

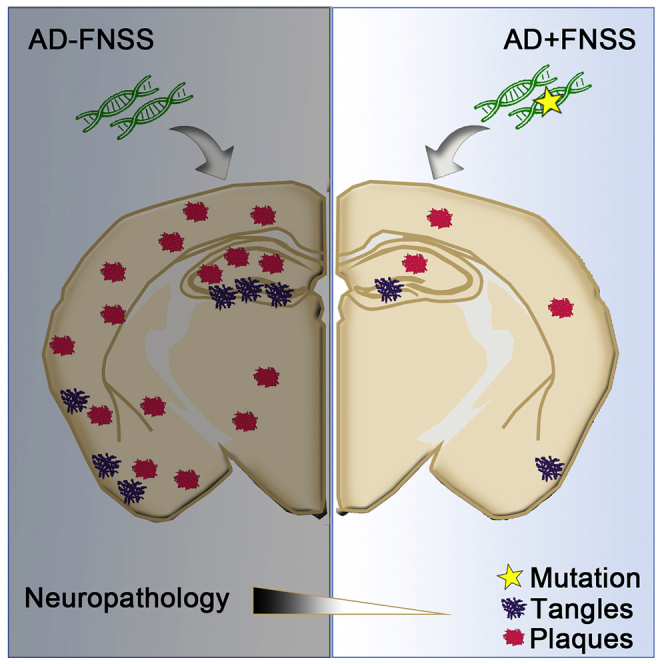
Summary
Although numerous studies have demonstrated that poor sleep increases the development of AD, direct evidence elucidating the benefits of good sleep on the AD pathogenesis is lacking. Familial Natural Short Sleepers (FNSS) are genetically wired to have lifelong reduction in nightly sleep duration without evident consequence on cognitive demise, implying that they may have better sleep quality. Here we investigated two FNSS mutations, DEC2-P384R and Npsr1-Y206H, on the development of tau and amyloid pathology in AD-like mouse models. We found that the development of tau pathology is attenuated in the hippocampus of tau mice carrying FNSS mutations. We also found that DEC2-P384R;5XFAD and female Npsr1-Y206H;5XFAD mice exhibit significantly less amyloid plaques than control mice at 6 months of age. Together, these results reveal that these two FNSS alleles are strong genetic modifiers of AD pathology and may confer resilience to the progression of tau pathology and amyloid plaque formation in neurodegeneration.
Subject areas: Behavioral neuroscience, Biological sciences, Molecular neuroscience
Graphical abstract
Highlights
-
•
Two FNSS mutations are strong genetic modifiers of AD-like pathology in mice
-
•
Mutant DEC2 and Npsr1 reduced tau pathology in PS19 mouse model of tauopathy
-
•
Mutant DEC2 and Npsr1 slowed down amyloid plaques in 5XFAD APP transgenic mouse model
-
•
Efficient sleep may be an exciting therapeutic target for ameliorating AD development
Behavioral neuroscience; Biological sciences; Molecular neuroscience
Introduction
Sleep is one of the most important factors necessary for human life, and dysregulation of sleep has been associated with increased risk of a plethora of human diseases (Mander et al., 2017; Scullin and Bliwise, 2015). Increasingly in recent years, research has found a link between sleep and the progression of neurodegeneration including Alzheimer’s disease (AD). The quality and duration of sleep appear to be important in stalling neurological decline, as older individuals with the most disturbed patterns tend to exhibit both the greatest likelihood of developing cognitive impairment and more rapid progression after its onset (Chang-Quan et al., 2012; Lee et al., 2020; Schmutte et al., 2007; Tafaro et al., 2007). In AD mouse models, canonical biomarkers of AD disease-load display characteristics of exacerbated pathology in response to sleep restriction (Holth et al., 2019; Kang et al., 2009; Roh et al., 2012; Rothman et al., 2013; Shi et al., 2017; Zhao et al., 2019). In turn, elevated levels of these molecules are associated with poorer sleep behaviors, together resulting in a positive feedback loop, where poor sleep and disease pathology each accelerate progression of the other (Chen et al., 2018; Ju et al., 2014; Lucey et al., 2019; Spira et al., 2013; Spira and Gottesman, 2017). Understanding the mechanism connecting sleep and AD pathology could provide insight into potential new approaches for delaying the onset of AD (Mander et al., 2016; Miyata et al., 2013; Tafaro et al., 2007).
Recent research has uncovered genetic influences upon human sleep duration through the identification of sleep phenotypes at the extremes of normal population variation (Kalmbach et al., 2017; Toh et al., 2001). Our team has reported a number of human mutations causing Familial Natural Short Sleep (FNSS) (He et al., 2009; Shi et al., 2019, 2021; Xing et al., 2019). These individuals sleep substantially fewer hours per night (<6.5) than most humans, and many are elderly- in the age range where AD is most likely to develop. Anecdotally, however, they do not appear to exhibit the cognitive symptoms associated with the development of these diseases. The reasons behind this phenomenon are entirely unknown and may support the possibility of enhanced sleep efficiency, where certain sleep features or individual genetic factors confer more of the positive benefits of sleep in a shorter period of time (Tkachenko and Dinges, 2018). To test this possibility, we crossed two previously validated FNSS mouse models individually onto two mouse lines commonly used to recapitulate the amyloid beta (Aβ) and tau components of the AD-like phenotype- 5XFAD (Oblak et al., 2021; Tkachenko and Dinges, 2018) and PS19 (Yoshiyama et al., 2007). Our results revealed that two different FNSS genes/mutations are strong genetic modifiers of Aβ and tau pathology, suggesting improved sleep may have potential as a therapeutic target for delaying dementia onset and progression.
Results
Tangles and plaques in FNSS;AD mice
As both tau tangles and Aβ plaques are considered hallmarks of AD pathology, we crossed our DEC2-P384R FNSS mouse line (He et al., 2009; Hirano et al., 2018) separately to PS19 and 5XFAD transgenic lines. Corresponding DEC2-WT;AD transgenic lines were also generated to serve as controls. To determine if mutant and WT DEC2 mice exhibited differences in AD-like neurodegeneration, we employed a number of well-characterized molecular metrics of disease progression. PS19 mice have been shown to develop tau aggregates consisting of hyperphosphorylated forms of tau as paired helical filaments (PHFs) by six months of age (Yoshiyama et al., 2007). Therefore, we performed immunohistological analysis of the dentate gyrus (DG) from six month old DEC2-P384R;PS19 and DEC2-WT;PS19 mice for phospho-tau aggregates and found significantly less deposition in DEC2-P384R;PS19 hippocampus compared with either DEC2-WT;PS19 or PS19 mice (Figures 1A and 1B).
Figure 1.

