

Abstract
The role of polymers has played a crucial role in developing templates that can promote regeneration as tissue-engineered matrices. The present study aims to develop functional matrices involving the protein mucin. The mucin used in this study is characterised using MALDI-TOF TOF and CD spectroscopy prior to conjugation. Thereupon, a hybrid scaffold comprising of a polysaccharide, chitosan, chemically conjugated to a protein, mucin, and encapsulated with montmorillonite is developed. Grafting of hydroxyethyl methacrylate (HEMA) is done to overcome the issue of mechanical weakness that mucin hydrogels usually undergo. It was observed that the presence of montmorillonite led to the stability of the hydrogels. The conjugations with varied ratios of the polysaccharide and protein were characterized using spectroscopic techniques. The prepared gels showed appreciable material properties in terms of water uptake and porosity. Hydrogels with different ratios of the polysaccharide and protein were evaluated for their biocompatibility. The biological evaluation of the hydrogels was performed with MC3T3E1 and C2C12 cell lines indicating their potential for wider tissue engineering applications.
A simple route to conjugate glycan to proteins with enhanced stabilization offered by montmorillonite leads to hydrogels with superior properties suitable for tissue engineering applications.
1. Introduction
The potential of hydrogel-based scaffolds in the field of tissue engineering is quite eminent. A few characteristics such as uniformly porous 3-D structure, surface properties, mechanical properties, non-toxicity and biocompatibility of scaffolds allow the materials to perform as suitable matrices. Generally, these factors can be modulated and are intended to facilitate the adhesion, growth and proliferation of cells. In the present work, we have developed a scaffold, which is a hybrid of a well-studied polysaccharide, chitosan, and a protein named mucin. While chitosan has established itself as a polymer in various tissue engineering applications ranging from bone to liver, mucin is a less explored protein in terms of developing as a biomaterial. Mucins are a family of huge glycoprotein polymers expressed as cell membrane-tethered molecules and as a prime component of the mucus gel when secreted by the goblet cells of the epithelium. Mucins have an important position within the offensive and defensive mechanisms of many species. The multiple and diverse physiological roles of mucins have been studied by a few without much detailed investigation on various tissues. The primary properties attributed to mucins consist of barrier, dynamicity, hydration, lubrication, and bioactivity. The aim of our research is to investigate and develop ways to present mucin glycoproteins as a useful building block that may be used to create new multifunctional biomaterials.1 In our recent publication related to mucin, we have observed its interaction with collagen in a simulated environment by in silico approaches, designed it chemically as a scaffold and thus investigate it as a suitable matrix specifically for bone tissue engineering.2 In an attempt to widen its usability, in other tissue engineering applications, we have designed a hydrogel combining it with a polysaccharide. The chosen polysaccharide has very well established its potential to be used as an extracellular matrix and also due to its ability to interact with mucin.3 Chitosan is a natural linear polysaccharide with repeating units of d-glucosamine and N-acetyl-d-glucosamine linked via β(1→4) glycosidic bond.4 Due to its antimicrobial nature, biocompatibility, high gelation rate and formation of porous scaffolds, chitosan has proven to be an excellent material for various tissue engineering applications.5 Chitosan has been used as a composite with proteins such as gelatin as a hydrogel for cartilage tissue engineering6 and as scaffolds by complexing with chitosan-beta glycerophosphate-hydroxyethyl cellulose.7 The limitation that was encountered in these systems was mostly the mechanical property of the materials. To overcome such limitations various synthetic polymers have come into use. Hydroxyethyl methacrylate (HEMA) is a water-soluble8,9 monomer and is widely accepted as a material used in the field of drug delivery.10–14 HEMA has been used as a scaffold in skin tissue engineering15–20 and as a hybrid hydrogel of xylan, HEMA and mesoporous silica has been studied as a scaffold for the growth and attachment of fibroblasts.21 In the present study, HEMA was used in the scaffold system to overcome the limitation of mechanical instability. The last component in our system is montmorillonite (MMT). Montmorillonite is a major component of bentonite belonging to the family of nanoclays, which is already approved by the FDA as an additive in various medicinal products.22 MMT is a layered silicate [(Na, Ca)0.33(Al, Mg)2Si4O10(OH)2·nH2O],23 belonging to the smectite group of minerals, with high specific surface area (up to 600 m2 g−1) and aspect ratio. The repeating structural unit of MMT consists of one alumina octahedral sheet sandwiched in between two silicon tetrahedral layers.23 MTT has many properties such as the biocompatibility, availability, and feasibility, due to which this particular mineral has gained significant attention in recent years. Broad research has been done to investigate the purpose of drug and gene delivery with natural or modified MMT.22,24,25 Many reports suggests that, fabrication of scaffolds by introduction of MMT in natural biomaterials, such as gelatin26,27 collagen,28 silk,29 and chitosan,30–32 improves interaction of cell-scaffold, cell proliferation, and increases cell differentiation.23 In our developed system poly-HEMA is grafted on the backbone of chitosan to form a hydrogel that is encapsulated with MMT for further assessment.
2. Materials and methodology
2.1. Materials
Mucin type-1S of the submaxillary gland from bovine (cat. no. M3895), chitosan medium molecular weight (cat no. 448877) and ammonium persulfate (APS) (cat no. A3678) was purchased from Sigma-Aldrich. 2-Hydroxyethyl methacrylate (HEMA) (cat no. 477028), nanoclay or montmorillonite, hydrophilic bentonite (cat no. 682659) purchased from sigma. The terms montmorillonite and nanoclay are interchangeably used in the manuscript. 1-(3-Dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (EDC) (cat. no. 49235), N-hydroxysuccinimide (NHS) (cat. no. 47913) and TEMED (cat no. 52145) were purchased from SRL.
2.2. Characterization of commercially available mucin type 1S
2.2.1. SDS PAGE analysis
Mucin type-1S purity was evaluated with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 8% polyacrylamide gel taking mucin in 1× PBS at the concentration of 50 mg mL−1. Then we diluted the mucin protein for SDS-PAGE gel run at the concentration of 30 µg and loaded 30 µL of mucin along with another protein, as experimental control, collagen, 30 µg of concentration with high molecular weight protein ladder. Molecular mass markers were purchased from (Thermo fisher scientific, # cat no. LC 5688) and the gel was stained with Coomassie blue R-250 (Himedia, cat no. MB153) using the manufacture's protocol.
2.2.2. MALDI-TOF TOF
The procedure involved here is cutting protein bands or spots from 2D PAGE gels, destaining the gel pieces, reducing and alkylating the protein, digesting with trypsin, analysing using MALDI-TOF TOF mass spectrometry (Sciex 5800) to determine the mass of the tryptic peptides and database searching with the PMF data to identify the protein.33
2.2.3. Circular dichroism (CD)
CD spectra were obtained using a Jasco 1500 spectrometer. Mucin protein was prepared 2 mg mL−1 in 1× PBS with pH 7.4 and 1 mL of protein was used for overnight dialysis with 3 times media change. The final concentration that was used for CD spectra analysis after dialysis was 0.1 mg mL−1. Samples (400 µL) were loaded into optically matched quartz cuvettes (0.1 cm path length). All ellipticity measurements were performed using the same cuvette for baselines and samples from 190–260 nm wavelength. Ellipticity measurements were corrected for buffer baseline, and when possible, ellipticity was converted to mean residue ellipticity (MRE) and molecular weights were used from the NCBI database.34 Measurement of wavelength regions 200–280 nm was done.
