

Abstract
Potassium ion homeostasis is essential for bacterial survival, playing roles in osmoregulation, pH homeostasis, regulation of protein synthesis, enzyme activation, membrane potential adjustment and electrical signaling. To accomplish such diverse physiological tasks, it is not surprising that a single bacterium typically encodes several potassium uptake and release systems. To understand the role each individual protein fulfills and how these proteins work in concert, it is important to identify the molecular details of their function. One needs to understand whether the systems transport ions actively or passively, and what mechanisms or ligands lead to the activation or inactivation of individual systems. Combining mechanistic information with knowledge about the physiology under different stress situations, such as osmostress, pH stress or nutrient limitation, one can identify the task of each system and deduce how they are coordinated with each other. By reviewing the general principles of bacterial membrane physiology and describing the molecular architecture and function of several bacterial K+-transporting systems, we aim to provide a framework for microbiologists studying bacterial potassium homeostasis and the many K+-translocating systems that are still poorly understood.
Keywords: K+ transport, Principles of K+ transporters and channels, Membrane potential, Bacterial physiology, Structural biology
Graphical Abstract
Introduction
Bacteria are true artists of survival. As motile cells or organized in biofilms, they can survive under very different environmental conditions with huge and sudden changes in pH, temperature and salinity. The partition of ions, especially K+ ions, across the microbial membrane is emerging as an important modulator of microbial physiology in both planktonic and multicellular niches. K+ uptake and release are involved in osmoregulation, pH homeostasis, regulation of protein synthesis, enzyme activation, membrane potential adjustment and electrical signaling. For these purposes, individual bacteria possess an array of different ion transport proteins. Some are passive channels that allow K+ to flow down its electrochemical gradient, others are active transporters that use ATP or the proton motive force to accumulate ions far above their electrochemical equilibrium setpoint. Each transport system possesses different molecular properties that influence how the protein responds to – or generates – stimuli, including conductances and transport rates, open probabilities, and ligand activation or deactivation. The actions of these various proteins integrate to establish the microbe’s membrane physiology.
For a discussion on potassium transport focused on cellular physiology we direct the reader to other recent reviews [1, 2]. Here, we provide a brief overview of the major principles that guide potassium-coupled membrane physiology in microbes. We then address the mechanisms of known bacterial K+ transport systems, primarily revealed through in vivo transport assays, electrophysiological approaches and structural biology, including recent structures of major bacterial potassium transport systems [3–8]. Understanding the most fundamental molecular properties of these proteins: whether transport is passive or active, the magnitude of the transport rate, and what stimuli are required for transport to occur, is an essential foundation to construct a coherent understanding of their physiological contributions and outcomes.
K+ transport and equilibrium ion distributions
In order to perform the processes essential to life, all cells regulate the partition of ions across the plasma membrane, expending energy to maintain a non-equilibrium ion distribution. For processes that induce a separation of charge, this non-equilibrium distribution contributes to an electrical potential across the membrane, Δψ. In respiring bacteria, the electron transport chain (ETC) is the major determinant of the cell’s membrane potential, due to the separation of charge across the membrane as protons are pumped from the cytosol to the outside or the periplasm. This electrical gradient, together with the chemical proton gradient, provide the major contribution to the “proton motive force” (pmf), which bacteria harness to drive the synthesis of ATP and various secondary active transport processes [9]. The membrane potential for a metabolizing bacterium is on the order of −150 mV [10, 11]. By some estimates, maintaining this inside-negative membrane potential is responsible for consumption of 50% of the bacteria’s total energy [12]. Other factors also contribute to the resting membrane potential, including the Donnan potential, which arises from impermeant, negatively charged macromolecules including DNA or RNA present in excess in the cytoplasm [13]. These fixed charges contribute to the membrane potential regardless of the metabolic activity of the bacterium.
When the rate of respiration is such that the electron transport chain defines the electrical gradient Δψ, other ions will distribute across that membrane according to this potential. The potential favors accumulation of membrane-permeant cations in the cytoplasm, as well as the expulsion of membrane-permeant anions, until the chemical free energy of any species balances this electrical potential, according to the Nernst equation. For K+, the equilibrium distribution is established by the membrane potential:
Where R is the gas constant, T is the temperature, F is the Faraday constant, and z is the charge of the ion. Simple diffusion of charged ions through the lipid bilayer is prohibitively slow; K+ permeability through the lipid bilayer is characterized by a logP of about −12, roughly 10 orders of magnitude slower than water [14]. Therefore, the equilibration of intracellular K+ concentrations requires proteins to establish ion selective pathways across the membrane.
Many bacteria maintain intracellular K+ concentrations of hundreds of millimolar, and some Gram-positive species accumulate K+ to half-molar concentrations [15]. These high potassium concentrations contribute to maintaining cell turgor, pH homeostasis, and are optimal for the activity of various enzymes, including the ribosome [16, 17]. Microbial environments are often comparatively lower in K+ (0.1–10 mM). For example, Luria-Bertani media commonly used to culture E. coli in the laboratory has a K+ concentration of about 7 mM [18, 19], and seawater has a concentration of roughly 10 mM (https://www.ncei.noaa.gov/access/ocean-carbon-data-system/oceans/DOE94.pdf), and so bacteria often maintain large K+ chemical gradients across the membrane. Multiple transporters, channels, pumps, and accessory systems with various transport mechanisms all play a role in K+ partition across the plasma membrane. The mechanism of transport is critically important to understanding the influence of a potassium transport protein on cellular potassium concentrations under different physiological circumstances.
Transporters couple the translocation of ions against their electrochemical gradient to the favorable hydrolysis of ATP or other established ion gradients, such as the proton gradient established by the ETC. This transport mechanism is characterized by an energy-coupled alternating access of the ion binding site. In a simplified way, this can be described as two gates opening only one at a time towards opposite sides of the membrane, facilitating substrate occlusion. Such proteins are essential for accumulating potassium, especially in K+-deficient environments (Figure 1a, 1b). The structures of both ATP- and H+-coupled microbial K+ transporters – KdpFABC and KimA, respectively – have been described in recent years [7, 8, 20].
Figure 1. Simplified scenarios showing the interplay between bacterial metabolism, K+ chemical gradient, and membrane potential.

Yellow arrows indicate proton movement and blue arrows indicate K+ movement. Darker shades of pink indicate higher K+ concentrations. Transporters (a, b) utilize energy derived from ATP hydrolysis (a) or the proton motive force (b) to accumulate intracellular K+ against its electrochemical gradient. Channels (c–f) facilitate the thermodynamically favorable movement of K+ down its electrochemical gradient. In the first case (c, d), an inside-negative membrane potential is established by the proton gradient due to the high relative permeability of protons from the electron transport chain (gETC). K+ can be accumulated to physiological levels in the cytoplasm even at low external concentrations (c), but the accumulation must be regulated to prevent uncontrolled K+ uptake causing K+-mediated cytotoxicity at higher external K+ concentrations (d). When the relative permeability of K+ (gK+) surpasses that of protons due to a decrease in metabolic activity or K+ channel opening (e,f), the degree of membrane depolarization is determined by the external K+ concentration.
In contrast, ion channels permit the flow of ions down their electrochemical gradient, which does not require an energy input. These proteins do not occlude the substrate but can simply be described as a gated pore that opens or closes to control ion passage. In classical mammalian physiological contexts, ion channels do not contribute to building potassium gradients; instead they exploit the potassium gradient built by the energy-coupled transporters for other physiological purposes. Thus it was a mechanistic surprise when the “potassium uptake” Trk system, which had been shown to play an important role in K+ accumulation in certain contexts [16, 21, 22], was shown via single-channel electrophysiology to be an ion channel [6]. However, an ion channel mechanism does not necessarily preclude ion transport against a chemical gradient, as long as the electrical gradient is favorable. For example, Ca2+ uptake into mitochondria is mediated by the Ca2+ channel MCU, and thermodynamically driven by the membrane potential established by the ETC in the energized mitochondrial membrane [23, 24]. Likewise, in bacteria, the maintenance of low intracellular fluoride (a bacterial toxin found at high concentrations in certain environmental niches) is managed by an ion channel. The expulsion of this anionic species is thermodynamically driven by the inside-negative electrical potential [25, 26].
Thus, the inside-negative membrane potential allows for bacteria to enrich positively charged ions within the cell relative to their external environment according to the Nernst potential. Assuming that other charged species were also able to equilibrate across the membrane via a slow process in order to compensate the accumulating charge, passive potassium diffusion across the membrane alone would lead to a cytoplasmic accumulation of intracellular potassium of ∼50-fold in a −100 mV potential, or 350-fold in a −150 mV potential, according to Eq. 1. Thus, in an environment with millimolar potassium concentrations, cytoplasmic accumulation of K+ to the physiological range of hundreds of millimolar could be achieved by ion channel-mediated passive diffusive mechanisms alone in a metabolically active cell (Figure 1c). These mechanisms must be tightly regulated, as unhindered K+ uptake would initiate processes of K+-mediated cytotoxicity (Figure 1d). Together, proteins with disparate mechanisms can work together to maintain the cytoplasmic K+ concentration.
K+ transport and dynamic changes to membrane potential
In the preceding section, we considered a scenario in which the bacterial membrane was only slightly permeable to potassium, such that the membrane potential was defined primarily by the rate of respiration and the electron transport chain. However, any transmembrane ion gradient can contribute to the membrane potential, with its relative impact defined by its relative membrane permeability. In the context of a bacterium, opening a selective, large conductance K+ channel would suddenly and transiently increase the membrane’s K+ permeability. This change would be accompanied by a change in the membrane potential according to the Goldman equation, which is a weighted average of the relative permeabilities of each ion:
If the rate of K+ movement is sufficiently high, the electrical gradient that evolves as it partitions across the membrane upon channel opening may outweigh other contributions to the membrane potential. There is substantial and detailed precedent for this in eukaryotic biology, where ionic gradients are maintained by Na+/K+ ATPase, but dynamically perturbed by the opening of different populations of ion channels, with dramatic consequences for the cell’s membrane potential [27].
Ion channels are typically expressed in low numbers in prokaryotic cell membranes [28, 29]. Could an ion channel influence the potential established by the ETC? The enzymes in the ETC, such as quinol oxidase, turn over at about ∼104 protons per second [30], and a rough calculation suggests a bacterial cell that dedicates 60% of its membrane to this enzyme could fit ∼100 molecules [31]. Thus, for a metabolically active bacterium, charge evolves across a microbial membrane via respiration on the order of 106 charges/sec. The opening of an electrodiffusive K+ channel could substantially influence Δψ if it permits the movement of ≥106 K+ ions per second. This is within the realm of physiological channel conductances, many of which can permeate 106 −108 ions per second. Thus, even for a rapidly metabolizing bacteria, opening a K+ permeation pathway could influence Δψ, and bacteria with low metabolic activity will be correspondingly more sensitive to membrane potential modulation through the opening of K+ channels.
Let’s compare two highly simplified scenarios: in the first, the resting bacterial membrane has low (but non-zero) potassium permeability. In this case, the membrane potential is dominated by contributions from the ETC. As long as a K+-permeable protein is expressed in the inner membrane, K+ will accumulate inside the cell, even in the absence of active transport, following the membrane potential established by the electron transport chain (Figure 1c). In fact, in this scenario, K+ permeation pathways must be tightly regulated to prevent K+ uptake causing cytotoxicity when environmental K+ is high (Figure 1d).
In the second scenario, imagine a high-conductance K+ channel opening, such that the transient high permeability of K+ dominates the electrical potential term (Figure 1e, 1f). Without the accompanying movement of a counterion, channel opening will not lead to drastic changes in the cellular K+ concentration, or K+ rushing out of the cell. Rather, the movement of just a few K+ ions will establish a new membrane potential that counterbalances the chemical potential and halts further ion translocation. This newly established membrane potential will depend on the relative magnitude of charge separation by the ETC and the ion translocation by the K+ channel, but regardless, will move towards the Nernst potential defined by the K+ gradient. If the Nernst potential for K+ is substantially different than the resting membrane potential, a transient perturbation in the membrane potential will occur, for as long as the high-conductance K+ channel is open. Single channel electrophysiological recordings of purified and reconstituted bacterial ion channels show that these proteins often exhibit “flickery” behavior, with frequent, rapid openings and closings and fast inactivation kinetics, meaning that the channel may only be open for millisecond timescales [4, 6, 32]. Once the channel closes (and K+ membrane permeability suddenly plummets), Δψ can return to the resting potential. In the scenario described here, the major physiological consequence of the channel is not to modulate potassium concentrations, but instead to modulate the membrane potential, which itself impacts a host of processes, such as magnesium influx and ribosome binding, motility, antibiotic efficacy, and bacterial proliferation [33–37]. The magnitude of the influence of the K+ gradient is modulated by the respiration rate of the bacterium [37], coupling the two processes. In addition, the membrane potential itself can regulate K+ channel activation or inactivation in a negative feedback mechanism. K+ channel activity has been linked to membrane potential changes in a variety of systems [38–41].
