

Abstract
Introduction:
Sickle cell disease (SCD), an inherited hemoglobinopathy, affects primarily African Americans in the US. In addition, about 15% African Americans carry sickle cell trait (SCT). Despite the risk associated with blood transfusions, SCD patients have lower risk of acquiring HIV-1 infection. SCT individuals might also have some protection from HIV-1 infection.
Areas covered:
Here, we will review recent and previous studies with the focus on molecular mechanisms that might underlie and contribute to the protection of individuals with SCD and SCT from HIV-1 infection. As both of these conditions predispose to hemolysis, we will focus our discussion on the effects of systemic and intracellular iron on HIV-1 infection and progression. We will also review changes in iron metabolism and activation of innate antiviral responses in SCD and SCT and their effects on HIV-1 infection.
Expert opinion:
previous studies, including ours, showed that SCD might protect from HIV-1 infection. This protection is likely due to the upregulation of complex protein network in response to hemolysis, hypoxia and interferon signaling. These findings are important not only for HIV-1 field but also for SCD cure efforts as antiviral state of SCD patients may adversely affect lentivirus-based gene therapy efforts.
Keywords: Sickle Cell Disease, sickle cell trait, HIV-1 infection, iron metabolism, anti-viral innate immune response.
1. Introduction
Sickle cell disease (SCD) is an inherited hemoglobinopathy in which mutations in the β-globin gene (HBB) lead to the production of mutated hemoglobins of which the most prevalent are hemoglobin S (HbS, Glu6Val mutation) and hemoglobin C (Glu6Lys mutation) [1]. HbS polymerizes under low oxygen conditions leading to sickling of red blood cells (RBCs) and development of sickle cell anemia (SCA) that is characterized by hemolysis, vaso-occlusion and ischemia [2]. Clinical manifestations of SCA include recurrent pain crises and development of chronic organ damage caused by stroke, osteonecrosis, nephropathy, pulmonary disease and retinopathy [3]. SCA has also been associated with the increased risk of pulmonary hypertension, renal dysfunction, proteinuria, stroke and the overall increased mortality [4–9]. Other risk factors of SCA include left ventricular diastolic dysfunction, asplenia, cholestatic hepatic dysfunction and systemic iron overload [4,10]. SCD affects approximately 100,000 people in the U.S., primarily African Americans [11], and several million Africans, primarily in sub-Saharan Africa [2]. Worldwide, over 300,000 newborns in 2010 had SCD (75% in Sub-Saharan Africa) and by 2050 this number is expected to reach 400,000 [12].
In the United States, an additional 3 million people carry sickle cell trait (SCT) [13]. We recently estimated the frequency of HbAS to be 9.8% and HbAC - 2.7% among individuals who were screened at Howard University Center for Sickle Cell Disease during the period 2009–2018 [14]. Worldwide, SCT individuals numbered over 78 million with 65 million living in sub-Saharan Africa [15].While SCT considered to be benign, individuals with SCT have increased chances of developing chronic kidney disease, atrial fibrillation and thromboembolism [16]. SCT RBCs are prone to sickling and may sickle and hemolyze when the SCT person is oxygen deprived or dehydrated [17].
Human immunodeficiency virus (HIV) epidemic affects over 36 million people worldwide primarily from countries in South Africa and Sub-Saharan Africa [18]. Thus, one would expect some degree of interaction between SCD and HIV. Surprisingly, previous studies discussed below indicated that individuals with SCD and SCT might be protected from HIV-1 infection. There are also several reports indicating increased chances of co-infections in SCD patients living with HIV-1. Several potential underlying molecular mechanisms might explain changes in HIV-1 infection among SCD patients and SCT individuals including induction of heme and iron regulatory pathways, expression of interferons and HIV-1 inhibitory factors. The aim of our report is to review these findings in non-exhaustive manner.
2. Effect of iron metabolism on HIV-1
2.1. Iron metabolism in SCD
SCD patients experience intravascular hemolysis and are often subjected to frequent blood transfusions which, cumulatively, lead to increased iron stores and elevated levels of systemic inflammation [3]. While, typically, macrophages recycle about 20 mg of iron daily, in SCD patients, over 100 mg of iron is recycled by macrophages [19]. SCD patient’s peripheral blood mononuclear cells (PBMCs) exhibit elevated expression levels of iron regulatory genes including ferritin light and heavy chains [20] and ferroportin (FPN) [21]. Release of hemoglobin and heme leads to induction of type I interferon (IFN-I) production and recruitment of monocytes to liver in SCD mouse model [22]. Increased iron metabolism is also linked to the increased inflammation in SCD [20] that is manifested by the increased levels of pro-inflammatory cytokines IL-1β, IL-6 and IL-17 and anti-inflammatory IL-10 [23]. While previous studies indicated association of SCD-related inflammation with TLR4 signaling [24], more recent analysis showed no effect of TRL4 knock out on heme-induced inflammation in mice [22]. Taken together, SCD is manifested by significantly upregulated iron metabolism which is a major contributory factor in SCD pathogenesis.
2.2. Effect of systemic iron on HIV-1 progression
Development of AIDS is accompanied by hemochromatosis - increasing iron accumulation in macrophages, microglia, endothelial cells, myocytes, bone marrow, brain white matter, muscle, and liver [25]. Iron is required for various steps in HIV-1 life cycle [26,27]. However, the role of extracellular iron is not fully understood. Several earlier and more recent studies reported a worse outcome in patients with higher iron load. Progression of HIV-1 toward AIDS was faster in thalassemia major patients who were treated with desferrioxamine and exhibited higher levels of serum ferritin (reviewed in [28]). Also administration of oral iron to HIV-1-positive patients with Pneumocystis carinii pneumonia associated with higher mortality rates [28]. Haptoglobin 2–2 polymorphism was associated with higher iron stores and reduced survival in people living with HIV-1 (PLWH) [28]. Elevated iron levels were predictive for higher mortality in PLWH in Gambia [29]. Non-anemic HIV-1+ Zimbabwean women with high serum ferritin levels exhibited increased viral load, indicating a correlation between higher iron stores and the severity of HIV-1 infection [30]. Retrospective study in Nigeria found that higher serum iron, increased total iron binding capacity (TIBC) and increased transferrin saturation correlated with reduced CD4+ T cell levels [31]. On the other hand, HIV-1 viral load dropped dramatically in a hemochromatosis patient who underwent venesection, suggesting that reduction in iron stores might be beneficial for the control of HIV-1 replication [32]. The cART-treated HIV-1+ individuals with mutation in the hemochromatosis (HFE) gene alleles HFE 845A and HFE 187G that predispose to iron loading showed higher levels of mitochondrial DNA (mtDNA) suggesting potential compensatory response to mitochondrial stress [33]. In a cohort of cART naïve Brazilian men, iron intake correlated with increased viral load although there was no correlation with CD4+ T cells counts [34]. Taken together, these findings point to the synergy between iron loading and HIV-1 replication and suggest that elevated iron stores might enhance HIV-1 infection.
2.3. Effect of systemic iron on HIV-1 co-infections
In South African PLWH, extremely high serum ferritin levels (>1500 μg/L) were associated with Mycobacterium tuberculosis (TB) co-infections whereas in HIV-1-negative individuals hereditary hemochromatosis associated with chronic renal failure [35]. Iron supplementation for anemic Malawian children living with HIV-1 increased their CD4+ levels but also increased the incidences of malaria underscoring difficulty in iron supplementation in malaria-endemic areas [36]. In PLWH cohort from Spain, higher ferritin levels (>200 μg/L) observed in 11% of the patients correlated with immunosuppression and increased levels of C-reactive proteins and HCV infection [37]. A cross-sectional study of thalassemia major Egyptian patients who were regularly transfused showed high rate (20.7%) of hepatitis C infection (HCV) which correlated with the frequency of transfusions; however, none of the patients were HIV-1 positive [38]. Taken together, HIV-1 co-infection might be promoted by higher iron load, especially with such infections as TB or malaria and less so with HCV. HCV infection might be enhanced by high iron load in the absence of HIV-1 co-infection.
