

Abstract
Anthrax Vaccine Adsorbed (AVA, BioThrax™) is the only Food and Drug Administration (FDA) approved vaccine for the prevention of anthrax in humans. Recent improvements in pre-exposure prophylaxis (PrEP) use of AVA include intramuscular (IM) administration and simplification of the priming series to three doses over 6 months. Administration IM markedly reduced the frequency, severity and duration of injection site reactions. Refinement of animal models for inhalation anthrax, identification of immune correlates of protection and cross-species modeling have created opportunities for reductions in the PrEP booster schedule and were pivotal in FDA approval of a post-exposure prophylaxis (PEP) indication. Clinical and nonclinical studies of accelerated PEP schedules and divided doses may provide prospects for shortening the PEP antimicrobial treatment period. These data may assist in determining feasibility of expanded coverage in a large-scale emergency when vaccine demand may exceed availability. Enhancements to the AVA formulation may broaden the vaccine’s PEP application.
Keywords: Anthrax, vaccines, BioThrax, Bacillus anthracis, antibody formation, post-exposure prophylaxis, pre-exposure prophylaxis, humans, nonhuman primate, correlates of protection
Introduction
Anthrax is a disease caused by the bacterium, Bacillus anthracis, that forms extremely resilient endospores which have the potential to be used as a biological weapon [1]. Anthrax Vaccine Adsorbed (AVA, BioThrax®, Emergent BioSolutions, Lansing, MI) is the only US FDA-approved vaccine in the United States for prevention of anthrax in humans. Licensed for preexposure prophylaxis (PrEP) in 1970 and for postexposure prophylaxis (PEP) in 2015, the AVA is prepared from undefined sterile culture filtrates of the toxigenic, nonencapsulated B. anthracis V770-NP1-R grown under microaerophilic conditions in a chemically defined protein-free medium. The final product formulation contains 1.2 mg/mL aluminum adjuvant (as aluminum hydroxide) in 0.85% sodium chloride with preservatives of 25 μg/mL benzethonium chloride and 100 μg/mL formaldehyde. The vaccine contains no live or dead bacteria. The primary immunogen in AVA is anthrax toxin protective antigen (PA) (http://www.fda.gov/downloads/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/LicensedProductsBLAs/UCM074923.pdf). The B. anthracis V770-NP1-R production strain secretes only low levels of anthrax toxin components edema factor and lethal factor in comparison to the PA – an aspect considered important in reducing reactogenicity [2].
The first large-scale use of AVA for PrEP was in 1991 for US military personnel deployed during the Persian Gulf War. In 1998, the Department of Defense (DoD) began the Anthrax Vaccine Immunization Program (AVIP), a mandatory anthrax PrEP vaccination program to protect US forces assigned to areas deemed to be at high risk for weaponized B. anthracis attack. Early in the AVIP, some members of the armed forces expressed concern that the vaccine itself might be responsible for potential later onset and chronic health effects, and that the mandatory program put them at unnecessary risk [3,4]. Facing concerns over both the need to protect military personnel against the threat of biological weapons and the fears about the vaccine, the US Congress directed the Centers for Disease Control and Prevention (CDC) to develop the Anthrax Vaccine Research Program (AVRP) to study the safety and immunogenicity of AVA [5] (Figure 1). Additionally, the AVRP was tasked with determining the immune correlates of protection (COP) of AVA and documenting vaccine efficacy. The AVRP expanded upon the DoD pilot studies of dose and schedule optimization by undertaking the largest ever prospective study of AVA safety and immunogenicity in a diverse study population [6]. A focus of the AVRP was a 43-month, randomized, double-blind, phase 4, placebo-controlled human clinical trial (clinicaltrials.gov identifier NCT00119067) [7,8]. The objectives of the clinical trial included assessing the safety and serological noninferiority of reduced schedules and a comparison of subcutaneous (SC) and intramuscular (IM) routes of administration. The AVRP human clinical trial started enrollment in 2002 and included as its minimum schedule an IM priming schedule of three doses, at Months 0, 1, and 6 (3-IM), with a 3-year (Month 42) booster (4-IM). CDC’s AVRP was initiated in 1999, before the intentional release of B. anthracis spores in the fall of 2001 which resulted in 5 deaths from inhalation anthrax and the possible exposure of more than 30,000 people [4,5]. The nation’s experience of civilian bioterrorism confirmed the urgency of the research that CDC had already planned and demonstrated the need for studies related to the possible PEP use of anthrax vaccine in the civilian population following exposure to B. anthracis spores.
Figure 1. Timeline of key activities and licensure changes for Anthrax Vaccine Adsorbed from 1955–2015.
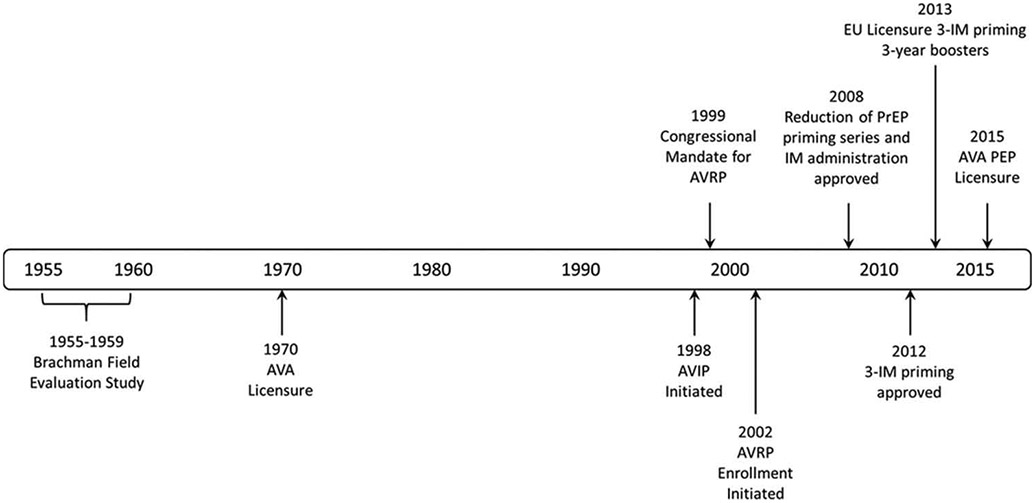
1955–1959 – Field evaluation of a predecessor anthrax vaccine by Brachman et al. [60]. Vaccine effectiveness to prevent both inhalation and cutaneous anthrax combined was 92.5% (lower 95%CI, 65%).
1962–1974 – CDC surveillance data on the occurrence of anthrax disease in at-risk industrial settings. Data were supportive of the effectiveness of AVA and an earlier version of anthrax vaccine. No cases of anthrax occurred in fully vaccinated subjects although the risk of infection continued (http://www.gpo.gov/fdsys/pkg/FR-2004-12-29/html/04-28322.htm).
1998 – Initiation of the DoD Anthrax Vaccine Immunization Program (AVIP), for mandatory vaccination of service members.
1999 – Announcement of congressional mandate for the CDC Anthrax Vaccine Research Program (AVRP).
2002 – Enrollment in the AVRP initiated.
2008 – AVRP interim analysis; FDA approved change of route of AVA administration to IM and reduction of PrEP priming series to 5 doses over 18 months, with annual boosters.
2012 – AVRP final analysis; FDA approved change to modify PrEP priming series to 3 IM doses over 6 months, with boosters at 12 and 18 months, and annually thereafter.
2013 – First approval to market AVA in the European Union under a 3-IM priming series and 3-yearly booster schedule (http://bioprepwatch.com/stories/510510714-emergent-biosolutions-receives-approval-to-market-anthrax-vaccine-in-germany).
2015 – AVA approved for PEP indication; first use of the ‘animal rule’ for licensure of a vaccine (http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm399217.pdf
Safety and reactogenicity
Short-term adverse events
Injection-site and systemic reactions have occurred with the AVA vaccine; however, these short-term reactions associated with AVA must be compared to other vaccines in common use today. Sources of AVA safety data include pre-licensure data, reports to the Vaccine Adverse Event Reporting System (VAERS), clinical trials, and observational studies that have examined immediate or short-term adverse events (AEs) (e.g. hours or days) that occurred after vaccination [9,10]. The majority of these AEs were local reactions (e.g. erythema, swelling, pain or tenderness, itching, and nodules) or mild, self-limited systemic symptoms (fever, chills, myalgia, arthralgia, and malaise). In 2002, the Institute of Medicine (IOM) committee found the most frequently reported AEs were reactions at the injection site such as redness, itching, swelling, or tenderness [4] and that a smaller number of individuals developed systemic AEs (fever and malaise). These reactions and the rates at which they occur were comparable to those observed with other aluminum-containing vaccines regularly administered to adults (e.g. tetanus and diphtheria toxoids adsorbed (Td); hepatitis A; hepatitis B) (http://www.fda.gov/BiologicsBloodVaccines/Vaccines/ApprovedProducts/ucm093830.htm). Importantly, the IOM committee found no evidence that AVA recipients experience an increased risk of life-threatening or permanently disabling AEs immediately after receiving AVA [4]. A contraindication to AVA (similar to other vaccinations) is a prior severe allergic reaction (e.g. anaphylaxis) after a previous dose of AVA or a component of the vaccine. Because anthrax is a potentially fatal disease, however, it may still be necessary to immunize individuals with contraindications such as prior hypersensitivity reactions if the perceived benefit outweighs the risk [11]. Precautions for AVA administration include (1) latex allergy – the stopper of the vaccine vial contains natural rubber latex and may cause allergic reactions in latex sensitive individuals, (2) history of anthrax disease – a history of anthrax disease may increase the potential for severe local AEs after AVA administration, (3) impaired immune response – patients with impaired immune response might not be adequately immunized after AVA administration, and (4) moderate or severe acute illness – in a standard preexposure vaccination program, vaccination of persons with moderate or severe acute illness should be postponed until after recovery. In a postevent setting, risk of administering the vaccine to a person who has been exposed to B. anthracis but has moderate or acute illness should be weighed against the benefits of vaccination [11]. Vaccine may be administered to persons who have a mild illness with or without a low-grade fever.
