

Abstract
A composite material of graphene (G) and polydopamine (PDA) on a copper (Cu) substrate (G/PDA@Cu) was fabricated successfully by sequential immersion deposition in a dopamine solution and an aqueous graphene oxide suspension before annealing. Optimum preparation conditions were explored by the orthogonal experimental method. The morphology and chemical composition of G/PDA@Cu were studied systematically by a series of characterization techniques. The thermal-conductive performance was evaluated by a laser flash thermal analyser. The thermal conductivity of G/PDA@Cu was 519.43 W m−1 K−1, which is ultrahigh and 30.50% higher than that of the Cu substrate. The adhesion force between G/PDA and the Cu substrate was 4.18 mN, which means that G bonds to the Cu substrate tightly. The model simulation also showed that G/PDA@Cu exhibits excellent thermal conductivity, allowing it to play a significant role in the thermal management of advanced electronic chips. The thermal-conductive devices using this material were prepared for practical applications.
A composite material of graphene (G) and polydopamine (PDA) on a copper (Cu) substrate (G/PDA@Cu) was fabricated successfully by sequential immersion deposition in a dopamine solution and an aqueous graphene oxide suspension before annealing.
1. Introduction
The increasing integration of electronic devices has led to increased heat generation in machines. In traditional designs, thermal control is achieved by three main strategies: increasing the fan speed, expanding the contact area, and developing new thermally conductive materials.1 The first two methods can go no further than certain limits due to design and safe operation aspects. Furthermore, thermally conductive materials, such as thermally conductive silicone and metal heat sinks, can no longer meet the safe device operation criteria. Thus, novel highly thermally conductive materials need to be rapidly developed. The goal of researchers is to obtain materials that are not brittle and have ultrahigh thermal conductivity (>429 W m−1 K−1).2
Copper is the most common thermally conductive metal, which has high thermal conductivity. Heat conduction in metals is mainly achieved through lattice vibration and electron transmission. High temperatures can cause the lattice to vibrate violently and affect the speed of electron transmission; thus, traditional metallic materials are not suitable for heat transmission.3 Graphene (G) is a new material that mainly transfers heat through lattice vibrations due to its unique honeycomb lattice structure. G lattice has three main vibration modes: longitudinal acoustic (LA)-mode phonons, transverse acoustic (TA)-mode phonons, and z-axis acoustic (ZA)-mode phonons. The ZA-mode phonons contribute the most to heat transfer, reaching 75%.4 G complexes have the property of high thermal conductivity even at high temperatures, unlike metals. Combining graphene with a metal substrate leads to the construction of a composite material with high thermal conductivity that is independent of temperature. H. Lee et al. reported a method to form multifunctional polymer coatings by simply dip-coating an object in an aqueous dopamine solution.5 No thermal conductivity tests were carried out on the composed material, and only the composition was analysed; additionally, the direction of G growth was not controlled. A. A. Balandin et al. prepared a single-layer thin-film of G, which was different from ordinary carbon-based materials and had an excellent thermal conductivity of 5780 W m−1 K−1.6 However, it could not be used, mainly because the G films had excellent thermal conductivity but were assembled on the surface. D. Smovzhet et al. used the chemical vapour deposition (CVD) method to connect G with a copper foil and obtained G/CVD@Cu. Although this material had a guaranteed number of G layers, there were problems in the growth direction and adhesion force. Furthermore, G/CVD@Cu could not be used as a thermally conductive material.7 To the best of our knowledge, there are few reports on the preparation of G and polydopamine (PDA) composites via chemical deposition on Cu sheets. However, it is challenging to prepare a material with high mechanical strength and good thermal conductivity on a Cu substrate without damaging its structure.8–10
In this paper, a composite of G and PDA (G/PDA@Cu) was fabricated on a Cu substrate by sequential immersion deposition in a dopamine solution and an aqueous graphene oxide (GO) suspension, followed by annealing treatment. The optimum preparation conditions were determined by the orthogonal experimental method. A highly thermally conductive material with reasonable bonding force was obtained by enhancing the adhesion force with PDA. To further explain this phenomenon, a model was developed for simulating the thermal conductive performance of this material. By comparing the previous experimental results with the simulation results, it has been concluded that the improved thermal conductivity of the composite is mainly due to PDA promoting the tight binding of G to the Cu substrate. Compared with the traditional thermal-conductive materials, the thermal conductivity of this material is better, and ultrahigh conductivity could be achieved. Additionally, a flexible thermally conductive device made was simulated for a ternary lithium battery, in which G/PDA@Cu was combined with foam wool to create a flexible heat sink component.