DEC2-P384R;AD and Npsr1-Y206H;AD mice have less tangles and plaques
(A) Immunostaining of PHFs tau located within the DG region of six-month-old PS19 mice (left), DEC2-WT;PS19 mice (middle) and DEC2-P384R;PS19 mice (right). PS19, n = 16; DEC2-WT;PS19, n = 10; DEC2-P384R;PS19, n = 17.
(B) Quantification of (A).
(C) Representative silver-stained slices for six-month-old female DEC2-WT or DEC2-P384R mice in 5XFAD background; female 5XFAD and Npsr1+/m;5XFAD mice.
(D) % area of silver-stained brain slice from hippocampus and cortex covered by plaques in six-month-old mice. Male hippocampus: DEC2-WT;5XFAD, n = 4 (N = 35 slices); DEC2-P384R;5XFAD, n = 6 (N = 49 slices); 5XFAD, n = 4 (N = 30 slices); Npsr1+/m;5XFAD, n = 8 (N = 58 slices). Female hippocampus: DEC2-WT;5XFAD, n = 3 (N = 21 slices); DEC2-P384R;5XFAD, n = 3 (N = 21 slices); 5XFAD, n = 5 (N = 35 slices); Npsr1+/m;5XFAD, n = 7 (N = 48 slices). Male cortex: DEC2-WT;5XFAD, n = 4 (N = 42 slices); DEC2-P384R;5XFAD, n = 7 (N = 61 slices); 5XFAD, n = 4 (N = 41 slices); Npsr1+/m;5XFAD, n = 8 (N = 68 slices). Female cortex: DEC2-WT;5XFAD, n = 3 (N = 21 slices); DEC2-P384R;5XFAD, n = 3 (N = 21 slices); 5XFAD, n = 4 (N = 35 slices); Npsr1+/m;5XFAD, n = 7 (N = 48 slices).
(E) Immunostaining of PHFs tau located within the DG region of six-month-old mice from left to right: PS19, n = 12; Npsr1+/m;PS19, n = 13; Npsr1+/m, n = 2; WT, n = 2.
(F) Quantification of (E). One-way ANOVA with Fisher’s LSD test (B). One-way ANOVA with post hoc Tukey’s multiple comparisons test (D). Two-way ANOVA with post hoc Tukey’s multiple comparisons test (F).
Data expressed as mean ± SEM. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; n.s., not significant. Scale bars, 50 μm (A and E) and 1mm (C).
We then measured Aβ plaque load in DEC2-P384R mice crossed with the 5XFAD line. Mice were sacrificed and assessed at 3 months of age, which previous research has indicated is when AD phenotypes are beginning to manifest in this model, and also at 6 months, when neurological decline is more evident (Jawhar et al., 2012). There was little Aβ plaque deposition in all genotypes at 3 months (Figure S1A). But at 6 months of age, 5XFAD mice showed obvious Aβ plaque deposition consistent with previous reports (Figure S1B). We found that plaque load in DEC2-P384R;5XFAD mice was significantly lower than that in DEC2-WT;5XFAD mice in both the cortex and hippocampus at 6 months of age (Figures 1C and 1D). Together, these data demonstrate that the DEC2 mutation reduces tau tangles and Aβ plaque formation in AD mice, suggesting DEC2-P384R;AD mice may be protected from disease progression.
To discern whether the apparent protective effect of DEC2-P384R is specific to this mutation or a broader phenomenon in FNSS, we examined the same metrics in PS19 and 5XFAD mice crossed with another FNSS mouse model harboring a mutation in a different FNSS gene (Npsr1-Y206H (Npsr1+/m)) (Xing et al., 2019). At 6 months of age and similar to the PS19 allele on a DEC2-P384R background, Npsr1-Y206H;PS19 (Npsr1+/m;PS19) mice also exhibited diminished tau phosphorylation in the DG (Figures 1E and 1F). We found that female Npsr1-Y206H;5XFAD mice showed significantly reduced plaque coverage in either hippocampus or cortex compared to 5XFAD controls, although the data for male did not reach significance (Figure 1D). This is not surprising because female 5XFAD mice have been shown to have more robust pathology than males (Bundy and Vied, 2019).
Tau and Aβ in brains of FNSS;AD mice
To shed some light on the process of reduced tangles and plaques observed in FNSS;AD double mutant mice, we next examined tau/phospho-tau and Aβ40/Aβ42 levels. Human tau protein in the brain lysates of three-month and 6-month-old WT and mutant DEC2;PS19 mice was sequentially obtained with buffers of increasing extraction strength (Julien et al., 2012). At 3 months of age, before the onset of tau pathology, DEC2-P384R;PS19 had comparable levels of total tau to DEC2-WT;PS19 in all tested fractions [labeled RAB (soluble), RIPA (less soluble), and FA (insoluble)] (Figures 2A and S2A), indicating that overexpression of tau from the transgene was maintained. We then detected the levels of phosphorylated tau (S202/T205) using the AT8 antibody and found that phospho-tau (S202/T205) in the soluble fraction was dramatically higher in DEC2-P384R;PS19 compared to DEC2-WT;PS19 (Figures 2B and S2B). Another antibody (AT270) that specifically recognizes phosphorylation at T181 displayed a similar pattern of distribution in all fractions to those detected with AT8 antibody (Figures 2C and S2C), confirming that a higher portion of phospho-tau is in the soluble form in the DEC2-P384R;PS19 than control mice at 3 months of age.
Figure 2.

Less tissue phospho-tau in DEC2-P384R;PS19 and Npsr1-Y206H;PS19
(A–C) Quantification of Western blot analysis for total tau (A), phospho-tau by AT8 (B), and phospho-tau by AT270 (C) in the hippocampus of three-month-old DEC2-WT;PS19 and DEC2-P384R;PS19 mice. n = 4 mice per group.
(D–F) Quantification of Western blot analysis for total tau (D), phospho-tau (E), and phospho-tau/total tau ratio (F) in the hippocampus of six-month-old DEC2-WT;PS19 and DEC2-P384R;PS19 mice. n = 10 mice per group.
(G–I) Quantification of Western blot analysis for total tau (G), phospho-tau by AT8 (H), and phospho-tau by AT270 (I) in the hippocampus of three-month-old PS19 and Npsr1+/m;PS19 mice. PS19, n = 6; Npsr1+/m;PS19, n = 7.
(J–L) Quantification of Western blot analysis for total tau (J), phospho-tau (K), and phospho-tau/total tau ratio (L) in the hippocampus of six-month-old PS19 and Npsr1+/m;PS19 mice. PS19, n = 10; Npsr1+/m;PS19, n = 9. Student’s two-tailed unpaired t-test was used for analysis.
