

SUMMARY
The mechanisms underlying subcellular targeting of cAMP-generating adenylyl cyclases and processes regulated by their compartmentalization are poorly understood. Here, we identify Ankmy2 as a repressor of the Hedgehog pathway via adenylyl cyclase targeting. Ankmy2 binds to multiple adenylyl cyclases, determining their maturation and trafficking to primary cilia. Mice lacking Ankmy2 are mid-embryonic lethal. Knockout embryos have increased Hedgehog signaling and completely open neural tubes showing co-expansion of all ventral neuroprogenitor markers, comparable to the loss of the Hedgehog receptor Patched1. Ventralization in Ankmy2 knockout is completely independent of the Hedgehog pathway transducer Smoothened. Instead, ventralization results from the reduced formation of Gli2 and Gli3 repressors and early depletion of adenylyl cyclase III in neuroepithelial cilia, implicating deficient pathway repression. Ventralization in Ankmy2 knockout requires both cilia and Gli2 activation. These findings indicate that cilia-dependent adenylyl cyclase signaling represses the Hedgehog pathway and promotes morphogenetic patterning.
Graphical Abstract
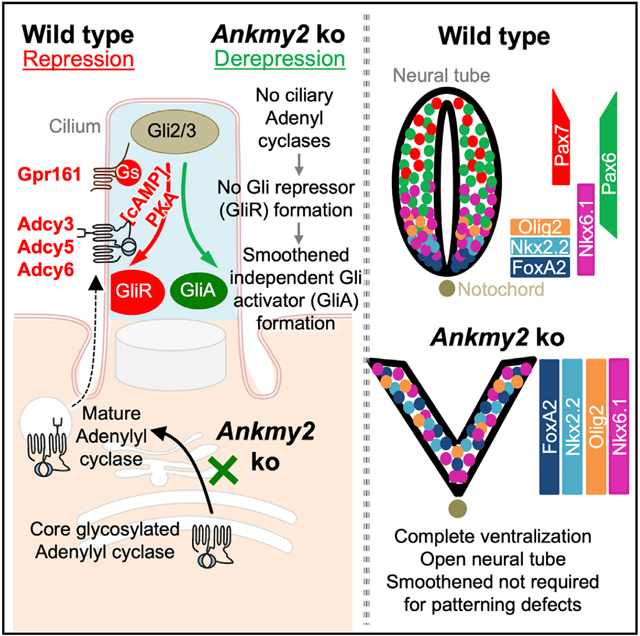
In Brief
Somatilaka et al. identify Ankmy2 as a repressor of the Hedgehog pathway. Ankmy2 knockout mouse show complete neural tube ventralization independently of Smoothened but requiring cilia and Gli2. Ankmy2 regulates maturation of adenylyl cyclases and trafficking to cilia, implicating cilia-dependent adenylyl cyclase signaling in Gli-repressor formation and morphogenetic patterning.
INTRODUCTION
Hedgehog (Hh) signaling in vertebrates is an excellent system to study subcellular cAMP signaling processes and the role of compartmentalized signaling. The primary cilium is a microtubule-based dynamic cellular appendage that mediates extra-cellular signaling, particularly with respect to vertebrate Hh signaling (Goetz and Anderson, 2010; Briscoe and Thérond, 2013; Kopinke et al., 2020). Binding of Hh to its receptor Patched (Ptch1) triggers removal of Ptch1 from cilia and promotes enrichment and activation of Smoothened (Smo)—the pathway transducer—in cilia, resulting in the generation of Gli transcriptional activator (GliA) (Corbit et al., 2005; Rohatgi et al., 2007). In contrast, the basal repression machinery of Hh signaling involves protein kinase A (PKA)-mediated partial proteolysis of full-length Gli2/3 into Gli2/3 repressor (GliR) forms in a cilia-dependent manner (Tuson et al., 2011; Mukhopadhyay and Rohatgi, 2014).
The neural tube is patterned during early embryonic development by Hh secreted from the notochord (Dessaud et al., 2008). Lack of cilia prevents ventral patterning and expansion of dorsal markers at the expense of ventral cell types or “dorsalization” (Huangfu et al., 2003), similar to lack of Hh (Chiang et al., 1996). Increased Hh signaling from lack of Ptch1 causes ectopic specification of ventral cell types at the expense of lateral and dorsal cell types or “ventralization” (Goodrich et al., 1997). Complete loss of both PKA catalytic subunits α/β (PKA null) also result in embryonic lethality by E9 and full ventralization of the neural tube (Tuson et al., 2011). Thus, high Hh signaling phenotypes can result from either increased pathway activation in Ptch1 knockouts or loss of PKA-mediated repression.
We previously described that the cilia-localized orphan GPCR, Gpr161, functions as a repressor of Hh signaling during early neural tube development in mice (Mukhopadhyay et al., 2013). Mice knockout for Gpr161 are embryonic lethal by embryonic day 10.5 (E10.5) and exhibit increased Hh signaling and ventralization throughout the rostrocaudal extent of the neural tube, albeit weaker than the loss of PKA (Hwang and Mukhopadhyay, 2015). Gpr161 determines Gli3R formation via cAMP signaling. Interestingly, Gpr161 C-tail has been proposed to be an A-kinase anchoring protein for PKA activation in cilia by binding to PKA regulatory subunit RIα and RIβ (Bachmann et al., 2016). In addition, the PKA regulatory subunit RIα localizes to cilia (Bachmann et al., 2016; Mick et al., 2015), whereas PKA regulatory RII subunits localize to centrosomes (Tuson et al., 2011; Saade et al., 2017; Barzi et al., 2010). As cAMP production regulates PKA activation, studying repressive pathways regulating Hh signaling might provide novel insights into both spatiotemporal regulation of cAMP signaling and Hh pathway derepression.
Other than GPCRs, another less-studied and plausible level of regulation of cAMP signaling might arise from spatiotemporal distribution of adenylyl cyclases (ACs). At least three of the ACs, types III, V, and VI (AC3/5/6 or ADCY3/5/6) are localized to cilia (Mick et al., 2015; Choi et al., 2011; Bishop et al., 2007). Overexpression of ADCY5/6 in the developing chicken neural tube only partially represses the Hh pathway (Vuolo et al., 2015), whereas individual knockdown or knockouts are only partially penetrant in increased Hh signaling phenotypes and are not embryonic lethal (Vuolo et al., 2015; Wang et al., 2009), unlike PKA null mutants (Tuson et al., 2011). Here, we identify the ankyrin-repeat and MYND-domain-containing protein, Ankmy2 in maturation and trafficking of ACs to primary cilia. By generating Ankmy2 mouse knockouts, we demonstrate that Ankmy2 is one of the strongest known repressors of Hh pathway during neural tube development. Our results suggest that repression of Hh signaling during neural tube development should not be considered as an inherent background state, but instead must be actively maintained by cilia-regulated AC signaling, lack of which causes Gli2 activation independent of Smo.
RESULTS
Identification of Ankmy2 as a Common Interacting Factor for Ciliary ACs
To identify factors that might regulate subcellular trafficking of ADCY3, we performed tandem affinity purification followed by mass spectrometry (TAP-MS) of stably expressed ACIII/ADCY3LAP in NIH 3T3 cells (C-term LAP, X-Stag-PreScission-EGFP). The ankyrin-repeat and MYND-domain-containing protein, Ankmy2, was identified among ADCY3LAP interacting partners (Figures 1A and 1B; Table S1). Reciprocal TAP-MS of LAPANKMY2 in NIH 3T3 cells (N-term LAP, EGFP-TEV-Stag-X) identified another AC (ADCY6) as an interacting partner, whereas TAP-MS of LAPADCY6 also detected Ankmy2 (Figure 1A; Table S1). Endogenous Ankmy2 could be detected by immunoblotting upon tandem affinity purification of LAP-tagged ADCY3, ADCY5, and ADCY6, suggesting Ankmy2 is a common interacting factor for these cilia-localized ACs (Figure 1C). As reported before (Saita et al., 2014), stably expressing LAPANKMY2 in NIH 3T3 cells was confirmed to be a predominantly cytosolic protein, but not enriched in cilia, using subcellular fractionation and immunofluorescence (Figures S1A and S1B). Endogenous Ankmy2 also fractionated into the cytosolic fraction (Figure S1A). Thus, Ankmy2 is a common interacting factor for multiple ACs.
Figure 1. Ankmy2 Knockout Mice Are Embryonic Lethal at 10 to 12-Somite Stage and Exhibit Completely Open Neural Tubes.
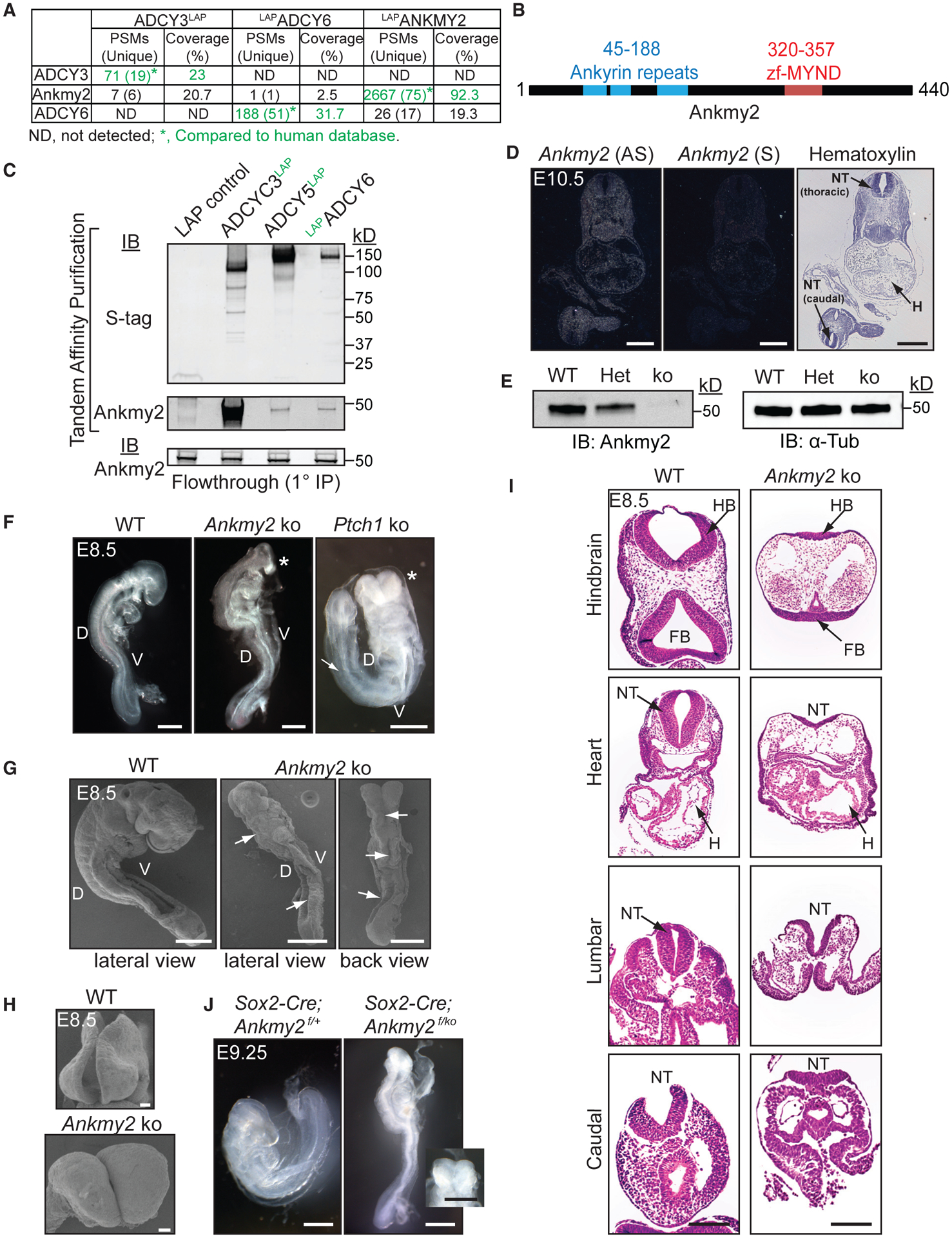
(A) TAP-MS of LAP-tagged ACs or ANKMY2 stably expressed in NIH 3T3 cells. ADCY3/6 and ANKMY2 clones were human ORFs, and peptides were compared to the human database.
(B) Ankmy2 protein domains. zf-MYND, zinc finger MYND domain.
(C) TAP of ADCY3/5/6 detects Ankmy2. Flow through (0.35%) from the initial anti-GFP IP below. n = 4 independent experiments.
(D) Radioisotopic RNA in situ hybridization using antisense or sense (AS or S) riboprobes for Ankmy2 in transverse sections of wild-type E10.5 embryos.
(E) Immunoblotting of whole-embryo lysates at E8.5 in wild-type (WT), heterozygote (Het), and Ankmy2 knockout (ko). α-tubulin, loading control. n = 3 embryos per genotype.
(F) Bright-field images of WT (16 somites), Ankmy2 ko (10–12 somites), and Ptch1 ko (14–16 somites) whole-mount embryos dissected at E8.5. Asterisks, rostral malformations. n > 10 (WT, Ankmy2 ko), 6 (Ptch1 ko).
(G and H) Scanning electron micrographs of WT (12–14 somites) (G) and Ankmy2 ko (12 somites) and en face views of head regions (H). Arrows, open neural tube. n = 3 (WT), 5 (ko).
(I) HE-stained transverse sections of WT (16 somites) and Ankmy2 ko (12 somites). n = 4 per genotype.
(J) Sox2-Cre; Ankmy2f/ko (12 somites) embryos dissected at E9.25 recapitulates Ankmy2 ko compared with control (20–22 somites) (inset: en face head view). n = 2 per genotype.
Scale: D, 500 μm; F and G, I and J (including inset), 200 μm; H, 20 μm.
Abbreviations: PSM, peptide-to-spectral matches; D, Dorsal; V, Ventral; HB, hindbrain; FB, Forebrain; H, Heart; NT, neural tube; WT, wild-type.
Ankmy2 Knockout Mice Are Embryonic Lethal at 10 to 12-Somite Stage
As we detected binding between ACs and Ankmy2, we hypothesized that Ankmy2 might regulate the Hh pathway. Ankmy2 was expressed in low abundance throughout the mid-gestational period of development (Figure S1C) and ubiquitously at E10.5 using radiometric RNA in situ hybridization (Figure 1D). We generated a knockout mouse model deleting exon 3 of Ankmy2, which is predicted to result in a frameshift after the initial 44 amino acids and truncation after a further 46 amino acids unrelated to the actual protein sequence (Figures 1B, S1D, and S1E). Ankmy2 protein and transcripts were absent in the homozygous knockout embryos (Ankmy2 ko) by immunoblotting and qRT-PCR, respectively at E8.5 (Figures 1E and S1F), confirming the present allele to be a null allele.
Interestingly, homozygous knockout embryos were arrested at 10–12 somites (E8.5) (Figure 1F). In contrast to wild-type littermates (Murdoch and Copp, 2010), the knockout embryos did not turn and had a completely open neural fold (craniorachischisis) at 10 to 12-somite stage (Figures 1F,1G, and 1I). Knockout embryos had extensive rostral malformations and open brain regions (Figures 1F–1H) that bore a striking resemblance to PKA null embryos (Tuson et al., 2011) or Ptch1 knockouts (Figure 1F) (Goodrich et al., 1997). However, Ptch1 knockout embryos arrest around E9-E9.5 (Goodrich et al., 1997; Bai et al., 2002) with at least 14–16 somites (Figure 1F), possibly due to the compensatory function of Ptch2 (Alfaro et al., 2014). The Ankmy2 heterozygous embryos had no characteristic defects and essentially resembled wild-type embryos.
To rule out trophoblast-intrinsic defects that could potentially affect embryogenesis (Perez-Garcia et al., 2018), we used Sox2-Cre to delete Ankmy2 conditionally in the epiblast (Figures 1J and S1G), while leaving expression intact in the trophoblast-derived cells of the placenta and the visceral yolk sac endoderm. The Sox2-Cre; Ankmy2f/ko embryos were embryonic lethal by 10 to 12-somite stage similar to the knockout, suggesting that Ankmy2 deletion in embryonic tissues caused the observed phenotypes (Figure 1J).