2. Experimental methods
2.1. Materials and reagents
Hydrochloric acid (HCl, 36–38%), sulfuric acid (H2SO4, 98%), phosphoric acid (H3PO4, 85%), ethanol (CH3CH2OH, 99.5%), acetone (CH3COCH3), ethyl ether (CH3CH2OCH2CH3), hydrogen peroxide (H2O2, 30%) and potassium permanganate (KMnO4, 99.5%) were obtained from Sinopharm Chemical Reagent Beijing Co., Ltd. Natural graphite powder (325 mesh, 99%) was obtained from Qingdao Hua Tai Lubricant Sealing S&T Co., Ltd. (Qingdao, China). Sodium hydroxide (NaOH, 99%) was obtained from Xi Long Chemical Co., Ltd. Dopamine hydrochloride [(OH)2C6H3CH2CH2NH2 HCl, 99%] was obtained from J&K Scientific Ltd. The above reagents were of analytical grade, and no further purification was performed before use. Copper sheets (99.95%) (10 mm × 10 mm × 0.1 mm) were obtained from Beijing Nonferrous Metal Research Institute. Thermally conductive silicone was obtained from Qingdao Hua Tai Lubricant Sealing S&T Co., Ltd. (Qingdao, China). Deionized water was used during the experiments.
2.2. Preparation of GO
GO was synthesized from graphite powder by an improved Hummer's method.11,12 A homogeneous aqueous GO suspension (0.5 mg mL−1) was achieved by dispersing GO in water with ultrasonication for 3 h.
2.3. Preparation of G/PDA@Cu
Cu sheets of size 10 mm × 10 mm × 0.1 mm were ultrasonically washed with ethanol, acetone, and deionized water for 5 min to remove organic contaminants from the surface. Dopamine hydrochloride was dissolved in deionized water to prepare the dopamine solution (20 mg mL−1), and then the pH was adjusted to approximately 8.5 using a 2 mol L−1 NaOH solution. Subsequently, a cleaned Cu sheet was set perpendicularly in the above-prepared dopamine solution for 1 h at room temperature. The immersed sample was rinsed with deionized water and then set horizontally in 4 mL of 0.5 mg mL−1 aqueous GO suspension for 1 h at room temperature. Finally, the sample was placed in a crucible and annealed at 200 °C in the air.
The optimal reaction conditions were obtained using the orthogonal experiment. See the orthogonal experimental data in the ESI† for more information on the material design based on the results of the orthogonal experiments.
To determine the optimum preparation conditions, the thermal conductivity of samples prepared under different conditions were investigated by varying the immersion time in the dopamine aqueous solution and GO aqueous suspension along with the annealing temperature and annealing time; the preparation conditions are shown in Table 1.
Preparation conditions of the samples.
| Sample | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| PDA | — | 1 h | 1 h | 1 h | 1 h | 1 h | 1 h |
| GO | — | — | — | 1 h | 1 h | 1 h | |
| Anneal (1 h) | — | — | 200 °C | 300 °C | — | 200 °C | 300 °C |
2.4. Characterization
The thermal diffusivity (α) of the sample was measured by using a laser flash thermal analyser (LFA447, NanoFlash, Germany) at room temperature. The specific heat (CP) used in this paper was measured by a physical property measurement system (PPMS-9T, Quantum Design, USA.) at room temperature. The density (ρ) was calculated according to eqn (1):
| ρ = m/V | 1 |
where m is the mass of the sample, and V is the volume of the sample.
The thermal conductivity (K) was evaluated based on eqn (2):
| K = α × CP × ρ | 2 |
where α is the thermal diffusivity of the sample, and CP is the specific heat of the sample.13
The adhesion force was measured by using a nano-scratch meter (Ti980, Brucker, Germany).
The surface morphology and chemical composition were characterized using scanning electron microscopy (SEM; NovaNano 430) with energy-dispersive X-ray spectroscopy (EDS). The crystal structure was investigated by X-ray powder diffraction (XRD, D8 Advance, Bruker, Germany) with a Cu Kα radiation source in the continuous scanning mode (40 kV, 40 mA, and λ = 0.15418 nm) at a scanning rate of 2° min−1. The surface chemical composition of the sample was analysed by X-ray photoelectron spectroscopy (XPS, Model PHI 5300, Physical Electronics, US.) using a 250 W Mg Kα (λ = 0.9891 nm) X-ray as the excitation source in the constant analyser energy mode, with C 1s at 284.8 eV as the reference. The infrared spectra of the samples were obtained using an attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectrometer (Nicolet 6700) equipped with a liquid nitrogen-cooled MCT detector. Raman spectroscopy was conducted using a micro-Raman system (LabRAM HR Evolution, France) equipped with a 532 nm Ar-ion incident laser. The vibrational Raman Stokes spectra were recorded from 200 to 3500 cm−1. Infrared imaging (Imi Tech, Korea) of the specially shaped materials was performed while heating them with a hairdryer (Xiaomi, China). Tensile strength test was performed using apparatus type LWD20 (Changchun Machinery Factory, China).
