

Abstract
Although it is known that the expression and activity of sirtuin 1 (SIRT1) significantly decrease in doxorubicin (DOX)-induced cardiomyopathy, the role of interaction between SIRT1 and sestrin 2 (SESN2) is largely unknown. In this study, we investigated whether SESN2 could be a crucial target of SIRT1 and the effect of their regulatory interaction and mechanism on DOX-induced cardiac injury. Here, using DOX-treated cardiomyocytes and cardiac-specific Sirt1 knockout mice models, we found SIRT1 deficiency aggravated DOX-induced cardiac structural abnormalities and dysfunction, whereas the activation of SIRT1 by resveratrol (RES) treatment or SIRT1 overexpression possessed cardiac protective effects. Further studies indicated that SIRT1 exerted these beneficial effects by markedly attenuating DOX-induced oxidative damage and apoptosis in a SESN2-dependent manner. Knockdown of Sesn2 impaired RES/SIRT1-mediated protective effects, while upregulation of SESN2 efficiently rescued DOX-induced oxidative damage and apoptosis. Most importantly, SIRT1 activation could reduce DOX-induced SESN2 ubiquitination possibly through reducing the interaction of SESN2 with mouse double minute 2 (MDM2). The recovery of SESN2 stability in DOX-impaired primary cardiomyocytes by SIRT1 was confirmed by Mdm2-siRNA transfection. Taken together, our findings indicate that disrupting the interaction between SESN2 and MDM2 by SIRT1 to reduce the ubiquitination of SESN2 is a novel regulatory mechanism for protecting hearts from DOX-induced cardiotoxicity and suggest that the activation of SIRT1-SESN2 axis has potential as a therapeutic approach to prevent DOX-induced cardiotoxicity.
Keywords: Doxorubicin, Cardiotoxicity, SIRT1, SESN2, Oxidative stress, Apoptosis
Graphical abstract
Highlights
-
•
DOX-induced cardiotoxicity is based on decreased SIRT1 and SESN2 levels.
-
•
SIRT1 activation improves DOX-induced cardiac oxidative stress and apoptosis.
-
•
The benefits of SIRT1 in DOX-impaired cardiac function require the activation of SESN2.
-
•
SIRT1 reduces ubiquitination and degradation of SESN2 via MDM2.
1. Introduction
Doxorubicin (DOX), an anthracycline chemotherapeutic agent, remains a widely used for the treatment of a variety of cancers owing to its potent therapeutic effects. However, its clinical application is largely compromised by cumulative and dose-dependent cardiotoxicity, which may ultimately lead to irreversible structural changes in the myocardium and progressive heart failure [1]. The pathogenesis of DOX-induced cardiotoxicity is complex and not clearly elucidated, including mitochondrial dysfunction, fibrosis, oxidative stress, endoplasmic reticulum stress, inflammatory response, apoptosis and dysregulation of autophagy [[2], [3], [4], [5], [6]]. Thus, it is highly important to clarify the mechanisms of DOX-induced cardiotoxicity for developing effective treatment strategies. Of particular interest, oxidative stress and myocardial cell apoptosis have been verified to be critical contributors to the development of DOX-induced cardiotoxicity [7]. Therefore, maintaining cardiac redox homeostasis and attenuating cell apoptosis are promising therapeutic strategies for DOX-induced cardiotoxicity.
Sirtuin 1 (SIRT1) is a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase that governs multiple cellular fates such as cardiomyocyte survival, metabolic homeostasis and senescence [[8], [9], [10]]. A clinical study indicated that cardiac SIRT1 was downregulated (54.92% ± 7.80% in advanced heart failure samples compared with healthy control hearts), which may be a contributing factor in the increase of proapoptotic molecules and decrease of antioxidants [11]. Moreover, increasing studies proved that the activation of SIRT1 prevented oxidative damage and controlled cardiomyocyte apoptosis in response to DOX [7,12]. AMP-activated protein kinase (AMPK) is a classical and key downstream target of the SIRT1 signaling, which exerts a beneficial role in the treatment of cardiac diseases, such as myocardial ischemia/reperfusion injury and angiotensin II-induced cardiac hypertrophy and dysfunction [13,14]. Our previous study demonstrated that DOX stimulation indeed decreased phosphorylation of AMPK, and when SIRT1 was activated by fibroblast growth factor 21, the AMPK phosphorylation was largely enhanced to protect against DOX-induced cardiotoxicity through reducing liver kinase B1 acetylation [7]. Recently, sestrin 2 (SESN2), a conserved stress-induced protein, has been proved to be a positive regulator of AMPK activation in the heart and decreased in both human hearts with DOX-induced chronic cardiomyopathy and dilated cardiomyopathy [15,16]. Wang et al. found that SESN2 inhibition was a primary contributor to DOX-triggered cardiomyocyte apoptosis, cardiac mitophagy and mitochondrial dysfunction, and the overexpression of SESN2 could prevent DOX-induced cardiotoxicity through improving mitochondrial function and rescuing mitophagy [17]. In contrast, genetic ablation of Sesn1 and Sesn2 could aggravate DOX-induced cardiac pathologies, which was associated with the suppression of the AMPK signaling [18]. However, whether SESN2 mediates the antioxidative and anti-apoptotic effects of SIRT1 on AMPK activation in cardiomyocytes has not been identified.
