

Abstract
The excelling role of organic chemistry in the medicinal field continues to be one of the main leads in the drug development process. Particularly, this industry requires organic chemists to discover small molecular structures with powerful pharmacological potential. Herein, a diverse range of chalcone (1–11) and aurone (12–22) derivatives was designed and synthesized and for the first time, and both motifs were evaluated as potent inhibitors of alkaline phosphatases (APs). Structural identification of the target compounds (1–22) was accomplished using common spectroscopic techniques. The effect of the nature and position of the substituent was interestingly observed and justified based on the detailed structure–activity relationship (SAR) of the target compounds against AP. It was concluded from the obtained results that all the newly synthesized compounds exhibit high inhibitory potential against the AP enzyme. Among them, compounds 12 (IC50 = 2.163 ± 0.048 μM), 15 (IC50 = 2.146 ± 0.056 μM), 16 (IC50 = 2.132 ± 0.034 μM), 18 (IC50 = 1.154 ± 0.043 μM), 20 (IC50 = 1.055 ± 0.029 μM) and 21 (IC50 = 2.326 ± 0.059 μM) exhibited excellent inhibitory activity against AP, and even better/more active than KH2PO4 (standard) (IC50 = 2.80 ± 0.065 μM). Remarkably, compound 20 (IC50 = 1.055 ± 0.029 μM) may serve as a lead structure to design more potent inhibitors of alkaline phosphatase. To the best of our knowledge, these synthetic compounds are the most potent AP inhibitors with minimum IC50 values reported to date. Furthermore, a molecular modeling study was performed against the AP enzyme (1EW2) to check the binding interaction of the synthesized compounds 1–22 against the target protein. The Lineweaver–Burk plots demonstrated that most potential derivative 20 inhibited h-IAP via a non-competitive pathway. Finally, molecular dynamic (MD) simulations were performed to evaluate the dynamic behavior, stability of the protein–ligand complex, and binding affinity of the compounds, resulting in the identification of compound 20 as a potential inhibitor of AP. Accordingly, excellent correlation was observed between the experimental and theoretical results. The pharmacological studies revealed that the synthesized analogs 1–22 obey Lipinski's rule. The assessment of the ADMET parameters showed that these compounds possess considerable lead-like characteristics with low toxicity and can serve as templates in drug design.
Aurones are the plant secondary metabolites belonging to the flavonoid’s family. The bioactivities of aurones are very promising, thus these heterocyclic compounds can be considered as an alluring scaffold for drug design and development.
Introduction
Alkaline phosphatases (APs, E.C. 3.1.3.1) are membrane-bound ectonucleotidases/hydrolase enzymes, which are ubiquitous in nature from bacteria to mammals, with the ability to perform significant physiological functions that accelerate the hydrolysis of esters containing phosphate groups.1–9 They are responsible for the effective removal of phosphate groups from alkaloids, nucleotides and proteins in alkaline medium.10 APs are dimeric non-specific enzymes with regard to the substrate, and thus hydrolyze various substrates. APs exhibit hydrolytic activity, phosphotransferase activity, and pyrophosphatase activity.11,12 The enzymatic activity of alkaline phosphatases is due to the presence of two Zn2+ ions and one Mg2+ ion in their catalytic site. In humans, three out of four isozymes are tissue-specific isozymes, for example, intestinal (IAP), placental (PLAP) and germ cells (GCAP) have 90–98% structural homology, while the fourth AP is tissue non-specific (TNAP) and is about 50% homologous to other isozymes.13–15 TNAP is expressed at elevated levels in bones, kidneys, liver and placenta in the 1st trimester of pregnancy, though lower levels are expressed in many other tissues. TNAP is essential for bone matrix mineralization and plays a vital role in the decomposition of extracellular inorganic pyrophosphate (PPi) and maintaining a specific level of PPi, which is important for normal bone mineralization.16a,b,17–21
APs have been reported to be overexpressed in solid and metastasized tumors including breast, esophageal, intestinal, prostate, liver, ovarian cancer and osteoporosis.22a–c,23a,b Thus, the presence of high levels of TNAP and IAP in many malignancies and other diseases makes them a promising molecular target for therapeutic development.8,24a,b The development of potent and selective inhibitors of AP isozymes is becoming increasingly popular. In recent years, several inhibitors of APs based on biaryl sulfonamide, chromone, triazole and coumarin sulfonate motifs have been reported.25–32 APs have become important target enzymes owing to their considerable pharmacological importance in medicinal chemistry.33,34a,b Some compounds were found to be effective inhibitors of AP, but most of them inhibited APs non-selectively.34c–f Biological investigations have revealed that the substitution of various groups on ring structures imparts different activities. Traditionally, levamisole and theophylline were the only available inhibitors of AP with Ki values of 16 and 82 μM, respectively.34g However, recently, several groups screened and optimized small molecules as efficient and selective inhibitors of AP isozymes. A comparative study among several heterocyclic classes showed varying degrees of inhibitory potential against AP (Fig. 1).29 Considering the literature findings and great interest in research on new bioactive heterocycles and our continuing interest in the development of potential enzyme inhibitors, we have explored aurone derivatives as a new class of AP inhibitors with enhanced inhibitory potential against human h-IAP. The bioassay results revealed that different scaffolds exhibit different AP inhibition, and thus, to the best of our knowledge, synthetic analogs of the aurone motif have not been explored against AP to date. Thus, owing to the biological significance of the aurone moiety, for the first time, herein, synthetic flavonoids (i.e., aurones) have been explored as potential AP inhibitors, and the obtained results revealed that among the reported classes of heterocyclic compounds, overall, this class exhibits better inhibitory activity against APs.
Fig. 1. Identified representative AP inhibitors.

With the aim to discover selective inhibitors of APs, in the present study, we investigated a series of synthetic molecules based on two pharmaceutically active scaffolds, namely chalcones (benzalacetophenone) and aurones (2-benzylidenebenzofuran-3(2H)-one).35–37 The chalcone scaffold is considered a privileged structure and represents the key structural motif in a plethora of bioactive synthetic and natural compounds\widely distributed in fruits, vegetables, and other plants.38
Chalcones (Fig. 2), a subfamily of flavonoids and their biosynthetic precursors, are unique among the flavonoids because they lack a central heterocyclic C-ring and exhibit a wide spectrum of biological activities including antileishmanial, anticancer, antiangiogenic, antioxidant, antibacterial, anti-infective and anti-inflammatory activities.39a,b They play a key role in yellow floral pigmentation. The growing interest in these compounds and their potential use in medicinal applications are evidenced by the number of publications concerning the synthesis and biological evaluation of chalcone analogs.40–42
Fig. 2. Chemical structures and numbering of (A) chalcone (1,3-diphenylprop-2-en-1-one) and (B) aurone (2-benzylidenebenzofuran-3(2H)-one) scaffolds.

Aurones represent another interesting structural motif. Aurones (Fig. 2) are plant secondary metabolites belonging to the flavonoid family, which give the bright yellow color to several essential ornamental blooms such as snapdragon, cosmos and dahlia and are biosynthesized from chalcones by the key enzyme aureusidin synthase.43–46 Structurally, aurones are a benzofuranone heterocyclic ring containing a phenyl group linked through a carbon–carbon exocyclic double bond. Importantly, aurone is one of the privileged structures in medicinal chemistry and is commonly associated with various pharmacologically active compounds.47–49 Aurones have been demonstrated as phytoalexins, which are used by plants in their defense mechanism against numerous infections. Perhaps due to their scarcity in nature, aurones have received less attention than other flavonoids and not been broadly studied for their biological activities.50,51 However, the present data on the biological activities of natural and synthetic aurones is very promising, and therefore these heterocyclic compounds can be considered as alluring scaffolds for drug design and development.52 As a member of the flavonoid family, thus far aurones have been reported to possess different bioactivities including antitumor, anticancer, antioxidant, metal ion-chelating, anti-tyrosinase, antiparasitic, antimicrobial, antiviral, anti-inflammatory, anti-diabetic, anti-hormonal and anti-obesity activity.53 They have been found to act as inhibitors for several enzymes such as monoamine oxidase A and B, histone deacetylase and cyclin-dependent kinase,54 and thus represent an overwhelming and rapidly developing field in modern heterocyclic chemistry.55
Considering the literature findings and in continuation of our previous studies47,48 aimed at developing new molecules as potent biological candidates, herein, we report the design and synthesis of a new series of chalcones and aurones. The synthesized compounds with different substitution patterns were evaluated, for the first time, for their alkaline phosphatase inhibitory activity, and remarkably these analogs were accessed as a potent heterocyclic class among the already reported AP inhibitors. Moreover, molecular docking and kinetic studies of these compounds were also carried out to gain further insights into their biological properties.
Material and methods
All reagents were obtained from Merck (Germany) and used as received. An electrothermal melting point apparatus was used to determine the melting points, which were uncorrected. iR spectra were measured on a Bio-Rad spectrophotometer. NMR spectra were measured on a Bruker DRX 300 instrument (1H, 300 MHz, 13C, 75 MHz) and Bruker DRX 600 instrument (1H, 600 MHz, 13C, 151 MHz). Absorption spectra were recorded on a Jasco UV-vis V-670 instrument using a quartz cell in very dilute solutions prepared in different solvents. Accurate mass measurements were carried out with a Fisons VG sector-field instrument (EI) and FT-ICR mass spectrometer.
General procedures for the synthesis of chalcones (1–11) and aurones (12–22)
In step 1, a mixture of substituted 2′-hydroxyacetophenone (1.0 mmol) and aqueous sodium hydroxide solution (30%, 5.0 mL) was dissolved in distilled methanol (20.0 mL) and stirred for 30 min at room temperature followed by the dropwise addition of substituted aryl aldehyde (1.0 mmol). Subsequently, the reaction mixture was stirred at the same temperature for 3–4 h. The progress of the formation of 2′-hydroxychalcone was examined by TLC using cyclohexane : ethyl acetate (3 : 1) as the mobile phase. After completion of the reaction (checked by TLC), the mixture was then acidified using dil. HCl (10%) and poured into the cold water. The separated solid was filtered, washed with H2O and the dried product was recrystallized from EtOH to obtain the purified compounds (1–11). In step 2, Hg(OAc)2 (1.0 mmol) was added to a solution of 2′-hydroxychalcone (1.0 mmol) in pyridine (10 mL) at room temperature and the mixture was refluxed for 2 h. Upon completion of the reaction, as indicated by TLC, the reaction mixture was left to cool, and then poured onto ice-cold water (30 mL) and acidified with dil. HCl to allow precipitation. The precipitated solid was filtered, washed with cold water, and dried to give the crude products, which were further purified by recrystallization using ethanol to afford the pure aurone product (12–22).
Enzyme inhibition assay
Alkaline phosphatase assay
The spectrophotometric test was used to determine the activity of CIAP, as previously described in the literature.21 The reaction mixture was made up of 50 mM Tris–HCl buffer (5 mM MgCl2, and 0.1 mM ZnCl2 pH 9.5), the compound (0.1 mM) and 1% DMSO (v/v) and pre-incubated for 10 min by adding 5 μL of CIAP (0.025 U mL−1). The assay mixture was then incubated for 30 min at 37 °C with 10 μL of the substrate (0.5 mM) p-NPP to start the reaction. Using a 96-well microplate reader, the change in absorbance of released p-nitrophenolate was observed at 405 nm (OPTIMAX, Tunable USA). All experiments were repeated in triplicate. KH2PO4 (potassium dihydrogen phosphate) was used as the reference inhibitor of calf AP.
