

Abstract
Transcriptional co-activator with PDZ-binding motif (TAZ), one of core modules of the Hippo pathway, involves inflammatory cell infiltration in the liver, but little information is available regarding its physiological function in the microglia-mediated inflammatory response. Here we revealed that activation of TAZ prevented microglia production of proinflammatory cytokines, indicating TAZ’s importance in anti-inflammation. After translocation into the nucleus, TAZ interacted with transcriptional enhanced associate domain (TEAD) and bound to the promoter of nuclear factor erythroid 2-related factor 2 (Nrf2), whose blockage caused inability of TAZ to improve inflammation, implying that Nrf2 is a direct target of TAZ. Further analysis showed that TAZ induced Nrf2 nuclear translocation to enhance antioxidant capacity with attenuation of oxidative stress and the inflammatory response. Under inflammatory conditions, TAZ impeded mitochondrial dysfunction, as indicated by amelioration of ATP levels, mtDNA copy numbers, and mitochondrial membrane potential with an obvious reduction in mitochondrial superoxide, but this impediment was neutralized by blockage of Nrf2. TAZ hindered opening of the mitochondrial permeability transition pore, restrained release of cytochrome c from mitochondria into the cytosol, and was sufficient to rescue microglia from apoptosis dependent on Nrf2. Nrf2 acted as a downstream target of TAZ to repress NF-κB activation by enhancing antioxidant capacity. Collectively, TAZ might ameliorate the microglia-mediated inflammatory response through the Nrf2-reactive oxygen species (ROS)-nuclear factor κB (NF-κB) pathway.
Keywords: oligonucleotide therapies and applications, TAZ, microglia, inflammation, oxidative stress, mitochondrial function, cell apoptosis, Nrf2-ROS-NF-κB pathway
Graphical abstract
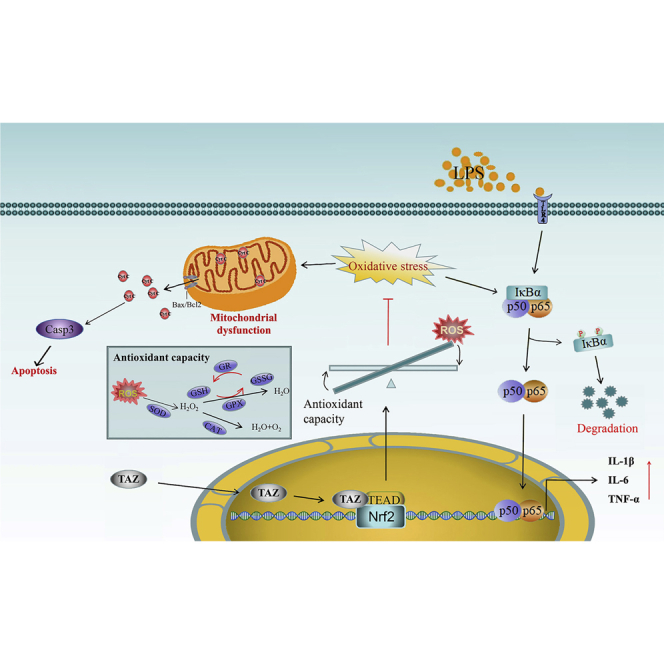
TAZ exhibits novel functions in amelioration of the microglia-mediated inflammatory response by preventing mitochondrial dysfunction and rescuing microglia from apoptosis dependent on the Nrf2-ROS-NF-κB pathway and acts as a promising therapeutical target for multiple neurodegenerative diseases.
Introduction
Microglial cells are the principal macrophages residing in the central nervous system and have significance in the innate immune response, neural development, and plasticity.1, 2, 3 Hyperactivation of microglia results in release of proinflammatory cytokines along with accelerated neuronal injury and is regarded as an inducer of neuroinflammation involved in the pathogenesis of various neurodegenerative diseases.1, 2, 3, 4 Activated microglia drive the production of excessive reactive oxygen species (ROS), which causes neurodegeneration and neuronal death concomitant with oxidative stress.1, 2, 3,5 Although attenuation of microglia-driven neuroinflammation has emerged as a potential therapeutic strategy for numerous neuropsychiatric diseases,3,6 there is a paucity of knowledge regarding its regulatory mechanism.
Transcriptional co-activator with PDZ-binding motif (TAZ), one of core modules of the Hippo pathway, is phosphorylated by large tumor suppressor kinases and retained in the cytoplasm for proteasomal degradation, whereas unphosphorylated TAZ is imported into the nucleus, where it orchestrates downstream gene transcription by cooperating with various DNA-binding transcriptional partners, such as transcriptional enhanced associate domain (TEAD) transcription factors.7,8 Accumulating evidence indicates the importance of TAZ in controlling the proliferation, differentiation, and myelination of Schwann cells and adjusting the development of the neural crest.9,10 TAZ is also involved in migration and invasion of glioma cells.11 Although TAZ deficiency leads to abnormal inflammatory cell infiltration in the liver from nonalcoholic steatohepatitis,12 its physiological function in the microglia-mediated inflammatory response remains unresolved.
Nuclear factor erythroid 2-related factor 2 (Nrf2), a member of the basic leucine zipper transcription factor family, plays a key role in regulation of cellular redox homeostasis by inducing various detoxifying and antioxidant enzymes and has been described as an important contributor to the etiology of neurodegenerative diseases.13,14 Nrf2 deficiency replicates pathological phenotypes of individuals with Alzheimer’s disease concurrent with microglial activation and worsened inflammatory response with an increase in intracellular ROS level, whereas activation of Nrf2 ameliorates the symptoms of Alzheimer’s disease in mice by attenuating oxidative stress and neuroinflammation.15, 16, 17, 18 There is still no clarity regarding the interplay between TAZ and Nrf2 in neuroinflammation.
The present study showed that constitutive activation of TAZ might ameliorate the inflammatory response of microglial cells via the Nrf2-ROS-nuclear factor κB (NF-κB) pathway. TAZ prevented mitochondrial dysfunction and rescued microglia from apoptosis by preventing opening of the mitochondrial permeability transition pore and repressing release of cytochrome c from mitochondria into the cytosol, dependent on Nrf2.
Results
TAZ alleviated the inflammatory response in microglia
To explore the relation between TAZ and inflammation, we first detected its expression after exposure to lipopolysaccharide (LPS) and found that TAZ mRNA and total protein levels as well as nuclear protein exhibited a transient elevation between 1 and 2 h, followed by a decline reaching a nadir by 8 h, whereas its phosphorylation remained more or less steady over different time courses (Figures 1A and 1B). In BV2 microglial cells, sustained activation of TAZ, which obviously enhanced the expression of TAZ mRNA as well as total protein and promoted its nuclear translocation with unaltered phosphorylation levels (Figures 1C and 1D), abrogated up-regulation of the proinflammatory cytokine interleukin-1 beta (IL-1β), IL-6 and tumor necrosis factor alpha (TNF-α) mRNA and impeded release of these cytokines elicited by LPS (Figures 1E and 1F).