Therefore, the aims of this study were (i) to determine whether SESN2, as a scaffold protein, plays a key role in SIRT1/AMPK-improved DOX-induced cardiotoxicity through inhibiting oxidative stress and apoptosis; (ii) if so, to study the likely mechanism by which SIRT1 regulates SESN2 activation in cardiomyocytes. By using transgenic mice with a cardiac-specific knockout of Sirt1 gene in combination with primary cardiomyocytes and H9c2 cells treated with SIRT1 and AMPK activators (resveratrol (RES), AICAR), and/or transfected with Sirt1-short hairpin RNA (Sirt1-shRNA), pcDNA3.1-Sirt1, Sesn2-small interfering RNA (Sesn2-siRNA), pCMV6-Sesn2, Ampk-shRNA and mouse double minute 2 (Mdm2)-siRNA, we have discovered for the first time that the activation of SIRT1 prevented DOX-induced cardiac oxidative stress and apoptosis in a SESN2-dependent manner. The mechanistic study further revealed that SIRT1 might decrease the recruitment of MDM2 to SESN2, thereby reducing its ubiquitin-dependent proteasomal degradation.
2. Materials and methods
2.1. Animals and treatments
Male cardiac-specific Sirt1 knockout (Sirt1flox/flox Myh6-creEsr1, Sirt1-CKO) mice on the C57BL/6J background were generated by crossing Sirt1flox/flox mice with Myh6-creEsr1 mice, followed by 1-week tamoxifen administration. Male Sirt1-CKO mice aged 8–10 weeks were applied to the study at 2 weeks after the last administration of tamoxifen. Age-matched male littermates (Sirt1flox/flox) were served as controls. All mice were raised at 22 °C with a 12:12-h light/dark cycle with free access to rodent standard diet and tap water. All experimental procedures for animal studies were approved by the Institutional Animal Care and Use Committee of Shandong University (permit number: 2020-D-07). Animals were acclimatized for 2 weeks before experimentation. For animal study, Sirt1-CKO mice and Sirt1flox/flox mice were randomly divided into four groups (six mice per group), respectively: (i) Control group (Ctrl); (ii) RES (MedChemExpress, NJ, USA) treatment group (RES); (iii) DOX (MedChemExpress) treatment group (DOX); (iv) DOX and RES co-treatment group (DOX/RES). DOX was intraperitoneally injected once a week with a dose of 5 mg/kg or the same volume of vehicle (saline) was conducted for 4 weeks (20 mg/kg cumulative dose of DOX). RES (10 mg/kg/day) or the same volume of vehicle (saline) was intraperitoneally injected every day for 5 weeks. The dosages of DOX and RES were chosen based on the previous studies [6,7,19,20]. There was no infection and mortality during medication administration. At the end of the experiments, all mice were anaesthetized by an intraperitoneal injection of sodium pentobarbital (50 mg/kg), and the effect of anesthesia was verified by the lack of a toe pinch withdrawal response. Then, mice were euthanized by exsanguination, and hearts were harvested for subsequent analyses.
2.2. Cell culture and treatments
Neonatal primary cardiomyocytes were isolated as previously described [21]. H9c2 cells (an embryonic rat myocardium-derived cell line) were purchased from the American Type Culture Collection (ATCC, VA, USA) and cultured in high-glucose Dulbecco's modified Eagle's medium (Macgene, Beijing, China) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA) and 1% antibiotics (10000U/ml penicillin and 10 mg/ml streptomycin, Gibco). Primary cardiomyocytes and H9c2 cells were incubated at 37 °C in the mixture of 95% atmospheric air and 5% CO2, and the medium was replaced every other day. Cells were treated with DOX (1 μM) in the presence or absence of RES (20 μM) for 24 h [6]. To study the regulatory effects of AMPK on SIRT1 and SESN2, primary cardiomyocytes were pretreated with AMPK activator AICAR (0.5 mM, Beyotime Biotechnology, Shanghai, China) for 30 min and subsequently stimulated with DOX (1 μM) for 24 h.
2.3. Transfection
Primary cardiomyocytes or H9c2 cells were transfected with negative control-shRNA (NC-shRNA), Sirt1-shRNA, Ampk-shRNA, nuclear factor erythroid 2-related factor 2 (Nrf2)-shRNA, pcDNA3.1-vector, pcDNA3.1-Sirt1, NC-siRNA, Sesn2-siRNA, pCMV6-Entry, pCMV6-Sesn2 (GenePharma, Shanghai, China) or Mdm2-siRNA (RiboBio, Guangzhou, China) by using Lipofectamine 3000 reagent (Invitrogen, Grand Island, NY, USA) according to the manufacturer's instructions.
2.4. Echocardiography
High-resolution transthoracic echocardiography (Vevo 2100, Visual Sonics, Toronto, ON, Canada) was performed on mice narcotized with isoflurane (RWD Life Science Co., Ltd., Shenzhen, China) as we previously reported [[22], [23], [24], [25]]. Chest hair was removed and bubble-free ultrasonic coupling agent was applied to acquire parasternal long-axis views. The main cardiac parameters include ejection fraction (EF), fractional shortening (FS), left ventricular internal dimension (LVID), LV posterior wall thickness (LVPW), interventricular septum thickness (IVS) and LV volume (LV vol).