Kinetic study
The enzyme inhibitory interaction mechanism of chalcones and aurones with the binding site of an enzyme was determined via Michaelis–Menten kinetic studies.21 Based on the IC50 value, the most potent inhibitor 20 was chosen for the study of the enzyme inhibition mechanism. The inhibitor concentrations used were 0.00, 1.15, 2.30, 4.60 and 9.20 μM. Similarly, the substrate p-NPP was used with concentrations of 10, 5, 2.5, 0.25 and 0.625 mM. For pre-incubation, the same conditions as that in the AP inhibition assay were maintained. The enzyme was injected at a rate of one per minute, and the maximum initial velocities were calculated from the linear component of the absorbance up to 10 min. The Lineweaver–Burk plot was used to determine the type of enzyme inhibition, where 1/V (inverse of velocities) vs. 1/[S] (inverse of substrate concentration) mM−1 was displayed on the graph.
Molecular modeling assay
Ligand preparation
The 2D structures of the synthesized compounds (1–22) were drawn using ChemDraw Professional 15.2 and prepared in their neutral form by LigPrep (Schrödinger) and their conformation optimized in the OPLS-3 force field. The prepared structures were further employed for the grid and docking analysis.
Protein preparation
Molecular docking simulations were performed to determine the possible binding orientations of the compounds (1–22) in the X-ray crystal structure of human alkaline phosphatase complexed with strontium. For this purpose, the highest resolution X-ray structure of the target protein was downloaded from the Protein Data Bank (RCSB PDB 2GLQ) (https://www.rcsb.org) and prepared using the “Protein Preparation Wizard” workflow in Maestro Schrödinger for adding hydrogens and setting the protonation states appropriate for pH 7. In this module, the bond orders were assigned, H-atoms were added and the water molecules 5 Å away from the het groups were removed from the protein structure. After the protein was processed, it was optimized using PROPKA at pH 7.0 and the OPLS3e force field was utilized to perform restrained minimization for energy minimization and geometry optimization of the protein structure.
Molecular docking and receptor grid generation
For grid generation preparation, the active site of the target protein was defined from the co-crystallized ligands from the Protein Data Bank. The grid was generated by specifying the Zn (2001, 2002) ions involved in the catalytic site of the target protein. The receptor grid box was defined as a 20 Å box (x = 38.92, y = 16.56, z = 12.50). The default docking setup parameters were employed for the ligand docking experiment.56 Glide (Schrödinger) was used to dock the ligands utilising XP precision with the default settings and glide scoring function, and the top 20 postures for each ligand were reported. The predicted binding energies (docking scores) and proper orientation of the ligands in the active site of the enzyme were also determined using Glide dock_XP. Finally, to predict the most favorable binding mode of the active compounds inside the binding pocket of the protein with the proper orientation in terms of docking score and binding energy, visual inspection and 3D graphical images of the binding pose and generation of the figure of the best-scored docking complex were also done with Maestro (Schrödinger). All the hydrogens of the ligands were omitted to get clear pictures of the ligand–protein complexes.
MD simulation
MD simulation of the selected compound 20 in complex with the protein was performed using the Desmond package of the Schrödinger suite.57 The initial binding pose of the ligand was obtained from the molecular docking studies, which provided only the static state. The dynamic behavior of the ligand with the protein was analyzed by MD simulation. The preparation of the protein–ligand complex was done using the Protein Preparation Wizard of Maestro.58,59 The preparation included preprocessing, optimization, and energy minimization of the complex. The OPLS_2005 force field was used to minimize the complex.60 The complex was then subjected to the System Builder tool to add the solvent model. TIP3P solvent61 with an orthorhombic box was added to the complex. Na+ and Cl− counter ions were used to neutralize the system. The temperature and pressure of the system were kept 300 K and 1 atm, respectively, by applying the NPT ensemble. The complex was relaxed before subjecting it to 100 ns simulation. The simulation trajectory was saved after every 50 ps for the analysis of RMSD, RMSF, and Rg.
Chemoinformatic ligand analysis
The Molsoft tool was used to estimate the drug-like characteristics of the envisaged compounds (1–22). Lipinski's RO5 established a framework for assessing the bioavailability and pharmacokinetics of a given molecule when administered orally using a set of physiochemical parameters.62,63
Results and discussion
Chemistry
A series of differently functionalized chalcones (1–11) and aurones (12–22) was synthesized starting from 2′-hydroxyacetophenone and aromatic benzaldehydes, as depicted in Scheme 1. 2′-Hydroxychalcones are significant key intermediates for the synthesis of aurone and were prepared via the base-catalyzed condensation of substituted 2′-hydroxyacetophenone and several benzaldehydes in the presence of aqueous sodium hydroxide as the base in MeOH. These intermediates were characterized via FTIR, UV-vis and NMR spectroscopy before proceeding to the next step. Subsequently, oxidative cyclization of the substituted 2′-hydroxychalcones (1–11) was performed using mercuric acetate (Hg(OAc)2) in refluxing pyridine to furnish aurones (12–22) in moderate to good yield. These compounds (1–22) were purified through recrystallization in EtOH. The structures of all the targeted compounds (1–22) were confirmed using various spectral techniques including UV-vis, FTIR, NMR and mass spectrometry. For instance, the iR spectra show the disappearance of the band attributed to the –OH group at around 3250 cm−1 and the appearance of a new iR band at around 1300 cm−1, corresponding to a new ether linkage (C–O–C). Similarly, the 1H NMR spectra of all the aurone derivatives exhibit a singlet at around δ 7.20 ppm, which is attributed to the characteristic olefinic hydrogen, in addition to aromatic signals. The two protons on ring A give two doublets at around δ 8.55 ppm for the proton at position 6 and around δ 8.35 ppm for the proton at position 8. The protons on the ring B show NMR signals in the range of δ 8.25–7.40 ppm. Similarly, all the 13C NMR spectra show distinguishing signals at around δ 180.0 ppm due to the ketonic carbon, in addition to other normal signals for the aromatic carbons. The structural identification of the desired final analogs (12–22) was further verified by electron ionization (EI) mass spectrometry. The accurate masses of all the aurones were determined by EI-MS. Thus, all the spectral data unequivocally corroborate the presented structures of all the newly synthesized compounds.
Scheme 1. Synthesis of chalcones (1–11) and aurones (12–22).

The spectral data of all the newly synthesized chalcones (1–11) and aurones (12–22) is presented below.
1-(3,5-Dibromo-2-hydroxyphenyl)-3-(furan-2-yl)prop-2-en-1-one (1)
Yellow powder; mp 151–153 °C; yield: 81%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.33; UV-vis λmax (MeOH) = 330 nm; FTIR (cm−1): 3061, 1637, 1584, 1547, 1279, 741, 682; 1H NMR (300 MHz, DMSO-d6): δ 13.12 (bs, 1H, OH), 8.36 (d, J = 3.0 Hz, 1H, Ar-H), 8.14 (d, J = 3.0 Hz, 1H, Ar-H), 8.01 (dd, J = 6.0, 3.0 Hz, 1H, Ar-H), 7.95 (m, 1H, Ar-H), 7.45 (dd, J = 6.0, 3.0 Hz, 1H, Ar-H), 7.28–7.24 (m, 2H, olefinic proton); 13C NMR (75 MHz, DMSO-d6): δ 191.1, 153.2, 142.7, 139.1, 137.0, 134.0, 131.8, 130.6, 129.8, 129.4, 128.8, 127.8, 122.8; accurate mass (EI-MS) of [M]˙+: calcd for C13H879Br2O3 369.8840; found 369.8832.
3-(4-Chlorophenyl)-1-(3,5-dibromo-2-hydroxyphenyl)prop-2-en-1-one (2)
Yellow solid; mp 196–198 °C; yield: 79%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.42; UV-vis λmax (MeOH) = 385 nm; FTIR (cm−1): 3073, 1632, 1593, 1557, 1299, 783, 694; 1H NMR (300 MHz, CDCl3): δ 13.08 (bs, 1H, OH), 8.25 (d, J = 3.0 Hz, 1H, Ar-H), 8.12 (d, J = 3.0 Hz, 1H, Ar-H), 7.95 (d, J = 15.0 Hz, 1H, olefinic proton), 7.67 (d, J = 9.0 Hz, 2H, Ar-H), 7.55 (d, J = 15.0 Hz, 1H, olefinic proton), 7.49 (d, J = 9.0 Hz, 2H, Ar-H); 13C NMR (75 MHz, DMSO-d6): δ 190.6, 154.8, 146.2, 137.8, 137.4, 135.7, 132.4, 130.3, 130.1, 130.0, 129.5, 126.1, 123.8, 121.1, (two carbons are isochronous); accurate mass (EI-MS) of [M]˙+: calcd for C15H979Br235ClO2 413.8658; found 413.8650.
1-(3,5-Dibromo-2-hydroxyphenyl)-3-(4-(trifluoromethyl)phenyl)prop-2-en-1-one (3)
Yellow amorphous solid; mp 151–153 °C; yield: 80%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.59; UV-vis λmax (MeOH) = 293 nm; FTIR (cm−1): 3067, 1642, 1573, 1434, 1279, 714, 693; 1H NMR (600 MHz, DMSO-d6): δ 12.36 (bs, 1H, OH), 8.20 (d, J = 6.0 Hz, 1H, Ar-H), 7.87–7.84 (m, 3H, Ar-H), 7.80 (d, J = 12.0 Hz, 2H, Ar-H), 6.00 (dd, J = 12.0, 6.0 Hz, 2H, –CH CH–); 13C NMR (151 MHz, DMSO-d6): δ 190.0, 156.8, 143.2, 141.2, 133.0, 130.5, 129.7, 128.6, 128.5, 127.6, 126.3, 126.2, 123.4, 113.8, 80.0, 42.7; accurate mass (EI-MS) of [M]˙+: calcd for C16H979Br2F3O2 447.8921; found 447.8910.
1-(5-Chloro-2-hydroxy-3-nitrophenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (4)
Yellow powder; mp 191–193 °C; yield: 77%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.47; UV-vis λmax (MeOH) = 256 nm; FTIR (cm−1): 3074, 1638, 1596, 1546, 1256, 780, 707, 637; 1H NMR (300 MHz, DMSO-d6): δ 13.81 (bs, 1H, OH), 8.65 (d, J = 3.0 Hz, 1H, Ar-H), 8.31 (d, J = 3.0 Hz, 1H, Ar-H), 7.92 (d, J = 9.0 Hz, 2H, Ar-H), 7.85 (s, 2H, –CH CH–), 7.04 (d, J = 9.0 Hz, 2H, Ar-H), 3.84 (s, 3H, OMe); 13C NMR (75 MHz, DMSO-d6): δ 192.7, 162.7, 154.5, 148.1, 139.1, 135.4, 132.3, 130.4, 127.2, 125.1, 122.5, 118.8, 115.0, 56.0 (two carbons are isochronous); accurate mass (EI-MS) of [M]˙+: calcd for C16H1235ClNO5 333.0404; found 333.0395.
