

Abstract
Repair of double-strand DNA breaks (DSBs) is important for preserving genomic integrity and stability. Break-induced replication (BIR) is a mechanism aimed to repair one-ended double-strand DNA breaks, similar to those formed by replication fork collapse or by telomere erosion. Unlike S-phase replication, BIR is carried out by a migrating DNA bubble and is associated with conservative inheritance of newly synthesized DNA. This unusual DNA synthesis leads to high level of mutagenesis and chromosomal rearrangements during BIR. Here we focus on several genetic and molecular methods to investigate BIR using our system in yeast Saccharomyces cerevisiae where BIR is initiated by a site-specific DNA break and the repair involves two copies of chromosome III.
Keywords: Break-Induced Replication, Double-Strand Break, Single-Strand DNA, Homologous Recombination, Contour-Clamped Homogenous Electric Field Electrophoresis, Gross Chromosomal Rearrangements, APOBEC
1. Introduction
Double-strand breaks (DSBs) are the most lethal type of DNA damage typically resulting from problems during DNA replication or chromosome segregation or due to a cell’s exposure to various chemicals or radiation (reviewed in [1]). DSBs can be repaired by non-homologous end joining (NHEJ) or by homologous recombination (HR). HR uses a homologous template to repair broken DNA and is believed to be more accurate than NHEJ, which proceeds via direct ligation of broken DNA ends. Several HR pathways have been described, including gene conversion (GC) that proceeds by gap repair or by synthesis–dependent strand annealing (SDSA), single-strand annealing (SSA), and break-induced replication (BIR). Here we focus on BIR, which is specific for situations where only one DSB end can find homology in the genome for invasion and repair. BIR starts with extensive 5’ to 3’ resection of a DSB end followed by strand invasion of a 3’ end into a homologous DNA donor (Fig. 1A, B). This leads to initiation of extensive DNA synthesis, which is carried out by a migrating DNA bubble, proceeding with asynchrony between leading and lagging strand synthesis and leading to conservative inheritance of newly synthesized DNA (Fig. 1B)[2–4]. This abnormal DNA synthesis is responsible for the increased level of mutagenesis and chromosome rearrangements associated with BIR [2, 5–8]. One important source of BIR-associated mutagenesis is long single-stranded DNA (ssDNA) resulting from extensive resection as well as from the asynchrony between leading and lagging strand synthesis. Such long ssDNA accumulates behind the BIR migrating bubble and can accumulate DNA damage, which leads to hyper-mutagenesis associated with BIR. In addition, ssDNA can promote formation of lethal recombination intermediates that can also form during BIR [9].
Figure 1. Yeast experimental system to investigate BIR.
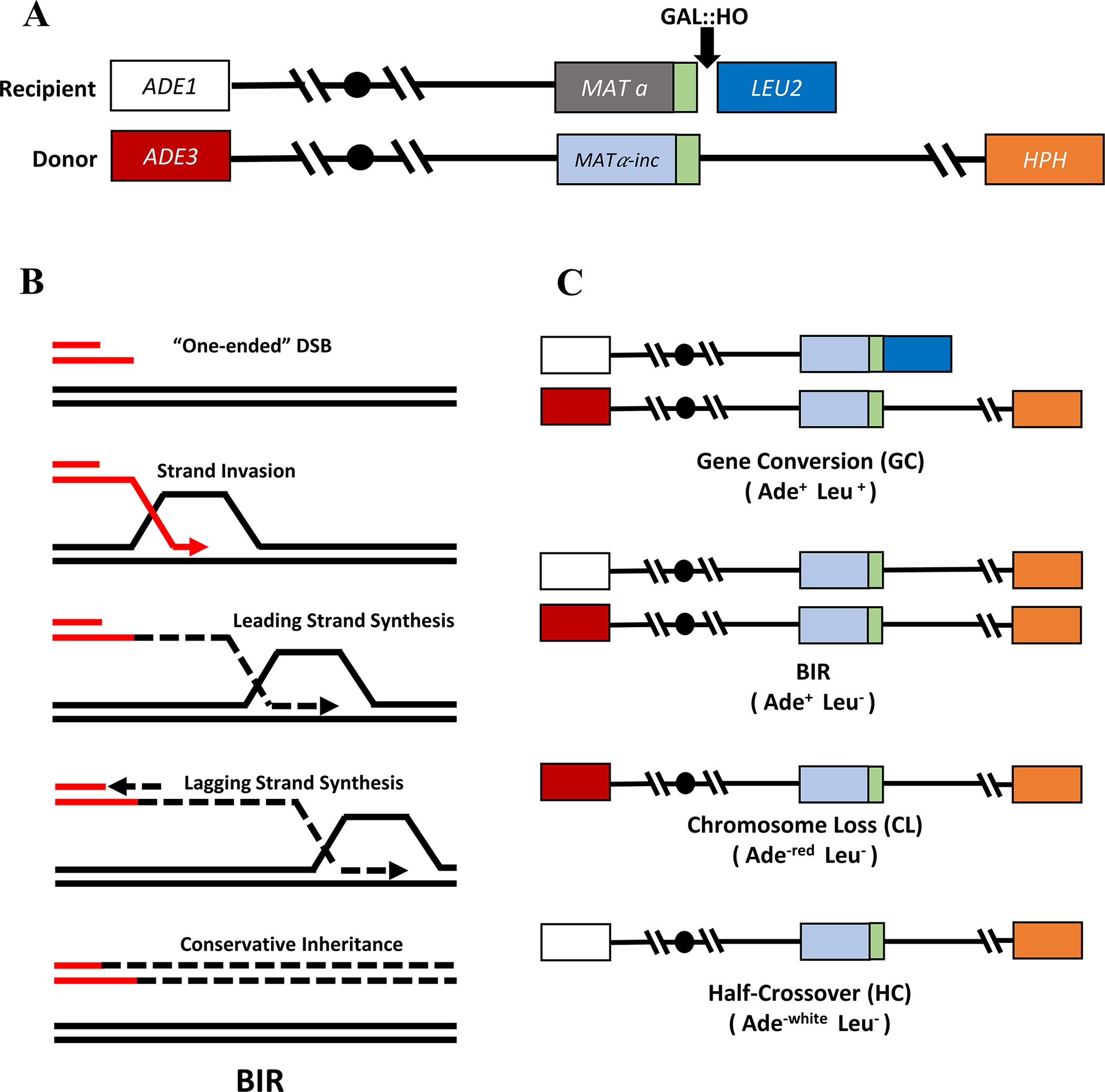
A. Schematic of the AM1003 experimental disome system that contains two copies of chromosome III. One copy contains a Gal::HO endonuclease recognition site (black arrow) at MATa and is truncated by insertion of LEU2 and telomere sequence next to MATa. The other copy of chromosome III is full-length and cannot be cut by HO due to presence of a MATα-inc (uncuttable) allele. B. Mechanism of BIR initiation and progression. BIR is initiated by 5’ to 3’ resection of the one-ended DSB, which leads to formation of exposed 3’ ssDNA. The ssDNA end invades into the homologous template initiating DNA synthesis. Leading strand synthesis progresses by a migrating bubble. ssDNA accumulates behind the migrating bubble due to uncoupled leading and lagging strand synthesis. Conservative inheritance of DNA. C. Repair outcomes following DSB initiation in AM1003. The following repair outcomes and their phenotypes are depicted: Gene conversion (Ade+ Leu+), BIR (Ade+ Leu−), Chromosome loss (Ade−red Leu−), and Half crossover (Ade−white Leu−).