Thus, potassium channels can influence membrane potential by eliciting large rapid changes in the relative permeability of potassium. Membrane potential changes could also be evoked by large rapid changes in the transmembrane ion gradients. Such events are unusual in eukaryotic cells, with their large volumes and carefully controlled extracellular potassium concentrations. But what about bacteria? The volume of a bacterium is very small compared to a eukaryotic cell, about one thousandth the size [1]. A bacterium with 100 mM cytoplasmic K+ would contain only 108 K+ ions. A respectable K+ channel can conduct that many ions in a few seconds. Would this be expected to correspond to a drastic reduction in the K+ concentration inside the cell? At least in the case of potassium, it’s unlikely: due to their small membrane surface area, bacteria also have a small capacitance, meaning that the transmembrane movement of just a few charges would quickly build up a large negative-inside membrane potential prohibiting further ion movement [42]. Without counterion movement, the cytoplasmic potassium would not be seriously depleted by ion channel opening, at least to a first approximation. Note that for different ions present at much lower concentrations, such as some divalent cations, this rough approximation fails since the movement of enough ions to change the membrane potential would be tightly coupled to chemical potential changes as well [1].
While intracellular K+ might be relatively stable, drastic and sudden changes in external ion concentrations are far from unusual. Sometimes, major shifts in external potassium are coupled to a bacterium’s particular lifestyle: for example, intracellular pathogens experience 4 mM external K+ in the bloodstream, but 150 mM K+ in the host’s cytosol [43, 44]. The potassium gradient, and thus the membrane potential change effected by the opening of a large-conductance potassium channel, would be extremely different in these two circumstances. In addition, in bacterial biofilms, oscillating potassium concentration gradients are coupled to nutrient limitation in the biofilm’s interior [45]. Such changes in K+ ion gradient could readily impact the membrane potential. Moreover, other stimuli could be integrated in the process by influencing K+ translocation, including high concentration of activating ligands or low concentrations of negative modulators, or perturbations in pH. This leads to the possibility that K+ transport systems could be involved in signaling pathways by dynamically altering membrane potentials, a scenario most thoroughly explored in the context of cell-to-cell communication in bacterial biofilms [40, 45, 46]. However, transient changes in membrane potential have been observed and linked to other microbial physiologies as well [35, 37, 47–49].
Consistent with diverse roles for K+ in signaling and physiology, several different potassium transport systems are typically found within one bacterial species. We will discuss the transport systems that are commonly found in most bacteria in different combinations in further detail. These transport systems exhibit various mechanisms for passage of K+ into and out of the cell, including ligand-gated channels, proton symport, and pH regulation. The presence of various regulatory modules in these K+ transport proteins allows for spatial and temporal control of K+ passage across the membrane and therefore could both induce and respond to dynamic changes in membrane potential upon some physical or biochemical stimulus. The rates at which these systems transport K+, the affinity for various ligands, how channels are influenced by voltage and the identity of co-transported molecules among other factors determine how these channels and transporters both generate and respond to chemical and electrical signals. A complete mechanistic understanding of the proteins that modulate K+ translocation can provide insight into how these ion gradients affect bacterial physiology. Here, we review studies of several K+-transporting proteins, highlighting insights into the mechanisms of K+ transport gained through structural and functional studies.
Molecular determinants of bacterial K+ channels and transporters
Current literature is ambiguous in identifying the transport modes of the different K+-translocating systems found in bacteria. Most assignments are relics of pre-structure times or are adapted from eukaryotic systems, not taking into account the different electrophysiology of pro- and eukaryotes. A few bacterial ion channels have been studied in structural and biophysical depth as models to understand eukaryotic K+ channels, but the mechanistic characterization of bacterial ion channels, for the sake of understanding distinctly microbial physiologies, has generally lagged. Partly this is because bacterial membranes are too small to apply the ion channel biophysicist’s most potent and versatile tool, patch clamp electrophysiology. And partly it is because the unique physiologies associated with microbial ion channel biology are only now being discovered. In the following section, we will discuss the transport mode and regulation of several K+-translocating systems based on structural and functional data. We will focus on those systems for which structural data of the complete systems is available. These are particularly the 2-TM potassium channels KcsA and MthK, the two-pored potassium channels Trk and Ktr, and the potassium transporters Kdp and Kup. A first overview of the assembly of the complexes and of the topology of the transmembrane subunits, which aims to highlight the differences and similarities between the different systems at a glance, is provided in Figure 2.
Figure 2. Assembly and topology of bacterial K+-translocating systems.

Overview of the assembly of the different systems discussed. Channel-like subunits/domains are shown in green, RCK subunits/domains in gray, the P-type ATPase KdpB in beige with blue, red and yellow, KdpC in purple, KdpF in cyan, the LeuT-fold domain in light blue, and the adenylyltransferase-like domain in orange. The topologies provide more molecular detail of the transmembrane domains. Characteristic are the M1PM2 domains of the channel-like proteins (green), the insertion of the cytosolic N, P and A domains (red, blue, and yellow, respectively) into loops of the transmembrane domain of KdpB (beige) and the 5+5 inverted repeat of the LeuT-fold domain (light blue) of KimA.
2-TM potassium channels
Almost every bacterium encodes at least one tetrameric potassium channel, most commonly of the 2-TM type. However, their physiological role is poorly understood. YugO from Bacillus subtilis was suggested to serve electrical signaling in biofilms [40], HpKch from Helicobacter pylori [50] and CglK from Corynebacterium glutamicum [51] are suggested to mediate K+ uptake, and Kch from Escherichia coli seems to be involved in membrane potential adjustment [52]. Unfortunately, sufficient structural and functional data to address the mechanisms of ion flux and regulation is not available for any of these channels. Instead, we will discuss KcsA from Streptomyces lividans and MthK from Methanothermobacter thermoautotrophicum, which are of unknown physiological role but have been studied in great molecular detail as model systems for eukaryotic potassium channels.
KcsA was first described in 1995 [53]. It is an ion channel highly selective for potassium. The channel was shown to be activated following a downshift of the intracellular pH [54]. Further, it is voltage-dependent, with an increased activity at depolarization (weak outward rectifier). Once activated, up to 108 ions per second flow through the channel for approximately 200 milliseconds, until a slow inactivation takes place [55]. In order to understand ion selectivity, activation and inactivation mechanisms of tetrameric potassium channels, massive amounts of data focusing on KcsA have been produced. This review does not aim to summarize all the details. Instead, we will briefly introduce the architecture of this archetypal 2-TM potassium channel and summarize the general principle of ion selectivity and of gating. Ion selectivity is required to ensure that potassium ions are the predominantly translocated species, while gating is needed to ensure that potassium flux only occurs in physiologically appropriate situations.
As a 2-TM potassium channel, KcsA forms a homotetramer, with each protomer consisting of two transmembrane helices and a pore domain in between them. This arrangement is called membrane-pore-membrane (M1PM2) motif (Figure 3). The pore domain contains a short pore helix followed by a pore loop. The orientation of four pore domains towards each other in the tetrameric assembly leads to the formation of the central pore. The pore loops form the major constriction of the pore and contain the selectivity filter, which is the basis for ion selectivity and mediates part of KcsA’s regulation. The large vestibule below the selectivity filter is separated from the cytosol by a helical bundle formed by the C-terminal end of the second transmembrane helices, functioning as a gate (Figure 3a). It can adopt open and closed conformations, regulating ion flux [56].
Figure 3. Gating of potassium channel KcsA.

(a) Structure (PDB 1J95) of the membrane-inserted domains of KcsA in its conductive, closed state with the inset highlighting the selectivity filter, (b) Bundle gate mechanism. Cartoons of closed, conductive and open, conductive states, respectively. For simplicity only two M1PM2 domains are shown.
The best-studied feature of KcsA is its selectivity filter, consisting of the highly conserved amino acid sequence TVGYG. The distances of the backbone carbonyls of these amino acids in the tetramer are ideal for replacing the hydration shell of potassium, allowing the translocation of a partially or fully dehydrated ion. The amino acids form four consecutive ion binding sites, named S1 to S4 (counted from the extracellular side), with eight coordinating oxygens for each potassium ion binding site (Figure 3a) [57]. The low energetic effort for K+ dehydration allows the high conductance of potassium ions resulting in the discrimination against other ions. This is particularly relevant for the naturally prevalent Na+, which is poorly translocated in a partially hydrated state [58]. When the potassium ion is released from the tunnel, it gets rehydrated in an energetically favorable event [59]. The long-proposed mechanism for potassium ion conduction suggested co-translocation of potassium ions and water molecules, where alternating positions (S1 and S3 or S2 and S4) were occupied by ions and surrounded by water. The rationale for this hypothesis is the high electrical repulsion between the dehydrated ions. In recent years this mechanism was challenged by a direct knock-on mechanism derived from molecular dynamics (MD) simulations. These suggest that it is rather a direct repulsion of the ions in consecutive sites that squeezes them through the pore, facilitating high conductance [53][60]. The voltage across the membrane is essential for this mechanism, as it drives the continuous flow of ions through the pore. The requirement of a direct knock-on mechanism hence might be the key to the observed voltage dependency of KcsA [61, 62].
To control the ion flux, KcsA has two gating points. The first is the selectivity filter, which can adopt two conformations. One is the conductive state, where potassium ions can pass through the filter, and the other is a collapsed state, adopted after a certain time of ion flux in a mechanism called C-type inactivation. The second gating point is the helical bundle, which widens the pore below the selectivity filter up to a diameter of 22 Å [63] in an open, active conformation and closes the pore restricting ion flux in the closed, inactive conformation.
In the resting state, KcsA’s helical bundle is closed, while the selectivity filter is in a conductive state (Figure 3a; inset). Upon a pH shift to a lower intracellular pH of 4, key residues in the helical bundle and the selectivity filter become protonated, triggering the opening of the helical bundle (Figure 3b) [56]. In this conformation, KcsA is in an open, conductive state; the flow of ions along the electrochemical gradient is possible for milliseconds, with increased ion flux at increased membrane voltages [64]. The flow is stopped by a C-type-like inactivation, in which the selectivity filter undergoes a conformational change due to allosteric coupling with the helical bundle gate. Conformational rearrangements along transmembrane helix 2 lead to the collapse of the selectivity filter, causing a constriction of the pore by 2 Å (8 Å to 6 Å diameter). . Once C-type inactivation is completed, the translocation of ions is stopped even though the helical bundle is still in an open conformation; the channel is thus in an active, non-conductive state. Upon an increase in pH, the helical bundle closes again. The recovery of the collapsed selectivity filter allows the restoration of the conductive state, while the closed helical bundle prevents ion translocation [63]. Besides serving C-type inactivation, conformational changes in the selectivity filter also preclude unspecific ion leaks. Normally, potassium is translocated through KcsA at high external potassium concentrations. At low potassium concentrations, the selectivity filter collapses to prevent the binding of unspecific ions, particularly sodium. This collapse does not occur when potassium ions are bound to the selectivity filter, thereby stabilizing it. Thus, this feature allows the channel to further distinguish between potassium and sodium ions [65].
All 2-TM potassium channels form a tetramer of M1PM2 domains with a central selectivity filter and share the same general mechanism of ion translocation, but differences have evolved in their regulation. While KcsA is regulated by pH, other potassium channels such as HpKchA from H. pylori, YugO from B. subtilis, and MthK from M. thermoautotrophicum have additional cytoplasmic regulatory domains. For the latter, structural data is available, although its physiological role remains elusive. All of these 2-TM potassium channels are regulated by so-called Regulator of Conductance of K+ (RCK) domains, which assemble to an octameric ring. The ring can result from different assemblies of the RCK domains: (1) a tandem pair of RCK domains covalently bound to the M1PM2 domain, (2) a single RCK domain each fused to the N and C terminus, (3) a single RCK domain covalently bound while the other is expressed separately via a second start codon, (4) a tandem soluble cytosolic RCK domain, and (5) a single RCK domain expressed solely as a soluble protein [66]. Independent of the assembly, all structurally characterized RCK gating rings interact directly with the pore and, through conformational changes triggered by the binding of ligands from the cytosol, are responsible for or involved in the opening and closing of the channel. Thus, the RCK domain couples the channel to the metabolic demands and status of the cell.
Every structurally known RCK gating ring consists of a tetramer of dimers. The dimers associate to form the two-layered octameric ring, with each dimer contributing an up and a down domain. Inside each layer, the single RCK domains are not in contact [67]. Each individual RCK domain consists of a conserved N and a less conserved C lobe. The N lobes form the inner ring, which is bound to or associates with the pore domain, while the C lobes are oriented to the periphery [67, 68]. The N lobes of each RCK domain contain a conserved Rossmann fold, which consists of alternating alpha helices and beta strands [69, 70]. RCK domains that bind nucleotides include a GxGxxG...D/E motif within the Rossmann fold. The C lobes are connected to the N lobes via a helix-turn-helix motif formed by so-called crossover helices of the dimer. Ligand binding sites can be located within the N lobe, at the interface between the N and the C lobes in the dimer interface, and within the C lobes. Ligands that bind to RCK domains vary from ADP [71], ATP [72], NAD+ [73] and cyclic di-AMP [74, 75] to ions like Na+ [76] or Ca2+ [77].