3. Analysis
3.1. Thermal conductivity test data and SEM
The thermal conductivity of each sample (Table 1) was tested separately according to the method described above. According to the tests, the thermal conductivity of pure Cu was 398 W m−1 K−1, while the thermal conductivity of the sample immersed in PDA and GO and annealed (sample 6) reached 519.43 W m−1 K−1, which is an improvement by 30.5%, making it a highly thermally conductive material that surpasses conventional thermally conductive metals. The high thermal conductivity of sample 6 is due to the in situ reduction of GO by PDA on the surface of the copper sheet, forming a thick graphene film on the Cu substrate. Additional performance tests were conducted on these samples to understand the reasons for their high thermal performance.14
The fabrication steps of the composites are illustrated in Fig. 1a; the G/PDA@Cu material is formed by converting dopamine to PDA on the surface of a copper sheet, followed by graphene loading and then annealing. The thermal conductivity of the individual samples is shown in Fig. 1b. One of the samples, sample 6, achieved ultrahigh thermal conductivity, the reasons for which are analyzed below. In Fig. 1c, it can be clearly observed that the surface of sample 4 had a large number of spherically distributed PDA particles. Fig. 1d and f show the electron microscope images of sample 6 at different magnifications, in which the dense deposition of graphene can be seen. Due to superconductivity, sample 6 was observed at low magnification. As shown in Fig. 1d, graphene was distributed in a network on the surface of sample 6, which is the main reason for its superconductivity. Therefore, in the overall reaction, it is believed that dopamine forms PDA on the surface of the copper sheet, which then adsorbs graphene oxide and forms a graphene thermal conductivity network after annealing. To further verify this idea, the samples were subjected to the next step of EDS testing. The surface elemental composition of the as-prepared samples was analysed by EDS, as shown in Fig. 2. Few C and O existed on the surface of the Cu sheet, as shown in Fig. 2a, and these elements probably belonged to the pollutants adsorbed on the sample or to CuO. In Fig. 2b, the C and O contents of sample 2 are greater than those of sample 1 (Cu substrate), which demonstrates that PDA was successfully adsorbed to the surface of the Cu substrate.
Fig. 1. (a) Experimental flow diagram, (b) thermal conductivity test date, (c) SEM images of sample 4 at high magnification, (d) sample 6 low magnification, (e) sample 6 at high magnification.

Fig. 2. EDS spectra of sample 1 (a), sample 2 (b), sample 5 (c) and sample 6 (d).

The presence of chlorine could be ascribed to dopamine hydrochloride. Fig. 2c shows that the C and O contents of sample 5 were significantly increased compared to those of sample 2, indicating that GO was adsorbed to the surface of the PDA film. After the annealing treatment, the C and O contents of sample 6 decreased slightly, as presented in Fig. 2d, indicating that some oxygen-containing functional groups were removed during the annealing process and that GO was reduced to form reduced graphene oxide (rGO).15 This is due to the in situ reduction of graphene oxide (rGO) by PDA. EDS can only perform the elemental analysis of the surface composition of a substance; therefore, other tests were required to analyse the internal design of the substance.
3.2. XRD analysis
Fig. 3 shows the XRD patterns of sample 1 (a), sample 3 (b), sample 5 (c), sample 6 (d), and sample 7 (e) in the 2θ region of 3–55°. No significant changes were found between the XRD patterns of sample 1 and sample 3, demonstrating that PDA did not affect the crystal structure of the Cu substrate. The peaks labelled as “Δ” at 43.3° and 50.4° were assigned to the extremes of Cu(111) and Cu(200) (JCPDS card no. 04-0836). New peaks located at 10.2° (♦) and 36.3° (*) in curve c could be attributed to the diffraction peaks of GO and Cu2+, respectively, indicating that Cu was oxidized. After annealing at 200 °C for 1 h, the diffraction peak of GO disappeared, which may indicate that GO was converted to an amorphous state. When the annealing temperature was 300 °C, new diffraction peaks, labelled as “*”, appeared at 29.4°, 36.3° and 42.1° corresponding to the (110), (111), and (200) planes of Cu2+. XRD determines the material composition, and next, the precise changes in the material were determined.16
Fig. 3. XRD patterns: sample 1 (a), sample 3 (b), sample 5 (c), sample 6 (d) and sample 7 (e).

3.3. ATR-FTIR spectroscopy
To further determine the structure of G/PDA@Cu, the ATR-FTIR spectra of sample 3 (a), sample 5 (b), and sample 6 (c) were obtained, as shown in Fig. 4. The broad peaks at 3387 cm−1 and 3350 cm−1 were attributed to the O–H and N–H stretching vibrations. Similarly, the peak at 1730 cm−1 corresponded to the C O stretching vibration. The C C stretching vibrations and N–H bending vibrations were discovered, and the peaks at 1618 cm−1 and 1575 cm−1 were the typical superimposed absorption peaks of the phenylic groups.17 Additionally, the C–O–H deformation vibration of phenols (1387 cm−1), C–OH stretching vibration (1223 cm−1), C–O stretching vibrations (1058 cm−1, 1110 cm−1), and C–H deformation vibration (approximately 775 cm−1) are revealed in Fig. 4. After the annealing treatment, the superimposed peaks of the phenylic C C stretching vibrations in sample 5 and sample 6 moved from 1618 cm−1 to 1575 cm−1, which is the result of the increase in the degree of conjugation compared with sample 3. The intensity of the peaks of the oxygen-containing functional groups in curves b and c had significantly decreased. The peak at 1387 cm−1 nearly vanished, which indicated that the C–O–H of phenols were removed during annealing, which is consistent with the EDS results. The intensity of the C O peak (curve b) became more intense than that of sample 6 (curve c) at 1730 cm−1, indicating an oxidation–reduction reaction between the hydroxyl group of PDA and the carboxyl group of GO during annealing. Thus, GO changes to rGO after annealing.18
Fig. 4. ATR-FTIR spectra: sample 3 (curve a), sample 5 (curve b), and sample 6 (curve c).