Ankmy2 Knockout Embryos Exhibit Increased Hh Signaling
Ankmy2 has been previously reported to positively regulate Hh signaling using knockdown and overexpression studies in cultured cells (Saita et al., 2014). However, the rostral malformations in Ankmy2 knockouts resembled PKA null and Ptch1 knockouts that exhibit high Hh signaling. Levels of the Gli1, Ptch1, and Ptch2 transcripts and Gli1 protein were increased in the knockout compared with those of wild-type littermates at E8.5 (Figures 2A–2D). In the Ankmy2 knockouts, Ptch1 expression was sharply elevated throughout the rostrocaudal extent in the ventral spinal cord, the ventral somatic and intermediate mesoderm, and the splanchic mesoderm compared with that of the wild-type littermates at E8.5 by RNA in situ hybridization in mid-trunk and caudal regions (Figures 2E and S2A). Ventral hindbrain also showed elevated levels of Ptch1 expression in the knockouts (Figure S2A). Similarly, Gli1 expression was expanded in the dorsal neural tube and increased in the somatic mesoderm in the mid-trunk and caudal regions, with respect to control littermates at E8.5 by RNA in situ hybridization (Figures 2F and S2B). Around this embryonic stage, Shh expression is restricted to the notochord. However, in Ptch1 knockouts, high expression of Shh is seen in the neural tube (Goodrich et al., 1997). In contrast, in Ankmy2 knockouts, Shh expression was not increased and confined to the notochord at E8.5 throughout the neural tube (Figures 2G, S2C, and S2D), with occasionally expanded domains only in the hindbrain (Figure S2C).
Figure 2. Ankmy2 Knockout Embryos Exhibit Increased Hh Signaling.
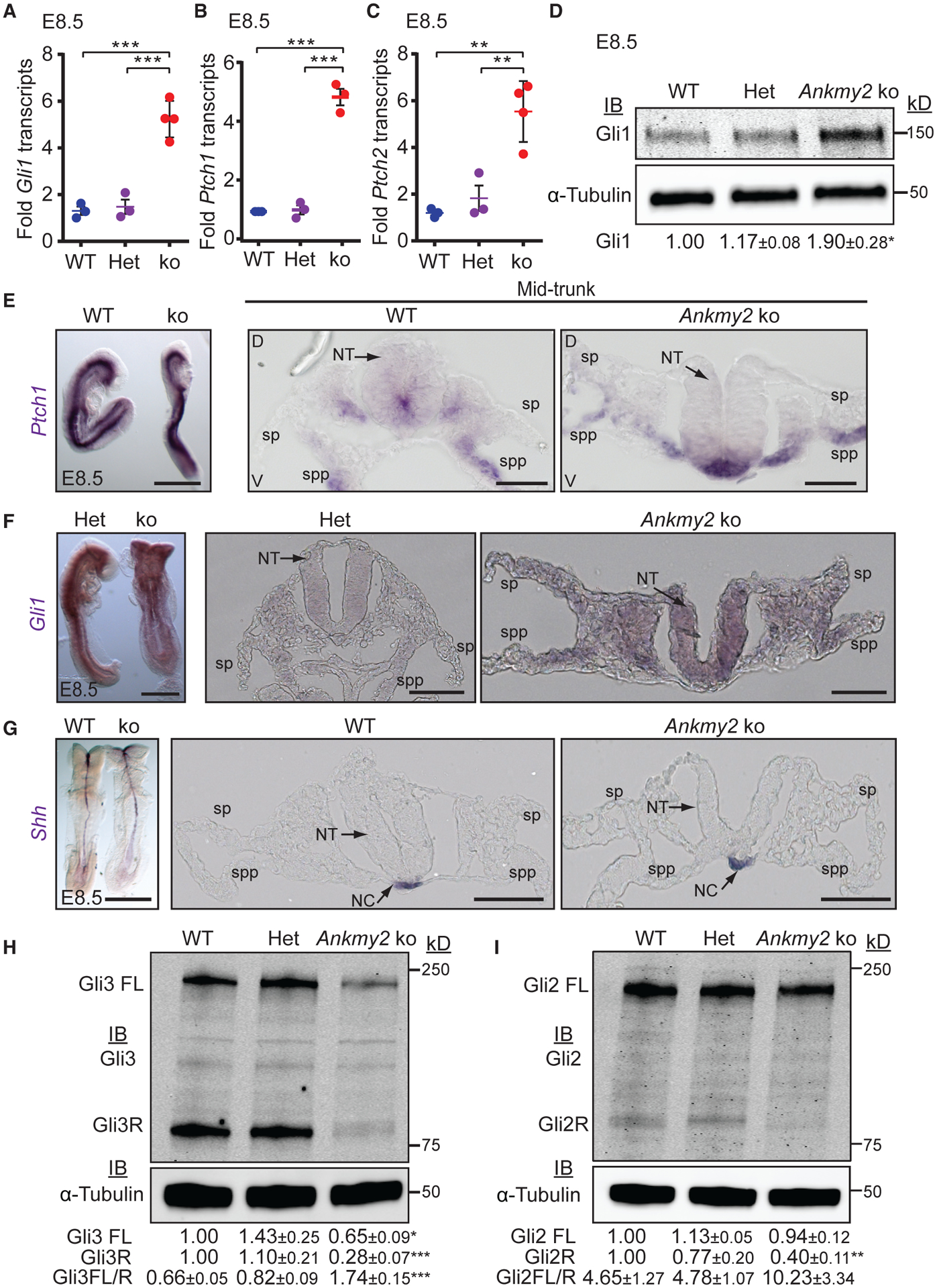
(A–C) qRT-PCR of Gli1 (A), Ptch1 (B), and Ptch2 (C) transcript levels in whole-embryo extracts at E8.5. n = 3–4 embryos each. Data shown as mean ± SEM.
(D) Immunoblotting in whole-embryo lysates at E8.5. n = 3 embryos per genotype in each experiment. Data shown as mean ± SEM (4 experiments) for Gli1 levels normalized to α-tubulin.
(E–G) RNA in situ hybridization for Ptch1 (E), Gli1 (F), Shh (G) showed increased expression in the Ankmy2 ko at E8.5. Left panel shows lateral or back views, whereas right panels are transverse sections of the mid-trunk region. n = 5 (control), 4 (ko) embryos (Ptch1); n = 3 (control), 2 (ko) embryos (Gli1); n = 4 (control), 5 (ko) embryos (Shh). Hindbrain and caudal regions in Figure S2.
(H and I) Immunoblotting for Gli3 (H) or Gli2 (I) and α-tubulin in whole-embryo lysates at E8.5 shows reduced Gli3R and Gli2R levels and increased Gli3FL to Gli3R ratio in ko. n = 3 embryos per genotype in each experiment. Data shown as mean ± SEM (4 [H] and 3 [I] experiments) normalized to α-tubulin.
Scale: (E–G) left panels, 500 μm; right panels, 50 μm.
Abbreviations: sp, somatopleure; spp, splanchnopleure; NT, neural tube; NC, notochord; FL, full length; R, repressor. *, p < 0.05; **, p < 0.01; ***, p < 0.001, as determined by unpaired t test with respect to wild-type embryos or as indicated. Also see Figure S2.
Ankmy2 Knockout Mice Exhibit Gli3/Gli2-Processing Defects
In the absence of Hh ligand, PKA-mediated phosphorylation of Gli3 and Gli2 results in partial proteolytic processing into Gli3R and Gli2R, respectively (Wang et al., 2000; Tempé et al., 2006; Niewiadomski et al., 2014). In Ankmy2 knockout embryos, the extent of Gli3 processing into Gli3R at E8.5 whole-embryonic extracts was strongly reduced compared with that of wild-type (Figure 2H), similar to Gpr161 (Mukhopadhyay et al., 2013) and PKA null knockouts (Tuson et al., 2011), suggesting that Ankmy2 regulates the processing of Gli3 into Gli3R. Levels of Gli2R were also reduced (Figure 2I). Upon high Hh signaling, activated full-length Gli proteins are formed that are unstable (Chen et al., 2009; Humke et al., 2010; Jia et al., 2009; Wang et al., 2010; Wen et al., 2010). The full-length form of Gli3 was also significantly reduced in Ankmy2 knockouts (Figure 2H), although levels of Gli2 full-length protein remained unchanged (Figure 2I). Gli2 transcripts were unaffected (Figure S2E). Gli3 full-length to Gli3R ratios were significantly increased in Ankmy2 knockout embryos with respect to wild-type (Figure 2G). A ~50% decrease in Gli3 transcript levels in Ankmy2 knockout embryos (Figure S2F), a likely consequence of activation of the Hh pathway (Wang et al., 2000; Bai et al., 2004), is not sufficient to explain the selective decrease in Gli3R levels. In summary, the Ankmy2 knockouts exhibit defects in GliR processing accompanying increased Hh signaling.
Ankmy2 Knockout Mice Exhibit a Ventralized Neural Tube
We next determined regions in the Ankmy2 knockout embryos that showed altered Hh signaling. When dissected at E9.5, Hh-dependent ventral cell types were ectopically specified at the expense of lateral and dorsal cell types (ventralization) in the Ankmy2 knockouts. Specifically, floor plate progenitors expressing FoxA2, p3 progenitors expressing Nkx2.2, pMN progenitors expressing Olig2, and p3/pMN/p2 progenitors expressing Nkx6.1 showed enlarged expression domains that expanded fully into dorsal regions throughout the rostrocaudal extent of the spinal cord and hindbrain (Figure 3A). Expression of these ventral progenitor markers predominantly overlapped with each other alongside some intermixing (Figures 3B and 3C). In contrast, the dorsolateral neural tube marker Pax6 was completely absent in the Ankmy2 knockouts (Figures 3A and S3). Neural tube ventralization and complete lack of closure were similar in extent and severity to Ptch1 knockouts (Figure S3) (Goodrich et al., 1997). The Sox2-Cre; Ankmy2ko/f embryos also had ventralization similar in extent and severity to the knockout, suggesting that Ankmy2 deletion in epiblast caused the observed phenotypes (Figure S3). Thus, Ankmy2 knockout embryos exhibit severely ventralized neural tube throughout the rostrocaudal extent and co-expression of most ventral markers.
Figure 3. Ankmy2 Knockout Mice Exhibit a Ventralized Neural Tube.
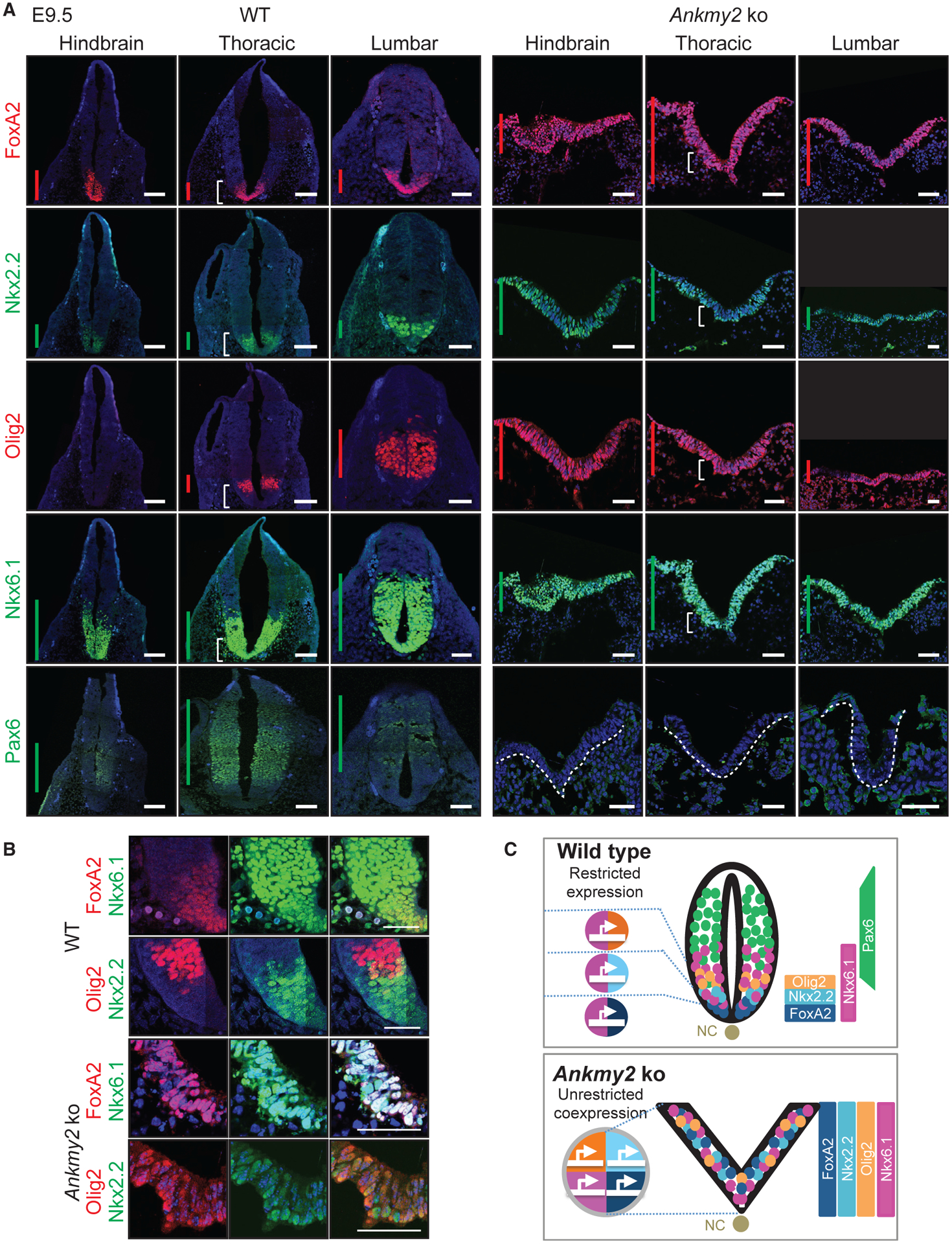
(A) Neural tube sections (hindbrain, thoracic, and lumbar regions) from WT (20 somites) and Ankmy2 ko (12 somites) dissected at E9.5 stained for designated neural tube markers. N > 10 embryos for Ankmy2 ko for FoxA2, Nkx6.1, and Pax6 immunostaining. n = 5 embryos for Ankmy2 ko for Nkx2.2 and Olig2 immunostaining. Vertical bars show the extent of dorsoventral expression of markers.
(B) Corresponding regions designated by white brackets in (A) show predominantly overlapping and some intermixing of ventral progenitor marker expression in the Ankmy2 ko.
(C) Cartoon showing unrestricted ventral progenitor transcription factor co-expression and complete ventralization of the open neural tube in the Ankmy2 ko. Abbreviations: NC, notochord.
All images were counterstained with DAPI. Scale: (A and B), 50 μm. Also see Figure S3.
Hh Pathway Activation in Ankmy2 Knockouts Is Independent of Smo
To test if the severe ventralization phenotype in Ankmy2 knockouts was dependent on Smo activation, we generated double knockouts of Ankmy2 and Smo. Mice knockout for Smo exhibit embryonic arrest by E9.5, defects in the ventral midline, including cyclopia (Zhang et al., 2001) and lack of expression of all ventral progenitor and floor plate markers (Figure 4A) (Caspary et al., 2007; Norman et al., 2009). Double knockouts of Smo and Ptch1 resemble Smo knockouts, suggesting that Smo is epistatic to Ptch1 (Zhang et al., 2001). However, double knockouts of Ankmy2 and Smo arrested by 10–12 somites and had rostral malformations similar to that of Ankmy2 knockouts (Figure 4A). Similarly, the expansion of ventral identity markers, FoxA2, Nkx2.2, Olig2, and Nkx6.1 occurred rostrocaudally as in Ankmy2 knockouts (Figures 4A and S4A). FoxA2 transcripts were similarly upregulated in Ankmy2 single and Ankmy2; Smo double knockout embryos by qRT-PCR (Figure 4B). Correspondingly, while the dorsolateral neural tube marker Pax6 was fully dorsalized rostrocaudally in Smo knockout, Pax6 was completely absent in Ankmy2; Smo double knockouts, similar to Ankmy2 knockouts (Figures 4A and S4A). Ankmy2 is fully epistatic to Smo even for maximal pathway activation, which suggests GliA generation independent of Smo (Figure 4C).
