

Abstract
Bacterial infections and antimicrobial resistance are one of the major public health problems and various strategies to prevent potential threats have been developed. Protonated polymers were proven as efficient agents against several microbial pathogens. Poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA) linear polymer and its copolymers represent one example of functional materials which inhibit the growth of both harmful Gram-negative and Gram-positive bacteria. However, the antimicrobial effect of positively charged PDMAEMA particles has been never tested. In this report, we deeply studied several parameters of free-radical polymerization, including the effect of crosslinking monomer, medium composition, solvency and polarity, and type and concentration of initiator and stabilizer, to fabricate high-quality poly[2-(dimethylamino)ethyl methacrylate-co-ethylene dimethacrylate] (PDMAEMA-EDMA) nanogel. We successfully found that dispersion polymerization in water/2-methoxyethanol medium (80/20 w/w), initiated with 0.2 wt% potassium persulfate (KPS) and stabilized with 0.5 wt% poly(vinyl alcohol) (PVA), produced a well-defined and sub-micron 167 nm PDMAEMA-EDMA nanogel. Bactericidal activity of the quaternized PDMAEMA-EDMA nanogel was assessed via time–kill curve assay against two Gram-positive and Gram-negative pathogenic bacteria, namely Staphylococcus aureus (S. aureus) and Acinetobacter baumannii (A. baumannii). The results illustrated that the quaternized PDMAEMA-EDMA nanogel acted as an effective bactericidal agent against both tested bacteria.
Quaternized 167 nm poly[2-(dimethylamino)ethyl methacrylate-co-ethylene dimethacrylate] nanogel by dispersion polymerization in water/2-methoxyethanol medium induced death of pathogenic Staphylococcus aureus and Acinetobacter baumannii.
1. Introduction
Functional polymer colloids have been widely used in a variety of fields, such as coatings, chromatography, biotechnology, biomedicine, and delivery systems. These particulate systems bear reactive anionic or cationic groups, such as carboxyl, hydroxyl, amine, sulpho, or quaternary ammonium to exhibit specific reactivity for the desired purpose.1 Among them, the cationic colloids are the group of positively charged polymers that find application, for example, in water and waste treatment, chromatography, drug and gene delivery, and as antimicrobial agents.2 Common methods for preparing positively charged colloids cover dispersion, precipitation, emulsion, multiple swelling and seeded polymerizations from various cationic monomers, such as 2-vinylpyridine, 4-vinylpyridine, 2-(diethylamino)ethyl methacrylate, 2-(dimethylamino)ethyl methacrylate (DMAEMA).3,4
Protonated polymer particles based on DMAEMA possess interesting properties, which endow them with a potential for various applications. Poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA) is a water-soluble polymer and, due to its positive charge, PDMAEMA and its copolymers are able to form electrostatic complexes with anionic biomacromolecules, such as DNA and RNA, and are often used for gene delivery.5–10 Another interesting feature of PDMAEMA is based on its ability to interact with the mucosal gel layer of a mucosal membrane, and thus Brannigan et al. synthesized a crosslinked PDMAEMA nanogel loaded with pilocarpine hydrochloride which served as an ocular drug delivery system.11 Thermosensitive and crosslinked PDMAEMA nanogel was also exploited in anticancer therapy as a drug delivery system of doxorubicin.12 Besides the cationic groups, PDMAEMA also contains hydrophobic alkyl. A combination of these properties renders PDMAEMA with antimicrobial activity against various pathogenic bacteria which may cause harmful infections.13–15 Especially, microbial contamination represents one of the major challenges and concerns in hospitals, food packaging, and water storage and treatment mainly due to the fact that these bacteria have developed multidrug resistance.14,16 Therefore, the new ways of control and reduction of these pathogens including, for example, Clostridium difficile, Acinetobacter baumannii (A. baumannii), Escherichia coli (E. coli), Pseudomonas aeruginosa, Staphylococcus aureus (S. aureus) have been developed and investigated.16,17 As mentioned above, it was found that cationic polymers have an affinity to the bacterial cytoplasmic membrane and the ability to disrupt the bacterial outer membrane and cause cell death.18 Thus, PDMAEMA-based polymers represent promising antimicrobial materials that are effective against harmful pathogens. For instance, Ward et al. reported the systematic evaluation of the antimicrobial activity of sulfobetaine copolymers derived from DMAEMA with bacteriostatic activity against E. coli and S. aureus which are notorious pathogens associated with nosocomial infections.19 Lu et al. synthetized various quaternary ammonium salts from DMAEMA including quaternized DMAEMA-based monomers and related linear polymers and they showed bactericidal activities against E. coli and S. aureus. Interestingly, they found that the polymer with quaternary ammonium groups exhibited greater bactericidal activities in comparison with the quaternized DMAEMA-based monomers.20 Rawlinson et al. published an investigation of antimicrobial activity of conjugated and unconjugated linear PDMAEMA derivatives showing that PDMAEMA inhibited the growth of Gram-negative bacteria and had a variable effect on Gram-positive bacteria and without inhibiting effect on the yeast.14,21 However, only one study by Kamlangmak et al. introduced a successful investigation of copolymer nanoparticles containing DMAEMA by emulsion iodine transfer polymerization via polymerization-induced self-assembly for inhibition of growth of E. Coli and S. aureus combining the effect of positively charged particles and the presence of an alkyl chain.22
According to the literature, we can find a few articles reporting the preparation of particles from DMAEMA and bifunctional monomers by various free-radical polymerizations including precipitation, emulsion, polymerization, or dispersion polymerization.7,11,12,23,24 Besides these free-radical polymerization techniques, Qian et al. successfully prepared pegylated PDMAEMA micelles by atom transfer radical polymerization for complexation with DNA. After the appropriate analyses, the resulting polyplexes had a diameter of less than 100 nm, exhibited a spherical shape and narrow particle size distribution.9 Fujii et al. reported the preparation of polystyrene/PDMAEMA-ethylene dimethacrylate (EDMA) core–shell particles by the seeded emulsion polymerization. These ∼200 nm high-quality latex particles were well characterized by various methods and shown on a TEM image depicting a sufficient number of the core–shell particles.25 Zhang et al. prepared various well-defined micelles from pH-sensitive cyclic statistical copolymers poly[oligo(ethylene glycol)-st-DMAEMA] by self-assembly with hydrodynamic diameters (Dh) from 140 to 190 nm.26 Thus, these alternative approaches can be considered as reliable and efficient procedures to obtain well-defined PDMAEMA-based particles or micelles. In contrast, the free-radical polymerizations for direct preparation of PDMAEMA-based nanogels seem to be more complicated due to the hydrophilic character of DMAEMA and the presence of tertiary amine groups which may reduce indispensable surface tension during free-radical polymerization. Only a few papers describing the preparation of PDMAEMA nanogels crosslinked with N,N′-methylenebis(acrylamide), EDMA, or with the photocleavable crosslinking agent can be found in literature.11,12,23,24,27 Unfortunately, these polymerizations had very low productivity in terms of number of particles and quality of final nanogels that is contrary to the efficacy of the conventional free-radical polymerizations.28–34 Due to the attractivity and use of PDMAEMA-based nanogels in various biotechnological and biomedical applications, we regard it as necessary to find optimal and effective conditions of free-radical polymerization for the production of a high-quality PDMAEMA-based nanogel.