Data expressed as mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
By 6 months of age, this pattern was lost. Instead, we found that both the total tau and phospho-tau in the insoluble FA fraction were significantly lower in DEC2-P384R;PS19 compared with DEC2-WT;PS19 mice (Figures 2D, 2E, S2D, and S2E). The phospho-tau in the less soluble RIPA fraction was also significantly lower (Figures 2E and S2E). To assess the degree of tau phosphorylation in relation to total tau levels, we calculated the ratios of phospho-tau to total tau. The ratio of phospho-tau to total tau was significantly lower in both less soluble (RIPA) and insoluble (FA) fractions of DEC2-P384R;PS19 vs. DEC2-WT;PS19 mice (Figure 2F). These results suggest that the DEC2-P384R mutation may play a role in suppressing the development of tau phosphorylation and subsequent accumulation.
We then performed the same experiment with the Npsr1-Y206H;PS19 mouse line. We found lower phospho-tau in both less soluble and insoluble fractions in Npsr1-Y206H;PS19 (Npsr1+/m;PS19) compared to control mice (Figures 2G–2I and S2F–S2H) at 3 months of age. At 6 months of age, the phospho-tau is significantly lower only in the insoluble fraction for Npsr1-Y206H;PS19 (Figures 2J–2L, S2I, and S2J). Overall, both DEC2-P384R;PS19 and Npsr1-Y206H;PS19 showed significantly lower relative phosphorylated tau/total tau ratios (relative to respective controls) in the less soluble and insoluble fractions. These results indicate that these FNSS mutations are genetic modifiers of the development of tau pathology possibly by suppressing tau phosphorylation.
In 5XFAD mice, we measured human Aβ42 in tissue by ELISA. The insoluble fraction of cortical and hippocampal tissues was found to have higher Aβ42 at 3 months of age in the cortex and hippocampus of DEC2-P384R;5XFAD vs. DEC2-WT;5XFAD mice, but this discrepancy was diminished or lost by 6 months of age (Figures 3A and 3B). Assessment of Aβ40 in soluble fractions showed that the same pattern was observed in the hippocampus, but there was no difference between conditions at either age in the cortex (Figure S3A). In the Npsr1-Y206H;5XFAD, we found no obvious difference in the Aβ42 and Aβ40 ELISA compared to the control 5XFAD (Figures 3A, 3B, and S3A). These results indicated that the FNSS;5XFAD mice have similar tissue Aβ42 and Aβ40 levels as control mice at a late stage of disease (6 months) when plaque differences were evident. We also examined the levels of amyloid precursor protein (APP) and one of the important enzymes in the APP processing pathway (Beta-secretase 1, BACE1) and found no difference between FNSS;5XFAD mice and their equivalent controls at both 3 and 6 months (Figure S4). Further investigation on the APP processing pathway is needed to reveal the mechanism contributing to these observations.
Figure 3.

Tissue Aβ42 and CSF Aβ42/tau in FNSS;AD mice
(A) Insoluble Aβ42 from cortex measured by ELISA (3 months from left to right: n = 10, n = 9, n = 11, n = 10; 6 months from left to right: n = 11, n = 12, n = 11, n = 7).
(B) Insoluble Aβ42 from hippocampus measured by ELISA (3 months from left to right: n = 9, n = 6, n = 11, n = 10; 6 months from left to right: n = 10, n = 12, n = 11, n = 8).
(C) CSF tau from indicated genotypes at 6 months measured by ELISA (from left to right: n = 12, n = 15; n = 2, n = 10, n = 12).
(D) Human CSF Aβ42 from indicated genotypes measured by ELISA (3 months from left to right: n = 9, n = 7, n = 13, n = 7; 6 months from left to right: n = 10, n = 8, n = 9, n = 10). One-way ANOVA with Tukey’s multiple comparisons test for (A), (B), and (D) Student’s two-tailed unpaired t-test for (C).
Data expressed as mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001; n.s., not significant.
CSF tau and Aβ in FNSS;AD mice
One possible explanation for the observed reduction in tangles and plaques is a faster clearance rate out of the brain via the glymphatic system (Rasmussen et al., 2018). Sleep also appears to reliably facilitate clearance of these metabolites into the cerebrospinal fluid (CSF) (Holth et al., 2019; Xie et al., 2013). We therefore collected CSF from the cisterna magna of FNSS;AD mice and measured tau and Aβ42 concentrations. There was no significant difference in CSF tau in 6 month old DEC2-WT;PS19 vs. DEC2-P384R;PS19 mice, though in Npsr1-Y206H;PS19 (Npsr1+/m;PS19) mice, we found reduced CSF tau compared to PS19 controls (Figure 3C).
In DEC2-P384R;5XFAD mice, we found that there was significantly more CSF Aβ42 than in DEC2-WT;5XFAD mice at 3 months of age, an effect lost by 6 months (Figure 3D), indicating increased clearance at 3 months. Interestingly, higher Aβ42 levels were also found by ELISA in extracts from the cortex and hippocampus at 3 months of age in mutant DEC2 mice. This difference was also lost by 6 months (Figures 3A and 3B). CSF Aβ42 is significantly higher in 6 month-old Npsr1-Y206H;5XFAD mice than the control mice, suggesting this might be a contributing factor to the lower plaque burden observed in these mice at this age (Figure 3D). There was no difference in plasma Aβ42 between any of the 5XFAD lines (Figure S3B).
Synaptosome tau in Npsr1-Y206H;PS19
We previously demonstrated that the mutant NPSR1 receptor is more active than its WT counterpart and that Npsr1-Y206H neurons are correspondingly hypersensitive to its ligand NPS (Neuropeptide S) (Xing et al., 2019). Interestingly, neuronal activity is known to contribute to the presence of extracellular tau (Pooler et al., 2013; Yamada et al., 2014), and the release of endogenous tau is mediated by activity at the presynapse (Sokolow et al., 2015), yet we found CSF tau levels to be lower in Npsr1-Y206H;PS19 than in control mice (Figure 3C). To reconcile this discrepancy, synaptosomes were prepared from the hippocampus of Npsr1-Y206H;PS19 and PS19 mice (Figure S5), and the extracellular tau levels were analyzed. At baseline, there was no difference in human tau release between Npsr1-Y206H;PS19 (Npsr1+/m;PS19) and PS19 mice (Figures 4A and 4B), though upon NPS treatment, tau secretion was increased only in the synaptosomes of Npsr1-Y206H;PS19 mice (Figures 4A and 4B). These observations, combined with the lower level of CSF tau in the Npsr1-Y206H;PS19 mice, suggests that alternative pathways in the mutant brain, such as degradation within glia, may participate in the clearance of extracellular tau.
Figure 4.

Less synaptic loss and astrogliosis in Npsr1-Y206H background mice
(A) Western blot analysis of human tau levels released from synaptosomes (top) and in the synaptosomes (bottom) with or without NPS treatment. PSD-95 and membranes (mem) stained with Coomassie brilliant blue (CBB) served as loading control. n = 4 mice per group.
(B) Quantification of (A).