Figure 4. Hh Pathway Activation in Ankmy2 Knockouts Is Independent of Smo.
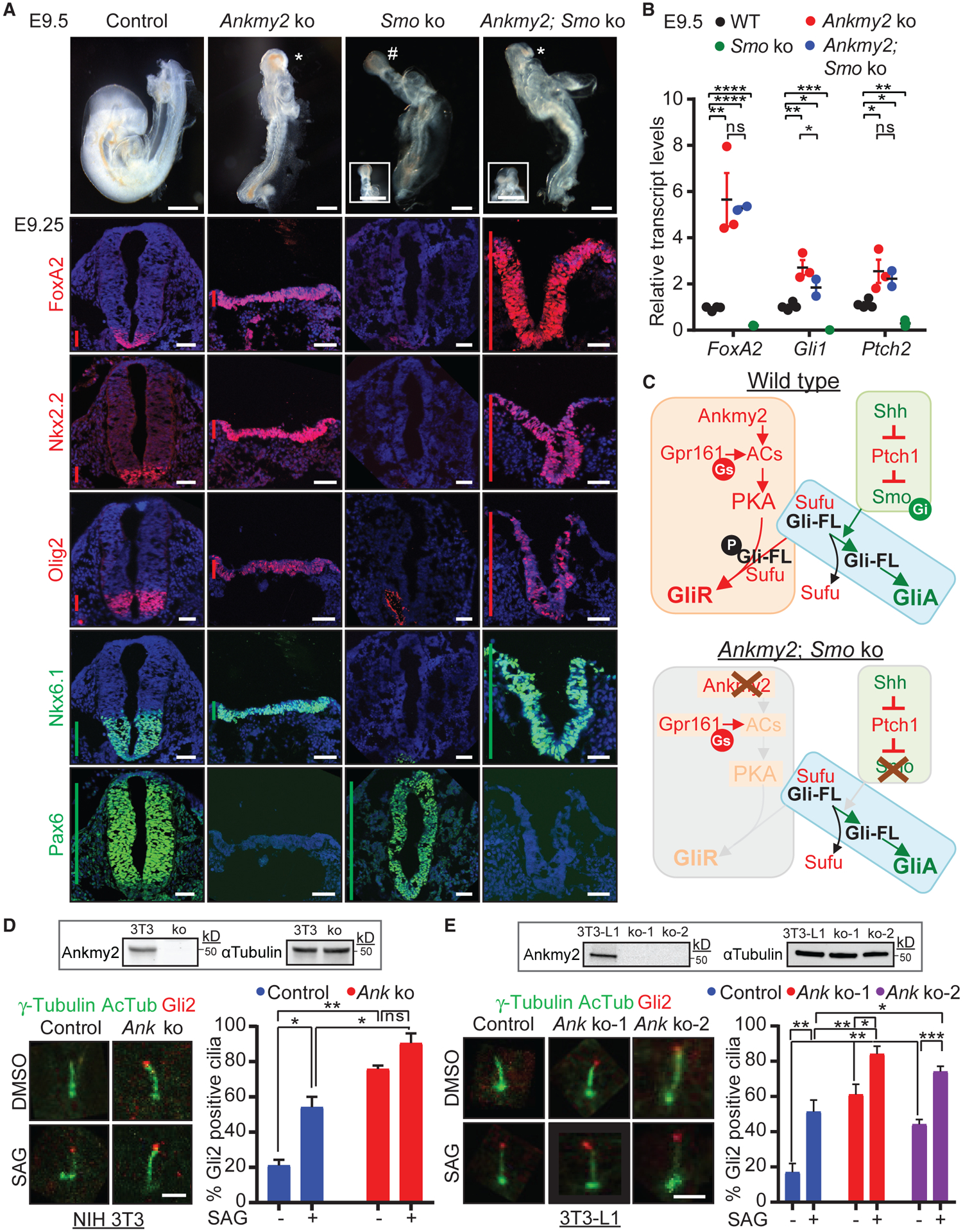
(A) Topmost panels show bright-field images of WT (18–20 somites), Ankmy2 ko (10–12 somites), Smo ko (10–12 somites), and Smo; Ankmy2 double ko (10–12 somites) whole-mount embryos dissected at E9.5. #, cyclopia; *, open cephalic neural tube. Inset shows en face views highlighting cyclopia or open cephalic neural tube. Bottom panels show thoracic neural tube horizontal sections immunostained for designated markers from control (Ankmy2WT/WT SmoWT/ko; 16–18 somites), Ankmy2 ko (10–12 somites), Smo ko (10 somites), and Smo; Ankmy2 double ko (10–12 somites) littermate embryos dissected at E9.25. Hindbrain and lumbar sections are shown in Figure S4A. n = 3 embryos for Smo ko and n = 5 embryos for double ko. Vertical bars show the extent of dorsoventral expression of markers.
(B) qRT-PCR of FoxA2, Gli1, and Ptch2 transcript levels normalized to Gapdh in whole-embryo extracts at E9.5. n = 2–4 embryos each. Data shown as mean ± SD.
(C) Cartoon showing model for GliA generation independent of Smo.
(D and E) Control and CRISPR-based Ankmy2 (Ank) ko NIH-3T3 cells (Figure S4) were starved for 24 h upon confluence and were treated for further 24 h ± SAG (500 nM) (D). Control and two independent CRISPR-based Ankmy2 (Ank) ko clones of 3T3-L1 cells (Figure S4) were treated upon confluence for 48 h ± SAG (500 nM) (E). After fixation, cells were immunostained for Gli2, γ-tubulin (centrosome), and acetylated tubulin (AcTub, cilia). Representative immunofluorescence images (left) and quantification (right) showing Gli2 enriched at the tips of cilia in untreated Ankmy2 ko cells. Immunoblotting shows a complete lack of Ankmy2 in the ko cells. Quantification from 60–100 (D) or 120–125 cells (E) total from 2 (D) or 4 (E) experiments. Data shown as mean ± SD.
Immunostained images in (A) were counterstained with DAPI. Scale: (A), bright-field images, 200 μm; insets, 250 μm; horizontal sections, 50 μm; (D and E), 2 μm. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 as determined by unpaired t test. ns, not significant. Also see Figure S4.
Gli2 Accumulation in Resting Ankmy2 Knockout Cells
Our genetic epistasis experiments between Ankmy2 and Smo suggested that Hh pathway hyperactivation occurs in Ankmy2 knockout in a Smo-independent manner. However, the predominant view in the field is that Hh triggers Gli2 activation in a Smo-dependent manner, which results in the accumulation of Gli2 in ciliary tips (Haycraft et al., 2005; Chen et al., 2009; Kim et al., 2009). We generated an Ankmy2 knockout clonal NIH 3T3 line and two independent Ankmy2 knockout clonal 3T3-L1 lines for testing Gli2 ciliary localization with respect to Hh signaling (Figures 4D, 4E, and S4B). While ~20% of cilia tips in control cells had detectable Gli2 staining, ~75% of Ankmy2 knockout cilia in NIH 3T3 cells and 45%–60% of Ankmy2 knockout cilia in 3T3-L1 cells had Gli2 staining in the absence of SAG (Figures 4D and 4E). Treatment with SAG further enhanced Gli2 accumulation in Ankmy2 knockout cilia in 3T3-L1 cells (Figure 4E). A similar accumulation of Gli2 in cilia has also been observed in resting PKA null MEFs (Tuson et al., 2011). Gli2 accumulation in ciliary tips of Ankmy2 knockout and PKA null cells suggests that Ankmy2 and PKA serve in the same pathway to prevent precocious Gli2 activation.
In contrast to knockout embryos (Figure 2), however, Ankmy2 knockout 3T3 and 3T3-L1 cells had no significant upregulation of basal Ptch1/Gli1 transcript or Gli1 protein levels and showed normal Gli3 processing in resting cells or upon pathway activation by SAG (Figures S4C and S4D). Persistence of Gli3R in Ankmy2 knockout cultured cells explains the lack of basal upregulation of Gli1 levels in these cell lines, despite Gli2 accumulation in ciliary tips (see Discussion).
Hh Pathway Activation in Ankmy2 Knockout Requires Gli2
As high Hh signaling in Ankmy2 knockout was Smo-independent, we tested if downstream factors in Hh pathway regulate ventralization by generating Ankmy2; Gli2 double knockouts. Gli2 single knockouts are perinatal lethal (Bai and Joyner, 2001). The Ankmy2; Gli2 double knockouts survived till the 26–28 somite stage, past the 10–12 somite stage embryonic lethality period for Ankmy2 single knockouts. The general morphology of Ankmy2; Gli2 double knockouts resembled that of Gli2 single knockouts, except that the embryos exhibited open neural tube in the brain (Figure 5A). At the thoracic and lumbar levels, by 26–28 somites, the neural tube was closed with sparse FoxA2 in floor plate as seen in Gli2 single knockouts (Figures 5A, S5A, and S5B). The dorsolateral markers Pax7 and Pax6 were restricted to their normal domains as in the wild-type, with some persistent but limited ventralization of Nkx6.1 domain (Figures 5A, S5A, and S5B). Such limited and intermediate-level Nkx6.1 ventralization in Ankmy2; Gli2 double knockout suggests a lack of Gli3R formation (Persson et al., 2002) contributing to the phenotype. In contrast, the hindbrain of the double knockout remained fully open (exencephaly) with ventralized Nkx6.1 and absent Pax7 domains similar to Ankmy2 knockout. However, unlike Ankmy2 knockout, FoxA2 levels were reduced (Figures 5A and S5A), while Pax6 was restored (Figure S5A). Thus, most of the ventralization and neural tube closure defects in the Ankmy2 knockouts were overcome in the absence of Gli2, while the brain defects persisted, suggesting a possible role for Gli3 in this region (Yu et al., 2009; Liu et al., 2015; Shimada et al., 2019). Similarly, exencephaly persisted in Ptch1 knockouts upon concomitant Gli2 loss, although spinal cord ventralization was rescued (Liu et al., 2015; Bai et al., 2002). Thus, strong ventralization in the spinal cord region of Ankmy2 knockout is dependent on Gli2.
Figure 5. Hh Pathway Activation in Ankmy2 Knockouts Requires Gli2 and Ift88.
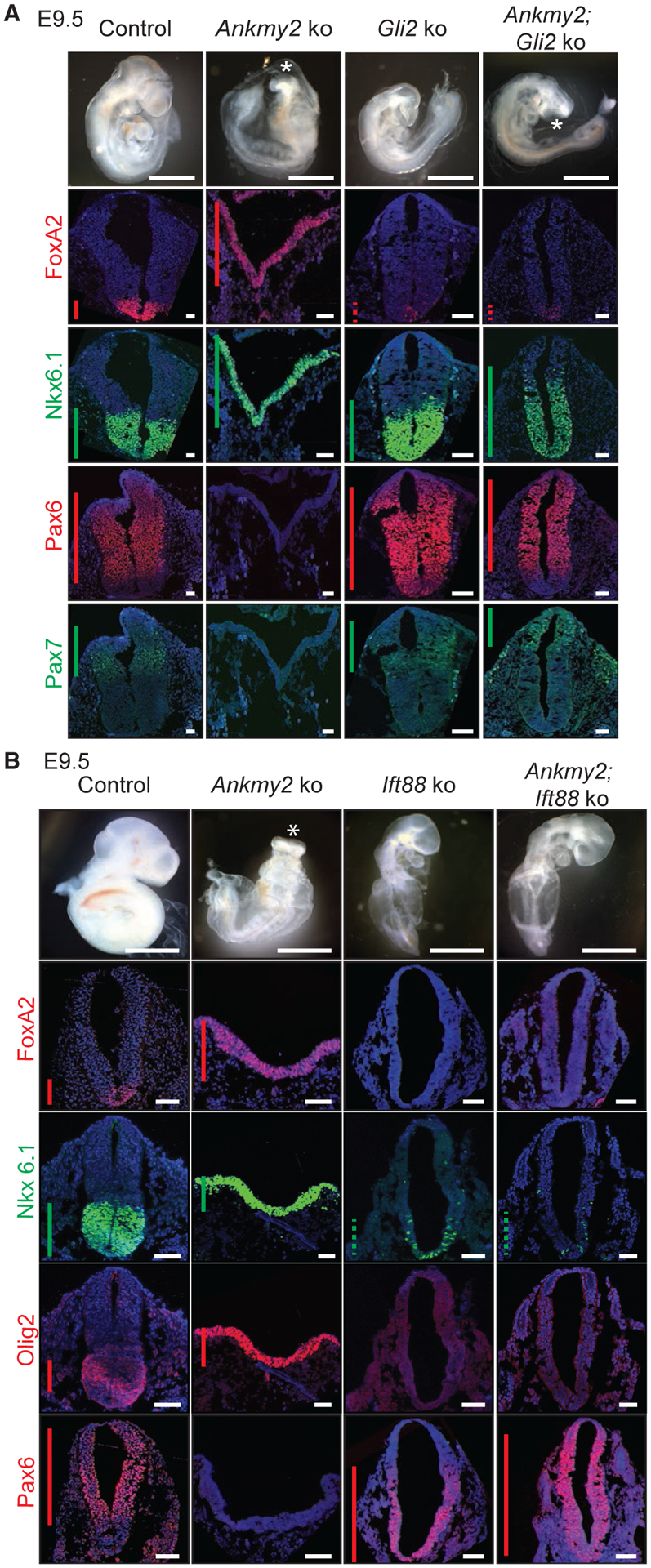
(A and B) Bright-field images of control (Ankmy2ko/WT Gli2ko/WT, 24–26 somites), Ankmy2 ko (12 somites), Gli2 ko (24–26 somites), and Gli2; Ankmy2 double ko (26–28 somites) whole-mount embryos dissected at E9.5 (A). Bright-field images of WT (26–28 somites), Ankmy2 ko (12 somites), Ift88 ko (20–22 somites), and Ift88; Ankmy2 double ko (20–22 somites) whole-mount embryos dissected at E9.5 (B). Neural tube sections (thoracic region) stained for designated markers. n = 3 embryos each for Gli2 ko and Ankmy2; Gli2 double ko stained for each marker (A). n = 4 embryos for Ift88 knockout and n = 5 embryos for Ankmy2; Ift88 double knockout stained for all markers except n= 2 each for Olig2 immunostaining (B). Hindbrain and lumbar regions are shown in Figure S5.
All images are counterstained with DAPI. Asterisks indicate rostral malformations. Vertical bars show the extent of expression of the transcription factor. Dotted bars depict weak expression. Scale: bright-field images, 200 μm; immunostained images, 50 μm. Also see Figure S5.
Hh Pathway Activation in Ankmy2 Knockouts Requires Cilia
We next investigated if the Ankmy2 knockout phenotype is dependent on cilia by generating Ankmy2; Ift88 double knockouts. Disruption of the anterograde IFT-B protein, Ift88, results in defective ciliogenesis and a loss of ventral cell types specified by high levels of Hh in the neural tube (Huangfu et al., 2003; Murcia et al., 2000). However, Suppressor of fused (Sufu), another negative regulator of mammalian Hh signaling, functions independent of cilia (Chen et al., 2009; Jia et al., 2009). Ift88 single knockouts arrested around E9.5, lacked cilia (Figure S5C), and exhibited pericardial effusion with heart looping defects (Figure 5B). Ankmy2; Ift88 double knockouts also lacked cilia and arrested at stages, and with phenotypes, similar to Ift88 single knockouts (Figures 5B and S5C). The rostral morphology of Ankmy2; Ift88 double knockouts resembled that of Ift88 single knockouts and lacked cilia while arresting at stages similar to Ift88 single knockouts (Figure 5B). Unlike Ankmy2 single knockouts, the neural tube of the double knockout was fully closed from hindbrain to lumbar levels by 14–16 somite stage (Figures 5B, S5D, and S5E). Throughout the hindbrain and spinal cord, FoxA2 was absent (Figures 5B, S5D, and S5E), Pax6 was dorsalized (Figures 5B, S5D, and S5E), whereas Nkx6.1 levels were sparse to absent similar to Ift88 knockouts by 20–22 somites (Figures 5B, S5D, and S5E). Thus, activation of the Hh pathway resulting from disruption of Ankmy2 depends on cilia.