2.3. Fabrication of the composite hydrogel
Synthesis of the hydrogel has been done in multiple steps starting from carbodiimide reaction, followed by redox polymerization and finally lyophilization. First, for the formation of an amide bond between mucin and chitosan, EDC and N-hydroxy succinimide (NHS) has been used. The molar ratio of both mucin and chitosan has been varied to understand the influence of the polysaccharide and the protein respectively in the final product. In a typical experiment, both chitosan and mucin were dissolved in 1% (v/v) acetic acid solution and phosphate buffer solution respectively. 0.25% (w/v) of mucin was treated with EDC, followed by NHS to form a stable ester. Next, a specific amount of 0.25% (w/v) chitosan was added to the system and incubated for half an hour. 500 µL of 12% (w/v) nanoclay and 300 µL of Milli Q water was added in all combinations of scaffold except in one (N scaffold), where 400 µL of Milli Q water was added instead of 300 µL. Now, 10 µL of 1 M APS followed by 1.5 µL TEMED was added to the system to generate free radical sites on the chitosan backbone. Immediately, 100 µL hydroxyethyl methacrylate (HEMA) was added, and after mixing well, the whole system was further incubated for the next 3 hours at 37 °C. The whole synthesis was done in a 24-well plate. After the incubation period, the purification of the material has been done by washing the scaffolds with Milli Q water three times each with the incubation period of 30 minutes and then washing it in acetone for 5 minutes. Next, the material was frozen at −80 °C for the next 24 hours followed by lyophilization in a freeze-dryer for another 24 hours. The nomenclature of the scaffolds has been done to the ratio of chitosan (C), mucin (M) and the presence of nanoclay (NC). Along with (mucin–chitosan)-g-poly HEMA, three more gels have been synthesized as controls. First, a scaffold without mucin (NC), the second one without chitosan (NM) and the last one without both mucin and chitosan (N), keeping other chemicals unaltered. Subsequently in all these three cases, the carbodiimide reaction has not been done as it is irrelevant to all of them. The synthesis details have been tabulated in Table 1.
Composition details of mucin and chitosan in the composite hydrogelsa.
| Sample name | Amount of mucin (µL) | Amount of chitosan (µL) |
|---|---|---|
| C1M1N | 50 | 50 |
| C1M2N | 66 | 33 |
| C2M1N | 33 | 66 |
| NC | 0 | 100 |
| NM | 100 | 0 |
| N | 0 | 0 |
Nanoclay-500 µL, Milli-Q water-300 µL, HEMA-100 µL, APS-10 µL, TEMED-1.5 µL are constant for all combinations, C1M1N, C1M2N and C2M1N represents the different combinations of chitosan and mucin, NC represents chitosan with nanoclay, NM represents mucin with nanoclay and N represents only nanoclay.
2.4. Gel fraction ratio and water content
Prepared scaffolds were kept in a culture plate with 30 mL Milli-Q water in a shaking incubator at 37 °C for 12 h. Initial weight was weighed before soaking in water (M0). After 12 h the scaffolds were taken out, dried with tissue paper followed by 30 min inside the incubator and weighed again (final weight M).6
The gel fraction ratio% was calculated using the following equation: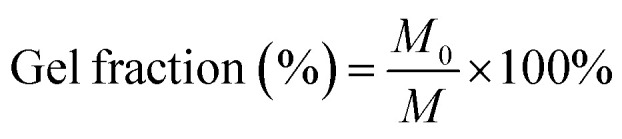
Freeze-dried scaffolds (approximately 50 mg) were prepared according to the procedure reported in the previous paragraph and used without any further purifications. The dry scaffolds were weighed and then immersed in distilled water (pH 7.4) at room temperature. The samples were weighed at regular time intervals after removal of excess surface liquid by blotting with a soft tissue. The water uptake of the scaffolds was established by calculating their percentage swelling degree (% SD) as: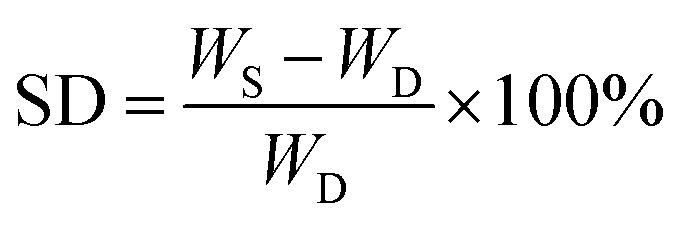 where WS represents the weight of the swollen sample at each time interval and WD represents the weight of the dry sample at the beginning of the experiment. The experiments were performed in triplicate and the SD% was reported as the mean value.
where WS represents the weight of the swollen sample at each time interval and WD represents the weight of the dry sample at the beginning of the experiment. The experiments were performed in triplicate and the SD% was reported as the mean value.
2.5. Surface morphology
Scanning electron microscopy (SEM) analysis was performed on an EVO 18 instrument. The samples were coated with a thin gold/palladium layer by using a plasma magnetron sputter-coater.
2.6. Spectroscopic analysis (FTIR and NMR)
FTIR is a non-destructive technique using a minimum amount of sample.35 The absorption spectra by the FTIR technique were obtained with the spectrum-GX (PerkinElmer) equipment. The wavelength ranged from 400 to 4000 cm−1 at room temperature. The samples were brought into intimate contact with the KBr by applying a loading pressure in reflectance mode. All the data were processed with Spectrum TM 10 (PerkinElmer) software and baseline correction was done.
In NMR spectra analysis, the 400 MHz IH-NMR experiments were performed on a Bruker AM-400 MHz device. Deuterated DMSO was used as a solvent to dissolve C2M1N scaffold and mucin protein at 10 mg mL−1 concentration.36
2.7. Physical and rheological properties
2.7.1. Mechanical measurements by rheology
Frequency sweep studies were conducted using a modular compact rheometer (MCR 702 Multi-Drive, Anton Paar). The sample specimens were analysed in parallel plate geometry with 8 mm diameter plates of aluminium. Before testing, the sample spilling out of the measuring plates was trimmed thoroughly and the force was normalized to 0 N. The frequency was varied between 500 rad s−1 to 0.1 rad s−1 at a constant shear strain of 0.1%. All the measurements were made at room temperature and 5 data points were recorded per each frequency decade. From the collected raw data, the software Rheoplus computes the corresponding storage modulus (G′), loss modulus (G″), loss factor and complex viscosity.
2.7.2. Thermogravimetric analysis/differential scanning calorimetry (TGA/DSC)
In order to characterize the thermal behaviour of the chitosan, mucin, nanoclay and a composite scaffold, C2M1N, TGA/DSC was performed by using the STA 449 F3 Jupiter® – NETZSCH equipped with a refrigerated cooling system (RCS).37,38 The system was calibrated by Al2O3 standard. Approximately 5 mg of sample was weighed in aluminium pans and sealed. The calibration pan was the hermetically sealed type, under argon atmosphere with flow rate 80 mL min−1 and heating (temperature rise) by 10 °C per min from room temperature to 600 °C both for TGA/DSC. The results were expressed as the mean of two independent measures.
2.8. Biological assay
2.8.1. Pre-osteoblast (MC3T3E1) and muscle cell (C2C12) culture
Biological assays to evaluate the cell biocompatibility response of 3D protein scaffolds were performed using MC3T3-E1 SUBCLONAL 4 cells (mouse calvaria preosteoclast, ATCC, CRL-2593) and C2C12 (ATCC® CRL-1772™ of Mus musculus, mouse muscle cells), cultured in alpha-minimal essential medium (α-MEM, HIMEDIA cat. no.-AL221A) and Dulbecco's Modified Eagle's Medium (DMEM, Gibco product code: 11574486) respectively and supplemented with 10% Fetal Bovine Serum (FBS, Gibco Invitrogen cat. no.-16000044), antibiotic solution (0.1% Pen.–Strep. Gibco cat. no. 15140-122). For all experimental procedures, MC3T3-E1 SUBCLONAL 4 and C2C12 cells at passages 10–15 were used and incubated at 37 °C with 5% CO2.