Figure 1.

TAZ alleviated the inflammatory response in microglia
(A) TAZ mRNA expression in BV2 microglial cells after treatment with LPS for 1, 2, 4, and 8 h. N = 3. (B) Western blot analysis of TAZ protein after exposure to LPS. N = 3. TAZ, total TAZ protein; p-TAZ, phospho-TAZ protein; nc, nuclear. (C and D) TAZ mRNA and protein expression after introduction of the TAZ overexpression plasmid. N = 3. Con, empty pcDNA3.1 vector; TAZ, TAZ overexpression plasmid. (E) Overexpression of TAZ down-regulated mRNA levels for IL-1β, IL-6, and TNF-α under inflammatory conditions. N = 6. (F) Activation of TAZ impeded release of IL-1β, IL-6, and TNF-α. N = 6. ∗p < 0.05 versus control in one-way ANOVA with Tukey’s post hoc test, #p < 0.05 versus LPS treatment in one-way ANOVA with Tukey’s post hoc test.
TAZ inhibited the inflammatory response by enhancing antioxidant capacity
It is well known that oxidative stress is directly linked with chronic inflammation.19 After exposure to LPS, the oxidative stress marker malondialdehyde (MDA) and 8-hydroxy-desoxyguanosine (8-OHdG) levels were dramatically boosted, whereas constitutive expression of TAZ hindered the occurrence of oxidative stress (Figures 2A and 2B), attributed to ROS overproduction beyond the scavenging capacity of the antioxidant system.20 In the context of inflammation, TAZ might weaken the accumulation of intracellular ROS and superoxide anions (O2−) (Figures 2C and 2D). TAZ strengthened the activities of the antioxidant enzyme superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPX), and glutathione reductase (GR), along with elevation of reduced glutathione (GSH) content and GSH/oxidized GSH (GSSG) ratio (Figures 2E–2H). Supplementation of the corresponding inhibitors of antioxidant enzymes or GSH synthesis prevented the reduction of MDA and 8-OHdG levels by TAZ overexpression and neutralized the rescue of TAZ upon release of the proinflammatory cytokines IL-1β, IL-6, and TNF-α, concomitant with the defective ability of recovering the expression of cytokine mRNA (Figures 2I–2P).
Figure 2.

TAZ inhibited the inflammatory response by enhancing antioxidant capacity
(A and B) Measurement of MDA and 8-OHdG levels after introduction of the TAZ overexpression plasmid and addition of LPS. N = 6. (C and D) Regulation of TAZ overexpression on intracellular ROS and O2− after exposure to LPS. N = 3. (E and F) Effects of TAZ overexpression on the activity of the antioxidant enzymes SOD, CAT, GPX, and GR in the context of LPS. N = 4. (G and H) GSH content and the GSH/GSSG ratio were determined after introduction of the TAZ overexpression plasmid and addition of LPS. N = 4. (I and J) Blockage of antioxidant enzymes and GSH synthesis by the corresponding inhibitors antagonized alleviation of TAZ on MDA and 8-OHdG levels. After introduction of the TAZ overexpression plasmid, MDA and 8-OHdG levels were analyzed in the presence or absence of the SOD inhibitor DDC, the CAT inhibitor 3AT, the GPX inhibitor MSA, the GR inhibitor BCNU, or the GSH synthesis inhibitor BSO in the context of LPS. N = 6. (K–M) Repression of antioxidant enzymes and GSH synthesis neutralized recovery of TAZ on the levels for IL-1β, IL-6, and TNF-α mRNA. N = 6. (N–P) Impediment of antioxidant enzymes and GSH synthesis counteracted rescue of TAZ on the release of IL-1β, IL-6 and TNF-α. N = 6. ∗p < 0.05 versus control in one-way ANOVA with Tukey’s post hoc test, #p < 0.05 versus LPS treatment in one-way ANOVA with Tukey’s post hoc test, &p < 0.05 versus transfection with the TAZ overexpression plasmid followed by addition of LPS in one-way ANOVA with Tukey’s post hoc test.
Nrf2 was a downstream target of TAZ
After translocation into the nucleus, TAZ orchestrates transcription of downstream genes by interaction with TEAD as a transcriptional co-activator.7,8 It has been reported previously that Nrf2 is an important transcription factor in regulating the cellular antioxidant defense and inflammation response.13,14 In BV2 microglial cells, overexpression of TAZ boosted the levels of Nrf2 mRNA and total protein as well as nuclear protein and increased its transcriptional activity, as indicated by elevated luciferase activity after introduction of the pARE-luc plasmid (Figures 3A–3C). Bioinformatics analysis of the Nrf2 promoter showed the existence of a TEAD binding site from −1,129 to −1,124 (Figure 3D). Sustained activation of TAZ enhanced the binding enrichment of TEAD on the promoter region of Nrf2 (Figure 3E). Concurrently, after co-transfection with the TAZ overexpression plasmid and Nrf2 promoter-luciferase reporter vector, luciferase activity exhibited an obvious increase, but mutation of the TEAD binding site caused failure of TAZ to up-regulate luciferase activity (Figure 3F). We next wanted to determine whether Nrf2 was involved in TAZ function in preventing the microglia-mediated inflammatory response. Replenishment of the Nrf2 inhibitor ML385 eliminated the restoration of TAZ overexpression on the proinflammatory cytokines IL-1β, IL-6, and TNF-α (Figures 3G and 3H).
Figure 3.

TAZ exerted anti-inflammation and antioxidant functions by directly targeting Nrf2
(A and B) Regulation of TAZ activation on expression of Nrf2 mRNA and protein. N = 4. (C) TAZ boosted the transcriptional activity of Nrf2. After co-transfection with the TAZ overexpression plasmid and pARE-luc vector, followed by addition of LPS, luciferase activity was measured. N = 5. (D) Schematic showing the TAZ/TEAD binding site and mutation in the Nrf2 promoter region. (E) ChIP analysis revealed the binding enrichment of TAZ/TEAD to the Nrf2 promoter region. N = 3. (F) Luciferase analysis indicating the binding site of TAZ/TEAD to the Nrf2 promoter. N = 3. (G and H) Blockage of Nrf2 by ML385 resisted the restoration of TAZ on the production of IL-1β, IL-6, and TNF-α. N = 4. (I and J) Suppression of Nrf2 antagonized attenuation of TAZ on MDA and 8-OHdG levels. N = 4. (K and L) Impediment of Nrf2 counteracted the improvement of TAZ on intracellular ROS and O2−. N = 3. (M–P) Blockage of Nrf2 antagonized rescue of TAZ on antioxidant enzyme activities, GSH content, and GSH/GSSG ratio. N = 4.