(C) Representative images of six-month-old WT, PS19, and Npsr1+/m;PS19 mice brain sections stained with synaptophysin and DAPI. WT, n = 8; PS19, n = 9; Npsr1+/m;PS19, n = 8.
(D) Quantification of (C).
(E) GFAP staining for astrocytes in six-month-old PS19 (top) and Npsr1+/m;PS19 (bottom) hippocampus.
(F) Quantification of (E). PS19, n = 3; Npsr1+/m;PS19, n = 4.
(G) Western blot analysis of GFAP protein levels in PS19 and Npsr1+/m;PS19 mouse hippocampus. β-actin served as loading control.
(H) Quantification of (G). n = 3 mice per group.
Data expressed as mean ± SEM, two-way ANOVA with Sidak’s multiple comparisons test (B), Student’s two-tailed unpaired t-test was used for analysis. ∗p < 0.05, ∗∗p < 0.01; n.s., not significant. Scale bars, 50 μm (C) and 100 μm (E).
Less synaptic loss and astrogliosis in Npsr1-Y206H;PS19
Synaptic deterioration and astrogliosis usually occur with the development of tau pathology in PS19 mice (Yoshiyama et al., 2007). We next chose to examine these properties in one of the mouse lines (Npsr1-Y206H;PS19) to further test the effects of FNSS mutations on tau pathology. We stained for synaptophysin in CA3 and found that the signal significantly decreased from WT controls in the PS19 mice but not Npsr1-Y206H;PS19 mice at 6 months of age (Figures 4C and 4D). Similarly, using GFAP as a measure of gliosis, we found reduced signal in the hippocampus of Npsr1-Y206H;PS19 compared with PS19 mice (Figures 4E and 4F), a result confirmed by Western blot (Figures 4G and 4H). These results demonstrate that the Npsr1-Y206H mutation attenuates synaptic loss and astrogliosis found in PS19 mice even though Npsr1 mutant neurons are more active and release more tau in response to its agonist.
Discussion
AD is the leading cause of age-related neurodegeneration, accounting for nearly 75% of all dementia cases. AD is incredibly complex, and its etiology is characterized by the interplay between many physiological, genetic, and environmental influences. Great effort has been put forward during the last few decades in seeking cures for AD, yet most of this has focused on deleterious genetic influences over preventive ones. FNSS individuals do not appear to suffer from increased risk of dementia despite lifelong shorter sleep duration, implying a potential protective effect stemming from these mutations. Because our database of FNSS families is not large enough to test this using epidemiology, we studied mice carrying human AD alleles on mouse models of FNSS that we have developed. Here, we described the pathological ramifications of two different FNSS mutations affecting upstream (sleep-related) phenotypes which have been tied to AD, providing a perspective into genetic influences that may curtail AD disease progression.
In the first mutation we described (DEC2-P384R), the variant leads to decreased deposition of tau with a significantly lower ratio of insoluble phospho-tau/total tau at 6 months of age when disease phenotypes are evident. Similarly, we found a robust reduction in amyloid plaque deposition for DEC2-P384R;5XFAD compared to control mice, although no difference was found for tissue Aβ42 at this age. We also observed that both tissue and CSF Aβ42 concentrations in DEC2-P384R;5XFAD were higher than control mice at 3 months of age. The higher CSF Aβ42 could be a consequence of the higher tissue Aβ42. Future studies on the APP processing pathways for the FNSS;5XFAD mice are necessary to understand the underlying mechanism.
Of note, orexin levels have been shown to directly modulate the dynamics of Aβ formation (Kang et al., 2009), and higher orexin is expected to increase Aβ plaques. We have previously shown that DEC2 regulates orexin levels by serving as a transcription repressor, and mutant DEC2 is a weaker repressor which leads to higher orexin, consistent with the higher Aβ42 in tissue of 3 months old DEC2-P384R;5XFAD. However, this phenomenon disappeared, and plaque was significantly reduced at 6 months old. Furthermore, soluble phospho-tau was also significantly higher in DEC2-P384R;PS19 than control mice at 3 months of age, which also disappeared at 6 months old. These observations together would suggest that mechanisms occurring between 3 and 6 months will be important targets for future investigation to reveal the underlying causes of this protective effect. Taken together, the evidence suggests that the DEC2-P384R mutation may already provide an environment less conducive for the aggregation of amyloid plaques or phospho-tau tangles at an age before manifestation of a severe neurodegeneration.
Interestingly, we found that plaque formation is significantly higher in DEC2-WT;5XFAD than 5XFAD alone, implying that WT DEC2 might contribute to the Aβ aggregation pathway. However, even with this caveat, DEC2-P384R;5XFAD has a significant effect on reduced plaque accumulation. Further studies on whether WT DEC2 participates in Aβ aggregation and how this single point mutation can lead to significant reduction of Aβ plaque formation will be important for understanding this effect.
For the second mutation (Npsr1-Y206H) and similar to DEC2-P384R, there was no difference found in tissue Aβ42 but the mutation likewise leads to reduced plaque pathology in double mutant mice, especially in females. This is not completely surprising because 5XFAD female mice have a more robust phenotype than male mice. Nonetheless, it is possible that the genetic protective effects of this FNSS mutation are sexually dymorphic. Contrary to DEC2-P384R;5XFAD, the CSF Aβ42 was increased in Npsr1-Y206H;5XFAD vs. control, suggesting the possibility of increased Aβ42 clearance in these mice. We found that the Npsr1-Y206H;PS19 mice, despite elevated neuronal activity and greater extracellular tau after stimulating the synapses, have fewer tangles, less phosphorylated tau, and less CSF tau than PS19 mice without the FNSS mutation. These results indicate that mechanisms in addition to the glymphatic system warrant further investigation for clearance of Aβ and tau to facilitate healthier brain aging. In addition, we verified the preservation of neurons and astrocytes as benefits of reduced tau aggregation in Npsr1-Y206H;PS19 mice.
Introduction of the PS19 allele onto two different FNSS genotypes has a notable effect on phospho-tau levels in less soluble and insoluble fractions. Both showed significantly lower phospho-tau/total tau ratios. Because total tau levels for different genotypes seem to be comparable, this observation suggests that reduced phospho-tau in the insoluble fractions is likely not caused by the production of total tau, but rather, in the production (and/or clearance) of phospho-tau.
Recent advances in basic research are revealing that AD is a multifaceted degenerative process including Aβ, tau, and other pathophysiological mechanisms (Gallardo and Holtzman, 2019; Johnson and Dammer, 2020). Our results support genetic interactions between FNSS alleles and dementia associated alleles (APP and tau). The phenotype of two different AD/AD-like mouse models was dramatically attenuated when crossed with two different FNSS alleles. We found significantly fewer amyloid plaques and tau tangles for both of these double mutants. However, further characterization on sleep and behavior features of these mice is necessary to confirm these protective effects. We found that the effect of the FNSS variants on AD pathology was not identical. Because the pathological mechanism of AD is complex, it is not surprising that there will be multiple mechanisms for slowing/preventing disease progression. Future investigation such as introducing multiple FNSS alleles into different AD mouse models to determine whether synergistic or additive effects are produced will shed additional light on these protective mechanisms.