2.8.2. Cell seeding and characterization of cell-scaffolds constructs
The hydrogels were sterilized using UV C (100–280 nm) exposure for 30 min on both upper and lower surfaces. Before seeding, the scaffolds were immersed in serum-free α-MEM placed in 24-well plates. After 24 h, the pre-osteoblast and muscle cells were seeded at a density of 1 × 106 cells per µL per scaffold and were allowed to adhere for 4 h. Alpha MEM and DMEM respectively complete medium (160 µL) was added to each well and the seeded scaffolds were further incubated overnight. After 24 h, the cell-scaffold constructs were placed in a 12 well-plate. For pre-osteoblast the cell scaffold construct was treated with osteogenic culture medium with 100 µg mL−1l-ascorbic acid (Sigma cat. no. A5960), 10 mM beta-glycerophosphate (Sigma cat. no. G9422) and 10 nm dexamethasone (Sigma cat. no. D4902) was added and the cell-scaffold constructs were cultured for 7 days at 37 °C with 5% CO2. Cell adhesion, proliferation, colonization and differentiation were evaluated after 7 days of post cell seeding.
2.8.3. Cell viability and proliferation assay
Cytotoxicity of nanoclay based hydrogel on MC3T3-E1 and C2C12 cell lines was studied using PrestoBlue® assay. For the study of cytotoxicity of nanoclay hydrogels, MC3T3-E1 and C2C12 cells (1 × 106 cells per µL per scaffold) were cultured in 12-well plates at 37 °C and 5% CO2 for 7 days. Nanoclay scaffolds without mucin-chitosan were used as controls and the cell viability was expressed. PrestoBlue® is ready to use the cell-permeable resazurin-based solution that functions as a cell viability indicator by using the reducing power of living cells to quantitatively measure the proliferation of cells. When added to cells, the PrestoBlue® reagent is modified by the reducing environment of the viable cell and turns red, becoming highly fluorescent. This colour change can be detected using fluorescence or absorbance measurements. 100 µL reagent of Prestoblue (A 13261 – THERMO) was added in 900 µL culture media. This reagent is added to cell per scaffold constructs and incubated for 2 h at 37 °C. The incubated medium is transferred to 96 well plates and the fluorescence intensity is measured with a multiplate reader at 535 nm (excitation)/615 nm (emission). Five replicates were analyzed from each sample at 7 days post-seeding.
2.8.4. Fluorescence microscopy
The in vitro qualitative analysis of cell viability was evaluated using live/dead assay by fluorescence microscopy. After being propagated for 7 days of post cell seeding on the hybrid hydrogels, cell-hydrogel constructs were washed with 1× PBS and a solution containing 500 µL 1× PBS supplemented with 2 µL (1 mg mL−1) calcein AM and 2 µL (1 mg mL−1) propidium iodide (live and dead staining kit – cat. no.-04511-1KT-F – Sigma) was added. Cultures were incubated for 10 min at room temperature, and washed twice with PBS. The cells/hydrogel constructs were analysed with inverted fluorescence microscopy (Invitrogen™ EVOS™ FL Digital Inverted Fluorescence Microscope AMF4300).
2.9. Biodegradability study of scaffolds
The degradation of chitosan scaffolds (n = 3) was performed in two media: phosphate buffer saline solution (PBS, pH = 7.4) water containing 800 mg L−1 of lysozyme39 and in glutathione solution (10 mM in PBS)40 at 37 °C in an orbital shaker at 50 rpm. Enzymatic degradation was monitored for four weeks, while the lysozyme solution was refreshed every third day. At a predetermined time (14 days) samples were removed from the medium, rinsed with distilled water and dried in the oven at 50 °C until constant mass. The samples were weighed before (m1) and after in vitro degradation (m2). The degradation degree (Δm) was determined as the weight loss with respect to the initial weight of the sample: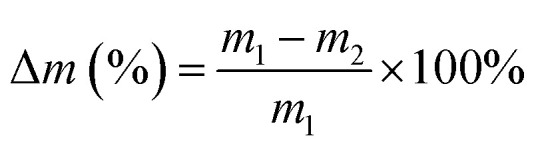
2.10. Statistical analysis
To determine the statistical significance of observed differences between the study groups, values are reported as the mean and standard deviation (SD), using one-way analysis of variance (ANOVA), and significance was defined at p < 0.05 (*), p < 0.01 (**).
3. Results and discussion
3.1. Characterization of commercial mucin
Prior to developing a composite scaffold involving mucin and chitosan, characterization of the commercially available mucin (Sigma-Aldrich) was done. The protein was detected with Coomassie blue staining Fig. 1(A). The bands for the mucin protein, behaved anomalously, migrating as three broad bands with an apparent molecular weight of 240 kDa, 62 kDa and 55 kDa in the SDS-PAGE (lane 3), as shown in Fig. 1(A). The SDS-PAGE gel of mucin with three different molecular sizes of bands was cut and processed for MALDI analysis to confirm the presence of mucin protein. Fig. 1B(i)–(iii) depicts the Mascot search results and Mascot score histogram along with the table as shown in Fig. 1B(iv) validates the best protein accession along with best protein score and best protein description. The nature and extent of the secondary structure in mucin were determined by using CD spectroscopy.41 The CD spectrum of mucin is characterized by a large negative minimum in between 200–210 nm (shown in Fig. 1(C)). Analysis of the spectrum predicts that mucin contains 21.8% helical structure with 35.8% antiparallel structure. These results indicate that a considerable portion of the mucin molecule is lacking a traditional secondary structure and that the confirmation of the native molecule is consistent with an extended non-globular structure.42
Fig. 1. Characterization of mucin protein. (A) SDS page gel run image showing mucin 3 bands marked within red box loaded in lane 3 along with ladder and collagen as experimental control in lanes 1 and 2 respectively. (B) MALDI TOF TOF analysis of SDS page gel image after gel digestion, Mascot score histogram of three different molecular weight (i) 55 kDa, (ii) 62 kDa, (iii) 240 kDa, (iv) table showing all the different molecular mass 2D gel analysis matched with Uniport database. (C) Circular dichroism spectrum of mucin showing the far-UV at around 200 nm wavelength, maximum with random coil secondary structure.