The CDC AVRP clinical trial provided comparative data on participants receiving AVA by either the SC or the IM route. An interim analysis of the trial [7] reported data for the first 1005 participants through study Month 7. Participants who received AVA by the IM route experienced a significant (p < 0.05) reduction in occurrence of moderate and severe injection-site AEs (warmth, tenderness, itching, erythema, induration, edema, and nodules) compared with participants who received the vaccine SC, as assessed at in-clinic visits (10.2% SC vs. 7.0% IM; p = 0.04). Percent reduction of these AEs ranged from 43% for tenderness to 95% for nodule. Frequency of injection-site bruising occurred significantly less frequently with IM administration (18.2% SC vs. 8.9% IM; p < 0.001); however, this was only demonstrated when summary (diary, telephone follow-up, and in-clinic combined) data were tallied. In the final trial analysis (1563 participants, through Month 43), injection-site AEs (warmth, itching, erythema, induration, edema, nodules, and bruise) occurred at lower proportions in the IM group compared to the SC group (ranging from 28% reduction for bruise to 93% reduction for nodule) [8]. There was a significant increase in reports of arm-motion limitation noted in IM compared to SC (Odds ratio [OR] = 1.80; 95% CI, 1.37–2.38) and there was no significant difference between the IM and SC groups for subjective pain/discomfort at the injection site (OR = 1.03; 95% CI, 0.84–1.26). However, the odds of experiencing pain immediately upon injection were reduced approximately 40% for the IM versus SC groups (OR = 0.61; 95% CI, 0.51–0.73) [8]. The route of administration did not have a significant influence on systemic AEs among clinical-trial participants, with the exception of a significantly higher occurrence of generalized muscle ache observed among the IM versus the SC groups (6.7% among IM compared to 5.3% for SC recipients; OR = 1.59; 95% CI, 1.13–2.23). No significant difference in fatigue was observed between IM and SC recipients (8.6% in IM compared to 10.6% in SC recipients; OR = 0.80; 95% CI, 0.62–1.03). Similar to other studies of vaccine AEs, and regardless of route of administration, women were noted to be almost twice as likely as men to experience any injection-site AE (OR = 1.90; 95% CI, 1.63–2.21). However, the absolute differences between males and females for warmth, tenderness, itching, pain, erythema, induration, edema, bruise, and nodules (all except arm-motion limitation) were largest amongst the SC recipients. Females were also significantly more likely to experience greater pain immediately upon injection (OR = 1.91; 95% CI, 1.64–2.22). When limited to participants’ in-clinic safety data, solicited systemic AEs occurred significantly more often in females compared to males including fatigue (OR = 1.39; 95% CI, 1.11–1.74), muscle ache (OR = 1.40; 95% CI, 1.10–1.77), and headache (OR = 1.87; 95% CI, 1.45–2.42) and these findings were not influenced by route of administration. The AE summary data also supported these findings and also demonstrated that occurrence of fever was not significantly different between males and females (OR = 1.26; 95% CI, 0.86–1.85). Serious AEs occurring in study participants were all considered unrelated or unlikely related to receipt of AVA. The safety profile of IM administered AVA was similar to other aluminum-containing vaccines such as Td, hepatitis A, and hepatitis B, with AEs typically limited to injection-site reactions such as erythema and nodule formation [8,12].
Long-term (Chronic) AEs
Safety concerns expressed for AVA included potential, rare, long-term (chronic) adverse health effects (e.g. cancer or infertility) after receiving the vaccine. However, the 2002 IOM committee’s review found no convincing evidence of elevated risk of developing chronic adverse health effects, although it indicated that data are limited in this regard, as they are for all vaccines [4]. DoD has published several studies of long-term health effects among vaccinated and unvaccinated military personnel [13-17]. Additional studies have assessed the long-term health of vaccinated researchers and fertility parameters for vaccinated males [18,19]. None of the studies found that the risk for adverse health effects or chronic diseases was higher after AVA vaccination.
In follow-up to the IOM committee’s recommendation that CDC analyze safety data on AVA contained in the Defense Medical Surveillance System (DMSS) database, CDC established the Vaccine Analytic Unit (VAU) in collaboration with DoD and with participation by FDA [20]. The DMSS database includes information on health outcomes and vaccines administered to active-duty and reserve service personnel. In a matched case-control study of US military personnel from January 1998 through December 2003, that assessed vaccine exposure in 6-, 12-, and 18-week intervals before disease onset, there were no significant associations found between optic neuritis and previous receipt of AVA, smallpox, hepatitis B, or influenza vaccines [21]. A retrospective study of an active US military personnel component cohort aged 17–35 years with a first-time diagnosis of type 1 diabetes mellitus between January 2002 and December 2008 was also completed. The study found no significant increased risk of type 1 diabetes mellitus after vaccination with AVA, smallpox vaccine, typhoid vaccine, hepatitis B vaccine, MMR, or yellow fever vaccine [22]. A recent VAU study to assess rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) after AVA vaccination found a statistically significant association with recent onset RA, but no increase in the risk of developing either RA in the long-term or SLE (Bardenheier et al., in press). While these observations may suggest that AVA is a potential trigger for RA disease onset, additional studies are needed to test this hypothesis, including assessing possible autoimmune pathways that can be triggered by epitopes present in the vaccine. Coadministering AVA and other vaccines is a common practice in the US military population. The VAU conducted a study of potential nonspecific AVA associations by comparing hospitalization risk in the active component from January 1998 through December 2003 [23]. Analysis of the automated study sample of 19,743 persons having concurrent receipt of multiple vaccinations did not identify an elevated risk of hospitalization. Additional studies in two separate civilian AVA-vaccinated populations assessed the potential for AVA vaccination to contribute to long-term impairment of physical and mental status. These study cohorts were (1) participants in the CDC Laboratory Response Network (437 vaccinated and 139 unvaccinated control subjects followed for 30 months) and (2) subjects enrolled in the AVRP clinical trial (1562 participants followed for 42 months). No evidence of an association between AVA vaccination and altered health-related quality of life was found in either evaluation [24,25].
After the 1991 Gulf War, many veterans reported chronic nonspecific multisymptom illness, with fatigue, neurocognitive complaints, and musculoskeletal symptoms, that they believed were associated with wartime exposure and remain unexplained. Multiple vaccine exposures, and in particular anthrax vaccination, was suggested as the potential etiology [26]. Mandatory anthrax vaccination also contributed to decisions by Air National Guard and Air Force Reserve personnel to leave military service or move to inactive status. More than 400 service personnel were reported to have refused vaccination with AVA and left military service voluntarily or involuntarily [4]. Speculation increased following the publication of an independent research study that suggested multisymptom illnesses were consistent with autoimmune disease presentations, likely triggered by exposure to squalene, although there was no evidence that squalene was added to either AVA or any other vaccine used in the military in the Gulf War Era [27]. The study was recognized as ‘inconclusive’ by the IOM [28]. Despite the US General Accounting Office publication of a detailed and specific account of AVA development and DoD policy regarding the use of adjuvants, concerns persisted regarding the potential for vaccine-induced illnesses (http://www.gao.gov/archive/1999/ns99005.pdf) [29]. Subsequently, assays for quantifying squalene antibodies were developed [30], and upon blinded testing of stored sera from veteran navy construction workers (Seabees) reporting multisymptom illness after Gulf War deployment, no association was found between squalene antibody status and chronic multisymptom illness [31].