2.5. Histological and cellular staining
Hearts of mice were fixed in 10% formalin, and then treated with dehydration, embedding and sectioning. Hematoxylin & eosin (H&E, Cat# G1005, Servicebio Technology, Wuhan, China) staining was performed to examine cardiac histological morphology. Fluorescein isothiocyanate-conjugated wheat germ agglutinin (FITC-conjugated WGA, excitation (Ex)/emission (Em) wavelengths: 490/520 nm, Cat# L4895, Sigma-Aldrich, St. Louis, MO, USA) staining was used to detect myocyte cross-sectional areas. Cardiac collagen deposition was examined by Masson's trichrome staining (Cat# G1006, Servicebio Technology) and Sirius red staining (Leagene Biotechnology, Beijing, China). To evaluate myocardial reactive oxygen species (ROS) production levels, sections were stained with dihydroethidium (DHE) fluorescence kit (Ex/Em wavelengths: 535/610 nm, Cat# S0063, Beyotime Biotechnology). Apoptosis was detected in tissue sections using Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) Cell Apoptosis Detection Kit (Cat# KGA704, Keygen Biotech, Jiangsu, China) or In Situ Cell Death Detection Kit from Sigma-Aldrich (Cat# 12156792910), and nuclei were counter-stained with DAPI (Cat# ab104139, Abcam, Cambridge, UK). Immunohistochemical (IHC) staining with anti-tumor necrosis factor-α (TNF-α, 1:300, Cat# ab6671, Abcam), anti-4-hydroxynonenal (4-HNE, 1:300, Cat# ab46545, Abcam) and anti-3-nitrotyrosine (3-NT, 1:300, Cat# AB5411, Millipore, Billerica, MA, USA) antibody and immunofluorescent staining with anti-nuclear factor κB p65 (NF-κB p65, 1:300, Cat# 8242, Cell Signaling Technology, Danvers, MA, USA), anti-NRF2 (1:200, Cat# ab31163, Abcam) and anti-NAD(P)H quinone dehydrogenase 1 (NQO1, 1:50, Cat# sc-32793, Santa Cruz Biotechnology, Dallas, TX, USA) antibody were performed as described previously [26]. DHE (final working concentration: 5 μM) and 2′,7′-dichlorofluorescein diacetate (DCFH-DA, final working concentration: 10 μM, Ex/Em wavelengths: 488/525 nm, Cat# S0033, Beyotime Biotechnology) probes were used for the detection of ROS levels in H9c2 cells. The co-localization of SIRT1 and SESN2 in H9c2 cells was detected by immunofluorescent staining with anti-SIRT1 (1:100, Cat# 8469, Cell Signaling Technology) and anti-SESN2 (1:100, Cat# 10795-1-AP, Proteintech, Chicago, IL, USA) antibody. Stained sections or cells were determined by the light microscope (Nikon, Tokyo, Japan) or fluorescence microscope (Nikon) and quantified using Image J software.
2.6. Real-time quantitative PCR
Gene expression was tested by real-time quantitative PCR (RT-qPCR) as previously described [27]. Briefly, total RNA was isolated from cardiac tissues using TRIzol reagent (Cwbio, Jiangsu, China), and used for the cDNA synthesis by reverse transcription using HiFiScript cDNA Synthesis Kit (Cwbio). Taking cDNA as template, RT-qPCR was performed with real-time PCR detection system (Bio-Rad, Hercules, CA, USA) using UltraSYBR Mixture (Cwbio). Primers used as follows: mouse atrial natriuretic peptide (Anp), mouse brain natriuretic peptide (Bnp), mouse connective tissue growth factor (Ctgf), mouse interleukin-1β (Il1b), mouse catalase (Cat), mouse superoxide dismutase (Sod), mouse glyceraldehyde-3-phosphate dehydrogenase (Gapdh), rat Tnfa, rat Il1b and rat Gapdh were purchased from Sangon Biotech (Shanghai, China). The primer sequences are described in the Supplementary Table S1.
2.7. Flow cytometry
ROS production levels were examined by DCFH-DA staining, and cell apoptosis was detected using FITC Annexin V Apoptosis Detection Kit (Ex/Em wavelengths: FITC: 490/520 nm, PI: 535/617 nm, Cat# 556547, BD Biosciences, NY, USA) according to the manufacturer's instructions. Finally, ROS levels and cell apoptosis were detected with a flow cytometer (CytoFLEX S, Beckman, USA).
2.8. Western blot analysis
Heart tissues, primary cardiomyocytes and H9c2 cells were homogenized in RIPA lysis buffer (Beyotime Biotechnology) including protease and phosphatase inhibitors (Beyotime Biotechnology). Then tissue or cell proteins were collected by centrifuging at 12000 rpm at 4 °C for 25 min. Nuclear proteins were extracted with the Nuclear-Cytosol Extraction Kit (Beyotime Biotechnology) according to the instructions. Protein samples, diluted with loading buffer and heated at 95 °C for 5 min, were subjected to electrophoresis and transferred to nitrocellulose membranes (GE Healthcare Life Sciences, Chicago, IL, USA). The membranes were blocked in 5% nonfat milk for 1 h, and then incubated overnight at 4 °C with the following antibodies: anti-SIRT1 (1:1000), anti-NF-κB p65 (1:1000), anti-NRF2 (1:1000), anti-Histone H3 (1:1500, Cat# BF9211, Affinity Biosciences, Cincinnati, OH, USA), anti-heme oxygenase-1 (HO-1, 1:1000, Cat# 10701-1-AP, Proteintech), anti-SESN2 (1:2000), anti-phosphorylated AMPKα (P-AMPKα, 1:1000, Cat# 2535, Cell Signaling Technology), anti-AMPKα (1:1000, Cat# 2532, Cell Signaling Technology), anti-P-mammalian target of rapamycin (mTOR) (1:1000, Cat# 5536, Cell Signaling Technology), anti-P-S6 Ribosomal Protein (P–S6, 1:1000, Cat# 2211, Cell Signaling Technology), anti-S6 (1:2000, Cat# 2217, Cell Signaling Technology), anti-cleaved caspase-3 (1:1000, Cat# 9664, Cell Signaling Technology), anti-MDM2 (1:1000, Cat# sc-965, Santa Cruz Biotechnology) and anti-GAPDH (1:1000, Cat# GB11002, Servicebio Technology). The immunoreactive bands were detected with the horseradish peroxidase-conjugated secondary antibody (1:1000, Proteintech), and visualized using an enhanced chemoluminescence detection kit (Millipore). Densitometric analysis was performed using Image Quant 4.2 software (Tanon, Shanghai, China).
2.9. Immunoprecipitation assay
Immunoprecipitation (IP) assays were performed as described previously [28]. Briefly, heart tissues, primary cardiomyocytes and H9c2 cells were lysed in IP buffer containing protease and phosphatase inhibitors. Then lysate was immunoprecipitated overnight at 4 °C with anti-SESN2 antibody and protein A/G PLUS-agarose beads (Santa Cruz Biotechnology). The immunocomplexes were detected by western blot with anti-SIRT1 (1:1000), anti-ubiquitin (1:1000, Cat# ab7780, Abcam) and anti-MDM2 (1:1000) antibody.