For several decades yeast Saccharomyces cerevisiae has been used as a model to investigate BIR. Several different experimental systems were developed that mainly differed in the way the double strand break was introduced [10–13]. Here we focus on our experimental system where BIR is initiated by a site specific HO endonuclease, and proceeds between two copies of chromosome III (Figure 1A) [14]. Using this system has allowed us to perform detailed study of the mechanism of BIR by employing various genetic and molecular approaches, identify proteins that carry out and regulate BIR, and investigate mutagenesis and gross chromosomal rearrangements associated with BIR [2, 5–7, 14]. Here we focus on determining the efficiency of BIR using genetic methods. In addition, we describe how to determine the amount of ssDNA formed during BIR by using APOBEC3A (A3A) -induced mutagenesis. In particular, APOBEC cytidine deaminase (reviewed in [15]) expressed in yeast converts cytidines in ssDNA accumulated during BIR into deoxyuridines (dUs) [16]. In ung1Δ mutants lacking uracil glycosylase these dUs are not repaired and all result in C to T mutations, which reveals the full mutagenic potential of A3A during BIR and can be used to characterize the amount of ssDNA formed during BIR. Finally, we describe investigation of BIR kinetics using CHEF gel electrophoresis and Southern blot hybridization.
2. Materials.
To investigate BIR we use our yeast strain AM1003 disomic for chromosome III (Fig. 1A) with the genotype hmlΔ::ADE1/hmlΔ::ADE3 MATa-LEU2-tel/MATα-inc hmrΔ::HPH FS2Δ::NAT/FS2 leu2/leu2-3,112 thr4 ura3-52 ade3::GAL::HO ade1 met13 [14] or its derivatives. In this strain, BIR is initiated by HO endonuclease regulated by a galactose-inducible promoter. A DSB is initiated at the MATa locus of the truncated copy of chromosome III and the break is repaired by BIR using another intact, full-length copy of chromosome III containing MATα-inc.
2.1. Yeast media.
All media, materials and solutions used for investigation of BIR by genetic methods need to be autoclaved (unless stated otherwise).
2.1.1.
Sc-Leu dropout liquid: 2% (w/v) D-glucose (RPI, # 50-99-7), 0.67% (w/v) yeast nitrogen base without amino acids (US Biological, #Y2025) 0.087% (w/v) Leu dropout amino acid mix, pH 5.5.
2.1.2.
YEP-lactate liquid: 3.15% (v/v) lactic acid (Fisher, #A162-500), 1% (w/v) yeast extract, 2% (w/v) peptone pH 5.5.
2.1.3.
20% galactose: 20g galactose dissolved in 100 mL of MilliQ water and filter sterilized.
2.1.4.
YEP-galactose plates: 2% (v/v) galactose (Sigma-Aldrich, # 59-23-4) 2.5 % (w/v) agar, 1% (w/v) yeast extract, 2% (w/v) peptone pH 5.5. (Autoclave everything except for galactose. Following autoclaving, add 20% filter-sterilized galactose (1:10 dilution to final concentration of 2%).
2.1.5.
Sc-Ade dropout plates: 2% D-glucose, 2.5% agar, 0.67% yeast nitrogen base without amino acids, 0.087% Ade dropout amino acid mix, pH 5.5.
2.1.6.
Sc-His dropout plates: 2% D-glucose, 2.5% agar, 0.67% yeast nitrogen base without amino acids, 0.087% His dropout amino acid mix, pH 5.5.
2.1.7.
Sc-Leu dropout plates: 2% D-glucose, 2.5% agar, 0.67% yeast nitrogen base without amino acids, 0.087% Leu dropout amino acid mix, pH 5.5.
2.1.8.
Sc-Ura dropout plates: 2% D-glucose, 2.5% Agar, 0.67% yeast nitrogen base without amino acids, 0.087% Sc-Ura dropout amino acid mix.
2.1.9.
Sc-Ura, Ade dropout plates: 2% D-glucose, 2.5% Agar, 0.67% yeast nitrogen base without amino acids, 0.087% Sc-Ura, Ade dropout amino acid mix.
2.1.10.
YEPD plates: 2% D-glucose, 2.5 % agar, 1% yeast extract, 2% peptone, pH 5.5.
2.1.11.
YEPD liquid: 2% D-glucose, 1% yeast extract, 2% peptone, pH 5.5.
2.1.12.
50mg/mL Hygromycin B stock solution (EMD Millipore #400052).
2.1.13.
YEPD + Hyg plates: 2% D-glucose, 2.5 % agar, 1% yeast extract, 2% peptone, pH 5.5, 0.05% (v/v) Hygromycin B. (add 10 mL of 50 mg/mL Hygromycin B stock solution per liter).
2.1.14.
Sc-Leu dropout + Hyg liquid: 2% D-glucose (RPI, # 50-99-7), 0.67% yeast nitrogen base without amino acids (US Biological, #Y2025), 0.087% Leu dropout amino acid mix, pH 5.5, 0.05% Hygromycin B (v/v). (add 10 mL of 50 mg/mL Hygromycin B stock solution per liter).
2.1.15.
YEP-lactate + Hyg liquid: 3.15% lactic acid (Fisher, #A162-500), 1% yeast extract, 2% peptone pH 5.5, 0.05% Hygromycin B (add 10mL of 50mg/mL Hygromycin B stock solution per liter).
2.1.16.
Petri dishes (100 × 15mm).
2.2. Yeast strains and plasmids.
2.2.1.
AM1003 [14] or its derivatives.
2.2.2.
AM3647 that is isogenic to AM1003, but ung1Δ ::KanMX, hmrΔ::ura3-29-HPH (containing insertion of ura3-29 reporter [17] at 90kb position centromere distal from MATα-inc).
2.2.3.
pSR435 and pSR419: centromeric plasmids [18]. pSR435 contains human APOBEC 3A gene under the control of the TetO7 promoter. pSR419 – empty vector control (similar to pSR435, but without A3A).
2.3. Detecting ssDNA accumulated on the course of BIR.
All solutions used for the molecular analysis of BIR need to be prepared by using MilliQ water.
2.3.1.
1M sorbitol (RPI, 10043-35-3): 18.217g dissolved in 100 mL MilliQ water.
2.3.2.
2 U/uL Zymolyase Solution: 0.1g of 20T Zymolyase (MP, #320921) in 1 mL of 50% glycerol.
2.3.3.
TE buffer: 10mM Tris,1mM EDTA, pH 7.6.
2.3.4.
20% SDS (Fisher, #BP166-5) 20g sodium dodecyl sulfate dissolved in 100 mL MilliQ water.
2.3.5.
5M potassium acetate (KAc)( RPI, #127-08-2): 49.1g potassium acetate dissolved in 100 mL MilliQ water.
2.3.6.
RNase A stock solution: 10mg RNaseA (ThermoFisher, #EN0531) dissolved in 1 mL MilliQ water.
2.3.7.
Phenol/Chloroform/Isoamyl alcohol (25:24:1) (Fisher # BP1752I).
2.4. Assessing BIR kinetics and BIR outcomes using CHEF gel electrophoresis.
2.4.1.
50 mL conical tubes.
2.4.2.
10% sodium azide solution: 10g sodium azide (Sigma Aldrich, #26628-22-8) dissolved in 100 mL MilliQ water.
2.4.3.
50mM EDTA, pH7.5: Diluted 1:10 from 0.5M EDTA in MilliQ water.
2.4.4.
Plug Molds (BioRad #1703713).
2.4.5.
SCE solution: 0.1M sorbitol, 1mM sodium citrate, 50mM EDTA pH7.5.
2.4.6.
1% LMP agarose (Fisher, 9012-36-6): in 0.75 mL water and 0.25 mL 0.5M EDTA pH7.5.
2.4.7.
Zymolyase buffer: 0.005g 20T zymolyase, 1 mL SCE solution, 50μL 2-mercaptoethanol.
2.4.8.
Incubation buffer 1: 50mM EDTA pH7.5, 0.01M Tris-HCl, pH7.5.
2.4.9.
Incubation buffer 2: 0.1M Tris-HCl pH7.5, 0.25M EDTA pH7.5, 1% sarkosyl.
2.4.10.
Wash Buffer: 10mM Tris pH7.5, 50mM EDTA pH7.5.
2.4.11.
Storage Buffer: 40mM EDTA pH7.5, 55% glycerol.
2.4.12.
Proteinase K: (RPI, #39450-01-6).
2.4.13.
Ethidium bromide (Sigma Aldrich, 1239-45-8).
2.4.14.