Sex- and race-related reactogenicity
Several reviews have noted that women report a higher proportion of certain AEs after AVA vaccination SC compared to men [4,10,32]. A comprehensive review of VAERS AVA data in 2004 demonstrated that women were 3 times more likely than men to have or report an AE [32]. Women were not more likely to be hospitalized than men because of AEs, although women accounted for a greater proportion of reported injection-site AEs and moderate or extensive injection-site inflammation. This phenomenon has also been documented with other vaccines [33]. The apparent sex differences in reactogenicity for vaccines remain unexplained. Klein et al. have suggested a potential role of sex-based differences in immunity and effects of sex steroids [34]. To assess potential biological reasons for this finding, a study conducted in the CDC’s VAU found in AVA-vaccinated women that obesity was associated with arm soreness and that decreased prevaccination serum progesterone levels were associated with an increased rate of arm swelling [35]. Elevated prevaccination anti-PA IgG was associated with an increased frequency of itching on the arm, and in obese women, an increase in the occurrence of arm swelling, lump or knot, redness, and warmth. The report also suggested that administering AVA according to a woman’s menstrual phase may reduce the occurrence of certain injection-site reactions. In a follow-up study, female AVRP clinical-trial participants were assessed for potential associations between postvaccination serum progesterone level, race, and body mass index on AEs and antibody responses [36]. While this study confirmed the earlier finding that female participants who received SC injections had significantly higher proportions of itching, redness, swelling, tenderness, and warmth compared to the IM group, it also found the proportions of redness, swelling, tenderness, and warmth were all significantly lower in black participants versus nonblack participants. In addition, arm-motion limitation, itching, pain swelling, and tenderness were more likely to occur in participants with the highest anti-PA IgG concentrations. In the SC study group, redness and swelling were more common for obese participants compared to non-obese participants. Females had significantly higher proportions of all AEs. In the follow-up study, menstrual phase was not associated with any AEs. Thus, female and nonblack participants had a higher proportion of AVA associated AEs and higher anti-PA IgG concentrations. The study concluded that antibody responses to other vaccines may also vary by sex and race. However, further studies may provide better understanding for higher proportions of AEs in women and nonblack participants [36].
Safety and immunogenicity in special populations
Children
AVA is not FDA approved for use in children under the age of 18 years, and the safety and effectiveness of the vaccine are not established in the pediatric population. A 2004 review demonstrated that children aged <18 months experienced more local erythema and induration after receipt of vaccines containing an aluminum hydroxide adjuvant than after receipt of non-adjuvanted vaccines [12]. Erythema and induration were not reported in children aged 10–18 years, although these older children experienced local pain lasting up to 14 days after administration of vaccines containing aluminum hydroxide. Because AVA contains aluminum hydroxide, local AEs are likely to be similar to those described in adults administered AVA and in children administered other vaccines with similar aluminum hydroxide concentrations. The concentration of aluminum per dose of AVA is similar to that in the diphtheria/tetanus/pertussis (DTaP) vaccine and is less than that in the combined DTaP/polio/hepatitis B vaccine (http://www.cdc.gov/vaccines/pubs/pinkbook/downloads/appendices/B/excipient-table-2.pdf). The CDC Advisory Committee on Immunization Practices (ACIP) concluded that no evidence suggests that the risk for serious AEs after receipt of anthrax vaccine is higher in children. The morbidity of inhalation anthrax should also be considered when making decisions regarding PEP for children who have been exposed to aerosolized B. anthracis spores [11].
There are substantial ethical considerations to conducting pediatric pre-event AVA studies. A Presidential Ethics Commission proposed a potential pathway for such studies utilizing an age de-escalation process comparing safety and immunogenicity data from 18- to 20-year olds to older adults and if acceptable, proceeding to evaluations of younger adolescents. A 2015 report evaluated available data on injection-site and systemic AEs by conducting summary reanalyses from prior US-government studies with AVA. Data from 18 to 20-year olds compared to adults aged 21–29 years were analyzed. No significant difference was found in rates of these types of AEs and both age groups demonstrated similar proportions with serological responses to vaccination [37]. Because there are no data on AVA use in children, during a public health emergency, vaccine would be offered to children less than 18 years of age under an Emergency Investigational New Drug (E-IND) protocol. The E-IND requirement for informed consent presents significant operational considerations for state and local public health officials who may distribute vaccine (http://www.fda.gov/RegulatoryInformation/Guidances/ucm126491.htm).
Pregnant and postpartum women
AVA is currently a Pregnancy Category D biologic. However, there is a paucity of safety data available regarding its use during pregnancy. Although the use of inactivated vaccines is considered safe during pregnancy, current ACIP recommendations are that pregnant women should not be vaccinated against anthrax unless the potential benefits of vaccination have been determined to outweigh the potential risk to the fetus [11,38-42]. In a pre-event setting, vaccination should be deferred until after pregnancy. Nonetheless, in settings where the potential benefits of vaccination outweigh the potential risks, such as during an anthrax emergency event, AVA vaccination is recommended [11,43]. DoD policy is to exempt pregnant military women from anthrax vaccination. Despite the exemption, some military women are inadvertently vaccinated with AVA while pregnant. A study of approximately 115,000 live births to military women suggested that infants (n = 3465) born to women who received anthrax vaccine in their first trimester of pregnancy had slightly higher odds of experiencing birth defects than infants born to never-vaccinated women (OR = 1.2; 95% CI, 1.02–1.42) or to women vaccinated only after pregnancy (OR = 1.02; 95% CI, 1.01–1.43) [42]. However, there were some important study limitations to consider. When infants born to women vaccinated during their first trimester were compared with infants born to women vaccinated outside the first trimester, no statistical association with birth defects was found (OR = 1.18; 95% CI, 1.00–1.41). Of the 10 specific defects assessed in this study, only atrial septal defect (ASD) demonstrated a significant increase among infants born to women vaccinated during the first trimester. These findings were also limited, partly because the code for ASD (per the International Classification of Diseases, Ninth Revision, Clinical Modification) is the same as the code for patent foramen ovale, a common finding in preterm infants. When preterm infants with ASD as the only reported birth defect were excluded from analyses, the association between ASD and vaccination in the first trimester was no longer statistically significant. In addition, late recognition of pregnancy, a moderate risk factor for many birth defects including ASD, might explain the number of women vaccinated during their first trimester. Following careful review of this study and other data, the ACIP concluded that AVA is safe to administer during pregnancy but recommended that pregnant women defer vaccination unless exposure to B. anthracis poses an immediate risk for disease [11]. Another military study analyzed automated health record data on 4092 servicewomen (3136 of whom received at least 1 dose of AVA) and identified a total 513 pregnancies (385 following at least 1 dose of AVA). Upon comparative analysis with AVA-unvaccinated women, these investigators found that AVA vaccination had no effect on pregnancy rate (pregnancy rate ratio = 0.94; 95% CI, 0.8–1.2), birth rate (birth OR = 0.9; 95% CI, 0.5–1.4), or adverse birth outcomes (OR = 0.9; 95% CI, 0.4–2.4); although power was limited to detect an increase in adverse birth outcomes [44]. Further reassurance of the safety of AVA is provided by a recent report of AVA-vaccinated women included in the National Smallpox Vaccine in Pregnancy Registry (NSVIPR) [45]. Although the registry’s primary function is to follow women inadvertently vaccinated with smallpox vaccine during or shortly before pregnancy to evaluate their reproductive health outcomes, approximately 65% of NSVIPR participants were found to also have inadvertently received AVA. Analyses of AVA-vaccinated and AVA-unvaccinated women who also received smallpox vaccine within 28 days prior were conducted and found that rates of adverse outcomes among the AVA-exposed group were similar to or lower than expected when compared with published reference rates and the AVA-unvaccinated population.
Breastfeeding is neither a precaution nor a contraindication to AVA vaccination and vaccination does not need to be deferred in a pre-event setting if the occupation of the breastfeeding mother poses a risk for exposure to B. anthracis [11]. No data have been collected on the use of AVA among breastfeeding women. Therefore, whether anti-anthrax antibodies are transferred through milk from mother to infant is unknown. However, data from similar vaccines indicate that this might occur, particularly for IgA antibodies [46]. No data suggest that breast feeding women or breastfed children have an increased risk for AEs after the mother receives AVA. Administration of other inactivated vaccines during breastfeeding is not medically contraindicated [47].
Other populations
AVA is not FDA approved for use in persons older than 65 years of age. Whether persons aged more than 65 have the same safety and immunogenicity profile to AVA as younger adults is unknown. In the AVRP cohort of 18–65 years of age, there was a trend in decreasing anti-PA IgG levels with increasing age, but these differences were not statistically significant [7]. Aging is associated with a decline in the normal function of the immune systems, both cellular and humoral, which often leads to a state of ‘immunosenescence’ [48,49]. In addition, specific immune responses differ in aging men and women and besides genetic factors, these differences are explained through the endocrine effects of aging in particular the greatly divergent and changing levels of sex steroid hormones and their interaction with the immune system [50]. The immunological effects of aging are due to age-related differences in both the innate and the adaptive immune systems. Recent studies have evaluated addition of the Toll-like receptor 9 (TLR9) agonist CPG 7909 to AVA as an enhancer of innate and adaptive immune responses [51,52]. CpG oligodeoxynucleotides (or CpG ODN) are short single-stranded synthetic DNA molecules that contain a cytosine triphosphate deoxynucleotide (“C”) followed by a guanine triphosphate deoxynucleotide (“G”). The “p” refers to the phosphodiester link between consecutive nucleotides. Unmethylated CpG motifs act as immunostimulants [83].