As stated above, TAZ enhanced the antioxidant capacity of BV2 microglial cells along with a reduction of intracellular ROS. We next dissected the requirement of Nrf2 for TAZ antioxidant function. After treatment with the Nrf2 inhibitor ML385, TAZ failed to block the reduction of intracellular ROS and O2− along with enhancement of MDA and 8-OHdG levels and rescue the activity of the antioxidant enzymes SOD, CAT, GPX, and GR (Figures 3I–3N). Simultaneously, ML385 antagonized the recovery of TAZ activation on the content of GSH as well as GSH/GSSG ratio (Figures 3O and 3P).
TAZ protected the mitochondrial function of microglia in the context of inflammation dependent on Nrf2
Mitochondria are necessary organelles for intracellular energy production.21 After exposure to LPS, adenosine triphosphate (ATP) content was distinctly reduced, followed by an increase in mitochondrial O2− levels, whereas overexpression of TAZ resisted the disorder of ATP and mitochondrial O2− levels (Figures 4A and 4B). As described above, TAZ might directly modulate transcription of Nrf2, which has an important role in maintaining mitochondrial function.22 Addition of the Nrf2 inhibitor ML385 counteracted the protective effects of TAZ on ATP content and mitochondrial O2− levels (Figures 4A and 4B). It has been reported previously that mitochondrial membrane potential (MMP) is a reliable indicator of mitochondrial function.21 As seen by flow cytometry analysis, sustained activation of TAZ antagonized the reduction of MMP in the context of inflammation, as evidenced by an elevation in the red/green fluorescence intensity ratio, whereas repression of Nrf2 by ML385 eliminated this antagonism (Figure 4C). To further confirm TAZ’s role in maintaining MMP, we added the fluorescent dye tetramethylrhodamine methyl ester (TMRM), another indicator of MMP. The result shows that TAZ recruited the fluorescence intensity of TMRM in LPS-treated BV2 microglial cells, whereas supplementation of ML385 led to an inability of TAZ to rescue fluorescence intensity (Figure 4D). Further analysis indicated that TAZ might counteract the aberration in mitochondrial DNA (mtDNA) copy number by targeting Nrf2 (Figure 5A).
Figure 4.

TAZ rescued the defects in ATP, mitochondrial O2−, and MMP via Nrf2
(A and B) TAZ resisted the disorder of ATP and mitochondrial O2− levels through Nrf2. After introduction of the TAZ overexpression plasmid, ATP (N = 9) and mitochondrial O2− (N = 3) levels were determined in the absence or presence of the Nrf2 inhibitor ML385 in the context of LPS. (C and D) TAZ improved the defective MMP via Nrf2. After treatment as outlined above, BV2 microglial cells were incubated with the JC-1 fluorescent probe or TMRM to analyze the change in MMP by flow cytometry or confocal microscopy in the context of LPS. N = 4.
Scale bar, 20 μm. Hoechst, Hoechst 33342.
Figure 5.

TAZ rescued the defects in mtDNA copy number, mPTP opening, and cytochrome c release
(A) TAZ counteracted the aberration in mtDNA copy number through Nrf2. After introduction of the TAZ overexpression plasmid, mtDNA copy number was analyzed in the absence or presence of the Nrf2 inhibitor ML385 in the context of LPS. N = 5. (B and C) TAZ prevented opening of the mPTP via Nrf2, as seen by flow cytometry or confocal microscopy. N = 3. (D) TAZ impeded release of cytochrome c from mitochondria into the cytosol via Nrf2. After co-transfection with the TAZ overexpression plasmid and the pCytochrome c-GFP vector, BV2 microglial cells were incubated with TMRM in the absence or presence of the Nrf2 inhibitor ML385 and stained with Hoechst 33342, followed by visualization under confocal microscopy. N = 3. Cyt C, cytochrome c.
The mitochondrial permeability transition pore (mPTP) perfectly reflects the integrity of mitochondrial function.23 After treatment with LPS, the mPTP was opened, as evidenced by the diminished fluorescence intensity of mitochondrial calcein, which was analyzed by flow cytometry and visualized by confocal microscopy (Figures 5B and 5C). Continuous expression of TAZ alleviated opening of the mPTP, whereas addition of the Nrf2 inhibitor ML385 prevented the recovery of TAZ on fluorescence intensity (Figures 5B and 5C). After opening of the mPTP, cytochrome c released from mitochondria into the cytosol.24,25 After transfection with the TAZ overexpression plasmid and replenishment of LPS, release of cytochrome c was blocked, but this blockage was of no avail along with supplementation of ML385 (Figure 5D).
TAZ prevented microglia apoptosis via Nrf2 in the context of inflammation
Under inflammatory conditions, BV2 microglial cells exhibited a higher apoptosis rate, which was measured by flow cytometry and imaged by fluorescence microscopy (Figures 6A and 6B). After introduction of the TAZ overexpression plasmid, cell apoptosis was obviously reduced, whereas administration of the Nrf2 inhibitor ML385 neutralized the defense of TAZ on microglia apoptosis (Figures 6A and 6B). To explore the potential mechanism of TAZ in anti-apoptosis, we examined its regulation of caspase-3 (Casp3), B cell leukemia/lymphoma 2 (Bcl2), and Bcl2-associated X protein (Bax), important modulators of cell apoptosis.21 The results revealed that TAZ attenuated Casp3 activity and repressed expression of Casp3 and Bax mRNA, accompanied by an increase in Bcl2 mRNA levels, whereas addition of ML385 hampered rescue of TAZ on Casp3 and Bax and counteracted the up-regulation of Bcl2 elicited by TAZ overexpression (Figures 6C and 6D).
Figure 6.

TAZ rescued microglia from apoptosis via Nrf2
(A and B) TAZ prevented microglia apoptosis via Nrf2. After introduction of the TAZ overexpression plasmid, cell apoptosis was evaluated in the absence or presence of the Nrf2 inhibitor ML385 by flow cytometry analysis or staining with Hoechst 33342, followed by visualization under a fluorescence microscope. N = 4. (C and D) TAZ attenuated Casp3 activity and reduced the levels of Casp3 and Bax mRNA along with recovery of Bcl2 expression through Nrf2. N = 5.