3.4. Raman spectra
The Raman spectra of sample 5 (curve a) and sample 6 (curve b) are shown in Fig. 5. The peaks centred at approximately 1349, 1597 and 2700 cm−1 could be attributed to the D, G, and 2D bands of carbon materials. The D band is related to the vibration of defects, representing the disorder of the material, and the G band arises from the pulse of the sp2-hybridized carbon atoms in the basal plane. The I2D/IG intensity ratios of sample 5 and sample 6 were 0.30 (a) and 0.22 (b), indicating the presence of two or more layers of G.19,20 The I2D/IG intensity ratios decreased after the annealing treatment, indicating more amorphous carbon particles in sample 6, as determined by XRD. The ID/IG intensity ratio is widely used to assess the defect density in graphite materials. The high intensity of the D bands in Fig. 5 is due to the disordered structures of the samples. The ID/IG intensity ratio of sample 6 (1.05) was lower than that of sample 5 (1.21), which demonstrated that the annealing treatment decreased the defect density of the sample, and GO had become rGO.21 The Raman tests illustrated the need for annealing, but the bonding energy between graphene and copper after annealing had to be investigated.
Fig. 5. Raman spectra of sample 5 (curve a) and sample 6 (curve b).

3.5. XPS analysis
X-ray photoelectron spectroscopy (XPS) was used to study the surface chemical composition of sample 4 and sample 5, as shown in Fig. 6. Fig. 6a shows the general XPS spectra of sample 4 (curve A) and sample 5 (curve B), which show a predominant C 1s peak at 285 eV, an N 1s peak at 398 eV, an O 1s peak at 531 eV and Cu peaks. The percentages of the elements in sample 4 and sample 5 are listed in Table 2.
Fig. 6. XPS spectra: (a) general XPS spectra of sample 5 (a) and sample 6 (b); (b) C 1s spectra of sample 5 (c) and sample 6 (d); (c) high-resolution C 1s spectrum of sample 5; (d) high-resolution C 1s spectrum of sample 6; (e) high-resolution Cu 2p spectrum of sample 6; and (f) high-resolution O 1 s spectrum of sample 6.

Percentage of elements in sample 5 and sample 6.
| C 1s | N 1s | O 1s | Cu 2p3 | |
|---|---|---|---|---|
| Sample 5 | 65.33 | 1.44 | 30.49 | 2.74 |
| Sample 6 | 54.75 | 1.21 | 30.64 | 13.4 |
Fig. 6b presents the C 1s spectra of sample 5 (curve A) and sample 6 (curve B), which show a significant difference. The high-resolution C 1s spectrum of sample 4 (Fig. 6c) could be deconvoluted into five main peaks at 283.7 eV, 284.6 eV, 285.5 eV, 286.2 eV, and 287.5 eV attributed to sp2-hybridized carbon, C–C, C–N, C–O and C O species,22 respectively. Fig. 6d exhibits the high-resolution C 1s spectrum of sample 5, which could be curve-fitted into five peaks at 284.6 eV (C–C), 285.2 eV (C–N), 286.3 eV (C–O), 287.9 eV (C O) and 288.6 eV (O–C O). The appearance of the O–C O peak (286.3 eV, 287.9 eV) indicated that an esterification reaction had occurred between the hydroxyl group of PDA and the carboxyl group of GO. The decrease in the C–O peak indicated the removal of the hydroxyl groups of PDA during annealing, verifying the oxidation–reduction reaction between the hydroxyl group of PDA and the carboxyl group of GO, which is consistent with the ATR-FTIR results. The high-resolution Cu 2p spectrum confirmed the valence state of Cu on the surface of sample 5, as seen in Fig. 6e. The peaks at approximately 933.1 eV and 952.9 eV could be assigned to Cu 2p3/2 and Cu 2p1/2 of Cu2+, and the peak at approximately 934.6 eV belonged to Cu2+.23,24 The appearance of Cu+ and Cu2+ suggested the partial oxidation of Cu, which is consistent with the XRD results. Fig. 6f depicts the high-resolution O 1s spectrum of sample 5, which could typically be deconvoluted into five peaks at 530.0 eV (O–Cu), 531.2 eV (O–C O), 531.9 eV (C O), 532.7 eV (C–O), and 533.5 eV (O–H). The presence of 530.0 eV (O–Cu) also confirmed the oxidation of Cu, which conforms with the above result.
3.6. Formation mechanism of G/PDA@Cu
PDA is formed by the self-polymerization of dopamine in an alkaline solution and adheres universally to materials, such as metals, polymers, G, and ceramics.25 Therefore, PDA has been used as an adhesive for Cu sheets and G films in this experiment. Oxygen-containing functional groups, such as hydroxyl and carboxyl groups, exist on the basal planes and edges of GO. GO adsorbs to the surface of the PDA film through an esterification reaction between the hydroxyl groups of PDA and the carboxyl group of GO. Then, GO is reduced to rGO through the annealing treatment, and a composite is formed. PDA is bonded to both G and the Cu substrate by an oxygen bond and acts as a bridge between them, as shown in Fig. 7, and oxygen-containing functional groups could also be seen in the XPS spectra, which agree with the experimental results. Here, the presence of oxygen bonding shortens the distance between the G and Cu sheets. Thus, the thermal conductivity of the material is greatly improved. Metals are isotropic materials, i.e. have the same thermal conductivity in all directions. On the other hand, graphene is anisotropic and has high thermal conductivity only along its face. PDA can control the adhesion direction of graphene so that its face is parallel to the substrate, thereby promoting high thermal conductivity.