In the following, we will focus on the potassium channel MthK and its regulation by an RCK domain. MthK is allosterically activated through the binding of Ca2+ ions to the RCK domains, which lead to conformational changes that allow the translocation of potassium ions with up to 108 molecules per second. The flux of potassium is stopped by an N-type inactivation [78] , also known as ball-and-chain inactivation, in which the N terminus plugs the pore. Like KcsA, MthK is pH dependent, but in this protein a shift to lower internal pH decreases activation of the channel because Ca2+ binding is reduced [79]. Voltage-dependent Ca2+ binding from the cytosolic side to the pore domain renders MthK an inward-rectifying channel [80].
The transmembrane domains of MthK show the same assembly as KcsA with the same conserved selectivity filter (TVGYG) (Figure 4a). However, instead of containing elongated helices following the helical bundle, an RCK domain is covalently linked. A second soluble copy of the RCK domain is produced from a second start codon within the mthK gene. This soluble domain binds to the tethered domain, forming the RCK dimer (Figure 4a). Subsequently, four dimers associate to form the octameric gating ring, which is bound to the M1PM2 domains. In its closed conformation, the gating ring has four-fold symmetry, and the TM2 helices of the transmembrane domains are straight and elongated by two helical turns compared to the open state. They form a helical bundle underneath the pore, constituting the intracellular gate. The helical bundle constricts the pore diameter immediately below the selectivity filter at the membrane-cytosol interface. This prevents ion translocation (Figure 4b). The C-linker between TM2 and the tethered RCK domain lies on top of the bound RCK domain, presumably stabilizing the closed state of the tunnel by interacting with the RCK domain and the membrane interface. A slight rocking of the RCK gating ring, caused by the rather flexible C-linker and interactions between the hydrophobic membrane and the RCK domains, can occur without leading to channel activation [4].
Figure 4. Ca2+-gating by K+ channel MthK.

(a) Structure (PDB 6U6H) of MthK in its closed, conductive state in the absence of Ca2+ with the inset highlighting the selectivity filter. (b) Helical bundle gating controlled by the cytosolic RCK domains (tethered RCK domains light grey, soluble RCK domains dark grey). Cartoons of closed, conductive and open, conductive state, respectively. For simplicity only two M1PM2 domains are shown.
Upon binding of Ca2+ to the RCK domains, MthK opens and potassium ions are translocated. The binding of Ca2+ to at least two of three known binding sites provides sufficient energy for a conformational change within the RCK domains, which in turn results in a loss of the four-fold symmetry of the gating ring. The ring breaks open by slight shifts at the four dimer-to-dimer interfaces, called assembly interfaces, resulting in two-fold symmetry. The structural change in the RCK gating ring results in a loosening of the C-linker and in a wrenching of the last 2 helical turns of TM2. This pulls the helical bundle open and causes the TM2 helices to kink at a highly conserved hinge glycine. Thus, potassium ion flux is enabled. The new conformational freedom allows the RCK gating ring to bend up to 20° away from the membrane plane and undergo an even stronger rocking motion (Figure 4b) [4]. Four to five seconds after activation, the channel is inactivated by an N-type inactivation [78], in which the N-terminal peptide of one protomer plugs into the pore and stops ion translocation while the helical bundle remains open. The previously flexible N terminus likely interacts with hydrophobic residues inside the pore and charged residues at the pore-cytoplasm interface [4]. To allow ion translocation again, the N-terminal peptide has to be released from the pore to restore the tunnel. This release probably coincides with the dissociation of Ca2+ from the RCK domains and the triggering of the closed conformation. In addition to the helical bundle gate, selectivity filter gating has been shown for MthK. At low potassium concentrations, the filter collapses into a non-conductive state, which at the same time stabilizes the closed helical bundle. However, C-type inactivation does not play a role in MthK, and the relevance of the non-conductive selectivity filter is unclear [81].
Both ion channels discussed here exhibit a key feature characteristic of channel and not transporter activity: none of the described states during ion translocation occlude the ion between two gates. Instead, the helical bundle presents the main gate, which opens upon a stimulus (pH shift, Ca2+ increase), while the selectivity filter is already conductive. To avoid extended open events that dissipate the membrane potential, a non-conductive state is established rather rapidly, either by filter gating (C-type inactivation) or N-type inactivation with an N-terminal plug. Further, the collapse of the selectivity filter at conditions where low external potassium concentrations are available precludes non-selective ion translocation. However, due to the different activation modes, it is also clear that both channels have to fulfil different physiological roles. Being strongly inward-rectifying, MthK might mainly serve K+ uptake at conditions with increased intracellular Ca2+ levels, while KcsA could be involved at pH homeostasis as it is activated at a rather low intracellular pH.
Potassium uptake by the multiligand-regulated potassium channels KtrAB and TrkAH
In most bacteria, KtrAB and TrkAH are the major K+ uptake systems for everyday challenges [16, 21, 82, 83]. With an apparent affinity of around 1 mM, they serve a role in osmoadapation, general potassium homeostasis at neutral to alkaline pH, pH homeostasis and, potentially, membrane potential adjustment [38]. Due to their Na+ or H+ dependency, KtrAB and TrkAH were initially described to function as active transporters [84–86], coupling potassium influx to either Na+ or H+ transport to accumulate K+ inside the cell against the concentration gradient. However, in 2013, Cao and colleagues demonstrated using single channel recordings that TrkAH is an ATP- and ADP-gated ion channel [6]. The measured single channel conductance for K+ of 550 pS at −60 mV is equivalent to an ion flow of 3.3*107 per second. Measurements at different voltages showed that TrkAH is slightly inward rectifying. The presence of ATP led to an increased open probability, while ADP decreased it. Similarly, flux assays and SSM-based electrophysiology revealed that KtrAB functions as a Na+- and ATP-dependent K+ channel, neither symporting nor antiporting sodium ions [5, 87].
The transport mode – channel activity – is also reflected by the structural data available for KtrAB from Vibrio alginolyticus and B. subtilis, and TrkAH from Vibrio parahaemolyticus. Both complexes contain two parallel pores regulated by an RCK gating ring. KtrAB consists of a dimer of the ion-translocating subunit KtrB and an octameric ring of the regulatory RCK protein KtrA (Figure 5a). In TrkAH, a dimer of the membrane-bound subunit TrkH forms a complex with a tetrameric ring of the regulatory RCK protein TrkA (Figure 5c). The ion-translocating subunits KtrB and TrkH are prokaryotic members of the Superfamily of K+ Transporters (SKT) [88–90]. SKT members most likely evolved from tetrameric 2-TM potassium channels like KcsA by gene duplication and gene fusion [91]. Consequently, each protomer consists of four nonidentical, covalently linked M1PM2 motifs, referred to as domains 1 to 4 (D1 to D4), which are arranged to a pseudo-fourfold symmetric pore for potassium translocation. TrkH contains two more N-terminal transmembrane helices of unknown function, which are named domain 0 and are located at the periphery of the pore [83, 90]. As in 2-TM potassium channels, the pore loops connecting the transmembrane helices at the extracellular side form the selectivity filter. Compared to the classical selectivity filter sequence T(X)GYG, KtrB and TrkH harbor a less conserved selectivity filter, reduced to the first glycine in each pore loop (Figure 5a) [92, 93]. Just below the selectivity filter, an intramembrane, pore-blocking loop, formed by the central part of broken helix D3M2 (consisting of D3M2a and D3M2b), together with a highly conserved arginine in helix D4M2, constitutes the channels’ gate [90, 93–95]. This loop seems to present a required adaptation compared to the helical bundle in 2-TM potassium channels, because two parallel pores cannot be gated by a helical bundle. Opening and closing of the gates in KtrB and TrkH is controlled by the soluble RCK proteins KtrA and TrkA, respectively. KtrA consists of only one RCK domain, eight of which assemble into a homooctameric ring [6, 67, 96]. In comparison, TrkA assembles as a tetrameric gating ring, in which each of the protomers comprises two homologous RCK domains, which are referred to as RCK1 and RCK2. Despite the different organization, each of the RCK domains consists of a ring-forming N lobe (called N1 and N2 in TrkA), which possesses the conserved Rossmann fold including a GxGxxG...D/E motif for nucleotide binding, and a flexible C lobe at the periphery [68].
Figure 5. Gating of nucleotide- and cation-gated two-pored K+ channels KtrAB and TrkAH.

(a) Structure (PDB 4J7C) of KtrAB in its ATP-bound (but still closed) state with the inset highlighting the selectivity filter, (b) Intramembrane loop gating of KtrAB controlled by the cytosolic RCK subunits. Cartoons of ADP-bound, closed and ATP-bound, open state, respectively, (c) Structure (PDB 4J9U) of TrkAH in its ADP-bound, closed state, (d) Intramembrane loop gating of TrkAH controlled by the cytosolic RCK subunits (RCK2 light grey, RCK1 dark grey). Cartoons of ADP-bound, closed and ATP- bound, open state, respectively.
To function as effective potassium channels in the context of osmoadaptation and pH homeostasis, KtrAB and TrkAH need to fulfill two tasks: First, they should selectively take up potassium even at a high excess of sodium. Second, they should be gated to grant rapid potassium flux when needed, while avoiding excessive potassium uptake causing cytotoxicity, or potassium efflux during membrane depolarization. At first sight, the poorly conserved selectivity filter seems to contradict the first requirement because it could result in a less selective ion translocation, as was shown for tetrameric potassium channels with degraded selectivity filter sequences [97, 98]. In line with this, single channel recordings of TrkAH revealed similar conductivity for K+, Rb+ and Cs+, whereas Na+ and Li+ showed lower but still significant rates [6]. Based on the apparent non-selective ion translocation, it was recently suggested that TrkAH might serve a particular role in membrane potential adjustment, rather than or in addition to potassium homeostasis [38]. By contrast, a significantly different channel profile has been described for KtrAB, revealing that it in fact functions as a selective K+ channel [87]. Like TrkAH, KtrAB binds all tested group 1 elements with a similar affinity, except Li+. However, ion translocation by channel subunit KtrB alone, without the KtrA gating domain, increases with decreasing ion size. Only K+ and Na+ are efficiently translocated, while the passage of bigger ions is blocked. This selection of translocated ions is mediated by the intramembrane gate, which apparently cannot open wide enough to accommodate the passage of the bigger ions. More important than the exclusion of the bigger ions, the channel also shows a significant preference for K+ over Na+: In the presence of Na+, the binding affinity for K+ increases at least 10-fold, resulting in the preferred binding of K+ over Na+. This consequently leads to the required selective accumulation of K+, as was shown in K+ uptake experiments at native-like mixed ion conditions [87].
With respect to gating, the pore-forming subunits, KtrB and TrkH, seem to permit potassium permeation independent of any ligands. The naturally occurring assemblies KtrAB and TrkAH, on the other hand, were both shown to be dependent on different ligands. First, Na+, in addition to its role in selectivity and affinity, is involved in the activation of the KtrAB complex. Na+ as ligand is plausible because internal Na+ levels probably transiently increase at hyperosmotic external conditions, when a maximal activity of KtrAB is required. In the presence of Na+, the Vmax of potassium uptake increases when compared to KtrB alone, while the KM decreases, approaching the determined KD for K+ binding. In the absence of Na+, KtrAB is completely inactive. It is hypothesized that Na+, in the presence of the KtrA gating domain, increases the gate’s open probability [86, 87, 92]. While the binding site and structural consequences of Na+ binding remain elusive, its effects on K+ binding affinity and gating suggest that it is located within KtrB [87]. Second, both KtrAB and TrkAH depend on nucleotides, which directly bind to the N lobe of KtrA and TrkA, respectively [5, 6]. While NAD+/NADH were initially suggested as ligands, extensive studies revealed the preferred binding of ATP and ADP to the N lobe, with ATP as an activating and ADP as an inactivating ligand [5, 6, 72, 99, 100]. In KtrAB, ATP stabilizes a round, in TrkAH a broken gating ring, while ADP promotes an oval-shaped ring spanning the width of the dimer interface. The basic principles of ATP- and ADP-dependent conformational changes in KtrA and TrkA, respectively, were reviewed by Levin and Zhou [93], while recent structures of both systems provide further details [38, 101]. Importantly, it was shown that divalent cations (Mg2+ or Ca2+) are necessary for the activation of KtrAB by ATP. Binding of the cations to a well-conserved site at the KtrA dimer interface tethers two ATP molecules via their γ-phosphate and thereby stabilizes the active round conformation of the octameric ring [101]. Similarly, divalent ions stabilize the active, ATP-bound conformation of KtrA homolog KtrC [102]. Third, KtrAB, TrkAH and KtrCD of Gram-positive bacteria (as shown for B. subtilis, Streptococcus agalactiae, Listeria monocytogenes, Staphylococcus aureus, Mycoplasma pneumoniae) are inactivated by c-di-AMP [103], which binds to the C lobe of KtrA, TrkA, and KtrC, respectively. A crystal structure of a KtrA C lobe dimer in the presence of c-di-AMP showed that it fosters a stable, compact interaction [75]. This interaction might prevent the flexibility of the C lobe observed in the absence of c-di-AMP [99, 100], which could result in the fixation of the gating ring in its closed conformation. Simultaneously, the stabilization of the ring might favor the binding of ADP over ATP. Whether a similar regulatory mechanism by second messengers exists in Gram-negative bacteria is unclear since the binding site for c-di-AMP is not conserved.