2.4. Effect of intracellular iron on HIV-1 replication
HIV-1 replication is dependent on host cell enzymes some of which require intracellular iron for their activity [26,27] (see schematics in Figure 1). HIV-1 reverse transcription, transcription, translation and viral assembly also require iron-containing enzymes [27]. HIV-1 reverse transcription requires intracellular deoxyribonucleotides (dNTPs) which are synthesized by ribonucleotide reductase 2 (RNR2) and degraded by SAM domain and HD domain-containing protein 1 (SAMHD1) [39–42] (Figure 1). RNR2 contains non-heme iron [43] and SAMHD1 active site contains iron metal ion [44]; thus both enzymes can be affected by intracellular iron levels. RNR2 downregulation in p21-expressing macrophages blocks HIV-1 reverse transcription [45]. Expression of p21 in HIV-1 elite controllers [46,47] was linked to a decrease in SAMHD1phosphorylation [48,49]. HIV-1 basal transcription is induced by NF-κB that binds to the HIV-1 LTR promoter [50]. Cellular iron efflux activates IκB kinase [51–53], inducing NF-κB release into nucleus and activation of HIV-1 transcription [54]. HIV-1 transcription is activated by HIV-1 Tat protein that recruits host CDK9/cyclin T1 kinase to HIV-1 TAR RNA [50] (Figure 1). Host CDK2/cyclin E activates HIV-1 transcription by phosphorylating CDK9 Ser-90 residue [56] and HIV-1 Tat Ser-16 residue [57,58]. Number of iron chelators inhibit HIV-1 transcription by inhibiting CDK2 and CDK9 activities [59,60] and also inducing p21 and IKBα expression levels and sequestering NF-κB [60,61]. HIV-1 Nef protein increases cellular iron levels by down regulating HFE and increasing HIV-1 Gag protein expression [62].Together, these studies indicate that down regulation of intracellular iron has negative effect on HIV-1 replication.
Fig. 1. Schematic representation of the effect of iron and anti-viral restriction factors on HIV-1 replication.

HIV-1 replication cycle steps (black arrows) include viral fusion and entry, reverse transcription, integration of the proviral DNA, transcription and export of viral RNA, translation, and viral assembly. Involvement of intracellular iron is indicated by black dotted arrows. HIV-1 reverse transcription regulation by SAMHD1 and RNR2, and upstream regulation of SAMHD1 by CDK2/cyclin E are shown. HIV-1 transcription activation by HIV-1 Tat protein that binds to HIV-1 TAR RNA and recruits host cell CDK9/cyclin T1 is shown. Also shown is the activation of CDK9/cyclin T1 by CDK2/cyclin E and induction of basal HIV-1 transcription by NF-κB. The inhibitory effect of iron chelators on CDK2/cyclin E, NF-kB, RNA export and HIV-1 translation and assembly are shown. Ferritin bound Fe3+ by transferrin receptor (TFR) and negative effect of HIV-1 Nef protein on HFE leading to the upregulation of TFR are shown. Hepcidin is shown to indicate its negative effect on FPN.
2.5. Inhibition of HIV-1 replication by increased intracellular iron
Several recent studies pointed to the inhibitory role of excessive levels of intracellular iron. Iron (II) supramolecular helicates inhibit Tat-TAR interaction in vitro suggesting that intracellular iron accumulation can inhibit HIV-1 transcription activation [64]. Iron loading of cultured cells leads to the downregulation of eIF5A expression and reduction in HIV-1 replication [65] suggesting an iron-induced inhibition of HIV-1. Deferiprone treatment reduced HIV-1 RNA in HIV-1 infected patients [66] suggesting a direct effect on viral RNA production. Deferiprone inhibits HIV-1 translation by preventing hypusine modification of eIF5α [67]. Thus, it seems that iron loading, in addition to the iron deprivation can be inhibitory for HIV-1 replication. While seemingly controversial, both low and high iron conditions are abnormal and thus might both have an adverse effect on HIV-1. Also, many of these observations need to be further validated in cultured cells and animal models.
2.6. Role of HO-1 and heme in HIV-1 replication
Heme oxygenase-1 (HO-1) plays an important role in iron recycling by macrophages that consume and degrade aged RBCs which results in the accumulation of heme in the cytoplasm and heme-activated transcription induction of host genes that include HO-1 [68]. HO-1 degrades heme into carbon monoxide (CO), biliverdin and iron (II) [68]. HO-1 expression induced by HbS and the release of CO modulate T cell activity and protects mice from cerebral malaria [69]. Thus SCT might protect against malaria in Africa [70,71]. Treatment of cultured cells with heme as well as injection of heme into humanized mice inhibit HIV-1 infection [72,73]. In contrast, hemin induces HIV-1 replication in latently infected lymphoblastic leukemia T cells, ACH-2 cells co-stimulated with TNF-alpha or ACH-2 cells co-cultured with monocyte-derived macrophages (MDM) [74]. HIV-1 activation was also associated with cytotoxicity that correlated with low levels of FPN expression, lack of HO-1 expression and iron accumulation in ACH-2 cells [74]. Ascorbate treatment of ACH-2 cells latently infected with HIV-1 facilitates Fe(II) release and activates HIV-1 provirus [75]. However, addition of CO-donor or bilirubin as well as iron chelation by desferrioxamine decreased p24 production in ACH-2 cells suggesting that HIV-1 activation required iron-mediated stress signal [75]. SIV infected myeloid cells in macaques undergo remodeling including hyper-SUMOylation and ubiquitin-mediated degradation of promyelocytic leukemia protein nuclear bodies in productively infected cells (PML NBs) [76]. PML NBs degradation is reversed upon decreasing cellular iron content [76] suggesting potential use of iron chelators as therapeutics targeting HIV-1 latency. Taken together, hemolysis and its products might prevent acute HIV-1 infection but might induce latent HIV-1 provirus, underscoring the complexity of HIV-1 regulation.
2.7. Effect of intracellular iron on HAND
HIV-1-associated neurological disorder (HAND) might be driven by the elevated iron levels in brain. Activation of HIV-1 transcription in cultured microglial cells by HIV-1 Tat added extracellularly in the presence of chloroquine was inhibited by iron accumulated in the endosomes that formed oligomers with HIV-1 Tat and prevented its release into cytoplasm [77]. HIV-1 gp120 protein induces endo lysosome de-acidification and leads to the iron efflux and increased levels of ROS in in U87MG glioblastoma cells [78]. Thus, iron accumulation in the brain of HIV-1+ individuals might promote HAND and will require careful approach for iron chelators treatment as endosome-targeted chelators could promote HIV-1 activation. As discussed above, further validation is needed with the use of animal models to confirm the in vitro observations and further clarify the effect of iron in HAND development.
Taken together, these studies highlight the importance of iron metabolism, hemolysisand intracellular iron levels for HIV-1 replication as many HIV-1 life cycle step are affected including reverse transcription, transcription, translation, and virus assembly.