Defective Trafficking of ACs to Cilia in Ankmy2 Knockouts
As we observed that Ankmy2 interacts with ACs and regulates Hh signaling in a cilia-dependent manner, we tested if Ankmy2 regulates ciliary trafficking of ACs. We generated stably expressing LAP-tagged cilia-localized ACs in control or Ankmy2 knockout NIH 3T3 cells (Figures S4B and S4C). Control experiments established that ADCY3LAP, ADCY5,LAP and LAPADCY6 were localized to cilia along with localization in the secretory pathway in wild-type cells (Figures 6A–6D). However, cilia positive for ACs in the Ankmy2 knockout cells were drastically reduced (Figures 6A–6D) along with a significant reduction in fluorescence intensities of ciliary AC pools (Figure S6A), despite persistent extraciliary localization in the secretory pathway (Figures 6A–6C). Importantly, upon stably expressing HAANKMY2 in the knockout cells, trafficking of the respective ACs to cilia was partially rescued (Figures 6A–6D). Endogenous levels of Adcy3 were also reduced in C3H10T1/2 cells upon CRISPR-mediated partial knockdown of Ankmy2 (Figures S6B and S6C). Ciliary levels of other integral membrane proteins, such as Smo (Figures S6D and S6E) and Gpr161 (Figures S6F and S6G) were unaffected in Ankmy2 knockout NIH 3T3 and IMCD3 cells, respectively. The morphology of cilia was not grossly affected in Ankmy2 knockout cells, as evidenced from acetylated tubulin immunostaining and measurements of cilia length (Figure S6H). Thus, defective trafficking of ACs to cilia upon Ankmy2 knockout was specific and not arising from a general and non-specific defect in ciliary membrane composition, as seen upon disruption of the transition zone (Chih et al., 2011).
Figure 6. ACs Traffic to Cilia in an Ankmy2-Dependent Manner.
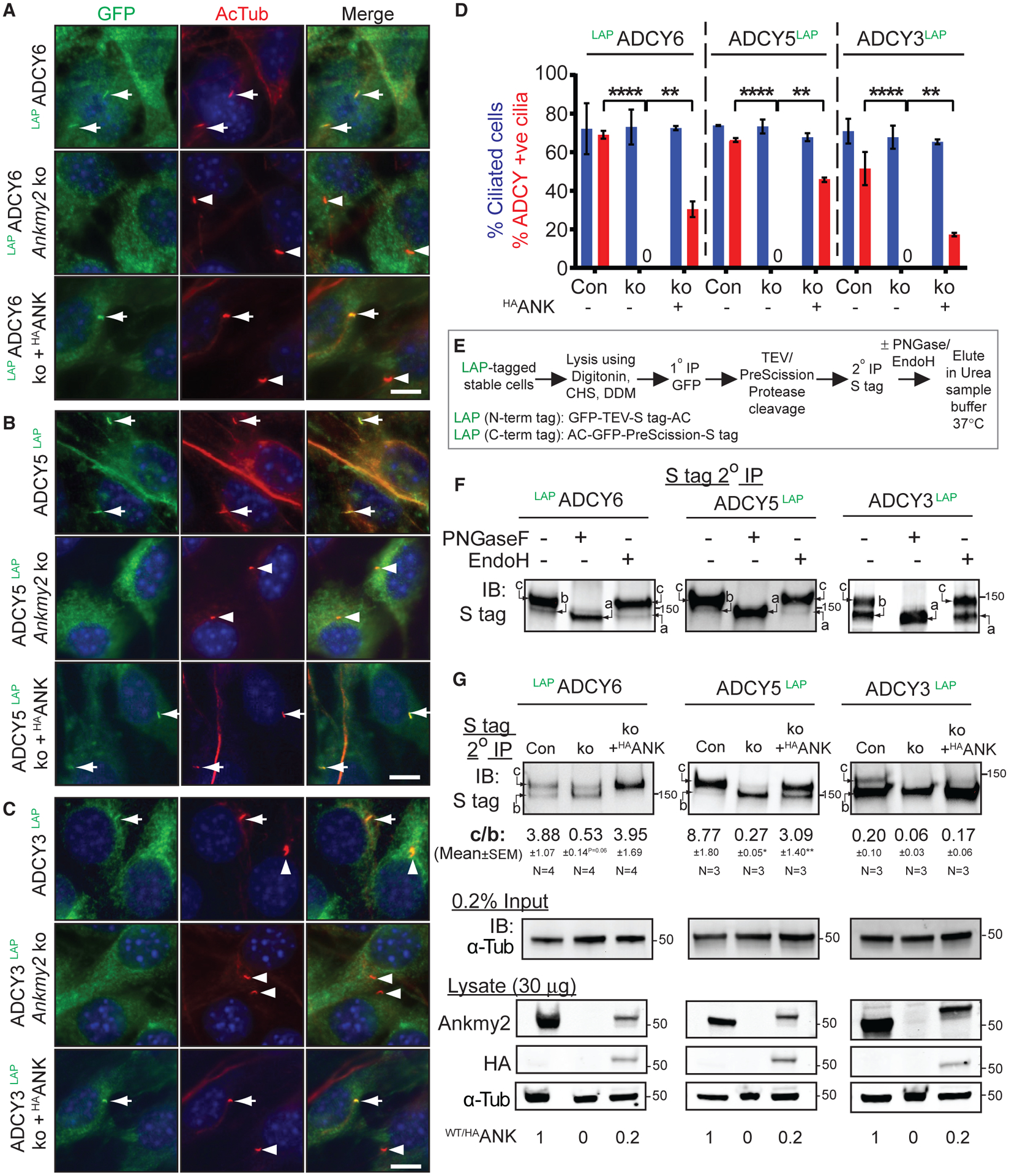
(A–C) LAPADCY6, ADCY5LAP, and ADCYC3LAP were localized to cilia in stably expressing NIH 3T3 cell lines and completely lacked them in cilia upon CRISPR-based Ankmy2 ko, as detected upon performing immunostaining with antibodies against GFP and acetylated tubulin. Ciliary levels of respective ACs were partially rescued upon stable expression of HAANKMY2 (HAANK).
(D) Quantification of localization of ADCYs to cilia as shown in (A)-(C). Data from 3 experiments shown as mean ± SEM. Total 250–500 cells counted per cell line in each experiment. Ciliary fluorescence intensity plots in Figure S6A.
(E) Flowchart showing procedure of tandem affinity purification and enzymatic digestion for assessing glycosylation state of stably expressed LAP-tagged ACs. Abbreviations: DDM, n-Dodecyl-β-D-Maltoside; CHS, Cholesteryl hemisuccinate.
(F) Immunoblots showing glycosylation state of stably expressed LAP-tagged ACs in NIH 3T3 cells after tandem affinity purification followed by Endo H or PNGase treatment. Form “c,” complex glycosylated; Form “b,” core glycosylated; Form “a,” non-glycosylated.
(G) Immunoblots showing different glycosylated states of stably expressed LAP-tagged ACs present in control and Ankmy2 ko NIH-3T3 cells after tandem affinity purification as shown in (E). Inputs from the same experiments shown below immunoblotted for α-tubulin. Data showing ratios of complex vs core glycosylated forms (C/B) from n = 3–4 experiments, expressed as mean ± SEM. p values with respect to control cells. Immunoblots for Ankmy2 and HAANKMY2 in lysates of the same cells and relative levels of Ankmy2 or HAANKMY2 are also shown.
Scale: (A–C) 10 μm.*, p < 0.05; **, p < 0.01; ****, p < 0.0001 as determined by unpaired t test. Also see Figure S6.
Decreased Maturation of ACs in Ankmy2 Knockout
Glycosylation stages serve as markers for the trafficking of transmembrane proteins through the secretory pathway. Specifically, core and complex carbohydrates demark ER-localized and post-trans Golgi localized forms, respectively, and are identifiable upon digestion with Endo H/PNGase treatment (Helenius and Aebi, 2001). We were unable to detect endogenous ACs by immunoblotting or immunoprecipitation using existing antibodies. Thus, we determined the glycosylation state of ACs stably expressed as LAP-tagged ADCYs (C-term LAP, X-Stag-PreScission-EGFP for ADCY3 and ADCY5; N-term LAP, EGFP-TEV-Stag-X for ADCY6) in NIH 3T3 cells by tandem affinity purification followed by Endo H/PNGase treatment (Figure 6E). To pre-serve the complex membrane topology of ACs during pull downs, detergents including digitonin, n-Dodecyl-β-D-Maltoside, and Cholesteryl hemisuccinate were used and proteins were eluted in urea sample buffer without heating (Pal et al., 2015). At steady state in control cells, the ACs existed in complex glycosylated state (“c” form) that was Endo H resistant but PNGase sensitive and core glycosylated form (“b” form) that was Endo H and PNGase sensitive (Figure 6F). In Ankmy2 knockout cells, the high MW “c” form was substantially reduced in these ACs (Figure 6G) and the core glycosylated “b” form was increased in ADCY5 and ADCY6 (Figure 6G). In addition, complex glycosylated pools of these ACs were restored upon HAANKMY2 expression (Figure 6G), suggesting a primary defect in maturation of these ACs upon loss of Ankmy2. Thus, Ankmy2 regulates the maturation of ADCY3/5/6 in the secretory pathway.
Defective Trafficking of Adcy3 to Cilia in Ankmy2 Knockouts during Neural Tube Development
As Ankmy2 determines the trafficking of multiple ACs to cilia (Figure 6), we next tested if ciliary levels of the ACs were affected upon Ankmy2 depletion during neural tube development. Adcy3 has been reported to localize to cilia throughout many regions of the adult mouse brain and to cilia of primary cultured neurons and glia from neonatal mice (Bishop et al., 2007). Adcy3 is also localized to cilia that project into the lumen of the mouse neural tube at E10.5 (Liem et al., 2012); however, the localization of Adcy3 during early embryogenesis is unknown. We detected Adcy3 to localize to cilia of neuroepithelial cells constituting the neural fold and that of surrounding mesenchymal cells during neurulation, as early as E7.5-E8.0 before somitogenesis (Figures 7A, 7B, 7D, and S7A; Video S1). By 6-somite stage, almost all cilia had Adcy3 in control embryos (Figures 7C, 7D, and S7A; Video S3). However, Ankmy2 knockout embryos were completely lacking in Adcy3 localization to cilia during all these stages (Figures 7B–7D; Videos S1, S2, S3, and S4). Despite absent ciliary levels, Adcy3 transcripts were unaffected in the E8.5 Ankmy2 knockout embryos (Figure S7B). Embryos at neurulation stages before somite formation or during early somitogenesis also had increased Hh signaling as evident from Ptch1 RNA in situ hybridization (Figure 7A) or ventral progenitor marker immunostaining (Figures 7C and S7C), suggesting that missing Adcy3 ciliary levels corresponded with high Hh pathway activity. Adcy5/6 are also expressed in the E8.5 embryos but at much lower levels than Adcy3 (Figure S7B) and were completely depleted in cilia of cultured Ankmy2 knockout cell lines (Figures 6A, 6B, and 6D). Further, ciliary targeting of membrane-associated palmitoylated protein, Arl13b, was unaffected in Ankmy2 knockout embryos (Figures 7B and 7C). Thus, depletion of ciliary Adcy3 pools in neuroepithelial and mesenchymal cells accompanied Hh pathway hyperactivation starting from early neurulation.
Figure 7. Adcy3 Trafficking during Neural Tube Development and Role of cAMP Signaling in Gli2 Activation.
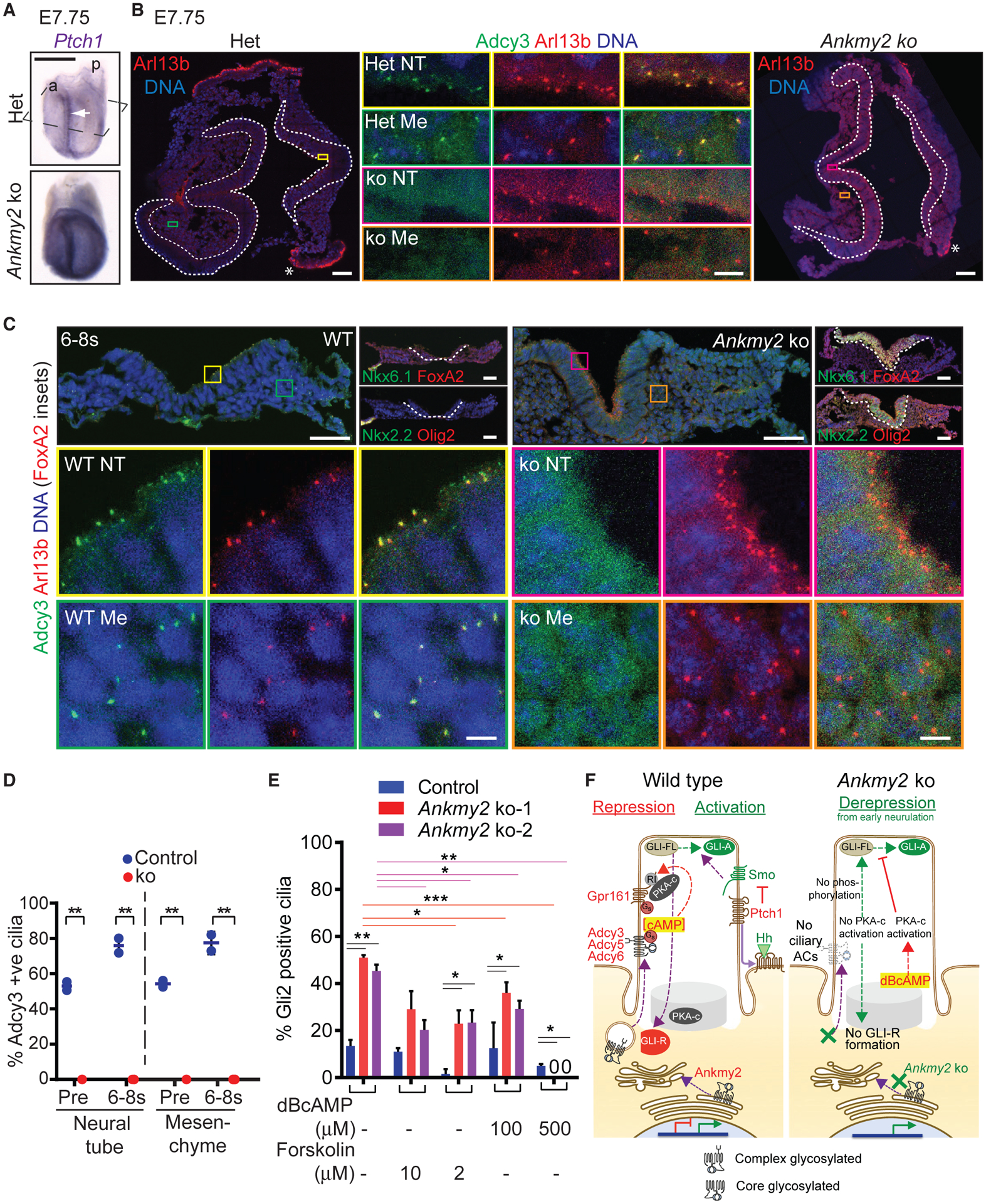
(A) En face ventral views of E7.75 Ankmy2 ko embryos after RNA in situ hybridization for Ptch1 showed increased expression with respect to heterozygous (Het) embryos (A). Rostral (a); caudal (p). Arrow, neural fold.
(B) Horizontal cryosections as shown in (A), immunostained for Arl13b and Adcy3. The neural fold of head (left) and of caudal regions (right) marked by white dotted lines. Colored boxed regions from immunostained neural fold (NT) and mesenchyme (Me) showing the absence of Adcy3 from Arl13b-positive cilia in Ankmy2 ko, in contrast to WT (middle insets). *, Non-specific staining in yolk sac endoderm.