2.10. Statistical analysis
Data were presented as means ± standard deviation (SD). Normal distribution of the data was assessed using Shapiro-Wilk test. If no normal distribution was found, we cased logarithmic transformation of the data. After the transformation, all data obeyed normal distribution, thus comparisons among different groups were performed by one-way analysis of variance (ANOVA) or two-way ANOVA followed by Tukey's or Sidak's post hoc test as appropriate with GraphPad Prism. P < 0.05 was considered statistically significant.
3. Results
3.1. Cardiac SIRT1 activation indicated the resistance to DOX-induced cardiac structural and functional abnormalities
To examine the cardioprotective effects of SIRT1 in vivo, Sirt1-CKO mice were generated by crossing Sirt1flox/flox mice with Myh6-creEsr1 mice, followed by 1-week tamoxifen administration. As shown in Fig. 1A, there was almost no expression of SIRT1 protein in cardiac tissues of Sirt1-CKO mice. Moreover, RES administration recovered DOX-reduced expression of SIRT1 in hearts of Sirt1flox/flox mice (Fig. 5A). To explore the role of SIRT1 in DOX-induced cardiotoxicity, echocardiography was performed to examine cardiac function. The results demonstrated that compared with Ctrl group, DOX application induced cardiac dysfunction without affecting the heart rate, as evidenced by decreased EF and FS and increased systolic LVID and systolic LV vol in Sirt1flox/flox mice. These cardiac function indexes were more pronounced in Sirt1-CKO mice compared with Sirt1flox/flox mice, whereas RES treatment significantly improved DOX-induced cardiac dysfunction in Sirt1flox/flox mice, but not in Sirt1-CKO mice (Fig. 1B–F and Supplementary Fig. S1).
Fig. 1.

The activation of sirtuin 1 (SIRT1) by resveratrol (RES) improved doxorubicin (DOX)-induced cardiac dysfunction and myocardial remodeling. (A) Western blot analysis of SIRT1 expression in different organs of Sirt1flox/flox and Sirt1-CKO mice. (B–F) Cardiac function was examined by echocardiography (n = 5–6). (G) FITC-conjugated WGA staining of cardiac tissues and quantification of myocytes cross-sectional areas (n = 6). (H, I) Representative (H) Masson's trichrome and (I) Sirius red staining images of cardiac tissues and quantification of cardiac fibrotic areas (n = 6). (J, K) RT-qPCR for measuring mRNA expression of hypertrophic markers atrial natriuretic peptide (Anp) and brain natriuretic peptide (Bnp) in cardiac tissues (n = 5–6). (L) Cardiac connective tissue growth factor (Ctgf) mRNA expression was measured by RT-qPCR (n = 5–6). Data were presented as mean ± SD. *P < 0.05, ns indicates no significance. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Fig. 5.

The activation of sestrin 2 (SESN2) and AMP-activated protein kinase α (AMPKα) by RES was dependent on SIRT1 in DOX-impaired mice hearts and cardiomyocytes. (A) The protein expression of SIRT1, SESN2, P-AMPKα, AMPKα, P-mammalian target of rapamycin (mTOR), P–S6 Ribosomal Protein (P–S6) and S6 was detected by western blot analysis with densitometric quantification in cardiac tissues of each group (n = 6). (B) Western blot analysis and densitometric quantification of SIRT1, SESN2, P-AMPKα, AMPKα, P-mTOR, P–S6 and S6 after transfection with NC-shRNA or Sirt1-shRNA in H9c2 cells of different groups. Three independent experiments were performed. (C) Primary cardiomyocytes were transfected with pcDNA3.1-vector or pcDNA3.1-Sirt1 for 24 h, and then treated with or without DOX (1 μM) for 24 h. The protein expression of SIRT1, SESN2, P-AMPKα and AMPKα was detected by western blot analysis with densitometric quantification of each group. Three independent experiments were performed. GAPDH as an internal control. Data were presented as mean ± SD. *P < 0.05, ns indicates no significance.
DOX-induced cardiotoxicity is characterized not only by cardiac dysfunction, but also by myocardial remodeling including myocardial hypertrophy and fibrosis [7,29]. H&E staining revealed obvious myocardial fibre disruption in DOX-impaired mice (Supplementary Fig. S2A). Cardiomyocyte size was increased in response to DOX by analysis of myocyte area via FITC-conjugated WGA staining (Fig. 1G). Furthermore, Masson's trichrome and Sirius red staining showed collagen accumulation in hearts of DOX-impaired mice (Fig. 1H and I and Supplementary Figs. S2B and C). Morphological hypertrophy and fibrosis of hearts were further confirmed by elevated mRNA levels of hypertrophic markers Anp and Bnp, and fibrotic marker Ctgf, respectively (Fig. 1J-L). All of these indexes were more significantly increased in Sirt1-CKO/DOX group than Sirt1flox/flox/DOX group and almost completely inhibited by RES treatment only in Sirt1flox/flox mice (Fig. 1G-L and Supplementary Fig. S2).
3.2. DOX-induced inflammatory response was improved by SIRT1 activation
Increasing studies have identified that a variety of cellular pathologies such as inflammatory response could participate in the development of cardiomyocyte hypertrophy. Meanwhile, DOX could directly cause myocardial fibrosis at the initial stage by exacerbation of proinflammatory events [30,31]. Therefore, cardiac expression of inflammatory cytokines was detected in our mouse model. The results showed that TNF-α protein and Il1b mRNA levels were increased in DOX groups of Sirt1flox/flox and Sirt1-CKO mice. RES treatment prevented DOX-associated inflammation only in Sirt1flox/flox mice (Fig. 2A and B). Moreover, these results were further confirmed by in vitro studies with H9c2 cells (Fig. 2C and D).