5x TBE: 64g Tris base (RPI, #77-86-1), 31g boric acid (RPI, #10043-35-3), 3.35g EDTA Disodium salt dihydrate (Fisher, BP120-1).
2.4.15.
1xTBE: Dilute 5xTBE by adding 200 mL 5xTBE in 1L of MilliQ water.
2.4.16.
1% Pulse-Field Certified agarose gel (Bio-Rad, #L004315B): 2g Pulse-Field Certified agarose in 200 mL 0.5x TBE.
2.4.17.
20x SSC pH 7: 175.3g NaCl, 88.2g sodium citrate, 900 mL MilliQ water
2.4.18.
Alkaline solution: 20g NaOH (RPI, #1310-73-2), 87.7g NaCl (RPI, #7647-14-5) in 1L of MilliQ water.
2.4.19.
Neutralization solution: 87.7g NaCl (RPI, #7647-14-5), 121.4g Tris Base (RPI, #77-86-1) in 1L of MilliQ water, pH 7.0
2.4.20.
QIAGEN gel extraction kit: (QIAGEN, # 28704).
2.4.21.
P32-dCTP (Perkin Elmer, #BLU513H250).
2.4.22.
RmT Random primer kit (Agilent, # 300392).
2.4.23.
Hybridization buffer: 50 mL of 0.5M sodium phosphate buffer pH 7.2 (Fisher, #S374-1), 25mL of 20% SDS (Fisher, #BP166-5), 200μL of 1M EDTA pH 8. Make up to 100 mL using MilliQ water.
2.4.24.
Low Stringency Wash Buffer: 2X SSC, 0.1% SDS
3. Methods.
3.1. Determining the efficiency of BIR.
3.1.1.
Inoculate a single colony of AM1003 or its derivative in liquid Sc-Leu dropout media (see Note 4.1) and grow for 24 hours at 30°C to approximately to 1× 108 cells/mL (to saturation).
3.1.2.
Transfer 5 mL of the saturated culture (inoculum) from 3.1.1 to 45 mL of YEP-lactate liquid media in 250 mL flask and grow at 30°C with constant aeration for 16 hours (see Note 4.2).
3.1.3.
Using a haemocytometer count the cells and calculate the cell density. The expected cell density is approximately 2×107 cells/mL.
3.1.4.
Perform serial dilutions of the cell culture to reach a density of approximately 5×102 cells/mL by first adding 5×105 cells to 1 mL of sterile water. Perform three serial 1:10 dilutions in sterile water.
3.1.5.
Plate 0.1 mL of the yeast cell suspension (5×102 cells/mL) on at least 3 YEPD plates of YEPD and 5–10 YEP-galactose plates.
3.1.6.
Incubate all plates (YEPD and YEP-galactose) for 3–5 days at 30°C. Usually, the colonies will form in approximately 3 days on YEPD, while it may take approximately 5 days for the colonies to form on YEP-galactose.
3.1.7.
Cell viability following DSB induction is determined by comparing the number of colonies grown on YEP-galactose to those grown on YEPD. Cell viability should be calculated using the formula:
(see Note 4.3)
3.1.8.
Replica plate the colonies grown on YEP-galactose onto Sc-Ade, Sc-Leu, and Sc-His dropout media plates.
3.1.9.
Score repair outcomes based on auxotrophic marker phenotypes (Fig.1C, 2A).
Figure 2.
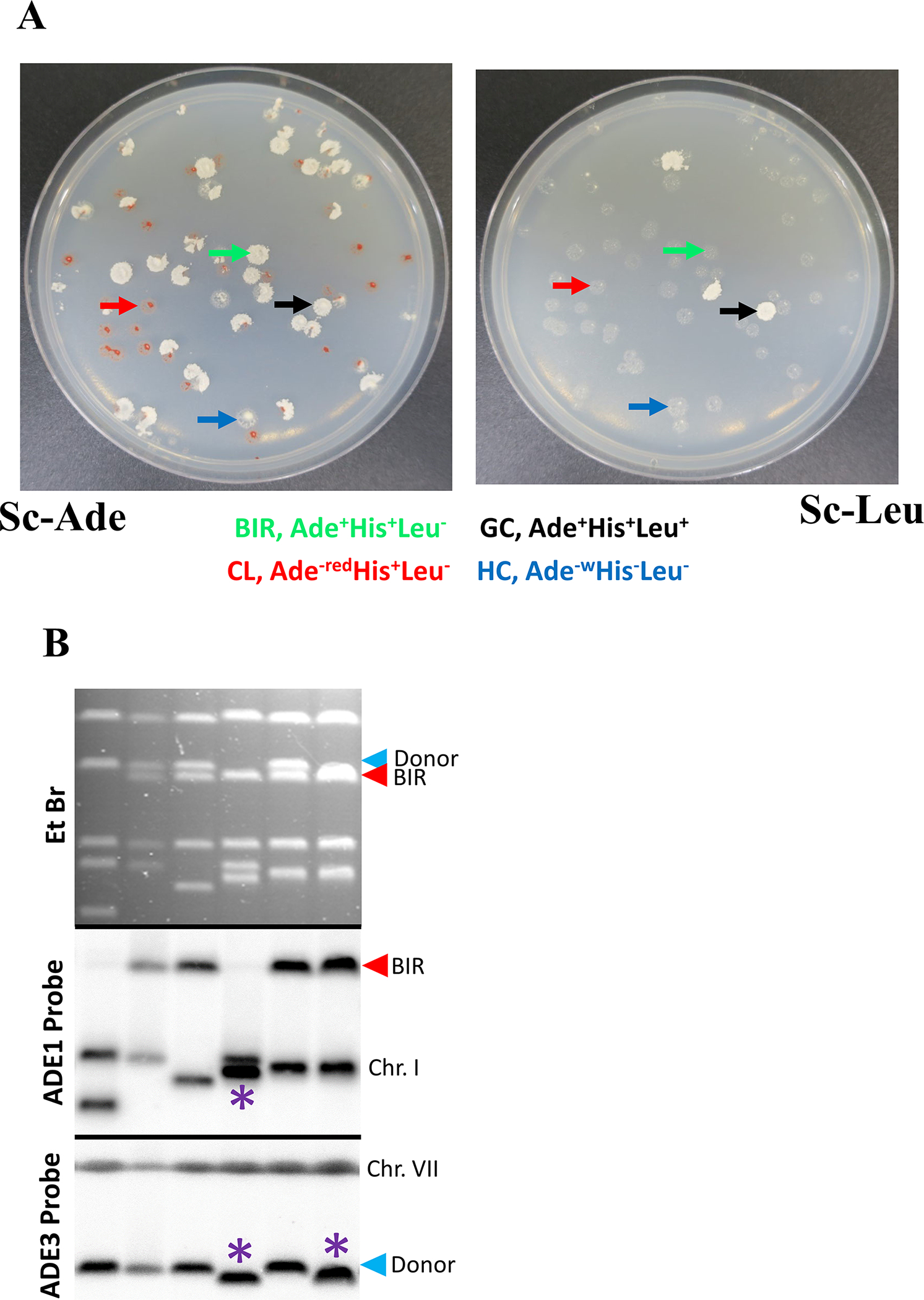
A. BIR repair plating assay. Cells were pre-grown in YEP-lactate liquid media followed by plating on YEP-galactose plates. The colonies were replica-plated on the Sc-Ade (left) and Sc-Leu (right) dropout plates. The rad9Δ derivative of AM1003 is shown that produces various repair outcomes. Arrows point to different repair outcomes, green arrows indicate a BIR outcome, blue arrows indicate a half crossover, black arrows indicate gene conversion, and red arrows indicate a chromosome loss outcome. In addition, the depicted mutant shows formation of many sectored colonies (containing sectors of several repair outcomes in the same colony, which can result from two sister chromatids repairing differently, or from delayed repair that occurs following one or several cellular divisions. The latter is frequent in DNA damage checkpoint mutants including rad9Δ depicted here. B. CHEF gel analysis of Ade+Leu− repair outcomes, including BIR repair outcomes and repair outcomes containing chromosomal rearrangements. Top: ethidium bromide-stained CHEF gels; middle: Southern blot hybridization with ADE1-specific probe and with ADE3-specific probe (bottom). Red arrow denotes BIR band. Blue arrow denotes Donor band. Asterisks denote rearranged recipient (ADE1-containing) and donor (ADE3-containing) chromosomes.