Recently, we report the investigation and optimization of free-radical polymerization of DMAEMA with crosslinking monomer EDMA in various solvents, resulting in a stable and well-defined 167 nm PDMAEMA-EDMA nanogel. We systematically studied the effects of polymerization media and concentrations of EDMA, initiator, and surfactant on the preparation and properties of PDMAEMA-EDMA nanogels. We varied EDMA concentration in the range from 1 to 20 wt%, concentration of KPS, and type and concentration of SDS or PVA stabilizer. The spherical PDMAEMA-EDMA nanogel was then prepared by dispersion polymerization in water/MetCel as polymerization medium, with thermal decomposition of KPS as initiator and stabilized with PVA as a stabilizer. Then, the prepared PDMAEMA-EDMA nanogel was quaternized with iodomethane and the bactericidal activity of PDMAEMA-EDMA nanogel at two different concentrations was successfully tested against pathogenic Gram-positive S. aureus and Gram-negative A. baumannii.
2. Experimental
2.1. Materials
2-(Dimethylamino)ethyl methacrylate (DMAEMA), ethylene dimethacrylate (EDMA), potassium persulfate (KPS), sodium chloride, and sodium dodecyl sulfate (SDS) were purchased from Sigma Aldrich (St. Louis, MO, USA). DMAEMA and EDMA were vacuum-distilled before use. Ethanol (EtOH) for UV spectroscopy and methanol (EtOH) were obtained from Lach-Ner (Neratovice, Czech Republic). 2-Methoxyethanol (MetCel) was purchased from VWR International (Stříbrná Skalice, Czech Republic). Poly(vinyl alcohol) 25/140 (PVA) was obtained from Wacker Chemie (München, Germany). Tryptone soya agar (TSA) was supplied by LabMediaServis Ltd. (Jaroměř, Czech Republic).
2.2. Free-radical polymerization of DMAEMA and EDMA
The polymerizations were carried out in a 100 ml glass reactor, equipped with an anchor-type stirrer. In a typical experiment, water (80 g), DMAEMA and EDMA monomers (in total 1.63 g; DMAEMA 99 wt%, 0.01026 mol; EDMA 1 wt%, 0.00008 mol), and KPS (0.12 g; 0.00044 mol; 0.15 wt% relative to the continuous phase) were placed in the reactor, purged with nitrogen for 15 min, and polymerization was allowed to proceed at 80 °C for 24 h under stirring (400 rpm). After polymerization, PDMAEMA-EDMA nanogel was removed by centrifugation, washed 10 times with water, and finally freeze-dried from water. In addition, SDS or PVA were used for stabilization of polymerizations and the amounts of both stabilizers were calculated relative to the continuous phase (Table 1) and dissolved in the solvent before the start of polymerization. In addition, water as the continuous phase was mixed with EtOH, MeOH, or MetCel. The total amount of the continuous phase was kept at 80 g. The mixtures of solvents are listed in Table 1.
Conditions of free-radical polymerizations and dispersion polymerizations of 2-(dimethylamino)ethyl methacrylate and ethylene dimethacrylate and properties of final poly[2-(dimethylamino)ethyl methacrylate-co-ethylene dimethacrylate] nanogels.
| Sample | EDMA (wt%) | Solvent (wt%) | KPS (wt%) | SDS (wt%) | PVA (wt%) | PVP (wt%) | D n (nm) | D w (nm) | Đ | Yield (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| PDMAEMA-EDMA01 | 1 | Water (100) | 0.15 | — | — | — | Aggregation | 6.4 | ||
| PDMAEMA-EDMA02 | 2 | Water (100) | 0.15 | — | — | — | Aggregation | 6.5 | ||
| PDMAEMA-EDMA03 | 3 | Water (100) | 0.15 | — | — | — | 360 | 401 | 1.11 | 3.9 |
| PDMAEMA-EDMA04 | 4 | Water (100) | 0.15 | — | — | — | 359 | 420 | 1.17 | 1.5 |
| PDMAEMA-EDMA05 | 5 | Water (100) | 0.15 | — | — | — | 359 | 395 | 1.10 | 2.2 |
| PDMAEMA-EDMA06 | 10 | Water (100) | 0.15 | — | — | — | 366 | 426 | 1.17 | 33.6 |
| PDMAEMA-EDMA07 | 20 | Water (100) | 0.15 | — | — | — | 292 | 332 | 1.14 | 41.2 |
| PDMAEMA-EDMA08 | 10 | Water (100) | 0.2 | — | — | — | Aggregation | 42.7 | ||
| PDMAEMA-EDMA09 | 20 | Water (100) | 0.2 | — | — | — | Aggregation | 38.5 | ||
| PDMAEMA-EDMA10 | 10 | Water (100) | 0.2 | 0.15 | — | — | Aggregation | 49.2 | ||
| PDMAEMA-EDMA11 | 20 | Water (100) | 0.2 | 0.15 | — | — | Aggregation | 62.1 | ||
| PDMAEMA-EDMA12 | 20 | Water/EtOH (90/10) | 0.2 | 0.15 | — | — | Aggregation | 43.8 | ||
| PDMAEMA-EDMA13 | 20 | Water/EtOH (80/20) | 0.2 | 0.15 | — | — | Aggregation | 47.0 | ||
| PDMAEMA-EDMA14 | 20 | Water/EtOH (90/10) | 0.2 | — | — | — | Aggregation | 35.1 | ||
| PDMAEMA-EDMA15 | 20 | Water/EtOH (80/20) | 0.2 | — | — | — | Aggregation | 50.1 | ||
| PDMAEMA-EDMA16 | 20 | Water/EtOH (80/20) | 0.2 | — | 1.0 | — | Aggregation | 29.6 | ||
| PDMAEMA-EDMA17 | 20 | Water/EtOH (80/20) | 0.2 | — | 0.5 | — | Aggregation | 30.3 | ||
| PDMAEMA-EDMA18 | 20 | Water/EtOH (70/30) | 0.2 | — | 0.5 | — | Aggregation | 9.7 | ||
| PDMAEMA-EDMA19 | 20 | Water/MeOH (90/10) | 0.2 | — | 0.5 | — | Aggregation | 8.3 | ||
| PDMAEMA-EDMA20 | 20 | Water/MeOH (80/20) | 0.2 | — | 0.5 | — | 256 | 292 | 1.14 | 38.7 |
| PDMAEMA-EDMA21 | 20 | Water/MeOH (75/25) | 0.2 | — | 0.5 | — | Aggregation | 28.3 | ||
| PDMAEMA-EDMA22 | 20 | Water/MeOH (80/20) | 0.25 | — | 0.5 | — | 168 | 180 | 1.07 | 30.2 |
| PDMAEMA-EDMA23 | 20 | Water/MeOH (80/20) | 0.3 | — | 0.5 | — | 201 | 216 | 1.08 | 22.6 |
| PDMAEMA-EDMA24 | 20 (38) | Water/MetCel (80/20) | 0.2 | 0.15 | — | — | 169 | 199 | 1.18 | 52.2 |
| PDMAEMA-EDMA25 | 20 (39) | Water/MetCel (80/20) | 0.2 | — | 0.5 | — | 167 | 193 | 1.16 | 51.0 |
The percent yield of polymerization was determined after freeze-drying of the PDMAEMA-EDMA nanogels, when the mass of PDMAEMA-EDMA nanogels was divided by the mass of monomers used and multiplied by 100.