Fig. 2.

The activation of SIRT1 by RES improved DOX-induced inflammation in mice hearts and H9c2 cells. (A) Cardiac inflammatory response was detected by IHC staining for tumor necrosis factor-α (TNF-α, brown considered positive staining) followed by a quantitative analysis of the positive stains (n = 6). (B) Relative myocardial interleukin-1β (Il1b) mRNA level was measured by RT-qPCR (n = 5–6). (C, D) H9c2 cells were transfected with NC-shRNA or Sirt1-shRNA for 24 h, and then treated with RES (20 μM) or DOX (1 μM) or both for 24 h. Relative Tnfa and Il1b mRNA levels were measured by RT-qPCR. Three independent experiments were performed. (E) Western blot analysis and densitometric quantification of nuclear nuclear factor κB p65 (NF-κB p65) expression in cardiac tissues of each group (n = 6). Histone H3 as an internal control. (F) Nuclear accumulation of NF-κB p65 (indicated by white arrows) determined by immunofluorescent staining with anti-NF-κB p65 antibody (red) in cardiac tissues. (G) Western blot analysis and densitometric quantification of nuclear NF-κB p65 expression in H9c2 cells of different groups. Three independent experiments were performed. Histone H3 as an internal control. Data were presented as mean ± SD. *P < 0.05, ns indicates no significance. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Among the intracellular signaling systems that regulate inflammatory and immune responses, NF-κB is of particular importance because it is recognized to be the master transcription factor controlling expression of a host of proinflammatory genes [32]. Therefore, we detected the regulatory effect of SIRT1 on NF-κB p65 expression by western blot and immunofluorescent staining both in vivo and in vitro. Clearly, DOX significantly increased the nuclear expression of NF-κB p65, and this effect was abolished by treatment with RES only in Sirt1flox/flox mice or H9c2 cells transfected with NC-shRNA (Fig. 2E–G).
3.3. DOX-caused redox imbalance and apoptosis were prevented by SIRT1 activation
Under pathological inflammatory conditions, the production of reactive species may significantly increase, some of which diffuse from phagocytes and induce oxidative stress [33]. Similarly, ROS production from damaged mitochondria could also activate NLR family pyrin domain containing 3 inflammasome to result in IL-1β secretion and localized inflammation [34]. Thus, to explore whether ROS elimination participated in the protective effects of SIRT1 against cardiotoxicity, ROS levels were examined in different groups using DHE probes. As shown in Fig. 3A and D, the fluorescence intensity of DHE was evident in cardiac tissues of Sirt1flox/flox/DOX group and further elevated in Sirt1-CKO/DOX group. Moreover, cardiac oxidative damage, assessed by examining the levels of 4-HNE and 3-NT, was noted in Sirt1flox/flox/DOX group and further increased in Sirt1-CKO/DOX group (Fig. 3B, C, E and F). RES treatment prevented all these effects in Sirt1flox/flox mice, but not in Sirt1-CKO mice (Fig. 3A–F).
Fig. 3.

The activation of SIRT1 attenuated DOX-induced cardiac oxidative stress. (A, D) Representative microphotographs and quantification of fluorescence intensity of DHE staining (red) in cardiac sections (n = 6). (B, C, E, F) IHC staining using (B, E) anti-4-hydroxynonenal (4-HNE) and (C, F) anti-3-nitrotyrosine (3-NT) antibody in cardiac tissues (brown considered positive staining) and related quantitative analysis (n = 6). (G–I) The protein expression of nuclear nuclear factor erythroid 2-related factor 2 (NRF2) and heme oxygenase-1 (HO-1) was analyzed by western blot with densitometric quantification of each group (n = 6). Histone H3 or GAPDH as an internal control. (J, K) Cardiac catalase (Cat) and superoxide dismutase (Sod) mRNA levels were tested by RT-qPCR (n = 6). (L) Nuclear accumulation of NRF2 (indicated by white arrows) determined by immunofluorescent staining with anti-NRF2 antibody (red) in cardiac tissues. Data were presented as mean ± SD. *P < 0.05, ns indicates no significance. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The transcription factor NRF2 plays a pivotal role in the cellular response to oxidative stress by upregulating multiple antioxidant enzymes [35]. NRF2 deficiency aggravated DOX-impaired cardiac function, whereas NRF2 activation protected against cardiotoxicity in response to DOX [35,36]. Here, we found that RES treatment blocked DOX-induced downregulation of nuclear NRF2 and preserved its transcriptional activity, as confirmed by increased mRNA levels of Cat and Sod and protein levels of HO-1 and NQO1. However, the beneficial effects of RES were abolished in the cardiac tissues of Sirt1-CKO mice (Fig. 3G-L and Supplementary Fig. S3). These findings indicated a causative role of NRF2-dependent antioxidant activity as a part of SIRT1-mediated protection against cardiotoxicity.
It is known that DOX-induced inflammation and excessive ROS generation could result in cardiomyocyte apoptosis and eventually aggravate cardiac injury [37,38]. As shown in Fig. 4A and B, increased numbers of TUNEL-positive cells were observed in hearts of Sirt1flox/flox/DOX group and further increased in Sirt1-CKO/DOX group as detected by TUNEL staining. RES treatment prevented this effect only in Sirt1flox/flox/DOX group.
Fig. 4.