This study employs an understudied approach for AD (both Aβ and tau) by investigating strong genetic factors that have potential in conferring resistance to onset and progression of AD-like pathology. This work was motivated by our idea that natural short sleepers are accomplishing restorative aspects of sleep more efficiently. The results indicate that there is great potential in identifying the means to use improved sleep as a target for protecting against neurodegeneration, thus decreasing the prevalence of AD, and potentially, other forms of neurodegeneration.
Limitations of the study
There are more than 100 different genetically engineered mouse lines reported to capture some aspects of AD disease and pathophysiology - so many that it has become impossible to exhaustively track them all (Jankowsky and Zheng, 2017). We chose the mouse models that were used in previous reports on sleep and AD to begin this line of investigation. Moreover, 5XFAD was recently demonstrated to be a good mouse model for studying AD pathology (Oblak et al., 2021). The fact that two independent FNSS genes/mutations are genetic modifiers suggests the possibility that the effect may be via sleep/restoration. Studies with additional AD and FNSS mouse models will be beneficial for further studying these genetic interactions. Another interesting question is when these effects are most important. It is attractive to think that increased efficiency of restoration over the lifespan could be protective. Generating Cre-dependent FNSS mouse models that can be modulated conditionally, temporally, and spatially will be necessary to address these possibilities. In addition, follow up studies in detailed sleep structure using EEG/EMG and behavioral cognitive analysis for these double mutant mice are necessary to confirm the phenotypical characterization of these effects.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AT8 | Thermo Scientific | Cat# MN1020, RRID:AB_223647 |
| AT270 | Thermo Scientific | Cat# MN1050, RRID:AB_223651 |
| HT7 | Thermo Scientific | Cat# MN1000, RRID:AB_2314654 |
| anti- β-actin | Cell Signaling Technology | Cat# 4967, RRID:AB_330288 |
| anti-PSD-95 | Abcam | Cat# ab18258, RRID:AB_444362 |
| anti-GFAP | Millipore | Cat# MAB3402, RRID:AB_94844 |
| anti-Synaptophysin antibody | Abcam | Cat# ab8049, RRID:AB_2198854 |
| Biological samples | ||
| Mouse brain tissues | UCSF | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| NPS | Tocris | 5857 |
| Critical commercial assays | ||
| human Amyloid beta 40 ELISA kit | Invitrogen | KHB3481 |
| human Amyloid beta 42 ELISA kit | Invitrogen | KHB3544 |
| Tau (Total) Human ELISA Kit | Invitrogen | KHB0041 |
| Experimental models: Organisms/strains | ||
| Mouse: 5XFAD line (on a C57B/6J background) | Jackson Labs | JAX: 34848 |
| Mouse: PS19 line (on a C57B/6J background) | Li Gan Lab | N/A |
| Mouse: DEC2-WT;PS19 | This paper | N/A |
| Mouse: DEC2-P384R;PS19 | This paper | N/A |
| Mouse: Npsr1-Y206H;PS19 | This paper | N/A |
| Mouse: DEC2-WT;5XFAD | This paper | N/A |
| Mouse: DEC2-P384R;5XFAD | This paper | N/A |
| Mouse: Npsr1-Y206H;5XFAD | This paper | N/A |
| Mouse: C57B/6J | Jackson Labs | JAX: 000664 |
| Software and algorithms | ||
| Fiji-ImageJ | National Institute of Health | https://imagej.net/Fiji |
| GraphPad Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ying-Hui Fu (Ying-Hui.Fu@ucsf.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
All experimental animals were singly housed on a light-dark cycle (LD 12:12) and given ad libitum access to food and water. When possible, littermates were used for studies comparing WT and transgenic mice. 3 month-old and 6 month-old male mice were used for all experiments unless otherwise noted, in order to control for the estrous cycle which can affect sleep-wake behaviors in female mice between experiments.
DEC2-WT and DEC2-P384R mice have been reported by our group (He et al., 2009; Hirano et al., 2018). We engineered a human BAC clone, CTD-2116MB, containing the entire DEC2 gene in a 125-kb genomic insert. The BAC clone was modified by homologous recombination to introduce the P384R mutation and to add a FLAG tag sequence to the C terminus of DEC2-P384R. All relevant segments generated by PCR and recombination were sequence-confirmed. BAC DNA was injected into C57BL/6J embryos following standard procedures. Npsr1-Y206H knock-in mice were generated via CRISPR/Cas9 (Xing et al., 2019). Briefly, the sgRNA targeting Npsr1 gene was transcribed in vitro, and then brought together with Cas9 protein and donor oligonucleotide and microinjected into pronucleus of fertilized C57BL/6J zygotes. The injected zygotes were implanted into oviducts of pseudopregnant CD1 female mice to create positive knock-in founders. Mice were backcrossed with C57BL/6J to dilute out potential off-target affects. The PS19 mice express the human 1N4R tau isoform carrying P301S mutation, driven by the mouse prion protein (Prnp) promoter. The 5XFAD line (on a C57B/6J background) was purchased from Jackson Labs (34848-JAX). Each mouse line was maintained by backcrossing to C57B/6J mice. Double transgenics were generated by crossing these FNSS mice with the 5XFAD or with the PS19 lines.
All experimental protocols were approved by the University of California, San Francisco Institutional Animal Care and Use Committee following the NIH’s Guide for the Care and Use of Laboratory.
Method details
Tau protein extraction
Tau protein in mouse brain was analyzed by immunoblot using a sequential extraction method as described with minor modifications (Yoshiyama et al., 2007). Briefly, the hippocampus of each brain was weighed and homogenized in 15 μL/mg RAB buffer [0.1 M MES, 1 mM EGTA pH 7.0, 0.5 mM MgSO4, 0.75 M NaCl, supplemented by protease inhibitor (cOmplete™, Sigma), phosphatase inhibitor (PhosSTOP, Roche) and 1 mM PMSF] and centrifuged at 35,000 g for 20 min at 4°C using an Optima MAX-TL Ultracentrifuge (Beckman). The supernatants were collected as RAB-soluble fractions and pellets were resuspended in 15 μL/mg 1 M Sucrose-RAB buffer and centrifuged at 35,000 g for 20 min at 4°C to remove myelin and related lipids. The supernatants were discarded and the resulting pellets were rehomogenized in 50 μL/mg RIPA buffer [50 mM Tris pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.5% Nadeoxycholate, 5 mM EDTA, 0.1% SDS, supplemented by protease inhibitor (cOmplete™, Sigma), phosphatase inhibitor (PhosSTOP, Roche) and 1 mM PMSF], and sonicated for 5s in ice-water mixture (0.5s on, 8s off, amplitude 10%), then centrifuged at 35,000 g for 20 min at 4°C to generate RIPA-soluble fractions. The pellets were further dissolved with 2 μL/mg 70% formic acid and incubated on ice for 1 h to recover highly insoluble protein. The samples were diluted with 1:5 (v/v) neutralization buffer (1 M Tris pH 8.0, 0.5 M NaH2PO4), mixed well and kept as FA-fractions. All fractions were stored at -80°C until analyzed.