2.3. Quaternization of PDMAEMA-EDMA nanogel
PDMAEMA-EDMA nanogel (0.25 g) was placed in a 50 ml round bottomed flask in excess of iodomethane (8 ml) and the nanogel was quaternized at ambient temperature for 24 h. Then, the mixture was heated to 50 °C to remove iodomethane by evaporation and final quaternized PDMAEMA-EDMA nanogel was obtained. The presence and quantity of quaternary ammonium cations in PDMAEMA-EDMA nanogel was studied with NMR analysis.
2.4. Characterization of PDMAEMA-EDMA nanogels
The size and distribution of dry nanogels were analyzed by scanning electron microscopy (SEM; JEOL JSM 6400). The number-average diameter (Dn), weight-average diameter (Dw), and uniformity characterized by dispersity (Đ = Dw/Dn) were calculated using ImageJ software, by counting at least 500 individual particles in the SEM micrographs.
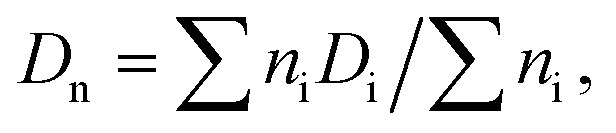 |
1 |
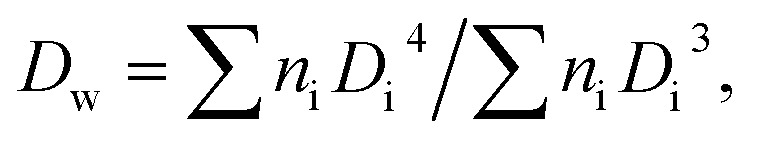 |
2 |
where ni and Di are the number and diameter of the i-th microsphere, respectively.
2.5. NMR analysis
Solid-state NMR (ssNMR) spectra were recorded at 500 MHz using a Bruker AVANCE III HD spectrometer. The 4 mm cross-polarization magic-angle spinning (CP/MAS) probe was used for 13C ssNMR experiments at Larmor frequency of ν(13C) =125.783 MHz. 13C NMR chemical shift was calibrated using α-glycine (176.03 ppm; carbonyl signal) as an external standard. The 13C MAS and CP/MAS spectra were acquired at the speed of the sample spinning of 11 kHz with 15 s and 3 s recycle delays, respectively. A cross-polarization (CP) contact time of 1.5 ms was used in 13C CP/MAS experiments. High-power 1H decoupling (SPINAL64) was used for the removal of heteronuclear coupling.
Samples were placed into ZrO2 rotors and subsequently kept at room temperature. All NMR experiments were conducted under active cooling in order to compensate for frictional heating caused by the rotation of the samples.35 All experiments were done at 298 K temperature. Bruker TopSpin 3.2 pl5 software package was used for the processing of the spectra.
2.6. Time–kill curve assay
Antimicrobial testing, carried out utilizing a time–kill curve assay, was performed using both A. baumannii (CCM 2355) and methicillin-resistant Staphylococcus aureus subsp. aureus (CCM 4750; S. aureus), grown on a tryptone soya agar at 37 °C for 18–24 h. Both cultures were supplied by the Czech Collection of Microorganisms (Brno, Czech Republic).
Time–kill curve analyses were carried out by incubating the test organisms in sterile saline (0.9% NaCl) in the presence of two concentrations of non- and quaternized PDMAEMA-EDMA nanogels, namely 1 and 5 mg ml−1. The bacterial inoculum was prepared by picking several single fresh colonies and their resuspending in saline; optical density was measured at 600 nm (CO8000 Cell Density Meter, Biochrom Ltd., Cambridge, UK). Individual Eppendorf tubes of 1 ml of the saline containing the fabricated non- and quaternized PDMAEMA-EDMA nanogels were then incubated with the inoculum of approximately 1 × 108 CFU ml−1 at 25 °C with continuous shaking (900 rpm). Also, a non-treated group (referred as to control), with inoculum but without the fabricated non- and quaternized PDMAEMA-EDMA nanogels, was included. At defined time points (namely 0, 1, and 4 h), plating of serial 10-fold dilutions (100 μl, in duplicate) on TSA was carried out to estimate the colony counts. The TSA plates were left to incubate overnight at 37 °C; bacterial colonies were counted manually. The assay was carried out in as three experiments.
2.7. Statistical analysis
Results are expressed as means ± SD. One-way ANOVA and Tukey's post hoc tests were used for analysis.
3. Results and discussion
Polymer nanogels based on DMAEMA belong to stimuli-responsive polymers which are widely utilized in biomedicine and biotechnology.3 For this reason, we investigated various polymerization parameters, with the aim of developing a reproducible and efficient procedure for the preparation of crosslinked PDMAEMA-EDMA nanogels by free-radical polymerization. This study describes the effects of polymerization media, initiator concentration, stabilizer type, and concentration, and crosslinking monomer on the morphology, particle size and particle size distribution of final PDMAEMA-EDMA nanogels summarized in Table 1. At first, the DMAEMA was copolymerized with various EDMA concentrations (1–20 wt%) in water as the polymerization medium, initiated with KPS (0.15 wt% relative to polymerization medium) and in the absence of stabilizer. As documented in Table 1, the free-radical copolymerization of DMAEMA with 1 wt% EDMA in water initiated with 0.15 wt% KPS (PDMAEMA-EDMA01) had a very low yield and a huge coagulum was produced (Fig. 1a). This result indicated that the free-radical polymerization was not stable to produce individual nanogels. In the case of PDMAEMA-EDMA02, the free-radical polymerization also failed with the production of coagulum and low yield (Table 1). With further increase of EDMA concentration (≥3 wt%), individual PDMAEMA-EDMA03-05 nanogels with diameters varying ∼360 nm started to be formed due to the increased covalently crosslinked polymer content. However, the polymerization yields still remained very low, varying from 1.5 to 3.9% (Table 1). One sample of PDMAEMA-EDMA05 containing 5 wt% EDMA is shown in Fig. 1b. We inferred from these results that the majority of formed PDMAEMA-EDMA remained in solution, without the requisite precipitation to form PDMAEMA-EDMA nanogel. The continuous increase in polymerization yield up to 41% (Table 1) was observed when EDMA concentration was raised to 10 wt% (PDMAEMA-EDMA06) and 20 wt% (PDMAEMA-EDMA07). This was explained by the fact that the more insoluble covalently crosslinked polymer was thereby formed. We also assumed that the higher EDMA concentration increased the hydrophobicity of the monomer phase. Thus, the shorter oligoradicals were precipitated out from the solution earlier leading to faster particle nucleation, in comparison with the free-radical polymerizations containing EDMA ≤5 wt%.28 This could also be supported by the increased monomer concentration in the case of free-radical polymerizations of DMAEMA with 10 and 20 wt% EDMA.36 Nevertheless, PDMAEMA-EDMA06 and 07 were partially aggregated, as is shown by the example of 292 nm PDMAEMA-EDMA07 in Fig. 1c. This result proved that the phase separation leading to a formation of individual PDMAEMA-EDMA nanogel was not complete, probably due to the high medium solvency for forming the polymer.3
Fig. 1. SEM micrographs of PDMAEMA-EDMA nanogels prepared in water with 1 (a), 5 (b), 20 (c) wt% of EDMA and initiated with 0.15 wt% KPS and 20 (d) wt% EDMA initiated with 0.2 wt% KPS.