The above experiments gave us a background on mucin as a protein that is obtained commercially before initiating scaffold design and preparation. The SDS-PAGE confirms the presence of bands when the mucin is loaded in SDS-PAGE at 240 kDa, which was further proved by MALDI TOF TOF experiments where the Mascot score histograms (Fig. 1B(i)–(iii)) and Mascot search result matched the mucin-like protein with best protein score of 84 (submaxillary mucin-like protein) (Fig. 1B(iv)). CD spectroscopy measurements gave an idea about the structure of mucin. Mucin shows a far-UV CD spectrum (Fig. 1(C)) that corresponds to a random coil structure as there are no detectable features of α-helices or β-sheets, the majority of the secondary structures.43 As the vast majority of the polypeptide backbone is heavily glycosylated, the results confirm the current understanding of the overall structure of mucin.44–46 However, whether refolding occurred or new random coil structures were formed was not possible to determine. The data presented here may indicate that the β-sheet is important for the functions of these globular regions of mucins, such as in the formation of intramolecular and intermolecular interactions that are paramount for mucin cross-linking and polymerization and thus the integrity of saliva and mucus gels.47 Furthermore, as previously reported, no tertiary structural features were detected for mucin.48 The spectra obtained from mucin was at around 200 nm wavelength as reported in many pieces of literature.43,48,49
3.2. Synthesis of (chitosan-co-mucin)-g-poly HEMA hydrogel scaffold
The whole reaction mechanism of the synthesis process can be separated into two distinct parts, the first is the coupling reaction assisted by carbodiimide between mucin and chitosan; the second is the free radical copolymerization reaction between chitosan and hydroxyethyl methacrylate (HEMA) monomers. EDC-NHS assisted reaction was done to form an amide bond between the carboxylic groups of mucin and the amine groups of chitosan. The first step of the mechanism goes through the deprotonation of carboxylic H-atom and the formation of a bond between the C-atom of the carbodiimide group and the O-atom of the carboxylic group. The pH of the chitosan solution is maintained at around 6 to enable the availability of the primary amines. With the addition of chitosan in the system, the primary amine group of chitosan further attacks the carbonyl center and substitutes the NHS group to form a C–N bond between the mucin and chitosan molecules. For simplicity, we only consider the carboxylic groups of mucin and amine groups of chitosan in place of their whole structure to represent the schematic of the mechanism shown in Fig. 2(A). The two hydroxyl groups in each unit of chitosan can act as a potential site for a free radical generation. Ammonium persulfate is an excellent material to initiate the formation of free radicals by cleaving the peroxy bond in it at elevated temperatures. After the initiation step when these free radicals get in contact with molecules of HEMA, immediately homolytic fission of double bond took place and one after another HEMA molecules attach to propagate the chain polymerization reaction. There are two possible ways to terminate the reaction, simply by the addition of hydrogen radical or by the addition of two similar long-chain polymers that leads to the formation of graft copolymer as the final product.50 Although there are several other possibilities for the grafting process to take place, as in the initiation step after the formation of sulphate radical anion, it also can take up H-atom from the amine group of chitosan or even can degrade the long chitosan chain into small ones by cleaving the C–O–C linkage.51
Fig. 2. (A) The detailed coupling mechanism of mucin and chitosan forming an amide bond and subsequently the grafting of the poly(HEMA) onto the chitosan backbone. (B) Pictorial representation of the final co-polymer (C) Representing diagram of the hydrogel formed with mucin, poly(HEMA) grafted chitosan stabilized by the presence of MMT.

The diagram of the final synthesized products has been shown in Fig. 2(B).
The formation of the hydrogel is hypothesized to be primarily initiated and done by MMT. MMT closely resembles LAPONITE®, which is a layered synthetic nanoclay with a chemical formula of Si8Mg5.45Li0.4O24Na0.7 and is already established as a promising rheology-modifier, or used as a mechanical reinforcing component and crosslinker with several hydrogel systems.52 In the same line of action, the gel-forming ability of MMT is hypothesized to be a multi-step mechanism. When particles react with hydroxide ions in the water, phosphate ions dissolve. After the ion dissolution, the MMT particles start to interact with each other while the sodium ions diffuse towards the surfaces within the galleries, resulting in an expanded gel-like behavior (schematic represented in Fig. 2(C)).53
3.3. Gel fraction and water retention ability
The crosslinked hydrogels were incubated in double-distilled water at 37 °C to determine the gel fraction of the crosslinked hydrogels (as shown in Fig. 3(A)). The gel fraction fundamentally determines the percentage of the polymer chains that are covalently linked into the network and do not leach out from the hydrogels by diffusion. The gel fraction results indicate crosslinking during which most of the crosslinkable moieties were consumed resulting in gel fractions of 70%. All the combinations resulted in similar gel fraction ratios.
Fig. 3. (A) Graphical representation of gel fraction. (B) Water retention ability expressed as a % of ratio when the hydrogels have attained swelling equilibrium.

Water absorption is an important property in biodegradable hydrogels. To determine the percentage of water absorption, swelling studies were performed by immersion of hydrogels in phosphate buffer solution at temperature of 37 °C. The dry weight of the scaffold was determined before immersion. Hydrogels were placed in distilled water and after a pre-determined time, i.e. 24 h, the scaffolds were taken out and surface adsorbed water was removed by filter paper and their wet weight was recorded. The swelling ratio is defined as the fractional increase in the weight of the hydrogel due to water absorption. The equilibrium swelling was reached at about 10 h of swelling, which was around 120% in the medium for all the samples (Fig. 3(B)). Amongst, the different combinations, the sample with higher mucin (C1M2N) exhibited higher swelling than the other chitosan:mucin combinations, however, the hydrogels containing only nanoclay absorbed and retained the maximum amount of water. The MMT hydrogels showed maximum swelling, thus confirming the hypothesis suggested above regarding the mechanism of MMT hydrogel formation.
3.4. Morphological characterization of the scaffolds
SEM images of the nanoclay hydrogel obtained (Fig. 4) showed a wide range of porosity in the structure. C1M2N showed some pores but surprisingly the mucin-nanoclay scaffolds did not have any pores. In general, the hydrogels did not demonstrate big pores but still exhibited reasonable water uptake capacity due to the presence of MMT.
Fig. 4. Scanning electron microscopy (SEM) images of top to bottom (clockwise) (i) C1M1N, (ii) C1M2N, (iii) C2M1N, (iv) NC, (v) NM and (vi) N. The arrows indicate the pores.

3.5. Spectroscopic analysis
In the FTIR spectrum of chitosan we can see a broad peak from 3408–3470 cm−1 which is due to the overlapping peaks of primary, secondary O–H stretching and the symmetrical stretching of N–H bonds (Fig. 5(A)). The sharp peaks at 2896 cm−1 and 2857 cm−1 can be attributed to C–H stretching. The stretching of the secondary amide group present in chitosan is seen in another sharp peak at 1640 cm−1. The deformation of the O–H bond and stretching of the C–O bond of alcohols are seen in 1418 cm−1 and 1076 cm−1 respectively. A sharp peak at 1153 cm−1 corresponds to the C–O–C stretching frequency.10 In the case of mucin, the whole spectrum is made of numerous overlapping peaks of N–H and O–H bonds present in all the amino acid units. This is why there is almost a flat region present in between 3554 cm−1 and 2955 cm−1. The synthesized hydrogel has a broad region from 3526 cm−1 to 3290 cm−1, which is proof of the coupling reaction and mucin to be present as a part of the material, as this region can be assigned to overlapping of multiple numbers of O–H and N–H bond stretching (Fig. 5(A)). There is one additional sharp peak at 1714 cm−1 which arises from the stretching of C O bonds of ester present in the synthesized material which comes from the addition of HEMA in the grafting process.54,55
Fig. 5. (A) FTIR image of chitosan, mucin and the composite scaffold C2M1N; (B) NMR spectra of C2M1N showing the peak assignment, the inset indicates the zoomed area from 2.00–4.00 ppm; (C) comparison of the NMR spectra of mucin and C2M1N.

The 400 MHz 1H NMR spectroscopy of mucin did not reveal much information. It is believed that sensitive NMR techniques would be required for getting relevant structural information regarding the protein. However, from the spectra of the C2M1N scaffold, the presence of chitosan and HEMA could be ascertained (Fig. 5 (B) and (C)).