3. Effect of FPN and hepcidin on HIV-1 infection.
Iron is absorbed by duodenal enterocytes and released into plasma by FPN which is expressed on the basolateral side of the duodenal enterocytes [79]. FPN is also expressed in macrophages and helps to control iron absorption and recycling [79]. While hemochromatosis in European populations is mostly associated with mutations in the HFE gene, the FPN Q248H mutation has been linked to hemochromatosis in African populations [80] and was associated with increased iron load in Zimbabwean children [81]. FPN mutations A77D, V162del and G490D showed reduced iron export which can lead to iron accumulation in macrophages [82,83]. Plasma levels of iron is regulated by hepcidin, a 25 amino acid long peptide secreted by hepatocytes [79]. Hepcidin interacts with FPN and leads to its internalization and degradation [79]. FPN is ubiquitinated by RNF217, an E3 ubiquitin ligase, which is independent from hepcidin [84]. FPN might also be ubiquitinated by UBA6, an E1 ubiquitin ligase and the adaptor protein NDFIP1, that prevents BMP6- and hepcidin- induced degradation of FPN [85]. FPN mutations Y64N, N144D, N144H and C326Y reduce its sensitivity to hepcidin, and lead to higher dietary iron uptake and high serum transferrin saturation [82]. Expression of FPN in human epithelial 293 cells leads to cellular iron deficiency and significant decrease of ferritin level and increased expression of transferrin receptor [86–88]. Rwandan women living with HIV who had FPN Q248H mutation (6% prevalence) showed higher serum ferritin, lower serum hepcidin and lower serum transferrin and also higher frequency of opportunistic infections [89]. Because FPN imports iron from duodenum into circulation and also exports iron from macrophages, its sustained expression might lead to higher serum iron predisposing to opportunistic infections. Conversely, sustained FPN expression will reduce intracellular iron storage decreasing chances of HIV-1 infection (Figure 1). In acute HIV-1 infection, hepcidin level is increased and plasma iron level is decreased [90]. Levels of hepcidin during the first two months of infection correlate with the later plasma viral load set-point [90]. Hepcidin levels remain elevated in individuals with untreated chronic HIV-1 infection and in subjects on ART, in contrast to the infections with HCV or HBV that do not induce hepcidin or hypoferremia during the primary viremia phase [90]. In a cohort of women living with HIV-1, serum hepcidin levels were lower comparing to the non-infected cohort controls, and positively correlated with levels of iron and ferritin [91]. In the same study, iron levels were found to be lower in patients with high HIV-1 viral load compared with patients with undetectable viral load, suggesting that hepcidin might be playing a role in patients with uncontrolled viral replication and that inhibitors of hepcidin can be used as supplemental therapy for HIV-1 [91]. In Chinese patients with HCV and HIV-1 co-infection, levels of hepcidin were higher than in HIV-1 infected group; however, both groups had hepcidin levels lower than in non-infected controls [92]. In vitro, expression of FPN inhibits HIV-1 transcription [93]. Also, hepcidin treatment reduces FPN expression, increases intracellular iron and induces HIV-1 replication in primary CD4+ T cells and MDM [93], further indicating that FPN expression negatively regulates HIV-1 transcription and replication. Taken together, iron metabolism is intrinsically connected to the HIV-1 infection and disease progression, and iron export by FPN and its control by hepcidin play important regulatory roles in HIV-1 pathogenesis.
4. HIV-1 infection in SCD
4.1. Lower frequencies of HIV-1 infection in SCD.
Several previous studies pointed to a possibility that SCD patients are protected from HIV-1 infection. The very first mention of SCD offering a protection from HIV was in a study of Kenyan children with SCD who had multiple blood transfusions and none of whom were HIV-1seropositive [94]. Another early study of 116 adults with SCD who were transfused with blood that was not screened for HIV-1, showed higher levels of antibodies against HTLV but not HIV-1 (2.7% versus 7.9%) [95]. Among small cohort of SCD patients with HIV-1 infection, 44% participants were long-term non-progressors who were also asplenic compared to 13.9% non-progressors in control group [96]. Spleen represents a major HIV-1 replication site, and splenectomy in HIV-1+ patients with thrombocytopenia led to sustained increase in CD4+ T cell counts with no acceleration of AIDS [97]. Analysis of 400,000 medical records ranging from 1997 to 2009 indicated lower odds of having HIV-1 infection and SCD disease comparing to other diagnoses and especially to hepatitis C infection that was higher among SCD patients [98]. A similar study found an estimated 686 hospitalizations of children with SCD and HIV infection in the United States in the 10-year period 1994–2003 with longer average hospital stay compared to children with SCD alone suggesting an adverse effect of the HIV-1 infection in SCD [99]. Potential protection of SCD from HIV-1 infection was observed among SCD patients in Democratic Republic of the Congo [100,101]. HIV-1 infected children in Nigeria showed absence of SCD and lower sickle cell trait prevalence among the study participants [102]. Recent analysis of potential SCD resistance to HIV showed that individuals with non-SCD congenital anemia had higher chances of HIV-1 infection acquisition compared to patients with SCD [103]. In the same study, analysis of 30 SCD cases and 30 non-SCD controls showed lower levels of CCR5 and CCR7 co-receptors and higher levels of CD4 receptor in CD4+ T cells from SCD patients [103]. Taken together, these studies point to a negative association between having SCD and acquiring HIV-1 infection.
4.2. Contributing factors for HIV-1 restriction in SCD
Several molecular mechanisms can explain the potential protection of SCD from HIV-1 infection including induction of heme and iron regulatory pathways [26] (detailed above), hypoxia [104], and chronic inflammation with higher levels of HIV-1 inhibitory cytokines such as IL-10 [23]. Gene expression analysis showed increased gene expression of HMOX-1 (HO-1), BLVRA (biliverdin reductase) and CDKNA1(p21) in PBMCs obtained from SCD patients in steady-state conditions [105]. Also iron-regulated genes such as GAPDH, FTL1 (ferritin light chain), ALDH1A1 (aldehyde dehydrogenase 1) and SAT2 (spermidine/spermine N1-acetyltransferase family member 2) were found to be upregulated in SCD patients [20]. HIF-1α is upregulated in hypoxia and in the cells treated with iron chelators and typically found upregulated in SCD patients [21]. We observed significant reduction of HIV-1 transcription and replication in the cells cultured under 3% oxygen concentration as compared to 21% oxygen [104]. The activity of CDK9/cyclin T1 critical for HIV-1 transcription activation (Figure 1) was reduced at 3% oxygen apparently due to the inability of CDK9 to associate with cyclin T1 [104] suggesting that hypoxia can also contribute to the HIV-1 suppression in SCD.
4.3. Inhibition of one round of HIV-1 infection in SCD-derived PBMC
To test whether SCD patients are refractory for HIV-1 infection ex vivo, we analyzed one-round HIV-1 infection in SCD PBMCs [21]. Significantly lower levels of one round HIV-1 infection, reduced p24 and HIV-1 Env mRNA levels indicated inhibition of HIV-1 replication. We also observed a marked 2.5-fold inhibition of early LTR production suggesting that HIV-1 reverse transcription was affected. Gene expression analysis of potential HIV-1 restriction factors as well selected heme and iron regulatory genes showed upregulation of NFKBIA (IκBα), HIF1A, CDKNA1 (p21), HMOX1 and SLC40A1 (FPN) [21] (Figure 2). Meta-analysis of genes expressed in SCD PBMCs indicates overexpression of RSAD2 (radical S-adenosyl methionine domain 2 protein), IFIT1 and IFIT2 (interferon induced protein with teratricopeptide repeats 1 and 2) that are known to affect HIV-1 replication [106,107]. Interestingly, treatment of SCD PBMCs by hepcidin reversed HIV-1 inhibition [21], suggesting that it was driven by FPN expression. Levels of circulating hepcidin in plasma of SCD patients were not elevated suggesting that higher levels of FPN expression could be sustained [21].
Fig. 2. Schematic representation of the SCD effect on HIV-1 replication at molecular level.

SCD conditions increase expression of host genes including NFKBIA (IκBα), HIF1A (HIF-1α), CDKNA1 (p21), HMOX1 (HO-1) and SLC40A1 (FPN). Expression of FPN leads to the reduction of intracellular iron. The lower iron levels stabilize transferring receptor (TFR) mRNA and HIF-1α and induce IκBα and p21 expression. Also, lower iron levels inhibit enzymatic activity of CDK2/cyclin E, reduces SAMHD1 phosphorylation and induces HIV reverse transcription inhibition by SAMHD1.