(C) Horizontal cryosections of E8.25 wild-type and Ankmy2 ko (ko) embryos (6–8 somite stage) immunostained for Arl13b and Adcy3. Colored boxed regions from immunostained NT/Me regions are enlarged below showing the absence of Adcy3 from Arl13b positive cilia in ko. Consecutive cryosections in top right show ventralization in the neural fold and predominantly overlapping ventral progenitor marker expression in the ko. Individual immunostained images in Figure S7.
(D) Quantification of Adcy3 in Arl13b positive cilia from different stages and regions from pre-somite stage (E7.75, Pre) and 6–8 somite stage (6–8 s) embryos. E7.75: WT, n = 2 (50–120 cells per region); ko, n = 2 (35–150 cells per region). 6–8 somite stage: WT, n = 1 (40–70 cells per region); Het, n = 1 (30–40 cells/region); ko, n = 3 (100–350 cells per region). Data shown as mean ± SD. Ciliary fluorescence intensities in Figure S7A.
(E) Control and Ankmy2 ko clones of 3T3-L1 cells (Figure 4E) were treated upon confluence ± indicated doses of dBcAMP and Forskolin for 48 h and immunostained as in Figure 4E. Representative immunofluorescence images shown in Figure S7D. n = 2 independent experiments, 50–75 total cilia counted per condition. Data shown as mean ± SD.
(F) Model of Ankmy2 function in repressing Hh pathway. Abbreviations: PKA-c, catalytic subunit; RI, PKARI subunit. *, p < 0.05; **, p < 0.01; ***, p < 0.001 as determined by unpaired t test. Scale: (A), 500 μm; (B), 50 μm (Middle insets, 5 μm); (C), Top panels, 50 μm (Bottom insets, 5 μm). Also see Figure S7; Videos S1, S2, S3, and S4 that show full horizontal sections of the respective sections in (B) and (C).
cAMP Prevents Gli2 Accumulation in Cilia of Ankmy2 Knockout Cells
If Hh-independent Gli2 activation arises from the loss of cAMP production as a result of defective AC maturation and trafficking, Gli2 accumulation in ciliary tips should be prevented by cAMP. Upon treatment with the cell-permeable cAMP analog, dBcAMP, we noticed near-complete reduction of Gli2 accumulation in two clonal Ankmy2 knockout lines in 3T3-L1 cells in a dose-dependent manner (Figures 7E and S7D). In comparison to dBcAMP treatment, we noticed only partial reduction of Gli2 accumulation upon treatment with the AC activator Forskolin in Ankmy2 knockout 3T3-L1 cells compared with untreated cells. Forskolin treatment also prevents Gli2 accumulation in ciliary tips of PKA null MEFs (Tuson et al., 2011), suggesting Forskolin functioning independent of PKA or even independent of AC activation (Hoshi et al., 1988) in such inhibition. Similar PKA/AC activation-independent role of Forskolin could account for the partial reduction of Gli2 accumulation in Ankmy2 knockout cells. Overall, dBcAMP-mediated rescue of Gli2 in ciliary tips of Ankmy2 knockout cells argues for a lack of AC-regulated cAMP signaling in causing premature Gli2 accumulation.
DISCUSSION
Ankmy2 Functions as a Strong Repressor of Hh Pathway via Cilia-Regulated AC Signaling
Factors regulating subcellular trafficking of ACs to cilia and their role in Hh pathway are unknown. Here, we report the identification of a cytosolic protein, Ankmy2 as a strong repressor of the Hh pathway. Ankmy2 functions in maturation and trafficking of multiple ACs to cilia. Ankmy2 knockout causes unrestricted ventral progenitor marker expression and the highest levels of Hh pathway hyperactivation in the neural tube. Based on the onset of embryonic lethality, neural tube defects, and extent of neural tube ventralization, Ankmy2 knockout has phenotypes severe or similar to Ptch1 knockout (Goodrich et al., 1997) that promotes Smo-mediated activation (Zhang et al., 2001) or PKA null mutants that prevent Gli3R generation (Tuson et al., 2011). The Ankmy2 knockout phenotypes are also much severe than Sufu (Svärd et al., 2006; Liu et al., 2015) or Gpr161 knockouts (Mukhopadhyay et al., 2013). Reduced Gli2 and Gli3 transcriptional repressor (GliR) levels in the Ankmy2 knockout implicates deficient pathway repression; however, ventralization is mostly Gli2-dependent, suggesting predominant role of Gli2A. In addition, by demonstrating complete independence of derepression from canonical activation by Smo, our results show GliA generation irrespective of Hh stimulation (Figure 4C). In contrast, Gli3 knockouts (that lack both Gli3 repressor and activator) show intermediate-level ventralization (Persson et al., 2002), whereas Gli2; Gli3 double knockouts (that lack both GliR and GliA) show dorsalization (Bai et al., 2004). Lack of GliR and simultaneous GliA generation in Ankmy2 knockout qualifies for derepression and establishes a critical role of Hh pathway repression during the neural tube patterning and closure.
Ankmy2 prevents the derepression of Hh signaling during neural tube development by regulating maturation and trafficking of multiple ACs to cilia. Missing Adcy3 ciliary levels in the neural fold and surrounding mesenchymal cells in Ankmy2 knockout embryos corresponded with high Hh pathway activity before somitogenesis and in later stages. Similarly, other ciliary ACs ADCY5/6 were also not trafficked to cilia in Ankmy2 knockout cell lines. The high Hh signaling phenotypes in the neural tube in Ankmy2 knockouts are completely rescued upon concurrent loss of cilia in Ift88 knockouts, suggesting that lack of AC signaling requires cilia for hyperactivation. While we cannot rule out the role of nonciliary ACs or nonciliary pools of ACs in additionally regulating Hh signaling in neural tube, ultimately the ventralization phenotype requires cilia. Such strict cilia dependence of the highest levels of ventralization argues for a lack of ACs and corresponding PKA activity in close proximity to cilia and centrosome. Furthermore, Gli2 accumulates in ciliary tips of Ankmy2 knockout cells irrespective of Smo activation. Rescue of Gli2 ciliary accumulation by dBcAMP in Ankmy2 knockout cells lends further credence to compromised AC trafficking and signaling contributing to Hh-independent Gli2 activation. Thus, juxtaciliary production of cAMP by ACs likely promotes Gli-repressor generation and prevents Smo-independent Gli2 activation (Figure 7F).
Complete Neural Tube Ventralization Irrespective of Smo Suggests Hh-Independent Gli Activator Generation
How do full-scale ventralization and Gli2 activation occur in Ankmy2 knockout in the absence of Smo? Complete lack of AC ciliary trafficking from early neurulation in Ankmy2 knockout could enable early and complete disruption of GliR gradient, unlike attenuated AC activity in Gpr161 knockout (Mukhopadhyay et al., 2013). Different activation states of Gli2 has been proposed to be encoded by different patterns of Gli phosphorylation at the C terminus, with full phosphorylation or lack of phosphorylation driving GliR or GliA formation, respectively (Niewiadomski et al., 2014). For e.g., overexpression of the activation domain dephosphorylated Gli2 has been shown to result in the ectopic specification of ventral progenitors, including expression of floor plate markers (Niewiadomski et al., 2014). Whereas GliR formation requires Gli-Sufu complex (Humke et al., 2010; Kise et al., 2009), dissociation of Sufu from Gli2/3 is a hallmark of Shh-stimulated signaling by Smo and requires cilia (Tukachinsky et al., 2010; Humke et al., 2010). Forskolin treatment prevents Gli-Sufu dissociation even upon Smo activation suggesting that PKA-mediated phosphorylation of either or both promotes binding (Humke et al., 2010). Similarly, lack of phosphorylation of Gli2 and Gli3 in Ankmy2 knockout, as evident from loss of Gli2R and Gli3R in the absence of juxtaciliary AC signaling, might enable Gli2A and Gli3A formation from Gli-Sufu dissociation even in the absence of Smo. Finally, our results showing that GliA can be formed independent of Smo necessitates reevaluation of the actual role of Smo activation in Gli-Sufu dissociation and suggests Smo’s role in promoting, rather than determining GliA generation (Figure 4C). Reduced PKA activation in cilia/centrosome complex by Smo-Gαi coupling (Barzi et al., 2011; Villanueva et al., 2015; Ogden et al., 2008) and Gpr161 removal from cilia (Pal et al., 2016) likely prevents further phosphorylation of full-length Gli proteins enabling Gli activator formation.
Hh Pathway Derepression from Loss of Ankmy2 or Gpr161 Is Not Modeled in NIH 3T3 Cells
Hh pathway hyperactivation is seen in Ankmy2 knockout embryos (this paper) and Gpr161 knockout embryos (Mukhopadhyay et al., 2013) or conditional knockouts in limb buds (Hwang et al., 2018), embryonic cerebellum (Shimada et al., 2018), and forebrain (Shimada et al., 2019). Hh hyperactivation phenotypes are also observed in zebrafish knockouts of GPR161 (Tschaikner et al., 2019). Predisposition to SHH-subtype medulloblastoma is also seen from loss of GPR161 in children (Begemann et al., 2020), whereas GPR161 mutations are detected in patients suffering from neural tube defects (Kim et al., 2019). Basal Hh pathway hyperactivation is, however, not recapitulated in Ankmy2 (Figures S4C and S4D) or Gpr161 knockouts in NIH 3T3 (Pusapati et al., 2018) and NIH3T3-L1 fibroblasts. CRISPR-based whole-genome screens for regulators of Hh pathway also did not detect Gpr161 or Ankmy2 as negative regulators (Breslow et al., 2018; Pusapati et al., 2018b). The lack of basal derepression in fibroblasts possibly result from intact Gli3R formation in either knockout (Figures S4C and S4D) (Pusapati et al., 2018) despite Gli2 accumulation in ciliary tips of Ankmy2 knockout cells (Figures 4D and 4E) similar to PKA null MEFs (Tuson et al., 2011). As PKA null knockout embryos still retain a small amount of Gli3R processing (Tuson et al., 2011), there could be additional PKA-independent mechanisms for Gli3 processing. Such alternative mechanisms for Gli3R formation might be activated in fibroblasts upon Gpr161 or Ankmy2 knockout, highlighting the importance of using in vivo approaches for studying the role of Hh pathway suppression during morphogenesis.
How Does Anmky2 Determine Maturation and Ciliary Localization of Multiple ACs?
The complex glycosylated forms of ACs are selectively reduced in Ankmy2 knockout cells, and at least, in the case of ADCY5 and ADCY6, such decrease is associated with a concomitant increase in core glycosylated forms. Reduction in complex glycosylated forms are restored upon HAANKMY2 expression, suggesting a primary defect in maturation of these ACs upon loss of Ankmy2 (Figure 7F). A distant MYND-domain protein DAF-25 determines the trafficking of guanylyl cyclases (GCs) to C. elegans chemosensory cilia (Fujiwara et al., 2010; Jensen et al., 2010). As Ankmy2 binds to both ACs and retinal-specific GC1 (Jensen et al., 2010), it is likely that such binding involves the highly homologous cyclase domains. Ankmy2 has been proposed to bind to Fkbp8, a member of the FK506-binding protein family (Saita et al., 2014). We also detect Fkbp8 in TAP-MS of ACs (Table S1). Both proteins were featured in quantitative chaperone interaction networks, where Fkbp8 was proposed to be a part of the core Hsp90 complex and Ankmy2 was proposed to be a peripheral interacting partner in facilitating specific chaperone-client interactions with transcription factors, possibly inferred so because of its MYND domain (Taipale et al., 2014). Interestingly, Fkbp8 mouse mutants exhibit high Hh signaling and ventralization in the lumbar neural tube, albeit weaker than Ankmy2 knockouts, in a Gli2-and cilia-dependent manner, and spina bifida (Bulgakov et al., 2004; Cho et al., 2008; Wong et al., 2008). Another MYND domain protein, Zmynd10, has been shown to confer specificity for the Fkbp8-Hsp90 chaperone complex toward axonemal dynein clients in motile cilia (Mali et al., 2018). Thus, Fkpb8 could play a more general role in the core Hsp90 chaperone in recruiting Ankmy2 as a client-specific factor during maturation of a subset of membrane-bound ACs.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Saikat Mukhopadhyay (saikat.mukhopadhyay@utsouthwestern.edu).
Materials Availability
The Ankmy2 knockout mouse allele will be deposited to EUCOMM and also will be made available to other researchers upon request.
Data and Code availability
This study did not generate/analyze datasets/code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
ES Cells
ES cells targeting Ankmy2 (NM_146033.3, MGI: 2144755; third exon flanked by LoxP sites) that was generated by homologous recombination in mouse ES cells of the C57BL/6 strain were from EUCOMM (Clone # HEPD0679–6-C03). ES cells were grown on SNL feeders with media containing 20% Serum, 6 mM L-glutamine, 1× Penicillin/Streptomycin, 1 mM β-mercaptoethanol, 1 mM Non-essential Amino Acids, 1× Nucleosides, 10 mg/L Sodium Pyruvate, ESGRO supplement 66 μl/L and incubated at 37°C in 5% CO2 (Dr. Robert Hammer lab, UT Southwestern Medical Center, Dallas).
Mouse Strains
The ES cells were injected into host embryos of the C57BL/6 albino strain by the transgenic core (Dr. Robert Hammer lab, UT Southwestern Medical Center, Dallas). The Ankmy2 knockout allele was generated by the deletion of third exon crossed with germline CAG-Cre recombinase strain (Sakai and Miyazaki, 1997). The double knockout analysis was performed using Ift88tm1.1Bky allele (Haycraft et al., 2007), Gli2tm1Alj allele (Mo et al., 1997), and Smotm1Amc allele (Zhang et al., 2001). Sox2-Cre (Hayashi et al., 2002) was crossed with the floxed allele of Ankmy2. Ptch1 allele was as described before (Goodrich et al., 1997). Yolk sac DNA was used for genotyping embryos. Mice were housed in standard cages that contained three to five mice per cage, with water and standard diet ad libitum and a 12 h light/dark cycle. Mice aged 8 weeks or older were used for breeding and timed pregnancies. Noon of the day on which a vaginal plug was found was considered E0.5. All the animals in the study were handled according to protocols approved by the UT Southwestern Institutional Animal Care and Use Committee, and the mouse colonies were maintained in a barrier facility at UT Southwestern, in agreement with the State of Texas legal and ethical standards of animal care.
Stable Cell Lines
NIH 3T3 Flp-In cells (Life Technologies) stably expressing LAP tag or LAP tagged ACs were generated by retroviral infection, antibiotic selection, and flow sorting. CRISPR/Cas9 knockout or knockdown lines for Ankmy2 were generated in NIH 3T3 Flp-In, 3T3-L1 (gift of Peter Michaely, UT Southwestern) or C3H10T1/2 (ATCC) cells by using guide RNA targeting sequences AAGGAACTGCTG GAAGTGAT (Exon 1) or GAATGTTCATGTCAACTGCT (Exon 2). Clonal lines were tested for knockout or knockdown by Sanger sequencing and immunoblotting for Ankmy2. ORF clones were as follows: ADCY3 (gift of Ron Taussig, UT Southwestern), ADCY5 (gift of Ron Taussig, UT Southwestern), ADCY6 (Life Technologies IOH40476), ANKMY2 (Origene RG206770; NM_020319 in PCMV6-AC-GFP vector from Origene). Gateway pENTR clones were generated by PCR cloning and BP reaction as necessary for N- or C-terminal tagging. Gatewaytized pBABE-LAP-N terminus and pBABE-LAP-C terminus plasmids were generated from LAP1 and LAP5 vectors (Addgene) and pBABE puro. We cloned 3×HA-ANKMY2 into pQXIN (Clontech), which was used for retroviral infection in knockout lines expressing LAP-tagged ACs. Antibiotic selection was used to generate rescue lines stably expressing 3×HA-ANKMY2. NIH 3T3 Flp-In cells were cultured in DMEM high glucose (Sigma-Aldrich) media with 10% Bovine calf serum, 0.05 mg/ml penicillin, 0.05 mg/ml streptomycin, and 4.5 mM glutamine. 3T3-L1 cells were cultured in DMEM high glucose (Sigma-Aldrich) media with 10% FBS, 0.05 mg/ml penicillin, 0.05 mg/ml streptomycin, 4.5 mM glutamine and 8 ng/ml Biotin. C3H10T1/2 cells were grown in DMEM high glucose (Sigma-Aldrich) media with 10% FBS, 0.05 mg/ml penicillin, 0.05 mg/ml streptomycin, 4.5 mM glutamine, 1 mM Sodium Pyruvate, and 1× MEM Non-essential Amino Acid solution.