In the next experiments, we tried to increase the polymerization yield by increasing the KPS concentration to 0.2 wt% relative to water as the polymerization medium. It was found that the free-radical polymerization of DMAEMA with 10 wt% EDMA (PDMAEMA-EDMA08), and 20 wt% EDMA (PDMAEMA-EDMA09) were not affected by the raising of KPS concentration in terms of higher yields, and the final products were also agglomerated (Table 1). The polymerization yields were ∼40%. We inferred from this result that the polymerization yield was mainly predetermined by copolymerization of DMAEMA with EDMA crosslinker and the formation of covalently crosslinked PDMAEMA-EDMA polymer. As an example, we provide a SEM microphotograph of PDMAEMA-EDMA09 with 20 wt% (Fig. 1d).
Due to the obvious tendency of the systems (PDMAEMA-EDMA01-09) to agglomerate during free-radical polymerizations, SDS was employed as a stabilizer of polymerization to avoid the undesired coagulation of PDMAEMA-EDMA nanogel. For example, Wu et al. showed that SDS successfully stabilized free-radical polymerization of N-isopropylacrylamide with N,N′-methylenebisacrylamide in water.37 The concentration of SDS was 0.15 wt% relative to the polymerization medium while varying EDMA concentration (10 and 20 wt%) and the polymerization medium composition and maintaining KPS concentration at 0.2 wt% relative to the polymerization medium (Table 1; PDMAEMA-EDMA10-13). However, our experiments revealed that, in the case of free-radical polymerization of DMAEMA with EDMA, SDS did not ensure necessary colloidal stability during particle nucleation.36 As illustrated in Fig. 2a, the free-radical polymerization of DMAEMA with 10 wt% EDMA in water stabilized with 0.15 wt% SDS (PDMAEMA-EDMA10) failed, because the coagulum was obtained. With the increase of EDMA concentration to 20 wt% (PDMAEMA-EDMA11), the free-radical polymerization also coagulated due to inadequate stabilization with SDS. On the other hand, it was found that the yield of polymerization increased up to 62% (Table 1) and it was profound with the increase of EDMA concentration up to 20 wt%. The higher content of more hydrophobic EDMA monomer in the polymerization mixture could affect the solubility of the monomer and polymer phase in the hydrophilic continuous phase. This resulted in faster polymerization velocity, more efficient phase separation, and higher polymerization yield.38
Fig. 2. SEM micrographs of PDMAEMA-EDMA nanogels prepared in water with 10 wt% EDMA and stabilized with 0.15 wt% SDS (a), in the mixture of water/EtOH (80/20 w/w) with 20 wt% EDMA and stabilized with 0.15 wt% SDS (b), and in the mixture of water/EtOH (80/20 w/w) with 20 wt% EDMA and without SDS (c). All polymerizations were initiated with 0.2 wt% KPS.

We supposed that the addition of polar organic solvent into the water as a polymerization medium could favor polymerization in the particle phase over polymerization in solution, increase the polymerization rate and facilitate the adsorption of stabilizer on to the forming PDMAEMA-EDMA nanogel.39,40 Therefore, we explored an effect of 10 (PDMAEMA-EDMA12) and 20 wt% EtOH (PDMAEMA-EDMA13) in a mixture with water (Table 1). The polymerizations were stabilized with 0.15 wt% SDS and initiated with 0.2 wt% KPS. The addition of alcohol (or MetCel in later experiments) caused the starting polymerization mixture to be homogeneous solution and subsequent polymerizations (PDMAEMA-EDMA12-13, and 16–25) run according to the mechanism of dispersion polymerization. However, the agglomeration of PDMAEMA-EDMA12 and 13 was not suppressed by the addition of EtOH (δ = 26 MPa1/2) into the polymerization medium, as documented in the example of PDMAEMA-EDMA13 (Fig. 2b). Our results revealed that SDS did not provide an effective steric stabilization for the preparation of high-quality PDMAEMA-EDMA nanogel.
Based on this result, the polymerization was carried out in the presence of 10 (PDMAEMA-EDMA14) and 20 wt% EtOH (PDMAEMA-EDMA15) without SDS as a stabilizer (Table 1). We found that the agglomeration of PDMAEMA-EDMA nanogel was not completely hindered in the mixture of water with 10 wt% EtOH (PDMAEMA-EDMA14), or 20 wt% (PDMAEMA-EDMA15). In spite of a slight decrease in yields, the final products did not contain such large irregular aggregates as could be seen in the polymerization in the presence of SDS as a stabilizer (Table 1; PDMAEMA-EDMA14 and 15). Fig. 2c illustrates an SEM microphotograph of PDMAEMA-EDMA15, showing the presence of agglomerated ∼550 nm nanogel. The persisting coagulation of both PDMAEMA-EDMA nanogel could be the result of the high solvency of PDMAEMA-EDMA polymer in continuous phase and the instability of polymerization.41,42
Subsequent dispersion polymerizations of DMAEMA with 20 wt% EDMA were performed in the presence of partially hydrolyzed PVA stabilizer, with different concentrations in water/EtOH mixture (80/20 w/w) initiated with 0.2 wt% KPS (Table 1; PDMAEMA-EDMA16-18). According to the SEM analyses (Fig. 3a–d), the steric stabilization effect of PVA was significantly efficient, compared to the previous results with the use of SDS because, PVA successfully stabilized the dispersion polymerization in water/alcohol polymerization media due to its low hydrophobic nature.42Fig. 3a displays that almost individual knobbly PDMAEMA-EDMA16 with Dn = 245 nm and Đ = 1.3 were produced in the presence of 1 wt% PVA. However, one can observe that the nanogel was still partially coagulated. Based on this result, the PVA concentration was decreased to 0.5 wt% relative to the polymerization media (Table 1). Even if the aggregation of PDMAEMA-EDMA17 (Fig. 3b) was not completely suppressed, 0.5 wt% PVA stabilized the polymerization more effectively in comparison with 1 wt% PVA (PDMAEMA-EDMA16, Fig. 3a), resulting in PDMAEMA-EDMA17 with Dn = 290 nm and Đ = 1.1. It was found that high PVA concentration destabilized polymer particles during polymerization and contributed to undesired flocculation.43 Further, we tried to change the medium solvency by increasing the EtOH content in the polymerization medium up to 30 wt% (Table 1; PDMAEMA-EDMA18) in favour of effective phase separation and particle formation. Nevertheless, the increased EtOH concentration had a deteriorating effect on the quality of the resulting PDMAEMA-EDMA18 that were aggregated and had small dimples in their surface (Fig. 3c). Our observation was supported by the work of Wang et al., who found that dispersion polymerization could be carried out at narrow EtOH or MeOH/water ratios without a deteriorating effect on the quality of the final nanogels.44
Fig. 3. SEM micrographs of PDMAEMA-EDMA nanogels prepared in a mixture of water/EtOH (80/20 w/w) with 20 wt% EDMA, stabilized with 1 wt% PVA and initiated with 0.2 wt% KPS (a); in a mixture of water/EtOH (80/20 w/w) with 20 wt% EDMA, stabilized with 0.5 wt% PVA and initiated with 0.2 wt% KPS (b); in a mixture of water/EtOH (70/30 w/w) with 20 wt% EDMA, stabilized with 0.5 wt% PVA and initiated with 0.2 wt% KPS (c); in a mixture of water/MeOH (80/20 w/w) with 20 wt% EDMA, stabilized with 0.5 wt% PVA and initiated with 0.2 wt% KPS (d).