The activation of SIRT1 ameliorated DOX-induced oxidative stress and apoptosis in mice and H9c2 cells. (A, B) Detection of apoptotic cells by TUNEL staining with quantification of apoptosis rate in cardiac tissues (n = 6 in B). (C, D) Representative images of (C) DCFH-DA (green) and (D) DHE (red) staining and quantification of the corresponding fluorescence intensity in H9c2 cells. Three independent experiments were performed. (E) H9c2 cells were stained with DCFH-DA and analyzed by flow cytometry. Three independent experiments were performed. (F) Apoptosis rate of H9c2 cells was determined by flow cytometry analysis using annexin V/PI double staining. Three independent experiments were performed. Data were presented as mean ± SD. *P < 0.05, ns indicates no significance. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To further confirm the antioxidative and anti-apoptotic effects of SIRT1 in vitro, ROS levels were examined using fluorescent probes DHE and DCFH-DA, and apoptosis rate was detected by flow cytometry analysis using annexin V/propidium iodide (PI) double staining after transfection with NC-shRNA or Sirt1-shRNA in different groups of H9c2 cells. The results indicated that compared with the NC-shRNA data, the ability of RES to attenuate DOX-induced excessive ROS production and cardiomyocyte apoptosis was significantly restricted after transfection with Sirt1-shRNA (Fig. 4C–F).
3.4. The activation of SESN2 improved DOX-induced cardiotoxicity in a SIRT1-dependent manner
AMPK is a crucial energy sensor that has been verified to be a target of SIRT1. As expected, the knockout of Sirt1 blocked RES-induced phosphorylation of AMPKα in hearts of DOX-treated mice (Fig. 5A). But the precise mechanism of the link between SIRT1 and AMPK in DOX-induced cardiotoxicity was still unclear. We next detected SESN2 protein expression, since it is regarded as an upstream activator of AMPK, and the activation of SESN2/AMPK pathway was involved in myocardial protection [39,40]. Western blot analysis showed that the expression of SESN2 was significantly inhibited following DOX stimulation. Moreover, the knockout of Sirt1 hindered RES-upregulated SESN2 expression in hearts of DOX-impaired mice (Fig. 5A).
In addition, mTOR is generally considered as a downstream target of AMPK. Meanwhile, the activation of AMPK by spermidine could downregulate mTOR expression to improve myocardial infarction-induced cardiac dysfunction [41]. Moreover, S6 is regarded as a downstream target of mTOR, and its phosphorylation status is commonly considered a functional readout of mTOR activity. To investigate whether SIRT1-mediated AMPKα activation could regulate mTOR in our model, the phosphorylation levels of mTOR and S6 were measured by western blot. Unexpectedly, P-mTOR and P–S6 levels were decreased in hearts of DOX-treated mice, and RES treatment did not apparently change DOX-suppressed mTOR activity both in hearts of Sirt1flox/flox and Sirt1-CKO mice (Fig. 5A).
To further verify the above findings in vitro, H9c2 cells were transfected with NC-shRNA or Sirt1-shRNA for 24 h, and then treated with RES (20 μM) or DOX (1 μM) or both for 24 h. Consistent with the data in vivo, we found that Sirt1 silencing abolished RES-mediated activation of SESN2 and AMPKα in DOX-treated H9c2 cells. Moreover, DOX-induced downregulation of P-mTOR and P–S6 was also not affected by RES treatment and Sirt1 knockdown (Fig. 5B). Subsequently, we confirmed the regulatory effects of SIRT1 on SESN2 and AMPKα by transfection with pcDNA3.1-Sirt1 plasmid. SIRT1 upregulation increased the expression of SESN2 and P-AMPKα in primary cardiomyocytes treated with DOX (Fig. 5C). Taken together, these data showed that RES reversed DOX-induced downregulation of SESN2 and P-AMPKα was SIRT1-dependent. This was also accompanied by almost complete improvement of DOX-impaired cardiac function via SIRT1 activation by RES in an mTOR-independent manner.
3.5. SESN2 is a scaffold protein for SIRT1 and AMPKα in response to DOX injury
Subsequently, to determine the pivotal role of SESN2 in the regulation of AMPK expression and cardiac-protection, we performed both knockdown of Sesn2 gene expression with Sesn2-siRNA and overexpression of SESN2 with pCMV6-Sesn2 plasmid (Fig. 6). Sesn2 knockdown not only abolished the ability of RES on AMPKα activation but also significantly blocked RES-exerted antioxidative role detected by fluorescent probe DHE and anti-apoptotic property examined by cleaved caspase-3 protein expression in DOX-treated cardiomyocytes (Fig. 6A and B), whereas overexpression of SESN2 by transfection with pCMV6-Sesn2 plasmid could largely prevent DOX-impaired AMPKα activation and DOX-induced oxidative stress and apoptosis (Fig. 6C and D). However, downregulation or upregulation of SESN2 had no apparent change on the expression of SIRT1 (Fig. 6A and C), suggesting that SIRT1 activated AMPKα to improve DOX-induced cardiac injury via upregulation of SESN2. Further mechanism study suggested that the antioxidative effect of SIRT1 was dependent on NRF2 activation via SESN2 (Supplementary Fig. S4).
Fig. 6.

The cross-talk among SIRT1, SESN2 and AMPKα in DOX-treated cardiomyocytes. (A) Primary cardiomyocytes were transfected with NC-siRNA or Sesn2-siRNA for 24 h, and then treated with DOX (1 μM) in the presence or absence of RES (20 μM) for 24 h. The protein expression of SIRT1, SESN2, P-AMPKα, AMPKα and cleaved caspase-3 was detected by western blot analysis with densitometric quantification of each group. Three independent experiments were performed. (B) Representative images of DHE (red) staining and quantification of the corresponding fluorescence intensity in H9c2 cells. Three independent experiments were performed. (C) The protein expression of SIRT1, SESN2, P-AMPKα, AMPKα and cleaved caspase-3 was detected by western blot analysis with densitometric quantification after transfection with pCMV6-Entry or pCMV6-Sesn2 in H9c2 cells of different groups. Three independent experiments were performed. (D) Representative images of DHE (red) staining and quantification of the corresponding fluorescence intensity in H9c2 cells. Three independent experiments were performed. GAPDH as an internal control. Data were presented as mean ± SD. *P < 0.05, ns indicates no significance. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Considering the fact that AMPK can also activate SIRT1 by increasing intracellular NAD+ levels [42], the regulatory effects of AMPK on SIRT1 and SESN2 were also tested in DOX-treated H9c2 cells and primary cardiomyocytes. The results demonstrated that silencing of Ampk with Ampk-shRNA abolished the activation of SIRT1 and SESN2 by RES in H9c2 cells treated with DOX (Supplementary Fig. S5A). Conversely, the activation of AMPKα by AICAR could recover DOX-decreased protein expression of SIRT1 and SESN2 in primary cardiomyocytes (Supplementary Fig. S5B). These data suggested that AMPK also promoted SESN2 expression by forming a positive feedback loop with SIRT1 in response to DOX damage.