1-(5-Chloro-2-hydroxy-3-nitrophenyl)-3-(4-fluorophenyl)prop-2-en-1-one (5)
Yellow powder; mp 148–150 °C; yield: 71%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.32; UV-vis λmax (MeOH) = 305 nm; FTIR (cm−1): 3095, 1646, 1595, 1567, 1300, 781, 742, 702; 1H NMR (300 MHz, DMSO-d6): δ 13.34 (bs, 1H, OH), 8.61 (d, J = 3.0 Hz, 1H, Ar-H), 8.32 (d, J = 3.0 Hz, 1H, Ar-H), 8.06–7.99 (m, 2H, Ar-H), 7.94–7.82 (m, 2H, Ar-H), 7.37–7.32 (m, 2H, –CH C–H); 13C NMR (75 MHz, DMSO-d6): δ 192.7, 166.0, 162.7, 154.4, 146.1, 139.3, 135.5, 132.5, 132.4, 131.3, 130.4, 126.1, 122.2, 116.6, 116.4; accurate mass (EI-MS) of [M]˙+: calcd for C15H935ClFNO4 321.0204; found 321.0200.
1-(5-Chloro-2-hydroxy-3-nitrophenyl)-3-(4-(dimethylamino)phenyl)prop-2-en-1-one (6)
Scarlet red powder; mp 158–160 °C; yield: 75%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.40; UV-vis λmax (MeOH) = 360 nm; FTIR (cm−1): 3079, 1633, 1602, 1522, 1306, 811, 737, 695; 1H NMR (300 MHz, DMSO-d6): δ 12.99 (bs, 1H, OH), 8.74 (d, J = 3.0 Hz, 1H, Ar-H), 8.35 (d, J = 3.0 Hz, 1H, Ar-H), 8.31–8.25 (m, 2H, Ar-H), 7.95–7.86 (m, 2H, Ar-H), 6.79–6.76 (m, 2H, olefinic proton), 3.08 (s, 3H, –N(CH3)3), 2.74 (s, 3H, –N(CH3)3); 13C NMR (75 MHz, DMSO-d6): δ 192.0, 155.0, 153.4, 150.0, 139.3, 136.2, 135.0, 132.9, 130.5, 125.0, 124.6, 122.6, 121.9, 114.0, 112.1, 28.8, 28.6; accurate mass (EI-MS) of [M]˙+: calcd for C17H1535ClN2O4 346.0720; found 346.0725.
1-(5-Chloro-2-hydroxy-3-nitrophenyl)-3-(4-chlorophenyl)prop-2-en-1-one (7)
Yellow powder; mp 171–173 °C; yield: 78%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.48; UV-vis λmax (MeOH) = 276 nm; FTIR (cm−1): 3083, 1643, 1559, 1523, 1262, 776, 656; 1H NMR (300 MHz, DMSO-d6): δ 13.10 (bs, 1H, OH), 8.62 (d, J = 3.0 Hz, 1H, Ar-H), 8.36 (d, J = 3.0 Hz, 1H, Ar-H), 7.98 (d, J = 15.0 Hz, 1H, olefinic proton), 7.72 (d, J = 6.0 Hz, 2H, Ar-H), 7.57 (d, J = 15.0 Hz, 1H, olefinic proton), 7.52 (d, J = 6.0 Hz, 2H, Ar-H); 13C NMR (75 MHz, DMSO-d6): δ 191.0, 155.5, 147.0, 138.1, 137.7, 136.6, 133.0, 131.5, 130.5, 129.8, 128.1, 127.0, 124.0, 121.3, (two carbons are isochronous); accurate mass (EI-MS) of [M]˙+: calcd for C15H935Cl2NO4 336.9908; found 336.9900.
1-(5-Chloro-2-hydroxy-3-nitrophenyl)-3-(2,4-dimethylphenyl)prop-2-en-1-one (8)
Yellow powder; mp 162–164 °C; yield: 65%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.62; UV-vis λmax (MeOH) = 256 nm; FTIR (cm−1): 3087, 1637, 1594, 1556, 1291, 789, 703, 676; 1H NMR (600 MHz, DMSO-d6): δ 13.42 (bs, 1H, OH), 8.64 (d, J = 6.0 Hz, 1H, Ar-H), 8.33 (d, J = 6.0 Hz, 1H, Ar-H), 8.12 (d, J = 12.0 Hz, 1H, Ar-H), 8.03 (d, J = 6.0 Hz, 1H, Ar-H), 7.89 (d, J = 12.0 Hz, 1H, Ar-H), 7.16–7.14 (m, 2H, olefinic protons), 2.43 (s, 3H, –CH3), 2.33 (s, 3H, –CH3); 13C NMR (151 MHz, DMSO-d6): δ 192.7, 154.1, 144.3, 142.1, 139.4, 135.6, 135.5, 132.1, 130.4, 130.3, 128.0, 127.6, 125.2, 122.5, 121.5, 21.5, 19.6; accurate mass (EI-MS) of [M]˙+: calcd for C17H1435ClNO4 331.0611; found 331.0602.
1-(5-Chloro-2-hydroxy-3-nitrophenyl)-3-(thiophen-2-yl)prop-2-en-1-one (9)
Yellow solid; mp 212–214 °C; yield: 69%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.41; UV-vis λmax (MeOH) = 348 nm; FTIR (cm−1): 3078, 1636, 1593, 1527, 1298, 789, 699, 675; 1H NMR (600 MHz, DMSO-d6): δ 10.88 (bs, 1H, OH), 8.64 (d, J = 6.0 Hz, 1H, Ar-H), 8.39 (d, J = 6.0 Hz, 1H, Ar-H), 7.99 (dd, J = 12.0, 6.0 Hz, 1H, Ar-H), 7.92 (dd, J = 12.0, 6.0 Hz, 1H, Ar-H), 7.36 (dd, J = 12.0, 6.0 Hz, 1H, Ar-H), 7.21–7.19 (m, 2H, olefinic proton); 13C NMR (151 MHz, DMSO-d6): δ 191.0, 140.4, 137.3, 135.4, 133.8, 131.5, 130.0, 129.5, 129.2, 128.5, 128.3, 127.5, 123.1; accurate mass (EI-MS) of [M]˙+: calcd for C13H835ClNO4S 308.9862; found 308.9850.
1-(5-Chloro-2-hydroxy-3-nitrophenyl)-3-(3-methoxy-4-propoxyphenyl)prop-2-en-1-one (10)
Orange powder; mp 199–201 °C; yield: 83%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.59; UV-vis λmax (MeOH) = 387 nm; FTIR (cm−1): 3087, 1662, 1595, 1551, 1233, 750, 702, 685, 619; 1H NMR (300 MHz, DMSO-d6): δ 10.2 (bs, 1H, OH), 7.98 (d, J = 3.0 Hz, 1H, Ar-H), 7.93 (m, 2H, Ar-H), 7.64 (d, J = 15.0 Hz, 1H, C C–H), 7.37 (m, 2H, Ar-H), 7.03 (d, J = 6.0 Hz, 1H, Ar-H), 4.01 (t, J = 6.0 Hz, 2H, –OCH2–), 3.86 (s, 3H, OMe), 1.83 (m, 2H, –CH2–), 1.04 (t, J = 6.0 Hz, 3H, –CH3); 13C NMR (75 MHz, DMSO-d6): δ 190.9, 162.8, 150.9, 149.5, 142.8, 140.9, 135.6, 132.7, 129.9, 128.2, 124.0, 123.6, 113.0, 111.7, 70.1, 56.1, 55.8, 22.50, 10.87; accurate mass (EI-MS) of [M]˙+: calcd for C19H1835ClNO6 391.0822; found 391.0815.
3-(4-(Benzyloxy)-3-methoxyphenyl)-1-(5-chloro-2-hydroxy-3-nitrophenyl)prop-2-en-1-one (11)
Reddish-brown powder; mp 218–220 °C; yield: 78%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.67; UV-vis λmax (MeOH) = 271 nm; FTIR (cm−1): 3100, 1710, 1653, 1584, 1248, 788, 727, 664, 625; 1H NMR (300 MHz, DMSO-d6): δ 9.87 (s, 1H, OH), 8.04 (d, J = 3.0 Hz, 1H, Ar-H), 7.84–7.82 (m, 4H, Ar-H), 7.62–7.25 (m, 6H, Ar-H and –CH CH), 7.13 (d, J = 9.0 Hz, 1H, Ar-H), 5.24 (s, 1H, –CH2–), 5.16 (s, 1H, –CH2–), 3.86 (s, 3H, OMe); 13C NMR (75 MHz, DMSO-d6): δ 191.8, 153.6, 150.0, 149.6, 137.3, 136.8, 135.7, 130.3, 129.0, 128.9, 128.5, 127.3, 126.3, 125.8, 122.8, 113.7, 113.1, 111.5, 110.2, 70.5, 70.3, 56.1, 56.0; accurate mass (EI-MS) of [M]˙+: calcd for C23H1835ClNO6 439.0822; found 439.0809.
4,6-Dibromo-2-(4-chlorobenzylidene)benzofuran-3(2H)-one (12)
Yellow powder; mp 206–208 °C; yield: 73%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.79; UV-vis λmax (CH2Cl2) = 379 nm; FTIR (cm−1): 3075, 2948, 1711, 1650, 1489, 1282, 733, 631; 1H NMR (600 MHz, DMSO-d6): δ 8.29 (s, 1H, Ar-H), 8.09 (d, J = 12.0 Hz, 2H, Ar-H), 7.41–6.85 (m, 4H, Ar-H); 13C NMR (151 MHz, DMSO-d6): δ 181.4, 161.3, 150.1, 148.2, 148.1, 144.56, 141.1, 126.3, 125.0, 124.4, 119.6, 116.6, 114.4, 107.2, 103.2; accurate mass (EI-MS) of [M]˙+: calcd for C15H779Br235ClO2 411.8501; found 411.8505.
4,6-Dibromo-2-(4-(trifluoromethyl)benzylidene)benzofuran-3(2H)-one (13)
Yellow powder; mp 184–186 °C; yield: 77%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.64; UV-vis λmax (CH2Cl2) = 350 nm; FTIR (cm−1): 3099, 2956, 1712, 1649, 1448, 1289, 773, 643; 1H NMR (300 MHz, DMSO-d6): δ 8.29 (d, J = 3.0 Hz, 1H, Ar-H), 8.17 (d, J = 6.0 Hz, 2H, Ar-H), 7.97 (d, J = 3.0 Hz, 1H, Ar-H), 7.89 (d, J = 6.0 Hz, 2H, Ar-H), 7.13 (s, 1H, –C CH–); 13C NMR (75 MHz, DMSO-d6): δ 182.3, 162.0, 147.5, 141.6, 135.9, 132.3, 130.4, 130.0, 126.6, 126.3, 126.2, 124.3, 116.9, 112.3, 107.3, 31.1; accurate mass (EI-MS) of [M]˙+: calcd for C16H779Br2F3O2 445.8764; found 445.8752.
4,6-Dibromo-2-(furan-2-ylmethylene)benzofuran-3(2H)-one (14)
Green solid; mp 201–203 °C; yield: 74%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.59; UV-vis λmax (CH2Cl2) = 390 nm; FTIR (cm−1): 3117, 2989, 1702, 1640, 1448, 1289, 770, 672; 1H NMR (600 MHz, DMSO-d6): δ 8.32 (d, J = 6.0 Hz, 1H, Ar-H), 8.04 (d, J = 12.0 Hz, 2H, Ar-H), 8.01 (d, J = 6.0 Hz, 1H, Ar-H), 7.64–7.62 (m, 1H, Ar-H), 7.12 (s, 1H, –C C–H); 13C NMR (151 MHz, DMSO-d6): δ 182.2, 161.8, 146.8, 141.5, 135.8, 133.6, 131.0, 129.8, 126.6, 124.5, 116.8, 113.2, 107.3; accurate mass (EI-MS) of [M]˙+: calcd for C13H679Br2O3 367.8683; found 367.8676.