According to the work by Wang et al., we mixed water with MeOH (δ = 36.2 MPa1/2) instead of EtOH, because polymerization in the water/MeOH mixture provided better conditions for the production of more colloidally stable nanogel.44 The MeOH concentration was varied from 10 to 25 wt% during dispersion polymerization of DMAEMA with 20 wt% EDMA initiated with 0.2 wt% KPS and stabilized with 0.5 wt% PVA (Table 1). The water/MeOH ratio 90/10 (w/w) was not optimal, due to the fact that ∼100 nm PDMAEMA-EDMA19 were aggregated and the polymerization produced a very low yield 8.3 wt% (Table 1).44 When the MeOH content was raised to 20 wt% in a mixture with water, the optimal water/MeOH ratio (80/20 w/w) was found for the production of 256 nm PDMAEMA-EDMA20 (Table 1) with broad particle size distribution Đ = 1.14 (Fig. 3d).44 The change in medium solvency by increased MeOH content to 20 wt% also resulted in a higher polymerization yield that was 39%. This indicated that the polymerization locus was shifted from solution to polymer phase.44 Also, MeOH is an inferior solvent to water for PDMAEMA-EDMA nanogels. Therefore, the stabilization of the nanogels with PVA was facilitated, leading to the formation of individual PDMAEMA-EDMA20.45 The next increase in MeOH content up to 25 wt% in polymerization medium yielded partial coagulation of PDMAEMA-EDMA21 indicating that the water/MeOH ratio 75/25 (w/w) was over the suitable range to obtain individual PDMAEMA-EDMA nanogel.44
Due to better colloidal stability of the resulting PDMAEMA-EDMA nanogels, we kept the water/MeOH ratio 80/20 (w/w) and PVA concentration 0.5 wt% for subsequent experiments. With the aim of increasing the polymerization yield, we investigated the effect of increased KPS concentration on polymerization yield. The dispersion polymerizations initiated with 0.25 (PDMAEMA-EDMA22) and 0.3 wt% KPS (PDMAEMA-EDMA23) led to a colloidally stable PDMAEMA-EDMA nanogels with diameters Dn = 168 nm and Dn = 201 nm (Fig. 4a), and dispersities Đ = 1.07 and Đ = 1.08 (Fig. 4b), respectively. However, we observed a slight decrease in polymerization yields in both cases (Table 1). We assumed that, at increased KPS concentrations, shorter polymer chains with higher solubility in the polymerization medium were produced, and mainly ended up in solution over particles.
Fig. 4. SEM micrographs of PDMAEMA-EDMA nanogels prepared in water/MeOH (80/20 w/w) with 20 wt% EDMA, stabilized with 0.5 wt% PVA and initiated with 0.25 wt% (a) and 0.3 wt% KPS (b).

In the next experiments, the polymerization medium solvency and polarity were adapted by mixing water with 20 wt% MetCel (δ = 23.3 MPa1/2). MetCel belongs to solvents of the cellosolve family and has a lower dielectric constant. In other words, it is less polar, in comparison with MeOH (εMetCel = 16.9 and εMeOH = 31). Lee et al. observed that the particle size and particle size distribution of poly(acrylamide) could be lowered and narrowed with the decrease of medium polarity during dispersion polymerization of acrylamide as a hydrophilic monomer.40 A reverse effect was observed for dispersion polymerization of hydrophobic monomers, due to better solubility of forming polymer in less polar medium.46 According to our previous results, the effects of 0.15 wt% SDS, and 0.5 wt% PVA on colloidal stability, particle size and particle size distribution of the resulting PDMAEMA-EDMA nanogels prepared in the polymerization medium of water/MetCel (80/20 w/w) were investigated. Fig. 5a documents that 169 nm PDMAEMA-EDMA24 (Table 1) was not effectively stabilized with 0.15 wt% SDS, because of obvious agglomeration of the nanogel.
Fig. 5. SEM micrographs of PDMAEMA-EDMA nanogels prepared in water/MetCel (80/20 w/w) with 20 wt% EDMA, initiated with 0.2 wt% KPS and stabilized with 0.15 wt% SDS (a), or with 0.5 wt% PVA (b).

On the other hand, the dispersion polymerization in water/MetCel (80/20 w/w) stabilized with 0.5 wt% PVA provided the ideal conditions for the production of individual and spherical 167 nm PDMAEMA-EDMA25 (Fig. 5b) with Đ = 1.16 (Table 1) as a result of PVA low hydrophobic nature.47 In both cases, the polymerization yield was higher (∼50%), and the size of PDMAEMA-EDMA24 and 25 decreased in comparison with dispersion polymerizations in water/MeOH mixture, due to the more suitable polarity of the polymerization medium.40 Moreover, dispersion polymerization in water/MetCel stabilized with 0.5 wt% PVA and initiated with 0.2 wt% KPS resulted in smaller nanogel (PDMAEMA-EDMA25) in comparison with water/MeOH (PDMAEMA-EDMA20).
Probably, oligomers with a shorter chain length were produced, which then coagulated in increased number of primary nuclei, leading to final nanogel with a smaller size.28 Tuned medium polarity by mixing of water with MetCel also contributed to better phase separation, because the resulting PDMAEMA-EDMA20 was spherical, compared to the PDMAEMA-EDMA nanogels prepared in the water/MeOH polymerization medium.48 In spite of average productivity, we found the optimal conditions for the preparation of colloidally stable PDMAEMA-EDMA25 by the combination of the suitable composition of polymerization medium, type and concentration of stabilizer, and concentration of initiator. This nanogel was subsequently quaternized with iodomethane and quaternized PDMAEMA-EDMA25Q was subjected to NMR analysis to confirm the presence and determine a content of quaternary ammonium groups.