3.6. SIRT1 reduced DOX-induced SESN2 ubiquitination possibly by disturbing the interaction of SESN2 and MDM2
To further determine the possible correlation of SIRT1with SESN2 in cardiomyocytes, the co-localization of SIRT1 and SESN2 was detected via immunofluorescence. We found that SIRT1 and SESN2 were predominantly localized in the cytoplasm in normal cardiomyocytes. DOX treatment led to translocation of cytosolic SIRT1 and SESN2 into the nucleus. Additionally, RES could further increase the expression of nuclear SIRT1 and SESN2 (Fig. 7A). To assess whether the same location of SITR1 and SESN2 in the nucleus reflected a physical protein interaction, primary cardiomyocyte lysates were immunoprecipitated with anti-SESN2 antibody, and followed by western blot analysis for the presence of SIRT1 under different treatment condition. Surprisingly, SIRT1 did not interact with SESN2 under any treatment condition (Fig. 7B), implying that SIRT1 did not directly increase SESN2 expression.
Fig. 7.

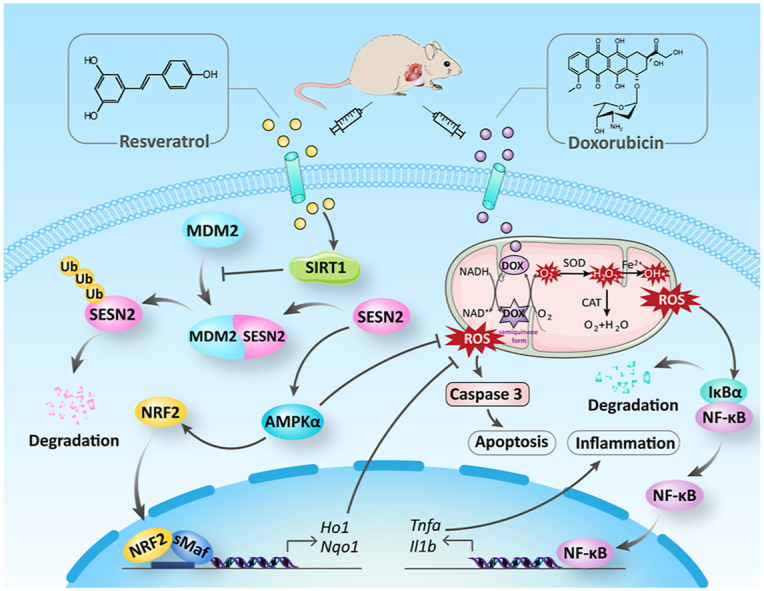
SIRT1 activation could reduce DOX-induced SESN2 ubiquitination possibly by disturbing the interaction of SESN2 and mouse double minute 2 (MDM2) in cardiomyocytes. (A) Co-localization of SIRT1 and SESN2 in H9c2 cells was determined by immunofluorescent staining. (B) Protein-protein interaction between SIRT1 and SESN2 in primary cardiomyocytes demonstrated by co-IP. (C) The immunoblotting of immunoprecipitated SESN2 with antibody recognized ubiquitin in H9c2 cells. (D, E) Protein-protein interaction between SESN2 and MDM2 in primary cardiomyocytes was demonstrated by co-IP with densitometric quantification. Three independent experiments were performed. (F) Western blot analysis and densitometric quantification of SIRT1 and SESN2 after transfection with NC-siRNA or Mdm2-siRNA in primary cardiomyocytes of different groups. Three independent experiments were performed. (G) Schematic illustration of a novel protective mechanism by SIRT1 activation to improve DOX-induced cardiotoxicity through SESN2/AMPKα pathway. GAPDH as an internal control. Data were presented as mean ± SD. *P < 0.05, ns indicates no significance.
Previous studies have shown that the stabilization of SESN2 was associated with ubiquitin-proteasome system [43,44]. Here, we immunoprecipitated SESN2 protein followed by the anti-ubiquitin antibody detection and observed that DOX stimulation increased SESN2 ubiquitination, which was restored by RES in H9c2 cells (Fig. 7C). This suggested that the activation of SIRT1 by RES reversed DOX-decreased SESN2 expression probably through inhibiting ubiquitination-mediated degradation. Under conditions of cellular stress, endogenous SIRT1 was reported to coimmunoprecipitate with MDM2, indicating that these two proteins may interact in cells [45]. Based on this study, we hypothesized that SIRT1-mediated SESN2 deubiquitination could be modulated by MDM2. Consistent with this hypothesis, co-IP analysis indicated that there was an interaction between SESN2 and MDM2, which was increased following DOX stimulation. As expected, overexpression of SIRT1 by RES and pcDNA3.1-Sirt1 plasmid could disturb DOX-enhanced interaction of SESN2 and MDM2 both in primary cardiomyocytes and H9c2 cells (Fig. 7D and E and Supplementary Fig. S6A). However, SIRT1 deficiency abolished RES-reduced interaction of SESN2 and MDM2 both in DOX-treated mice hearts and H9c2 cells (Supplementary Figs. S6B and C). To further explore the regulatory effect of MDM2 on SESN2, Mdm2 was efficiently knockdown in primary cardiomyocytes transfected with Mdm2-siRNA (Supplementary Fig. S6D). Knockdown of Mdm2 reversed DOX-induced SESN2 downregulation (Fig. 7F), suggesting that MDM2 acts as a key E3 ligase to ubiquitinate SESN2. Taken together, all these results implied the activation of SIRT1 could reduce DOX-induced SESN2 ubiquitination possibly through disturbing the interaction of SESN2 and MDM2 in cardiomyocytes.