Silver staining for plaque load
Silver stains were performed using the Campbell-Switzer method and kit from Neuroscience Associates (Braak et al., 1989). Sections from embedded 4% paraformaldehyde and 30% sucrose treated mouse brains were cut in 40 μM sections and washed 3 times for 10 min in ddH2O then treated for 5 min with 2% NH4OH. After 2 × 1 min ddH2O washes, slices were placed in Silver-Pyridine-Carbonate solution for 40 min. Next, the slices were washed in 1% Citric Acid for 3 min, then placed in Neuroscience Associates’ proprietary ABC developer solution until developed. The development was then halted with pH 4.99 Acetate buffer, which was replaced multiple times. Finally, after one subsequent 30 s ddH2O wash, the slices were placed in 0.5% Sodium Thiosulfate for 45 s before 3 × 2 min ddH2O washes and cover-slipping. At least seven consecutive slices (bregma -1.34 mm to -2.18 mm) were analyzed from 4-8 mice each using ImageJ (https://imagej.net/Auto_Threshold).
Brain protein extraction and Western blot
Samples were isolated with RIPA buffer supplemented by protease inhibitor (cOmplete™, Sigma), phosphatase inhibitor (PhosSTOP, Roche) and 1 mM PMSF] then separated by 4–12% NuPAGE (Invitrogen) gel with MOPS buffer then transferred to PVDF (Millipore) membrane. Primary antibodies used in this study were AT8 (Thermo Scientific, MN1020), AT270 (Thermo Scientific, MN1050), HT7 (Thermo Scientific, MN1000), anti- β-actin (Cell Signaling Technology, 4967S), anti-PSD-95 (Abcam, ab18258), and anti-GFAP (Millipore, MAB3402). Membranes were developed using Chemiluminescent HRP Substrate (Millipore) and exposed to CL-XPosure™ Film (Thermo Scientific).
Synaptosome preparation
The crude synaptosome pellet was prepared from fresh mouse brain tissues (Pooler et al., 2013). Briefly, fresh mouse hippocampus was gently homogenized in 15X volume of Syn-PER™ Synaptic Protein Extraction Reagent (Thermo Scientific, 87793) containing protease and phosphatase inhibitors. The homogenate was centrifuged at 1,200 g for 10 min at 4°C. The supernatant was transferred to a new tube and centrifuged at 15,000 g for 20 min at 4°C to obtain the synaptosomes.
Freshly prepared synaptosome pellets were resuspended in Syn-PER reagent (500 μL for 200–400 mg of tissue). One-half of the synaptosomes were treated with 0.4 mM NPS (Tocris, 5857). The other half were treated with saline as control. The samples were incubated for 1 h at 37°C and centrifuged at 2,000 g for 4 min at room temperature. Supernatants were collected for analysis, and the remaining pellet was resuspended in RIPA buffer for the identification of synaptosomal components.
Immunohistological staining
The brain of each mouse was stored in 4% PFA overnight at 4°C and then incubated in a 30% sucrose solution for 24 h at 4°C. Samples were sectioned coronally at 40 μm using a freezing sliding microtome. Two to three sections (bregma -1.4 mm to -2.0 mm) from each mouse were used for free-floating p-tau staining. Brain sections were washed three times in 1X PBS followed by blocking with 5% donkey serum 1% BSA in 0.5% PBS-X (Triton X-100) for 1 h. The sections were incubated with the primary antibody AT8 (Thermo Scientific, MN1020, 1:1000) or anti-GFAP (Chemicon, MAB360, 1:1000) overnight at 4°C. The next day, the sections were washed three times in 1X PBS, and incubated with fluorescence-labeled secondary antibody Goat Anti-Mouse Alexa Fluor 488 (Abcam, ab150113, 1:1000) for 2 h at room temperature. The slices were then washed three times in 1X PBS and placed in mounting medium with DAPI and DABCO™ (EMS). For analysis of synapses, the brain sections were incubated with primary anti-Synaptophysin antibody (Abcam, ab8049, 1:50) for 48 h at 4°C and then incubated with secondary antibody (JIR, anti-mouse Cy™3, 1:500). Data analysis was performed with blinding to animal genotypes.
CSF surgeries
Mice were anesthetized with isoflurane for surgery (Lim et al., 2018). Following head-fixation and testing for withdrawal response, the skin and overlaying muscles were removed from the mouse’s posterior skull region until the cisterna magna was revealed. Positive pressure and expulsion of contaminants was achieved by pumping a syringe connected to a sharpened glass capillary tube fixed within a micromanipulator device. This device was used to puncture the cisterna magna at a 30–45° angle with the capillary tube while carefully avoiding puncturing nearby blood vessels or the underlying brain tissue. CSF was allowed to accumulate in the tube before removal and expulsion into a tube immediately placed in liquid nitrogen. The mouse was then sacrificed by cervical dislocation and decapitation, when blood for plasma analysis was immediately collected in an additional tube. To avoid the fluctuation of CSF Aβ/tau caused by circadian rhythm and sleep-wake cycle, we started the CSF collection at ZT4 for each batch and completed within 2 h.
ELISA
The solid-phase sandwich ELISAs on brain tissue for human Amyloid Beta 40 and 42 were performed using an Invitrogen (Cat Number EZBRAIN-SET) kit according to their recommended protocol. In brief, brain tissue was lysed on ice with protease inhibitor, and equal weights of sample were extracted and homogenized with a pestle. CSF samples skipped the pestle and lysis steps. Samples rotated for 2 h at 4°C before centrifugation and clearance of the lysate. Lysate, representing the sample containing soluble Amyloid beta, was saved for subsequent steps. The pellet was resuspended in formic acid, sonicated for 30 min, and neutralized in 1M Tris. The resulting supernatant after centrifugation contained insoluble amyloid beta. Both lysates were diluted evenly according to previous calibration and applied to the ELISA plate (in quadruplicate) in solution with provided diluents and allowed to develop as described in supplier protocols. Amyloid beta 40 was measured in the soluble fraction, and Amyloid beta 42 in the insoluble fraction. All sample values were normalized to total protein levels, whose values were derived using the kit’s provided standard curve protocol and adjusted by dilution. ELISAs on CSF and plasma for human Amyloid beta 40 and 42 were performed using an Invitrogen (Cat Number KHB3481 and KHB3544, respectively) kit according to their recommended protocol. Finally, concentrations were calculated from colorimetric measurements made using a Synergy H4 Hybrid Multi-Mode Microplate Reader (Biotek).