TAZ repressed NF-κB by enhancing antioxidant capacity dependent on Nrf2
NF-κB is a pivotal modulator of the inflammation response.26,27 After LPS stimulation, phosphorylated IκBα and nuclear NF-κB subunit p65 were notably strengthened, but IκBα protein did not exhibit any obvious change (Figure 7A). Sustained activation of TAZ attenuated up-regulation of IκBα phosphorylation and nuclear p65 levels by LPS, whereas replenishment of the Nrf2 inhibitor ML385 disrupted this attenuation (Figure 7A). To visualize p65 expression in the nucleus, we transfected the EGFP-p65 plasmid, followed by nuclear staining with Hoechst 33342, and found that overexpression of TAZ alleviated the nuclear translocation of p65 elicited by LPS, whereas ML385 neutralized this alleviation (Figure 7B). Further analysis showed that, after introduction of the pNFκB-luc plasmid, which might reflect NF-κB transcriptional activity, TAZ impeded induction of luciferase activity by LPS via Nrf2 (Figure 7C). As mentioned above, TAZ strengthened the antioxidant capacity of BV2 microglial cells. We next wanted to determine whether TAZ might restrain NF-κB by enhancing its antioxidant capacity. Replenishment of the corresponding inhibitors of antioxidant enzymes or GSH synthesis mitigated the resistance of TAZ on IκBα phosphorylation and nuclear p65 levels, hindered the improvement of TAZ on nuclear translocation of p65, and abrogated rescue of TAZ on NF-κB transcriptional activity (Figures 7A–7C).
Figure 7.

TAZ repressed NF-κB by enhancing antioxidant capacity dependent on Nrf2
(A) Western blot analysis of IκBα, phosphorylated IκBα, and p65 expression after transfection with the TAZ overexpression plasmid, followed by addition of the corresponding inhibitor for antioxidant enzymes, GSH synthesis, and Nrf2. p-IκBα, phosphorylated IκBα. N = 3. (B) Visualization of nc p65 after introduction of the TAZ overexpression plasmid and EGFP-p65 vector, followed by nc staining with Hoechst 33342 in the presence or absence of different inhibitors or the NF-κB activator BA. N = 3. (C) Determination of NF-κB transcriptional activity after introduction of the TAZ overexpression plasmid and pNFκB-luc vector in the presence or absence of different inhibitors or the NF-κB activator BA. N = 5. (D) Effects of the NF-κB activator BA on expression of IκBα, phosphorylated IκBα, and p65 after introduction of the TAZ overexpression plasmid. N = 3. (E) Activation of NF-κB by BA resisted the recruitment of TAZ on the levels for IL-1β, IL-6, and TNF-α mRNA. N = 5. (F) Activation of NF-κB by BA counteracted the rescue of TAZ on the release of IL-1β, IL-6 and TNF-α. N = 5.
To dissect whether NF-κB was involved in the defense of TAZ in the microglia-mediated inflammatory response, we guided the TAZ overexpression plasmid into BV2 microglial cells, followed by addition of the NF-κB activator betulinic acid (BA) in the context of inflammation. The results revealed that supplementation of BA, which raised IκBα phosphorylation and nuclear p65 levels along with an obvious accumulation of p65 in the nucleus and heightened NF-κB transcriptional activity, resulted in a failure of TAZ to defend against release of the proinflammatory cytokines IL-1β, IL-6, and TNF-α and antagonized recruitment of TAZ on the above cytokine mRNA levels (Figures 7B–7F).
Discussion
TAZ, one of the core modules of the Hippo pathway, is involved in inflammatory cell infiltration in the liver from nonalcoholic steatohepatitis,12 but little evidence is available regarding its physiological function in the inflammatory response, especially neuroinflammation. The present study found that overexpression of TAZ impeded release of the proinflammatory cytokines IL-1β, IL-6, and TNF-α, followed by a reduction of their transcription levels in LPS-treated BV2 microglial cells, indicating the importance of TAZ in resisting the inflammatory response. It is known that, after shuttling into the nucleus, TAZ interacts with the DNA-binding transcriptional factor TEAD to drive transcription of downstream genes.7,8 In BV2 microglial cells, TAZ enhanced mRNA expression of Nrf2, whose deficiency brought about abnormal microglial activation and an exacerbated inflammatory response, whereas activation of Nrf2 relieved neuroinflammation in Alzheimer’s disease mice.16,17 Further analysis revealed that the Nrf2 promoter region exhibited a TAZ/TEAD binding site whose mutation abrogated binding of TAZ/TEAD to the Nrf2 promoter. Replenishment of the Nrf2 inhibitor ML385 neutralized the improvement of TAZ on the production of proinflammatory cytokines. These data reveal that Nrf2 is a direct target of TAZ in anti-inflammatory function.
Oxidative stress is regarded as a trigger of chronic inflammation, one of the most prevalent hallmarks of several neurodegenerative diseases.19,28 Sustained activation of TAZ hinders the occurrence of oxidative stress, which is attributed to ROS generation in excess of the scavenging capacity of the antioxidant system.20 TAZ might reduce the accumulation of intracellular ROS, whose removal by dendrimer-N-acetyl-L-cysteine attenuated neuroinflammation followed by an improvement of oxidative stress and neuronal injury in a rabbit model of cerebral palsy.29 The defective antioxidant capacity persists under conditions of inflammation. Dietary antioxidant intake reduces the incidence of multiple inflammatory diseases.30 In BV2 microglial cells, TAZ enhances the activity of the antioxidant enzyme SOD, which catalyzes dismutation of O2− to hydrogen peroxide (H2O2) and oxygen.24,31 Mutation of SOD1 promotes neuroinflammation, along with motor neuron death and microglial hyperactivation.32 Repression of SOD by diethyldithiocarbamate (DDC) antagonized the defense of TAZ on the inflammatory response. Further analysis showed that TAZ increased the content of intracellular GSH, a non-enzymatic antioxidant, and its supplementation decreased inflammation.19,33 GPX might oxidize GSH and cause GSSG as a by-product of H2O2 to water, whereas GR converted GSSG back to GSH.24,31 TAZ also increased the activity of the antioxidant enzymes GPX and GR in the context of inflammation. Blockage of GPX, GR, and GSH synthesis by the corresponding inhibitors counteracted the defense of TAZ on microglia-mediated inflammation. TAZ exerts its anti-inflammatory function via CAT, an important antioxidant enzyme in decomposing H2O2 to water.31 These observations suggest that TAZ may alleviate the inflammatory response by reducing intracellular ROS and enhancing antioxidant capacity. It has been reported previously that, after translocation into the nucleus, Nrf2 binds to an antioxidant-responsive element in the regulatory region to drive transcription of the gene encoding the antioxidant enzymes SOD, CAT, GPX, and GR as well as the antioxidant GSH.31,34 In BV2 microglial cells, TAZ promoted nuclear translocation of Nrf2 and strengthened its transcriptional activity. Inhibition of Nrf2 by ML385 resisted rescue of TAZ on antioxidant capacity and disrupted the improvement of intracellular ROS by TAZ with elevated oxidative stress, indicating a Nrf2 requirement for TAZ antioxidant function.