13C CP/MAS NMR spectrum of PDMAEMA-EDMA25 (Fig. 6a) confirms the formation of the PDMAEMA-EDMA copolymer. The broad peak at 19 ppm was assigned to methyl groups (–CH3) from the copolymer backbone, peak at 45 ppm was assigned to methyl groups attached to the nitrogen atom (–N(CH3)2) and quaternary carbons (>C<). The peaks at 57 and 62 ppm were assigned to methylene groups near the nitrogen atom (>N–CH2–), methylene groups from the copolymer backbone (–CH2–) and methylene groups adjacent to the oxygen atom (–O–CH2–), respectively. Last signal at 177 ppm corresponds to the carbonyl carbons (>C O). The 13C CP/MAS NMR spectrum of the quaternized PDMAEMA-EDMA25Q (Fig. 6b) reveals the appearance of a distinct intensive signal (at 55 ppm) which was assigned to the methyl groups attached to the quaternary nitrogen atom (–N+(CH3)3). However, this signal is overlapped with the signals from backbone methylene groups (–CH2–). The peak assignment of both 13C CP/MAS SNMR spectra was accomplished based on the literature data.49–51
Fig. 6. Experimental 13C CP/MAS NMR (solid line) and 13C MAS NMR (dashed line) spectra of PDMAEMA-EDMA25 (a) and quaternized PDMAEMA-EDMA25Q (b) samples recorded at 11 kHz spinning speed on 500 MHz NMR spectrometer.

In order to calculate the extent of quaternization (of the amine nitrogen atom), the 13C MAS NMR spectrum of the quaternized PDMAEMA-EDMA25Q sample was recorded (Fig. 6b, dashed line). The amount of quaternized polymer was calculated by comparison of the integral intensity of the signals at 45 and 55 ppm corresponding to the methyl groups attached to ternary and quaternary nitrogen atoms respectively. As both signals contain the same contribution from the copolymer backbone (signal at 45 ppm is overlapped with quaternary carbon, whereas signal at 55 ppm is overlapped with methylene carbon from the backbone), their ratio was used for content calculation of quaternized polymer chains. Taking into the account that ternary and quaternary nitrogen atoms have two and three adjacent methyl groups, respectively, the content of the quaternized polymer (CQP) can be calculated by the following eqn (3):
 |
3 |
The extent of quaternization of the PDMAEMA-EDMA25Q was calculated to 54%.
Finally, we innovatively evaluated bactericidal activities of the fabricated 167 nm non-quaternized PDMAEMA-EDMA25 and quaternized PDMAEMA-EDMA25Q nanogels at two different concentrations, 1 and 5 mg ml−1, against Gram-positive S. aureus and Gram-negative A. baumannii via a time–kill curve assay. It was found that non-quaternized PDMAEMA-EDMA25 nanogel at both concentrations (1 and 5 mg ml−1) inhibited the growth of the bacterial population only negligibly in the case of both S. aureus (Fig. 7a) and A. baumannii (Fig. 7c). In contrast, quaternized PDMAEMA-EDMA25Q nanogel demonstrated an elevated antimicrobial activity. At 1 mg ml−1, the S. aureus population was reduced by ca. 1.9 log units after 4 hours of the treatment. Compared to both non-quaternized PDMAEMA-EDMA25 and the non-treated control, the log unit reductions were found to be statistically significant (p <0.001 and p <0.001, respectively). Notably, at 5 mg ml−1, the S. aureus population was reduced by ca. 2.3 log units after the 4 hour treatment (Fig. 7a and b) showing that quaternized PDMAEMA-EDMA25Q nanogel effectively killed harmful Gram-positive methicillin-resistant S. aureus pathogen what proved their bactericidal activity. This was found to be significant when compared to both non-quaternized PDMAEMA-EDMA25 and the non-treated control (p <0.001 and p <0.001, respectively). These results were in good accordance with work by Ward et al. about the bacteriostatic activity (preventing of the growth of bacteria) of the various DMAEMA-based copolymers with ethyl, butyl, octyl, cyclohexyl methacrylates against Gram-positive S. aureus and Gram-negative E. coli. They determined the minimum inhibitory concentrations (MICs) of these copolymers ranging from 1.125 to 2 mg ml−1 and the more hydrophobic copolymers demonstrated higher activity in inhibition of growth of these bacterial pathogens, especially in the case of Gram-positive S. aureus due to simpler wall structure which is more porous and permeable in comparison with the cell wall of Gram-negative E. coli.19 Besides, Rawlinson et al. studied the MICs of PDMAEMA linear polymer by transfer radical polymerization which were >3.2 mg ml−1 against to 11 strains of S. aureus. Moreover, they also tested the antibacterial activity of PDMAEMA against S. aureus biofilm, and surprisingly no effect was observed even at a concentration up to 50 mg ml−1.52 Interestingly, another methacrylate polymers with tertiary amine groups, poly[2-(dimethylamino)ethyl acrylate] and poly[2-(diethylamino)ethyl acrylate] demonstrated even lower MICs ranging from 0.088 to 0.5 mg ml−1 in comparison with PDMAEMA.53
Fig. 7. Antibacterial testing of the non-quaternized PDMAEMA-EDMA25 and quaternized PDMAEMA-EDMA25Q nanogels. Time-kill curve assay was carried out to estimate the effect of the nanogels on both S. aureus (a) and A. baumannii (c) counts after 1 and 4 h of the treatment at room temperature. The data indicate mean values ± SD. Asterisks represent a significant difference (***p < 0.001) when compared to non-treated control. (b and d) Representative photographs of corresponding agar plates with S. aureus ((b), 5 mg ml−1, representative suspension dilution of 10−5) and A. baumannii ((d), 5 mg ml−1, representative suspension dilution of 10−4) colonies including non-treated controls (NT).

Similar results were obtained from the testing with A. baumannii with rather elevated susceptibility to the PDMAEMA-EDMA25Q. Also, the revealed effects were in a time- and concentration-dependent manner. Considering the initial number of bacteria, at the lower tested PDMAEMA-EDMA25Q concentration (1 mg ml−1), A. baumannii population was reduced by ca. 1.4 log unit after 4 hours of the treatment (statistically significant compared to non-treated control, p <0.001). At 5 mg ml−1, A. baumannii number was reduced by ca. 4 log units (Fig. 7c, d). Similar to the S. aureus time-killing, this reduction of bacterial population was found to be statistically significant when compared to the non-treated control (p <0.001) and the PDMAEMA-EDMA25 testing (p <0.001). Note the killing (bactericidal) activity was notably elevated than in the case of S. aureus; the log unit decrease was ca. 2 log more in the case of A. baumannii. Our observation was in contrary with results by Ward et al. who demonstrated better bacteriostatic activity against S. aureus compared to E. coli due to more simpler wall structure of Gram-positive bacteria.19 The observed effect could be explained by the fact that positively charged PDMAEMA-EDMA25Q nanogel with quaternary ammonium groups easily interacted with negatively charged cell surface of A. baumannii facilitating thus the damaging of cell wall and cytoplasmic membrane.15
Overall, the bactericidal properties of the developed PDMAEMA-EDMA25Q nanogel with quaternary ammonium groups against two harmful pathogens, S. aureus and A. baumannii, were proven with a time–kill curve assay. The observed bactericidal effects against both bacteria were comparable, even at a concentration 5 mg ml−1 of the nanogel, elevated activity was found against A. baumannii, which becomes one of the major nosocomial pathogens.