Human tau ELISAs were performed according to the manufacturer's instructions for a human tau ELISA kit (Invitrogen, KHB0041) with modifications. The ELISA plates were incubated with 100 μL of diluted tau standard or 50 times diluted CSF samples overnight at 4°C. The next morning, plates were washed four times with 1X wash buffer. The human tau Biotin conjugated solution was added and incubated for 1 h at room temperature. After washing four times with 1X wash buffer, 1X Streptavidin-HRP solution was added and incubated for 30 min at room temperature. Chromogen solution was used as a substrate of peroxidase and absorbance was read at 450 nm with a Synergy H4 Hybrid Multi-Mode Microplate Reader (BioTek).
Quantification and statistical analysis
Statistical analysis
All mouse data is shown as mean ± SEM unless explicitly stated. Statistical differences between groups were assessed by one-way ANOVA or two-way ANOVA with post hoc multiple comparisons, and unpaired t-test. GraphPad Prism 8 software was used for analyzing the data and generating graphs.
Acknowledgments
We thank all members in the Ptáček and Fu labs for helpful discussions. This work was supported by NIH grants NS117929, NS072360, and NS104782 (L.J.P. and Y-H.F.), and by the William Bowes Neurogenetics Fund (L.J.P. and Y.H.F.).
Author contributions
Q.D., N.W.G., L.G., L.J.P., and Y.-H.F., conceived and designed the study. Q.D., T.M., N.W.G., T.W., and L.B.R. performed experiments. M.Y. and Q.D. managed mouse colony resources and upkeep. Q.D., T.M., and N.W.G. statistically analyzed and interpreted data. Q.D., N.W.G., and Y.-H.F. wrote and organized the manuscript. Q.D., N.W.G., T.M., L.J.P., L.G., and Y.-H.F. edited the manuscript. All authors approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: March 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103964.
Contributor Information
Louis Ptáček, Email: ljp@ucsf.edu.
Ying-Hui Fu, Email: Ying-Hui.Fu@ucsf.edu.
Supplemental information
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Braak H., Braak E., Ohm T., Bohl J. Alzheimer's disease: mismatch between amyloid plaques and neuritic plaques. Neurosci. Lett. 1989;103:24–28. doi: 10.1016/0304-3940(89)90479-5. [DOI] [PubMed] [Google Scholar]
- Bundy J.L., Vied C. Sex-biased hippocampal pathology in the 5XFAD mouse model of Alzheimer's disease: a multi-omic analysis. J. Comp. Neurol. 2019;527:462–475. doi: 10.1002/cne.24551. [DOI] [PubMed] [Google Scholar]
- Chang-Quan H., Bi-Rong D., Yan Z. Association between sleep quality and cognitive impairment among Chinese nonagenarians/centenarians. J. Clin. Neurophysiol. 2012;29:250–255. doi: 10.1097/WNP.0b013e3182570f2e. [DOI] [PubMed] [Google Scholar]
- Chen D.W., Wang J., Zhang L.L., Wang Y.J., Gao C.Y. Cerebrospinal fluid amyloid-β levels are increased in patients with insomnia. J. Alzheimer's Dis.: JAD. 2018;61:645–651. doi: 10.3233/jad-170032. [DOI] [PubMed] [Google Scholar]
- Gallardo G., Holtzman D.M. Amyloid-β and tau at the crossroads of Alzheimer's disease. Adv. Exp. Med. Biol. 2019;1184:187–203. doi: 10.1007/978-981-32-9358-8_16. [DOI] [PubMed] [Google Scholar]
- He Y., Jones C.R., Fujiki N., Xu Y., Guo B., Holder J.L., Jr., Rossner M.J., Nishino S., Fu Y.H. The transcriptional repressor DEC2 regulates sleep length in mammals. Science (New York, N.Y.) 2009;325:866–870. doi: 10.1126/science.1174443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A., Hsu P.K., Zhang L., Xing L., McMahon T., Yamazaki M., Ptáček L.J., Fu Y.H. DEC2 modulates orexin expression and regulates sleep. Proc. Natl. Acad. Sci. U S A. 2018;115:3434–3439. doi: 10.1073/pnas.1801693115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holth J.K., Fritschi S.K., Wang C., Pedersen N.P., Cirrito J.R., Mahan T.E., Finn M.B., Manis M., Geerling J.C., Fuller P.M., et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science (New York, N.Y.) 2019;363:880–884. doi: 10.1126/science.aav2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky J.L., Zheng H. Practical considerations for choosing a mouse model of Alzheimer's disease. Mol. Neurodegener. 2017;12:89. doi: 10.1186/s13024-017-0231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawhar S., Trawicka A., Jenneckens C., Bayer T.A., Wirths O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer's disease. Neurobiol. Aging. 2012;33 doi: 10.1016/j.neurobiolaging.2010.05.027. 196.e29-40. [DOI] [PubMed] [Google Scholar]
- Johnson E.C.B., Dammer E.B. Large-scale proteomic analysis of Alzheimer's disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020;26:769–780. doi: 10.1038/s41591-020-0815-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Y.E., Lucey B.P., Holtzman D.M. Sleep and Alzheimer disease pathology--a bidirectional relationship. Nat. Rev. Neurol. 2014;10:115–119. doi: 10.1038/nrneurol.2013.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien C., Bretteville A., Planel E. Biochemical isolation of insoluble tau in transgenic mouse models of tauopathies. Methods Mol. Biol. (Clifton, N.J.) 2012;849:473–491. doi: 10.1007/978-1-61779-551-0_32. [DOI] [PubMed] [Google Scholar]
- Kalmbach D.A., Schneider L.D., Cheung J., Bertrand S.J., Kariharan T., Pack A.I., Gehrman P.R. Genetic basis of chronotype in humans: insights from three landmark GWAS. Sleep. 2017;40:zsw048. doi: 10.1093/sleep/zsw048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J.E., Lim M.M., Bateman R.J., Lee J.J., Smyth L.P., Cirrito J.R., Fujiki N., Nishino S., Holtzman D.M. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science (New York, N.Y.) 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W., Gray S.L., Barthold D., Maust D.T., Marcum Z.A. Association between informant-reported sleep disturbance and incident dementia: an analysis of the national Alzheimer's coordinating center uniform data set. J. Appl. Gerontol. 2020;41:285–294. doi: 10.1177/0733464820967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim N.K., Moestrup V., Zhang X., Wang W.A., Møller A., Huang F.D. An improved method for collection of cerebrospinal fluid from anesthetized mice. J. Vis. Exp.: JoVE. 2018;(133):56774. doi: 10.3791/56774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucey B.P., McCullough A., Landsness E.C., Toedebusch C.D., McLeland J.S., Zaza A.M., Fagan A.M., McCue L., Xiong C., Morris J.C., et al. Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer's disease. Sci. Transl. Med. 2019;11:eaau6550. doi: 10.1126/scitranslmed.aau6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mander B.A., Winer J.R., Jagust W.J., Walker M.P. Sleep: a novel mechanistic pathway, biomarker, and treatment target in the pathology of Alzheimer's disease? Trends Neurosci. 2016;39:552–566. doi: 10.1016/j.tins.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mander B.A., Winer J.R., Walker M.P. Sleep and human aging. Neuron. 2017;94:19–36. doi: 10.1016/j.neuron.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata S., Noda A., Iwamoto K., Kawano N., Okuda M., Ozaki N. Poor sleep quality impairs cognitive performance in older adults. J. Sleep Res. 2013;22:535–541. doi: 10.1111/jsr.12054. [DOI] [PubMed] [Google Scholar]