6-Chloro-2-(4-(dimethylamino)benzylidene)-4-nitrobenzofuran-3(2H)-one (15)
Maroon solid; mp 207–209 °C; yield: 71%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.65; UV-vis λmax (CH2Cl2) = 412 nm; FTIR (cm−1): 3059, 2940, 1690, 1617, 1430, 1278, 777, 648; 1H NMR (600 MHz, DMSO-d6): δ 8.59 (d, J = 6.0 Hz, 1H, Ar-H), 8.07 (d, J = 6.0 Hz, 1H, Ar-H), 7.70 (d, J = 12.0 Hz, 2H, Ar-H), 6.80 (d, J = 12.0 Hz, 2H, Ar-H), 6.78 (s, 1H, olefinic proton), 3.06 (s, 6H, –N(CH3)2); 13C NMR (151 MHz, DMSO-d6): δ 190.3, 154.6, 153.2, 140.7, 140.5, 132.7, 132.2, 128.1, 127.6, 127.0, 125.0, 122.0, 112.1, 111.5, 80.3, 78.4, 42.5; accurate mass (EI-MS) of [M]˙+: calcd for C17H1335ClN2O4 344.0563; found 344.0554.
6-Chloro-2-(4-chlorobenzylidene)-4-nitrobenzofuran-3(2H)-one (16)
Yellow powder; mp 205–207 °C; yield: 82%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.42; UV-vis λmax (CH2Cl2) = 455 nm; FTIR (cm−1): 3108, 2934, 1711, 1656, 1451, 1289, 778, 684; 1H NMR (300 MHz, DMSO-d6): δ 8.60 (d, J = 3.0 Hz, 1H, Ar-H), 8.37 (d, J = 3.0 Hz, 1H, Ar-H), 8.14 (d, J = 9.0 Hz, 2H, Ar-H), 7.67 (d, J = 9.0 Hz, 2H, Ar-H); 7.24 (s, 1H, –C C–H), 13C NMR (75 MHz, DMSO-d6): δ 181.0, 156.7, 146.3, 136.2, 134.7, 133.8, 131.7, 131.0, 130.6, 129.8, 128.3, 126.7, 114.4, 112.7, 31.1; accurate mass (EI-MS) of [M]˙+: calcd for C15H735Cl2NO4 334.9752; found 334.9741.
6-Chloro-2-(4-fluorobenzylidene)-4-nitrobenzofuran-3(2H)-one (17)
Light yellow powder; mp 221–223 °C; yield: 78%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.52; UV-vis λmax (CH2Cl2) = 429 nm; FTIR (cm−1): 3102, 2970, 1717, 1660, 1470, 1290, 773, 689; 1H NMR (600 MHz, DMSO-d6): δ 8.59 (d, J = 6.0 Hz, 1H, Ar-H), 8.36 (d, J = 6.0 Hz, 1H, Ar-H), 8.21 (dd, J = 12.0, 6.0 Hz, 2H, Ar-H), 7.47 (dd, J = 12.0, 6.0 Hz, 2H, Ar-H), 7.26 (s, 1H, –C C–H); 13C NMR (151 MHz, DMSO-d6): δ 181.0, 164.7, 163.0, 156.6, 145.8, 143.8, 134.8, 131.6, 128.4, 128.3, 128.2, 126.7, 117.0, 116.9, 114.7; accurate mass (EI-MS) of [M]˙+: calcd. for C15H735ClFNO4 319.0047; found 319.0040.
6-Chloro-2-(2,4-dimethylbenzylidene)-4-nitrobenzofuran-3(2H)-one (18)
Yellow solid; mp 213–215 °C; yield: 80%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.48; UV-vis λmax (CH2Cl2) = 380 nm; FTIR (cm−1): 3078, 2929, 1710, 1646, 1498, 1303, 777, 661, 620; 1H NMR (300 MHz, DMSO-d6): δ 8.55 (d, J = 3.0 Hz, 1H, Ar-H), 8.34 (d, J = 3.0 Hz, 1H, Ar-H), 8.22 (d, J = 9.0 Hz, 1H, Ar-H), 7.21 (m, 2H, Ar-H), 7.12 (s, 1H, –C C–H), 2.46 (s, 3H, –Me), 2.34 (s, 3H, –Me); 13C NMR (75 MHz, DMSO-d6): δ 180.7, 156.5, 145.8, 141.9, 140.4, 134.6, 132.1, 131.4, 131.3, 131.0, 128.2, 127.9, 127.0, 126.7, 112.2, 21.6, 20.0; accurate mass (EI-MS) of [M]˙+: calcd for C17H1235ClNO4 329.0454; found 329.0448.
6-Chloro-4-nitro-2-(thiophen-2-ylmethylene)benzofuran-3(2H)-one (19)
Green solid; mp 262–265 °C; yield: 78%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.47; UV-vis λmax (CH2Cl2) = 410 nm; FTIR (cm−1): 3087, 2921, 1713, 1641, 1448, 1302, 775, 647, 612; 1H NMR (600 MHz, DMSO-d6): δ 8.59 (d, J = 6.0 Hz, 1H, Ar-H), 8.35 (d, J = 6.0 Hz, 1H, Ar-H), 8.15–8.13 (m, 1H, Ar-H), 7.91 (dd, J = 12.0, 6.0 Hz, 1H, Ar-H), 7.68 (s, 1H, –C C–H), 7.34 (dd, J = 12.0, 6.0 Hz, 1H, Ar-H); 13C NMR (151 MHz, DMSO-d6): δ 180.0, 161.7, 149.5, 135.6, 134.7, 131.3, 130.8, 129.2, 128.1, 127.5, 123.6, 122.5, 110.8; accurate mass (EI-MS) of [M]˙+: calcd for C13H635ClNO4S 306.9706; found 306.9700.
6-Chloro-2-(4-methoxybenzylidene)-4-nitrobenzofuran-3(2H)-one (20)
Yellow powder; mp 212–214 °C; yield: 69%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.71; UV-vis λmax (CH2Cl2) = 473 nm; FTIR (cm−1): 3071, 2841, 1705, 1644, 1510, 1321, 777, 683, 665; 1H NMR (300 MHz, DMSO-d6): δ 8.56 (d, J = 3.0 Hz, 1H, Ar-H), 8.32 (d, J = 3.0 Hz, 1H, Ar-H), 8.11 (d, J = 9.0 Hz, 2H, Ar-H), 7.20 (s, 1H, –C C–H), 7.15 (d, J = 9.0 Hz, 2H, Ar-H), 3.88 (s, 3H, OMe); 13C NMR (75 MHz, DMSO-d6): δ 180.4, 162.2, 156.2, 145.0, 134.6, 134.4, 131.2, 130.9, 128.0, 127.0, 124.1, 116.5, 115.4, 56.0 (remaining carbons are isochronous); accurate mass (EI-MS) of [M]˙+: Calcd. for C16H1035ClNO5 331.0247; found 331.0236.
6-Chloro-2-(3-methoxy-4-propoxybenzylidene)-4-nitrobenzofuran-3(2H)-one (21)
Orange powder; mp 200–202 °C; yield: 74%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.58; UV-vis λmax (CH2Cl2) = 374 nm; FTIR (cm−1): 2968, 2876, 1710, 1647, 1473, 1301, 777, 661, 627, 607; 1H NMR (600 MHz, DMSO-d6): δ 8.55 (d, J = 6.0 Hz, 1H, Ar-H), 8.31 (d, J = 6.0 Hz, 1H, Ar-H), 7.94 (d, J = 6.0 Hz, 1H, Ar-H), 7.57 (d, J = 12.0 Hz, 1H, Ar-H), 7.17 (s, 1H, –C
C–H), 7.11 (d, J = 12.0 Hz, 1H, Ar-H), 4.03 (t, J = 6.0 Hz, 2H, 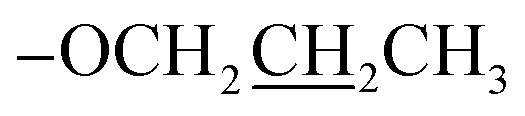 ), 3.92 (s, 3H, OMe), 1.80–1.75 (m, 2H,
), 3.92 (s, 3H, OMe), 1.80–1.75 (m, 2H, 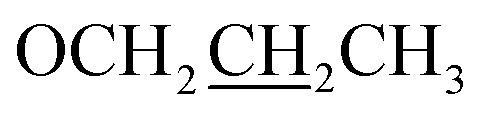 ), 1.01 (t, J = 6.0 Hz, 3H,
), 1.01 (t, J = 6.0 Hz, 3H, 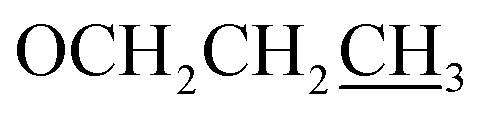 ); 13C NMR (151 MHz, DMSO-d6): δ 180.2, 156.1, 151.6, 149.4, 145.0, 134.6, 131.2, 131.0, 128.1, 128.0, 127.1, 124.1, 117.1, 114.0, 113.0, 70.1, 55.7, 22.4, 10.8; accurate mass (EI-MS) of [M]˙+: calcd for C19H1635ClNO6 389.0666; found 389.0653.
); 13C NMR (151 MHz, DMSO-d6): δ 180.2, 156.1, 151.6, 149.4, 145.0, 134.6, 131.2, 131.0, 128.1, 128.0, 127.1, 124.1, 117.1, 114.0, 113.0, 70.1, 55.7, 22.4, 10.8; accurate mass (EI-MS) of [M]˙+: calcd for C19H1635ClNO6 389.0666; found 389.0653.
2-(4-(Benzyloxy)-3-methoxybenzylidene)-6-chloro-4-nitrobenzofuran-3(2H)-one (22)
Yellow solid; mp 231–233 °C; yield: 75%; Rf (cyclohexane : ethyl acetate 3 : 1) = 0.49; UV-vis λmax (CH2Cl2) = 440 nm; FTIR (cm−1): 3085, 2967, 1716, 1650, 1492, 1301, 778, 668, 624, 612 (C–OC7); 1H NMR (300 MHz, DMSO-d6): δ 8.58 (d, J = 3.0 Hz, 1H, Ar-H), 8.34 (d, J = 3.0 Hz, 1H, Ar-H), 7.99 (d, J = 3.0 Hz, 1H, Ar-H), 7.62–7.23 (m, 7H, Ar-H), 7.20 (s, 1H, –C
C–H), 5.24 (m, 2H, 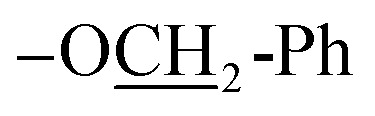 ), 3.95 (s, 3H, OMe); 13C NMR (75 MHz, DMSO): δ 181.8, 155.6, 151.1, 149.8, 145.0, 137.0, 131.3, 130.2, 128.5, 128.4, 128.0, 127.9, 127.1, 126.3, 124.5, 116.9, 114.1, 113.7, 113.1, 110.2, 108.5, 70.5, 56.0; accurate mass (EI-MS) of [M]˙+: calcd for C23H1635ClNO6 437.0666; found 437.0651.
), 3.95 (s, 3H, OMe); 13C NMR (75 MHz, DMSO): δ 181.8, 155.6, 151.1, 149.8, 145.0, 137.0, 131.3, 130.2, 128.5, 128.4, 128.0, 127.9, 127.1, 126.3, 124.5, 116.9, 114.1, 113.7, 113.1, 110.2, 108.5, 70.5, 56.0; accurate mass (EI-MS) of [M]˙+: calcd for C23H1635ClNO6 437.0666; found 437.0651.