3.6. Thermal and rheological properties
The thermogravimetric analysis of the combination scaffold C2M1N showed four temperature intervals of weight loss. The first weight loss corresponds to the loss occurring due to moisture, the second weight loss occurred at around 200 °C, the third at 310 °C and the fourth one occurred at around 430 °C (Fig. 6(A)). In the case of chitosan, a more documented pattern was observed with the first weight loss, corresponding to the evaporation of physically adsorbed and strongly hydrogen-bonded water to chitosan.56,57 The second weight losses, occurring due to depolymerisation/decomposition of polymer chains through deacetylation and cleavage of glycosidic linkages.58 The last stage, for the thermal destruction of the pyranose ring and the decomposition of the residual carbon. Mucin also showed a peak corresponding to loss of moisture and another degradation peak at 250 °C. The unavailability of much information regarding the protein limits its discussion. MMT being a crystalline material had a degradation at 190 °C and the highest residual mass (Fig. 6(A)).
Fig. 6. Analysis of chitosan, mucin, nanoclay and composite hydrogel. (A) Thermogravimetric analysis; (B) differential scanning calorimetry; (C) rheological analysis of the hydrogels; comparison of G′ and G″ with respect to angular frequency; (D) comparison of the tan δ with respect to the angular frequency.

Fig. 6(B) shows the DSC thermograms of the samples, pure chitosan exhibited an endothermic peak at 75.4 °C associated with the evaporation of absorbed water and an exothermic peak at about 300.5 °C which is attributed to the polymer degradation, including saccharide ring dehydration, depolymerisation and decomposition of deacetylated and acetylated chitosan unit. This is in line with previously reported results.59 The other samples show more complicated thermograms caused by the overlapping. Mucin also shows an endothermic peak that is correlated with the loss of water associated with hydrophilic groups of the protein while a clear exothermic peak was not evident although TGA revealed a degradation peak at 250 °C. The composite hydrogel thermograms revealed a similar pattern showing a broad endothermic peak at 100 °C which can be ascribed to the coalescence of isolated endothermic dehydration peaks resulting from the individual contribution of both polymers. The formation of the new chemical bonds is expected due to the interaction and complexation between mucin and chitosan.
The hydrogels have a higher G′ than G″, irrespective of their combination or native composition, indicating that they are more gel-like (Fig. 6(C)). The G′ increased in all the hydrogels as a function of angular frequency. As a comparison between the different samples, chitosan hydrogels displayed the maximum G′ and the MMT scaffolds without any other component showed the least G′. The combined effects of the samples on the storage and loss moduli are shown in Fig. 6(D), by comparing tan δ = G″/G′. The loss factor reflects the difference in strength between the loss and storage moduli. The loss factor value suggested that the elastic behavior (solid-like) was more prevalent than the viscous (liquid-like) nature of the material. However, no correlation could be observed amongst the various samples in terms of possessing more or less solid-like behavior.
3.7. Biodegradation of scaffold
When designing hydrogels for various tissue engineering applications, it is crucial to consider their degradation properties, as it will affect the release of hydrogel load (cells or therapeutic compounds) and the replacement of the hydrogel with tissue.40 To this end, degradation properties of the hydrogels were evaluated in vitro under conditions that mimic the in vivo. Lysozyme was used as the enzyme for degradation and the average overall rate of degradation by lysozyme was around 45% in 14 days. Fig. 7 shows the degradation profile of the different combinations of scaffolds after 14 days incubation in PBS containing lysozyme and glutathione. A specific molecule of interest for hydrogel degradation in vivo is glutathione, an intracellular and circulating reducing agent.60 Hence, the degradation of these hydrogels are tested in the presence of glutathione along with lysozyme. The hydrogels were found to slowly degrade over a period of 14 days when treated with glutathione solution along with lysozyme. The graph shows notable difference in the degradation rate of the composite scaffolds compared to the control scaffolds, NC and NM and blank scaffold N. The C2M1N scaffold showed slower degradation and C1M2N showed maximum degradation amongst composites. C1M2N had a more porous structure compared to the other samples, which could be the reason behind an enhanced dissolution followed by degradation. NC also has a higher degradation with chitosan being the major component, thus being prone to lysozyme degradation.
Fig. 7. Degradation profiles of the hydrogels in the presence of enzymes lysozyme and glutathione activity.

3.8. Biological evaluation of the scaffold
3.8.1. Cell viability and proliferation on the scaffolds
Implantable biomaterials should be primarily non-toxic to the cells.61 The three key requirements in tissue engineering include signals, cells that can respond to morphogens and scaffolds that are biomimetic thus mimicking the extracellular matrix.62–64 The biocompatibility of the developed hydrogels was validated with two cell lines preosteoblast, MC3T3 E1 and muscle cell line C2C12 to indicate the applicability of the developed scaffolds for wider tissue engineering applications. The rationale of chosing the cell lines also in accordance with well supported evidence from literature.65–68
The viability of cells as determined by LIVE/DEAD® cytotoxicity assay is shown in Fig. 8(A) and 9(A). On day 7, the live cells layer (green fluorescence) can be observed on all composite hydrogels, which reveals that the chitosan–mucin matrix and nanoclay combination has no negative effect on the cytocompatibility. All the hydrogels promoted and indicated proliferation of cells.
Fig. 8. (A) Fluorescence images of MC3T3-E1 cells on nanoclay based hydrogel after being cultured for 7 days determined by live/dead assay. (B) The proliferation of MC3T3-E1 after being cultured for 7 days on nanoclay hydrogels. *(p < 0.05) and **(p < 0.01) indicate statistical significance.

Fig. 9. (A) Fluorescence images of C2C12 cells on nanoclay based hydrogel after being cultured for 7 days determined by live/dead assay. (B) The proliferation of C2C12 cells after cultured for 7 days on nanoclay hydrogels. *(p < 0.05) and **(p < 0.01) indicate statistical significance.

The cell proliferation results via PrestoBlue assay are shown in Fig. 8(B) and 9(B) for MC3T3-E1 and C2C12 cells respectively. There is no statistically significant difference between samples on the 7th day of cell seeding. From the live and dead assay, it could be validated that the amount of cell proliferation was in appreciable amount in all the hydrogels, which was also further confirmed by PrestoBlue assay. In Fig. 8 and 9, it was observed that C2M1N has the highest cell viability as compared to other ratios of chitosan : mucin hydrogel. However, upon comparing all the composite hydrogels, the combination of chitosan and mucin along with nanoclay showed good result as compared to the control groups (NC, NM and blank N). The result indicates that the nanoclay with the combination of chitosan and mucin content can exhibit better cell adhesion and proliferation, which should be preferentially chosen for future in vivo investigation and application.
4. Conclusion
In the performed study mucin is explored for its suitability in combining with a polysaccharide chitosan for wider tissue engineering applications. Different combination of the protein and the polysaccharide are developed, analyzed and compared with their native counterparts. An important observation made during the study is the stabilization provided by MMT which leads to the formation of stable hydrogels. Based on the assays conducted, it is concluded that the composite scaffold, C2M1N seems to have better performance with good gel fraction%, porosity and rheological performance. The developed hydrogels show biocompatibility in two cell line MC3T3E1 and C2C12, thus showing promise for different tissue engineering applications.
Conflicts of interest
The authors declare no conflicts of interest.