As elaborated above, the complex assembly of KtrAB and TrkAH is unique and differs from tetrameric ligand-gated K+ channels. The RCK domains are separately expressed proteins, which interact with the two pores while lacking the covalent tether [5, 6, 104]. In consequence, these unique systems require an adapted mechanism to transmit the conformational changes of the regulatory rings to the channel subunits.
For the KtrAB complex, structural analysis revealed two different kinds of contacts between the KtrB dimer and the octameric KtrA ring: The lateral contact involves the C terminus of KtrB, which on the one hand interacts with neighboring KtrB subunit, stabilizing dimer formation, and on the other hand interacts with the KtrA ring. Deletion of the last 4 residues of the C terminus led to a non-functional channel and abolished the assembly of the complex, while a deletion of the last 10 residues even prevented dimer formation [5]. The second contact of the KtrAB complex was initially described as tip contact, because in the ATP-bound conformation a cytoplasmic loop between a helical hairpin formed by D1M2b and helix D2M1 was observed to form a direct but minor interaction with the KtrA ring. However, in the ADP-bound cryo-EM map, this interaction became much more significant due to a rearrangement of the helical hairpin into an elongated subunit-interconnecting helix reaching from the KtrB protomer into the oval-shaped KtrA ring [100]. Hence, the tip contact is not only required for complex formation but appears to be the major element for controlling gating by allosteric coupling at a distance. Together with the adjacent D4M2b helix, which is also extended in the ADP-bound state to span the entire membrane, extended helix D1M2b narrows the pore, reduces water accessibility and likely locks the gate, the intramembrane loop, in a rigid, closed conformation [100]. Consequently, the current working model for channel activation is that, upon stimulating conditions, e.g. hyperosmotic stress where the ATP level and possibly also the internal Na+ concentration transiently increase, ADP is replaced by ATP in a competitive manner, inducing a conformational rearrangement of KtrA to a round gating ring [5, 67]. The elongated helices D1M2b form helical hairpins and helices D4M2b partially unfold, unlocking the intramembrane loop to, in principle, allow K+ permeation. Na+ might bind in the KtrB protomers, further stabilizing an open gate conformation. If sufficient intracellular K+ concentrations are reached, the corresponding ATP to ADP ratio and the internal Na+ concentration decrease, inducing the adoption of the closed conformation (Figure 5b). Further, in Gram-positive bacteria, c-di-AMP is produced, stabilizing the inactive state. However, it needs to be mentioned that this working model has various weaknesses. First, in the published ATP-bound structure of KtrAB, the intramembrane loop remains in the closed conformation, although the crystallizing condition contained a high Na+ concentration [5]. It remains to be investigated whether there is an additional stimulus/ligand required for channel opening. Further, the resolution of the ADP-bound KtrAB map of 6.6 A is too low to reveal any details on the stabilization of the closed channel by ADP. Additionally, a functional proof of complex inactivation upon ADP binding is still missing, and the detailed role of c-di-AMP upon closing needs to be elucidated. Furthermore and conceptually most important, it is puzzling to understand under which conditions ADP would ever bind to KtrA, because, even under non-stressed conditions, the cellular ATP concentration is always in excess compared to the ADP concentration, while the binding affinities for both nucleotides appear to be similar [72, 100].
When compared to KtrAB, both the organization of the TrkA ring and the interaction surfaces between the TrkH and T rkA differ. Therefore, despite the similarities of both systems, the two channels are modulated by different mechanisms. In TrkH, the C terminus is much shorter than in KtrB and is not involved in any interactions [90]. Instead, interactions between each TrkH protomer and the TrkA N1 and N2 domains, respectively, are established by a cytosolic loop attached to the broken D3M2b helix, which is directly linked to the gate, the intramembrane loop, and lies parallel to the membrane plane. The interactions are named HN1 and HN2, respectively. Importantly, the TrkA gating ring comprises two distinct subunit interfaces between the N lobes of the RCK1 domain and the N lobes of the RCK2 domain, which are described as N1-N1 and N2-N2, respectively. In the presence of ADP, each N2 domain of TrkA binds one ADP molecule, while the N1 domain remains unoccupied [6, 38]. The pores of the dimeric TrkH are blocked by the intramembrane loops constricting ion permeation, as observed in KtrAB (cf. [99, 100]). In TrkAH, the closure of the gate by ADP has been confirmed by patch clamp measurement [6]. A recently solved structure of partially open TrkAH with bound ATP allows the proposal of a nucleotide-dependent gating mechanism: In contrast to ADP, ATP binds to both the N1 and the N2 domain, which is consistent with previously solved structures of isolated T rkA [6, 38]. Detailed analysis of the nucleotide binding sites revealed that the N1 domain has conserved residues selecting for ATP by γ-phosphate recognition, while the N2 domain can coordinate both ADP and ATP. ATP binding causes the N2 domain of each TrkA protomer to rotate relative to the N1 domain, breaking the N2-N2 interfaces. This results in a splitting of the tetrameric gating ring into two dimers. While the N2 domain remains attached to TrkH, the HN1 interface is completely abolished, which likely results in a pulling on the D3M2b helix. The rearrangement of D3M2b consequently moves the intramembrane loop towards the intracellular side resulting in an unlocked intramembrane loop and an open pore (Figure 5d)[38]. Thus, the current assumption is that the binding of ATP to the N1 domain is crucial for the conformational changes in TrkA and the opening of the pore [38]. This hypothesis is supported by the observation that ATP-stimulated channel opening is abolished upon mutation of the ATP binding site in the N1 domain [6]. The role of nucleotide binding in the N2 domain is less clear. ADP binding to this domain stabilizes the closed state, possibly by more efficiently stabilizing the HN2 interface. While the gating mechanism of potassium channel TrkAH is much better understood than that of KtrAB, here, too, it remains to be established why TrkAH would ever be closed under physiological conditions with an excess of ATP. A rather low affinity for ATP binding in the N1 domain could pose a possible explanation.
Despite the many details that are missing to fully understand the regulatory mechanism of TrkAH and particularly KtrAB, it is clear from the presented data that both systems, like other RCK-gated K+ channels, function as ligand-gated ion channels. Importantly, the observed pmf dependency described for both systems as well as the determined low Km values and the rather high minimal external K+ concentrations required for potassium accumulation are in line with the channel activity of these proteins. Channel activity also explains how the systems can play different physiological roles. When activated, scenarios in which they serve osmoadaptation, pH homeostasis, and/or membrane potential adjustment are conceivable.
K+ pump KdpFABC
In some circumstances, especially when the external potassium concentration is too low to accumulate enough K+ with the given membrane potential, an active transporter is required to maintain cytoplasmic K+ levels. In many bacteria facing potassium limitation in their natural environment, the KdpFABC (K+-dependent P-type ATPase) system is found, which has been best characterized for E. coli. KdpFABC is a heterotetrameric K+ pump, which is unique for combining P-type ATPase and SKT subunits, resulting in an active transporter with high apparent affinity for K+ of 2 μM. It actively pumps potassium ions with a turnover rate of ∼100 per second, fueled by the hydrolysis of ATP [105]. The complex consists of the P-type ATPase KdpB, the SKT protein KdpA, and the supplementary subunits KdpF and KdpC. Each of these subunits has its own characteristics and functions within the KdpFABC heterotetramer.
Subunit KdpA has 10 TM helices and, like TrkH and KtrB, belongs to the SKT superfamily [106]. As such, it contains the structural motifs D1 to D4, forming a pore-like structure, with an additional transmembrane helix each fused N- and C-terminally. The KdpA selectivity filter, as in TrkH and KtrB, features a reduced selectivity filter motif, in which ion coordination has been observed in only two sites [7, 20]. Mutations of the central glycine residue of the D2 pore loop result in an altered ion selectivity: exchanging it for serine allows for stimulation of ATPase activity by NH4+, while a mutation to aspartate enables ATPase stimulation by Rb+ [107, 108]. Insertions of positively charged residues in the site corresponding to S1 of the KcsA selectivity filter result in a reduced affinity for K+ [109]. The first crystal structure of KdpFABC suggested that the positively charged side chain occupies the coordination site of K+, thus competing for binding [7]. The selectivity filter is probably the main mediator of the high ion specificity and affinity required for the physiological function of KdpFABC, and thus could be the main reason for the inclusion of a channel-like subunit in an active transporter. Below the selectivity filter, an essential arginine was identified by mutagenesis [110]. Moreover, KdpA also contains a broken D3M2 helix, forming an intramembrane loop in the potential translocation pore below the selectivity filter [7]. Of note, this loop is much less polar than those of KtrB and TrkH. Furthermore, the pore below the intramembrane loop appears to be too narrow to accommodate the translocation of potassium ions. In line with this, KdpA alone is unable to facilitate K+ uptake [20].
KdpB is a 7 TM P-type ATPase, responsible for ATP hydrolysis driving K+ translocation by KdpFABC. Like all P-type ATPases, KdpB consists of a transmembrane domain and three cytosolic domains (N – nucleotide binding, P – phosphorylation, A – actuator), which are responsible for ATP binding and hydrolysis. During the transport cycle, known as the Post-Albers cycle, P-type ATPases undergo inward- and outward-facing conformations, canonically known as E1 and E2, with the phosphorylated intermediates E1-P and E2-P [111–114]. Energy-coupled alternating access is one of the main criteria allowing active transport against an electrochemical potential. In the phosphorylated intermediates, an aspartate in the P domain is phosphorylated. Dephosphorylation is mediated by a conserved TGES sequence in the A domain. The transmembrane domain contains the canonical substrate binding site (CBS), formed around a kink in transmembrane helix 4 induced by a conserved proline [7]. Mutagenesis of an aspartate within the transmembrane domain uncouples ATP hydrolysis from K+ transport, while mutating a lysine abolishes ATPase activity [110, 115]. These two residues form a dipole in the immediate vicinity of the CBS [7].
KdpF, the smallest subunit in the KdpFABC complex, consists of only a single transmembrane helix. Based on in vitro complementation of KdpF by lipids and its absence in some kdp operons, it is assumed to function as a lipid-like stabilizer of the complex [116, 117]. KdpC is a subunit of unassigned classification, consisting of a single transmembrane helix and a periplasmic soluble domain that lies on top of the channel-like subunit KdpA [7]. It is essential to the KdpFABC complex [118]. Although its function remains unclear, sequence alignments have suggested a role similar to β-subunits of other P-type ATPases, which are involved in substrate binding and affinity [119, 120].
In the assembled complex, KdpB shares a large interface with KdpA while KdpC is associated with the other side of KdpA (Figure 6a). The complex contains several notable features: The cytosolic P domain of KdpB is associated with KdpA by a salt bridge with an arginine in KdpA that lies at the bottom of a helix coupling to the intramembrane loop (coupling helix). A hydrated tunnel between KdpA and KdpB was observed, ranging from below the selectivity filter in KdpA to the CBS in KdpB within the membrane plane [7]. In the inward-open E2 conformation, this tunnel appears to close at the subunit interface, with a new half-channel opening from the KdpB CBS to the cytosol between the P domain and KdpA [20]. Because KdpB is the smallest member of the P-type ATPase family, it was long assumed not to have an intrinsic transport function, but rather to solely provide the energy for active transport through KdpA [121, 122]. However, ATP hydrolysis and K+ transport are strictly co-dependent, raising the question how these processes are coupled [123]. This requires an intricate interplay between the pore and ATPase subunits. Recently, two different transport models have been proposed that attempt to unify previous functional data with new structural insights.
Figure 6. Ion transport models through K+ pump KdpFABC.

(a) Structure (PDB 5MRW) of KdpFABC in its E1 state. (b) Transport cycle of the coupling helix model. (c) Transport cycle of the intersubunit transport model.