4.4. Reduction of intracellular iron levels, CDK2 activity and SAMHD1 phosphorylation in SCD PBMCs
Analysis of intracellular iron levels in SCD PBMCs showed reduced intracellular labile iron pool (LIP) [21], further supported by increased levels of TFR mRNA (Figure 2). Elevated expression levels of HIF1A, NFKBIA, HMOX1 and SLC40A1 (Figure 2) further indicated upregulation of hypoxia and hemolytic pathways. CDK2/cyclin E activity in SCD PBMC was reduced by more than 2-fold [21]. Accordingly, SAMHD1 phosphorylation on Thr-592, that is known to be phosphorylated by CDK2 [108], was significantly reduced in SCD PBMC [21]. SAMHD1 phosphorylation was also reduced in hemin-treated promonocytic THP-1 cells, suggesting that hemolysis can induce levels of non-phosphorylated SAMHD1 and inhibit HIV-1. While SAMHD1 knock down only partially rescued HIV-1 replication in the hemin-treated THP-1 cells, FPN knock down completely rescued HIV-1 replication in the hemin-treated THP-1 cells [21]. These observations suggest that while SAMHD1 is not the only factor counteracting HIV-1, FPN is clearly functions as an overall upstream restriction trigger controlling HIV-1 replication in SCD PBMCs (summarized in Figure 2).
4.5. Expression of pro- and anti-inflammatory factors in SCD.
SCD patients have higher levels of iron-regulatory, hypoxia-related and inflammatory proteins including significantly upregulated (>100-fold) FTL1, TLR4 (toll-like receptor 4), IL6 (interleukin-6) gene expression levels and higher levels of C-reactive protein [20]. SCD patients also have higher levels of anti-inflammatory IL-10 and pro-inflammatory IFN-γ, IL-6, IL-8 and RANTES cytokines [23]. As described in the previous section, SCD PBMCs express many interferon-inducible proteins, many of which can be induced by type 1 interferons (IFN-α/β). Recently, plasma heme levels in SCD patients were positively correlated with circulating levels of IFN-α and upregulation of the IFN-α/β inducible genes in circulating monocytes [22]. In mice treated with hemin, IFN-α/β expression occurred in liver monocyte and macrophages suggesting a novel IFN-α/β activating pathway mediated by hemolysis [22]. Among 12 subtypes of IFN-α, only IFN-α14 inhibits HIV-1 and induces expression of BST2, MX2 and APOBEC3G genes [109]. Expression of IFN-β by HIV-1 infected macrophages restricted HIV-1 infection [110]. Earlier studies also showed that transduction of macrophages with IFN-β - expressing retroviral vector led to HIV-1 resistance linked to the elevated expression of RANTES and downregulation of CCR5 co-receptor [111]. Interestingly, elevated levels of RANTES were detected in SCD patients’ plasma [23]. Taken together, SCD patients express both pro- and anti-inflammatory cytokines including type 1 and 2 interferons. Expression of some of the HIV-1 restriction factors that we and others observed in SCD PBMCs might be driven in part by IFN-α/β expression.
5. HIV-1 infection in sickle cell trait (SCT)
In the United States, around 3 million people are SCT carriers [13]. SCT children observed in a pediatric HIV program in Newark showed less frequent HIV-1 infection compared to HbAA type [112]. A study in France enrolled HIV-1+ patients who came from various sub Saharan Africa countries and found no difference in the HIV-1 clinical outcome between participants with and without SCT [113]. Also a study in Uganda that screened over 150,000 blood samples did not find any influence of SCT on HIV-1 infection [114]. In contrast, a study in Nigeria found lower SCT prevalence in individuals with HIV-1 infection suggesting SCT protection [102].
In our recent study, we analyzed ex vivo HIV-1 infection of PBMCs obtained from SCT individuals and observed moderate suppression of HIV-1 infection; the affected steps including reverse transcription, integration, and transcription [115]. This finding paralleled our pervious findings with HIV-1 infection in SCD PBMCs. Analysis of protein expression in SCT PBMCs pointed to elevated levels of HO-1 (Figure 3), while there was no changes in the expression of IKBα, p21 and FPN [115]. CDK2 activity and SAMHD1 phosphorylation were not altered. Analysis of 42 HIV-1 restriction factors only showed decreased expression of APOBEC3F, APOBEC3H, IFITM2 and TRIM5α [115]. To further understand the reason for reverse transcription inhibition, we analyzed RNR2 expression and found it be significantly reduced (Figure 3). Analysis of gene expression in HIV-1 positive SCT participants among Howard University cohort showed upregulated expression of HMOX1 and SAMHD1 mRNAs (Figure 3) [115].
Fig. 3. Schematic representation of the sickle cell trait (SCT) effect on HIV-1 replication at molecular level.
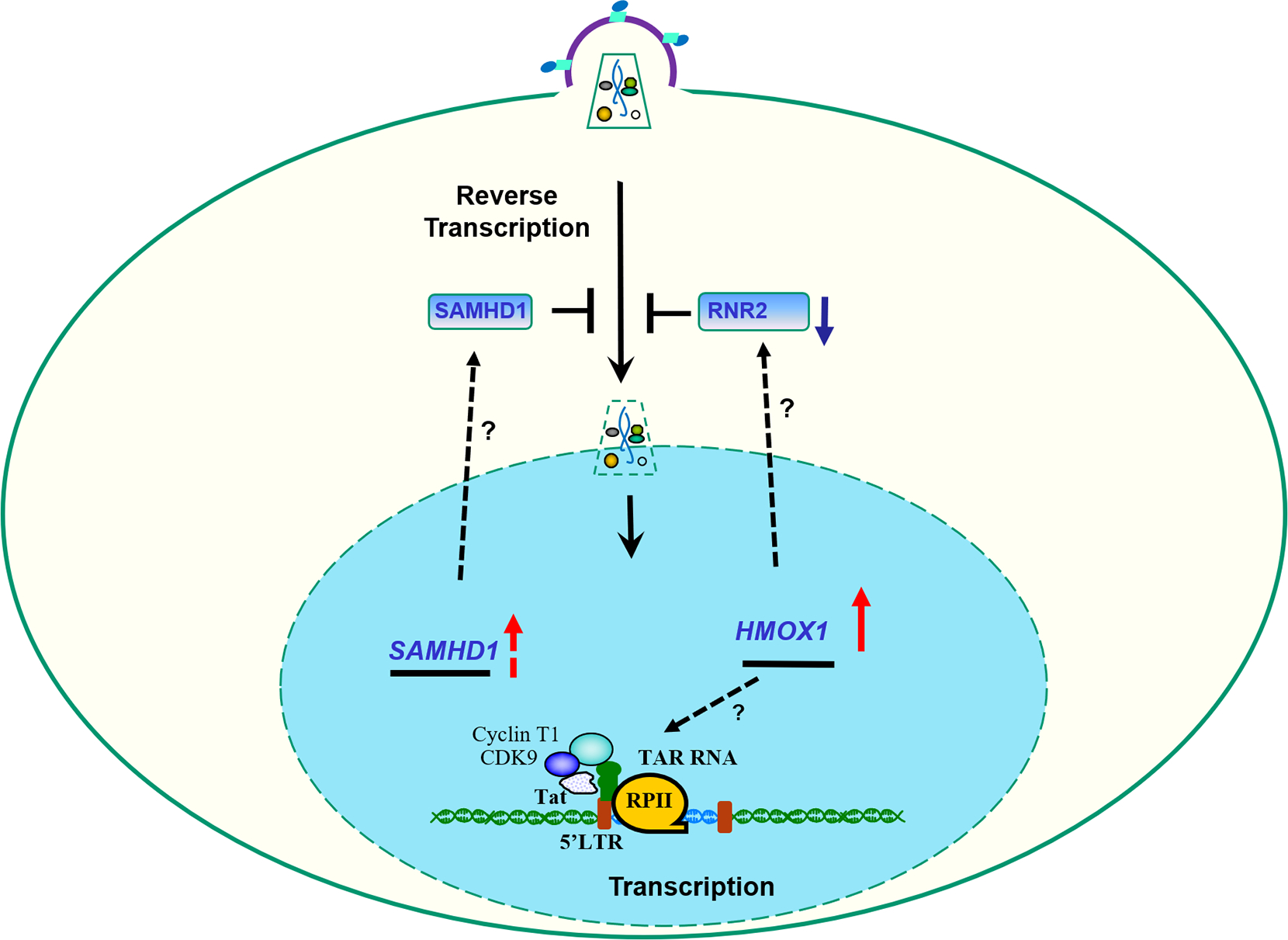
In SCT condition, increased expression of HMOX-1 and SAMHD1 was observed. Protein levels of RNR2 were downregulated.