The mitochondrion is a necessary organelle for intracellular energy production, and its dysfunction increases cellular susceptibility to proinflammatory mediators, resulting in exacerbation of inflammatory responsiveness.30,31 Under the condition of inflammation, TAZ ameliorated the defect of ATP level, an important indicator of mitochondrial function.21 Because of the lack of protective histones, mtDNA is prone to oxidative injury, leading to excessive mitochondrial ROS generation followed by mitochondrial membrane depolarization.25,31,35 Sustained activation of TAZ might reduce the accumulation of mitochondrial O2− and antagonize the reduction of MMP along with an improvement of mtDNA copy number by targeting Nrf2. Further analysis demonstrated that mitochondrial impairment led to opening of the mPTP, which might trigger release of cytochrome c from mitochondria into the cytosol.24,25 Bax opened the mPTP and promoted release of cytochrome c, whereas overexpression of Bcl2 exhibited the contrary, with an obvious improvement of mitochondrial dysfunction and inflammatory response.36, 37, 38 In BV2 microglial cells, activation of TAZ impeded opening of the mPTP and alleviated release of cytochrome c by repression of Bax and induction of Bcl2 dependent on Nrf2. Following delivery of cytochrome c, the caspase cascade was activated and subsequently facilitated cell death.38 TAZ prevented microglia apoptosis by restraining Casp3 activity, but this was abrogated by the Nrf2 inhibitor ML385. This shows that TAZ may ameliorate the microglia-mediated inflammatory response by hindering mitochondrial dysfunction and cellular apoptosis dependent on Nrf2.
NF-κB is a pivotal modulator of the inflammation response.26,27 In the context of inflammation, TAZ obstructed phosphorylation of IκBα, which underwent ubiquitylation and proteasomal degradation, resulting in release of the NF-κB subunit p65 and then migration into the nucleus to initiate transcription of the proinflammatory cytokines IL-1β, IL-6, and TNF-α by binding to specific DNA sequences of the promoter region.34,39 In BV2 microglial cells, TAZ might alleviate nuclear translocation of p65 and suppress the transcriptional activity of NF-κB, whose activation by BA disturbed the defense of TAZ on the inflammatory response, implying that NF-κB is a downstream target of TAZ in anti-inflammatory function. Further analysis showed that Nrf2 deficiency in mouse embryonic fibroblasts led to activation of NF-κB.40 Blockage of Nrf2 by ML385 hampered regulation of TAZ on NF-κB, suggesting Nrf2’s importance in the cross-talk between TAZ and NF-κB. Mutation of SOD1 caused activation of NF-κB, whose repression rescued motor neurons from death by improving proinflammatory microglial activation in SOD1-G93A transgenic mice.41 Repression of SOD by DDC disrupted recruitment of TAZ on NF-κB. Supplementation of the corresponding inhibitors for CAT, GPX, GR, or GSH synthesis neutralized recovery of NF-κB by TAZ activation. These data demonstrate that TAZ may restrain activation of NF-κB by strengthening antioxidant capacity.
In conclusion, TAZ might ameliorate the microglia-mediated inflammatory response. After translocation into the nucleus, TAZ initiated transcription of Nrf2 by interacting with TEAD and induced nuclear translocation of Nrf2 to enhance antioxidant capacity along with a reduction of intracellular ROS, resulting in improvement of mitochondrial function, rescue of microglia from apoptosis, and prevention of NF-κB activation (Figure 8).
Figure 8.

Schematic of TAZ regulation in the inflammatory response
After shuttling into the nucleus, TAZ might drive transcription of Nrf2 by interacting with the TEAD transcription factor and induce nc translocation of Nrf2 to enhance antioxidant capacity with reduction of intracellular ROS, resulting in impediment of NF-κB activation to ameliorate the microglia-mediated inflammatory response. Simultaneously, TAZ rescued microglia from apoptosis by preventing mitochondrial dysfunction, followed by blockage of mPTP opening and Cyt C release from mitochondria into the cytosol.
Materials and methods
Cell culture
BV2 microglial cells were kindly provided by Prof. Shou-Peng Fu (Jilin University, China) and then cultured in DMEM/high-glucose medium supplemented with 10% fetal bovine serum for further treatment.
Plasmid construction and transfection
Full-length cDNA fragments for the TAZ variants TAZa and TAZb were amplified by the corresponding primer (Table 1) and inserted into the pGEM-T vector. After enzyme digestion, fragments were ligated into pcDNA3.1 to construct the TAZ overexpression plasmid. After introduction of the TAZ overexpression plasmid, BV2 microglial cells were treated with LPS (1 μg/mL, Sigma) for 4 or 12 h in the presence or absence of the SOD inhibitor DDC (10 mM, Sigma), the CAT inhibitor 3-amino-1,2,4 triazole (3AT; 10 mM, Sigma), the GPX inhibitor mercaptosuccinic acid (MSA; 50 μM, Sigma), the GR inhibitor 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU; 50 μM, Sigma), the GSH synthesis inhibitor buthionine sulfoximine (BSO; 100 μM, Sigma), the Nrf2 inhibitor ML385 (MedChemExpress; 10 μΜ), and the NF-κB activator BA (10 μM, Selleck).
Table 1.