The first transport model, known as the coupling helix model, proposes ion transport through the channel subunit KdpA powered by ATP hydrolysis in KdpB, as was long assumed [7]. Two separate communication pathways between the subunits are the basis for this suggested mechanism (Figure 6b). When ions bind in the selectivity filter in KdpA, the charge induces a proton dislocation via Grotthuss mechanism along a water wire that fills the tunnel between KdpA and KdpB. This process is supported by a dipole at each end of the water wire (the essential arginine and an aspartate in KdpA, and the aspartate/lysine dipole previously identified in KdpB), and the terminal acceptor is a water molecule in the KdpB CBS, whose protonation mimics a positive substrate, inducing ATP hydrolysis. The conformational shift in the P domain pulls on the coupling helix via the salt bridge between the two elements, displacing the intramembrane loop and allowing ion passage through the KdpA channel. The proposed gating function of the intramembrane loop is comparable to that in KtrB [95]. When the ion is released, the proton is transferred back to KdpA and the P domain moves the coupling helix back, returning the complex to its initial state. KdpC was suggested to act as a periplasmic gate facilitating alternating access. New structural data, including the first E2 structure of KdpFABC, cast doubt on this model [20]. Notably, no movement of the proposed gating elements, the intramembrane loop and KdpC, was observed, while individual densities possibly representing ions were observed along the tunnel connecting KdpA and KdpB in cryo-EM maps. Moreover, a new half-channel opening from the KdpB CBS to the cytosol in the E2 state, which includes the breaking of the salt bridge between the P domain and the coupling helix, suggests an alternating access mechanism for the canonical P-type ATPase transport site. This led to the proposition of K+ transport through the intersubunit tunnel rather than through the KdpA pore. In this model, ions entering the complex through the KdpA are diverted into the intersubunit tunnel by the positive charge of the conserved arginine common to SKT proteins. Ion binding in the CBS induces ATP hydrolysis and the conformational shift to the E2 state, in which ion release through the inward-open half-channel is facilitated by a rearrangement of the side chains of the lysine/aspartate dipole in the KdpB CBS (Figure 6c). Although the intersubunit transport model addresses many of the issues of the coupling helix model, it remains to be proven, particularly because the assignment of densities as ions with the cryo-EM approaches used in these structural studies is difficult. Both models highlight the chimeric nature of the KdpFABC complex, which unifies the affinity and selectivity of ion channels with the active energy coupling of P-type ATPases to fill a highly important niche in the spectrum of prokaryotic K+ uptake systems.
Being an active transporter, it is particularly important that KdpFABC is regulated in its activity because otherwise it would continuously pump potassium ions into the cytosol to the point of toxicity. KdpFABC synthesis is controlled by the two-component system KdpDE depending on the internal and external potassium concentration [124, 125]. However, a faster protein-based regulatory mechanism is required to prevent excessive K+ uptake by KdpFABC already in the membrane. A regulatory phosphorylation of the serine in the conserved TGES motif of the A domain was observed in all structural studies, and shown to inhibit ATPase and transport activity by the complex [7, 20]. Although the kinase responsible for the phosphorylation remains unknown, it was shown to be induced by high K+ concentrations in a time-dependent manner, indicating a fast adaptation mechanism to a rise in K+ levels [126]. The initial inhibition mechanism was proposed to lock the A domain via a salt bridge with the phosphate, but subsequent functional studies suggested that the phosphate stalls the protein in the E1-P state, preventing the transition to the E2-P conformation and the dephosphorylation of the protein [126]. Thus, active K+ uptake by KdpFABC is regulated on the transcriptional level, and for a more acute regulation after the normalization of K+ levels, on a protein level by phosphorylation.
K+/H+ symporter family KUP
Like KtrAB and TrkAH, members of the KUP family, Kup and KimA, serve the potassium uptake at moderate external potassium concentrations, with a particularly high activity at acidic pH. They have been studied in different detail in E. coli, B. subtilis, L. monocytogenes, S. aureus and Lactococcus lactis [103, 127, 128]. The apparent affinities for K+ vary from micro- to millimolar concentrations depending on the organism the systems originate from; KimA from B. subtilis has a medium affinity for K+ with a Km value of 215 μM [8]. The KUP family is a member of the APC (amino acid-polyamine-organocation) superfamily, whose members are solute/cation symporters or solute/solute antiporters [129]. KimA and Kup are K+/H+ symporters and thus facilitate the accumulation of K+ against its concentration gradient by the transport of protons along the concentration gradient [8, 127, 130].
The first structural insight into how KUP transporters shuttle K+ across the membrane was gained recently by solving a cryo-EM structure of inward-occluded B. subtilis KimA [8]. KimA forms a homodimer in the membrane, in which each protomer consists of a transmembrane domain formed by 12 transmembrane helices and a C-terminal cytosolic domain (Figure 7a). Dimer formation is mediated by the interaction of the loop connecting these two domains with the cytosolic domain of the adjacent protomer. In the dimer, the cytosolic domains are swapped with respect to the transmembrane domain, so each cytoplasmic domain sits underneath the transmembrane domain of the opposite protomer.
Figure 7. K+/H+ symport by KimA.

(a) Structure (PDB 6S3K) of KimA in its inward, occluded state, rainbow-colored from N terminus (blue) to C terminus (red). (b) Cartoon of the alternating access transport by KimA.
In the transmembrane domain, the first ten helices are arranged in a 5+5 inverted repeat with discontinuous transmembrane helices 1 and 6. This packing is called the LeuT fold. The additional helices 11 and 12 connect the transmembrane domain to the cytosolic domain, which presumably has a regulatory function. In general, the substrate binding site in proteins adopting the LeuT fold is positioned in the broken region of helices 1 and 6, which form a cavity. In this region, non-protein densities assigned to three potassium ions were observed in the cryo-EM structure of KimA. Functional assays showed that only one of these potassium ions is required for efficient transport, with access to its binding site controlled by a tyrosine and an aspartate in TM1, serving as external and internal gates, respectively (Figure 7b). Importantly, these residues are conserved among KUP members. With a pKa of 8, a glutamate residue in TM6 located slightly below the substrate binding site is likely responsible for proton binding. The other two ions captured in the cryo-EM structure probably present a first regulatory mechanism for KimA’s activity. The binding of potassium ions in the inward-facing tunnel leads to trans-inhibition, probably hindering the further K+ uptake at elevated intracellular potassium concentrations (Figure 7b). Based on these first structural data, an alternating access transport cycle for KimA, and by extrapolation Kup, was proposed, where H+ and K+ can enter the tunnel freely from the extracellular space when KimA is in the outward-open conformation. In this state, the inward-facing exit tunnel is closed. Upon binding of both ions, the gating tyrosine moves to close the external gate. At the same time, TM1 and TM6 (broken helices) close the extracellular tunnel and open up the intracellular pathway. In this inward-occluded state (as captured in the structure), the pathway for K+ towards the cytosol is still blocked. The proton-binding glutamate needs to be deprotonated to allow opening of the gate towards the cell interior and K+ release (Figure 7b) [8].
Like for KdpFABC, a regulatory mechanism is particularly important for KUP proteins to avoid an excessive accumulation of potassium ions. One mechanism is the described transinhibition. Another level of regulation is probably mediated by the cytoplasmic domain, which is conserved in KimA and Kup. The structure showed that it consists of β-strand-α-helix motifs. A 5-stranded β-sheet is surrounded by 4 helices in each protomer. This motif extends upon dimer formation to a β-sheet with 10 strands [8]. This structure is comparable to that of many other soluble proteins, such as the phosphopantetheine adenylyltransferase (PPAT, PDB 1GN8). It has been shown that these proteins can bind ADP and ATP [131]. KimA and Kup from Gram-positive bacteria were shown to bind and be inactivated by c-di-AMP [132–134]. It seems reasonable to speculate that the cytosolic domain is involved in the binding, yet structural data is missing.
Towards understanding the molecular physiology of potassium homeostasis
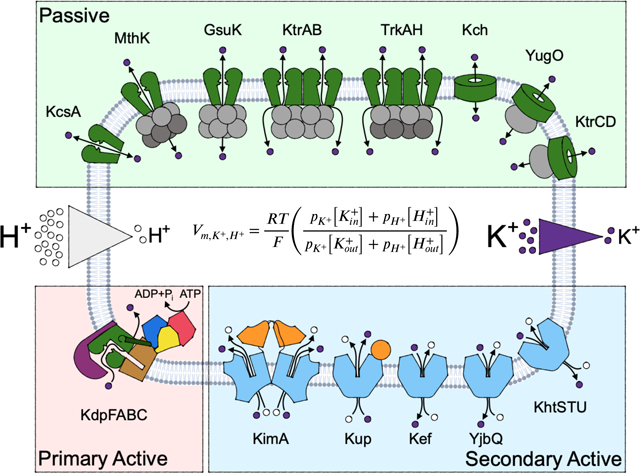
So far, the molecular characteristics of individual potassium transport systems as identified in specific organisms have been described. However, these systems are not limited to these organisms, but are widely spread among different prokaryotic species. Moreover, these proteins do not work in isolation. Most bacteria have a number of K+ transport systems, which are present in different combinations. (Table 1). Together, the action of these various potassium channels and transporters influences each bacterium’s unique physiology [16]. For example, E. coli possesses three identified potassium uptake systems, Trk, Kup and Kdp [85, 128, 135]. Further uptake with low affinity is proposed to be mediated by different, non-specific systems. Similarly, potassium efflux is mediated by mechanosensitive channels, which expel K+ together with other osmotically active compounds [136], and by the glutathione-controlled potassium/proton antiporter Kef [137, 138]. In addition to these systems of known function, E. coli encodes the ligand-gated tetrameric potassium channel Kch. Like most tetrameric potassium channels, which are found in almost every prokaryote, its physiological role is unknown. It does not seem to participate in K+ uptake or efflux under tested conditions [52]. Instead, it was hypothesized that it might contribute to the adjustment of the membrane potential. Likewise, B. subtilis encodes numerous potassium-transporting systems, the channels KtrAB, KtrCD, YugO, the K+/H+ symporter KimA, and the K+/H+ antiporters KhtSTU and potentially CpaA (formerly YjbQ) [40, 82, 132, 139, 140]. Additionally, B. subtilis also harbors mechanosensitive channels, which mediate K+ efflux. KtrAB and KtrCD are homologs of each other and of TrkAH from E. coli and other organisms, but interestingly KtrCD has a tenfold lower affinity for K+ [82], which is modulated by glutamate [141].
Table 1. Bacterial K+-translocating systems.
Summary of known bacterial K+-translocating systems highlighting the individual composition, hypothesized transport mode and direction, and representative structures. Putative transport modes are indicated with *. No transport direction is indicated for channels, as by definition ion fluxes are dictated by the electrochemical gradient.
| System | Subunit | Description, family | Transport mode (incl. dependency, direction) | Structures (PDB/EMDB) |
|---|---|---|---|---|
| Ion channel | ||||
| KcsA [150] | 2-TM potassium channel | Voltage-dependent channel | 3EFF (full-length, closed) 1K4C (closed, conductive) 1K4D (closed, inactive) 5VK6 (open, conductive) 5VKE (open, inactive) |
|
| MthK [77, 151] | 2-TM potassium channel with fused and soluble RCK domain | Ca2+-gated channel | 6U6D (Ca2+-free closed) 6UX7,6UXA,6UXB (Ca2+-bound open) 6U68,6U6E,6U6H (Ca2+-bound open-inactivated) |
|
| GsuK [71] | 2-TM potassium channel with dual fused RCK domain | Ca2+- & ADP/NAD+-gated channel | 4GX5 (Ca2+-bound closed) | |
| EcKch | 6-TM potassium channel with fused and soluble RCK domain | Ligand-gated channel* | 1ID1 (RCK domain) | |
| HpKch | 2-TM potassium channel with fused and soluble RCK domain | Ligand-gated channel* | ||
| YugO | 2-TM potassium channel with fused RCK domain | Ligand-gated channel* | ||
| TrkAH [6] | TrkA | RCK subunit (cytosolic) | ATP-, proton- (& c-di-AMP-) gated channel | 4J9U (ADP-bound) 6V4K (ATP-bound) |
| TrkH | Translocating subunit (TM), SKT family | |||
| KtrAB [5, 100] | KtrA | RCK subunit (cytosolic) | ATP-, Na+- (& c-di-AMP-) gated, pmf-dependent channel | 4J7C (ATP-bound) EM-3405 (ADP-bound) 4XTT (c-di-AMP-bound KtrA C-term) |
| KtrB | Translocating subunit (TM), SKT family | |||
| KtrCD [82, 141, 152] | KtrC | RCK subunit (cytosolic) | ATP- (& c-di-AMP) gated, glutamate-dependent channel | 6I8V (ATP-bound KtrC) |
| KtrD | Translocating subunit (TM), SKT family | |||
| Primary active transporter | ||||
| KdpFABC [7, 20] | KdpF | Small lipid-like subunit | K+ pump (uptake) | 5MRW (E1 state) 6HRA (E1 state) 6HRB (E2 state) |
| KdpA | Channel-like subunit, SKT family | |||
| KdpB | P-type ATPase subunit | |||
| KdpC | Periplasmatically oriented single TM subunit | |||
| Secondary active transporter | ||||
| KimA [8, 132, 152–154] | KUP family | K+/H+ symporter (uptake) | 6S3K | |
| Kup (TrkD) [127, 155] | KUP family | K+/H+ symporter (uptake) | ||
| Kef system [138, 140] | KefB/C | Cation:Proton antiporter 2 with fused RCK/KTN domain | Glutathione-gated K+/H+ antiporter (export) |
3L9W (KefC_RCK-KefF) |
| KefG/F | Flavoprotein | |||
| YjbQ(CpaA)[132, 156] |
Cation:Proton antiporter 2 | K+/H+ antiport (export) | 5F29 (RCK domain) | |
| KhtSTU [140, 157, 158] | KhtS | Antiporter modulator of KhtU | Ligand-gated K+/H+ antiport* (export) | |
| KhtT | C-terminal RCK protein | |||
| KhtU | Cation:Proton antiporter 2 | |||
The individual systems are active under different physiological conditions (which can be partly deduced from their functional characterization) and work together in concert to play their physiological roles, many of which are still being discovered. The intertwining roles of multiple K+ transport systems in osmoadaptation and maintaining or adapting the internal pH have been characterized for decades but are still being expanded upon. For example, c-di-AMP emerged somewhat recently as an overarching regulator that controls the activity of different uptake and efflux systems involved in osmoadaptation in Gram-positive bacteria [103].