BIR: Ade+ Leu−
Gene conversion: Ade+ Leu+
Half crossover: Ade−white Leu−
Chromosome loss: Ade−red Leu−
3.1.10.
Calculate the frequency of BIR outcomes (FBIR) using the following formula:
3.2. Determining the amount of ssDNA formed during BIR.
3.2.1.
Inoculate a single colony of AM3647 (Fig. 3A) transformed with pSR435 (containing A3A gene) into 1mL of Sc-Leu dropout + Hyg liquid media, and one colony of AM3647 transformed with pSR419 (empty vector control) into 1mL of Sc-Leu dropout + Hyg liquid media and incubate for 24 hours at 30°C with constant aeration until it reaches saturation (to the density approximately 1×108 cells/mL).
Figure 3. Mutations accumulating in ssDNA formed during BIR.
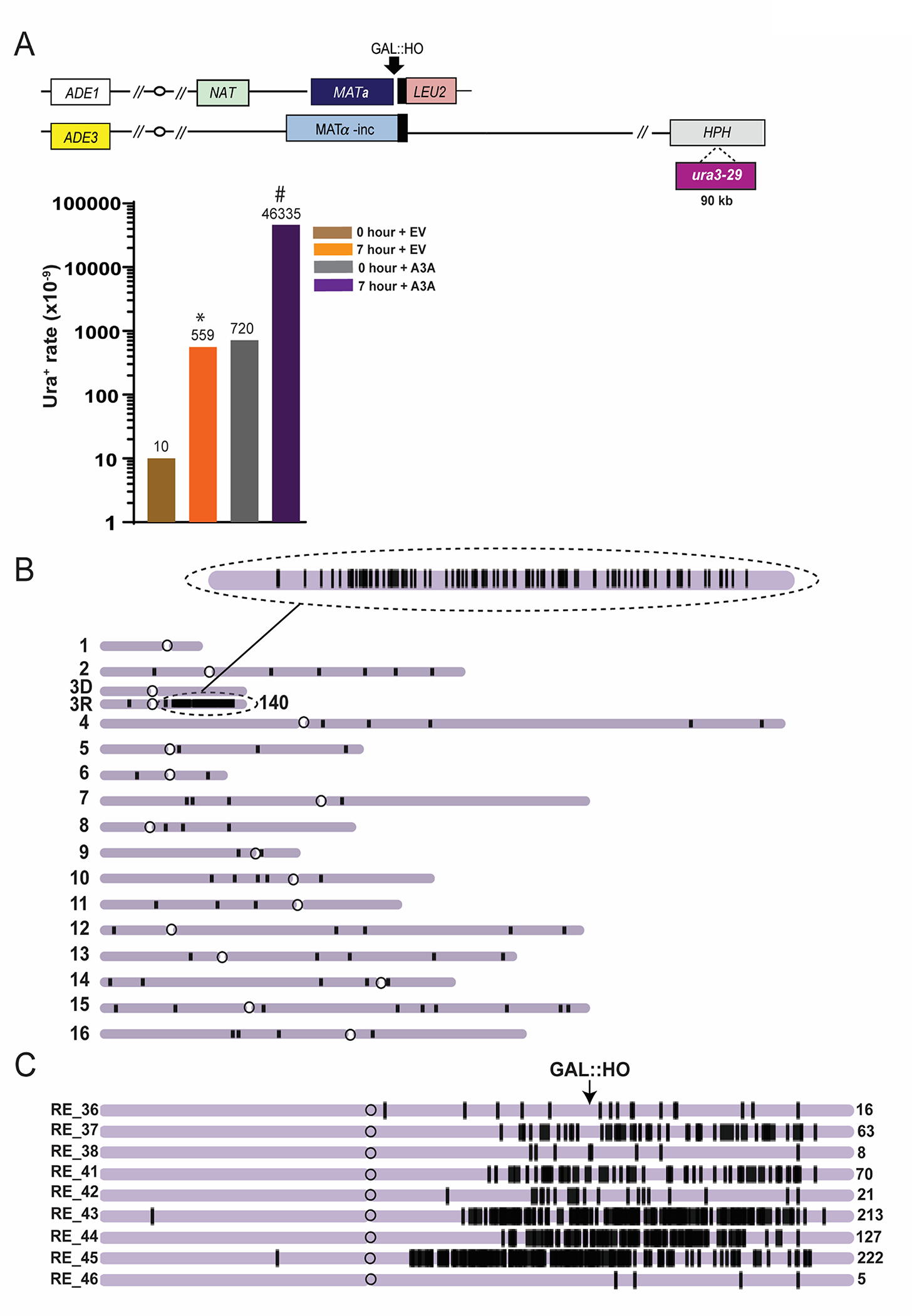
(From data in Elango et al [16] published under Oxford University Press’s open access and a Creative Commons Attribution 4.0 International License). A. The increase of Ura+ reversions resulting from BIR in the presence of expression of APOBEC3A. Top: experimental disomic system (similar to that shown in Fig. 1A, but containing ura3-29 mutation reporter inserted at position 90kb centromere distal to MATα-inc. Bottom: Ura+ rates for ura3-29 inserted at 90kb Chr. III position following BIR in the presence of APOBEC3A containing plasmid (A3A) or empty vector (EV). Asterisk and pound-symbol indicate statistically significant differences from No-DSB (0h) and from no-A3A (EV), respectively. B. A3A-induced mutations (black vertical lines) in one BIR outcome in the progeny of ung1Δ derivative of AM1003 formed in the presence of A3A. Enlarged: mutation cluster on the track of BIR C. Clustered mutations in BIR isolates formed in the presence of A3A. Positions of mutations (black lines) are depicted along the chromosome III reference. The total number of mutations in each cluster is indicated by a number (on the right).
3.2.2.
For the culture transformed with the A3A gene (pSR435), transfer 0.5 mL of inoculum into 4.5 mL YEP-lactate + Hyg media (in a 125-mL flask). For the culture transformed with the empty vector control (pSR419), transfer 1 mL of inoculum into 9 mL YEP-lactate + Hyg media (in a 125-mL flask). Both should have a starting cell density of ~1×106 cells/mL. Incubate all cultures at 30°C for ~16–20 hours with constant aeration, to a final concentration of approximately 1–2 × 107 cells/mL.
3.2.3.
From the A3A transformed cultures, collect 0.5 mL of culture from step 3.2.2 into a conical tube, and likewise from the empty vector transformed cultures collect 3 mL of the culture from step 3.2.2 into a conical tube. Use these as the “0 hour” time point to assess the level of mutagenesis associated with S-phase replication (as a control).
3.2.4.
Take an additional 0.5 mL from all cultures from step 3.2.2 and serially dilute them by transferring 0.1 mL of culture into 0.9 mL sterile water. Typically, four 10-fold dilutions are made to obtain a concentration of approximately 1×103 cells/mL.
3.2.5.
Plate each suspension from 3.2.4 with a concentration of approximately 1×103 cells/mL by plating 0.1 mL per Sc-Ade dropout media (3 Sc-Ade dropout plates total with approximately 100 cells per plate). From these plates, the concentration of cells in the culture will be determined.
3.2.6.
To estimate the number of mutations accumulated in the culture at 0 hour (before induction of the DSB by HO) centrifuge the samples collected in 3.2.3. at 5000 rpm for 2 minutes at 25°C. Decant the supernatant and keep the pellet. Re-suspend the pellet in 0.5 mL of sterile water and spread onto 5 Sc-Ura, Ade dropout plates (0.1 mL per plate). Be cautious not to plate >1×108 cells per plate to avoid formation of a lawn due to residual growth.
3.2.7.