Biological activities
Alkaline phosphatase inhibitory activity
Chalcone and aurone analogs (1–22) were designed and synthesized as AP inhibitors. Table 1 demonstrates that the key factor influencing their AP inhibitory activity is the chromone scaffold, benzofuranone and –Cl, –OCH3, or –N(CH3)2 benzyloxy moieties on the benzene ring at the para position. Compounds 1, 4 and 6 having IC50 = 4.311 ± 0.154, 3.141 ± 0.176 and 3.023 ± 0.087 μM showed good activity due to their five-membered heterocyclic ring (furan), –OCH3 and –N(CH3)2 groups, respectively. This reveals that the –OCH3-substituted phenyl moiety plays a vital role in the AP inhibitory activity. Excitingly, analog 21 bearing an –OCH3 group on the phenyl ring at the para position exhibited the most potent activity, having IC50 = 1.055 ± 0.029 μM, compared to compounds 12, 15, 16, 18 and 21 having IC50 = 2.163 ± 0.048, 2.146 ± 0.056, 2.132 ± 0.034, 1.154 ± 0.043 and 2.326 ± 0.059 μM, respectively. Furthermore, all these analogs showed excellent/potent activity compared to the standard KH2PO4 (IC50 = 2.80 ± 0.065 μM). These derivatives possessed –Cl, –OCH3, or –N(CH3)2 benzyloxy moieties attached to ring B of the aurone derivatives. The substitution pattern of the –Cl and –OCH3 functionalities at the aryl ring in compounds 12 and 20 is the crucial factor for enzyme inhibition in these compounds. Compound 16 with a –Cl group present at the para position of benzene ring B and –Cl and –NO2 groups attached to ring A has a much better IC50 value than that of analog 12, where the –Cl group is present at the p-position of phenyl ring B and –Br groups are attached to ring A. The derivatives with electron-donating groups attached to the phenyl ring have better AP inhibitory activity than the analogs with electron-withdrawing groups attached to phenyl rings (Table 1 and Fig. 3).
AP inhibitory activity of chalcones (1–11) and aurones (12–22)a.
| Compound no. | Alkaline phosphatase | Compound no. | Alkaline phosphatase |
|---|---|---|---|
| IC50 ± SEM (μM) | IC50 ± SEM (μM) | ||
| 1 | 4.311 ± 0.154 | 12 | 2.163 ± 0.048 |
| 2 | 6.231 ± 0.098 | 13 | 5.786 ± 0.081 |
| 3 | NAb | 14 | 4.657 ± 0.051 |
| 4 | 3.141 ± 0.176 | 15 | 2.146 ± 0.056 |
| 5 | 7.547 ± 0.265 | 16 | 2.132 ± 0.034 |
| 6 | 3.023 ± 0.087 | 17 | 4.876 ± 0.086 |
| 7 | NA | 18 | 1.154 ± 0.043 |
| 8 | 12.563 ± 0.298 | 19 | 5.876 ± 0.083 |
| 9 | 8.453 ± 0.245 | 20 | 1.055 ± 0.029 |
| 10 | 6.967 ± 0.097 | 21 | 2.326 ± 0.059 |
| 11 | 9.563 ± 0.263 | 22 | 5.543 ± 0.079 |
| KH2PO4 (standard) | 2.80 ± 0.065 μM | ||
Values are presented as mean ± SEM (standard error of the mean).
NA = No activity.
Fig. 3. Graphical representation of IC50 values of analogs 1–22 based on AP enzyme.

Kinetic study results
To understand the inhibitory mechanism of the synthetic compounds on AP, an inhibition kinetic study was performed. Based on the IC50 results, the most potent compound 20 was used to determine the inhibition type and inhibition constant. The kinetic study showed the concentration-dependent inhibition of alkaline phosphatase by the inhibitors. Continuous monitoring of the reaction showed a marked decrease in the reaction rate in the presence of the inhibitors, ultimately indicating a decrease in the final absorbance compared with the controls containing no inhibitor. The potency of inhibition exhibited by these compounds varied depending on the presence and position of the different substitutions and the class of compounds. The inhibition kinetics was analyzed using the Lineweaver–Burk plot.
According to the kinetic analyses, the Lineweaver–Burk plot of 1/Vmaxvs. 1/[S] in the presence of various concentrations of compound 20 was plotted, and the Km value remained the same, while the value of 1/Vmax changed (Fig. 4). This behavior shows that derivative 20 is a non-competitive inhibitor and the Michaelis–Menten constant (Km) is 0.027.
Fig. 4. Lineweaver–Burk plots for the inhibition of alkaline phosphatase activity in the presence of different concentrations of compound 20, i.e., 0.00, 1.15, 2.30, 4.60 and 9.20 μM.

Structure–activity relationship based on alkaline phosphatase inhibition and docking study
All the synthetic analogs (1–22) were subjected to an in vitro alkaline phosphatase inhibitory activity and in silico molecular modelling study. The inhibitory activity data presented in Table 1 allowed us to generate an initial SAR model to investigate the effect of substituent on the phenyl ring, and type of substituent on the activity profile could be explored.
Fascinatingly, compound 20 incorporating a p-methoxy substituent on ring B produced remarkable inhibition potential against human alkaline phosphatase with an IC50 value of 1.055 ± 0.029 μM, which is ∼2.6-fold higher inhibition compared to KH2PO4 (IC50 = 2.80 ± 0.065 μM). The high inhibition potential may be due to the nucleophilic nature of the methoxy group at the para of the phenyl ring (ring B), which suitably interacts with the enzyme active site. The next most potent derivative is compound 18 (IC50 = 1.154 ± 0.043 μM), which has chloro and nitro groups present on ring A and dimethyl (–CH3) groups at the o- & p-positions of ring B.
Furthermore, the next most active compound of this series is compound 16 (IC50 = 2.132 ± 0.034 μM), which has chloro and nitro groups present on ring A and chloro (–Cl) group at p-position of ring B. Next, compound 21 (IC50 = 2.326 ± 0.059 μM) having 3-OCH3 and 4-OC3H7 groups on ring B shows outstanding activity compared to the other derivatives and the standard KH2PO4 (IC50 = 2.80 ± 0.065 μM) due to the presence of two EWG (–Cl and –NO2) substituents on ring A.
Moreover, compound 6 (IC50 = 3.023 ± 0.087 μM) having Cl, –NO2 substitutions on ring A and a dimethylamino substituent on aryl ring B showed better inhibitory activity compared to the standard KH2PO4. Similarly, compound 4 (IC50 = 3.141 ± 0.176 μM) bearing p-OCH3 on the aryl ring resulted in decreased inhibition due to its poor interaction with the active pockets of the enzyme.
Additionally, compounds 1 (IC50 = 4.311 ± 0.154 μM), 2 (IC50 = 6.231 ± 0.098 μM), 5 (IC50 = 7.547 ± 0.265 μM), 8 (IC50 = 12.563 ± 0.298 μM), 9 (IC50 = 8.453 ± 0.245 μM), 10 (IC50 = 6.967 ± 0.097 μM), 11 (IC50 = 9.563 ± 0.263 μM), 13 (IC50 = 5.786 ± 0.081 μM), 14 (IC50 = 4.657 ± 0.051 μM), 17 (IC50 = 4.876 ± 0.086 μM), 19 (IC50 = 5.876 ± 0.083 μM) and 22 (IC50 = 5.543 ± 0.079 μM) were found to be less active in the chalcone and aurone series against the targeted enzyme. Thus, it can be concluded from the whole study that the main skeleton of the molecules having either EWGs or EDGs plays a vital role. However, the slight difference in their potential was greatly affected by the number, positions, and nature of the substituents (Fig. 5).
Fig. 5. Combined structure–activity relationship (SAR) based on IC50 values and docking study.

Finally, it is noteworthy to mention that the aurone series is comparatively more active against AP than simple 2-hydroxychalcones. This factor sheds light on the importance of ring C in the aurone scaffold, and thus motivates the development of AP inhibitors based on the benzofuranone moiety.
To recognize the binding interactions of the potent analogues, a molecular docking study was carried out. The docking study indicates that compounds 8 and 20 are potential derivatives compared to KH2PO4 (standard), which was also used in the inhibition assays for ALP. The minimum binding energy of analog 8 is −6.038 kcal mol−1, which was discovered to be a unique inhibitor vs. the specific biological specimen 2GLQ. The two –Cl and –NO2 functionalities present on ring A at positions 3 and 5 activate this ring to produce hydrogen-bonding connections with the Asn167 and Tyr169 amino acid residues within the active pocket of 2GLQ, respectively. The docking score of −5.211 kcal mol−1 for analog 20 depicts its speculating repressive potential for the 2GLQ protein. The –NO2 group of analog 20 shows hydrogen bond interaction with Lys87 (1.87 Å) and its methyl group shows interaction with the HOH2338 (1.73 Å) water molecule of the specific biological specimen 2GLQ. The presence of a halogen atom (–Cl) on ring A of the chalcone and aurone scaffolds boosts the electron density and hydrophobicity of the molecules, which favor their strong bonding interactions (Fig. 5). To verify the interaction of the newly synthesized compounds, the predicted enzymatic pocket was mutated by replacing Asn167 with His and a variation in the docking score and interaction pattern and orientation of the ligands inside the active site of 2GLQ (Table 2) was found, which confirms the functionality of the active site.
Binding energies (kcal mol−1) of compounds with 1EW2 calculated using Maestro Schrodingera.
| Code | Docking score | Mutated docking score | ΔGCoulomb | ΔGH-bond | ΔGvdW | ΔGbinding |
|---|---|---|---|---|---|---|
| 1 | −4.25 | −3.316 | −1.095 | −1.18 | −24.829 | −29.638 |
| 2 | −4.204 | −3.305 | −1.716 | −1.107 | −27.319 | −35.359 |
| 3 | −4.248 | −2.759 | −9.001 | −0.278 | −24.221 | −36.678 |
| 4 | −5.434 | −3.346 | −10.275 | −1.871 | −19.788 | −34.555 |
| 5 | −5.138 | −3.733 | −4.752 | −2.105 | −24.858 | −36.747 |
| 6 | −5.233 | −2.587 | −6.938 | −1.31 | −26.177 | −38.324 |
| 7 | −5.877 | −4.155 | −8.947 | −2.305 | −23.89 | −44.234 |
| 8 | −6.038 | −2.873 | −8.513 | −2.35 | −22.432 | −34.942 |
| 9 | −4.862 | −4.30 | −5.893 | −1.895 | −23.563 | −36.743 |
| 10 | −5.263 | −3.178 | −8.988 | −1.738 | −25.298 | −45.752 |
| 11 | −5.862 | −3.823 | −9.303 | −2.416 | −27.037 | −50.023 |
| 12 | −3.456 | −2.538 | −0.671 | 0.000 | −22.593 | −26.501 |
| 13 | −3.273 | −1.570 | −2.883 | −0.7 | −21.538 | −29.806 |
| 14 | −3.512 | −2.425 | −3.716 | −0.47 | −19.181 | −27.928 |
| 15 | −3.47 | −2.252 | −4.177 | −0.7 | −21.932 | −32.72 |
| 16 | −3.896 | −2.529 | −3.487 | −1.53 | −21.798 | −32.733 |
| 17 | −4.606 | −2.553 | −4.753 | −0.83 | −23.993 | −33.802 |
| 18 | −3.843 | −2.136 | −4.626 | −1.198 | −22.927 | −34.969 |
| 19 | −4.355 | −2.659 | −5.127 | −1.536 | −22.751 | −32.449 |
| 20 | −5.211 | −2.312 | −7.151 | −0.7 | −18.916 | −35.206 |
| 21 | −4.113 | −3.128 | −4.271 | −1.334 | −26.051 | −30.134 |
| 22 | −4.907 | −2.936 | −7.534 | −2.087 | −28.329 | −48.103 |
ΔGb inding: binding free energy; ΔGCoulomb: Coulomb binding energy; ΔGH-bond: hydrogen bonding energy; and ΔGvdW: van der Waals forces. All these energies contribute to the binding free energy.