Further analysis of HIV-1 infection in a cohort of women living with HIV-1 (WIHS) showed SCT prevalence among HIV-1 infected versus non-infected participants [115].While HIV viral load did not differ between HIV-1 positive women with and without SCT, the individuals with SCT had higher levels of CD4/CD8, suggesting milder disease progression for participants with SCT.
6. Potential implications of HIV-1 restriction in SCD gene therapy.
SCD gene therapy came to a spotlight in 2018 when NIH launched an initiative to accelerate genetic therapies for SCD. First vectors for SCD gene therapy were developed over 30 years ago when locus control region for β-hemoglobin was discovered [116,117]. The original gamma-retroviral vectors eventually replaced by HIV-derived lentiviral vectors (LVs) that also included insulators to prevent non-specific expression of neighboring genes (reviewed in [118]). Several trials are currently being conducted in the USA and worldwide including LentiGlobin trail that employs BB305 lentiviral vector encoding HbAT87Qmodified hemoglobin that is not capable of sickling [119]. Three other clinical trials with BB305 vector are being conducted in France, Australia, and Thailand [118]. Additional cure approached include downregulation of elements that suppress fetal hemoglobin production such as inactivation of BCL11A gene [118]. In these trials, patients CD34+ cells are typically collected after myeloablation, transduced in vitro and then reintroduced for bone marrow engraftment. Success of these trials gave rise to the hope that the SCD cure is achievable for wider patient population. Future application of SCD cure in Sib Saharan Africa where the majority of SCD patients reside, will likely require direct delivery of hemoglobin expressing or gene-modifying vectors in vivo. As safety of genome-editing technologies (and base editing in particular) is fast improving, the near future editing approaches will likely replace LVs-based gene therapy strategies. To achieve the success, antiviral state of SCD patients’ needs to be overcome. Thus, the antiviral restriction mechanisms described above might of interest for future application of the gene therapy for SCD and development of pharmacological approaches to alleviate HIV-1 restriction and achieve optimized vector delivery and expression in vivo.
7. Expert opinion
In conclusion, previous studies, including ours, showed that SCD might offer a protection from HIV-1 infection and that this protection might be due to the upregulation of complex protein network that include hemolytic, hypoxia and interferon signaling factors. In contrast, SCT individuals might have a moderate protection that appeared to be mediated by the increased expression levels of HMOX1 and SAMHD1 and decreased expression of RNR2. These finding strongly implicate hemolysis as major contributor for HIV-1 inhibition in SCD patients and individuals with SCT. While we learned many mechanistic details of HIV-1 restriction in SCD and SCT, many gaps still exist. For example, the role of IFN-α/β needs further investigation. Also, of interest would be to learn whether HIV-2 is able to infect SCD PBMCs as HIV-2 can bypass SAMHD1 restriction. Mouse model of HIV-1 infection could clarify whether SCD affects HIV-1 infection in vivo and might help to identify additional restriction factors that contribute to the resistance phenotype. It will be of interest to learn about SCD protection having an effect on additional HIV-1 subtypes, such as subtype A or C. This analysis will be of value for better understanding of HIV-1 infection in African countries where major HIV-1 subtypes are different compared to the United States. Finally, additional mechanistic knowledge is needed for the understanding of HIV-1 suppression in SCT. Whether HO-1 is the sole mediator of HIV-1 restriction in SCT remains to be determined. As our recent study was only carried in HIV-1+ women, analysis of man with SCT who are living with HIV-1 would be of interest. These findings will be important not only for HIV-1 field but also for SCD cure efforts as discussed above as antiviral state of SCD patients may adversely affect lentivirus-based future gene therapy efforts.
Article highlights.
Systemic iron load positively correlates with severity of HIV-1 infection and co-infections
Reduced intracellular iron inhibits HIV-1 reverse transcription and transcription in part through downregulation of host CDK2 activity and activation of SAMHD1 and inhibition of CDK9 activity.
Patients with SCD are protected from HIV-1 infection. This protection is mediated by the expression of antiviral factors induced by hemolysis and interferon production.
Expression of iron export protein, ferroportin, inhibits HIV-1 replication in cultured cells and in SCD PBMCs
Patients with SCT have lower chances of acquiring HIV-1 infection. The mechanism of protection is driven by HO-1 expression.
Funding:
This paper was supported in part by extramural NIH grants (1R01HL125005, 5U54MD007597, 1UM1AI26617 and P30AI087714).
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
REFERENCES
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- 1.Bender MA. Sickle Cell Disease. In: Adam MP, Ardinger HH, Pagon RA, et al. , editors. GeneReviews((R)). Seattle (WA)1993. [Google Scholar]
- 2.Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nature reviews Disease primers. 2018. Mar 15;4:18010. [DOI] [PubMed] [Google Scholar]
- 3.Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017. Mar 1;127(3):750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004. Feb 26;350(9):886–95. [DOI] [PubMed] [Google Scholar]
- 5.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007. Jan;21(1):37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kato GJ, Hsieh M, Machado R, et al. Cerebrovascular disease associated with sickle cell pulmonary hypertension. Am J Hematol. 2006. Jul;81(7):503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato GJ, McGowan V, Machado RF, et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood. 2006. Mar 15;107(6):2279–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris CR, Kato GJ, Poljakovic M, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA. 2005. Jul 6;294(1):81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002. Dec;8(12):1383–9. [DOI] [PubMed] [Google Scholar]
- 10.Sachdev V, Machado RF, Shizukuda Y, et al. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. 2007. Jan 30;49(4):472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brousseau DC, Panepinto JA, Nimmer M, et al. The number of people with sickle-cell disease in the United States: national and state estimates. American journal of hematology. 2010. Jan;85(1):77–8. [DOI] [PubMed] [Google Scholar]
- 12.Piel FB, Hay SI, Gupta S, et al. Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS medicine. 2013;10(7):e1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hassell KL. Population estimates of sickle cell disease in the U.S. American journal of preventive medicine. 2010. Apr;38(4 Suppl):S512–21. [DOI] [PubMed] [Google Scholar]
- 14.Niu X, Parry CS, Mason A, et al. Prevalence of Sickle Cell Trait and Rare Hemoglobin Variants in the Metropolitan Washington DC Area. Journal of hematology. 2020. Sep;9(3):93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization. 2008. Jun;86(6):480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wood JC. Sickle cell trait: A sigh of relief? EClinicalMedicine. 2019. May-Jun;11:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu X, Chaudhury A, Higgins JM, et al. Oxygen-dependent flow of sickle trait blood as an in vitro therapeutic benchmark for sickle cell disease treatments. American journal of hematology. 2018. Oct;93(10):1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. https://www.who.int/news-room/fact-sheets/detail/hiv-aids.