Add 440 uL of 20% galactose (filter sterilized) to the remaining 4 mL of the A3A culture from step 3.2.2 and 715 uL of 20% galactose (filter sterilized) to the remaining 6.5 mL of the empty vector culture from step 3.2.2, to obtain a final concentration of 2% galactose in all cultures. Addition of galactose initiates formation of the DSB. Incubate with constant aeration at 3000 rpm at 30°C for 7 hours.
3.2.8.
For A3A cultures, collect 25 uL of culture from step 3.2.7, and from empty vector cultures, collect 2 mL of culture from step 3.2.7 (see Note 4.5). Centrifuge at 5000 rpm for 2 minutes at 25°C. Decant the supernatant (or remove with a pipette for small volumes) and re-suspend each pellet in 0.5mL of sterile water and plate onto five Sc-Ura dropout plates (0.1 mL of suspension per plate). Be cautious to not plate >1×108 cells per plate to avoid formation of a lawn.
3.2.9.
Take an additional 0.5 mL from all cultures from step 3.2.7 and serially dilute them by transferring 0.1 mL of culture into 0.9 mL sterile water. Typically, four 10-fold dilutions are made to obtain a concentration of approximately 1×103 cells/mL.
3.2.10.
Plate each suspension from 3.2.9 with a concentration of approximately 1×103 cells/mL by plating 0.1 mL per YEPD plate (3 YEPD plates total with approximately 100 cells per plate). From these plates, the concentration of cells in the culture will be determined.
3.2.11.
Incubate all plates for 5 days at 30°C and count the colonies on each plate.
3.2.12.
Calculate the rate of Ura+ reversions (see Note 4.6) associated with BIR using the following formula:
It is expected that the Ura+ BIR rate (at 7 hours) will significantly (more than 10 times) exceed the Ura+ rate at 0hr for both cells transformed with A3A plasmid and with empty vector. In addition, Ura+ BIR rate in cells with A3A vector is expected to be more than 100 times higher than in cells transformed with empty vector [16] (Fig. 3A) (see Note 4.8).
3.2.13.
Pick Ura+ colonies from Sc-Ura dropout plates (7 hours, formed after BIR) from the culture containing the A3A expression plasmid, and streak on SC-Ura dropout plates to isolate single colonies.
3.2.14.
Take the singles from the last step and patch them on YEPD, grow at 30°C.
3.2.15.
Replica plate patches on Sc-Leu, Sc-Ade, Sc-Ura, and also on YEPD + Hyg plates to confirm that the colonies have completed BIR repair, that they are Ura+, and that they lost the A3A-containing plasmid. (They should be HygsAde+ Ura+Leu−).
3.2.16.
Use the patch from 3.2.14 to start a culture in 50 mL YEPD, grow to saturation (approximately 18–24 hours).
3.2.17.
Pellet the culture from 3.2.16. by centrifugation at 3000 rpm for 5 minutes.
3.2.18.
Resuspend yeast pellet in 5 mL of 1M sorbitol.
3.2.19.
Add 500 uL of 2U/uL zymolyase solution. Mix by vortexing.
3.2.20.
Incubate for at least 1 hour at 37°C and periodically agitate gently.
3.2.21.
Centrifuge at 3000 rpm for 7 minutes. Discard as much as possible of the supernatant.
3.2.22.
Resuspend the cells in 10 mL of TE buffer.
3.2.23.
Add 1.1 mL of 20% SDS. Invert the tube quickly 20 times to mix.
3.2.24.
Incubate at 65°C for 30 minutes. (see Note 4.9).
3.2.25.
Add 11 mL 5 M KAc. Invert the tube gently 10 times to mix. Set the tube on ice for 30–60 minutes.
3.2.26.
Centrifuge for 15 minutes at 15,000 rpm. Transfer 10–15 mL of supernatant into a fresh centrifuge tube. (see Note 4.10).
3.2.27.
Add equal volume of room temperature isopropanol to the supernatant. Invert the tube 10 times to mix.
3.2.28.
Allow the tube to sit at room temperature for 15 minutes. (see Note 4.11).
3.2.29.
Centrifuge for 10 min at 15,000 rpm. Pour off the supernatant.
3.2.30.
Partially air-dry the pellet for 15 minutes. Place the tube upside down on a napkin.
3.2.31.
Add 20 mL of 70% ethanol (EtOH). Invert the tube 10 times to mix.
3.2.32.
Allow the tube to sit overnight at 4°C.
3.2.33.
Centrifuge for 2 minutes at 15,000 rpm. Pour the supernatant away immediately once it is removed from centrifuge. (see Note 4.12). Allow the tube to sit with its lid open to dry further.
3.2.34.
Re-suspend yeast genomic DNA in 2 mL TE. Transfer to a 15mL polypropylene centrifuge tube.
3.2.35.
Add 30 uL of RNase A stock solution. Incubate at 37°C for 30 minutes.
3.2.36.
Add 2 mL of phenol/chloroform/isoamyl alcohol mix. Vortex and spin tubes at 3000–4000 rpm for 10 minutes.
3.2.37.
Take the top phase (try not to get white protein at phase interface; about 1.8 mL).
3.2.38.
Add 100 uL of 5 M KAc and mix gently by inverting the tube.
3.2.39.
Split into 5 aliquots in 1.5mL microcentrifuge tubes (400–500 uL per tube). Precipitate with 2x volume (800–1000 uL) of 100% ethanol. Spin all aliquots in a microcentrifuge at 15,000 rpm for 10 minutes. (see Note 4.13.).
3.2.40.
Wash twice with 70% ethanol, then leave the tubes open to allow the DNA to dry.
3.2.41.
Dissolve dry DNA in a total of 0.5 mL TE (100 uL per DNA pellet).
3.2.42.
Submit ≥2 ug of purified yeast genomic DNA for library preparation/Illumina sequencing. Library preparation can be performed using KAPA DNA Hyper kit (Kapa Biosystems). Whole genome sequencing can be performed with the HiSeq4000 PE 2×150 (Illumina)sequencing platform.
3.2.43.
Once sequencing reads are received, perform read alignment to a reference and mutation calling using the CLC Genomic Workbench version 7.5 software or later, specifically:
Trim (if poor sequencing quality necessitates) and align the sequence reads to a reference genome of AM1003 that was created for the [5] study, but was tailored to the AM3617 strain using the “Trim Reads” and “Map Reads to Reference” functions of CLC software.
Use the aligned read track for mutation calling using the “Fixed Ploidy Variant Detection tool” with ploidy parameter set to 2, requiring SNVs to have greater than 23% allele frequency, and having a read coverage greater than 10% of the mean coverage across the samples.
Filter out pre-existing variants and SNVs due to mapping artifacts. Parental variants can be removed with the “Remove Variants Present in Control Reads” function. Alternatively, remove mutations that occur in more than one sample (including a list of SNVs generated for an untreated parental control strain).
Use the filtering tool to remove all types of mutations from the resulting list except for single nucleotide variants (SNVs). Unless indicated otherwise, all the parameters are kept at default values. (see Note 4.14)
3.2.44.
Identify mutation clusters using the Cluster Finder software developed by Dmitry Gordenin’s lab [19, 20] (see Note 4.15.). The length of mutation clusters identified in ung1Δ serves as an estimate of the ssDNA length formed during BIR [16] (see Figure 3B, CF for examples).
3.3. Investigation of BIR kinetics and outcomes using CHEF gel electrophoresis.
3.3.1.
Inoculate a single colony of AM1003 or its derivative in 50 mL of Sc-Leu dropout media, and incubate for 18– 24 hours at 30°C with constant aeration until the culture reaches a density of ~108 cells/mL.
3.3.2.
Add the inoculum from 3.3.1 (50mL) to 500 mL of YEP-lactate in a 2L-flask. Incubate the culture at 30°C with constant aeration to reach a cell density of ~2×107 cells/mL (should take approximately 16 hours).
3.3.3.
Collect 50 mL of culture for a 0 hour sample (sample taken before galactose addition) in a 50 mL conical tube, then add 0.5 mL of 10% sodium azide to this aliquot. Mix thoroughly.