Fig. 7. Formation mechanism of Gr@PDA/Cu.

3.7. Adhesion force test
The binding force between the prepared G film and substrate in this study was improved compared with similar materials. In the designed experiment, the substrate surface was evaluated by a nano-scratch meter by making a linear scratch with a sphere-conical indenter. The test data and images are shown in Fig. 8a. The beginning load was 0.3 mN, the end load was 10 mN, the loading rate was 19.4 mN min−1, and the scanning load was 0.3 mN. In Fig. 8, curve 1 is the pre-scanned surface height. Curves 2 and 3 are the residual depth and penetration depth, respectively, and curve 4 is the load pressure line. The upper part of Fig. 8 is the image of the sample. It can be clearly seen that the scratch penetrated through G, exposing the metal substrate underneath. The lower part of Fig. 8 shows the load condition. The loading force and adhesion force between the G film and the Cu substrate were determined. The G film ruptured at the load limit, exposing the metal substrate.26 As the force gradually deepened, the G film broke at 4.18 mN, exposing the metal layer. The corresponding loading force was 4.18 mN. To test the tensile properties of the material, tensile tests were carried out on the copper sheet and G@PDA/Cu, as shown in Fig. 8b and c. The tensile properties of G@PDA/Cu had improved by 12.4%. The copper sheet had no structural damage during the preparation, and G@PDA/Cu met the target requirements.
Fig. 8. (a) Adhesion force between the membrane and substrate. (b) Tensile strength of copper sheet. (c) Tensile strength of G@PDA/Cu.

The intermembrane bonding force of the composite material was tested, and the results proved that G was strong. The test also measured the thickness of the G film as 3 μm. According to previous research, the thermal conductivity of the G film decreases to 500 W m−1 K−1 when the thickness of the film is greater than 5 nm.22 The main reason for this is that when the G volume is too large, the folded layers become larger. G accumulates in a disorderly manner on the film; thus, the more significant the G nanoribbon proportion, the greater is the decrease in thermal conductivity.22 In contrast, with the use of PDA, G undergoes orderly growth while forming self-assembled layers, thereby still showing an increase in thermal conductivity. The following theoretical calculations are based on film thickness. PDA shows a small change in elastic modulus as the temperature changes, and this can control the formation of G aggregates during annealing, making it easier to form thermally conductive networks.
3.8. Model simulation
The optimal thermal conductivity of the G/PDA@Cu composite material was modelled by a mathematical method. Model A was based on a previous tandem model and considered G, PDA, and Cu sheets in an up- and down superimposed structure. Model A and a structural diagram of the thermal conductive directions are shown in Fig. 9. PDA is reduced after the annealing treatment, leaving only the oxygen bond connecting the Cu sheet and G; thus, the established model was a sandwich-like structure, as shown in Fig. 9a. A heat transfer system is formed in both directions parallel to the elongated Cu layer and across the Cu layer, as shown in Fig. 9b and c, respectively.
Fig. 9. Model A (a) and the structural diagram of the thermal conduction directions: parallel to the elongated Cu layer (b) and across the Cu layer (c).

According to model A, the thermal conductivity of the composite can be calculated according to the parameters of lamellar composites in relevant studies. The volume fraction of the composite material (Vv) was obtained by eqn (3).
According to the adhesion force test result, the volume fraction (Vv) of graphene in the composite was 1%. The thermal conductivity calculation of G/CVD@Cu was performed with the same Vv value.27
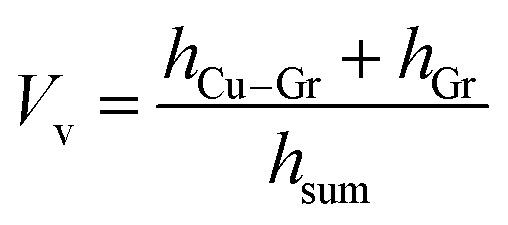 |
3 |
where hCu–G is the thickness of the Cu–G interface, hG is the thickness of the G layer, and hsum is the thickness of the composite material. The hsum value was obtained by eqn (4).
| hsum = hGr + hCu + 2hCu–Gr [μm] | 4 |
where hCu is the thickness of the copper layer.
The thermal conductivity parallel to the elongated Cu layer (ε∥) was obtained using eqn (5).
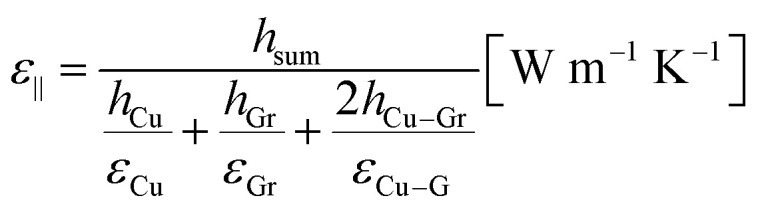 |
5 |
where εCu, εG, and εCu–G are the thermal conductivities of the copper layer, G layer, and Cu–G interface, respectively.