- 19.Cazzola M, Pootrakul P, Huebers HA, et al. Erythroid marrow function in anemic patients [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Blood. 1987. Jan;69(1):296–301. [PubMed] [Google Scholar]
- 20. van Beers EJ, Yang Y, Raghavachari N, et al. Iron, inflammation, and early death in adults with sickle cell disease. Circ Res. 2015. Jan 16;116(2):298–306. ** A seminal study showing expression upregulation of iron meatbolisma and inflammation genes in SCD.
- 21. Kumari N, Ammosova T, Diaz S, et al. Increased iron export by ferroportin induces restriction of HIV-1 infection in sickle cell disease. Blood Adv. 2016. Dec 27;1(3):170–183. ** First mechanistical study explaining molecular details of HIV-1 restriction in SCD.
- 22. Liu Y, Pal M, Bao W, et al. Type I interferon is induced by hemolysis and drives antibody-mediated erythrophagocytosis in sickle cell disease. Blood. 2021. Sep 30;138(13):1162–1171. ** A seminal study explaining the mechanism of interferon production in SCD.
- 23.Niu X, Nouraie M, Campbell A, et al. Angiogenic and inflammatory markers of cardiopulmonary changes in children and adolescents with sickle cell disease. PLoS One. 2009. Nov 23;4(11):e7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Belcher JD, Chen C, Nguyen J, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. 2014. Jan 16;123(3):377–90. **A groundbreaking study explaining the mechanims of heme mediated inflammation as a ersult of TRL4 singalling in SCD.
- 25.Boelaert JR, Weinberg GA, Weinberg ED. Altered iron metabolism in HIV infection: mechanisms, possible consequences, and proposals for management. Infect Agents Dis. 1996. Jan;5(1):36–46. [PubMed] [Google Scholar]
- 26.Nekhai S, Kumari N, Dhawan S. Role of cellular iron and oxygen in the regulation of HIV-1 infection. Future virology. 2013. Mar;8(3):301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Drakesmith H, Prentice A. Viral infection and iron metabolism. Nature reviews Microbiology. 2008. Jul;6(7):541–52. **A comprehensive review on the role of iron metabolism in HIV-1 infection.
- 28.Gordeuk VR, Delanghe JR, Langlois MR, et al. Iron status and the outcome of HIV infection: an overview. J Clin Virol. 2001. Feb;20(3):111–5. [DOI] [PubMed] [Google Scholar]
- 29.McDermid JM, Jaye A, Schim van der Loeff MF, et al. Elevated iron status strongly predicts mortality in West African adults with HIV infection. J Acquir Immune Defic Syndr. 2007. Dec 1;46(4):498–507. [DOI] [PubMed] [Google Scholar]
- 30.Rawat R, Humphrey J, Ntozini R, et al. Elevated iron stores are associated with HIV disease severity and mortality among postpartum women in Zimbabwe. Public Health Nutr. 2008. Nov 12:1–9. [DOI] [PubMed] [Google Scholar]
- 31.Banjoko SO, Oseni FA, Togun RA, et al. Iron status in HIV-1 infection: implications in disease pathology. BMC clinical pathology. 2012. Dec 17;12:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greaves DE, Griffiths WJ, Lever AM. Does venesection reduce HIV viral load in patients with hereditary haemochromatosis? Antiviral therapy. 2013;18(1):135–8. [DOI] [PubMed] [Google Scholar]
- 33.Kallianpur AR, Gerschenson M, Hulgan T, et al. Hemochromatosis (HFE) Gene Variants Are Associated with Increased Mitochondrial DNA Levels During HIV-1 Infection and Antiretroviral Therapy. AIDS research and human retroviruses. 2018. Nov;34(11):942–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goncalves JL, Silva MCA, Roma EH, et al. Iron intake is positively associated with viral load in antiretroviral naive Brazilian men living with HIV. Memorias do Instituto Oswaldo Cruz. 2020;114:e190350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Visser A, Mostert C. Causes of hyperferritinaemia classified by HIV status in a tertiary-care setting in South Africa. Epidemiology and infection. 2013. Jan;141(1):207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esan MO, van Hensbroek MB, Nkhoma E, et al. Iron supplementation in HIV-infected Malawian children with anemia: a double-blind, randomized, controlled trial. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2013. Dec;57(11):1626–34. [DOI] [PubMed] [Google Scholar]
- 37.Lopez-Calderon C, Palacios R, Cobo A, et al. Serum ferritin in HIV-positive patients is related to immune deficiency and inflammatory activity. International journal of STD & AIDS. 2015. May;26(6):393–7. [DOI] [PubMed] [Google Scholar]
- 38.Atwa ZT, Abdel Wahed WY. Transfusion transmitted infections in frequently transfused thalassemic children living in Fayoum Governorate, Egypt: Current prevalence and risk factors. Journal of infection and public health. 2017. Nov - Dec;10(6):870–874. [DOI] [PubMed] [Google Scholar]
- 39.Berger A, Sommer AF, Zwarg J, et al. SAMHD1-deficient CD14+ cells from individuals with Aicardi-Goutieres syndrome are highly susceptible to HIV-1 infection. PLoS pathogens. 2011. Dec;7(12):e1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldstone DC, Ennis-Adeniran V, Hedden JJ, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011. Nov 6;480(7377):379–82. [DOI] [PubMed] [Google Scholar]
- 41.Hrecka K, Hao C, Gierszewska M, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011. Jun 29;474(7353):658–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laguette N, Sobhian B, Casartelli N, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011. May 25;474(7353):654–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsimberidou AM, Alvarado Y, Giles FJ. Evolving role of ribonucleoside reductase inhibitors in hematologic malignancies. Expert review of anticancer therapy. 2002. Aug;2(4):437–48. [DOI] [PubMed] [Google Scholar]
- 44.Morris ER, Caswell SJ, Kunzelmann S, et al. Crystal structures of SAMHD1 inhibitor complexes reveal the mechanism of water-mediated dNTP hydrolysis. Nature communications. 2020. Jun 23;11(1):3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allouch A, David A, Amie SM, et al. p21-mediated RNR2 repression restricts HIV-1 replication in macrophages by inhibiting dNTP biosynthesis pathway. Proceedings of the National Academy of Sciences of the United States of America. 2013. Oct 15;110(42):E3997–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Pablo A, Bogoi R, Bejarano I, et al. Short Communication: p21/CDKN1A Expression Shows Broad Interindividual Diversity in a Subset of HIV-1 Elite Controllers. AIDS research and human retroviruses. 2016. Mar;32(3):232–6. [DOI] [PubMed] [Google Scholar]
- 47.Chen H, Li C, Huang J, et al. CD4+ T cells from elite controllers resist HIV-1 infection by selective upregulation of p21. The Journal of clinical investigation. 2011. Apr;121(4):1549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pauls E, Ruiz A, Riveira-Munoz E, et al. p21 regulates the HIV-1 restriction factor SAMHD1. Proceedings of the National Academy of Sciences of the United States of America. 2014. Apr 8;111(14):E1322–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pauls E, Ruiz A, Badia R, et al. Cell cycle control and HIV-1 susceptibility are linked by CDK6-dependent CDK2 phosphorylation of SAMHD1 in myeloid and lymphoid cells. Journal of immunology. 2014. Aug 15;193(4):1988–97. [DOI] [PubMed] [Google Scholar]
- 50.Lin X, Ammosova T, Kumari N, et al. Protein Phosphatase-1 -targeted Small Molecules, Iron Chelators and Curcumin Analogs as HIV-1 Antivirals. Current pharmaceutical design. 2017;23(28):4122–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen L, Xiong S, She H, et al. Iron causes interactions of TAK1, p21ras, and phosphatidylinositol 3-kinase in caveolae to activate IkappaB kinase in hepatic macrophages. J Biol Chem. 2007. Feb 23;282(8):5582–8. [DOI] [PubMed] [Google Scholar]
- 52.Xiong S, She H, Sung CK, et al. Iron-dependent activation of NF-kappaB in Kupffer cells: a priming mechanism for alcoholic liver disease. Alcohol. 2003. Jun;30(2):107–13. [DOI] [PubMed] [Google Scholar]