Overall, it can be concluded that the theoretical results have good correlation with the experimental data obtained from the in silico studies. Compound 20 exhibits the highest potential in the series with IC50 = 1.055 ± 0.029 μM (standard KH2PO4 IC50 = 2.80 ± 0.065 μM) and docking score of −5.211. Compound 20 was found to be a non-competitive inhibitor, which is in accordance with the in vitro results.
In silico studies
Docking analysis
Docking calculations are helpful to predict the ligand pose within the binding site of a target protein. Thus, we studied all the compounds (1–22) using molecular docking simulations to determine their possible binding orientations in the enzymatic pocket of the human alkaline phosphatase enzyme (PDB ID: 2GLQ) (Table 2). According to the docking results, it can be concluded that all the compounds possess good binding energy values, and interestingly no significant energy difference was observed among the docked molecules (Fig. 6). The docking results predicted that compound 8 is the most active, which exhibited the most favorable energy value (−6.038 kcal mol−1) among the derivatives. Moreover, the other chalcone analogues (1–7 and 9–11) also exhibited good binding energy values (−4.204 to −5.877). The aurone derivatives (12–22) also presented significant energy values (−3.273 to −5.211) against the receptor molecule. However, all the compounds have the same basic chemical nucleus, and therefore the majority of the ligands showed efficient energy values.
Fig. 6. Graphical depiction of the docking score (kcal mol−1) of the synthesized compounds (1–22).

Binding pocket analysis of derivatives 8 and 20 against alkaline phosphatase
To deeply recognize the binding interaction behavior of the ligand against the target protein, compound 8 was predicted to have the lowest docking score (−6.038 kcal mol−1) among the chalcone derivatives. The docking study showed that compound 8 was actively confined inside the receptor active binding region of the molecule (Fig. 7a and b). The chromone-type ring structure intruded in the inner part of the binding pocket, whereas the bulky dimethyl moiety remained outside in the opening region of the binding pocket. This dimethyl may cause slight steric hindrance, which may restrict this part to gain entry inside the binding pocket. It was observed that the ΔGvdW contribution for ligand 8 is −22.432 kcal mol−1 and its hydrogen bond (ΔGH-bond) energy is −2.35 kcal mol−1. Furthermore, four hydrogen bond interactions were predicted. The phenolic hydroxyl forms an H-bond interaction with active site residue Asn167 (2.73 Å) and the oxygen of the nitro group forms H-bond interactions with the side chain residue Tyr169 (2.08 Å), whereas the α,β unsaturated carbonyl of ligand 8 interacts with Gln108 (1.86 Å) and the dimethyl substituted phenyl ring is stabilized by π–π stacking with Hid432.
Fig. 7. Ball and stick model before mutation (a) and ball and stick model of the docking pose of 8 in the active site of mutated 2GLQ (b). The inhibitor binds in the active site closer to Asn167. The amino acid residues forming the binding pocket of the 2GLQ protein are illustrated as sticks. Zn2+ and Mg2+ ions are shown in blue and pink and labeled, respectively.

Similarly, compound 20 was predicted to have the best docking score (−5.211 kcal mol−1) among the aurone derivatives. It was observed that the ΔGvdW contribution for 20 is −18.916 kcal mol−1 and the hydrogen bond (ΔGH-bond) energy is −0.7 kcal mol−1. Furthermore, the hydrogen bond interaction is found between –NO2 of 20 and side-chain Lys87 (1.87 Å) and methoxy oxygen shows an interaction with the HOH2338 active site (1.73 Å) (Fig. 8a and b). The large size and planner and rigid structure of 20 prevent it from entering the binuclear zinc-binding site, and thus it is bound to a shallow area on the surface of the enzyme pocket. The described binding mode will allow the substrate to enter the pocket, providing a useful non-competitive model, which is in accordance with our in vitro enzyme inhibitory kinetic assay.
Fig. 8. Ball and stick model before mutation (a) and ball and stick model of mutated protein 2GLQ with the docking pose of 20 in the active site (b). The inhibitor binds in the active site closer to HOH2338. Amino acid residues forming the binding pocket of the 2GLQ protein are labeled as sticks. Zn2+ and Mg2+ ions are shown in blue and pink and labeled, respectively.

MD simulation results
To check the stability of the complex, a root mean square deviation (RMSD) analysis was performed during simulation. Fig. 9a exhibits the RMSD plot of the protein when compound 20 was bound to it. The RMSD plot shows that the RMSD of the conformation is ∼1.5 Å at the start of the simulation. The RMSD value increased to ∼3.5 Å at 5 ns. According to this plot, it can be observed that the RMSD became stable after 5 ns and remained stable up to 60 ns with a minor change. From 5 to 60 ns, the RMSD value remained in the range of ∼3 to 3.5 Å. From 60 to 85 ns, the average RMSD was in the range of ∼3.25 to 3.5 Å. After 85 ns, the RMSD dropped to ∼2.5 Å towards the end of simulation. The average RMSD value of the protein during whole simulation was 2.93 ± 0.012. Similarly, to check the stability of the ligand with the protein, the RMSD of the ligand was also analyzed. Fig. 9b shows the RMSD plot of the ligand, which shows that the RMSD value remained below 1 Å during the whole simulation. The average RMSD of the ligand was 0.68 ± 0.004.
Fig. 9. (a) RMSD plot of protein when compound 20 was bound to it, (b) RMSD plot of the ligand, (c) RMSF plot of the protein and (d) Rg plot of the protein.

To examine the fluctuations of the amino acid residues of the protein during the simulation, the root mean square fluctuation (RMSF) was computed. A high value of RMSF shows that the residues are flexible or in the loop region, while a low value of RMSF shows that the respective residues are rigid or located in the helices or sheets. Fig. 9c shows the RMSF plot of the protein. The plot shows that the initial 50 residues had the highest RMSF value, which indicates that these residues showed maximum flexibility during the simulation. The remaining residues showed a lower RMSF value than the initial 50 residues, indicating that they were rigid during simulation.
The radius of gyration (Rg) shows the compactness of the protein when a certain ligand was bound to it. A low Rg value shows that the protein remained compact during simulation, whereas a high value shows that there were some unfolding events during simulation. Fig. 9d shows the Rg plot of the protein. The Rg value at the start was ∼23 Å, which increased to ∼23.2 Å at 5 ns, and then dropped to ∼22.75 Å after 5 ns. From 5 to 15 ns, the Rg value remained in the range of ∼22.75 to 23 Å. After 20 ns, the Rg value increased to ∼23.1 Å and remained in this range up to 70 ns. The Rg value dropped to ∼22.9 Å at 80 ns and then remained in this range until the end of the simulation. The average Rg value during the simulation was 23.04 ± 0.002.
To determine the stability of the complex, RMSD analysis was performed for the protein and ligand. The trend of the RMSD value for the protein showed that the protein remained stable when compound 20 was bound to it for 100 ns. Similarly, the RMSD of the ligand showed that the ligand was tightly bound to the protein and did not leave the protein, which shows the stability of the complex. The RMSF showed that the protein residues did not exhibit high fluctuations except for the loop region residues. According to the Rg plot, it was observed that the protein remained compact during simulation and did not show major unfolding events. This, compound 20 was stably bound to it and can be treated as drug candidate in wet lab studies.
Chemoinformatics and Lipinski's rule
The predicted chemoinformatic properties such as polarizability, solubility and polar surface area were computationally evaluated. A study of the literature established the standard value for molar molecular weight (160–480) and number of atoms (20–70).62,63 The results revealed that the predicted values of 8, 9, 19 and 20 are much better than the standard values and all the other synthesized compounds. Moreover, Lipinski's rule of five (RO5) does not indicate anything about the specific chemistry or structural features found in drugs or non-drugs. The computational results predicted that 8, 9, 19 and 20 possesses 4, 5, 5 and 5 HBA (≤10), 1, 1, 0 and 0 HBD (≤5), 4.31, 2.68, 2.72 and 3.22 log P (<5), 10.58, 142.36, 65.60 and 25.75 mg mL−1 log S and 66.65, 67.67, 60.34 and 66.87 Å2 PSA (≤120 Å2) values, respectively, which significantly justify their drug-like behavior with drug like model scores of 0.17 and 0.82. Moreover, their molecular weight (308.99 g mol−1) was also much better than the standard value (Table 3 and Fig. 10). The findings show that several analogs have drug-like effects without violating any of the rules governing their medicinal potential.
Pharmacokinetic assessment of the synthesized chalcones and aurone analogs (1–22)a.
| Compound no. | Mol. formula | Mol wt. (g mol−1) | No. HBAb | No. HBDc | Mol. log Pd | Mol. log Se (mg L−1) | Mol. PSAf Å2 | Drug-likeness model score | Lipinski's rule of 5 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | C13H8Br2O3 | 369.88 | 3 | 1 | 4.42 | 17.32 | 36.44 | −0.24 | Yes |
| 2 | C15H9Br2ClO2 | 413.87 | 2 | 1 | 5.85 | 0.50 | 28.69 | −0.06 | No |
| 3 | C16H9Br2F3O2 | 447.89 | 2 | 1 | 6.19 | 1.06 | 28.69 | −0.42 | No |
| 4 | C16H12ClNO5 | 333.04 | 5 | 1 | 3.26 | 77.48 | 74.20 | −0.20 | Yes |
| 5 | C15H9ClFNO4 | 321.02 | 4 | 1 | 3.37 | 49.51 | 66.65 | −0.20 | Yes |
| 6 | C17H15ClN2O4 | 346.07 | 4 | 1 | 3.37 | 49.29 | 69.46 | −0.35 | Yes |
| 7 | C15H9Cl2NO4 | 336.99 | 4 | 1 | 3.90 | 11.30 | 66.65 | −0.10 | Yes |
| 8 | C17H14ClNO4 | 331.06 | 4 | 1 | 4.31 | 10.58 | 66.65 | −0.55 | Yes |
| 9 | C13H8ClNO4S | 308.99 | 5 | 1 | 2.68 | 142.36 | 67.67 | −0.17 | Yes |
| 10 | C19H18ClNO6 | 391.08 | 6 | 1 | 4.11 | 9.72 | 81.79 | 0.04 | Yes |
| 11 | C23H18ClNO6 | 439.08 | 6 | 1 | 4.61 | 6.00 | 81.60 | −0.25 | Yes |
| 12 | C15H7Br2ClO2 | 411.85 | 2 | 0 | 5.75 | 0.09 | 21.36 | −0.56 | No |
| 13 | C16H7Br2F3O2 | 445.88 | 2 | 0 | 6.09 | 0.39 | 21.36 | −0.92 | No |
| 14 | C13H6Br2O3 | 367.87 | 3 | 0 | 4.31 | 6.64 | 29.11 | −0.83 | Yes |
| 15 | C17H13Br2NO2 | 420.93 | 2 | 0 | 5.22 | 2.03 | 24.17 | −0.81 | No |
| 16 | C15H7Cl2NO4 | 334.98 | 4 | 0 | 3.86 | 2.65 | 59.32 | −0.66 | Yes |
| 17 | C15H7ClFNO4 | 319.00 | 4 | 0 | 3.33 | 18.97 | 59.32 | −0.70 | Yes |
| 18 | C17H12ClNO4 | 329.05 | 4 | 0 | 4.25 | 2.11 | 59.32 | −1.01 | Yes |
| 19 | C13H6ClNO4S | 306.97 | 5 | 0 | 2.72 | 65.60 | 60.34 | −0.82 | Yes |
| 20 | C16H10ClNO5 | 331.02 | 5 | 0 | 3.22 | 25.75 | 66.87 | −0.77 | Yes |
| 21 | C19H16ClNO6 | 389.07 | 6 | 0 | 3.89 | 3.49 | 74.46 | −0.35 | Yes |
| 22 | C23H16ClNO6 | 437.07 | 6 | 0 | 4.39 | 2.59 | 74.27 | −0.59 | Yes |
The table above depicts all the Lipinski's RO5 components, i.e.