The thermal conductivity of parallel to the elongated Cu layer (ε∥) can be obtained by eqn (5).
The thermal conductivity of across the Cu layer (ε⊥) was obtained using eqn (6).
 |
6 |
Along with data from previous studies, the parameters of G/CVD@Cu are shown in Table 3.
Parameters of the lamellar composite fabricated in this studya.
| Model A | ε Cu | ε Cu–G∥ | ε Cu–G | ε G∥ | ε G⊥ | N G | h Cu |
|---|---|---|---|---|---|---|---|
| 398 | 50–350 | 1000 | 0.03–0.3 | 0.67 | 2 | 0.3 |
N G is the number of layers.
The thermal conductivity enhancement (TCE) was obtained by applying eqn (7):28
| TCE = ε − εCu [W m−1 K−1] | 7 |
The macroscopic thermal conductivity enhancement parallel to the elongated Cu layer (TCE∥) and that across the Cu layer (TCE⊥) were obtained using eqn (8) and (9), respectively:
| TCE∥ = ε∥ − εCu [W m−1 K−1] | 8 |
| TCE⊥ = ε⊥ − εCu [W m−1 K−1] | 9 |
According to Table 4, the overall thermal conductivity of copper did not significantly improve when only G was used to form the film, and the longitudinal improvement of G/CVD@Cu was only 0.05 W m−1 K−1; thus, the TCE of G/CVD@Cu was 10.6 W m−1 K−1. The adhesion force significantly decreased the interface thickness hCu–G in G/PDA@Cu and conventional G/CVD@Cu, which is the main reason for the improved thermal conductivity. Briefly, hCu–G decreases, leading to the rapid and barrier-free propagation of phonons between spaces and resulting in a substantial increase in thermal conductivity. Moreover, the reason for the difference in thermal conductivity between G/PDA@Cu and G/CVD@Cu can be seen in Fig. 10a. This result is almost the same as the data obtained for a conventional thermally conductive G film.29 The high thermal conductivity of the G/PDA@Cu material is because the PDA component controls the growth direction of graphene and forms layers of self-assembled graphene oxide films, which are reduced after annealing to form graphene films. The graphene films in the G/PDA@Cu material are more robust compared with those formed by conventional growing methods. Due to the self-assembled layered structure, the thermal conductivity network is more complete, and the phonon-free range is increased, allowing fast transfer and thereby achieving super thermal conductivity.
Calculated values of the lamellar composite fabricated in this study.
| Material | h | V v | ε ∥ | ε ⊥ | TCE⊥ | TCE∥ |
|---|---|---|---|---|---|---|
| Cu | — | 0 | 398 | 0.23 | — | — |
| G/CVD@Cu | 100 | 0.1% | 408 | 0.24 | 0.01 | 8 |
| G/PDA@Cu | 100 | 0.1% | 519 | 0.29 | 0.06 | 90 |
Fig. 10. (a) Diagram showing the reasons for the difference in thermal conductivity between Gr/CVD@Cu and Gr/PDA@Cu, (b) comparison of ε between Gr/PDA@Cu and other works.

Fig. 10(b) summarizes the Vv of previously reported graphene/metal composites. In similar research works, high thermal conductivity is often achieved by adding large amounts of matrix material to the substrate material. A higher Vv ratio means that the matrix materials are closer to each other, making it easier to achieve a thermally conductive network. However, in the design of composite materials, lower Vv means a more rational approach is necessary for the construction of the thermal network. Notably, the comparative results indicated that G/PDA@Cu exhibited an excellent thermal conductivity of 519.43 W m−1 K−1, which is enhanced by 30.5% compared with that of the Cu substrate; this is among the highest values reported for graphene/metal bulk composites till date and comparable to the best value reported for graphene/metal-film composites.30–34 The literature summarized here has used similar tests and preparation methods, thereby providing comparative values.
3.9. Construction of flexible devices with high thermal conductivity
The main types of batteries are lead-acid batteries, ternary lithium batteries, and lithium-iron-phosphate batteries. Due to their unique structure, ternary lithium batteries generate a large amount of heat during the charge/discharge process and undergo volume change. Additionally, a break in a ternary lithium battery causes intense thermal changes, with severe cases leading to vehicle incineration. Traditionally, most of these batteries use thermally conductive silicone to stabilize and control the temperature. Based on this application, a flexible heat-conducting device was constructed using G/PDA@Cu in combination with a heat-insulating sponge that allows heat to travel quickly without causing damage to the battery. The structure of the flexible heat-conducting device is shown in Fig. 11.
Fig. 11. Flexible device. (a) Structure of a flexible thermally conductive device. (b) The device is crushed and deformed. (c) The flexible thermally conductive device under battery expansion.

Fig. 12. Infrared imaging; (a) photograph of the device consisting of 5 × 5 arrays of individually addressable pixels with an area of 10 × 10 mm2. Y is for Gr@PDA/Cu; N is for copper sheet. (b) Schematic diagram of the test structure. The hot air temperature is 30 °C. (c) Infrared imaging of the device.