Supplementary Material
Acknowledgments
Mamoni Dash acknowledges the Ramalingaswami Fellowship 2016–17 (D.O.NO.BT/HRD/35/02/2006), Department of Biotechnology, Government of India, Institute of Life Sciences (ILS), Debyashreeta Barik acknowledges INSPIRE, Department of Science and Technology Government of India, Institute of Life Sciences, Priyadarshi Rai is acknowledged for his help during SEM, Pradipta Mukherjee from Anton Paar is highly acknowledged for his assistance for Rheological experiments, Dr Amaresh Panda is acknowledged for providing C2C12 cell line. We highly acknowledge Ms Swagatika Mohanty and CSIR-Institute of minerals and materials technology, Bhubaneswar is acknowledged for conducting FTIR, TGA-DSC experiments. Dr Nivedita Jena and Ravenshaw University are acknowledged for their help in conducting NMR experiments. R. Vishwa is acknowledged for his support in the SDS-PAGE and CD spectra experiments. Dr Amol Ratnakar Suryawanshi, Central Proteomics Facility, Institute of Life Sciences, Bhubaneswar is acknowledged for conducting Maldi experiments. Dr Dileep Vasudevan and his group are acknowledged for helping with conducting CD spectra analysis.
References
- Bansil R. Turner B. S. Mucin structure, aggregation, physiological functions and biomedical applications. Curr. Opin. Colloid Interface Sci. 2006;11(2):164–170. doi: 10.1016/j.cocis.2005.11.001. [DOI] [Google Scholar]
- Barik D. Bejugam P. R. Nayak C. Mohanty K. T. Singha A. Declercq H. A. Dash M. Polymer-Protein Hybrid Network Involving Mucin: A Mineralized Biomimetic Template for Bone Tissue Engineering. Macromol. Biosci. 2021;6:e2000381. doi: 10.1002/mabi.202000381. [DOI] [PubMed] [Google Scholar]
- Collado-González M. González Espinosa Y. Goycoolea F. M. Interaction Between Chitosan and Mucin: Fundamentals and Applications. Biomimetics. 2019;(2):32. doi: 10.3390/biomimetics4020032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M. Zeng X. Ma C. Yi H. Ali Z. Mou X. Li S. Deng Y. He N. Injectable hydrogels for cartilage and bone tissue engineering. Bone Res. 2017;5(1):17014. doi: 10.1038/boneres.2017.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash M. Samal S. K. Douglas T. E. L. Schaubroeck D. Leeuwenburgh S. C. Van Der Voort P. Declercq H. A. Dubruel P. Enzymatically biomineralized chitosan scaffolds for tissue-engineering applications. J. Tissue Eng. Regener. Med. 2017;(5):1500–1513. doi: 10.1002/term.2048. [DOI] [PubMed] [Google Scholar]
- Van Nieuwenhove I. Salamon A. Peters K. Graulus G.-J. Martins J. C. Frankel D. Kersemans K. De Vos F. Van Vlierberghe S. Dubruel P. Gelatin- and starch-based hydrogels. Part A: hydrogel development, characterization and coating. Carbohydr. Polym. 2016;152:129–139. doi: 10.1016/j.carbpol.2016.06.098. [DOI] [PubMed] [Google Scholar]
- Naderi-Meshkin H. Andreas K. Matin M. M. Sittinger M. Bidkhori H. R. Ahmadiankia N. Bahrami A. R. Ringe J. Chitosan-based injectable hydrogel as a promising in situ forming scaffold for cartilage tissue engineering. Cell Biol. Int. 2014;(1):72–84. doi: 10.1002/cbin.10181. [DOI] [PubMed] [Google Scholar]
- Abasi S. Davis R. Podstawczyk D. A. Guiseppi-Elie A. Distribution of water states within Poly(HEMA-co-HPMA)-based hydrogels. Polymer. 2019;185:121978. doi: 10.1016/j.polymer.2019.121978. [DOI] [PubMed] [Google Scholar]
- Spicer C. D. Hydrogel scaffolds for tissue engineering: the importance of polymer choice. Polym. Chem. 2020;11(2):184–219. doi: 10.1039/C9PY01021A. [DOI] [Google Scholar]
- Rani G. U. Dey K. P. Bharti S. Mishra S. Controlled drug release of 5-amino salicylic acid by poly(2-hydroxyethylmethacrylate) grafted agar. Front. Chem. Sci. Eng. 2014;8(4):465–470. doi: 10.1007/s11705-014-1452-8. [DOI] [Google Scholar]
- Roointan A. Farzanfar J. Mohammadi-Samani S. Behzad-Behbahani A. Farjadian F. Smart pH responsive drug delivery system based on poly(HEMA-co-DMAEMA) nanohydrogel. Int. J. Pharm. 2018;552(1):301–311. doi: 10.1016/j.ijpharm.2018.10.001. [DOI] [PubMed] [Google Scholar]
- Chatterjee S. Upadhyay P. Mishra M. Dhara S. M. Akshara M. R. Pal K. N. Zaidi Z. S. Iqbal S. F. Misra S. K. Advances in chemistry and composition of soft materials for drug releasing contact lenses. RSC Adv. 2020;10(60):36751–36777. doi: 10.1039/D0RA06681H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D. Das R. Ghosh P. Dhara S. Panda A. B. Pal S. Dextrin cross linked with poly(HEMA): a novel hydrogel for colon specific delivery of ornidazole. RSC Adv. 2013;3(47):25340–25350. doi: 10.1039/C3RA44716B. [DOI] [Google Scholar]
- Durán-Lobato M. Carrillo-Conde B. Khairandish Y. Peppas N. A. Surface-Modified P(HEMA-co-MAA) Nanogel Carriers for Oral Vaccine Delivery: Design, Characterization, and In Vitro Targeting Evaluation. Biomacromolecules. 2014;15(7):2725–2734. doi: 10.1021/bm500588x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury P. Kumar S. Singh A. Kumar A. Kaur N. Sanyasi S. Chawla S. Goswami C. Goswami L. Hydroxyethyl methacrylate grafted carboxy methyl tamarind (CMT-g-HEMA) polysaccharide based matrix as a suitable scaffold for skin tissue engineering. Carbohydr. Polym. 2018;189:87–98. doi: 10.1016/j.carbpol.2018.01.079. [DOI] [PubMed] [Google Scholar]
- Radhakumary C. Nandkumar A. M. Nair P. D. Hyaluronic acid-g-poly(HEMA) copolymer with potential implications for lung tissue engineering. Carbohydr. Polym. 2011;85(2):439–445. doi: 10.1016/j.carbpol.2011.03.007. [DOI] [Google Scholar]
- Arica T. A. Guzelgulgen M. Yildiz A. A. Demir M. M. Electrospun GelMA fibers and p(HEMA) matrix composite for corneal tissue engineering. Mater. Sci. Eng., C. 2021;120:111720. doi: 10.1016/j.msec.2020.111720. [DOI] [PubMed] [Google Scholar]
- Potorac S. Popa M. Verestiuc L. Le Cerf D. New semi-IPN scaffolds based on HEMA and collagen modified with itaconic anhydride. Mater. Lett. 2012;67(1):95–98. doi: 10.1016/j.matlet.2011.09.039. [DOI] [Google Scholar]
- Gao Q. Hu B. Ning Q. Ye C. Xie J. Ye J. Gao C. A primary study of poly(propylene fumarate)–2-hydroxyethyl methacrylate copolymer scaffolds for tarsal plate repair and reconstruction in rabbit eyelids. J. Mater. Chem. B. 2015;3(19):4052–4062. doi: 10.1039/C5TB00285K. [DOI] [PubMed] [Google Scholar]