Current ACIP guidance is that all inactivated vaccines can be administered safely to persons with altered immunocompetence and the usual doses and schedules are recommended [47]. However, except for inactivated influenza vaccine, vaccination during chemotherapy or radiation therapy should be avoided if possible because antibody response may be suboptimal. Because of the risk for hematoma formation after IM injections among persons with bleeding disorders, the SC route has been retained as an option for AVA PrEP administration [11]. The limited available data concerning the effect of obesity on vaccine immunogenicity and efficacy suggest that body mass index is a factor that increases the likelihood of a poor vaccine–immune response and increased reactogenicity [35]. Currently, it is estimated that over one-third of the US population is obese and this trend is predicted to continue. It is likely that vaccines deposited into fat pads are less immunogenic and may lead to greater injection-site AEs than those deposited deeper into muscle tissue [36].
Genetic analysis of the immune response
The complexities and variations in human immune responses to vaccines are in part due to genetic polymorphisms in the major histocompatibility complex (MHC) class II and other genes [53,54]. The CDC AVRP facilitated analysis of relationships between host genetics and the PA-specific cell-mediated and humoral antibody responses. Significant race associated differences in anti-PA IgG responses have been reported, with African-Americans having lower responses than European-Americans [7,8]. Pajewski et al. evaluated associations between humoral anti-PA IgG responses and polymorphisms at MHC class I (HLA-A, -B, and -C) and class II (HLA-DRB1, -DQA1, -DQB1, -DPB1) loci [55]. Among European-Americans (n = 794), genes from tightly linked HLA-DRB1, -DQA1, -DQB1 haplotypes displayed significant overall associations with anti-PA IgG persistence at 4, 8, 26, and 30 weeks from baseline in response to vaccination with three or four doses of AVA. In particular, carriage of the DRB1–DQA1–DQB1 haplotypes (*)1501–(*)0102–(*)0602, (*)0101–(*)0101–(*)0501, and (*)0102–(*)0101–(*)0501 was associated with significantly lower anti-PA IgG levels. In carriers of two copies of these haplotypes, anti-PA IgG responses remained consistently lower, even in response to subsequent booster vaccinations. No significant associations were observed amongst African-Americans (n = 200) or for any HLA class I allele/haplotype. The involvement of HLA DR-DQ haplotypes involving component HLA-DRB1 alleles of *15:01, *01:01, or *01:02 in the magnitude of and persistence of anti-PA IgG responses was affirmed in a follow-up analysis of single nucleotide polymorphisms (SNPs) within the same cohorts [56]. Specifically, tentative associations were identified for SNPs in or near the MHC class II region, in the promoter region of SPSB1, and adjacent to allele MEX3C. Genomic copy number variant (CNV) analysis by Falola et al. explored the central role of the MHC region and identified that in African-Americans, segmental deletions spanning PRR20, PCDH17, and PCH68 genes on chromosome 13 were associated with elevated early antibody production. The CNV analysis additionally identified a genomic insertion on chromosome 17 – containing NSF, RL17, and LRRC37A genes – that was associated with elevated anti-PA IgG responses in both the European-American and African-American populations [57].
Relationships between PA-specific cell-mediated immunity (CMI) responses and HLA genotypes, haplotypes, and homozygosity were evaluated by Ovsyannikova et al. using lymphocyte proliferation (LP) as a model [58]. Limited associations were determined between individual HLA alleles or haplotypes and variable LP responses. However, the authors hypothesized ‘heterozygote advantage’ such that Individuals who were homozygous for any HLA locus demonstrated significantly lower PA-specific LP responses than subjects who were heterozygous at the eight loci assessed. In agreement with the humoral antibody studies, MHC class II (HLA-DQA1 and HLA-DQB1) homozygosity was significantly associated with an overall decrease in LP responses compared with heterozygosity at those loci. The inferences of these studies were that HLA homozygosity may adversely influence immune responses to bacterial protein antigens.
Immunological memory and duration of protection
It is frequently cited that annual AVA boosters are required to maintain immunity to anthrax [11]. However, numerous studies by Pittman and coworkers have consistently demonstrated that increasing the interval between booster vaccinations and resumption of booster vaccinations following a lapse in the schedule resulted in rapid and high anamnestic antibody responses in humans [6,59-61]. In the AVRP human clinical study at Month 42, the time point with the lowest level of serum anti-PA IgG in the group receiving only the 3-dose (3-IM) priming series, 66% of study group participants had anti-PA IgG concentrations above the lower limit of quantification of the assay (3.7 μg/mL). Consistent with Pittman et al., increasing time intervals between boosters in the AVRP generated faster and statistically superior antibody responses to the final Month 42 (4-IM) vaccination [8]. The persistence of quantifiable anti-PA IgG confirmed that 3-IM priming established long-term (42 months) antibody-secreting plasma-cell populations in humans. Furthermore, the rapid and high anamnestic antibody responses to booster doses, whether administered at either Month 18 or Month 42, were indicative of a stable population of memory B cells. In addition, in nonhuman primates (NHP) the 3-IM schedule provided statistically significant levels of protection (60–100%) at dilutions of up to 1/10 AVA for at least 4 years after the first vaccination [62]. A striking aspect of that study was that 3-IM priming elicited sustained production of functional PA-specific interferon gamma (IFN-ɣ)- and interleukin-4 (IL-4)-secreting T cells, LP responses, and memory B cells for the study duration, even at later time points when serum anti-PA IgG levels were low or undetectable. Irrespective of the humoral antibody titers at the time of infection, vaccinated NHP were able to mount rapid and protective anamnestic responses after aerosol exposure to high doses of B. anthracis spores (median, 504 LD50 equivalents; approximately 28 × 106 spores). Immunological profiles in NHP indicated a mixed Th1/Th2 response with a Th2 dominance. Analogous CMI profiles and functional persistence with rapid, high magnitude anamnestic anti-PA IgG responses were also evident in human AVA vaccinees in the AVRP (Quinn et al., in press). Collectively, these data indicate that maintaining high circulating anti-PA IgG levels by administration of annual boosters were not an absolute requirement for maintaining immunity to high infectious dose B. anthracis spore challenges in NHP, and by extension, for continued protection of humans against inhalation anthrax.
Schedule optimization – achieving more with less
Vaccine effectiveness
A major focus of AVA research in the past decade has been the reduction and optimization of immunization schedules. The study of Brachman et al. with a predecessor vaccine to AVA – the Fort Detrick formulation – used a PrEP booster schedule of 0, 2, and 4 weeks (primary series) with boosters at Months 6 and 12 and annual boosters thereafter [63]. The study cohort (n = 1330) comprised mill workers in the northeastern United States with ongoing occupational exposure to B. anthracis spores from processing raw imported goat hair. The mill environments experienced annual case rates of 0.6–1.8% (mean 1.2%), with rates as high as 6.1% in persons categorized as ‘high risk.’ Exposures in those environments were estimated at 21–2100 infectious particles per 8-h day. The study demonstrated vaccine effectiveness of 92.5% in those environments, with no reported cases of inhalation anthrax in any participants who completed the vaccination at Month 6, analogous to completing the current 3-IM priming series [8]. The challenge for schedule optimization using fewer or less frequent booster doses is therefore to demonstrate comparable effectiveness or efficacy post-priming.
Vaccine efficacy and immune COP
B. anthracis is highly clonal. In particular, the pagA gene coding for PA is highly conserved across the isolates evaluated [64]. From 124 B. anthracis isolates of diverse geographical origin there are currently 11 reported alleles of pagA, and 6 of these have mutations coding for differences in amino acid sequence [65]. This clonality is an indication that AVA, and other PA-based anthrax vaccines, will likely have broad efficacy. Indeed, AVA has been demonstrated to protect a variety of animal genera and species (guinea pigs, rabbits, cynomolgus macaques, and rhesus macaques) against challenge with geographically diverse B. anthracis isolates [66,67]. Because human anthrax has extremely low incidence in the United States, the primary efficacy data for improved schedules or optimized formulations of AVA are therefore dependent on these animal models. Most animal models of inhalation anthrax have adopted B. anthracis Ames as the challenge strain, with a target inhaled dose of 200 LD50 equivalents, the actual delivered dose being dependent on the animal genus used. The recent approval of AVA for a PEP indication was the first using this strategy [68].
In the CDC AVRP, the immunological data generated by the NHP nonclinical study were used to generate immune COP for inhalation anthrax that could be applied to humans. Vaccinated NHP received only the 3-IM priming series. The study design included a range of AVA doses from a full human dose to a 1/40 diluted dose. The objective was to modulate immunogenicity and generate a graded range of immunological data to identify COP. Groups of NHP were challenged with high levels of aerosolized B. anthracis Ames spores at study Months 12 and 30 to correspond with time points in the AVRP human booster schedule at which vaccinations were replaced by saline injection. Groups of NHP were also challenged at study Month 52; 10 months later than the final booster time point in the AVRP 4-IM human schedule.