3.3.4.
Induce the DSB by adding 50 mL of 20% galactose to the YEP-lactate culture (see Note 4.16).
3.3.5.
Use a tabletop centrifuge to pellet the sample collected at time point “0h” (3000 rpm for 5 minutes).
3.3.6.
Discard the supernatant into hazardous waste container, and re-suspend the pellet in 50 mL of 50 mM EDTA pH 7.5 solution.
3.3.7.
Pellet the sample using a tabletop centrifuge (spin at 3000 rpm for 5 minutes). Discard supernatant. Pellets can be stored in −80°C freezer until needed for plug preparation.
3.3.8.
Collect 50 mL of culture every hour (or every two hours) and repeat steps 3.3.5 – 3.3.7. (see Note 4.17.).
3.3.9.
When ready to prepare DNA plugs, thaw all pellets from the time course assay on ice.
3.3.10.
Prepare LMP agarose by adding 0.01g of LMP agarose to 0.75 mL of MilliQ water, and then adding 0.25 mL 0.5M EDTA pH7.5. Heat LMP agarose on heating block to 100° C to dissolve the LMP agarose.
3.3.11.
Prepare zymolyase buffer by adding 50 μL of 2-mercaptoethanol and 0.005g 20T zymolyase to 1 mL of SCE. Keep on ice.
3.3.12.
Equilibrate LMP agarose to 40° C by placing in 40° C water bath (see Note 4.18).
3.3.13.
Use a vortex to loosen the pellet. Equilibrate the pellet in a 40° C water bath.
3.3.14.
Quickly mix 0.085 mL zymolyase buffer with 0.415 mL of agarose, and then add 0.25 mL cells to this mixture.
3.3.15.
Transfer quickly the cell agarose mixture into plug molds by pipetting (see Note 4.19.).
3.3.16.
Chill plug molds containing cell agarose mixture in refrigerator at 4° C for approximately 5 minutes.
3.3.17.
Prepare Incubation Buffer 1 mix by adding 750μl mercaptoethanol and 10μl 10mg/mL RNase into 10 mL of incubation Buffer 1. Dispense 2.5 mL of incubation buffer 1 mixture into 50 mL conical tubes.
3.3.18.
Remove solidified plugs from the plug molds into conical tubes containing incubation buffer 1 by a toothpick. Incubate at 37° C in a water bath for 1 hour (see Note 4.20).
3.3.19.
Discard the incubation buffer from conical tubes; use a scoopula to keep the plugs in place.
3.3.20.
Rinse plugs with MilliQ water, and dump the water. Use a scoopula to keep plugs in place.
3.3.21.
Prepare incubation buffer 2 mix by adding 0.005g proteinase K into 10 mL of incubation buffer 2.
3.3.22.
Add 2.5 mL of incubation buffer 2 mixture into each conical tube with plugs. Incubate at 50° C for 16 hours.
3.3.23.
Rinse plugs with MilliQ water. Use a scoopula to keep the plugs in place
3.3.24.
Rinse plugs with 2.5 mL of wash buffer.
3.3.25.
Plugs can be stored in wash buffer for two days at 4° C before they are used for CHEF gel electrophoresis. For long term storage, decant wash buffer, use scoopula to keep the plugs. Add 50 mL of storage buffer, keep at −20° C for up to 6 months (see Note 4.21).
3.3.26.
Make 3L of 0.5x TBE by adding 300 mL of 5x TBE and 2.7L of MilliQ water in a 4L beaker.
3.3.27.
Assemble gel casting tray (a part of CHEF BioRad equipment) with black base plate on the bottom. Make sure to tighten screws on each side.
3.3.28.
Remove plugs using a scoopula and rinse in wash buffer. Place each plug against the longer side of the gel casting tray.
3.3.29.
Prepare 1% pulse-field agarose gel by melting 2g agarose in 200 mL of the previously made 0.5x TBE in a microwave oven. Allow the gel to cool to about 60°C before pouring it into gel casting tray (see Note 4.22).
3.3.30.
Transfer the remaining 0.5x TBE buffer into CHEF electrophoresis cell. Circulate and cool the buffer to 14°C.
3.3.31.
Once the gel solidifies, disassemble the gel-casting tray while leaving the gel adhered to the black plate (see Note 4.23).
3.3.32.
Place the gel and the black base plate into frame in the middle of the CHEF electrophoresis cell. Close the lid of the CHEF electrophoresis cell.
3.3.33.
Enter the parameters for the initial and final switch times, the duration of the run, and the voltages on the control module. These parameters will vary depending on the sizes of yeast chromosomes that you aim to separate. If you are aiming to separate the donor chromosome III (355 kb) from the repaired by BIR chromosome III (345 kb) of AM1003, and using CHEF DRII apparatus, the parameters should be as follows: initial switch time of 10 seconds, final switch time of 35 seconds, running time of 40 hours, and the voltage is 6 Volts/cm (see Note 4.24).
3.3.34.
When the CHEF run is finished, add 300 mL 0.5x TBE into a glass baking dish.
3.3.35.
Remove the CHEF gel from the CHEF apparatus and place into the glass baking dish that contains 0.5x TBE.
3.3.36.
Add ethidium bromide to a final concentration of 0.5 μg/mL into the glass dish containing the CHEF gel and 0.5x TBE. Incubate for 30 minutes with constant gentle shaking.
3.3.37.
Carefully remove gel from black base plate. Take a picture of the CHEF gel with separated chromosomes using a UV gel imager.
3.3.38.
Carefully remove buffer with ethidium bromide, and rinse the gel with water. Dispose the EtBr containing solution appropriately. Place the gel into UV cross-linker and set the exposure to 600μJ/cm2 at 254nm wavelength. Expose the gel to this UV dose.
3.3.39.
Submerge the gel into alkaline solution, and incubate for 30 min with constant gentle shaking. Decant alkaline solution.
3.3.40.
Submerge gel in neutralization solution and incubate for 30 min with constant gentle shaking.
3.3.41.
To transfer gel, construct gel transfer sandwich in the following order from bottom to top: glass tray containing 10x SSC, plexiglass surface, filter paper wick cut to the width of the gel that contacts the SSC below and is wetted with 10x SSC, CHEF gel (DNA side up), Nylon transfer membrane wetted with 2x SSC, filter paper cut to the size of the gel wetted in 10x SSC, two stacks of paper towels (~20 cm high), second plexiglass surface, 500 g weight. Capillary transfer methods further described in [21]. Let the transfer occur for 20 hours.
3.3.42.
Place the membrane (DNA side up) in the UV crosslinker. Expose the membrane to UV (254nm wavelength) using the auto-crosslink function (120 mJ/cm2).
3.3.43.
Prepare the DNA probes for Southern blot hybridization.
-
ADE1-specific DNA probe targets not only the recipient copy of chromosome III (before and after the DSB, and also following DSB repair) but also chromosome I containing a native ADE1 gene (see Fig. 4A). This probe is prepared by PCR amplification of the ADE1 fragment from the yeast genome (using genomic DNA prepared from AM1003) and using the following PCR primers:
FP: 5’-GGTTTGAAACAACCTCAAGGACTT-3’
RP: 5’-AAGTCCTTGAGGTTGTTTCAAACC-3’
-
ADE3-specific DNA probe targets the donor copy of chromosome III (before and after the DSB, and also following DSB repair) but also chromosome VII containing the native ADE3 gene. This probe is prepared by PCR amplification of the ADE3 fragment from the yeast genome (using genomic DNA prepared from AM1003) and using the following PCR primers:
FP: 5’- GCAGGGTTCGATTTCACTATGGGT −3’
RP: 5’- ACCGTCATCATCGACTTCCACGGC - 3’
Figure 4. BIR kinetics assay using CHEF gel electrophoresis.