Primers used in this study
| Gene | Sequence of forward primer | Sequence of reverse primer | Application |
|---|---|---|---|
| TAZ | 5′-CGACTCAGAACCAACCCACA-3′ | 5′-CATGAGCTCCTCTTGACGCA-3′ | real-time PCR |
| IL-1β | 5′-GTTCCCATTAGACAACTGCACTACAG-3′ | 5′-GTCGTTGCTTGGTTCTCCTTGTA-3′ | real-time PCR |
| IL-6 | 5′-CCAGAAACCGCTATGAAGTTCC-3′ | 5′-GTTGGGAGTGGTATCCTCTGTGA-3′ | real-time PCR |
| TNF-α | 5′-GCAACTGCTGCACGAAATC-3′ | 5′-CTGCTTGTCCTCTGCCCAC-3′ | real-time PCR |
| Nrf2 | 5′-CCCAGCACATCCAGACAGAC-3′ | 5′-TATCCAGGGCAAGCGACTCA-3′ | real-time PCR |
| Bcl2 | 5′-TCAGAGCGAGAAGGTAGGGA-3′ | 5′-CTGTGGGGTAACAAGAAGGTC-3′ | real-time PCR |
| Bax | 5′-CCGGCGAATTGGAGATGAACT-3′ | 5′-CCAGCCCATGATGGTTCTGAT-3′ | real-time PCR |
| Casp3 | 5′-CTGGACTGTGGCATTGAGAC-3′ | 5′-GCAAAGGGACTGGATGAACC-3′ | real-time PCR |
| Gapdh | 5′-GCCTTCCGTGTTCCTACCC-3′ | 5′-TGCCTGCTTCACCACCTTC-3′ | real-time PCR |
| TAZa | 5′-GATATCTGGCTCCGGCCAGGAGGTCAT-3′ | 5′-CTCGAGGTGATTACAGCCAGGTTAGA-3′ | overexpression |
| TAZb | 5′-GATATCCCGAGTCCCCAGAAAGATG-3′ | ||
| mtDNA | 5′-CCTATCACCCTTGCCATCAT-3′ | 5′-GAGGCTGTTGCTTGTGTGAC-3′ | real-time PCR |
| ncDNA | 5′-ATGGAAAGCCTGCCATCATG-3′ | 5′-TCCTTGTTGTTCAGCATCAC-3′ | real-time PCR |
| Nrf2 | 5′-AAGCTTGGTTTTCAACTATTAACACAT-3′ | 5′-CTCGAGGATGCTTGGGGTGGCTCTCC-3′ | promoter analysis |
| Nrf2 | 5′-TCGGGGGCAGTTAAAGAAGTAT-3′ | 5′-GATGCTTGGGGTGGCTCTC-3′ | ChIP |
ELISA
After introduction of the TAZ overexpression plasmid followed by treatment with LPS for 12 h in the presence or absence of the abovementioned inhibitors and activators, supernatants were collected to determine the secretion of IL-1β, IL-6, and TNF-α protein using an ELISA kit (BioLegend). Briefly, 96-well plates were coated with diluted capture antibody, blocked with 1% BSA, and then samples were added. After incubation with a biotinylated detection antibody against IL-1β, IL-6, or TNF-α, Avidin-horseradish peroxidase (HRP) solution was supplemented in each well. After replenishment of TMB substrate solution, absorbance was read at 450 nm, and protein concentration was determined in accordance with a standard curve.
Real-time PCR
After total RNA extraction with TRIPURE reagent followed by synthesis of cDNA, the expression levels of different genes were assessed by real-time PCR analysis as described previously.37 The corresponding primers are listed in Table 1.
Western blotting
After total and nuclear proteins were isolated and quantified using the kit, western blotting was performed with primary antibodies against TAZ (1:1,000, Thermo Fisher Scientific), phospho-TAZ (Ser89, 1:1,000, Cell Signaling Technology), Nrf2 (1:1,000, Proteintech), histone H3 (1:5,000, Proteintech), Gapdh (1:5,000, Proteintech), IκBα (1:1,000, Proteintech), phospho-IκBα (1:1,000, Cell Signaling Technology), and NF-κB subunit p65 (1:1,000, Cell Signaling Technology), as described previously.42
Measurement of antioxidant enzyme activity and GSH content
After different treatments, proteins were extracted to assess the activity of the antioxidant enzymes SOD, CAT, GPX, and GR and resolve GSH content as well as the GSH/GSSG ratio in accordance with the corresponding assay kit (Beyotime). After samples were mixed with WST-8/enzyme solution and then added reaction-started working solution, absorbance was calculated at 450 nm to determine SOD activity using the corresponding formula. For CAT activity, samples were blended with 5 mM hydrogen peroxide for 5 min, followed by addition of substrate solution, and absorbance was measured at 520 nm to estimate CAT activity basing on a standard curve. For GPX activity, samples were incubated with working solution for 15 min, followed by addition of 3 mM Cum-OOH, and absorbance was measured at 340 nm to analyze GPX activity according to the corresponding formula. For GR activity, samples were mixed with GSSG and NADPH solution concomitant with supplementation of DTNB substrate solution, and absorbance was evaluated at 412 nm to determine GR activity according to the corresponding formula. Additionally, samples were incubated with working solution containing GR and DTNB for 5 min, followed by addition of NADPH solution, and absorbance was read at 412 nm every 5 min for a total of 25 min to calculate GSH and GSSG content according to a standard curve.
Determination of intracellular and mitochondrial ROS
After various treatments, BV2 microglial cells were incubated with the fluorescent probe DCFH-DA (10 μM, Beyotime), dihydroethidium (5 μM, Beyotime), or MitoSOX Red mitochondrial superoxide indicator (5 μM, Invitrogen) and then analyzed by flow cytometry to determine the levels of intracellular ROS, O2−, and mitochondrial O2−.
Measurement of MMP
After incubation with the JC-1 fluorescent probe (Beyotime), BV2 microglial cells were analyzed by flow cytometry to determine MMP change by monitoring the ratio of red to green fluorescence. To visualize MMP change, BV2 microglial cells were incubated with TMRM (1:1,000, Thermo Fisher Scientific) and then stained with Hoechst 33342. Images from 20 random fields in a confocal dish were captured by an observer blinded to the experimental condition under an Olympus FluoView FV1000 confocal microscope using a ×60 oil immersion objective.
Measurement of MDA, 8-OHdG, and ATP content
After treatment as outlined above, proteins were extracted to evaluate the content of MDA, 8-OHdG, and ATP using the corresponding kit. After samples were mixed with thiobarbituric acid solution (Beyotime) and then heated for 15 min, absorbance was read at 532 nm to assess MDA content based on standard curve. For 8-OHdG measurement (Cusabio), samples were added to microtiter plate wells and then incubated with an HRP-conjugated antibody for 30 min. After addition of TMB substrate solution, absorbance was read at 450 nm to determine 8-OHdG content in accordance with a standard curve. Additionally, samples were blended with detection solution (Beyotime), and luminescence was measured to calculate intracellular ATP levels according to a standard curve.
Determination of mtDNA copy number
After treatment as outlined above, total DNA was extracted and then used to perform real-time PCR. mtDNA copy number was determined by analyzing the ratio of mtDNA/nuclear DNA (ncDNA) as described previously.43
Opening of mPTP
After incubation with Calcein AM (Beyotime) followed by addition of the fluorescence quencher cobalt chloride, BV2 microglial cells were analyzed by flow cytometry to determine opening of the mPTP. Simultaneously, cells were stained with Hoechst 33342, and images were randomly captured under a confocal microscopy equipped with a ×60 oil immersion objective to visualize the change of the mPTP.