To identify and select the optimal COP from the AVRP data, Chen et al. performed a comprehensive analysis of 21 NHP immune-response variables and ratios at multiple time points [69]. The anti-PA IgG level at the last available time point before challenge and lymphocyte stimulation index at study Months 2 and 6 were identified consistently as COP. Anti-PA IgG levels and anthrax lethal toxin neutralization activity (TNA) at Months 6 and 7 (peak) and the frequency of IFN-ɣ-secreting cells at Month 6 also had statistically significant positive correlations with survival. The ratio of PA-specific in vitro stimulated IL-4 mRNA to IFN-ɣ mRNA in peripheral blood mononuclear cells at Month 6 had a statistically significant negative correlation with survival. TNA titers had lower accuracy as a COP than did anti-PA IgG levels. Anti-PA IgG levels at both the time of exposure and at Month 7 were practicable and accurate metrics for correlating vaccine-induced immunity with protection against inhalation anthrax. The model indicated that even very low levels of circulating antibody at the time of challenge correlated with significant levels of protection.
Predicted probability of survival in humans
To assess the feasibility of COP bridging from NHP and other animal model data to humans, Fay et al. performed a meta-analysis of animal challenge studies [70]. The analysis included data from rabbits, cynomolgus macaques, and rhesus macaques. TNA-assay data were applied as a potential species-neutral assay. The analysis indicated that there was a significant effect of genus and species on bridging, which will have an unknown contribution when bridging between an animal model and humans. Several additional factors were identified that had significant impact on cross-species survival predictions and the authors suggested that these be matched as closely as possible. These factors were vaccine formulation, vaccination schedule, time of immune-response measurement, and time of infectious challenge.
Schiffer et al. applied the same cross-species logistic regression method using both TNA titers and anti-PA IgG levels – the highest accuracy COP identified by Chen et al. – to predict probability of survival in humans receiving reduced booster schedules in the 43-month AVRP study [71]. The models used included the antibody levels at the time of challenge, as well as multi-correlate models incorporating peak (Month 7) IgG or TNA plus last IgG. All models predicted high human survival probabilities for the reduced schedules from Months 7 to 43. The lowest estimates of predicted survival probabilities for the reduced schedules in humans occurred when antibody levels were lowest at Month 42, but still were 86.8% (3-IM priming only) and 95.8% (3-IM priming, Month 18 booster), compared to 98.1% in the annual booster group at the same time point. Similar to the 88% survival reported for the approved PEP indication, these data indicated that a 3-yearly and a 1- and 2-yearly booster schedule both have potential as viable alternatives to annual boosters [67].
PEP
In 2015, under the ‘animal rule,’ FDA approved a PEP indication for AVA in adults 18–65 years of age [68]. The schedule is three doses administered SC at 2-week intervals (0, 2, and 4 weeks), in conjunction with a 60-day course of antimicrobials (http://www.fda.gov/downloads/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/LicensedProductsBLAs/UCM074923.pdf). The duration of the PEP antimicrobial course derives in part from the observation of disease resulting from late germination or the persistence of spores in the lungs of surviving animals as late as 58 days post-challenge. Henderson et al. reported that 15–20% of the original spore content was still present in the lungs 42 days post-low-level exposure, decreasing to 2% by 50 days and to 0.5–1% by 75 days [72]. The Sverdlovsk anthrax outbreak of 1979 included at least 10 reported cases more than 34 days after the hypothesized release, including one case of disease onset more than 6 weeks after the release [73]. In the PEP scenario, it is considered that germinated spores would be killed by antimicrobial therapy while disease resulting from latent spore germination, occurring after cessation of antimicrobial therapy, would be prevented by a vaccination-acquired protective immune response.
The immunogenicity and safety of the PEP regimen administered SC concurrently with antimicrobials in healthy human volunteers (n = 200), 18–65 years of age, were reported by Hopkins et al. [74]. AVA was well tolerated. No related serious AEs occurred during the study, and no subjects withdrew from the study because of an AE. Tenderness and pain at the injection site were recorded most often in subject diaries following vaccination. For immunogenicity, 82% of the subjects met or exceeded the prechallenge TNA titer associated with a 70% probability of survival in rabbits and 88% probability of survival in NHPs, a survival estimate based on data from animal PrEP studies [67]. The data supported the ability of AVA to provide added protection against B. anthracis compared to antimicrobials alone.
The success of the animal models in establishing an approved PEP indication for AVA has found additional application in studies to optimize the use of AVA during an emergency response (http://emergency.cdc.gov/bioterrorism/pdf/AVA-Post-Event-Prioritization-Guidance.pdf). In 2014, the Public Health Emergency Medical Countermeasures Enterprise (PHEMCE) announced an intention to develop the animal model to support assessment of a shortened antimicrobial PEP duration following B. anthracis exposure (http://www.phe.gov/Preparedness/mcm/phemce/Documents/2014-phemce-sip.pdf). The overarching hypothesis was that once vaccine-induced protection has been achieved, antimicrobial therapy may no longer be required. The resulting study evaluated short-term efficacy of a 2-dose AVA schedule (0, 2 weeks) in cynomolgus macaques challenged with high levels of B. anthracis spores at study Week 4 (2 weeks postvaccination) – the time point at which humans would receive the third injection of the PEP schedule (Sivko et al., submitted for publication). In addition, to evaluate multifold expansion of the current AVA stockpile, the PHEMCE established a parallel-designed human clinical trial to determine the safety and immunogenicity of a 2-dose PEP schedule and the use of half-doses of vaccine. The clinical study evaluated two, two-injection SC AVA regimens (0, 2 weeks or 0, 4 weeks) with the full antigen dose and a three-injection regimen (0, 2, 4 weeks) with full and half the standard antigen amount. As anticipated using the SC route of administration, a high proportion of subjects (46–64%) developed moderately severe local reactions, with 3.3% severe reactions. The 0, 2 weeks schedule effected a more rapid onset of anti-PA IgG responses, but a 0, 4 week schedule induced higher peak levels of antibody that persisted longer. Half-dose regimens induced lower antibody responses in humans [75]. The corresponding NHP study demonstrated a vaccine dose-dependent immune response that correlated with protection against a high exposure dose B. anthracis challenge 2 weeks after the second vaccination. Cross-species analysis of these data to the human clinical trial indicated that the 0, 2-week full-dose schedule had estimated probabilities of survival at Week 4 (time of third human dose) of approximately 95% and 90% based on anti-PA IgG and TNA models, respectively. For the half-dose group the estimated probabilities of survival were approximately 91% and 83%, respectively. The corresponding lower bounds of the 95% bootstrap confidence intervals for survival based on those Week 4 immune responses all indicated greater than 70% probability of survival for the half-dose regimen. In an anthrax emergency response that required vaccination of large numbers of people, a half-dose regimen was calculated to provide 80–95% greater vaccination coverage than the full-dose PEP regimen (Stark et al., submitted for publication).
Enhanced AVA as a next generation anthrax vaccine
The logistical and programmatic challenges of deploying and administering a 3-dose PEP schedule during an emergency are significant [76]. An anthrax vaccine that required fewer doses and could accelerate the onset of protective and long-term immunity would be of considerable public health impact. Hopkins et al. and Minang et al. have reported an enhanced version of AVA designed to address these specific requirements. NuThrax™ (AV7909; Emergent BioSolutions, Lansing, MI) is AVA with the addition of CPG 7909 adjuvant [51,77]. CPG 7909 is an immunostimulatory TLR9 agonist oligodeoxynucleotide that has been demonstrated to augment Th1 type responses in humans and enhance innate and adaptive immunity [51,52,78]. AV7909 is under development as a PEP candidate vaccine and has been reported in two Phase 1 trials to enhance immune responses beyond those elicited by AVA alone [51,52,77]. In the larger of these Phase 1 studies, the safety and immunogenicity of 2 IM doses (Days 0 and 14) of 4 formulations of AV7909 with 2 IM doses of AVA and 2 IM doses of saline placebo were compared in 105 healthy adults, 18–50 years of age. The mean peak normalized TNA responses (NF50) in the AV7909 recipients were higher than for AVA. Differences between AV7909 formulations evaluated were not statistically significant. Subjects who received AV7909 (n = 73) reached peak titers 1 week sooner (Day 28) compared to AVA (Day 35). The most common AEs assessed as related to vaccination were injection-site reactions. Transient lymphopenia was observed after one dose of AV7909. No AEs of special interest (e.g. autoimmune events) were reported [77].
Minang et al. [51] confirmed that the enhanced effects of AV7909 were demonstrable in biomarkers of innate and adaptive immunity. Levels of innate immune response markers (IP-10, IL-6, and C-reactive protein (CRP)) were elevated 24–48 h after administration of AV7909, returning to baseline by Day 7. AVA alone resulted in elevated IL-6 and CRP, but not IP-10. Absolute lymphocyte counts correlated with transiently increased IP-10. PA-specific CMI IFN-γ-secreting cell frequencies were significantly higher in AV7909 recipients compared to AVA after the second immunization. The elevated anti-PA IgG responses in recipients of AV7909 compared to AVA had no direct correlations with elevated CMI responses [51]. The study concluded that early biomarkers correlated with subsequent adaptive humoral immunity but not cellular immunity. AV7909 administered IM was reported to be commonly associated with local injection-site reactions. The most common reported systemic events were headache, muscle ache, and fatigue. A nonsignificant trend to a greater frequency and severity of AEs was noted [52]. Although no serious AEs occurred that were determined to be causally related to the AV7909, one participant was withdrawn after receiving the second vaccine dose due to development of a vaccination-associated grade 2 generalized rash. Potential advantages of AV7909 may include a reduction in antigen levels required for immunization and a more rapid onset of protective immunity. This would be helpful for pre-deployment vaccination of service personnel and in the setting of PEP may shorten the currently recommended 60-day treatment with antimicrobial agents, itself leading to cost saving, conservation of the national stockpile of antimicrobials, reducing side effects, and increasing antimicrobial adherence. AV7909 is in Phase 2 human clinical studies (clinicaltrials.gov identifier NCT01770743).