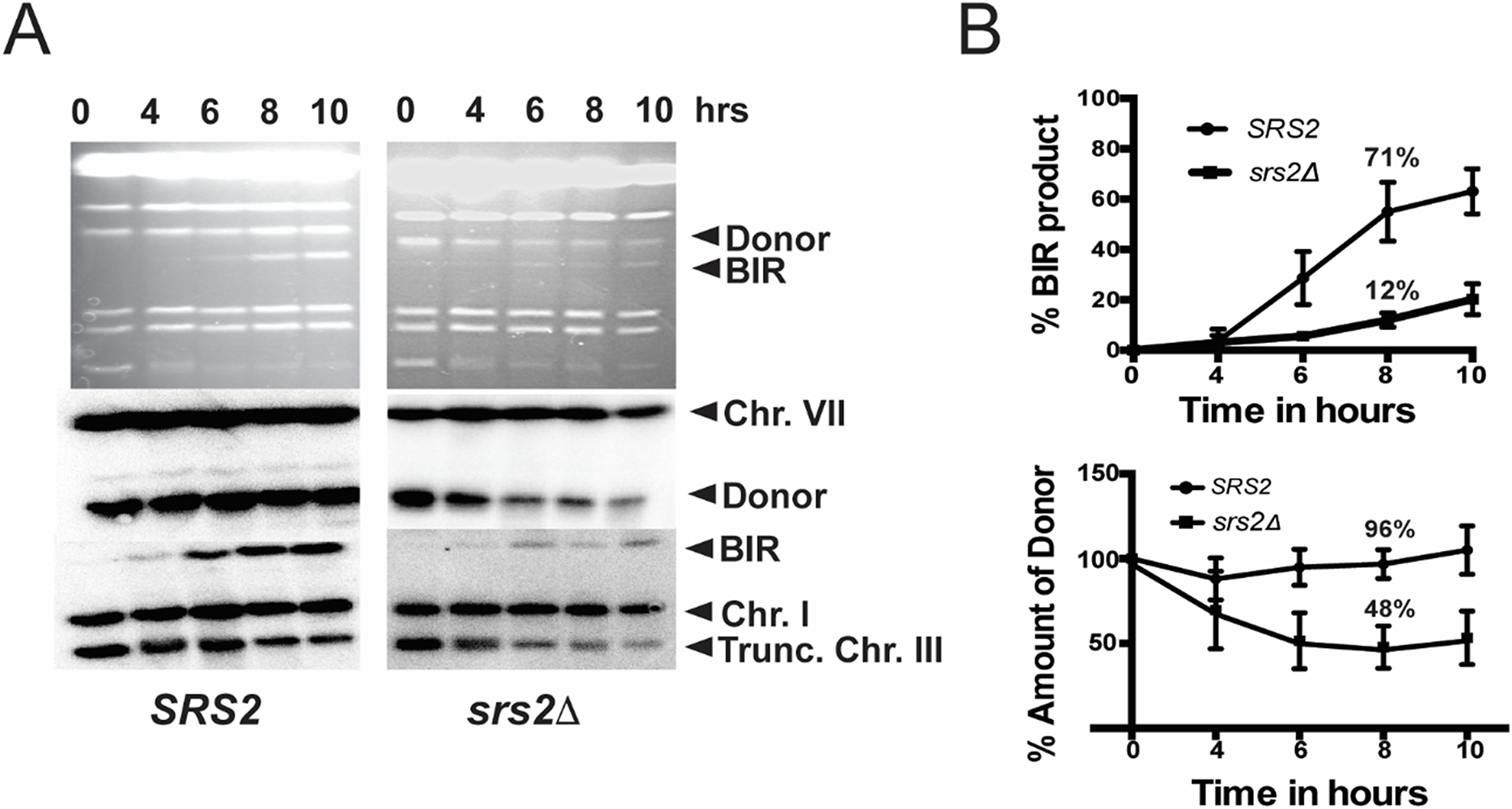
(From data in Elango et al [9] published under open access and a Creative Commons Attribution 4.0 International License). A. The BIR repair kinetics in wild type (SRS2) strain isogenic to AM 1003 (left) and BIR-defective srs2Δ derivative. The aliquots were taken from the liquid cultures undergoing BIR repair before (0h) and following the addition of galactose to the media at indicated time points. The genomic DNA was separated by CHEF. The upper panels represent ethidium bromide stained gels of the separated chromosomes. The middle panels represent the results of Southern blot analysis using an ADE3-specific probe that highlights the donor chromosome III. The bottom panels represent the results of Southern blot analysis using an ADE1-specific probe highlighting the recipient chromosome III. Chr.: chromosome; Trunc. Chr III: truncated chromosome III. B. Quantification of BIR product (top) and of donor chromosome entering the gel (bottom) in wild type (SRS2) and srs2Δ strains from the results of analysis shown in A.
3.3.44.
Run the ADE1-specific or ADE3-specific probe fragment(s) on a 1% agarose 1xTBE gel and excise the fragment(s).
3.3.45.
Using the QIAGEN Gel extraction kit extract the probe PCR fragment.
3.3.46.
Resuspend and dissolve the contents of one tube of the RmT Random primer kit in 37μL of MilliQ water, and transfer to a 1.5 mL microcentrifuge tube.
3.3.47.
Add 5μL (~50ng) of the gel extracted PCR probe fragment.
3.3.48.
Incubate RmT Random primer kit and probe fragment mixture at 100°C for 5 minutes. Be sure to add a microcentrifuge tube locker to keep the lid closed.
3.3.49.
After incubation period, place reaction mixture on ice for 1 minute and briefly spin down (to collect all the liquid on the bottom of the tube).
3.3.50.
Add 5μL of P32-dCTP to the cooled reaction mixture, and add 3μL of Magenta polymerase from the RmT Random primer kit, and incubate at 37°C dry bath for 10 minutes.
3.3.51.
Add 2μL of stop mix from the RmT Random primer kit to stop the reaction.
3.3.52.
Add 200μL of MilliQ water to the reaction mixture and denature at 100°C dry bath for 10 minutes and snap cool on ice for 1 minute.
3.3.53.
Pre-hybridize the crosslinked membrane from step 14 in 25 mL of hybridization buffer at 65°C for at least 10 minutes.
3.3.54.
Hybridize the membrane at 65°C with 50 uL of the labeled probe diluted in 25 mL of hybridization buffer for 24 hours. [22].
3.3.55.
Use low stringency wash buffer preheated to 65°C to wash the membrane in an open tray. Gently rock the wash buffer and membrane for 10 minutes. Discard the waste in a special radioactive waste container.
3.3.56.
Repeat the same wash two more times or until background is significantly diminished, as being checked using a Geiger counter.
3.3.57.
Using 20cm × 25cm GE Phosphor screen (or similar screen), expose membrane for 1 day and image on a Typhoon Phosphorimager FLA7000 (or similar brand). (See Fig. 4A for examples of images).
3.3.58.
To calculate the amount of BIR product present in the culture at each time point, the intensity of BIR product (345-kb- band) hybridizing to ADE1-specific probe should be measured using ImageQuant (GE Healthcare Life Sciences). In addition, the intensity of an intact, truncated copy of chromosome III (243-kb long) that also hybridizes to ADE1-specific probe and that is present at the time “0” should be also calculated. To account for variation in DNA loads, intensities of the bands corresponding to the intact chromosome III, as well as to the repaired chromosome III, should be normalized to intensities of the bands corresponding to chromosome I, which also hybridizes to the ADE1-specific probe. The efficiency of BIR repair, presented as the percentage of truncated chromosome III that was converted to BIR product, should be calculated by dividing the normalized intensity of a repair band by the normalized intensity of uncut, truncated chromosome III. Results of three time-course experiments can be used to calculate the average +/− SD BIR efficiency at each time point for each strain. The intensities of the template (donor) Chr. III molecule ((355-kb- band) at each time point (visualized by hybridization with radioactively labeled ADE3 probe) should be normalized to intensities of the bands corresponding to chromosome VII, which also hybridizes to the ADE3-specific probe. In wild-type cells the amount of the donor DNA should remain constant throughout the course of BIR. However, in some mutants, for example in srs2Δ, the amount of the donor is drastically decreased following the initiation of BIR (for example, at 8h time point following the initiation of BIR) (Fig. 4A, B)[9]. This decrease of donor molecules in the agarose gel in srs2Δ is indicative of the accumulation recombination intermediates as branched DNA structures. The percent decrease of the donor DNA (Fig. 4B) can be used to estimate the amount of branched lethal intermediates formed in the course of BIR.