4. Conclusions
In summary, conventional dispersion polymerization was successfully found as a facile, effective, and reproducible procedure for the production of sub-micron PDMAEMA-EDMA nanogel with bactericidal activity. In our study, we explored the effect of EDMA crosslinking monomer concentration (1–20 wt%), polymerization medium solvency and polarity, concentration of KPS initiator, and type and concentration of stabilizers, such as SDS and PVA, to produce high-quality and well-defined PDMAEMA-based nanogel. As is typical for conventional free-radical polymerization, our results showed that the formation of hydrophilic PDMAEMA-EDMA nanogel was largely dependent on the medium polarity and solvency, and in addition, it was affected by initiator concentration and the selection of a suitable stabilizer with the proper concentration. Despite the productivity ∼50%, tuning of medium solvency and polarity represented by the water/MetCel mixture (80/20 w/w), efficient stabilization with 0.5 wt% PVA, and initiation of dispersion polymerization of DMAEMA with 20 wt% EDMA resulted in the hydrophilic 167 nm PDMAEMA-EDMA nanogel. After quaternization of the prepared PDMAEMA-EDMA nanogel to introduce quaternary ammonium groups, the bactericidal properties were tested against two bacterial pathogens, namely S. aureus and A. baumannii. The time–kill curve assays proved that the application of the quaternized PDMAEMA-EDMA nanogel at concentrations 1 and 5 mg ml−1 displayed bactericidal activity against both pathogenic bacteria. Our findings can be of great value for novel antibacterial interventions using functional polymeric nanomaterials.
Author contributions
Petr Šálek: conceptualization, methodology, preparation of PDMAEMA-EDMA nanogels, evaluation of results, writing of article. Jiří Trousil: conceptualization and methodology of biological experiments, antimicrobial testing. Jitka Nováčková: preparation of PDMAEMA-EDMA nanogels. Jiřina Hromádková: SEM analyses of PDMAEMA-EDMA nanogels. Andrii Mahun – NMR analysis and data evaluation. Libor Kobera – NMR analysis and data evaluation.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors thank the Czech Academy of Sciences (CAS), Research Project RP10 “Molecules and Materials for Life” for financial support.
References
- Voorn D. J. Ming W. van Herk A. M. Macromolecules. 2005;38:3653–3662. doi: 10.1021/ma0475030. [DOI] [Google Scholar]
- Natu A. M. Wiggins M. van de Mark M. R. Colloid Polym. Sci. 2015;293:1191–1204. doi: 10.1007/s00396-015-3508-9. [DOI] [Google Scholar]
- Ramos J. Forcada J. Hidalgo-Alvarez R. Chem. Rev. 2014;114:367–428. doi: 10.1021/cr3002643. [DOI] [PubMed] [Google Scholar]
- Liu Q. Li Y. Duan Y. Zhou H. Polym. Int. 2012;61:1593–1602. doi: 10.1002/pi.4347. [DOI] [Google Scholar]
- Samal S. K. Dash M. van Vlierberghe S. Kaplan D. L. Chiellini E. van Blitterswijk C. Moroni L. Dubruel P. Chem. Soc. Rev. 2012;41:7147–7194. doi: 10.1039/C2CS35094G. [DOI] [PubMed] [Google Scholar]
- You Y. Z. Manickam D. S. Zhou Q. H. Oupický D. J. Controlled Release. 2007;122:217–225. doi: 10.1016/j.jconrel.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L. Zhai Y. Guo S. Jin F. Xie Z. He X. Dong A. J. Nanopart. Res. 2009;11:365–374. doi: 10.1007/s11051-008-9391-2. [DOI] [Google Scholar]
- Cheng H. Deng H. Zhou L. Su Y. Yu S. Zhu X. Zhou Y. Yan D. J. Controlled Release. 2011;152:e187–e188. doi: 10.1016/j.jconrel.2011.08.080. [DOI] [PubMed] [Google Scholar]
- Qian Y. Zha Y. Feng B. Pang Z. Zhang B. Sun X. Ren J. Zhang C. Shao X. Zhang Q. Jiang X. Biomaterials. 2013;34:2117–2129. doi: 10.1016/j.biomaterials.2012.11.050. [DOI] [PubMed] [Google Scholar]
- Zhu C. Zheng M. Meng F. Mickler F. M. Ruthardt N. Zhu X. Zhong Z. Biomacromolecules. 2012;13:769–778. doi: 10.1021/bm201693j. [DOI] [PubMed] [Google Scholar]
- Brannigan P. R. Khutoryanskiy V. V. Colloids Surf., B. 2017;155:538–543. doi: 10.1016/j.colsurfb.2017.04.050. [DOI] [PubMed] [Google Scholar]
- Maiti D. Chao Y. Dong Z. Yi X. He J. Liu Z. Yang K. Nanoscale. 2018;10:13976. doi: 10.1039/C8NR03986K. [DOI] [PubMed] [Google Scholar]
- Yang Y. Cai Z. Huang Z. Tang X. Zhang X. Polym. J. 2018;50:33–44. doi: 10.1038/pj.2017.72. [DOI] [Google Scholar]
- Rawlinson L. A. B. Ryan S. M. Mantovani G. Syrett J. A. Haddleton D. M. Brayden D. J. Biomacromolecules. 2010;11:443–453. doi: 10.1021/bm901166y. [DOI] [PubMed] [Google Scholar]
- Lu G. Wu D. Fu R. React. Funct. Polym. 2007;67:355–366. doi: 10.1016/j.reactfunctpolym.2007.01.008. [DOI] [Google Scholar]
- Peleg A. Y. Hooper D. C. N. Engl. J. Med. 2010;362:1804–1813. doi: 10.1056/NEJMra0904124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agaba P. Tumukunde J. Tindimwebwa J. V. B. Kwizera A. BMC Res. Notes. 2017;10:349. doi: 10.1186/s13104-017-2695-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S. J. Isufi K. Wilkins L. E. Lipecki J. Fullam E. Gibson M. I. Biomacromolecules. 2018;19:256–264. doi: 10.1021/acs.biomac.7b01561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward M. Sanchez M. Elasri M. O. Lowe A. B. J. Appl. Polym. Sci. 2006;101:1036–1041. doi: 10.1002/app.23269. [DOI] [Google Scholar]