METHOD DETAILS
Mouse Genotyping
To genotype Ankmy2 mice, the following primers were used. For floxed allele with or without deletion: 3F (5’-CTG TCT CCA TAT TCA CAC ATT GAA TAG C-3’), 4R (5’-GCT GCA TGC ATC AAA GGA GTC ATT CC-3’) and 2R (5’-TGA ACT GAT GGC GAG CTC AGA CC-3’) gave 508 bp for wild type, 732 bp for floxed allele, and 289 bp for deleted floxed allele (cko). For KO allele: 3F, 4R, and 5R (5’-CAA CGG GTT CTT CTG TTA GTC C-3’) gave 508 bp and 316 bp for wild type and knockout bands, respectively (Figures S1D, S1E, and S1G). Cre allele was genotyped with Cre-F (5’-AAT GCT GTC ACT TGG TCG TGG C-3’) and Cre-R (5’-GAA AAT GCT TCT GTC CGT TTG C-3’) primers (100 bp amplicon). To genotype Ptch1 mice, P1 (5’-TGG GGT GGG ATT AGA TAA ATG CC-3’), P2 (5’-TGT CTG TGT GTG CTC CTG AAT CAC-3’), P3 (5’-CTG CGG CAA GTT TTT GGT TG-3’) and P4 (5’-AGG GCT TCT CGT TGG CTA CAA G-3’) primers were used. Wild type and knockout bands were 400 bp and 100 bp, respectively. To genotype Smo mice, Smo WT-F (5’-CCT GCG CTG CTC AAC ATG G-3’), Smo WT-R (5’-GGT CAC GTT CCC GCT CAA G-3’), and Smo KO-R (5’-ACT TCC ATT TGT CAC GTC CTG CAC-3’) primers were used. Wild type and knockout bands were 340 bp and 490 bp, respectively. To genotype Gli2 mice, Gli2 sense (5’-AAA CAA AGC TCC TGT ACA CG-3’), Gli2 antisense (5’-CAC CCC AAA GCA TGT GTT TT-3’) and pPNT (5’-ATG CCT GCT CTT TAC TGA AG-3’) primers were used. Wild type and knockout bands were 300 bp and 600 bp, respectively. To genotype Ift88 mice, Ift88 Flox-F (5’-GAC CAC CTT TTT AGC CTC CTG-3’), Ift88 Flox-R (5’-AGG GAA GGG ACT TAG GAA TGA-3’) and Ift88 KO-R (5’-CAT TTT TGG CTT CCA AAG G-3’) primers were used. Wild type and knockout bands were 365bp and 319bp respectively.
Tissue Processing, Antibodies, Immunostaining, and Microscopy
Mouse embryos were fixed in 4% paraformaldehyde (PFA) overnight at 4°C (except for Adcy3 immunostaining, see below), and processed for cryosectioning or paraffin embedding and sectioning. For cryosectioning, the embryos were incubated in 30% sucrose at 4°C until they were submerged in the solution. Embryos were mounted with OCT compound. For Adcy3 immunostaining, presomite stage and 6–8 somite stage embryos were fixed for 30 min on ice with freeze-thawed chilled 4% PFA, transferred into a 30% sucrose solution on ice until the embryos submerged, and then mounted in OCT. Embryos in OCT were cut into 15 μm frozen sections. The sections were incubated in PBS for 15 min to dissolve away the OCT. Sections were then blocked in blocking buffer (1% normal donkey serum [Jackson immunoResearch, West Grove, PA] in PBS) for 1 h at room temperature. Sections were incubated with primary antibodies against the following antigens; overnight at 4°C: FoxA2 (1:1000, ab40874, Abcam or 1:1000, ab108422; Abcam), Nkx6.1 (1:100, F55A10-s; DSHB), Nkx2.2 (1:10, 74.5A5-s; DSHB), Olig2 (1:500, MABN50; Millipore Corp), Pax6 (1:2000, 901301; Biolegend), Pax7 (1:10; DSHB) and Adcy3 (1:500, LS-C204505; LifeSpan BioSciences). An earlier available antibody against Adcy3 (sc-588) was used for immunofluorescence studies only in C3H10T1/2 cells (Figures S6C and S6D). This antibody has currently been discontinued and for all embryo cryosections (Figures 7, S7, and Supplemental Videos 1–4), the anti-Adcy3 antibody from LifeSpan BioSciences (LS-C204505) was used at a dilution of 1:500. After three PBS washes, the sections were incubated in secondary antibodies (Alexa Fluor 488-, 555-, 594-, 647- conjugated secondary antibodies, 1:500; Life Technologies, Carlsbad, CA or Jackson ImmunoResearch) for 1 h at room temperature. Cell nuclei were stained with DAPI (Sigma). Slides were mounted with Fluoromount-G (0100–01; Southern Biotech) and images were acquired with a Zeiss AxioImager.Z1 microscope and a Zeiss LSM780/LSM880 confocal microscope using a 40x oil immersion objective lens with tile stitching feature. For paraffin sections, embryos were processed over a 12 h period using a Thermo-Shandon Hypercenter Automated Tissue Processor (A78400001D; ThermoFisher Scientific) which dehydrated the brains through 6 ethanol concentrations, from 50% ethanol to 100% ethanol, cleared through 3 changes of xylene, and infiltrated with wax through 3 Paraplast Plus paraffin baths (39602004; Leica). Samples were embedded in Paraplast Plus using paraffin-filled stainless steel base molds and a Thermo-Shandon Histocenter 2 Embedding Workstation (6400012D; ThermoFisher Scientific). The embryos were then cut in 5 μm thick sections. For hematoxylin and eosin staining, paraffin sections were stained by hematoxylin and eosin (Hematoxylin 560; 3801575; Leica and Alcoholic Eosin Y 515; 3801615; Leica) using a Sakura DRS-601 x-y-z robotic-stainer (DRS-601; Sakura-FineTek, Torrance, CA). Slides were dehydrated and mounted with Permount (SP15–100; ThermoFisher Scientific). For immunofluorescence experiments in cell lines, cells were cultured on coverslips until confluent and starved for indicated periods before fixation. Cells were fixed with 4% PFA. After blocking with 5% normal donkey serum, the cells were incubated with primary antibody solutions for 1 h at room temperature followed by treatment with secondary antibodies for 30 min along with DAPI. Primary antibodies used were against the following antigens: GFP (Abcam ab13970 (1: 5000, Figure 6), ab290 (1:300, Figure S1B)), Arl13b (1:500, gift from Tamara Caspary) Caspary et al., 2007, acetylated tubulin (1:5000, T6793; Sigma), Gpr161 (1:200, custom-made) (Pal et al., 2016), Smo (1:500, gift from Kathryn Anderson) Ocbina and Anderson, 2008, Gli2 (1:500, gift from Jonathan Eggenschwiler) and γ-tubulin (1:500, ab11316; Abcam). Coverslips were mounted with Fluoromount-G and images were acquired with a Zeiss AxioImager.Z1 microscope.
Reverse Transcription and Quantitative PCR
RNA was extracted using the GenElute mammalian total RNA purification kit (RTN350; Sigma). Genomic DNA was eliminated by DNase I (D5307; Sigma). qRT-PCR was performed with SYBR Green Quantitative RT-qPCR Kit (QR0100; Sigma) or Kicqstart One-Step Probe RT-qPCR ReadyMix (KCQS07; Sigma). Gli1 TaqMan probes for qRT-PCR were published before (Wen et al., 2010), and inventoried probes for FoxA2 and Gapdh were from Applied Biosystems. Primer sequences are in Table S2. Reactions were run in CFX96 Real-time System (Bio Rad). Gapdh was used for normalization of Taqman assays (Figures 2, 4, and S4). αTubulin (Figures 2 and S2) or Hprt (Figures S4 and S7) were used for normalization with SYBR Green Quantitative RT-qPCR Kit.
In Situ Hybridization (ISH)
Antisense riboprobes were made using the following templates: Ptch1, Gli1, Shh (gifts from Andrew McMahon lab and Deanna Grant, Andrew Peterson lab). For Ankmy2 sense and antisense probes, 900bp of Ankmy2 cDNA initiating from the start site was cloned into pcDNA3.1 in both directions using Ankmy2 whole cDNA from Origene (MR204185) as template. Whole-mount in situ hybridization using digoxigenin-labeled probes was performed on embryos using standard protocols. Images were acquired using a Leica stereomicroscope (M165 C) with a digital camera (DFC500) or Zeiss stereomicroscope (Discovery.V12) and AxioCam MRc.
Radiolabeled sense and antisense probes were generated using T7 RNA polymerases and 35S-UTP (>1,000 Ci/mmol; NEG039H, PerkinElmer LAS Canada, Inc.) using linearized cDNA templates by in-vitro transcription using the Maxiscript kit (AM1324M, Life technologies). Radioisotopic in situ hybridization was performed as described below (Shelton et al., 2000). Briefly, 5 μm thick sections were deparaffinized, permeabilized, and acetylated prior to hybridization at 70°C with riboprobes diluted in a mixture containing 50% formamide, 0.75 M NaCl, 20 mM Tris-HCl, pH 8.0, 5 mM EDTA, 10 mM NaPO4, 14% dextran sulfate, 1× Denhardt’s, and 0.5 mg/ml tRNA. Following hybridization, the sections were rinsed with increasing stringency washes, subjected to RNAse A (2μg/ml, 30 min at 37°C), and dehydrated prior to dipping in K.5 nuclear emulsion gel (AGP9281; Ilford, UK). Following autoradiographic exposure for 21–35 days, photographic development was carried out with D-19 Developer Substitute and Kodak Fixer (26920–4; 26942, Ted Pella Inc.). Sections were counterstained with hematoxylin, dehydrated with ethanol, cleared with xylene, and coverslipped with synthetic mounting media (SP15, Fisher Chemical). Radioisotopic in-situs were analyzed using dark-field and bright-field microscopy. Sense (control) riboprobes established the level of background signal. Review and photography of all radioisotopic in situ hybridizations were carried out on a Leica DM2000 photomicroscope equipped with brightfield, and incident-angle darkfield illumination. Photomicrography was achieved using this microscope and an Optronics Microfire digital CCD color camera using PictureFrame 3.0 acquisition software (Optronics, Inc. Goleta, CA, USA). Resulting ISH silver-grain signal was imaged with camera settings to produce near binary intensity and contrast.
Scanning EM
Embryos were fixed in ½ Karnovsky’s fixative (2% PFA, 2.5% Glutaraldehyde in 0.1M Sodium Cacodylate buffer, pH 7.4) overnight and post fixed in 1% OsO4 for 1 h. They were dehydrated through a series of ethanol. After three washes of hexamethyldesilazane, the samples were air-dried at room temperature. The embryos were oriented and mounted on carbon tape on aluminum stubs. They were then sputter-coated with 10 nm of gold/palladium mixture and viewed on a FEI XL30 SEM at 10 kV.
Tandem Affinity Purification and Immunoblotting
NIH 3T3 cells stably expressing LAP tag or LAP tagged ACs were lysed in buffer containing 50 mM Tris-HCl, pH 7.4, 200 mM KCl, 1 mM MgCl2, 1mM EGTA, 10 % glycerol, 1 mM DTT, 1% digitonin, 0.05% n-Dodecyl-β-D-Maltoside, 0.25% Cholesteryl hemisuccinate, 1 mM of AEBSF, 0.01 mg/mL of Leupeptin, pepstatin and chymostatin (Pal et al., 2015). Lysates were centrifuged at 12000 × g for 10 min followed by tandem IPs (Cheeseman and Desai, 2005). Briefly, the GFP immunoprecipitates were first digested with TEV (N terminal LAP) or PreScission (for C terminal LAP) protease for 16 h at 4°C. The supernatants were subjected to secondary IPs with S-protein agarose. Treatment with Endo H and PNGase F (NEB) was performed on the IP-ed proteins on S-protein agarose beads in NEB designated buffers at 37°C for 2 h. The resulting secondary IPs were eluted in 2× urea sample buffer (4 M urea, 4% SDS, 100 mM Tris, pH 6.8, 0.2% bromophenol blue, 20% glycerol, and 200 mM DTT) at 37°C for 30 min and analyzed by immunoblotting (Pal et al., 2015). For detection of different glycosylation forms of the LAP-tagged ACs by S-tag immunoblotting and based on the stable expression levels, we required ~0.75 ml packed cell pellet for finally eluting secondary IPs in 30–40 μl of 2× urea sample buffer from 30–40 μl S-protein agarose beads. Tandem-IPs were mostly run on 4–20% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad).
Immunoblots from tandem affinity purifications were probed with antibodies against S-tag (mouse monoclonal MAC112; EMD Millipore), Ankmy2 (HPA067100, Sigma), α-tubulin (clone DM1A, T6199, Sigma), HA tag (clone 3F10, Roche) followed by visualization using IRDye-tagged secondary antibodies.
Embryos were prepared by lysing in RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate) containing freshly added phosphatase and protease inhibitors on ice for 1 h and shredded using Qiashredder spin-columns (Qiagen). Insoluble debris was removed by centrifugation at 16,000 × g for 20 min at 4°C. Lysate concentrations were determined with the bicinchoninic acid (BCA) kit (Pierce, Rockford, IL) and compared to BSA standards in RIPA buffer. Samples were boiled at 95°C for 5 minutes with 5× Laemmli sample buffer and kept on ice. 30 μg of total protein lysates were analyzed using antibodies against Gli3 (AF3690, R&D, 1ug/ml), Gli2 (AF3635, R&D, 1ug/ml), Gli1 (L42B10, Cell Signaling; 1:1000) and α-tubulin (clone DM1A, T6199, Sigma; 1:5000). For mass spectrometry, total protein was cut out from the gel and analyzed as mentioned below.
Mass Spectrometry
In the UT Southwestern proteomics core, protein in gel bands was reduced and alkylated with DTT and iodoacetamide (Sigma), then digested overnight with trypsin (Pierce). Following solid-phase extraction cleanup with Oasis HLB plates (Waters), the resulting peptides were reconstituted in 10 μl of 2% (v/v) acetonitrile (ACN) and 0.1% trifluoroacetic acid in water. 2 μl was injected and analyzed by LC/MS/MS using a Q Exactive mass spectrometer (Thermo) coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system (Dionex). Samples were separated on a 50-cm length 75 μm ID EasySpray column (Thermo) and eluted with a gradient from 1–28% buffer B over 60 min at a 400 nl/min flow rate. Buffer A contained 2% (v/v) acetonitrile (ACN) and 0.1% formic acid in water, and buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.08% formic acid in water. The mass spectrometer was operated in positive ion mode with a source voltage of 2.4 kV, the capillary temperature of 250°C, and S-lens RF level at 50.0%. MS scans were acquired at 70,000 resolution and up to 10 MS/MS spectra were obtained for each full spectrum acquired using higher-energy collisionally induced dissociation (HCD) for ions with charge ≥2. Raw MS data files were converted to a peak list format and analyzed using the central proteomics facilities pipeline (CPFP), version 2.0.3 (Trudgian and Mirzaei, 2012; Trudgian et al., 2010). Peptide identification was performed using the TANDEM (Craig and Beavis, 2004) and open MS search algorithm (OMSSA) (Geer et al., 2004) search engines against the mouse protein database from Uniprot and the sequence of the tagged proteins, with common contaminants and reversed decoy sequences appended (Elias and Gygi, 2007). Fragment and precursor tolerances of 20 ppm and 0.5 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification, with the oxidation of Met set as a variable modification. An additional requirement of two unique peptide sequences per protein was used for protein identification.