Assessment of cytochrome c release
After introduction of the TAZ overexpression plasmid and the pCytochrome C-GFP vector (Addgene), followed by addition of LPS in the presence or absence of the Nrf2 inhibitor ML385, BV2 microglial cells were incubated with TMRM and stained with Hoechst 33342. Images were randomly captured under a confocal microscopy equipped with a ×100 oil immersion objective.
Measurement of cell apoptosis
After treatment as outlined above, cell apoptosis was evaluated by flow cytometry and fluorescence microscope using an Annexin V- fluorescein isothiocyanate (FITC) apoptosis detection kit (Beyotime). Briefly, after incubation with Annexin V-FITC and propidium iodide (PI), BV2 microglial cells were analyzed by flow cytometry or stained with Hoechst 33342, followed by visualization under a Nikon 80i fluorescence microscope equipped with a ×20 objective. Cells were lysed and mixed with Ac-DEVD-pNA substrate solution (Beyotime). Absorbance was read at 405 nm to determine Casp3 activity based on a standard curve.
Chromatin immunoprecipitation (ChIP)
After introduction of the TAZ overexpression plasmid and addition of LPS, a ChIP assay was performed using the corresponding kit (Beyotime). Briefly, cells were cross-linked with 1% formaldehyde, and chromatin from lysed nuclei was fragmented by sonication. Sheared DNA was precipitated with an antibody against TAZ or control immunoglobulin G (IgG), followed by incubation with protein A/G beads. Then, enriched chromatin DNA was purified and subjected to real-time PCR using the specific primer (Table 1).
Dual luciferase analysis
The Nrf2 promoter sequence (−1,238 to −1,023) contained TEAD binding site was amplified by the specific primer (Table 1) and corresponding segment included mutant site was synthesized by Sangon Biotech. After enzyme digestion, the fragment was inserted into the pGL6 vector. After co-transfection with the TAZ overexpression plasmid and the pGL6-Nrf2 or pGL6-Nrf2-mutant vector, followed by addition of LPS, luciferase activity was measured using a dual luciferase reporter gene assay kit (Beyotime). To determine the transcriptional activity of Nrf2 and NF-κB, the pARE-luc or pNFκB-luc plasmid (Beyotime) was co-transfected with the TAZ overexpression vector. The pRL-SV40 plasmid (Beyotime) was used for data normalization.
Visualization of nuclear p65 location
After introduction of the TAZ overexpression plasmid and EGFP-p65 vector (Addgene), followed by addition of LPS in the presence or absence of the corresponding inhibitor or the NF-κB activator BA, BV2 microglial cells were stained with Hoechst 33342. Images were randomly captured under a confocal microscopy equipped with a ×100 oil immersion objective.
Statistical analysis
All tests were independently repeated at least three times. The significance of statistical difference data was analyzed by one-way ANOVA with Tukey’s post hoc test using SPSS 11.0 software. Data are shown as means ± SEM. p < 0.05 was considered statistically significant.
Acknowledgments
We thank Prof. Shou-Peng Fu for generously providing BV2 microglial cells. This work was financially supported by the National Key Research and Development Program of China Stem Cell and Translational Research (2017YFA0105101) and the National Natural Science Foundation of China (31873003 and 31472158).
Author contributions
J.-C.H. and Z.-P.Y. performed the experiments and analyzed the data. J.-C.H., Z.-P.Y., and B.G. wrote and edited the manuscript. H.-F.Y., Z.-Q.Y., and Y.-S.W. provided technical assistance. B.G. designed and conducted the research. All authors read and approved the final manuscript.
Declaration of interest
The authors declare no competing interests.
Data and code availability
The datasets generated during the current study are available from the corresponding author upon reasonable request.
References
- 1.Wolf S.A., Boddeke H.W., Kettenmann H. Microglia in physiology and disease. Annu. Rev. Physiol. 2017;79:619–643. doi: 10.1146/annurev-physiol-022516-034406. [DOI] [PubMed] [Google Scholar]
- 2.Voet S., Prinz M., van-Loo G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol. Med. 2019;25:112–123. doi: 10.1016/j.molmed.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Prinz M., Jung S., Priller J. Microglia biology: one century of evolving concepts. Cell. 2019;179:292–311. doi: 10.1016/j.cell.2019.08.053. [DOI] [PubMed] [Google Scholar]
- 4.Madore C., Yin Z., Leibowitz J., Butovsky O. Microglia, lifestyle stress, and neurodegeneration. Immunity. 2020;52:222–240. doi: 10.1016/j.immuni.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheignon C., Tomas M., Bonnefont-Rousselot D., Faller P., Hureau C., Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 2018;14:450–464. doi: 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta N., Shyamasundar S., Patnala R., Karthikeyan A., Arumugam T.V., Ling E.A., Dheen S.T. Recent progress in therapeutic strategies for microglia-mediated neuroinflammation in neuropathologies. Expert Opin. Ther. Targets. 2018;22:765–781. doi: 10.1080/14728222.2018.1515917. [DOI] [PubMed] [Google Scholar]
- 7.Dey A., Varelas X., Guan K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020;19:480–494. doi: 10.1038/s41573-020-0070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rausch V., Hansen C.G. The Hippo pathway, YAP/TAZ, and the plasma membrane. Trends Cell Biol. 2020;30:32–48. doi: 10.1016/j.tcb.2019.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Deng Y., Wu L.M.N., Bai S., Zhao C., Wang H., Wang J., Xu L., Sakabe M., Zhou W., Xin M., et al. A reciprocal regulatory loop between TAZ/YAP and G-protein Galphas regulates Schwann cell proliferation and myelination. Nat. Commun. 2017;8:15161. doi: 10.1038/ncomms15161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J., Xiao Y., Hsu C.W., Martinez-Traverso I.M., Zhang M., Bai Y., Ishii M., Maxson R.E., Olson E.N., Dickinson M.E., et al. Yap and Taz play a crucial role in neural crest-derived craniofacial development. Development. 2016;143:504–515. doi: 10.1242/dev.126920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ji J., Xu R., Zhang X., Han M., Xu Y., Wei Y., Ding K., Wang S., Huang B., Chen A., et al. Actin like-6A promotes glioma progression through stabilization of transcriptional regulators YAP/TAZ. Cell Death Dis. 2018;9:517. doi: 10.1038/s41419-018-0548-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X., Zheng Z., Caviglia J.M., Corey K.E., Herfel T.M., Cai B., Masia R., Chung R.T., Lefkowitch J.H., Schwabe R.F., et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2016;24:848–862. doi: 10.1016/j.cmet.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osama A., Zhang J., Yao J., Yao X., Fang J. Nrf2: a dark horse in Alzheimer's disease treatment ageing. Res. Rev. 2020;64:101206. doi: 10.1016/j.arr.2020.101206. [DOI] [PubMed] [Google Scholar]