Number of hydrogen-bond acceptors.
Number of hydrogen-bond donors.
Octanol–water partition coefficient.
Measured solubility.
Total polar surface area.
Fig. 10. Drug-likeness score bar chart.

Conclusions
In this work, a series of new chalcones (1–11) and their oxidative cyclization aurone products (12–22) were synthesized using precedent methodologies starting from substituted 2′-hydroxyacetophenone and various substituted aryl aldehydes over two steps and characterized using common spectroscopic techniques (FTIR, UV-vis, NMR and mass spectrometry, etc.). All the synthetic compounds (1–22) were evaluated in vitro against the human alkaline phosphatase enzyme. To explore the activities of the synthesized chalcones and aurones in terms of potential human intestinal AP inhibitors, herein, for the first-time, we reported that chalcones and aurones are potential h-IAP inhibitors. Among the synthesized analogs, compounds 12, 15, 16, 18, 20 and 21 exhibited excellent inhibitory activity against AP, and were more active than the reference standard KH2PO4 (IC50 = 2.80 ± 0.065 μM). The higher inhibitory potential of compounds 18 (IC50 = 1.154 ± 0.043 μM) and 20 (IC50 = 1.055 ± 0.029 μM) than the standard KH2PO4 (IC50 = 2.80 ± 0.065 μM) may prove that they are effective inhibitors of AP. Molecular docking studies were also performed to examine their binding interactions with IAP human proteins. The binding interactions were noteworthy compared with the interactions shown by the reference standard in the active site of h-IAP. Moreover, the kinetic studies illustrated that compound 20 had a non-competitive inhibition mechanism. The SAR results revealed the involvement of the active inhibitors in H-bonding at various sites with different amino acid residues in addition to secondary metal ion interactions with the Zn2+ ions inside the active pocket of the enzyme. Moreover, the comparative results of the aurones and chalcones showed that the aurones are potent AP inhibitors. Owing to their low toxicity and high inhibition activity, this may represent a milestone on the path toward new valuable agents in the medical field, where it was recently suggested that AP can play key roles. However, it is recommended that the selective inhibitors of h-IAP should be further subjected to detailed analysis to explore their inhibitory effects. The comparative binding energy analysis revealed that 20 is the most active compound and confined in a compatible position in binding pocket of AP. Therefore, compound 20 is the most potent derivative according to the in vitro (IC50 = 1.055 ± 0.029 μM) and in silico (docking score = −5.211) results. Finally, the MD simulations revealed the stability of the docked structures at the binding sites. The pharmacokinetics disclosed that compounds 1–22 all obey Lipinski's rule and show promising drug scores. The ADMET parameters revealed that these compounds have good lead-like efficiency with no irritating effect. Thus, our research group is focusing on the skeletal modification of chalcones and aurones in the design of new and novel materials with diverse applications. Hence, chalcone and aurones are innovative motifs and can play a significant role in drug discovery. The results revealed that the newly synthesized scaffolds may be the interesting molecules and drug targets for future studies.
Abbreviations
- APs
Alkaline phosphatases
- CIAP
Calf intestinal alkaline phosphatase
- GCAP
Germ cell alkaline phosphatase
- IAP
Intestinal alkaline phosphatase
- PLAP
Placental alkaline phosphatase
- p-NPP
para-Nitrophenylphosphate disodium salt
- TLC
Thin-layer chromatography
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors would like to extend their sincere appreciation to Taif University Researchers Supporting Project number (TURSP-2020/312), Taif University, Taif, Saudi Arabia. Dr Mughal gratefully acknowledges the financial support by Higher Education Commission of Pakistan (HEC) under NRPU research project no. 6484.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07379f
References
- Millán J. L., Mammalian alkaline phosphatases: from biology to applications in medicine and biotechnology, John Wiley & Sons, 2006 [Google Scholar]
- al-Rashida M. Raza R. Abbas G. Shah M. S. Kostakis G. E. Lecka J. Sévigny J. Muddassar M. Papatriantafyllopoulou C. Iqbal J. Eur. J. Med. Chem. 2013;66:438–449. doi: 10.1016/j.ejmech.2013.06.015. [DOI] [PubMed] [Google Scholar]
- Ardecky R. J. Bobkova E. V. Kiffer-Moreira T. Brown B. Ganji S. Zou J. Pass I. Narisawa S. Iano F. G. Rosenstein C. Bioorg. Med. Chem. Lett. 2014;24:1000–1004. doi: 10.1016/j.bmcl.2013.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I. Shah S. J. A. Ejaz S. A. Ibrar A. Hameed S. Lecka J. Millán J. L. Sévigny J. Iqbal J. RSC Adv. 2015;5:64404–64413. doi: 10.1039/C5RA12455G. [DOI] [Google Scholar]
- Lanier M. Sergienko E. Simão A. M. Su Y. Chung T. Millán J. L. Cashman J. R. Bioorg. Med. Chem. 2010;18:573–579. doi: 10.1016/j.bmc.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miliutina M. Ejaz S. A. Khan S. U. Iaroshenko V. O. Villinger A. Iqbal J. Langer P. Eur. J. Med. Chem. 2017;126:408–420. doi: 10.1016/j.ejmech.2016.11.036. [DOI] [PubMed] [Google Scholar]
- Younus H. A. Hameed A. Mahmood A. Khan M. S. Saeed M. Batool F. Asari A. Mohamad H. Pelletier J. Sévigny J. Bioorg. Chem. 2020;100:103827. doi: 10.1016/j.bioorg.2020.103827. [DOI] [PubMed] [Google Scholar]
- Zaher D. M. El-Gamal M. I. Omar H. A. Aljareh S. N. Al-Shamma S. A. Ali A. J. Zaib S. Iqbal J. Arch. Pharm. 2020;353:2000011. doi: 10.1002/ardp.202000011. [DOI] [PubMed] [Google Scholar]
- Mumtaz A. Saeed K. Mahmood A. Zaib S. Saeed A. Pelletier J. Sévigny J. Iqbal J. Bioorg. Chem. 2020;101:103996. doi: 10.1016/j.bioorg.2020.103996. [DOI] [PubMed] [Google Scholar]
- Chirambo G. van Niekerk C. Crowther N. J. Int. J. Exp. Pathol. 2017;98:260–268. doi: 10.1111/iep.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal J. El-Gamal M. I. Ejaz S. A. Lecka J. Sévigny J. Oh C.-H. J. Enzyme Inhib. Med. Chem. 2018;33:479–484. doi: 10.1080/14756366.2018.1428193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal Z. Ashraf Z. Hassan M. Abbas Q. Jabeen E. Bioorg. Chem. 2019;90:103108. doi: 10.1016/j.bioorg.2019.103108. [DOI] [PubMed] [Google Scholar]
- Zalatan J. G. Fenn T. D. Herschlag D. J. Mol. Biol. 2008;384:1174–1189. doi: 10.1016/j.jmb.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Balcerzak M. Radisson J. Thouverey C. Pikula S. Azzar G. Buchet R. J. Biol. Chem. 2005;280:37289–37296. doi: 10.1074/jbc.M504260200. [DOI] [PubMed] [Google Scholar]
- Sergienko E. A. Millán J. L. Nat. Protoc. 2010;5:1431. doi: 10.1038/nprot.2010.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Sharma U. Pal D. Prasad R. Indian J. Clin. Biochem. 2014;29:269–278. doi: 10.1007/s12291-013-0408-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sidique S. Ardecky R. Su Y. Narisawa S. Brown B. Millán J. L. Sergienko E. Cosford N. D. Bioorg. Med. Chem. Lett. 2009;19:222–225. doi: 10.1016/j.bmcl.2008.10.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosyan A. Ghochikyan T. V. Ejaz S. A. Mardiyan Z. Z. Khan S. U. Grigoryan T. Gevorgyan A. Samvelyan M. A. Galstyan A. S. Parpart S. ChemistrySelect. 2017;2:5677–5683. doi: 10.1002/slct.201700339. [DOI] [Google Scholar]
- Yegutkin G. G. Biochim. Biophys. Acta, Mol. Cell Res. 2008;1783:673–694. doi: 10.1016/j.bbamcr.2008.01.024. [DOI] [PubMed] [Google Scholar]
- Whitehouse J. S. Riggle K. M. Purpi D. P. Mayer A. N. Pritchard Jr K. A. Oldham K. T. Gourlay D. M. J. Surg. Res. 2010;163:79–85. doi: 10.1016/j.jss.2010.04.048. [DOI] [PubMed] [Google Scholar]
- Salar U. Khan K. M. Iqbal J. Ejaz S. A. Hameed A. Al-Rashida M. Perveen S. Tahir M. N. Eur. J. Med. Chem. 2017;131:29–47. doi: 10.1016/j.ejmech.2017.03.003. [DOI] [PubMed] [Google Scholar]
- Saeed A. Saddique G. Channar P. A. Larik F. A. Abbas Q. Hassan M. Raza H. Fattah T. A. Seo S.-Y. Bioorg. Med. Chem. 2018;26:3707–3715. doi: 10.1016/j.bmc.2018.06.002. [DOI] [PubMed] [Google Scholar]