The understanding of the molecular basis of other K+-linked physiologies is still in its infancy. For example, potassium fluxes were recently shown to mediate electrical communication between cells within biofilms [40], the synchronization of metabolic behavior between two or more biofilms, and the attraction of motile bacteria to established biofilms [142]. Interestingly, electrical signaling even permits interspecies interaction because potassium ions are essential currencies for all cells [33]. However, the molecular principles, including when and how the involved ion channels, such as YugO from B. subtilis, are activated, which ligands or stimuli control activity, and whether other K+-transporting systems are involved remains elusive.
K+ homeostasis has additionally been implicated in the virulence of various pathogenic bacteria. A deletion of the Trk system in Mycobacterium tuberculosis results in an attenuation in host colonization in a murine model [44]. Similarly, Trk channels are critical to the virulence of Salmonella enterica, and have been implicated in multiple characteristic processes of growth and virulence in vitro, including protein secretion, motility and invasion of epithelial cells, and in the pathogenesis of Salmonella in mice and chicken. A deletion of Trk results in a severe defect in the type III secretion system of Salmonella pathogenicity island 1, which is not based on a failure to regulate intracellular pH or ATP but rather on a disturbed K+ homeostasis [143, 144]. Staphylococcus aureus exhibits an unusually high level of osmotolerance and Na+ tolerance, properties that support its capacity for human colonization, pathogenesis, and growth in food. These tolerances are mediated by potassium uptake systems Ktr and Kdp. Deleting the Ktr system results in sensitivity to hyperosmotic conditions, a hyperpolarized plasma membrane, and an increased susceptibility to aminoglycoside antibiotics and cationic antimicrobials. In a mouse competitive index model of bacteraemia, the ΔktrAB mutant was significantly outcompeted by the parental strain [145, 146]. In general, many pathogenic bacteria, including S. aureus, S. typhimurium, Yersinia pestis, Acinetobacter baumannii, S. pneumoniae, and M. tuberculosis, encode the high-affinity potassium pump Kdp, which reflects their need to accumulate potassium ions in low-potassium surroundings such as those faced when invading a host [145, 147–149]. Finally, 2-TM potassium channel HpKchA, the only identified K+-translocating system of Helicobacter pylori, is essential for the colonization of the murine stomach [50].
Thus, K+ homeostasis plays a role in an expanding collection of microbial physiologies. Meanwhile, a host of situation-dependent processes such as energy state (reflected in ATP concentrations), second messengers (such as c-di-AMP), the internal and external pH, the membrane permeability of other common environmental ions like Cl-, the respiration rate, and the changing environmental K+ concentrations all influence K+ movement across the microbial membrane. Teasing out a mechanistic understanding of how K+ homeostasis contributes to physiology first requires revealing the mysteries of the individual proteins – do they transport K+ with or against the chemical gradient, do they themselves influence the membrane potential, and what ligand, environmental conditions, or co-transported substrates are required for their activity? While molecular details for some systems have been gained in recent years, highlighted in this review, we are still lacking this important information for many others.
Acknowledgments
I.H. acknowledges funding from the Deutsche Forschungsgemeinschaft (HA6322/3–1, HA6322/4–1), and the SFB 807 ‘Transport and Communication across Biological Membranes’. RBS is supported by the Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Disease.
References
- [1].Benarroch JM, Asally M. The Microbiologist’s Guide to Membrane Potential Dynamics. Trends Microbiol. 2020;28:304–14. [DOI] [PubMed] [Google Scholar]
- [2].Beagle SD, Lockless SW. Unappreciated Roles for K(+) Channels in Bacterial Physiology. Trends Microbiol. 2020. [DOI] [PMC free article] [PubMed]
- [3].Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001;411:657–61. [DOI] [PubMed] [Google Scholar]
- [4].Fan C, Sukomon N, Flood E, Rheinberger J, Allen TW, Nimigean CM. Ball-and-chain inactivation in a calcium-gated potassium channel. Nature. 2020;580:288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vieira-Pires RS, Szollosi A, Morais-Cabral JH. The structure of the KtrAB potassium transporter. Nature. 2013;496:323–8. [DOI] [PubMed] [Google Scholar]
- [6].Cao Y, Pan Y, Huang H, Jin X, Levin EJ, Kloss B, et al. Gating of the TrkH ion channel by its associated RCK protein TrkA. Nature. 2013;496:317–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Huang CS, Pedersen BP, Stokes DL. Crystal structure of the potassium-importing KdpFABC membrane complex. Nature. 2017;546:681–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tascon I, Sousa JS, Corey RA, Mills DJ, Griwatz D, Aumuller N, et al. Structural basis of proton-coupled potassium transport in the KUP family. Nat Commun. 2020;11:626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Poolman B, Konings WN. Secondary solute transport in bacteria. Biochim Biophys Acta. 1993;1183:5–39. [DOI] [PubMed] [Google Scholar]
- [10].Zilberstein D, Schuldiner S, Padan E. Proton electrochemical gradient in Escherichia coli cells and its relation to active transport of lactose. Biochemistry. 1979;18:669–73. [DOI] [PubMed] [Google Scholar]
- [11].Felle H, Porter JS, Slayman CL, Kaback HR. Quantitative measurements of membrane potential in Escherichia coli. Biochemistry. 1980;19:3585–90. [DOI] [PubMed] [Google Scholar]
- [12].Milo R, Phillips R. Cell biology by the numbers. New York, NY: Garland Science, Taylor & Francis Group. 2016. [Google Scholar]
- [13].Eisenberg DS, Crothers DM. Physical chemistry: with applications to the life sciences. Menlo Park, California: Benjamin/Cummings Publishing Company; 1979. [Google Scholar]
- [14].Paula S, Volkov AG, Van Hoek AN, Haines TH, Deamer DW. Permeation of protons, potassium ions, and small polar molecules through phospholipid bilayers as a function of membrane thickness. Biophys J. 1996;70:339–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Martinac B, Saimi Y, Kung C. Ion channels in microbes. Physiol Rev. 2008;88:1449–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Epstein W The roles and regulation of potassium in bacteria. Prog Nucleic Acid Res Mol Biol. 2003;75:293–320. [DOI] [PubMed] [Google Scholar]
- [17].Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289:920–30. [DOI] [PubMed] [Google Scholar]
- [18].Mm Kuo, Haynes WJ Loukin SH, Kung C, Saimi Y. Prokaryotic K(+) channels: from crystal structures to diversity. FEMS Microbiol Rev. 2005;29:961–85. [DOI] [PubMed] [Google Scholar]
- [19].Kuo MM, Saimi Y, Kung C. Gain-of-function mutations indicate that Escherichia coli Kch forms a functional K+ conduit in vivo. EMBO J. 2003;22:4049–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stock C, Hielkema L, Tascon I, Wunnicke D, Oostergetel GT, Azkargorta M, et al. Cryo-EM structures of KdpFABC suggest a K(+) transport mechanism via two inter-subunit half-channels. Nat Commun. 2018;9:4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schlösser A, Meldorf M, Stumpe S, Bakker EP, Epstein W. TrkH and its homolog, TrkG, determine the specificity and kinetics of cation transport by the Trk system of Escherichia coli. J Bacteriol. 1995;177:1908–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dosch DC, Helmer GL, Sutton SH, Salvacion FF, Epstein W. Genetic analysis of potassium transport loci in Escherichia coli: evidence for three constitutive systems mediating uptake potassium. J Bacteriol. 1991;173:687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–4. [DOI] [PubMed] [Google Scholar]
- [24].Bernardi P Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–55. [DOI] [PubMed] [Google Scholar]
- [25].Ji C, Stockbridge RB, Miller C. Bacterial fluoride resistance, Fluc channels, and the weak acid accumulation effect. J Gen Physiol. 2014;144:257–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stockbridge RB, Robertson JL, Kolmakova-Partensky L, Miller C. A family of fluoride-specific ion channels with dual-topology architecture. Elife. 2013;2:e01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hille B Ion Channels of Excitable Membranes. 3rd ed. Thunderland, USA: Sinauer Associates; 2001. [Google Scholar]
- [28].Wisniewski JR, Rakus D. Multi-enzyme digestion FASP and the ‘Total Protein Approach’-based absolute quantification of the Escherichia coli proteome. J Proteomics. 2014;109:322–31. [DOI] [PubMed] [Google Scholar]
- [29].Papanastasiou M, Orfanoudaki G, Kountourakis N, Koukaki M, Sardis MF, Aivaliotis M, et al. Rapid label-free quantitative analysis of the E. coli BL21(DE3) inner membrane proteome. Proteomics. 2016;16:85–97. [DOI] [PubMed] [Google Scholar]
- [30].Borisov VB, Murali R, Verkhovskaya ML, Bloch DA, Han H, Gennis RB, et al. Aerobic respiratory chain of Escherichia coli is not allowed to work in fully uncoupled mode. Proc Natl Acad Sci U S A. 2011;108:17320–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Szenk M, Dill KA, de Graff AMR. Why Do Fast-Growing Bacteria Enter Overflow Metabolism? Testing the Membrane Real Estate Hypothesis. Cell Syst. 2017;5:95–104. [DOI] [PubMed] [Google Scholar]
- [32].Cuello LG, Jogini V, Cortes DM, Perozo E. Structural mechanism of C-type inactivation in K(+) channels. Nature. 2010;466:203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Humphries J, Xiong L, Liu J, Prindle A, Yuan F, Arjes HA, et al. Species-Independent Attraction to Biofilms through Electrical Signaling. Cell. 2017;168:200–9 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lee DD, Galera-Laporta L, Bialecka-Fornal M, Moon EC, Shen Z, Briggs SP, et al. Magnesium Flux Modulates Ribosomes to Increase Bacterial Survival. Cell. 2019;177:352–60 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bruni GN, Kralj JM. Membrane voltage dysregulation driven by metabolic dysfunction underlies bactericidal activity of aminoglycosides. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bryan LE, Kwan S. Roles of ribosomal binding, membrane potential, and electron transport in bacterial uptake of streptomycin and gentamicin. Antimicrob Agents Chemother. 1983;23:835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stratford JP, Edwards CLA, Ghanshyam MJ, Malyshev D, Delise MA, Hayashi Y, et al. Electrically induced bacterial membrane-potential dynamics correspond to cellular proliferation capacity. Proc Natl Acad Sci U S A. 2019;116:9552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang H, Pan Y, Hu L, Hudson MA, Hofstetter KS, Xu Z, et al. TrkA undergoes a tetramer-to-dimer conversion to open TrkH which enables changes in membrane potential. Nat Commun. 2020;11:547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ochrombel I, Ott L, Krämer R, Burkovski A, Marin K. Impact of improved potassium accumulation on pH homeostasis, membrane potential adjustment and survival of Corynebacterium glutamicum. Biochim Biophys Acta. 2011;1807:444–50. [DOI] [PubMed] [Google Scholar]
- [40].Prindle A, Liu J, Asally M, Ly S, Garcia-Ojalvo J, Suel GM. Ion channels enable electrical communication in bacterial communities. Nature. 2015;527:59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kashket ER, Barker SL. Effects of potassium ions on the electrical and pH gradients across the membrane of Streptococcus lactis cells. J Bacteriol. 1977;130:1017–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Booth IR. Regulation of cytoplasmic pH in bacteria. Microbiol Rev. 1985;49:359–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xue T, You Y, Hong D, Sun H, Sun B. The Staphylococcus aureus KdpDE two-component system couples extracellular K+ sensing and Agr signaling to infection programming. Infect Immun. 2011;79:2154–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].MacGilvary NJ, Kevorkian YL, Tan S. Potassium response and homeostasis in Mycobacterium tuberculosis modulates environmental adaptation and is important for host colonization. PLoS Pathog. 2019;15:e1007591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liu J, Prindle A, Humphries J, Gabalda-Sagarra M, Asally M, Lee DY, et al. Metabolic co-dependence gives rise to collective oscillations within biofilms. Nature. 2015;523:550–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Martinez-Corral R, Liu J, Prindle A, Suel GM, Garcia-Ojalvo J. Metabolic basis of brain-like electrical signalling in bacterial communities. Philos Trans R Soc Lond B Biol Sci. 2019;374:20180382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kralj JM, Hochbaum DR, Douglass AD, Cohen AE. Electrical spiking in Escherichia coli probed with a fluorescent voltage-indicating protein. Science. 2011;333:345–8. [DOI] [PubMed] [Google Scholar]