- 14.Cuadrado A., Rojo A.I., Wells G., Hayes J.D., Cousin S.P., Rumsey W.L., Attucks O.C., Franklin S., Levonen A.L., Kensler T.W., et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019;18:295–317. doi: 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- 15.Zweig J.A., Caruso M., Brandes M.S., Gray N.E. Loss of NRF2 leads to impaired mitochondrial function, decreased synaptic density and exacerbated age-related cognitive deficits. Exp. Gerontol. 2020;131:110767. doi: 10.1016/j.exger.2019.110767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rojo A.I., Pajares M., García-Yagüe A.J., Buendia I., Van-Leuven F., Yamamoto M., López M.G., Cuadrado A. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. 2018;18:173–180. doi: 10.1016/j.redox.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rojo A.I., Pajares M., Rada P., Nuñez A., Nevado-Holgado A.J., Killik R., van-Leuven F., Ribe E., Lovestone S., Yamamoto M., et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer's patients and worsens APP and TAU pathology. Redox Biol. 2017;13:444–451. doi: 10.1016/j.redox.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uruno A., Matsumaru D., Ryoke R., Saito R., Kadoguchi S., Saigusa D., Saito T., Saido T.C., Kawashima R., Yamamoto M. Nrf2 suppresses oxidative stress and inflammation in App knock-in Alzheimer's disease model mice. Mol. Cell. Biol. 2020;40:e00467–e00519. doi: 10.1128/MCB.00467-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hussain T., Tan B., Yin Y., Blachier F., Tossou M.C., Rahu N. Oxidative stress and inflammation: what polyphenols can do for us? Oxid. Med. Cell Longev. 2016;2016:7432797. doi: 10.1155/2016/7432797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schieber M., Chandel N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu H.F., Duan C.C., Yang Z.Q., Wang Y.S., Yue Z.P., Guo B. HB-EGF ameliorates oxidative stress-mediated uterine decidualization damage. Oxid. Med. Cell Longev. 2019;2019:6170936. doi: 10.1155/2019/6170936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bento-Pereira C., Dinkova-Kostova A.T. Activation of transcription factor Nrf2 to counteract mitochondrial dysfunction in Parkinson's disease. Med. Res. Rev. 2021;41:785–802. doi: 10.1002/med.21714. [DOI] [PubMed] [Google Scholar]
- 23.Forbes J.M., Thorburn D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018;14:291–312. doi: 10.1038/nrneph.2018.9. [DOI] [PubMed] [Google Scholar]
- 24.Sabharwal S.S., Schumacker P.T. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat. Rev. Cancer. 2014;14:709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zorov D.B., Juhaszova M., Sollott S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Catrysse L., van-Loo G. Inflammation and the metabolic syndrome: the tissue-specific functions of NF-kB. Trends. Cell. Biol. 2017;27:417–429. doi: 10.1016/j.tcb.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 27.Sun S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017;17:545–558. doi: 10.1038/nri.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang Y., Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016;53:1181–1194. doi: 10.1007/s12035-014-9070-5. [DOI] [PubMed] [Google Scholar]
- 29.Kannan S., Dai H., Navath R.S., Balakrishnan B., Jyoti A., Janisse J., Romero R., Kannan R.M. Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Sci. Transl. Med. 2012;4:130ra46. doi: 10.1126/scitranslmed.3003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.López-Armada M.J., Riveiro-Naveira R.R., Vaamonde-García C., Valcárcel-Ares M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion. 2013;13:106–118. doi: 10.1016/j.mito.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Bhargava P., Schnellmann R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017;13:629–646. doi: 10.1038/nrneph.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Philips T., Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 2011;10:253–263. doi: 10.1016/S1474-4422(11)70015-1. [DOI] [PubMed] [Google Scholar]
- 33.Mittal M., Siddiqui M.R., Tran K., Reddy S.P., Malik A.B. Reactive oxygen species in inflammation and tissue injury. Antioxidants Redox Signal. 2014;20:1126–1167. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sivandzade F., Prasad S., Bhalerao A., Cucullo L. NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019;21:101059. doi: 10.1016/j.redox.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chow J., Rahman J., Achermann J.C., Dattani M.T., Rahman S. Mitochondrial disease and endocrine dysfunction. Nat. Rev. Endocrinol. 2017;13:92–104. doi: 10.1038/nrendo.2016.151. [DOI] [PubMed] [Google Scholar]
- 36.Shimada K., Crother T.R., Karlin J., Dagvadorj J., Chiba N., Chen S., Ramanujan V.K., Wolf A.J., Vergnes L., Ojcius D.M., et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Autret A., Martin S.J. Bcl-2 family proteins and mitochondrial fission/fusion dynamics. Cell Mol. Life Sci. 2010;67:1599–1606. doi: 10.1007/s00018-010-0286-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bock F.J., Tait S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020;21:85–100. doi: 10.1038/s41580-019-0173-8. [DOI] [PubMed] [Google Scholar]
- 39.Afonina I.S., Zhong Z., Karin M., Beyaert R. Limiting inflammation-the negative regulation of NF-қB and the NLRP3 inflammasome. Nat. Immunol. 2017;18:861–869. doi: 10.1038/ni.3772. [DOI] [PubMed] [Google Scholar]
- 40.Thimmulappa R.K., Lee H., Rangasamy T., Reddy S.P., Yamamoto M., Kensler T.W., Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frakes A.E., Ferraiuolo L., Haidet-Phillips A.M., Schmelzer L., Braun L., Miranda C.J., Ladner K.J., Bevan A.K., Foust K.D., Godbout J.P., et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81:1009–1023. doi: 10.1016/j.neuron.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu H.F., Duan C.C., Yang Z.Q., Wang Y.S., Yue Z.P., Guo B. Malic enzyme 1 is important for uterine decidualization in response to progesterone/cAMP/PKA/HB-EGF pathway. FASEB J. 2020;34:3820–3837. doi: 10.1096/fj.201902289R. [DOI] [PubMed] [Google Scholar]
- 43.Chen H., Vermulst M., Wang Y.E., Chomyn A., Prolla T.A., McCaffery J.M., Chan D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during the current study are available from the corresponding author upon reasonable request.