ACIP
The current ACIP recommendations for AVA use were published in 2010, prior to some of the recent significant updates in the FDA-approved use of the vaccine [11]. PrEP and PEP were two scenarios considered by ACIP in which an anthrax vaccine would be used. The ACIP approach is that decisions for pre-event vaccination should be made on the basis of a calculated risk assessment or else vaccination may be considered based on an estimated/presumed risk-benefit assessment. The risk–benefit profile for pre-event vaccination for the general public was presumed to be low, and pre-event vaccination was not recommended. Based on the data available in 2009, the ACIP recommendations were for (1) PrEP – five doses 0.5 mL administered IM (0 weeks, 4 weeks, 6 months, 12 months, and 18 months) and an annual booster to maintain immunity and (2) PEP – three doses 0.5 mL SC (0, 2, and 4 weeks) administered under an Investigational New Drug protocol or an Emergency Use Authorization in conjunction with a 60-day course of antimicrobials. The latter recommendation is in agreement with the recently FDA-approved indication. Vaccination of pregnant women and children, in particular, is not recommended unless there is an assessment of high risk of exposure to B. anthracis [11]. A reevaluation by the ACIP to incorporate recent data on the use of anthrax vaccine in the United States is merited.
Expert commentary
AVA is safe and effective for the approved indications. It is the only approved anthrax vaccine for human use in the United States. Since the mandatory DoD vaccination policy of the late 1990s and the intentional release of B. anthracis spores in the fall of 2001, enhancements in the safety profile and a better understanding of AVA-induced immunity have been actively pursued. Beginning in 2008, the approved use of AVA in humans has undergone substantial data-driven improvements; the first changes since its licensure in 1970. The PrEP schedule changes include the use of the IM route of administration to reduce the frequency, duration, and severity of injection-site AEs; elimination of a dose at Week 2 in the priming series; and completion of immunological priming using three doses over 6 months. The changes were based on serological noninferiority analyses at key time points in the PrEP vaccination schedule in humans. Accurate COP have subsequently been determined in NHP models of inhalation anthrax. These models have been applied for estimation of predicted probability of survival in humans and were factors in the FDA approval for a PEP indication. Acceptance of animal model data for FDA approval of human indications may lead the way to further reductions in the PrEP booster schedule. The focus on priming series optimization, IM administration, and reduction of booster frequency for PrEP, together with assessment of using fewer doses, half-doses, and the inclusion of CpG 7909 adjuvant for PEP, has demonstrated the potential for achieving rapid, effective, and lasting immunity using less vaccine, or fewer doses.
Five-year view
The resurgence of interest in anthrax vaccine research and development since 2001 has resulted in many innovative technologies and new approaches to create an anthrax vaccine that is safe, effective, defined, and that has both rapid onset and long-duration immunity against inhalation anthrax (reviewed in [79]). However, improvements in the safety and immunogenicity of an anthrax vaccine in the United States have thus far been the purview of AVA; investments in next generation anthrax vaccines have yet to generate a new FDA-approved product. The increased body of knowledge on 3-IM AVA priming and duration of protection in NHP, comparative immunogenicity in humans and animals, and the application of cross-species models of vaccine efficacy to estimate survival probabilities in humans are noteworthy advances. Preparation for a large-scale anthrax emergency continues to be a priority activity for the PHEMCE. AVA research over the next 5 years is likely to focus on simplification of the PrEP booster schedule and further optimization of vaccine and antimicrobial use for PEP. Emergency preparedness for special populations will continue to be of significance; priorities include pediatric populations and pregnant and post-partum women (http://pediatrics.aappublications.org/content/early/2015/12/31/peds.2015-4273) [37,43,80,81]. Development of next-generation anthrax vaccines continues. Important objectives for AVA and next-generation anthrax vaccines for emergency-response applications must include assessment of single-dose schedules, perhaps with adjuvants such as CPG 7909, minimizing the cost per dose and establishing a ‘just-in-time’ capability for rapid large-scale manufacturing. Significant steps toward at least some of these objectives may already include at least one viral vector-based anthrax vaccine currently in clinical evaluation (ClinicalTrials.gov identifier: NCT01979406) and recent investments in high-yield recombinant PA produced in Pseudomonas fluorescens [82].
Key issues.
AVA (BioThrax™) is safe and effective for the approved indications.
AVA is the only FDA-approved anthrax vaccine in the United States.
Congressionally mandated CDC AVRP provided data for the first FDA-approved changes in AVA PrEP since 1970.
IM administration reduces AVA reactogenicity and improves the safety profile.
Duration of protection of 3-IM priming demonstrated to 4 years in rhesus macaques.
Immune COP in NHP identified by rigorous statistical analysis.
Cell-mediated and humoral antibody responses to AVA in humans are analogous to protective immunity profiles in NHP.
Cross-species modeling of immune COP developed and applied to human data.
Data from human and nonclinical studies indicate the feasibility of PrEP booster schedule reduction.
PEP approval of AVA in 2015 demonstrates first use of the ‘animal rule’ for vaccine approval.
There is a paucity of AVA safety and efficacy data in special populations.
There is a need for a low-cost, single-dose anthrax vaccine for emergency response.
Next-generation anthrax vaccines are still in development.
Footnotes
Declaration of Interests
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Papers of special note have been highlighted as:
•of interest
•of considerable interest
- 1.Inglesby TV, O’Toole T, Henderson DA, et al. Anthrax as a biological weapon, 2002: updated recommendations for management. JAMA. 2002;287(17):2236–2252. [DOI] [PubMed] [Google Scholar]
- 2.Johnson AD, Spero L. Comparison of growth and toxin production in two vaccine strains of Bacillus anthracis. Appl Environ Microbiol. 1981;41(6):1479–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mazzuchi JF, Claypool RG, Hyams KC, et al. Protecting the health of U.S. military forces: a national obligation. Aviat Space Environ Med. 2000;71(3):260–265. [PubMed] [Google Scholar]
- 4.Committee to Assess the Safety and Efficacy of the Anthrax Vaccine, Medical Follow-up Agency. The anthrax vaccine: is it safe? Does it work? (Joellenbeck L, Zwanziger L, Durch J, Strom BE, editors). Washington, DC: Institute of Medicine, National Academy Press; 2002. [PubMed] [Google Scholar]
- 5.Committee to Assess the Safety and Efficacy of the Anthrax Vaccine, Medical Follow-up Agency. An Assessment of the CDC anthrax vaccine safety and efficacy research program. (Joellenbeck L, Zwanziger L, Durch J, Kazmerzak K, Bailey P, Miller RE, editors). Washington, DC: Institute of Medicine, National Academy Press; 2002. [Google Scholar]
- 6. Pittman PR, Kim-Ahn G, Pifat DY, et al. Anthrax vaccine: immunogenicity and safety of a dose-reduction, route-change comparison study in humans. Vaccine. 2002;20(9–10):1412–1420. ••A pivotal pilot study demonstrating the feasibility of schedule optimization and dose reduction. This study provided the basis for the congressionally mandated CDC AVRP.
- 7. Marano N, Plikaytis BD, Martin SW, et al. Effects of a reduced dose schedule and intramuscular administration of anthrax vaccine adsorbed on immunogenicity and safety at 7 months: a randomized trial. JAMA. 2008;300(13):1532–1543. • An interim analysis of the congressionally mandated CDC Anthrax Vaccine Research Program (AVRP) that provided data for the first FDA-approved changes in AVA use for 38 years; the change in route of administration from subcutaneous to intramuscular and elimination of the priming dose at week 2.
- 8. Wright JG, Plikaytis BD, Rose CE, et al. Effect of reduced dose schedules and intramuscular injection of anthrax vaccine adsorbed on immunological response and safety profile: a randomized trial. Vaccine. 2014;32(8):1019–1028. •• Final report on congressionally mandated CDC study of safety and immunogenicity of AVA in humans provided data for regulatory action and establishes 3-dose intramuscular priming series with protection achieved at 6 months.