- [48].Bruni GN, Weekley RA, Dodd BJT, Kralj JM. Voltage-gated calcium flux mediates Escherichia coli mechanosensation. Proc Natl Acad Sci U S A. 2017;114:9445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sirec T, Benarroch JM, Buffard P, Garcia-Ojalvo J, Asally M. Electrical Polarization Enables Integrative Quality Control during Bacterial Differentiation into Spores. iScience. 2019;16:378–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Stingl K, Brandt S, Uhlemann EM, Schmid R, Altendorf K, Zeilinger C, et al. Channel-mediated potassium uptake in Helicobacter pylori is essential for gastric colonization. EMBO J. 2007;26:232–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Follmann M, Becker M, Ochrombel I, Ott V, Krämer R, Marin K. Potassium transport in Corynebacterium glutamicum is facilitated by the putative channel protein CglK, which is essential for pH homeostasis and growth at acidic pH. J Bacteriol. 2009;191:2944–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ungar D, Barth A, Haase W, Kaunzinger A, Lewitzki E, Ruiz T, et al. Analysis of a putative voltage-gated prokaryotic potassium channel. Eur J Biochem. 2001;268:5386–96. [DOI] [PubMed] [Google Scholar]
- [53].Schrempf H, Schmidt O, Kummerlen R, Hinnah S, Muller D, Betzler M, et al. A prokaryotic potassium ion channel with two predicted transmembrane segments from Streptomyces lividans. EMBO J. 1995;14:5170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hirano M, Onishi Y, Yanagida T, Ide T. Role of the KcsA channel cytoplasmic domain in pH-dependent gating. Biophys J. 2011;101:2157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Xu Y, McDermott AE. Inactivation in the potassium channel KcsA. J Struct Biol X. 2019;3:100009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Baker KA, Tzitzilonis C, Kwiatkowski W, Choe S, Riek R. Conformational dynamics of the KcsA potassium channel governs gating properties. Nat Struct Mol Biol. 2007;14:1089–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. [DOI] [PubMed] [Google Scholar]
- [58].Morais-Cabral JH, Zhou Y, MacKinnon R. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 2001;414:37–42. [DOI] [PubMed] [Google Scholar]
- [59].Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414:43–8. [DOI] [PubMed] [Google Scholar]
- [60].Öster C, Hendriks K, Kopec W, Chevelkov V, Shi C, Michl D, et al. The conduction pathway of potassium channels is water free under physiological conditions. Sci Adv. 2019;5:eaaw6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Köpfer DA, Song C, Gruene T, Sheldrick GM, Zachariae U, de Groot BL. Ion permeation in K(+) channels occurs by direct Coulomb knock-on. Science. 2014;346:352–5. [DOI] [PubMed] [Google Scholar]
- [62].Kopec W, Köpfer DA, Vickery ON, Bondarenko AS, Jansen TLC, de Groot BL, et al. Direct knock-on of desolvated ions governs strict ion selectivity in K(+) channels. Nat Chem. 2018;10:813–20. [DOI] [PubMed] [Google Scholar]
- [63].Cuello LG, Cortes DM, Perozo E. The gating cycle of a K(+) channel at atomic resolution. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cordero-Morales JF, Cuello LG, Zhao Y, Jogini V, Cortes DM, Roux B, et al. Molecular determinants of gating at the potassium-channel selectivity filter. Nat Struct Mol Biol. 2006;13:311–8. [DOI] [PubMed] [Google Scholar]
- [65].Valiyaveetil FI, Leonetti M, Muir TW, Mackinnon R. Ion selectivity in a semisynthetic K+ channel locked in the conductive conformation. Science. 2006;314:1004–7. [DOI] [PubMed] [Google Scholar]
- [66].Schrecker M, Wunnicke D, Hanelt I. How RCK domains regulate gating of K+ channels. Biol Chem. 2019;400:1303–22. [DOI] [PubMed] [Google Scholar]
- [67].Albright RA, Ibar JL, Kim CU, Gruner SM, Morais-Cabral JH. The RCK domain of the KtrAB K+ transporter: multiple conformations of an octameric ring. Cell. 2006;126:1147–59. [DOI] [PubMed] [Google Scholar]
- [68].Jiang Y, Pico A, Cadene M, Chait BT, MacKinnon R. Structure of the RCK domain from the E. coli K+ channel and demonstration of its presence in the human BK channel. Neuron. 2001;29:593–601. [DOI] [PubMed] [Google Scholar]
- [69].Rao ST, Rossmann MG. Comparison of super-secondary structures in proteins. J Mol Biol. 1973;76:241–56. [DOI] [PubMed] [Google Scholar]
- [70].Roosild TP, Miller S, Booth IR, Choe S. A mechanism of regulating transmembrane potassium flux through a ligand-mediated conformational switch. Cell. 2002;109:781–91. [DOI] [PubMed] [Google Scholar]
- [71].Kong C, Zeng W, Ye S, Chen L, Sauer DB, Lam Y, et al. Distinct gating mechanisms revealed by the structures of a multi-ligand gated K(+) channel. Elife. 2012;1:e00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kröning N, Willenborg M, Tholema N, Hänelt I, Schmid R, Bakker EP. ATP binding to the KTN/RCK subunit KtrA from the K+ -uptake system KtrAB of Vibrio alginolyticus: its role in the formation of the KtrAB complex and its requirement in vivo. J Biol Chem. 2007;282:14018–27. [DOI] [PubMed] [Google Scholar]
- [73].Tamsett TJ, Picchione KE, Bhattacharjee A. NAD+ activates KNa channels in dorsal root ganglion neurons. J Neurosci. 2009;29:5127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Corrigan RM, Campeotto I, Jeganathan T, Roelofs KG, Lee VT, Grundling A. Systematic identification of conserved bacterial c-di-AMP receptor proteins. Proc Natl Acad Sci U S A. 2013;110:9084–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kim H, Youn SJ, Kim SO, Ko J, Lee JO, Choi BS. Structural Studies of Potassium Transport Protein KtrA Regulator of Conductance of K+ (RCK) C Domain in Complex with Cyclic Diadenosine Monophosphate (c-di-AMP). J Biol Chem. 2015;290:16393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hite RK, Yuan P, Li Z, Hsuing Y, Walz T, MacKinnon R. Cryo-electron microscopy structure of the Slo2.2 Na(+)-activated K(+) channel. Nature. 2015;527:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–22. [DOI] [PubMed] [Google Scholar]
- [78].Kuo MM, Maslennikov I, Molden B, Choe S. The desensitization gating of the MthK K+ channel is governed by its cytoplasmic amino terminus. PLoS Biol. 2008;6:e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Pau VP, Abarca-Heidemann K, Rothberg BS. Allosteric mechanism of Ca2+ activation and H+-inhibited gating of the MthK K+ channel. J Gen Physiol. 2010;135:509–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Li Y, Berke I, Chen L, Jiang Y. Gating and inward rectifying properties of the MthK K+ channel with and without the gating ring. J Gen Physiol. 2007;129:109–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Boiteux C, Posson DJ, Allen TW, Nimigean CM. Selectivity filter ion binding affinity determines inactivation in a potassium channel. Proc Natl Acad Sci USA. 2020;117:29968–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Holtmann G, Bakker EP, Uozumi N, Bremer E. KtrAB and KtrCD: two K+ uptake systems in Bacillus subtilis and their role in adaptation to hypertonicity. J Bacteriol. 2003;185:1289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Nakamura T, Yuda R, Unemoto T, Bakker EP. KtrAB, a new type of bacterial K(+)-uptake system from Vibrio alginolyticus. J Bacteriol. 1998;180:3491–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Matsuda N, Kobayashi H, Katoh H, Ogawa T, Futatsugi L, Nakamura T, et al. Na+-dependent K+ uptake Ktr system from the cyanobacterium Synechocystis sp. PCC 6803 and its role in the early phases of cell adaptation to hyperosmotic shock. J Biol Chem. 2004;279:54952–62. [DOI] [PubMed] [Google Scholar]
- [85].Rhoads DB, Epstein W. Energy coupling to net K+ transport in Escherichia coli K-12. J Biol Chem. 1977;252:1394–401. [PubMed] [Google Scholar]
- [86].Tholema N, Vor der Brüggen M, Mäser P, Nakamura T, Schroeder JI, Kobayashi H, et al. All four putative selectivity filter glycine residues in KtrB are essential for high affinity and selective K+ uptake by the KtrAB system from Vibrio alginolyticus. J Biol Chem. 2005;280:41146–54. [DOI] [PubMed] [Google Scholar]
- [87].Mikusevic V, Schrecker M, Kolesova N, Patino-Ruiz M, Fendler K, Hänelt I. A channel profile report of the unusual K(+) channel KtrB. J Gen Physiol. 2019;151:1357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hänelt I, Tholema N, Kröning N, Vor der Brüggen M, Wunnicke D, Bakker EP. KtrB, a member of the superfamily of K+ transporters. Eur J Cell Biol. 2011. ;90:696–704. [DOI] [PubMed] [Google Scholar]
- [89].Corratge-Faillie C, Jabnoune M, Zimmermann S, Very AA, Fizames C, Sentenac H. Potassium and sodium transport in non-animal cells: the Trk/Ktr/HKT transporter family. Cell Mol Life Sci. 2010;67:2511–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Cao Y, Jin X, Huang H, Derebe MG, Levin EJ, Kabaleeswaran V, et al. Crystal structure of a potassium ion transporter, TrkH. Nature. 2011;471:336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Sr Durell, Hao Y, Nakamura T Bakker EP, Guy HR Evolutionary relationship between K(+) channels and symporters. Biophysical Journal. 1999;77:775–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Tholema N, Bakker EP, Suzuki A, Nakamura T. Change to alanine of one out of four selectivity filter glycines in KtrB causes a two orders of magnitude decrease in the affinities for both K+ and Na+ of the Na+ dependent K+ uptake system KtrAB from Vibrio alginolyticus. FEBS Lett. 1999;450:217–20. [DOI] [PubMed] [Google Scholar]
- [93].Levin EJ, Zhou M. Recent progress on the structure and function of the TrkH/KtrB ion channel. Curr Opin Struct Biol. 2014;27:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Hänelt I, Wunnicke D, Müler-Trimbusch M, Vor der Brüggen M, Kraus I, Bakker EP, et al. Membrane region M2C2 in subunit KtrB of the K+ uptake system KtrAB from Vibrio alginolyticus forms a flexible gate controlling K+ flux: an electron paramagnetic resonance study. J Biol Chem. 2010;285:28210–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Hänelt I, Löchte S, Sundermann L, Elbers K, Vor der Brüggen M, Bakker EP. Gain of function mutations in membrane region M2C2 of KtrB open a gate controlling K+ transport by the KtrAB system from Vibrio alginolyticus. J Biol Chem. 2010;285:10318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Vieira-Pires RS, Szollosi A, Morais-Cabral JH. The structure of the KtrAB potassium transporter. Nature. 2013;496:323–8. [DOI] [PubMed] [Google Scholar]
- [97].Zhou M, MacKinnon R. A mutant KcsA K(+) channel with altered conduction properties and selectivity filter ion distribution. J Mol Biol. 2004;338:839–46. [DOI] [PubMed] [Google Scholar]
- [98].Derebe MG, Sauer DB, Zeng W, Alam A, Shi N, Jiang Y. Tuning the ion selectivity of tetrameric cation channels by changing the number of ion binding sites. Proc Natl Acad Sci U S A. 2011;108:598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Szollosi A, Vieira-Pires RS, Teixeira-Duarte CM, Rocha R, Morais-Cabral JH. Dissecting the Molecular Mechanism of Nucleotide-Dependent Activation of the KtrAB K+ Transporter. PLoS Biol. 2016;14:e1002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Diskowski M, Mehdipour AR, Wunnicke D, Mills DJ, Mikusevic V, Bärland N, et al. Helical jackknives control the gates of the double-pore K(+) uptake system KtrAB. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Teixeira-Duarte CM, Fonseca F, Morais-Cabra JH Activation of a nucleotide-dependent RCK domain requires binding of a cation cofactor to a conserved site. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Rocha R, Teixeira-Duarte CM, Jorge JMP, Morais-Cabral JH. Characterization of the molecular properties of KtrC, a second RCK domain that regulates a Ktr channel in Bacillus subtilis. J Struct Biol. 2019;205:34–43. [DOI] [PubMed] [Google Scholar]