- 53.Xiong S, She H, Tsukamoto H. Signaling role of iron in NF-kappa B activation in hepatic macrophages. Comparative hepatology. 2004. Jan 14;3 Suppl 1:S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pereira LA, Bentley K, Peeters A, et al. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res. 2000. Feb 1;28(3):663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nekhai S, Petukhov M, Breuer D. Regulation of CDK9 activity by phosphorylation and dephosphorylation. BioMed research international. 2014;2014:964964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Breuer D, Kotelkin A, Ammosova T, et al. CDK2 regulates HIV-1 transcription by phosphorylation of CDK9 on serine 90. Retrovirology. 2012. Nov 9;9:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ivanov A, Lin X, Ammosova T, et al. HIV-1 Tat phosphorylation on Ser-16 residue modulates HIV-1 transcription. Retrovirology. 2018. May 23;15(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ammosova T, Berro R, Jerebtsova M, et al. Phosphorylation of HIV-1 Tat by CDK2 in HIV-1 transcription. Retrovirology. 2006. Nov 3;3:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Debebe Z, Ammosova T, Jerebtsova M, et al. Iron chelators ICL670 and 311 inhibit HIV-1 transcription. Virology. 2007. Oct 25;367(2):324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Debebe Z, Ammosova T, Breuer D, et al. Iron chelators of the di-2-pyridylketone thiosemicarbazone and 2-benzoylpyridine thiosemicarbazone series inhibit HIV-1 transcription: identification of novel cellular targets--iron, cyclin-dependent kinase (CDK) 2, and CDK9. Molecular pharmacology. 2011. Jan;79(1):185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumari N, Iordanskiy S, Kovalskyy D, et al. Phenyl-1-Pyridin-2yl-ethanone-based iron chelators increase IkappaB-alpha expression, modulate CDK2 and CDK9 activities, and inhibit HIV-1 transcription. Antimicrobial agents and chemotherapy. 2014. Nov;58(11):6558–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drakesmith H, Chen N, Ledermann H, et al. HIV-1 Nef down-regulates the hemochromatosis protein HFE, manipulating cellular iron homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2005. Aug 2;102(31):11017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koppensteiner H, Hohne K, Gondim MV, et al. Lentiviral Nef suppresses iron uptake in a strain specific manner through inhibition of Transferrin endocytosis. Retrovirology. 2014. Jan 2;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malina J, Hannon MJ, Brabec V. Iron(II) supramolecular helicates interfere with the HIV-1 Tat-TAR RNA interaction critical for viral replication. Scientific reports. 2016. Jul 12;6:29674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mancone C, Grimaldi A, Refolo G, et al. Iron overload down-regulates the expression of the HIV-1 Rev cofactor eIF5A in infected T lymphocytes. Proteome science. 2017;15:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saxena D, Spino M, Tricta F, et al. Drug-Based Lead Discovery: The Novel Ablative Antiretroviral Profile of Deferiprone in HIV-1-Infected Cells and in HIV-Infected Treatment-Naive Subjects of a Double-Blind, Placebo-Controlled, Randomized Exploratory Trial. PloS one. 2016;11(5):e0154842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoque M, Hanauske-Abel HM, Palumbo P, et al. Inhibition of HIV-1 gene expression by Ciclopirox and Deferiprone, drugs that prevent hypusination of eukaryotic initiation factor 5A. Retrovirology. 2009. Oct 13;6:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beaumont C Multiple regulatory mechanisms act in concert to control ferroportin expression and heme iron recycling by macrophages. Haematologica. 2010. Aug;95(8):1233–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ferreira A, Marguti I, Bechmann I, et al. Sickle hemoglobin confers tolerance to Plasmodium infection. Cell. 2011. Apr 29;145(3):398–409. [DOI] [PubMed] [Google Scholar]
- 70.Williams TN, Mwangi TW, Wambua S, et al. Sickle cell trait and the risk of Plasmodium falciparum malaria and other childhood diseases. J Infect Dis. 2005. Jul 1;192(1):178–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aidoo M, Terlouw DJ, Kolczak MS, et al. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet. 2002. Apr 13;359(9314):1311–2. [DOI] [PubMed] [Google Scholar]
- 72.Devadas K, Dhawan S. Hemin activation ameliorates HIV-1 infection via heme oxygenase-1 induction. Journal of immunology. 2006. Apr 1;176(7):4252–7. [DOI] [PubMed] [Google Scholar]
- 73.Devadas K, Hewlett IK, Dhawan S. Lipopolysaccharide suppresses HIV-1 replication in human monocytes by protein kinase C-dependent heme oxygenase-1 induction. Journal of leukocyte biology. 2010. May;87(5):915–24. [DOI] [PubMed] [Google Scholar]
- 74.Huang H, Zhou ZH, Adhikari R, et al. Defective iron homeostasis in human immunodeficiency virus type-1 latency. Current trends in immunology. 2016;17:125–131. [PMC free article] [PubMed] [Google Scholar]
- 75.Shankaran P, Madlenakova M, Hajkova V, et al. Effects of heme degradation products on reactivation of latent HIV-1. Acta virologica. 2017;61(1):86–96. [DOI] [PubMed] [Google Scholar]
- 76.Shytaj IL, Lucic B, Forcato M, et al. Alterations of redox and iron metabolism accompany the development of HIV latency. The EMBO journal. 2020. May 4;39(9):e102209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khan N, Halcrow PW, Lakpa LK, et al. Endolysosome iron restricts Tat-mediated HIV-1 LTR transactivation by increasing HIV-1 Tat oligomerization and beta-catenin expression. Journal of neurovirology. 2021. Sep 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Halcrow PW, Lakpa KL, Khan N, et al. HIV-1 gp120-Induced Endolysosome de-Acidification Leads to Efflux of Endolysosome Iron, and Increases in Mitochondrial Iron and Reactive Oxygen Species. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2021. Apr 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ganz T Molecular control of iron transport. J Am Soc Nephrol. 2007. Feb;18(2):394–400. [DOI] [PubMed] [Google Scholar]
- 80.Pietrangelo A Hereditary hemochromatosis. Biochim Biophys Acta. 2006. Jul;1763(7):700–10. [DOI] [PubMed] [Google Scholar]
- 81.Kasvosve I, Gomo ZA, Nathoo KJ, et al. Effect of ferroportin Q248H polymorphism on iron status in African children. Am J Clin Nutr. 2005. Nov;82(5):1102–6. [DOI] [PubMed] [Google Scholar]
- 82.Drakesmith H, Schimanski LM, Ormerod E, et al. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood. 2005. Aug 1;106(3):1092–7. [DOI] [PubMed] [Google Scholar]
- 83.Schimanski LM, Drakesmith H, Merryweather-Clarke AT, et al. In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood. 2005. May 15;105(10):4096–102. [DOI] [PubMed] [Google Scholar]
- 84.Jiang L, Wang J, Wang K, et al. RNF217 regulates iron homeostasis through its E3 ubiquitin ligase activity by modulating ferroportin degradation. Blood. 2021. Aug 26;138(8):689–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Traeger L, Wiegand SB, Sauer AJ, et al. UBA6 and NDFIP1 regulate the degradation of ferroportin. Haematologica. 2021. Jul 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000. Jun 30;275(26):19906–12. [DOI] [PubMed] [Google Scholar]
- 87.McKie AT, Marciani P, Rolfs A, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000. Feb;5(2):299–309. [DOI] [PubMed] [Google Scholar]
- 88.Donovan A, Brownlie A, Zhou Y, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000. Feb 17;403(6771):776–81. [DOI] [PubMed] [Google Scholar]
- 89.Masaisa F, Breman C, Gahutu JB, et al. Ferroportin (SLC40A1) Q248H mutation is associated with lower circulating serum hepcidin levels in Rwandese HIV-positive women. Annals of hematology. 2012. Jun;91(6):911–6. [DOI] [PubMed] [Google Scholar]
- 90. Armitage AE, Stacey AR, Giannoulatou E, et al. Distinct patterns of hepcidin and iron regulation during HIV-1, HBV, and HCV infections. Proceedings of the National Academy of Sciences of the United States of America. 2014. Aug 19;111(33):12187–92. ** A seminal study exploring markers of iron metabolism in HIV-1 infection
- 91.Malvoisin E, Makhloufi D, Livrozet JM. Serum hepcidin levels in women infected with HIV-1 under antiviral therapy. Journal of medical virology. 2014. Oct;86(10):1656–60. [DOI] [PubMed] [Google Scholar]
- 92.Liu Y, Lv Q, Gao J, et al. Coinfection with HIV-1 alleviates iron accumulation in patients with chronic hepatitis C virus infection. PloS one. 2014;9(6):e98039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu M, Kashanchi F, Foster A, et al. Hepcidin induces HIV-1 transcription inhibited by ferroportin. Retrovirology. 2010. Dec 2;7:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Waweru SE, Meme JS, Kinuthia DM, et al. Waweru SE, Meme JS, Kinuthia DM, Kitonyi GW. “Absence of HIV seropositivity in children with sickle cell anaemia at Kenyatta National Hospital, Nairobi, Kenya.”. 1987. 1987. [Google Scholar]
- 95.Castro O, Saxinger C, Barnes S, et al. Prevalence of antibodies to human immunodeficiency virus and to human T cell leukemia virus type I in transfused sickle cell disease patients. J Infect Dis. 1990. Sep;162(3):743–5. [DOI] [PubMed] [Google Scholar]
- 96. Bagasra O, Steiner RM, Ballas SK, et al. Viral burden and disease progression in HIV-1-infected patients with sickle cell anemia. American journal of hematology. 1998. Nov;59(3):199–207. *First comprehensive study pointing to the protection of SCD pateints from HIV-1 infection.