In the Taplin proteomics core (LAPADCY6), excised gel bands were cut into approximately 1 mm3 piece. Gel pieces were then subjected to a modified in-gel trypsin digestion procedure (Shevchenko et al., 1996). Gel pieces were washed and dehydrated with acetonitrile for 10 min followed by removal of acetonitrile. Pieces were then completely dried in a speed-vac. Rehydration of the gel pieces was with 50 mM ammonium bicarbonate solution containing 12.5 ng/μl modified sequencing-grade trypsin (Promega, Madison, WI) at 4°C. After 45 min, the excess trypsin solution was removed and replaced with 50 mM ammonium bicarbonate solution to just cover the gel pieces. Samples were then placed in a 37°C room overnight. Peptides were later extracted by removing the ammonium bicarbonate solution, followed by one wash with a solution containing 50% acetonitrile and 1% formic acid. The extracts were then dried in a speed-vac (~1 h). The samples were then stored at 4°C until analysis. On the day of analysis, the samples were reconstituted in 5–10 μl of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid). A nano-scale reverse-phase HPLC capillary column was created by packing 2.6 μm C18 spherical silica beads into a fused silica capillary (100 μm inner diameter x ~30 cm length) with a flame-drawn tip (Peng and Gygi, 2001). After equilibrating the column each sample was loaded via a Famos autosampler (LC Packings, San Francisco CA) onto the column. A gradient was formed and peptides were eluted with increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid). As peptides eluted they were subjected to electrospray ionization and then entered into an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences (and hence protein identity) were determined by matching protein databases with the acquired fragmentation pattern by the software program, Sequest (Thermo Fisher Scientific, Waltham, MA) (Eng et al., 1994). All databases include a reversed version of all the sequences and the data was filtered to between a one and two percent peptide false discovery rate.
Cell Fractionation
Cells were washed with PBS and homogenization buffer (1 mM HEPES pH 7.4, 1 mM EDTA and 250 mM sucrose, 1 mM AEBSF, 1 μM Leupeptin, 1 μM Pepstatin, 1 μM Chymostatin, and 1 mM DTT) and lysed using a ball-bearing homogenizer with a clearance of 10 μm (Balch and Rothman, 1985). The homogenate was centrifuged for 5 min at 4°C at 1000 × g. The post-nuclear supernatant was again centrifuged for 20 min at 5000 × g for separating the mitochondrial pellet, followed by 30 min at 4°C at 100,000 × g. The supernatant was collected as the cytosolic fraction and the pellet was collected as the microsomal fraction. Both mitochondrial and microsomal pellets after washing once with homogenization buffer (without disrupting the pellet) were finally resuspended in homogenization buffer, which was 1/10th of final cytosolic fraction in volume. Equal volumes of cytosolic, mitochondrial, and microsomal fractions were loaded for immunoblotting using antibodies against S-tag, Ankmy2 (Sigma HPA067100), Pgk1 (GeneTex GTX107614) and Timm50 (Epitomics EPR5785). The mitochondrial and microsomal fractions were concentrated 10× compared to a cytosolic fraction.
QUANTIFICATION AND STATISTICAL ANALYSIS
AC positive cilia in LAP-tagged ADCY cell lines and Smo/Gpr161 positive cilia in Ankmy2 CRISPR knockout cell lines were quantified as follows. Three independent immunostaining experiments were performed for each cell line. After immunostaining, images of 5 independent fields per cell line were acquired. Number of DAPI positive nuclei and acetylated tubulin positive cilia were counted for each field and expressed as the % of ciliated cells. Cilia positive for acetylated tubulin and ACs/Smo/Gpr161 were counted next and expressed as % of ADCY/Smo/Gpr161 positive cilia. To quantify ciliary pools of ACs, fluorescence levels were measured using the “Measure” tool of Fiji software. Fluorescence levels of neighboring background areas were subtracted from that of the selected ciliary areas and expressed as “corrected fluorescence”. For Adcy3 staining in embryo sections, cilia that could be identified for Adcy3 signal independent of Arl13b ciliary staining were considered to be positive for Adcy3. Such cilia were counted in neural fold and mesenchyme and expressed as a percentage of Arl13b positive cilia. Statistical analyses were performed using Student’s t-test for comparing two groups using GraphPad Prism.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Acetylated tubulin | Sigma | Cat# T6793 RRID: AB_477585 |
| Rabbit polyclonal anti-Arl13b | Gift from Tamara Caspary, Emory University School of Medicine (Caspary et al., 2007) | N/A |
| Mouse monoclonal anti-Arl13b | NeuroMab Facility | Cat# N295B/66 RRID: AB_2750771 |
| Rabbit polyclonal anti-FoxA2 | Abcam | Cat# ab40874 RRID: AB_732411 or Cat# ab108422 RRID: AB_11157157 |
| Mouse monoclonal anti-Nkx6.1 | DSHB | Cat# F55A10-s RRID: AB_532378 |
| Mouse monoclonal anti-Nkx2.2 | DSHB | Cat# 74.5A5 RRID: AB_531794 |
| Mouse monoclonal anti-Olig2 | Millipore Corp | Cat# MABN50 RRID: AB_10807410 |
| Rabbit polyclonal anti-Pax6 | Biolegend | Cat# 901302 RRID: AB_2749901 |
| Mouse monoclonal anti-Pax7 | DSHB | RRID: AB_528428 |
| Rabbit polyclonal anti-Adcy3 (Figure 7) | LifeSpan BioSciences | LS-C204505 |
| Rabbit polyclonal anti-Adcy3 (only in Figures S6C and S6D) | Santa Cruz Biotechnologies | Cat# sc-588 RRID: AB_630839 |
| Rabbit polyclonal anti-Smo | Gift from Kathryn Anderson, Memorial Sloan Kettering Cancer Center (Ocbina and Anderson, 2008) | N/A |
| Guinea pig anti-Gli2 | Gift from Jonathan Eggenschwiler, University of Georgia (Cho et al., 2008) | N/A (Figures 4 and 7) |
| Mouse monoclonal anti-γ-tubulin | Abcam | Cat# ab11316 RRID: AB_297920 |
| Chicken anti-GFP | Abcam | Cat# ab13970 RRID: AB_300798 (Figure 6) |
| Rabbit anti-GFP | Abcam | Cat# ab290 RRID: AB_303395 (Figure S1) |
| Rabbit polyclonal anti-Gpr161 | Custom-made (Pal et al., 2016) | N/A |
| Mouse monoclonal anti-S-tag | EMD Millipore | MAC112 |
| Rabbit polyclonal anti-Ankmy2 | Sigma | Cat# HPA067100 RRID: AB_2685777 |
| Mouse monoclonal anti-α-tubulin (clone DM1A) | Sigma | Cat# T6199 RRID: AB_477583 |
| Rat monoclonal anti-HA tag (clone 3F10) | Roche | Cat# 11867423001 RRID: AB_390918 |
| Goat polyclonal anti-Gli3 | R&D Systems | AF3690 RRID: AB_2232499 |
| Goat polyclonal anti-Gli2 | R&D Systems | AF3635 (Figure 2) RRID: AB_2111902 |
| Mouse monoclonal anti-Gli1 | Cell Signaling | Cat# 2643 RRID: AB_2294746 |
| Rabbit polyclonal anti-Pgk1 | GeneTex | GTX107614 RRID: AB_2037666 |
| Timm50 | Epitomics (EPR5785) | Cat# ab23938 RRID: AB_778461 |
| Goat polyclonal anti-Flag | Abcam | Cat# ab1257 RRID: AB_299216 |
| Alexa Fluor 488-, 555-, 594-, 647- conjugated secondary antibodies | Life Technologies | N/A |
| anti-mouse IgG isotype-specific secondary antibodies | Life Technologies | N/A |
| IRDye tagged secondary antibodies | LI-COR | N/A |
| IRdye tagged neutravidin | LI-COR | 926–68079 |
| Rabbit polyclonal anti-Arl13b | Gift from Tamara Caspary, Emory University School of Medicine (Caspary et al., 2007) | N/A |
| Mouse monoclonal anti-γ-tubulin | Abcam | Cat# ab11316 RRID:AB_297920 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Penicillin | Sigma | P4333 |
| Streptomycin | Sigma | P4333 |
| Glutamine | Sigma | G7513 |
| Polyfect | Qiagen | 301107 |
| DMEM | Sigma | D5796 |
| Bovine calf serum | Sigma | 12133C |
| FBS | Sigma | F6178 |
| Biotin | Sigma | B4639 |
| Sodium Pyruvate | Sigma | S8636 |
| MEM Non-essential Amino Acid solution | Sigma | M7145 |
| Dibutryl cAMP | Sigma | D0627 |
| Paraformaldehyde | Electron microscopy solutions | 15710 |
| Normal donkey serum | Jackson ImmunoResearch | 017-000-121 |
| Fluoromount-G | Southern Biotech | 0100–01 |
| Hematoxylin 560 | Leica | 3801575 |
| Alcoholic Eosin Y 515 | Leica | 3801615 |
| Permount | Thermo Fisher Scientific | SP15–100 |
| GenElute mammalian total RNA purification kit | Sigma | RTN350 |
| DNase I | Sigma | D5307 |
| SYBR Green Quantitative RT-qPCR Kit | Sigma | QR0100 |
| M-MLV Reverse Transcriptase M1427 | Sigma | M1427 |
| Kicqstart One-Step Probe RT-qPCR ReadyMix | Sigma | KCQS07 |
| 35S-UTP | PerkinElmer | NEG039H |
| Maxiscript kit | Life Technologies | AM1324M |
| K.5 nuclear emulsion gel | Ilford, UK | AGP9281 |
| Kodak Fixer | Ted Pella Inc | 26920–4; 26942 |
| Synthetic mounting media | Fisher Chemical | SP15 |
| OCT Compound | Tissue-Tek | 4583 |
| Mini-PROTEAN TGX Precast Protein Gels | Bio-Rad | N/A |
| DAPI | Sigma | D9542 |
| QIAshredder | Qiagen | 79654 |
| BCA kit | Pierce | 23225 |
| Experimental Models: Organisms/Strains | ||
| ES cells targeting the third exon of Ankmy2 (used to generate Ankmy2 knockout and conditional alleles in the transgenic core, UT Southwestern Medical Center, Dallas). | EUCOMM | EUCOMM (HEPD0679–6-C03) |
| Mouse: CAG-Cre recombinase | Sakai and Miyazaki, 1997 | N/A |
| Mouse: Ift88tm1Bky | Jackson Laboratory, Bar Harbor, ME Haycraft et al., 2007 | Strain #022409 |
| Mouse: Gli2tm1Ali | Mo et al., 1997 | N/A |
| Mouse: Smotm1Amc | Zhang et al., 2001 | N/A |
| Mouse: Sox2-Cre | Hayashi et al., 2002 | N/A |
| Mouse: Ptch1tm1Mps | Jackson Laboratory, Bar Harbor, ME Goodrich et al., 1997 | Strain #003081 |
| Experimental Models: Cell Lines | ||
| Ankmy2 stable knockout cell lines in NIH-3T3 Flp-In and 3T3-L1 | This study (methods) | N/A |
| NIH-3T3 Flp-In | Life Technologies | R76107 |
| 3T3-L1 | Gift of Peter Michaely, UT Southwestern | N/A |
| C3H10T1/2 cells | ATCC | CCL-226 |
| IMCD3 cells | ATCC | CRL-2123 |
| Recombinant DNA | ||
| ADCY3 ORF clone | Gift of Ron Taussig, UT Southwestern | N/A |
| ADCY5 ORF clone | Gift of Ron Taussig, UT Southwestern | N/A |
| ADCY6 ORF clone | Life Technologies | IOH40476 |
| ANKMY2 ORF clone in PCMV6-AC-GFP vector | Origene | RG206770 |
| pgLAP1 vector | Addgene | 19702 |
| pgLAP5 vector | Addgene | 19706 |
| pQXIN vector | Clontech | 631514 |
| Ankmy2 whole cDNA | Origene | MR204185 |
| Oligonucleotides | ||
| Primers for genotyping, Taqman assays, and qRT-PCR, See Table S2 | Sigma | N/A |
| Inventoried Taqman probes for FoxA2 | Applied Biosystems | Mm01976556_s1 |
| Inventoried Taqman probes for Gapdh | Applied Biosystems | Mm99999915_g1 |
| Ankmy2 guide RNA targeting sequence: AAGGAACTGCTGGAAGTGAT (Exon 1) | Sigma | N/A |
| Ankmy2 guide RNA targeting sequence: GAATGTTCATGTCAACTGCT (Exon 2) | Sigma | N/A |
| Software and Algorithms | ||
| ImageJ software | National Institutes of Health, Bethesda, MD | https://imagej.nih.gov/ij/ |
| GraphPad Prism | GraphPad, La Jolla, CA | https://www.graphpad.com/scientific-software/prism/ |
| aequest3.0 acquisition software | Optronics, Inc. Goleta, CA, USA | N/A |
| Sequest | Thermo Fisher Scientific, Waltham, MA | N/A |
| Other | ||
| Thermo-Fisher Excelsior Automated Tissue Processor | Thermo Fisher Scientific | A82300001 |
| Paraplast Plus paraffin bath | Leica | 39602004 |
| Thermo-Shandon Histocenter 2 Embedding Workstation | Thermo Fisher Scientific | 6400012D |
| Leica stereomicroscope (M165 C) with digital camera (DFC500) | Leica | N/A |
| Zeiss stereomicroscope (Discovery.V12) | Zeiss | N/A |
| Zeiss LSM780 confocal microscope | Zeiss | N/A |
| Leica DM2000 photomicroscope | Leica | N/A |
| Sakura DRS-601 x-y-z robotic-stainer | Sakura-FineTek, Torrance, CA | DRS-601 |
| CFX96 thermocycler | Bio-Rad | N/A |
| Superfrost® Plus slides | Fisher Scientific | 12-550-15 |
| Kodak BioMaxMR x-ray film | Kodak | N/A |
| PrimeHisto XE slide scanner | Pacific Imaging, Inc. | N/A |
| Vitros 250 chemistry analyzer | GMI Inc, Ramsey, MN | N/A |
| FEI XL30 Scanning Electron Microscope | N/A | |
| Q Exactive mass spectrometer | Thermo | N/A |
| Ultimate 3000 RSLC-Nano liquid chromatography system | Dionex | N/A |
Highlights.
