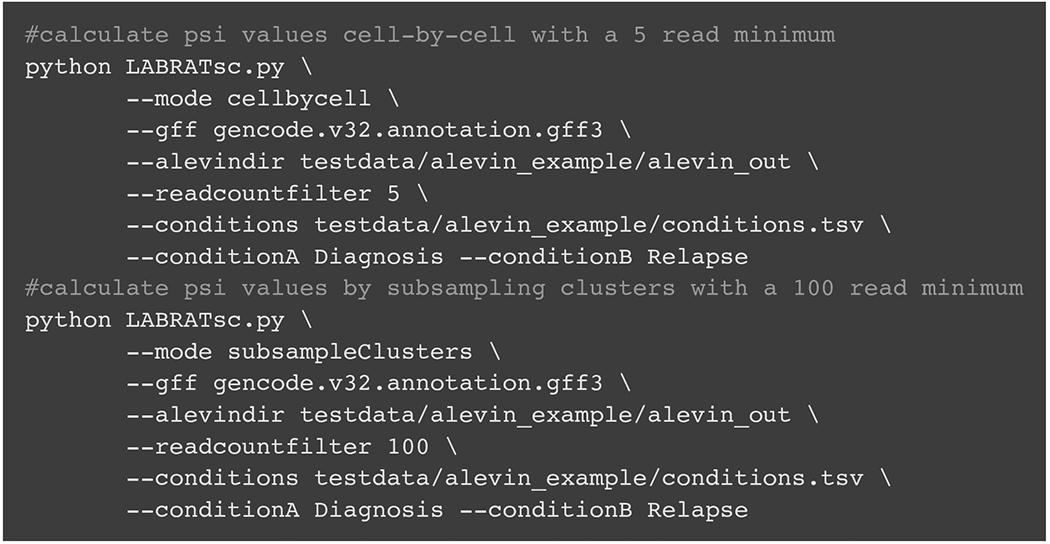
Example code showing the use of LABRATsc to calculate psi and delta psi values in both cellbycell and subsampleClusters modes.
Following quantification, ψ values for all genes in all conditions as well as raw and Benjamini-Hochberg corrected p-values are reported in files named “results.subsampleclusters.txt” (subsampleClusters mode) or “results.cellbycell.txt” (cellbycell mode). Differences in mean ψ values across the conditions are also reported. In cellbycell mode, the results file additionally includes the number of cells in each condition passing read depth filters for each gene. Finally, per-cell psi values are reported for each gene when run in cellbycell mode in a file named “psis.cellbycell.txt.gz.” These results can be used to annotate existing single-cell analyses, as demonstrated by the example in Fig. 9.
Fig. 9.
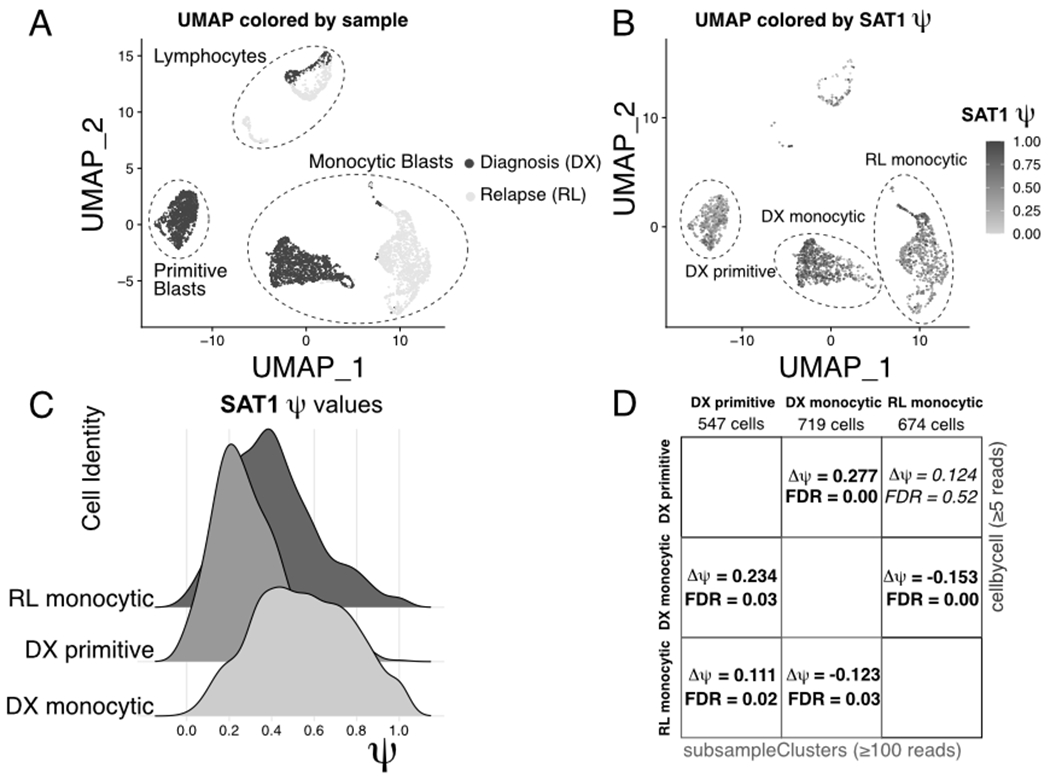
Alternative polyadenylation of SAT1 in acute myeloid leukemia. (A) UMAP projection showing the diagnosis and relapse samples from GSE143363. Major cell types are indicated with dashed circles. (B) UMAP projection from (A), with cells colored by SAT1 ψ value. Important clusters are indicated with dashed circles. (C) Ridge plot showing distributions of SAT1 ψ values in the clusters highlighted in (C). (D) Table comparing SAT1 pairwise delta-ψ tests between the clusters highlighted in (C) using “--mode subsampleClusters” and “--mode cellbycell.”