- Oblak A.L., Lin P.B., Kotredes K.P., Pandey R.S., Garceau D., Williams H.M., Uyar A., O'Rourke R., O'Rourke S., Ingraham C., et al. Comprehensive evaluation of the 5XFAD mouse model for preclinical testing applications: a MODEL-AD study. Front. Aging Neurosci. 2021;13:713726. doi: 10.3389/fnagi.2021.713726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler A.M., Phillips E.C., Lau D.H., Noble W., Hanger D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14:389–394. doi: 10.1038/embor.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M.K., Mestre H., Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17:1016–1024. doi: 10.1016/s1474-4422(18)30318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh J.H., Huang Y., Bero A.W., Kasten T., Stewart F.R., Bateman R.J., Holtzman D.M. Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer's disease pathology. Sci. Transl. Med. 2012;4:150ra122. doi: 10.1126/scitranslmed.3004291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman S.M., Herdener N., Frankola K.A., Mughal M.R., Mattson M.P. Chronic mild sleep restriction accentuates contextual memory impairments, and accumulations of cortical Aβ and pTau in a mouse model of Alzheimer's disease. Brain Res. 2013;1529:200–208. doi: 10.1016/j.brainres.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutte T., Harris S., Levin R., Zweig R., Katz M., Lipton R. The relation between cognitive functioning and self-reported sleep complaints in nondemented older adults: results from the Bronx aging study. Behav. Sleep Med. 2007;5:39–56. doi: 10.1207/s15402010bsm0501_3. [DOI] [PubMed] [Google Scholar]
- Scullin M.K., Bliwise D.L. Sleep, cognition, and normal aging: integrating a half century of multidisciplinary research. Perspect. Psychol. Sci.: J. Assoc. Psychol. Sci. 2015;10:97–137. doi: 10.1177/1745691614556680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Yamada K., Liddelow S.A., Smith S.T., Zhao L., Luo W., Tsai R.M., Spina S., Grinberg L.T., Rojas J.C., et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523–527. doi: 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G., Xing L., Wu D., Bhattacharyya B.J., Jones C.R., McMahon T., Chong S.Y.C., Chen J.A., Coppola G., Geschwind D., et al. A rare mutation of β(1)-Adrenergic receptor affects sleep/wake behaviors. Neuron. 2019;103:1044–1055.e7. doi: 10.1016/j.neuron.2019.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G., Yin C., Fan Z., Xing L., Mostovoy Y., Kwok P.Y., Ashbrook L.H., Krystal A.D., Ptáček L.J., Fu Y.H. Mutations in metabotropic glutamate receptor 1 contribute to natural short sleep trait. Curr. Biol.: CB. 2021;31:13–24.e4. doi: 10.1016/j.cub.2020.09.071. [DOI] [PubMed] [Google Scholar]
- Sokolow S., Henkins K.M., Bilousova T., Gonzalez B., Vinters H.V., Miller C.A., Cornwell L., Poon W.W., Gylys K.H. Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer's disease. J. Neurochem. 2015;133:368–379. doi: 10.1111/jnc.12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira A.P., Gottesman R.F. Sleep disturbance: an emerging opportunity for Alzheimer's disease prevention? Int. Psychogeriatr. 2017;29:529–531. doi: 10.1017/s1041610216002131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira A.P., Gamaldo A.A., An Y., Wu M.N., Simonsick E.M., Bilgel M., Zhou Y., Wong D.F., Ferrucci L., Resnick S.M. Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol. 2013;70:1537–1543. doi: 10.1001/jamaneurol.2013.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafaro L., Cicconetti P., Baratta A., Brukner N., Ettorre E., Marigliano V., Cacciafesta M. Sleep quality of centenarians: cognitive and survival implications. Arch. Gerontol. Geriatr. 2007;44(Suppl 1):385–389. doi: 10.1016/j.archger.2007.01.054. [DOI] [PubMed] [Google Scholar]
- Tkachenko O., Dinges D.F. Interindividual variability in neurobehavioral response to sleep loss: a comprehensive review. Neurosci. Biobehav. Rev. 2018;89:29–48. doi: 10.1016/j.neubiorev.2018.03.017. [DOI] [PubMed] [Google Scholar]
- Toh K.L., Jones C.R., He Y., Eide E.J., Hinz W.A., Virshup D.M., Ptácek L.J., Fu Y.H. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science (New York, N.Y.) 2001;291:1040–1043. doi: 10.1126/science.1057499. [DOI] [PubMed] [Google Scholar]
- Xie L., Kang H., Xu Q., Chen M.J., Liao Y., Thiyagarajan M., O'Donnell J., Christensen D.J., Nicholson C., Iliff J.J., et al. Sleep drives metabolite clearance from the adult brain. Science (New York, N.Y.) 2013;342:373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L., Shi G., Mostovoy Y., Gentry N.W., Fan Z., McMahon T.B., Kwok P.Y., Jones C.R., Ptacek L.J., Fu Y.H. Mutant neuropeptide S receptor reduces sleep duration with preserved memory consolidation. Sci. Transl. Med. 2019;11:eaax2014. doi: 10.1126/scitranslmed.aax2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K., Holth J.K., Liao F., Stewart F.R., Mahan T.E., Jiang H., Cirrito J.R., Patel T.K., Hochgräfe K., Mandelkow E.M., Holtzman D.M. Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 2014;211:387–393. doi: 10.1084/jem.20131685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y., Higuchi M., Zhang B., Huang S.M., Iwata N., Saido T.C., Maeda J., Suhara T., Trojanowski J.Q., Lee V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zhao B., Liu P., Wei M., Li Y., Liu J., Ma L., Shang S., Jiang Y., Huo K., Wang J., Qu Q. Chronic sleep restriction induces Aβ accumulation by disrupting the balance of Aβ production and clearance in rats. Neurochem. Res. 2019;44:859–873. doi: 10.1007/s11064-019-02719-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.