Ankmy2 represses the Hh pathway via targeting of adenylyl cyclases to cilia
Ankmy2 knockout mouse shows complete neural tube ventralization
Ventralization is completely independent of Smoothened but requires cilia and Gli2
Reduced Gli repressors in knockout mice cause Gli2 activation and accumulation in ciliary tips
ACKNOWLEDGMENTS
This project was funded by Alex’s Lemonade Foundation (A-grant to S.M.), Welch Foundation (grant #I-1906 to S.M.), and National Institutes of Health (1R01GM113023 to S.M. and S10RR029731 to Live Cell Imaging Facility, UT Southwestern). We thank UT Southwestern’s transgenic, molecular pathology, electron microscopy and mass spectrometry cores, and mouse animal care facility. We thank Ross Tomaino (Taplin Mass Spectrometry Facility) and Andrew Lemoff (UT Southwestern proteomics core) for assistance with mass spectrometry and Cameron Perry for paraffin sectioning. We acknowledge gifts of reagents from Tamara Caspary, Kathryn Anderson, Jonathan Eggenschwiler, Ron Taussig, Andrew McMahon, Deanna Grant, Andrew Peterson, Thomas Carroll, Peter Michaely, Rhonda Bassel-Duby, and Eric Olson. We thank Sandra Schmid and Sandii Constable for comments on the manuscript and Karel Liem for advice on immunostaining of ACs. Monoclonal antibodies developed by T.M. Jessell/S. Brenner-Morton (Nkx2.2), O.D. Madsen (Nkx6.1), and A. Kawakami (Pax7) were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the Department of Biological Sciences, the University of Iowa, Iowa City, IA 52242, USA.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.devcel.2020.06.034.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alfaro AC, Roberts B, Kwong L, Bijlsma MF, and Roelink H (2014). Ptch2 mediates the Shh response in Ptch1−/− cells. Development 141, 3331–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann VA, Mayrhofer JE, Ilouz R, Tschaikner P, Raffeiner P, Röck R, Courcelles M, Apelt F, Lu TW, Baillie GS, et al. (2016). Gpr161 anchoring of PKA consolidates GPCR and cAMP signaling. Proc. Natl. Acad. Sci. USA 113, 7786–7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai CB, Auerbach W, Lee JS, Stephen D, and Joyner AL (2002). Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 129, 4753–4761. [DOI] [PubMed] [Google Scholar]
- Bai CB, and Joyner AL (2001). Gli1 can rescue the in vivo function of Gli2. Development 128, 5161–5172. [DOI] [PubMed] [Google Scholar]
- Bai CB, Stephen D, and Joyner AL (2004). All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev. Cell 6, 103–115. [DOI] [PubMed] [Google Scholar]
- Balch WE, and Rothman JE (1985). Characterization of protein transport between successive compartments of the Golgi apparatus: asymmetric properties of donor and acceptor activities in a cell-free system. Arch. Biochem. Biophys 240, 413–425. [DOI] [PubMed] [Google Scholar]
- Barzi M, Berenguer J, Menendez A, Alvarez-Rodriguez R, and Pons S (2010). Sonic-hedgehog-mediated proliferation requires the localization of PKA to the cilium base. J. Cell Sci 123, 62–69. [DOI] [PubMed] [Google Scholar]
- Barzi M, Kostrz D, Menendez A, and Pons S (2011). Sonic Hedgehog-induced proliferation requires specific Galpha inhibitory proteins. J. Biol. Chem 286, 8067–8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begemann M, Waszak SM, Robinson GW, Jäger N, Sharma T, Knopp C, Kraft F, Moser O, Mynarek M, Guerrini-Rousseau L, et al. (2020). Germline GPR161 mutations predispose to pediatric medulloblastoma. J. Clin. Oncol 38, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop GA, Berbari NF, Lewis J, and Mykytyn K (2007). Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J. Comp. Neurol 505, 562–571. [DOI] [PubMed] [Google Scholar]
- Breslow DK, Hoogendoorn S, Kopp AR, Morgens DW, Vu BK, Kennedy MC, Han K, Li A, Hess GT, Bassik MC, et al. (2018). A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nat. Genet 50, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J, and Thérond PP (2013). The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol 14, 416–429. [DOI] [PubMed] [Google Scholar]
- Bulgakov OV, Eggenschwiler JT, Hong DH, Anderson KV, and Li T (2004). FKBP8 is a negative regulator of mouse sonic hedgehog signaling in neural tissues. Development 131, 2149–2159. [DOI] [PubMed] [Google Scholar]
- Caspary T, Larkins CE, and Anderson KV (2007). The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell 12, 767–778. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, and Desai A (2005). A combined approach for the localization and tandem affinity purification of protein complexes from metazoans. Sci. STKE 2005, pl1. [DOI] [PubMed] [Google Scholar]
- Chen MH, Wilson CW, Li YJ, Law KK, Lu CS, Gacayan R, Zhang X, Hui CC, and Chuang PT (2009). Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev 23, 1910–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, and Beachy PA (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407–413. [DOI] [PubMed] [Google Scholar]
- Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, and Peterson AS (2011). A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat. Cell Biol 14, 61–72. [DOI] [PubMed] [Google Scholar]
- Cho A, Ko HW, and Eggenschwiler JT (2008). FKBP8 cell-autonomously controls neural tube patterning through a Gli2- and Kif3a-dependent mechanism. Dev. Biol 321, 27–39. [DOI] [PubMed] [Google Scholar]
- Choi YH, Suzuki A, Hajarnis S, Ma Z, Chapin HC, Caplan MJ, Pontoglio M, Somlo S, and Igarashi P (2011). Polycystin-2 and phosphodi-esterase 4C are components of a ciliary A-kinase anchoring protein complex that is disrupted in cystic kidney diseases. Proc. Natl. Acad. Sci. USA 108, 10679–10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, and Reiter JF (2005). Vertebrate smoothened functions at the primary cilium. Nature 437, 1018–1021. [DOI] [PubMed] [Google Scholar]
- Craig R, and Beavis RC (2004). TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467. [DOI] [PubMed] [Google Scholar]
- Dessaud E, Mcmahon AP, and Briscoe J (2008). Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development 135, 2489–2503. [DOI] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Eng JK, Mccormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Fujiwara M, Teramoto T, Ishihara T, Ohshima Y, and Mcintire SL (2010). A novel zf-MYND protein, CHB-3, mediates guanylyl cyclase localization to sensory cilia and controls body size of Caenorhabditis elegans. PLoS Genet 6, e1001211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, and Bryant SH (2004). Open mass spectrometry search algorithm. J. Proteome Res 3, 958–964. [DOI] [PubMed] [Google Scholar]
- Goetz SC, and Anderson KV (2010). The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet 11, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, and Scott MP (1997). Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109–1113. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Lewis P, Pevny L, and Mcmahon AP (2002). Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev 119, S97–S101. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, and Yoder BK (2005). Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet 1, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, and Yoder BK (2007). Intraflagellar transport is essential for endochondral bone formation. Development 134, 307–316. [DOI] [PubMed] [Google Scholar]
- Helenius A, and Aebi M (2001). Intracellular functions of N-linked glycans. Science 291, 2364–2369. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Garber SS, and Aldrich RW (1988). Effect of forskolin on voltage-gated K+ channels is independent of adenylate cyclase activation. Science 240, 1652–1655. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, and Anderson KV (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87. [DOI] [PubMed] [Google Scholar]
- Humke EW, Dorn KV, Milenkovic L, Scott MP, and Rohatgi R (2010). The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev 24, 670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SH, and Mukhopadhyay S (2015). G-protein-coupled receptors and localized signaling in the primary cilium during ventral neural tube patterning. Birth Defects Res. A Clin. Mol. Teratol 103, 12–19. [DOI] [PubMed] [Google Scholar]
- Hwang SH, White KA, Somatilaka BN, Shelton JM, Richardson JA, and Mukhopadhyay S (2018). The G protein-coupled receptor Gpr161 regulates forelimb formation, limb patterning and skeletal morphogenesis in a primary cilium-dependent manner. Development 145, 10.1242/dev.154054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen VL, Bialas NJ, Bishop-Hurley SL, Molday LL, Kida K, Nguyen PA, Blacque OE, Molday RS, Leroux MR, and Riddle DL (2010). Localization of a guanylyl cyclase to chemosensory cilia requires the novel ciliary MYND domain protein DAF-25. PLoS Genet 6, e1001199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Kolterud A, Zeng H, Hoover A, Teglund S, Toftgård R, and Liu A (2009). Suppressor of Fused inhibits mammalian Hedgehog signaling in the absence of cilia. Dev. Biol 330, 452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kato M, and Beachy PA (2009). Gli2 trafficking links Hedgehog-dependent activation of smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. USA 106, 21666–21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SE, Lei Y, Hwang SH, Wlodarczyk BJ, Mukhopadhyay S, Shaw GM, Ross ME, and Finnell RH (2019). Dominant negative GPR161 rare variants are risk factors of human spina bifida. Hum. Mol. Genet 28, 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kise Y, Morinaka A, Teglund S, and Miki H (2009). Sufu recruits GSK3beta for efficient processing of Gli3. Biochem. Biophys. Res. Commun 387, 569–574. [DOI] [PubMed] [Google Scholar]
- Kopinke DM, Norris A, and Mukhopadhyay S (2020). Developmental and regenerative paradigms of cilia regulated hedgehog signaling. Semin Cell Dev Biol 10.1016/j.semcdb.2020.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liem KF, Ashe A, He M, Satir P, Moran J, Beier D, Wicking C, and Anderson KV (2012). The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J. Cell Biol 197, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zeng H, and Liu A (2015). The loss of Hh responsiveness by a nonciliary Gli2 variant. Development 142, 1651–1660. [DOI] [PubMed] [Google Scholar]
- Mali GR, Yeyati PL, Mizuno S, Dodd DO, Tennant PA, Keighren MA, Zur Lage P, Shoemark A, Garcia-Munoz A, Shimada A, et al. (2018). ZMYND10 functions in a chaperone relay during axonemal dynein assembly. eLife 7, 10.7554/eLife.34389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mick DU, Rodrigues RB, Leib RD, Adams CM, Chien AS, Gygi SP, and Nachury MV (2015). Proteomics of primary cilia by proximity labeling. Dev. Cell 35, 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, Chik KW, Shi XM, Tsui LC, Cheng SH, et al. (1997). Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 124, 113–123. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, and Rohatgi R (2014). G-protein-coupled receptors, Hedgehog signaling and primary cilia. Semin. Cell Dev. Biol 33, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, and Jackson PK (2013). The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 152, 210–223. [DOI] [PubMed] [Google Scholar]
- Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, and Woychik RP (2000). The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development 127, 2347–2355. [DOI] [PubMed] [Google Scholar]
- Murdoch JN, and Copp AJ (2010). The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res. A Clin. Mol. Teratol 88, 633–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiadomski P, Kong JH, Ahrends R, Ma Y, Humke EW, Khan S, Teruel MN, Novitch BG, and Rohatgi R (2014). Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep 6, 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman RX, Ko HW, Huang V, Eun CM, Abler LL, Zhang Z, Sun X, and Eggenschwiler JT (2009). Tubby-like protein 3 (TULP3) regulates patterning in the mouse embryo through inhibition of Hedgehog signaling. Hum. Mol. Genet 18, 1740–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocbina PJ, and Anderson K (2008). Intraflagellar transport, cilia, and mammalian Hedgehog signaling: analysis in mouse embryonic fibroblasts. Dev Dyn 237, 2030–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden SK, Fei DL, Schilling NS, Ahmed YF, Hwa J, and Robbins DJ (2008). G protein Galphai functions immediately downstream of smoothened in Hedgehog signalling. Nature 456, 967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal K, Badgandi H, and Mukhopadhyay S (2015). Studying G protein-coupled receptors: immunoblotting, immunoprecipitation, phosphorylation, surface labeling, and cross-linking protocols. Methods Cell Biol 127, 303–322. [DOI] [PubMed] [Google Scholar]
- Pal K, Hwang SH, Somatilaka B, Badgandi H, Jackson PK, Defea K, and Mukhopadhyay S (2016). Smoothened determines beta-arrestin-mediated removal of the G protein-coupled receptor Gpr161 from the primary cilium. J. Cell Biol 212, 861–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, and Gygi SP (2001). Proteomics: the move to mixtures. J. Mass Spectrom 36, 1083–1091. [DOI] [PubMed] [Google Scholar]
- Perez-Garcia V, Fineberg E, Wilson R, Murray A, Mazzeo CI, Tudor C, Sienerth A, White JK, Tuck E, Ryder EJ, et al. (2018). Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature 555, 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson M, Stamataki D, Te Welscher P, Andersson E, Böse J, Rüther U, Ericson J, and Briscoe J (2002). Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev 16, 2865–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusapati GV, Kong JH, Patel BB, Gouti M, Sagner A, Sircar R, Luchetti G, Ingham PW, Briscoe J, and Rohatgi R (2018a). G protein-coupled receptors control the sensitivity of cells to the morphogen Sonic Hedgehog. Sci. Signal 11, eaao5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusapati GV, Kong JH, Patel BB, Krishnan A, Sagner A, Kinnebrew M, Briscoe J, Aravind L, and Rohatgi R (2018b). CRISPR screens uncover genes that regulate target cell sensitivity to the morphogen sonic hedgehog. Dev. Cell 44, 113–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, and Scott MP (2007). Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372–376. [DOI] [PubMed] [Google Scholar]
- Saade M, Gonzalez-Gobartt E, Escalona R, Usieto S, and Martí E (2017). Shh-mediated centrosomal recruitment of PKA promotes symmetric proliferative neuroepithelial cell division. Nat. Cell Biol 19, 493–503. [DOI] [PubMed] [Google Scholar]
- Saita S, Shirane M, Ishitani T, Shimizu N, and Nakayama KI (2014). Role of the ANKMY2-FKBP38 axis in regulation of the Sonic hedgehog (Shh) signaling pathway. J. Biol. Chem 289, 25639–25654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, and Miyazaki J.i. (1997). A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem. Biophys. Res. Commun 237, 318–324. [DOI] [PubMed] [Google Scholar]
- Shelton JM, Lee MH, Richardson JA, and Patel SB (2000). Microsomal triglyceride transfer protein expression during mouse development. J. Lipid Res 41, 532–537. [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, and Mann M (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem 68, 850–858. [DOI] [PubMed] [Google Scholar]
- Shimada IS, Hwang SH, Somatilaka BN, Wang X, Skowron P, Kim J, Kim M, Shelton JM, Rajaram V, Xuan Z, et al. (2018). Basal suppression of the sonic hedgehog pathway by the G-protein-coupled receptor Gpr161 restricts medulloblastoma pathogenesis. Cell Rep 22, 1169–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada IS, Somatilaka BN, Hwang SH, Anderson AG, Shelton JM, Rajaram V, Konopka G, and Mukhopadhyay S (2019). Derepression of sonic hedgehog signaling upon Gpr161 deletion unravels forebrain and ventricular abnormalities. Dev. Biol 450, 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svärd J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergström A, Ericson J, Toftgård R, and Teglund S (2006). Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev. Cell 10, 187–197. [DOI] [PubMed] [Google Scholar]
- Taipale M, Tucker G, Peng J, Krykbaeva I, Lin ZY, Larsen B, Choi H, Berger B, Gingras AC, and Lindquist S (2014). A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 158, 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempé D, Casas M, Karaz S, Blanchet-Tournier MF, and Concordet JP (2006). Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol. Cell. Biol 26, 4316–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudgian DC, and Mirzaei H (2012). Cloud CPFP: a shotgun proteomics data analysis pipeline using cloud and high performance computing. J. Proteome Res 11, 6282–6290. [DOI] [PubMed] [Google Scholar]
- Trudgian DC, Thomas B, Mcgowan SJ, Kessler BM, Salek M, and Acuto O (2010). CPFP: a central proteomics facilities pipeline. Bioinformatics 26, 1131–1132. [DOI] [PubMed] [Google Scholar]
- Tschaikner P, Regele D, Salvenmoser W, Geley S, Stefan E, and Aanstad P (2019). Zebrafish GPR161 contributes to basal hedgehog repression in a tissue-specific manner. bioRxiv616482 10.1101/616482. [DOI] [PubMed] [Google Scholar]
- Tukachinsky H, Lopez LV, and Salic A (2010). A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol 191, 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuson M, He M, and Anderson KV (2011). Protein kinase A acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development 138, 4921–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva H, Visbal AP, Obeid NF, Ta AQ, Faruki AA, Wu MF, Hilsenbeck SG, Shaw CA, Yu P, Plummer NW, et al. (2015). An essential role for Galpha(i2) in smoothened-stimulated epithelial cell proliferation in the mammary gland. Sci. Signal 8, ra92. [DOI] [PubMed] [Google Scholar]
- Vuolo L, Herrera A, Torroba B, Menendez A, and Pons S (2015). Ciliary adenylyl cyclases control the Hedgehog pathway. J. Cell Sci 128, 2928–2937. [DOI] [PubMed] [Google Scholar]
- Wang B, Fallon JF, and Beachy PA (2000). Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434. [DOI] [PubMed] [Google Scholar]
- Wang C, Pan Y, and Wang B (2010). Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 137, 2001–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li V, Chan GC, Phan T, Nudelman AS, Xia Z, and Storm DR (2009). Adult type 3 adenylyl cyclase-deficient mice are obese. PLoS One 4, e6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Lai CK, Evangelista M, Hongo JA, De Sauvage FJ, and Scales SJ (2010). Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol. Cell. Biol 30, 1910–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RL, Wlodarczyk BJ, Min KS, Scott ML, Kartiko S, Yu W, Merriweather MY, Vogel P, Zambrowicz BP, and Finnell RH (2008). Mouse Fkbp8 activity is required to inhibit cell death and establish dorsoventral patterning in the posterior neural tube. Hum. Mol. Genet 17, 587–601. [DOI] [PubMed] [Google Scholar]
- Yu W, Wang Y, Mcdonnell K, Stephen D, and Bai CB (2009). Patterning of ventral telencephalon requires positive function of Gli transcription factors. Dev. Biol 334, 264–275. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Ramalho-Santos M, and Mcmahon AP (2001). Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R symmetry by the mouse node. Cell 106, 781–792. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.