- [103].Stulke J, Kruger L. Cyclic di-AMP Signaling in Bacteria. Annu Rev Microbiol. 2020;74:159–79. [DOI] [PubMed] [Google Scholar]
- [104].Chakrapani S, Perozo E. How to gate an ion channel: lessons from MthK. Nat Struct Mol Biol. 2007;14:180–2. [DOI] [PubMed] [Google Scholar]
- [105].Pedersen BP, Stokes DL, Apell HJ. The KdpFABC complex – K(+) transport against all odds. Mol Membr Biol. 2019;35:21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Durell SR, Bakker EP, Guy HR. Does the KdpA subunit from the high affinity K(+)- translocating P-type KDP-ATPase have a structure similar to that of K(+) channels? Biophysical Journal. 2000;78:188–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Schrader M, Fendler K, Bamberg E, Gassel M, Epstein W, Altendorf K, et al. Replacement of glycine 232 by aspartic acid in the KdpA subunit broadens the ion specificity of the K(+)-translocating KdpFABC complex. Biophys J. 2000;79:802–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].van der Laan M, Gassel M, Altendorf K. Characterization of amino acid substitutions in KdpA, the K+-binding and -translocating subunit of the KdpFABC complex of Escherichia coli. J Bacteriol. 2002;184:5491–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Buurman ET, Kim KT, Epstein W. Genetic evidence for two sequentially occupied K+ binding sites in the Kdp transport ATPase. J Biol Chem. 1995;270:6678–85. [DOI] [PubMed] [Google Scholar]
- [110].Becker D, Fendler K, Altendorf K, Greie JC. The conserved dipole in transmembrane helix 5 of KdpB in the Escherichia coli KdpFABC P-type ATPase is crucial for coupling and the electrogenic K+-translocation step. Biochemistry. 2007;46:13920–8. [DOI] [PubMed] [Google Scholar]
- [111].Albers RW. Biochemical aspects of active transport. Annu Rev Biochem. 1967;36:727–56. [DOI] [PubMed] [Google Scholar]
- [112].Post RL, Hegyvary C, Kume S. Activation by adenosine triphosphate in the phosphorylation kinetics of sodium and potassium ion transport adenosine triphosphatase. J Biol Chem. 1972;247:6530–40. [PubMed] [Google Scholar]
- [113].Post RL, Kume S. Evidence for an aspartyl phosphate residue at the active site of sodium and potassium ion transport adenosine triphosphatase. J Biol Chem. 1973;248:6993–7000. [PubMed] [Google Scholar]
- [114].Puppe W, Siebers A, Altendorf K. The phosphorylation site of the Kdp-ATPase of Escherichia coli: site-directed mutagenesis of the aspartic acid residues 300 and 307 of the KdpB subunit. Mol Microbiol. 1992;6:3511–20. [DOI] [PubMed] [Google Scholar]
- [115].Bramkamp M, Altendorf K. Single amino acid substitution in the putative transmembrane helix V in KdpB of the KdpFABC complex of Escherichia coli uncouples ATPase activity and ion transport. Biochemistry. 2005;44:8260–6. [DOI] [PubMed] [Google Scholar]
- [116].Gassel M, Möllenkamp T, Puppe W, Altendorf K. The KdpF subunit is part of the K(+)-translocating Kdp complex of Escherichia coli and is responsible for stabilization of the complex in vitro. J Biol Chem. 1999;274:37901–7. [DOI] [PubMed] [Google Scholar]
- [117].Bramkamp M, Altendorf K, Greie JC. Common patterns and unique features of P-type ATPases: a comparative view on the KdpFABC complex from Escherichia coli (Review). Mol Membr Biol. 2007;24:375–86. [DOI] [PubMed] [Google Scholar]
- [118].Gassel M, Altendorf K. Analysis of KdpC of the K(+)-transporting KdpFABC complex of Escherichia coli. Eur J Biochem. 2001;268:1772–81. [PubMed] [Google Scholar]
- [119].Shull GE, Lane LK, Lingrel JB. Amino-acid sequence of the beta-subunit of the (Na+ + K+)ATPase deduced from a cDNA. Nature. 1986;321:429–31. [DOI] [PubMed] [Google Scholar]
- [120].Axelsen KB, Palmgren MG. Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol. 1998;46:84–101. [DOI] [PubMed] [Google Scholar]
- [121].Biology Kühlbrandt W., structure and mechanism of P-type ATPases. Nat Rev Mol Cell Biol. 2004;5:282–95. [DOI] [PubMed] [Google Scholar]
- [122].Greie JC, Altendorf K. The K+-translocating KdpFABC complex from Escherichia coli: a P-type ATPase with unique features. J Bioenerg Biomembr. 2007;39:397–402. [DOI] [PubMed] [Google Scholar]
- [123].Damnjanovic B, Weber A, Potschies M, Greie JC, Apell HJ. Mechanistic analysis of the pump cycle of the KdpFABC P-type ATPase. Biochemistry. 2013;52:5563–76. [DOI] [PubMed] [Google Scholar]
- [124].Schramke H, Tostevin F, Heermann R, Gerland U, Jung K. A Dual-Sensing Receptor Confers Robust Cellular Homeostasis. Cell Rep. 2016;16:213–21. [DOI] [PubMed] [Google Scholar]
- [125].Laermann V, Cudic E, Kipschull K, Zimmann P, Altendorf K. The sensor kinase KdpD of Escherichia coli senses external K+. Mol Microbiol. 2013;88:1194–204. [DOI] [PubMed] [Google Scholar]
- [126].Sweet ME, Zhang X, Erdjument-Bromage H, Dubey V, Khandelia H, Neubert TA, et al. Serine phosphorylation regulates the P-type potassium pump KdpFABC. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Trchounian A, Kobayashi H. Kup is the major K+ uptake system in Escherichia coli upon hyper-osmotic stress at a low pH. FEBS Lett. 1999;447:144–8. [DOI] [PubMed] [Google Scholar]
- [128].Schleyer M, Bakker EP. Nucleotide sequence and 3’-end deletion studies indicate that the K(+)-uptake protein Kup from Escherichia coli is composed of a hydrophobic core linked to a large and partially essential hydrophilic C terminus. J Bacteriol. 1993;175:6925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Vastermark A, Wollwage S, Houle ME, Rio R, Saier MH, Jr. Expansion of the APC superfamily of secondary carriers. Proteins. 2014;82:2797–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Zakharyan E, Trchounian A. K+ influx by Kup in Escherichia coli is accompanied by a decrease in H+ efflux. FEMS Microbiol Lett. 2001;204:61–4. [DOI] [PubMed] [Google Scholar]
- [131].Izard T The crystal structures of phosphopantetheine adenylyltransferase with bound substrates reveal the enzyme’s catalytic mechanism. J Mol Biol. 2002;315:487–95. [DOI] [PubMed] [Google Scholar]
- [132].Gundlach J, Krüger L, Herzberg C, Turdiev A, Poehlein A, Tascon I, et al. Sustained sensing in potassium homeostasis: Cyclic di-AMP controls potassium uptake by KimA at the levels of expression and activity. J Biol Chem. 2019. [DOI] [PMC free article] [PubMed]
- [133].Quintana IM, Gibhardt J, Turdiev A, Hammer E, Commichau FM, Lee VT, et al. The KupA and KupB proteins of Lactococcus lactis IL1403 are novel c-di-AMP receptor proteins responsible for potassium uptake. J Bacteriol. 2019. [DOI] [PMC free article] [PubMed]
- [134].Pham HT, Turner MS. Onward and [K(+)]Upward: a New Potassium Importer under the Spell of Cyclic di-AMP. J Bacteriol. 2019;201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Epstein W, Kim BS. Potassium transport loci in Escherichia coli K-12. J Bacteriol. 1971;108:639–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Sukharev SI, Blount P, Martinac B, Blattner FR, Kung C. A large-conductance mechanosensitive channel in E. coli encoded by mscL alone. Nature. 1994;368:265–8. [DOI] [PubMed] [Google Scholar]
- [137].Bakker EP, Booth IR, Dinnbier U, Epstein W, Gajewska A. Evidence for multiple K+ export systems in Escherichia coli. J Bacteriol. 1987;169:3743–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Roosild TP, Castronovo S, Healy J, Miller S, Pliotas C, Rasmussen T, et al. Mechanism of ligand-gated potassium efflux in bacterial pathogens. Proc Natl Acad Sci U S A. 2010;107:19784–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Gundlach J, Herzberg C, Kaever V, Gunka K, Hoffmann T, Weiss M, et al. Control of potassium homeostasis is an essential function of the second messenger cyclic di-AMP in Bacillus subtilis. Sci Signal. 2017;10. [DOI] [PubMed] [Google Scholar]
- [140].Fujisawa M, Ito M, Krulwich TA. Three two-component transporters with channel-like properties have monovalent cation/proton antiport activity. Proc Natl Acad Sci U S A. 2007;104:13289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Krüger L, Herzberg C, Warneke R, Poehlein A, Stautz J, Weiss M, et al. Two ways to convert a low- to a high-affinity potassium channel: Control of Bacillus subtilis KtrCD by glutamate. J Bacteriol. 2020. [DOI] [PMC free article] [PubMed]
- [142].Liu J, Martinez-Corral R, Prindle A, Lee DD, Larkin J, Gabalda-Sagarra M, et al. Coupling between distant biofilms and emergence of nutrient time-sharing. Science. 2017;356:638–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Su J, Gong H, Lai J, Main A, Lu S. The potassium transporter Trk and external potassium modulate Salmonella enterica protein secretion and virulence. Infect Immun. 2009;77:667–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Liu Y, Ho KK, Su J, Gong H, Chang AC, Lu S. Potassium transport of Salmonella is important for type III secretion and pathogenesis. Microbiology. 2013;159:1705–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Price-Whelan A, Poon CK, Benson MA, Eidem TT, Roux CM, Boyd JM, et al. Transcriptional profiling of Staphylococcus aureus during growth in 2 M NaCl leads to clarification of physiological roles for Kdp and Ktr K+ uptake systems. MBio. 2013;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Gries CM, Bose JL, Nuxoll AS, Fey PD, Bayles KW. The Ktr potassium transport system in Staphylococcus aureus and its role in cell physiology, antimicrobial resistance and pathogenesis. Mol Microbiol. 2013;89:760–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Freeman ZN, Dorus S, Waterfield NR. The KdpD/KdpE two-component system: integrating K(+) homeostasis and virulence. PLoS Pathog. 2013;9:e1003201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Samir R, Hussein SH, Elhosseiny NM, Khattab MS, Shawky AE, Attia AS. Adaptation to Potassium-Limitation Is Essential for Acinetobacter baumannii Pneumonia Pathogenesis. J Infect Dis. 2016;214:2006–13. [DOI] [PubMed] [Google Scholar]
- [149].Yost S, Duran-Pinedo AE, Krishnan K, Frias-Lopez J. Potassium is a key signal in host-microbiome dysbiosis in periodontitis. PLoS Pathog. 2017;13:e1006457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Cortes DM, Cuello LG, Perozo E. Molecular architecture of full-length KcsA: role of cytoplasmic domains in ion permeation and activation gating. J Gen Physiol. 2001;117:165–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Ye S, Li Y, Chen L, Jiang Y. Crystal structures of a ligand-free MthK gating ring: insights into the ligand gating mechanism of K+ channels. Cell. 2006;126:1161–73. [DOI] [PubMed] [Google Scholar]
- [152].Gundlach J, Herzberg C, Hertel D, Thurmer A, Daniel R, Link H, et al. Adaptation of Bacillus subtilis to Life at Extreme Potassium Limitation. MBio. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Gibhardt J, Hoffmann G, Turdiev A, Wang M, Lee VT, Commichau FM. c-di-AMP assists osmoadaptation by regulating the Listeria monocytogenes potassium transporters KimA and KtrCD. J Biol Chem. 2019;294:16020–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Watson PY, Fedor MJ. The ydaO motif is an ATP-sensing riboswitch in Bacillus subtilis. Nat Chem Biol. 2012;8:963–5. [DOI] [PubMed] [Google Scholar]
- [155].Sato Y, Nanatani K, Hamamoto S, Shimizu M, Takahashi M, Tabuchi-Kobayashi M, et al. Defining membrane spanning domains and crucial membrane-localized acidic amino acid residues for K(+) transport of a Kup/HAK/KT-type Escherichia coli potassium transporter. J Biochem. 2014;155:315–23. [DOI] [PubMed] [Google Scholar]
- [156].Chin KH, Liang JM, Yang JG, Shih MS, Tu ZL, Wang YC, et al. Structural Insights into the Distinct Binding Mode of Cyclic Di-AMP with SaCpaA_RCK. Biochemistry. 2015;54:4936–51. [DOI] [PubMed] [Google Scholar]
- [157].Chandrangsu P, Dusi R, Hamilton CJ, Helmann JD. Methylglyoxal resistance in Bacillus subtilis: contributions of bacillithiol-dependent and independent pathways. Mol Microbiol. 2014;91:706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Fujisawa M, Wada Y, Ito M. Modulation of the K+ efflux activity of Bacillus subtilis YhaU by YhaT and the C-terminal region of YhaS. FEMS Microbiol Lett. 2004;231:211–7. [DOI] [PubMed] [Google Scholar]