The thermally conductive material was constructed by cutting a 10 mm × 10 mm × 0.1 mm composite of G/PDA@Cu and bonding it to a thermal-resistant sponge through PET to form a flexible device. Its composition is shown in Fig. 11(a). The device had high thermal conductivity and deformed under pressure without damaging the structure of the device, as shown in Fig. 11(b). Therefore, this device can be used in the thermal management of ternary lithium batteries to solve heat generation and volume expansion during operation. The operating principle of the device is shown in Fig. 11(c). According to tests, this device had a thermal conductivity of 519.43 W m−1 K−1. The thermal conductivity of the original product was 200 W m−1 K−1; thus, the prepared device showed a vast improvement relative to the actual device.
3.10. Simulation of heat diffusion
Briefly, G/PDA@Cu and a Cu sheet were placed apart from each other to form 50 mm × 50 mm squares. At room temperature, the two materials were indistinguishable by an infrared camera. When heated with hot air, the temperature of the copper sheet increased to 30 °C, while the temperature of G/PDA@Cu was only 25 °C. This result was due to the difference in thermal conductivity between the copper sheet and G/PDA@Cu and showed different patterns in the infrared camera image. After 30 s of fan heating, the difference in temperature between the two kinds of sheets was 5 °C.
The reason for conducting this experiment was to visualise the thermal conductivity of the two materials. Generally, pure copper is a common metallic material with close-to-ultrahigh thermal conductivity in its own right, and the thermal conductivity of the newly constructed material is ultrahigh. Using this discrepancy in thermal conductivity combined with image recognition technology, this composite can be thought of as a simple temperature sensor. That is, a discrepancy pattern can be observed in the infrared image when abnormal heat is generated in the environment. Thus, this device can be used as an emergency alarm.
4. Conclusions
The graphene/polydopamine composite material on a copper substrate (G/PDA@Cu), with ultrahigh thermal conductivity, was obtained by the adsorption of graphene oxide on polydopamine and annealing. Graphene was self-assembled on the surface of the copper sheet by the adsorption of polydopamine. The thermal conductivity of the composite prepared by this method was found to be significantly higher than those synthesized by conventional methods, such as CVD. The composite showed an ultrahigh thermal conductivity of 519.43 W m−1 K−1, which is an increase of 30.5% relative to that of the Cu substrate. The adhesion force and tensile strength of G/PDA@Cu were tested in order to verify its applicability. The adhesion force was 4.18 mN, and the tensile strength of the composite was increased by 12.4% than that of copper. Using the ultrahigh thermal conductivity of this material, thermo-responsive devices were constructed, which presented patterns in infrared images when unexpected heat was encountered. Also ternary lithium batteries under charge and discharge were accompanied by heat release and volume expansion. Flexible thermally conductive devices were prepared using the composite material to test the practical application of the material.
Conflicts of interest
The authors declare there is no conflict of interest.
Supplementary Material
Acknowledgments
We gratefully acknowledge the National Natural Science Foundation of China (no. 21271027) for their financial support of this work.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05252g
Notes and references
- Pe M. I. S., Surface Production Operations, 3rd edn, 2014, pp. 39–97 [Google Scholar]
- Marcano D. C. Kosynkin D. V. Berlin J. M. Sinitskii A. Tour J. M. ACS Nano. 2010;4:4806–4814. doi: 10.1021/nn1006368. [DOI] [PubMed] [Google Scholar]
- Zhao C. Y. Lu W. Tian Y. Sol. Energy. 2010;84:1402–1412. doi: 10.1016/j.solener.2010.04.022. [DOI] [Google Scholar]
- Balandin A. A. Nat. Mater. 2011;10:569–581. doi: 10.1038/nmat3064. [DOI] [PubMed] [Google Scholar]