- 9.Niu MT, Ball R. Adverse events after anthrax vaccination reported to the Vaccine Adverse Event Reporting System (VAERS), 1990-2007. Vaccine. 2009;27(2):6654–6655. [DOI] [PubMed] [Google Scholar]
- 10.Niu MT, Ball R, Woo EJ, et al. Adverse events after anthrax vaccination reported to the vaccine adverse event reporting system (VAERS), 1990-2007. Vaccine. 2009;27(2):290–297. [DOI] [PubMed] [Google Scholar]
- 11.Wright JG, Quinn CP, Shadomy S, et al. Use of anthrax vaccine in the United States: recommendations of the advisory committee on immunization practices (ACIP), 2009. MMWR Recomm Rep. Recommendations and Reports/Centers for Disease Control. 2010;59(Rr–6):1–30. [PubMed] [Google Scholar]
- 12.Jefferson T, Rudin M, Di Pietrantonj C. Adverse events after immunisation with aluminium-containing DTP vaccines: systematic review of the evidence. Lancet Infect Dis. 2004;4(2):84–90. [DOI] [PubMed] [Google Scholar]
- 13.Downing J, Greig TW, Quattlebaum MD, et al. Assessing the safety of anthrax immunization in US army aircrew members via physical examination. J Occupational Environ Med/American Coll Occup Environ Med. 2007;49(10):1079–1085. [DOI] [PubMed] [Google Scholar]
- 14.Rehme PA, Williams R, Grabenstein J. Ambulatory medical visits among anthrax-vaccinated and unvaccinated personnel after return from southwest Asia. Mil Med. 2002;167(3):205–210. [PubMed] [Google Scholar]
- 15.Sato PA, Reed RJ, Smith TC, et al. Monitoring anthrax vaccine safety in US military service members on active duty: surveillance of 1998 hospitalizations in temporal association with anthrax immunization. Vaccine. 2002;20(17–18):2369–2374. [DOI] [PubMed] [Google Scholar]
- 16.Smith B, Leard CA, Smith TC, et al. Anthrax vaccination in the millennium cohort: validation and measures of health. Am J Prev Med. 2007;32(4):347–353. [DOI] [PubMed] [Google Scholar]
- 17.Sulsky SI, Grabenstein JD, Delbos RG. Disability among U.S. army personnel vaccinated against anthrax. J Occupational Environ Med/American Coll Occup Environ Med. 2004;46(10):1065–1075. [DOI] [PubMed] [Google Scholar]
- 18.Catherino WH, Levi A, Kao TC, et al. Anthrax vaccine does not affect semen parameters, embryo quality, or pregnancy outcome in couples with a vaccinated male military service member. Fertil Steril. 2005;83(2):480–483. [DOI] [PubMed] [Google Scholar]
- 19.Pittman PR, Coonan KM, Gibbs PH, et al. Long-term health effects of repeated exposure to multiple vaccines. Vaccine. 2004;23(4):525–536. [DOI] [PubMed] [Google Scholar]
- 20.Payne DC, Franzke LH, Stehr-Green PA, et al. Development of the vaccine analytic unit’s research agenda for investigating potential adverse events associated with anthrax vaccine adsorbed. Pharmacoepidemiol Drug Saf. 2007;16(1):46–54. [DOI] [PubMed] [Google Scholar]
- 21.Payne DC, Rose CE Jr., Kerrison J, et al. Anthrax vaccination and risk of optic neuritis in the United States military, 1998-2003. Arch Neurol. 2006;63(6):871–875. [DOI] [PubMed] [Google Scholar]
- 22.Duderstadt SK, Rose CE Jr., Real TM, et al. Vaccination and risk of type 1 diabetes mellitus in active component U.S. military, 2002-2008. Vaccine. 2012;30(4):813–819. [DOI] [PubMed] [Google Scholar]
- 23.Payne DC, Aranas A, McNeil MM, et al. Concurrent vaccinations and U.S. military hospitalizations. Ann Epidemiol. 2007;17(9):697–703. [DOI] [PubMed] [Google Scholar]
- 24.Stewart B, Rose CE, Tokars JI, et al. Health-related quality of life in the CDC anthrax vaccine adsorbed human clinical trial. Vaccine. 2012;30(40):5875–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart B, Zhang Y, Rose CE Jr., et al. Health-related quality of life in the anthrax vaccination program for workers in the laboratory response network. Vaccine. 2012;30(10):1841–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Unwin C, Blatchley N, Coker W, et al. Health of UK servicemen who served in Persian Gulf War. Lancet. 1999;353(9148):169–178. [DOI] [PubMed] [Google Scholar]
- 27.Asa PB, Wilson RB, Garry RF. Antibodies to squalene in recipients of anthrax vaccine. Exp Mol Pathol. 2002;73(1):19–27. [DOI] [PubMed] [Google Scholar]
- 28.Institute of Medicine Committee on Health Effects Associated with Exposures During the Gulf W. In: Fulco CE, Liverman CT, Sox HC, editors. Gulf war and health: volume 1, Chap. 7. Depleted uranium, sarin, pyridostigmine bromide, vaccines. Washington (DC): National Academies Press (US); 2000. [PubMed] [Google Scholar]
- 29.Steele L. Prevalence and patterns of Gulf War illness in Kansas veterans: association of symptoms with characteristics of person, place, and time of military service. Am J Epidemiol. 2000;152(10):992–1002. [DOI] [PubMed] [Google Scholar]
- 30.Matyas GR, Rao M, Pittman PR, et al. Detection of antibodies to squalene: III. Naturally occurring antibodies to squalene in humans and mice. J Immunol Methods. 2004;286(1–2):47–67. [DOI] [PubMed] [Google Scholar]
- 31.Phillips CJ, Matyas GR, Hansen CJ, et al. Antibodies to squalene in US Navy Persian Gulf War veterans with chronic multisymptom illness. Vaccine. 2009;27(29):3921–3926. [DOI] [PubMed] [Google Scholar]
- 32.Sever JL, Brenner AI, Gale AD, et al. Safety of anthrax vaccine: an expanded review and evaluation of adverse events reported to the Vaccine Adverse Event Reporting System (VAERS). Pharmacoepidemiol Drug Saf. 2004;13(12):825–840. [DOI] [PubMed] [Google Scholar]
- 33.Cook IF. Sex differences in injection site reactions with human vaccines. Hum Vaccin. 2009;5(7):441–449. [DOI] [PubMed] [Google Scholar]
- 34.Klein SL, Jedlicka A, Pekosz A. The Xs and Y of immune responses to viral vaccines. Lancet Infect Dis. 2010;10(5):338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Martin SW, Rose CE Jr., et al. Evaluation of body mass index, pre-vaccination serum progesterone levels and anti-anthrax protective antigen immunoglobulin G on injection site adverse events following anthrax vaccination in women. Pharmacoepidemiol Drug Saf. 2008;17(11):1060–1067. [DOI] [PubMed] [Google Scholar]
- 36.Pondo T, Rose CE Jr., Martin SW, et al. Evaluation of sex, race, body mass index and pre-vaccination serum progesterone levels and post-vaccination serum anti-anthrax protective immunoglobulin G on injection site adverse events following anthrax vaccine adsorbed (AVA) in the CDC AVA human clinical trial. Vaccine. 2014;32(28):3548–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.King JC Jr., Gao Y, Quinn CP, et al. Evaluation of anthrax vaccine safety in 18 to 20 year olds: a first step towards age de-escalation studies in adolescents. Vaccine. 2015;33(21):2470–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cavalcanti DP, Salomao MA, Lopez-Camelo J, et al. Early exposure to yellow fever vaccine during pregnancy. Trop Med Int Health. 2007;12(7):833–837. [DOI] [PubMed] [Google Scholar]
- 39.Czeizel AE, Rockenbauer M. Tetanus toxoid and congenital abnormalities. Int J Gynaecol Obstet. 1999;64(3):253–258. [DOI] [PubMed] [Google Scholar]
- 40.Grabenstein JD. Pregnancy and lactation in relation to vaccines and antibodies. Pharm Pract Manag Q. 2001;20(3):1–10. [PubMed] [Google Scholar]
- 41.Munoz FM, Englund JA. Vaccines in pregnancy. Infect Dis Clin North Am. 2001;15(1):253–271. [DOI] [PubMed] [Google Scholar]
- 42.Ryan MA, Smith TC, Sevick CJ, et al. Birth defects among infants born to women who received anthrax vaccine in pregnancy. Am J Epidemiol. 2008;168(4):434–442. [DOI] [PubMed] [Google Scholar]
- 43.Meaney-Delman D, Zotti ME, Creanga AA, et al. Special considerations for prophylaxis for and treatment of anthrax in pregnant and postpartum women. Emerg Infect Dis. 2014;20(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiesen AR, Littell CT. Relationship between prepregnancy anthrax vaccination and pregnancy and birth outcomes among US army women. JAMA. 2002;287(12):1556–1560. [DOI] [PubMed] [Google Scholar]
- 45.Conlin AM, Bukowinski AT, Gumbs GR. Analysis of pregnancy and infant health outcomes among women in the national smallpox vaccine in pregnancy registry who received anthrax vaccine adsorbed. Vaccine. 2015;33(36):4387–4439. [DOI] [PubMed] [Google Scholar]
- 46.Healy CM, Baker CJ. Prospects for prevention of childhood infections by maternal immunization. Curr Opin Infect Dis. 2006;19(3):271–276. [DOI] [PubMed] [Google Scholar]
- 47.CDC. General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practice (ACIP). MMWR Recomm Rep. 2011;60(2):1–64. [PubMed] [Google Scholar]