- (a) Saito K. Shinozuka T. Nakao A. Kiho T. Kunikata T. Shiiki T. Nagai Y. Naito S. Bioorg. Med. Chem. Lett. 2019;29:1769–1773. doi: 10.1016/j.bmcl.2019.05.014. [DOI] [PubMed] [Google Scholar]; (b) Hassan S. Ejaz S. A. Saeed A. Shehzad M. Khan S. U. Lecka J. Sévigny J. Shabir G. Iqbal J. Bioorg. Chem. 2018;76:237–248. doi: 10.1016/j.bioorg.2017.11.013. [DOI] [PubMed] [Google Scholar]; (c) Hessle L. Johnson K. A. Anderson H. C. Narisawa S. Sali A. Goding J. W. Terkeltaub R. Millán J. L. Proc. Natl. Acad. Sci. U. S. A. 2002;99:9445–9449. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Le Du M.-H. Millán J. L. J. Biol. Chem. 2002;277:49808–49814. doi: 10.1074/jbc.M207394200. [DOI] [PubMed] [Google Scholar]; (b) Saeed A. Khurshid A. Shabir G. Mahmood A. Zaib S. Iqbal J. Bioorg. Med. Chem. Lett. 2020;30:127238. doi: 10.1016/j.bmcl.2020.127238. [DOI] [PubMed] [Google Scholar]
- (a) Matsuoka I. Ohkubo S. J. Pharmacol. Sci. 2004;94:95–99. doi: 10.1254/jphs.94.95. [DOI] [PubMed] [Google Scholar]; (b) Khurshid A. Saeed A. Ashraf Z. Abbas Q. Hassan M. Mol. Diversity. 2020:1–15. doi: 10.1007/s11030-020-10136-9. [DOI] [PubMed] [Google Scholar]
- Al-Rashida M. Ejaz S. A. Ali S. Shaukat A. Hamayoun M. Ahmed M. Iqbal J. Bioorg. Med. Chem. 2015;23:2435–2444. doi: 10.1016/j.bmc.2015.03.054. [DOI] [PubMed] [Google Scholar]
- Ejaz S. A. Saeed A. Siddique M. N. un Nisa Z. Khan S. Lecka J. Sévigny J. Iqbal J. Bioorg. Chem. 2017;70:229–236. doi: 10.1016/j.bioorg.2017.01.003. [DOI] [PubMed] [Google Scholar]
- Ashraf A. Ejaz S. A. Rahman S. U. Siddiqui W. A. Arshad M. N. Lecka J. Sévigny J. Zayed M. E. M. Asiri A. M. Iqbal J. Eur. J. Med. Chem. 2018;159:282–291. doi: 10.1016/j.ejmech.2018.09.063. [DOI] [PubMed] [Google Scholar]
- Aziz H. Mahmood A. Zaib S. Saeed A. El-Seedi H. R. Pelletier J. Sévigny J. Iqbal J. J. Biomol. Struct. Dyn. 2020:1–14. doi: 10.1080/07391102.2020.1802336. [DOI] [PubMed] [Google Scholar]
- Ibrar A. Zaib S. Khan I. Jabeen F. Iqbal J. Saeed A. RSC Adv. 2015;5:89919–89931. doi: 10.1039/C5RA14900B. [DOI] [Google Scholar]
- Iqbal Z. Iqbal A. Ashraf Z. Latif M. Hassan M. Nadeem H. Drug Dev. Res. 2019;80:646–654. doi: 10.1002/ddr.21542. [DOI] [PubMed] [Google Scholar]
- Channar P. A. Irum H. Mahmood A. Shabir G. Zaib S. Saeed A. Ashraf Z. Larik F. A. Lecka J. Sévigny J. Bioorg. Chem. 2019;91:103137. doi: 10.1016/j.bioorg.2019.103137. [DOI] [PubMed] [Google Scholar]
- Khan I. Hanif M. Hussain M. T. Khan A. A. Aslam M. A. S. Rama N. H. Iqbal J. Aust. J. Chem. 2012;65:1413–1419. doi: 10.1071/CH12134. [DOI] [Google Scholar]
- Coburn S. P. Mahuren J. D. Jain M. Zubovic Y. Wortsman J. J. Clin. Endocrinol. Metab. 1998;83:3951–3957. doi: 10.1210/jcem.83.11.5288. [DOI] [PubMed] [Google Scholar]
- (a) Harris H. Clin. Chim. Acta. 1990;186:133–150. doi: 10.1016/0009-8981(90)90031-M. [DOI] [PubMed] [Google Scholar]; (b) Sergienko E. A. Millán J. L. Nat. Protoc. 2010;5:1431. doi: 10.1038/nprot.2010.86. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fishman W. H. Green S. Inglis N. I. Nature. 1963;198:685–686. doi: 10.1038/198685b0. [DOI] [PubMed] [Google Scholar]; (d) Khan I. Ibrar A. Ejaz S. A. Khan S. U. Shah S. J. A. Hameed S. Simpson J. Lecka J. Sévigny J. Iqbal J. RSC Adv. 2015;5:90806–90818. doi: 10.1039/C5RA14836G. [DOI] [Google Scholar]; (e) Al-Rashida M. Ejaz S. A. Ali S. Shaukat A. Hamayoun M. Ahmed M. Iqbal J. Bioorg. Med. Chem. 2015;23:2435–2444. doi: 10.1016/j.bmc.2015.03.054. [DOI] [PubMed] [Google Scholar]; (f) Miliutina M. Ejaz S. A. Iaroshenko V. O. Villinger A. Iqbal J. Langer P. Org. Biomol. Chem. 2016;14:495–502. doi: 10.1039/C5OB01350J. [DOI] [PubMed] [Google Scholar]; (g) Khan I. Shah S. J. A. Ejaz S. A. Ibrar A. Hameed S. Lecka J. Millán J. L. Sévigny J. Iqbal J. RSC Adv. 2015;5:64404–64413. doi: 10.1039/C5RA12455G. [DOI] [Google Scholar]
- Kiffer-Moreira T. Sheen C. R. da Silva Gasque K. C. Bolean M. Ciancaglini P. Van Elsas A. Hoylaerts M. F. Millán J. L. PLoS One. 2014;9:89374. doi: 10.1371/journal.pone.0089374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallès J. P. Nutr. Rev. 2014;72:82–94. doi: 10.1111/nure.12082. [DOI] [PubMed] [Google Scholar]
- Miliutina M. Ejaz S. A. Iaroshenko V. O. Villinger A. Iqbal J. Langer P. Org. Biomol. Chem. 2016;146:495–502. doi: 10.1039/C5OB01350J. [DOI] [PubMed] [Google Scholar]
- Rammohan A. Reddy J. S. Sravya G. Rao C. N. Zyryanov G. V. Environ. Chem. Lett. 2020;18:433–458. doi: 10.1007/s10311-019-00959-w. [DOI] [Google Scholar]
- (a) Xue Y. Liu Y. Zhang L. Wang H. Luo Q. Chen R. Liu Y. Li Y. Int. J. Quantum Chem. 2019;119:25808. doi: 10.1002/qua.25808. [DOI] [Google Scholar]; (b) Chen Z.-H. Zheng C.-J. Sun L.-P. Piao H.-R. Eur. J. Med. Chem. 2010;45:5739–5743. doi: 10.1016/j.ejmech.2010.09.031. [DOI] [PubMed] [Google Scholar]
- Zhuang C. Zhang W. Sheng C. Zhang W. Xing C. Miao Z. Chem. Rev. 2017;117:7762–7810. doi: 10.1021/acs.chemrev.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaonkar S. L. Vignesh U. Res. Chem. Intermed. 2017;43:6043–6077. doi: 10.1007/s11164-017-2977-5. [DOI] [Google Scholar]
- Singh P. Anand A. Kumar V. Eur. J. Med. Chem. 2014;85:758–777. doi: 10.1016/j.ejmech.2014.08.033. [DOI] [PubMed] [Google Scholar]
- Sui G. Li T. Zhang B. Wang R. Hao H. Zhou W. Bioorg. Med. Chem. 2020:115895. doi: 10.1016/j.bmc.2020.115895. [DOI] [PubMed] [Google Scholar]
- Zwergel C. Gaascht F. Valente S. Diederich M. Bagrel D. Kirsch G. Nat. Prod. Commun. 2012;7:322. doi: 10.1177/1934578X1200700322. [DOI] [PubMed] [Google Scholar]
- Irshad M. Ali Q. Iram F. Ahamad S. Saleem M. Saadia M. Batool M. Kanwal A. Tabassum S. Russ. J. Gen. Chem. 2019;89:1519–1527. doi: 10.1134/S1070363219070235. [DOI] [Google Scholar]
- Popova A. V. Bondarenko S. P. Frasinyuk M. S. Chem. Heterocycl. 2019;55:285–299. doi: 10.1007/s10593-019-02457-x. [DOI] [Google Scholar]
- Mughal E. U. Sadiq A. Khan B. A. Zafar M. N. Ahmed I. Zubair M. Lett. Drug Des. Discovery. 2017;14:1035–1041. [Google Scholar]
- Mughal E. U. Sadiq A. Murtaza S. Rafique H. Zafar M. N. Riaz T. Khan B. A. Hameed A. Khan K. M. Bioorg. Med. Chem. 2017;25:100–106. doi: 10.1016/j.bmc.2016.10.016. [DOI] [PubMed] [Google Scholar]
- Chintakrindi A. S. Gohil D. J. Chowdhary A. S. Kanyalkar M. A. Bioorg. Med. Chem. 2020;28:115191. doi: 10.1016/j.bmc.2019.115191. [DOI] [PubMed] [Google Scholar]
- Hassan G. S., Georgey H. H., George R. F. and Mohamed E. R., Bulletin of Faculty of Pharmacy, Cairo University, 2018, vol. 56, pp. 121–127 [Google Scholar]
- Kumar S. Green Chem. Lett. Rev. 2014;17:95–99. doi: 10.1080/17518253.2014.895867. [DOI] [Google Scholar]
- Zwick V. Chatzivasileiou A. O. Deschamps N. Roussaki M. Simões-Pires C. A. Nurisso A. Denis I. Blanquart C. Martinet N. Carrupt P.-A. Bioorg. Med. Chem. Lett. 2014;24:5497–5501. doi: 10.1016/j.bmcl.2014.10.019. [DOI] [PubMed] [Google Scholar]
- Nguyen P. T. V. Huynh H. A. Truong D. V. Tran T.-D. Vo C. V. T. Molecules. 2020;25:4657. doi: 10.3390/molecules25204657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar G. Lathwal E. Saroha B. Kumar S. Chauhan N. S. Kumar T. ChemistrySelect. 2020;5:3539–3543. doi: 10.1002/slct.201904912. [DOI] [Google Scholar]
- Haudecoeur R. Boumendjel A. Curr. Med. Chem. 2012;19:2861–2875. doi: 10.2174/092986712800672085. [DOI] [PubMed] [Google Scholar]
- Friesner R. A. Murphy R. B. Repasky M. P. Frye L. L. Greenwood J. R. Halgren T. A. Sanschagrin P. C. Mainz D. T. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Bowers K. J., Chow D. E., Xu H., Dror R. O., Eastwood M. P., Gregersen B. A., Klepeis J. L., Kolossvary I., Moraes M. A. and Sacerdoti F. D., Proceedings of the ACM/IEEE Conference on Supercomputing, 2006, pp. 43–45 [Google Scholar]
- Adane L. Bharatam P. V. J. Mol. Model. 2011;17:657–667. doi: 10.1007/s00894-010-0756-y. [DOI] [PubMed] [Google Scholar]
- Bhachoo J. and Beuming T., Modeling Peptide–Protein Interactions, 2017, pp. 235–254 [DOI] [PubMed] [Google Scholar]
- Shivakumar D. Williams J. Wu Y. Damm W. Shelley J. Sherman W. J. Chem. Theory Comput. 2010;6:1509–1519. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- Calandrini V. Dreyer J. Ippoliti E. Carloni P. J. Chem. Phys. 2014;141:1622–1624. doi: 10.1063/1.4901111. [DOI] [PubMed] [Google Scholar]
- Ashraf J. Mughal E. U. Alsantali R. I. Obaid R. J. Sadiq A. Naeem N. Ali A. Massadaq A. Javed Q. Javid A. Bioorg. Med. Chem. 2021;35:116057. doi: 10.1016/j.bmc.2021.116057. [DOI] [PubMed] [Google Scholar]
- Ashraf J. Mughal E. U. Sadiq A. Naeem N. Muhammad S. A. Qousain T. Zafar M. N. Khan B. A. Anees M. J. Mol. Struct. 2020;12:128458. doi: 10.1016/j.molstruc.2020.128458. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.