- Dragusin D.-M. Van Vlierberghe S. Dubruel P. Dierick M. Van Hoorebeke L. Declercq H. A. Cornelissen M. M. Stancu I.-C. Novel gelatin–PHEMA porous scaffolds for tissue engineering applications. Soft Matter. 2012;8(37):9589–9602. doi: 10.1039/C2SM25536G. [DOI] [Google Scholar]
- García-Uriostegui L. Delgado E. Meléndez-Ortiz H. I. Camacho-Villegas T. A. Esquivel-Solís H. Gatenholm P. Toriz G. Spruce xylan/HEMA-SBA15 hybrid hydrogels as a potential scaffold for fibroblast growth and attachment. Carbohydr. Polym. 2018;201:490–499. doi: 10.1016/j.carbpol.2018.08.066. [DOI] [PubMed] [Google Scholar]
- Jayrajsinh S. Shankar G. Agrawal Y. K. Bakre L. Montmorillonite nanoclay as a multifaceted drug-delivery carrier: a review. J. Drug Delivery Sci. Technol. 2017;39:200–209. doi: 10.1016/j.jddst.2017.03.023. [DOI] [Google Scholar]
- Cui Z. K. Kim S. Baljon J. J. Wu B. M. Aghaloo T. Lee M. Microporous methacrylated glycol chitosan-montmorillonite nanocomposite hydrogel for bone tissue engineering. Nat. Commun. 2019;10(1):3523. doi: 10.1038/s41467-019-11511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koç Demir A. Elçin A. E. Elçin Y. M. Strontium-modified chitosan/montmorillonite composites as bone tissue engineering scaffold. Mater. Sci. Eng., C. 2018;89:8–14. doi: 10.1016/j.msec.2018.03.021. [DOI] [PubMed] [Google Scholar]
- Thakur G. Singh A. Singh I. Chitosan-Montmorillonite Polymer Composites: Formulation and Evaluation of Sustained Release Tablets of Aceclofenac. Sci. Pharm. 2015;84(4):603–617. doi: 10.3390/scipharm84040603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haroun A. A. Gamal-Eldeen A. Harding D. R. Preparation, characterization and in vitro biological study of biomimetic three-dimensional gelatin-montmorillonite/cellulose scaffold for tissue engineering. J. Mater. Sci.: Mater. Med. 2009;20(12):2527–2540. doi: 10.1007/s10856-009-3818-x. [DOI] [PubMed] [Google Scholar]
- Kevadiya B. D. Rajkumar S. Bajaj H. C. Chettiar S. S. Gosai K. Brahmbhatt H. Bhatt A. S. Barvaliya Y. K. Dave G. S. Kothari R. K. Biodegradable gelatin-ciprofloxacin-montmorillonite composite hydrogels for controlled drug release and wound dressing application. Colloids Surf., B. 2014;122:175–183. doi: 10.1016/j.colsurfb.2014.06.051. [DOI] [PubMed] [Google Scholar]
- Nistor M. T. Vasile C. Chiriac A. P. Hybrid collagen-based hydrogels with embedded montmorillonite nanoparticles. Mater. Sci. Eng., C. 2015;53:212–221. doi: 10.1016/j.msec.2015.04.018. [DOI] [PubMed] [Google Scholar]
- Mieszawska A. J. Llamas J. G. Vaiana C. A. Kadakia M. P. Naik R. R. Kaplan D. L. Clay enriched silk biomaterials for bone formation. Acta Biomater. 2011;7(8):3036–3041. doi: 10.1016/j.actbio.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z.-K. Kim S. Baljon J. J. Wu B. M. Aghaloo T. Lee M. Microporous methacrylated glycol chitosan-montmorillonite nanocomposite hydrogel for bone tissue engineering. Nat. Commun. 2019;10(1):3523. doi: 10.1038/s41467-019-11511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu S.-h. Wang M.-C. Lin J.-J. Biocompatibility and antimicrobial evaluation of montmorillonite/chitosan nanocomposites. Appl. Clay Sci. 2012;56:53–62. doi: 10.1016/j.clay.2011.09.016. [DOI] [Google Scholar]
- Katti K. S. Katti D. R. Dash R. Synthesis and characterization of a novel chitosan/montmorillonite/hydroxyapatite nanocomposite for bone tissue engineering. Biomed. Mater. 2008;3(3):034122. doi: 10.1088/1748-6041/3/3/034122. [DOI] [PubMed] [Google Scholar]
- Webster J. Oxley D. Protein identification by MALDI-TOF mass spectrometry. Methods Mol. Biol. 2012;800:227–240. doi: 10.1007/978-1-61779-349-3_15. [DOI] [PubMed] [Google Scholar]
- Drzewiecki K. E. Grisham D. R. Parmar A. S. Nanda V. Shreiber D. I. Circular Dichroism Spectroscopy of Collagen Fibrillogenesis: A New Use for an Old Technique. Biophys. J. 2016;111(11):2377–2386. doi: 10.1016/j.bpj.2016.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanelli S. Zimmermann A. Totóli E. G. Salgado H. R. N. FTIR Spectrophotometry as a Green Tool for Quantitative Analysis of Drugs: Practical Application to Amoxicillin. J. Chem. 2018;2018:3920810. [Google Scholar]
- Naganagowda G. A. Gururaja T. L. Satyanarayana J. Levine M. J. NMR analysis of human salivary mucin (MUC7) derived O-linked model glycopeptides: comparison of structural features and carbohydrate-peptide interactions. J. Pept. Res. 1999;54(4):290–310. doi: 10.1034/j.1399-3011.1999.00102.x. [DOI] [PubMed] [Google Scholar]
- Momoh M. A. Adikwu M. U. Ibezim C. E. Ofokansi K. C. Attama A. A. Thermal characterisation of PEGylated mucin. Asian Pac. J. Trop. Med. 2010;3(6):458–460. doi: 10.1016/S1995-7645(10)60110-1. [DOI] [Google Scholar]
- Tamburic S. Craig D. Q. M. Thermorheological and thermogravimetric analysis of bioadhesive polymer/mucin mixtures. Thermochim. Acta. 1997;294(1):99–106. doi: 10.1016/S0040-6031(96)03149-8. [DOI] [Google Scholar]
- Andrea L. Marica I. Anamarija R. Lysozyme-Induced Degradation of Chitosan: The Characterisation of Degraded Chitosan Scaffolds. J. Tissue Repair Regen. 2017;1(1):12–22. [Google Scholar]
- Pupkaite J. Ahumada M. McLaughlin S. Temkit M. Alaziz S. Seymour R. Ruel M. Kochevar I. Griffith M. Suuronen E. J. Alarcon E. I. Collagen-Based Photoactive Agent for Tissue Bonding. ACS Appl. Mater. Interfaces. 2017;9(11):9265–9270. doi: 10.1021/acsami.7b01984. [DOI] [PubMed] [Google Scholar]
- Kelly S. M. Jess T. J. Price N. C. How to study proteins by circular dichroism. Biochim. Biophys. Acta. 2005;1751(2):119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Eckhardt A. E. Timpte C. S. Abernethy J. L. Toumadje A. Johnson, Jr W. C. Hill R. L. Structural properties of porcine submaxillary gland apomucin. J. Biol. Chem. 1987;262(23):11339–11344. doi: 10.1016/S0021-9258(18)60964-0. [DOI] [PubMed] [Google Scholar]
- Madsen J. B. Svensson B. Abou Hachem M. Lee S. Proteolytic Degradation of Bovine Submaxillary Mucin (BSM) and Its Impact on Adsorption and Lubrication at a Hydrophobic Surface. Langmuir. 2015;31(30):8303–8309. doi: 10.1021/acs.langmuir.5b01281. [DOI] [PubMed] [Google Scholar]
- Strous G. J. Dekker J. Mucin-type glycoproteins. Crit. Rev. Biochem. Mol. Biol. 1992;27(1–2):57–92. doi: 10.3109/10409239209082559. [DOI] [PubMed] [Google Scholar]