- Lu G. Wu D. Fu R. React. Funct. Polym. 2007;67:355–366. doi: 10.1016/j.reactfunctpolym.2007.01.008. [DOI] [Google Scholar]
- Keely S. Rawlinson L. A. B. Haddleton D. M. Brayden D. J. A. Pharm. Res. 2008;25:1193. doi: 10.1007/s11095-007-9501-3. [DOI] [PubMed] [Google Scholar]
- Kamlangmak N. Eiamprasert U. Chaiyasat P. Chaiyasat A. ACS Appl. Polym. Mater. 2021;3:3549–3559. doi: 10.1021/acsapm.1c00444. [DOI] [Google Scholar]
- Ghavami S. Bardajee G. R. Mirshokraie A. Didehban K. A. Polym. Sci., Ser. B. 2019;61:376–386. doi: 10.1134/S1560090419030047. [DOI] [Google Scholar]
- Hayashi H. Iijima M. Kataoka K. Nagasaki Y. Macromolecules. 2004;37:5389–5396. doi: 10.1021/ma049199g. [DOI] [Google Scholar]
- Fujii S. Randalla D. P. Armes S. P. Langmuir. 2004;20:11329–11335. doi: 10.1021/la048473x. [DOI] [PubMed] [Google Scholar]
- Zhang M. Liu Y. Peng J. Liu Y. Liu F. Ma W. Ma L. Yu C. Y. Wei H. Polym. Chem. 2020;11:6139–6148. doi: 10.1039/D0PY01076F. [DOI] [Google Scholar]
- Cao Z. Zhou X. Wang G. ACS Appl. Mater. Interfaces. 2016;8:28888–28896. doi: 10.1021/acsami.6b10360. [DOI] [PubMed] [Google Scholar]
- Macková H. Horák D. J. Polym. Sci., Part A: Polym. Chem. 2006;44:968–982. doi: 10.1002/pola.21223. [DOI] [Google Scholar]
- Macková H. Králová D. Horák D. J. Polym. Sci., Part A: Polym. Chem. 2007;45:5884–5898. doi: 10.1002/pola.22341. [DOI] [Google Scholar]
- Zasońska B. A. Šálek P. Procházková J. Müllerová S. Svoboda J. Petrovský E. Proks V. Horák D. Šafařík I. Sci. Rep. 2019;9:1543. doi: 10.1038/s41598-018-38012-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. Yang X. Wang Y. Polym. Int. 2007;56:905–913. doi: 10.1002/pi.2223. [DOI] [Google Scholar]
- Vaihinger D. Landfester K. Kräuter I. Brunner H. Tovar G. E. M. Macromol. Chem. Phys. 2002;203:1965–1973. doi: 10.1002/1521-3935(200209)203:13<1965::AID-MACP1965>3.0.CO;2-C. [DOI] [Google Scholar]
- Fehrenbacher U. Ballauff M. Macromolecules. 2002;35:3653–3661. doi: 10.1021/ma011985n. [DOI] [Google Scholar]
- De La Vega J. C. Elischer P. Schneider T. Häfeli U. O. Nanomedicine. 2013;8:265–285. doi: 10.2217/nnm.12.210. [DOI] [PubMed] [Google Scholar]
- Brus J. Solid State Nucl. Magn. Reson. 2000;16:151–160. doi: 10.1016/S0926-2040(00)00061-8. [DOI] [PubMed] [Google Scholar]
- Lee K. C. Lee S. E. Song B. K. Macromol. Res. 2002;10:140–144. doi: 10.1007/BF03218263. [DOI] [Google Scholar]
- Wu X. Pelton R. H. Haielec A. E. Woods D. R. McPhee W. Colloid Polym. Sci. 1994;272:467–477. doi: 10.1007/BF00659460. [DOI] [Google Scholar]
- Qum H. Gong F. Ma G. Su Z. J. Appl. Polym. Sci. 2007;105:1632–1641. doi: 10.1002/app.26199. [DOI] [Google Scholar]
- Ye Q. He W. Ge X. Jia H. Liu H. Zhang Z. J. Appl. Polym. Sci. 2002;86:2567–2573. doi: 10.1002/app.11170. [DOI] [Google Scholar]
- Lee K. C. Lee S. E. Song B. K. Macromol. Res. 2002;10:140–144. doi: 10.1007/BF03218263. [DOI] [Google Scholar]
- Thomson B. Rudin A. Lajoie G. J. Polym. Sci., Part A: Polym. Chem. 1995;33:345–357. doi: 10.1002/pola.1995.080330301. [DOI] [Google Scholar]
- Okaya T. Kikuchi K. Suzuki A. Ikeda N. Polym. Int. 2005;54:143–148. doi: 10.1002/pi.1658. [DOI] [Google Scholar]
- Lee A. Tsai H. Y. Yates M. Z. Langmuir. 2010;26:18055–18060. doi: 10.1021/la1039128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D. Dimonie V. L. Sudol E. D. El-Asser M. S. J. Appl. Polym. Sci. 2002;84:2692–2709. doi: 10.1002/app.10592. [DOI] [Google Scholar]
- Ye Q. Ge X. Liu H. Jia H. He W. Zhang Z. J. Macromol. Sci., Part A: Pure Appl. Chem. 2002;39:545–556. doi: 10.1081/MA-120004246. [DOI] [Google Scholar]
- Sáenz J. M. Asua J. M. J. Polym. Sci., Part A: Polym. Chem. 1996;34:1977–1992. doi: 10.1002/(SICI)1099-0518(19960730)34:10<1977::AID-POLA16>3.0.CO;2-F. [DOI] [Google Scholar]
- Kim O. H. Lee K. Kim K. Lee B. H. Choe S. Polymer. 2006;47:1953–1959. doi: 10.1016/j.polymer.2006.01.025. [DOI] [Google Scholar]
- Thompson B. Rudin A. Lajoie G. J. Appl. Polym. Sci. 1996;59:2009–2028. doi: 10.1002/(SICI)1097-4628(19960328)59:13<2009::AID-APP6>3.0.CO;2-L. [DOI] [Google Scholar]
- Souto-Maior R. M. Tavares M. I. B. Monteiro E. E. C. Annals of Magnetic Resonance. 2005;4:69–72. [Google Scholar]
- Silverstein M. R., Webster F. X. and Kiemple J. D., Chapter 4 – Carbon 13NMR Spectroscopy in Spectrometric identification of organic compounds, Wiley, 7th edn, 2005, p. 226 [Google Scholar]
- Fairchild E. H. J. Am. Oil Chem. Soc. 1982;59:305–308. doi: 10.1007/BF02662232. [DOI] [Google Scholar]
- Rawlinson L. A. B. ÓGara J. P. Jones D. S. Brayden D. J. J. Med. Microbiol. 2011;60:968–976. doi: 10.1099/jmm.0.025619-0. [DOI] [PubMed] [Google Scholar]
- Grace J. L. Huang J. X. Cheah S. E. Truong N. P. Cooper M. A. Li J. Davis T. P. Quinn J. F. Velkov T. Whittaker M. R. RSC Adv. 2016;6:15469–15477. doi: 10.1039/C5RA24361K. [DOI] [PMC free article] [PubMed] [Google Scholar]