- 48.Haq K, McElhaney JE. Immunosenescence: influenza vaccination and the elderly. Curr Opin Immunol. 2014;29:38–42. [DOI] [PubMed] [Google Scholar]
- 49.Pawelec G, Goldeck D, Derhovanessian E. Inflammation, ageing and chronic disease. Curr Opin Immunol. 2014;29:23–28. [DOI] [PubMed] [Google Scholar]
- 50.Giefing-Kroll C, Berger P, Lepperdinger G, et al. How sex and age affect immune responses, susceptibility to infections, and response to vaccination. Aging Cell. 2015;14(3):309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minang JT, Inglefield JR, Harris AM, et al. Enhanced early innate and T cell-mediated responses in subjects immunized with anthrax vaccine adsorbed plus CPG 7909 (AV7909). Vaccine. 2014;32(50):6847–6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rynkiewicz D, Rathkopf M, Sim I, et al. Marked enhancement of the immune response to BioThrax(R) (Anthrax Vaccine Adsorbed) by the TLR9 agonist CPG 7909 in healthy volunteers. Vaccine. 2011;29(37):6313–6320. [DOI] [PubMed] [Google Scholar]
- 53.Garman L, Dumas EK, Kurella S, et al. MHC class II and non-MHC class II genes differentially influence humoral immunity to Bacillus anthracis lethal factor and protective antigen. Toxins. 2012;4(12):1451–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poland GA, Ovsyannikova IG, Jacobson RM, et al. Heterogeneity in vaccine immune response: the role of immunogenetics and the emerging field of vaccinomics. Clin Pharmacol Ther. 2007;82(6):653–664. [DOI] [PubMed] [Google Scholar]
- 55.Pajewski NM, Parker SD, Poland GA, et al. The role of HLA-DR-DQ haplotypes in variable antibody responses to anthrax vaccine adsorbed. Genes Immun. 2011;12(6):457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pajewski NM, Shrestha S, Quinn CP, et al. A genome-wide association study of host genetic determinants of the antibody response to anthrax vaccine adsorbed. Vaccine. 2012;30(32):4778–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Falola MI, Wiener HW, Wineinger NE, et al. Genomic copy number variants: evidence for association with antibody response to anthrax vaccine adsorbed. Plos One. 2013;8(5):e64813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ovsyannikova IG, Pankratz VS, Vierkant RA, et al. Human leukocyte antigens and cellular immune responses to anthrax vaccine adsorbed. Infect Immun. 2013;81(7):2584–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pittman PR, Cavicchia MA, Kingsbury JL, et al. Anthrax vaccine adsorbed: further evidence supporting continuing the vaccination series rather than restarting the series when doses are delayed. Vaccine. 2014;32(39):5131–5139. [DOI] [PubMed] [Google Scholar]
- 60. Pittman PR, Fisher D, Quinn X, et al. Effect of delayed anthrax vaccine dose on Bacillus anthracis protective antigen IgG response and lethal toxin neutralization activity. Vaccine. 2013;31(44):5009–5014. • A first demonstration of enhanced anamnestic responses to decreasing the frequency of booster vaccinations.
- 61.Pittman PR, Mangiafico JA, Rossi CA, et al. Anthrax vaccine: increasing intervals between the first two doses enhances antibody response in humans. Vaccine. 2000;19(2–3):213–216. [DOI] [PubMed] [Google Scholar]
- 62.Quinn CP, Sabourin CL, Niemuth NA, et al. A three-dose intramuscular injection schedule of anthrax vaccine adsorbed generates sustained humoral and cellular immune responses to protective antigen and provides long-term protection against inhalation anthrax in rhesus macaques. Clin Vaccine Immunol. 2012;19(11):1730–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brachman PS, Gold H, Plotkin SA, et al. Field evaluation of a human anthrax vaccine. Am J Public Health Nations Health. 1962;52(4):632–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Price LB, Hugh-Jones M, Jackson PJ, et al. Genetic diversity in the protective antigen gene of Bacillus anthracis. J Bacteriol. 1999;181(8):2358–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sue D, Marston CK, Hoffmaster AR, et al. Genetic diversity in a Bacillus anthracis historical collection (1954 to 1988). J Clin Microbiol. 2007;45(6):1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fellows PF, Linscott MK, Ivins BE, et al. Efficacy of a human anthrax vaccine in guinea pigs, rabbits, and rhesus macaques against challenge by Bacillus anthracis isolates of diverse geographical origin. Vaccine. 2001;19(23–24):3241–3247. [DOI] [PubMed] [Google Scholar]
- 67. Ionin B, Hopkins RJ, Pleune B, et al. Evaluation of immunogenicity and efficacy of anthrax vaccine adsorbed for postexposure prophylaxis. Clin Vaccine Immunol. 2013;20(7):1016–1026. •• A pivotal cross-species study supporting the PEP licensure indication.
- 68.Burns DL. Licensure of vaccines using the animal rule. Curr Opin Virol. 2012;2(3):353–356. [DOI] [PubMed] [Google Scholar]
- 69. Chen L, Schiffer JM, Dalton S, et al. Comprehensive analysis and selection of anthrax vaccine adsorbed immune correlates of protection in rhesus macaques. Clin Vaccine Immunol. 2014;21(11):1512–1520. • A rigorous statistical analysis to identify AVA immune correlates of protection in rhesus macaques.
- 70. Fay MP, Follmann DA, Lynn F, et al. Anthrax vaccine-induced antibodies provide cross-species prediction of survival to aerosol challenge. Sci Transl Med. 2012;4(151):151ra126. •• Establishes precedent for interspecies modeling of immune responses in animal models to generate estimates of human probability of survival.
- 71.Schiffer JM, Chen L, Dalton S, et al. Bridging non-human primate correlates of protection to reassess the anthrax vaccine adsorbed booster schedule in humans. Vaccine. 2015;33(31):3709–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Henderson DW, Peacock S, Belton FC. Observations on the prophylaxis of experimental pulmonary anthrax in the monkey. J Hyg. 1956;54(1):28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meselson M, Guillemin J, Hugh-Jones M, et al. The Sverdlovsk anthrax outbreak of 1979. Science. 1994;266(5188):1202–1208. [DOI] [PubMed] [Google Scholar]
- 74.Hopkins RJ, Howard C, Hunter-Stitt E, et al. Phase 3 trial evaluating the immunogenicity and safety of a three-dose BioThrax(R) regimen for post-exposure prophylaxis in healthy adults. Vaccine. 2014;32(19):2217–2224. [DOI] [PubMed] [Google Scholar]
- 75. Bernstein DI, Jackson L, Patel SM, et al. Immunogenicity and safety of four different dosing regimens of anthrax vaccine adsorbed for post-exposure prophylaxis for anthrax in adults. Vaccine. 2014;32(47):6284–6293. • An evaluation of AVA reactogenicity and immunogenicity in humans when using reduced schedules and divided doses for the AVA PEP schedule.
- 76.Bowman CS, Arino J, Moghadas SM. Evaluation of vaccination strategies during pandemic outbreaks. Math Biosci Eng. 2011;8(1):113–122. [DOI] [PubMed] [Google Scholar]
- 77. Hopkins RJ, Daczkowski NF, Kaptur PE, et al. Randomized, double-blind, placebo-controlled, safety and immunogenicity study of 4 formulations of anthrax vaccine adsorbed plus CPG 7909 (AV7909) in healthy adult volunteers. Vaccine. 2013;31(30):3051–3058. • Demonstrates the safety and efficacy in humans of an enhanced AVA formulation (AV7909) containing a CPG adjuvant.
- 78.Krieg AM, Efler SM, Wittpoth M, et al. Induction of systemic TH1-like innate immunity in normal volunteers following subcutaneous but not intravenous administration of CPG 7909, a synthetic B-class CpG oligodeoxynucleotide TLR9 agonist. J Immunother (Hagerstown, Md.: 1997). 2004;27(6):460–471. [DOI] [PubMed] [Google Scholar]
- 79.Saile E, Quinn CP. Anthrax vaccines. In: Bergman N, editor. Bacillus anthracis and anthrax. Hoboken (NJ): Wiley-Blackwell; 2010. p.269–293. [Google Scholar]
- 80.Meaney-Delman D, Rasmussen SA, Beigi RH, et al. Prophylaxis and treatment of anthrax in pregnant women. Obstet Gynecol. 2013;122(4):885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bradley JS, Peacock G, Krug SE, et al. Pediatric anthrax clinical management: executive summary. Pediatrics. 2014;133(5):940–942. [DOI] [PubMed] [Google Scholar]
- 82.Reed MD, Wilder JA, Mega WM, et al. Immunization with a recombinant, pseudomonas fluorescens-expressed, mutant form of Bacillus anthracis-derived protective antigen protects rabbits from anthrax infection. Plos One. 2015;10(7):e0130952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM. CpG DNA as a vaccine adjuvant. Expert Rev Vaccines. 2011. Apr; 10(4):499–511. doi: 10.1586/erv.10.174. [DOI] [PMC free article] [PubMed] [Google Scholar]