Fig. 9 shows the significant alternative polyadenylation event at the SAT1 gene in bone marrow mononuclear cells from a published acute myeloid leukemia dataset (Pei et al., 2020) (GEO accession: GSE143363). Fig. 9A shows a UMAP projection of the cells labeled with cluster names and colored by sample (Diagnosis or Relapse). Fig. 9B shows the per-cell calculated ψ values for the SAT1 gene on the same projection, with higher ψ values observed in the “DX monocytic” cluster when compared to the “DX primitive” and “RL monocytic” clusters. This pattern is also evident when plotting distributions of ψ values in the three major clusters (Fig. 9C). Notably, this event is detectable using both the cellbycell and subsampleClusters modes, which report virtually identical ψ value changes (Fig. 9D). These results demonstrate that LABRATsc is able to identify significant APA events between different cell types (“DX primitive” vs “DX monocytic”) and in closely related cells after in vivo therapy (“DX monocytic” vs “RL monocytic”).
Acknowledgments
We thank Krysta Engel for helpful comments and suggestions. This work was funded by the National Institutes of Health (R35-GM133885) (JMT), the Boettcher Foundation (Webb-Waring Early Career Investigator Award AWD-182937), a Predoctoral Training Grant in Molecular Biology (NIH-T32-GM008730) (RG) and the RNA Bioscience Initiative at the University of Colorado Anschutz Medical Campus (RG and JMT).
References
- Benjamini Y, & Hochberg Y (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B, Statistical Methodology, 57(1), 289–300. [Google Scholar]
- Bray NL, Pimentel H, Melsted P, & Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology, 34(5), 525–527. [DOI] [PubMed] [Google Scholar]
- Cho PF, Poulin F, Cho-Park YA, Cho-Park IB, Chicoine JD, Lasko P, et al. (2005). A new paradigm for translational control: Inhibition via 5′-3’ mRNA tethering by Bicoid and the eIF4E cognate 4EHP. Cell, 121(3), 411–423. [DOI] [PubMed] [Google Scholar]
- Goering R, Engel KL, Gillen AE, Fong N, Bentley DL, & Matthew Taliaferro J (2020). LABRAT reveals association of alternative polyadenylation with transcript localization, RNA binding protein expression, transcription speed, and cancer survival. Cold Spring Harbor Laboratory. 2020.10.05.326702 10.1101/2020.10.05.326702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi E, Mariella E, Lembo A, Molineris I, & Provero P (2016). Roar: Detecting alternative polyadenylation with standard mRNA sequencing libraries. BMC Bioinformatics, 17(1), 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha KCH, Blencowe BJ,& Morris Q (2018). QAPA: A new method for the systematic analysis of alternative polyadenylation from RNA-seq data. Genome Biology, 19(1), 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. (2018). RNA velocity of single cells. Nature, 560(7719), 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, & Bartel DP (2009). Widespread shortening of 3’UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell, 138(4), 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai AA, Baharian G, Pagé Sabourin A, Brinkworth JF, Nédélec Y, Foley JW, et al. (2016). Widespread shortening of 3’ untranslated regions and increased exon inclusion are evolutionarily conserved features of innate immune responses to infection. PLoS Genetics, 12(9), e1006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, & Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods, 14(4), 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei S, Pollyea DA, Gustafson A, Stevens BM, Minhajuddin M, Fu R, et al. (2020). Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discovery, 10(4), 536–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, & Burge CB (2008). Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science, 320(5883), 1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, & Robinson MD (2015). Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research, 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Malik L, Smith T, Sudbery I, & Patro R (2019). Alevin efficiently estimates accurate gene abundances from dscRNA-seq data. Genome Biology, 20(1), 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaferro JM, Vidaki M, Oliveira R, Olson S, Zhan L, Saxena T, et al. (2016). Distal alternative last exons localize mRNAs to neural projections. Molecular Cell, 61(6), 821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Donehower LA, Cooper TA, Neilson JR, Wheeler DA, Wagner EJ, et al. (2014). Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nature Communications, 5, ncomms6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C, & Shi Y (2014). Global and quantitative profiling of polyadenylated RNAs using PAS-seq. In Rorbach J, & Bobrowicz AJ (Eds.), Polyadenylation: Methods and protocols (pp. 179–185). Humana Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Liu X, & Tian B (2016). 3’READS+, a sensitive and accurate method for 3′ end sequencing of polyadenylated RNA. RNA, 22(10), 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]