- Bansil R. Stanley E. Lamont J. T. Mucin Biophysics. Annu. Rev. Physiol. 1995;57(1):635–657. doi: 10.1146/annurev.ph.57.030195.003223. [DOI] [PubMed] [Google Scholar]
- Lee S. Müller M. Rezwan K. Spencer N. D. Porcine Gastric Mucin (PGM) at the Water/Poly(Dimethylsiloxane) (PDMS) Interface: Influence of pH and Ionic Strength on Its Conformation, Adsorption, and Aqueous Lubrication Properties. Langmuir. 2005;21(18):8344–8353. doi: 10.1021/la050779w. [DOI] [PubMed] [Google Scholar]
- Davies H. S. Singh P. Deckert-Gaudig T. Deckert V. Rousseau K. Ridley C. E. Dowd S. E. Doig A. J. Pudney P. D. A. Thornton D. J. Blanch E. W. Secondary Structure and Glycosylation of Mucus Glycoproteins by Raman Spectroscopies. Anal. Chem. 2016;88(23):11609–11615. doi: 10.1021/acs.analchem.6b03095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen J. B. Pakkanen K. I. Duelund L. Svensson B. Hachem M. A. Lee S. A simplified chromatographic approach to purify commercially available bovine submaxillary mucins (BSM) Prep. Biochem. Biotechnol. 2015;45(1):84–99. doi: 10.1080/10826068.2014.887583. [DOI] [PubMed] [Google Scholar]
- Madsen J. B. Pakkanen K. I. Lee S. Thermostability of bovine submaxillary mucin (BSM) in bulk solution and at a sliding interface. J. Colloid Interface Sci. 2014;424:113–119. doi: 10.1016/j.jcis.2014.03.006. [DOI] [PubMed] [Google Scholar]
- Sutirman Z. A. Sanagi M. M. Naim A. A. Karim K. J. A. Ibrahim W. J. S. M. Ammonium persulfate-initiated graft copolymerization of methacrylamide onto chitosan: synthesis, characterization and optimization. Sains Malays. 2017;46:2433–2440. doi: 10.17576/jsm-2017-4612-19. [DOI] [Google Scholar]
- Mochalova A. E. Smirnova L. A. State of the Art in the Targeted Modification of Chitosan. Polym. Sci., Ser. B. 2018;60(2):131–161. doi: 10.1134/S1560090418020045. [DOI] [Google Scholar]
- Dávila J. L. d'Ávila M. A. Rheological evaluation of Laponite/alginate inks for 3D extrusion-based printing. Int. J. Adv. Manuf. Technol. 2019;101(1):675–686. doi: 10.1007/s00170-018-2876-y. [DOI] [Google Scholar]
- Jatav S. Joshi Y. M. Chemical stability of Laponite in aqueous media. Appl. Clay Sci. 2014;97–98:72–77. doi: 10.1016/j.clay.2014.06.004. [DOI] [Google Scholar]
- Subramanian K. G. Vijayakumar V. Synthesis and evaluation of chitosan-graft-poly (2-hydroxyethyl methacrylate-co-itaconic acid) as a drug carrier for controlled release of tramadol hydrochloride. Saudi Pharm. J. 2012;20(3):263–271. doi: 10.1016/j.jsps.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H. Li L. Leong W. C. Gan L. H. Thermo-Responsive Association of Chitosan-graft-Poly(N-isopropylacrylamide) in Aqueous Solutions. J. Phys. Chem. B. 2010;114(32):10666–10673. doi: 10.1021/jp105041z. [DOI] [PubMed] [Google Scholar]
- Franca E. F. Lins R. D. Freitas L. C. G. Straatsma T. P. Characterization of Chitin and Chitosan Molecular Structure in Aqueous Solution. J. Chem. Theory Comput. 2008;4(12):2141–2149. doi: 10.1021/ct8002964. [DOI] [PubMed] [Google Scholar]
- Corazzari I. Nisticò R. Turci F. Faga M. G. Franzoso F. Tabasso S. Magnacca G. Advanced physico-chemical characterization of chitosan by means of TGA coupled on-line with FTIR and GCMS: thermal degradation and water adsorption capacity. Polym. Degrad. Stab. 2015;112:1–9. doi: 10.1016/j.polymdegradstab.2014.12.006. [DOI] [Google Scholar]
- Moussout H. Ahlafi H. Aazza M. Bourakhouadar M. Kinetics and mechanism of the thermal degradation of biopolymers chitin and chitosan using thermogravimetric analysis. Polym. Degrad. Stab. 2016;130:1–9. doi: 10.1016/j.polymdegradstab.2016.05.016. [DOI] [Google Scholar]
- Dudek G. Turczyn R. New type of alginate/chitosan microparticle membranes for highly efficient pervaporative dehydration of ethanol. RSC Adv. 2018;8(69):39567–39578. doi: 10.1039/C8RA07868H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni K. S. Desale S. S. Bronich T. K. Nanogels: an overview of properties, biomedical applications and obstacles to clinical translation. J. Controlled Release. 2016;240:109–126. doi: 10.1016/j.jconrel.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przekora A. The summary of the most important cell-biomaterial interactions that need to be considered during in vitro biocompatibility testing of bone scaffolds for tissue engineering applications. Mater. Sci. Eng., C. 2019;97:1036–1051. doi: 10.1016/j.msec.2019.01.061. [DOI] [PubMed] [Google Scholar]
- Becerra J. Andrades J. A. Guerado E. Zamora-Navas P. López-Puertas J. M. Reddi A. H. Articular Cartilage: Structure and Regeneration. Tissue Eng., Part B. 2010;16(6):617–627. doi: 10.1089/ten.teb.2010.0191. [DOI] [PubMed] [Google Scholar]
- Ambre A. H. Katti D. R. Katti K. S. Biomineralized hydroxyapatite nanoclay composite scaffolds with polycaprolactone for stem cell-based bone tissue engineering. J. Biomed. Mater. Res., Part A. 2015;103(6):2077–2101. doi: 10.1002/jbm.a.35342. [DOI] [PubMed] [Google Scholar]
- Ambre A. H., Katti D. R. and Katti K. S., Nanoclays mediate stem cell differentiation and mineralized ECM formation on biopolymer scaffolds, J. Biomed. Mater. Res., Part A, 2013, 101, 2644–2660 [DOI] [PubMed] [Google Scholar]
- Tripathy J., Polymer Nanocomposites for Biomedical and Biotechnology Applications, in Properties and Applications of Polymer Nanocomposites: Clay and Carbon Based Polymer Nanocomposites, ed. D. K. Tripathy and B. P. Sahoo, Springer Berlin Heidelberg, Berlin, Heidelberg, 2017, pp. 57–76 [Google Scholar]
- Quint J. P. Mostafavi A. Endo Y. Panayi A. Russell C. S. Nourmahnad A. Wiseman C. Abbasi L. Samandari M. Sheikhi A. Nuutila K. Sinha I. Tamayol A. Vivo Printing of Nanoenabled Scaffolds for the Treatment of Skeletal Muscle Injuries. Adv. Healthcare Mater. 2021;10(10):2002152. doi: 10.1002/adhm.202002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Chen M. Fan X. Zhou H. Recent advances in bioprinting techniques: approaches, applications and future prospects. J. Transl. Med. 2016;14(1):271. doi: 10.1186/s12967-016-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cidonio G. Cooke M. Glinka M. Dawson J. I. Grover L. Oreffo R. O. C. Printing bone in a gel: using nanocomposite bioink to print functionalised bone scaffolds. Mater. Today Bio. 2019;4:100028. doi: 10.1016/j.mtbio.2019.100028. [DOI] [PMC free article] [PubMed] [Google Scholar]