Acknowledgements
A.M is supported by R35GM127006 grant from NIGMS, and R03ES029306 R21 ES030307 from NIEHS. S.A.R is supported by R01 award CA218112 from the National Cancer Institute.
4. Notes
Sc-Leu dropout media is used for the inoculum to eliminate cells that underwent spontaneous BIR and therefore became Leu− prior to the beginning of experiment.
The lack of glucose in the media enables the efficient galactose uptake and the maximum HO induction, and therefore DSB initiation. It is expected that more than 90% of cells will undergo HO-induced DSB in less than 20 minutes following galactose addition.
Loss of viability after the DSB induction is rare due to the design of the disomic system that contains two chromosomes III (Fig. 2A). These cells remain viable even when repair of the one (broken) chromosome is unsuccessful and it is lost. However, the loss of viability can occur due to more complex events affecting both recombining chromosomes [9].
It is important to mention that not all Ade+ Leu− events have been proven to be BIR. Some Ade+ Leu− outcomes contain gross chromosome rearrangements (GCRs). BIR can be distinguished from GCR outcomes using CHEF gel electrophoresis. In particular, BIR events without chromosomal rearrangements contain two copies of chromosome III: a 355kb chromosome that hybridize to ADE3-specific probe and a 345kb chromosome that hybridize to an ADE1-specific probe. BIR outcomes with rearrangements are defined as having two copies of Chr. III where at least one of the copies deviate from its expected size (see Fig. 2B for examples). The fraction of GCRs among Ade+Leu− should be taken into account when calculating BIR efficiency (see in [6] for details).
Typically for wild-type strains, 5 mL samples are collected at 7 hours. However, for certain mutants, more cells need to be collected due to a lower mutation rate.
The ura3-29 mutation located at 90kb position centromere distal from MATα-inc can revert to a Ura+ phenotype via C to T, C to G, or C to A base substitutions [17].
The best way to determine mutation rate during S-phase is to use a no-DSB control strain. A no-DSB strain has a genotype similar to AM1003 except it contains Matα-inc-LEU2-tel and in these strains DSBs cannot be induced by HO endonuclease.
Due to the accumulation of mutations over several generations, the spontaneous mutagenesis rate (the rate of mutations during S-phase replication) is calculated using Drake equation: μ=0.4343 f/log (Nμ), where μ is the rate of spontaneous mutagenesis, f is mutation frequency, and N is the number of cells in yeast culture [23]. However, during BIR, the cells are arrested at G2/M by DSB induction until they are repaired, and then they recover from the arrest. This means that there are no cell divisions between 0 hour and 7 hour time points following galactose addition. This is why the mutation rate during BIR can be calculated using a modified Drake equation. (Mutation Rate during BIR = Mutation frequency (7 hour)- Mutation frequency (0 hour)).
It is expected that in ung1Δ cells transformed with pSR419 (empty vector), the S-phase Ura+ rate will be approximately 1 × 10−7, while the BIR Ura+ rate will be approximately 1 × 10−6, while in the cells containing pSR435 (A3A vector), the S-phase and BIR Ura+ rates are expected to be approximately 1 × 10−5 and 5 × 10−4 respectively (Fig. 3A).
The tubes can also be kept overnight at ambient temperature. In addition, ice bath can be prepared at this point.
Don’t disturb pellet. It may help to filter through sterile cheesecloth.
During this time, you can prepare napkins on lab bench.
Be careful not to lose the pellet. Use pipette to remove rest of the liquid. Place the tip of the pipette on the bottom of the tube next to the pellet.
You may need to split the sample before spinning –For example, split into 4–1.5 Eppendorf tubes ~ 0.5mL in each).
The procedure can be streamlined by assembling all the steps into a single workflow. It is also possible to perform all the steps with some tools available in open access, including Bowtie (for the alignments), and GATK (for mutation calling).
The Cluster Finder program uses the filtered mutation calls list from the previous step as an input. The mutation table needs to be reformatted first according to author’s instructions. Enter the path to the modified table into the control script and then execute the control script to launch the cluster search program.
DSB formation should occur within the first hour following addition of galactose.
BIR repair products appear usually at 4–6 hours following galactose addition (DSB induction).
To prevent degradation of zymolyase, the LMP agarose mix should not be hot at the time of mixing with zymolyase.
While transferring cell mixture, with LMP agarose and zymolase, into plug molds avoid air bubbles in the mold so as to have a solid plug.
A pressurized stream of air can also be used to remove plugs from the plug mold as an alternative to a toothpick.
For long term storage of the plugs, the volume of storage solution should be at least half of the storage tube to prevent freezing of the plugs as well as to protect against breaking of the plugs.
While you are pouring the gel into the casting tray, make sure that the holes in each corner of the black plate are filled without air bubbles.
When removing the gel from the casting tray, make sure to remove as much excess gel from the edges and bottom of the black plate.
For different CHEF machines the optimal running parameters to separate the chromosomes of interest vary.
References:
- 1.Mehta A and Haber JE, Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol, 2014. 6(9): p. a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saini N, et al. , Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature, 2013. 502(7471): p. 389–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson MA, et al. , Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature, 2013. 502(7471): p. 393–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donnianni RA and Symington LS, Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci U S A, 2013. 110(33): p. 13475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakofsky CJ, et al. , Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep, 2014. 7(5): p. 1640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vasan S, et al. , Cascades of genetic instability resulting from compromised break-induced replication. PLoS Genet, 2014. 10(2): p. e1004119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakofsky CJ, et al. , Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Mol Cell, 2015. 60(6): p. 860–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith CE, Lam AF, and Symington LS, Aberrant double-strand break repair resulting in half crossovers in mutants defective for Rad51 or the DNA polymerase delta complex. Mol Cell Biol, 2009. 29(6): p. 1432–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elango R, et al. , Break-induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat Commun, 2017. 8(1): p. 1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malkova A, Ivanov EL, and Haber JE, Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc Natl Acad Sci U S A, 1996. 93(14): p. 7131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malkova A, et al. , RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol Cell Biol, 2005. 25(3): p. 933–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrow DM, Connelly C, and Hieter P, “Break copy” duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics, 1997. 147(2): p. 371–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis AP and Symington LS, RAD51-dependent break-induced replication in yeast. Mol Cell Biol, 2004. 24(6): p. 2344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deem A, et al. , Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics, 2008. 179(4): p. 1845–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts SA and Gordenin DA, Clustered and genome-wide transient mutagenesis in human cancers: Hypermutation without permanent mutators or loss of fitness. Bioessays, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elango R, et al. , Repair of base damage within break-induced replication intermediates promotes kataegis associated with chromosome rearrangements Nucleic Acids Res, 2019. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shcherbakova PV and Pavlov YI, 3’-->5’ exonucleases of DNA polymerases epsilon and delta correct base analog induced DNA replication errors on opposite DNA strands in Saccharomyces cerevisiae. Genetics, 1996. 142(3): p. 717–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoopes JI, et al. , Avoidance of APOBEC3B-induced mutation by error-free lesion bypass. Nucleic Acids Res, 2017. 45(9): p. 5243–5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan K, et al. , An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet, 2015. 47(9): p. 1067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saini N, et al. , The Impact of Environmental and Endogenous Damage on Somatic Mutation Load in Human Skin Fibroblasts. PLoS Genet, 2016. 12(10): p. e1006385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook J and Russell DW, Southern blotting: capillary transfer of DNA to membranes. CSH Protoc, 2006. 2006(1). [DOI] [PubMed] [Google Scholar]
- 22.Church GM and Gilbert W, Genomic sequencing. Proc Natl Acad Sci U S A, 1984. 81(7): p. 1991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deem A, et al. , Break-induced replication is highly inaccurate. PLoS Biol, 2011. 9(2): p. e1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]