- Lee H. Dellatore S. M. Miller W. M. Messersmith P. B. Science. 2007;318:426–430. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balandin A. A. Ghosh S. Bao W. Calizo I. Teweldebrhan D. Miao F. Lau C. N. Nano Lett. 2008;8:902. doi: 10.1021/nl0731872. [DOI] [PubMed] [Google Scholar]
- Chu K. Wang X. H. Wang F. Li Y. B. Huang D. J. Liu H. Ma W. L. Liu F. X. Zhang H. Carbon. 2017;127:102–112. doi: 10.1016/j.carbon.2017.10.099. [DOI] [Google Scholar]
- Chen L. Y. Konishi H. Fehrenbacher A. Ma C. Xu J. Q. Choi H. Xu H. F. Pfefferkorn F. E. Li X. C. Scr. Mater. 2012;67:29–32. doi: 10.1016/j.scriptamat.2012.03.013. [DOI] [Google Scholar]
- Firkowska I. Boden A. Vogt A. M. Reich S. J. Mater. Chem. 2011;21:17541. doi: 10.1039/C1JM12671G. [DOI] [Google Scholar]
- Muhammad R., Pan F., Tang A. and Muhanmmad A., in, Natural Science:Materials International, ProGess, 2014, vol. 24, pp. 101–108 [Google Scholar]
- Dong X. Cheng J. Li J. Wang Y. Anal. Chem. 2010;82:6208–6214. doi: 10.1021/ac101022m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei Z. Hou C. Zhang Q. Wang H. Li Y. Mater. Chem. Phys. 2012;135:826–831. doi: 10.1016/j.matchemphys.2012.05.065. [DOI] [Google Scholar]
- Mu Q. Feng S. Diao G. Polym. Compos. 2010;28:125–130. doi: 10.1002/pc.20276. [DOI] [Google Scholar]
- Pop E. Varshney V. Roy A. K. Bull. 2012;37:1273–1281. [Google Scholar]
- Porwal H. Tatarko P. Gasso S. Khaliq J. Dlouhy I. Reece M. J. Carbon. 2013;64:359–369. doi: 10.1016/j.carbon.2013.07.086. [DOI] [Google Scholar]
- Mattevi C. Kim H. Chhowalla M. J. Mater. Chem. 2011;21:3324–3334. doi: 10.1039/C0JM02126A. [DOI] [Google Scholar]
- Pavithra C. L. P. Sarada B. V. Rajulapati K. V. Rao T. N. Sundararajan G. Sci. Rep. 2014;4:2045–2322. doi: 10.1038/srep04049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L. Q. Yang W. J. Neoh K. G. Kang E. T. Fu G. D. Macromolecules. 2010;43:8336–8339. doi: 10.1021/ma101526k. [DOI] [Google Scholar]
- Kim Y. Lee J. Yeom M. S. Shin J. W. Kim H. Cui Y. Kysar J. W. Hone J. Jung Y. Jeon S. Nat. Commun. 2013;4:2114. doi: 10.1038/ncomms3114. [DOI] [PubMed] [Google Scholar]
- Goli P. Ning H. Li X. Lu C. Y. Balandin A. A. Nano Lett. 2014;14:1497–1503. doi: 10.1021/nl404719n. [DOI] [PubMed] [Google Scholar]
- Jiehe S. Cheng Z. Jing L. Zhiliang Y. Wei and C. Mater. Lett. 2012;75:158–160. doi: 10.1016/j.matlet.2012.02.007. [DOI] [Google Scholar]
- Chang H. Wu H. Energy Environ. Sci. 2013;6:3483–3507. doi: 10.1039/C3EE42518E. [DOI] [Google Scholar]
- Cha S. I. Kim K. T. Arshad S. N. Mo C. B. Hong S. H. Adv. Mater. 2005;17:1377–1381. doi: 10.1002/adma.200401933. [DOI] [PubMed] [Google Scholar]
- Wan M. Ming L. Li J. Liu Z. J. Appl. Polym. Sci. 1994;53:131–139. doi: 10.1002/app.1994.070530201. [DOI] [Google Scholar]
- Kumar S. V. Huang N. M. Lim H. N. Zainy M. Harrisond I. Chiae C. H. Sensor.Actuator. B Chem. 2013;181:885–893. doi: 10.1016/j.snb.2013.02.045. [DOI] [Google Scholar]
- Park S. Lee K. Bozoklu G. Cai W. Nguyen S. Ruoff' R. Acs Nano. 2008;2:572–578. doi: 10.1021/nn700349a. [DOI] [PubMed] [Google Scholar]
- Wejrzanowski M. G. T. Chmielewski M. Pietrzak K. Kurzydlowski K. J. Strojny-Nedza A. Mater. Des. 2016;99:163–173. doi: 10.1016/j.matdes.2016.03.069. [DOI] [Google Scholar]
- Zhu M. Du Z. Yin Z. Zhou W. Liu Z. Tsang S. H. Teo E. H. T. ACS Appl. Mater. Interfaces. 2015;13:502–510. doi: 10.1021/acsami.5b09453. [DOI] [PubMed] [Google Scholar]
- King W. P. Saxena S. Nelson B. A. Weeks B. L. Pitchimani R. Nano Lett. 2015;6:2145–2149. doi: 10.1021/nl061196p. [DOI] [PubMed] [Google Scholar]
- Andrey M. A. Sergey V. K. Fedor M. S. Appl. Therm. Eng. 2012;48:72–80. doi: 10.1016/j.applthermaleng.2012.04.063. [DOI] [Google Scholar]
- Chu K. Wang X. H. Wang F. Li Y. B. Huang D. J. Liu H. Ma W. L. Liu F. X. Zhang H. Carbon. 2017;127:102–112. doi: 10.1016/j.carbon.2017.10.099. [DOI] [Google Scholar]
- Gao X. Yue H. Guo E. Zhang H. Lin X. Yao L. Wang B. Powder Technol. 2016;301:601–607. doi: 10.1016/j.powtec.2016.06.045. [DOI] [Google Scholar]
- Hwang J. Yoon T. Jin S. H. Lee J. Kim T. S. Hong S. H. Jeon S. Adv. Mater. 2013;25:6724–6729. doi: 10.1002/adma.201302495. [DOI] [PubMed] [Google Scholar]
- Liu Q. He X. B. Ren S. B. Zhang C. Ting-Ting L. Qu X. H. J. Alloys Compd. 2014;587:255–259. doi: 10.1016/j.jallcom.2013.09.207. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.