- 97.Kemeny MM, Cooke V, Melester TS, et al. Splenectomy in patients with AIDS and AIDS-related complex. Aids. 1993. Aug;7(8):1063–7. [DOI] [PubMed] [Google Scholar]
- 98.Nouraie M, Nekhai S, Gordeuk VR. Sickle cell disease is associated with decreased HIV but higher HBV and HCV comorbidities in U.S. hospital discharge records: a cross-sectional study. Sexually transmitted infections. 2012. Nov;88(7):528–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kourtis AP, Bansil P, Johnson C, et al. Children with sickle cell disease and human immunodeficiency virus-1 infection: use of inpatient care services in the United States. The Pediatric infectious disease journal. 2007. May;26(5):406–10. [DOI] [PubMed] [Google Scholar]
- 100.Batina A, Kabemba S, Malengela R. [Infectious markers among blood donors in Democratic Republic of Congo (DRC)]. Rev Med Brux. 2007. May-Jun;28(3):145–9. [PubMed] [Google Scholar]
- 101.Batina Agasa S, Dupont E, Kayembe T, et al. Multiple transfusions for sickle cell disease in the Democratic Republic of Congo: the importance of the hepatitis C virus. Transfus Clin Biol Oct;17(4):254–9. [DOI] [PubMed] [Google Scholar]
- 102.David AN, Jinadu MY, Wapmuk AE, et al. Prevalence and impact of sickle cell trait on the clinical and laboratory parameters of HIV infected children in Lagos, Nigeria. The Pan African medical journal. 2018;31:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kelly S, Jacobs ES, Stone M, et al. Influence of sickle cell disease on susceptibility to HIV infection. PloS one. 2020;15(4):e0218880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Charles S, Ammosova T, Cardenas J, et al. Regulation of HIV-1 transcription at 3% versus 21% oxygen concentration. J Cell Physiol. 2009. Nov;221(2):469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jison ML, Munson PJ, Barb JJ, et al. Blood mononuclear cell gene expression profiles characterize the oxidant, hemolytic, and inflammatory stress of sickle cell disease. Blood. 2004. Jul 1;104(1):270–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Raposo RA, Gupta R, Abdel-Mohsen M, et al. Antiviral gene expression in psoriasis. Journal of the European Academy of Dermatology and Venereology : JEADV. 2015. Oct;29(10):1951–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zahoor MA, Xue G, Sato H, et al. HIV-1 Vpr induces interferon-stimulated genes in human monocyte-derived macrophages. PLoS One. 2014;9(8):e106418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yan J, Hao C, DeLucia M, et al. CyclinA2-Cyclin-dependent Kinase Regulates SAMHD1 Protein Phosphohydrolase Domain. The Journal of biological chemistry. 2015. May 22;290(21):13279–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lavender KJ, Gibbert K, Peterson KE, et al. Interferon Alpha Subtype-Specific Suppression of HIV-1 Infection In Vivo. J Virol. 2016. Jul 1;90(13):6001–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gessani S, Puddu P, Varano B, et al. Role of endogenous interferon-beta in the restriction of HIV replication in human monocyte/macrophages. Journal of leukocyte biology. 1994. Sep;56(3):358–61. [DOI] [PubMed] [Google Scholar]
- 111.Cremer I, Vieillard V, De Maeyer E. Retrovirally mediated IFN-beta transduction of macrophages induces resistance to HIV, correlated with up-regulation of RANTES production and down-regulation of C-C chemokine receptor-5 expression. Journal of immunology. 2000. Feb 1;164(3):1582–7. [DOI] [PubMed] [Google Scholar]
- 112.Oleske JM, Spiegel HMI, Makani J, et al. Potential Protective Effect of Sickle Cell Gene Allele on HIV Academic Journal of Pediatrics and Neonatology. 2017;2(5):555600. [Google Scholar]
- 113.Sellier P, Masson E, Zini JM, et al. Disease progression in HIV-1-infected patients heterozygous for the sickle hemoglobin gene. Aids. 2009. Nov 13;23(17):2362–4. [DOI] [PubMed] [Google Scholar]
- 114.Kiyaga C, Hernandez AG, Ssewanyana I, et al. Sickle cell screening in Uganda: High burden, human immunodeficiency virus comorbidity, and genetic modifiers. Pediatric blood & cancer. 2019. Aug;66(8):e27807. [DOI] [PubMed] [Google Scholar]
- 115. Kumari N, Nouraie SM, Ahmad A, et al. Restriction of HIV-1 Infection in Sickle Cell Trait. Blood advances. 2021. Sep 8. ** First mechanistical study exploring potential protection effect and underlying molecular mechanisms of SCT in HIV-1 infection.
- 116.Chang JC, Liu D, Kan YW. A 36-base-pair core sequence of locus control region enhances retrovirally transferred human beta-globin gene expression. Proc Natl Acad Sci U S A. 1992. Apr 1;89(7):3107–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Plavec I, Papayannopoulou T, Maury C, et al. A human beta-globin gene fused to the human beta-globin locus control region is expressed at high levels in erythroid cells of mice engrafted with retrovirus-transduced hematopoietic stem cells. Blood. 1993. Mar 1;81(5):1384–92. [PubMed] [Google Scholar]
- 118.Magrin E, Miccio A, Cavazzana M. Lentiviral and genome-editing strategies for the treatment of beta-hemoglobinopathies. Blood. 2019. Oct 10;134(15):1203–1213. [DOI] [PubMed] [Google Scholar]
- 119.Kanter J, Walters MC, Krishnamurti L, et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N Engl J Med. 2021. Dec 12. [DOI] [PubMed] [Google Scholar]