4. Discussion
Accumulative evidence has demonstrated that DOX treatment resulted in severe cardiac oxidative stress and apoptosis both in clinical patients and preclinical mice models, which plays a critical role in the development of cardiac dysfunction and complications. In spite of its critical importance, the mechanisms for impaired redox homeostasis and excessive cardiomyocyte apoptosis during chemotherapy remain largely unknown. The present work provided three new findings implicating SIRT1-initiated pathway in DOX-induced cardiotoxicity. The first finding is to confirm the relationship between SIRT1 levels and DOX-induced morphological and functional abnormalities: cardiac morphological structure, function and SIRT1 levels were impaired in DOX-treated mice, which could be largely improved by RES treatment. But these favorable effects on the myocardium were blunted in mice with cardiac-specific deletion of Sirt1. The second innovative finding is that the benefits of upregulated SIRT1 expression in DOX-induced cardiotoxicity required the activation of SESN2-mediated anti-apoptosis and antioxidant signaling. Our third novel finding is that SIRT1 increased the expression of SESN2 by interfering with the ubiquitination and degradation of SESN2 via MDM2 in DOX-treated cardiomyocytes.
Emerging evidence demonstrated that SIRT1 activation is essential for the regulation of multiple cellular functions. Our previous study also indicated that the activation of SIRT1 prevented DOX-induced cardiotoxicity [7]. Consistent with previous study, DOX stimulation indeed decreased myocardial levels of SIRT1, and the activation of SIRT1 by RES improved cardiac dysfunction and alleviated myocardial fibre disruption, fibrosis, hypertrophy and inflammation in DOX-treated mice and cardiomyocytes (Fig. 1B-L, 2 and Supplementary Fig. S2). Sufficient evidence revealed that DOX-induced cardiotoxicity is closely associated with increased oxidative stress in cardiomyocytes. DOX accumulation rapidly increases mitochondrial ROS production in cardiomyocytes by redox cycling, which damages mitochondrial function and then leads to more sustained oxidative stress [46,47]. Here, we observed that SIRT1 activation obviously alleviated oxidative stress by promoting nuclear NRF2 accumulation and function in DOX-treated cardiomyocytes, whereas the deletion of Sirt1 exacerbated DOX-induced oxidative stress (Figs. 3, 4C-E and Supplementary Fig. S3). However, in the present study, only unspecific ROS formation was assessed but no specific read-out for superoxide was measured. Apoptosis is regarded as another major contributor to DOX-induced cardiotoxicity. Indeed, activating SIRT1 by RES could decrease DOX-induced cardiomyocyte apoptosis, and the deletion of Sirt1 abolished this protective effect provided by RES (Fig. 4A, B and F). Collectively, these results indicated that SIRT1 activation could inhibit oxidative stress and reduce cardiomyocyte apoptosis to ameliorate DOX-induced cardiac morphological and functional abnormalities.
The second innovative finding of this study is that the benefits of increased SIRT1 expression in DOX-induced cardiotoxicity required the activation of SESN2-mediated antioxidant and anti-apoptotic signaling. The importance of SESN2 to cardiac function has been proved in earlier study with Sesn2-knockout mice accompanied with exacerbated postischemic contractile dysfunction [15]. SESN2 has also been verified to play a protective role in DOX-induced cardiomyopathy through rescuing mitophagy and improving mitochondrial function [17]. In this study, we provided direct evidence that the overexpression of SIRT1 restored DOX-induced downregulation of SESN2. In contrast, the inhibition of SIRT1 blocked the effect of RES on SESN2 upregulation in response to DOX injury both in vivo and in vitro (Fig. 5). We also demonstrated that SESN2 downregulation abolished RES-increased AMPKα activation and cardiac-protective properties, whereas overexpression of SESN2 could rescue AMPKα activation and prevent DOX-induced oxidative stress and apoptosis in cardiomyocytes (Fig. 6). These results confirmed that SESN2 is a key mediator of SIRT1 action on AMPKα activity to protect against oxidative stress and cell apoptosis. Of note, we further found that the knockdown of Ampk could abolish RES-induced SIRT1 and SESN2 activation in DOX-treated cardiomyocytes (Supplementary Fig. S5A). However, the activation of AMPKα by AICAR could increase the expression of SIRT1 and SESN2 in DOX-treated cardiomyocytes (Supplementary Fig. S5B). These findings can be partly explained by the fact that AMPK can enhance SIRT1 activity by increasing NAD+ levels [42]. However, more studies are necessary to explore the detailed mechanism of SESN2 regulation by AMPK.
Our third important new finding of this study is that SIRT1-mediated increased SESN2 expression was related to the inhibition of MDM2-mediated SESN2 ubiquitination in DOX-treated cardiomyocytes. The stability of SESN2 is closely associated with the ubiquitin-proteasome system. Previous studies have shown that E3 ubiquitin ligases ring box protein 1 and ring finger protein 186 are interactive partners for SESN2 which mediate its ubiquitination [43,44]. The function of another E3 ubiquitin ligase MDM2 expressed in hearts was proved to be modulated by SIRT1 through deacetylating Lys182 and Lys185 of MDM2 [45]. Based on these findings, we hypothesized that SIRT1 upregulated SESN2 expression possibly through inhibiting MDM2-mediated SESN2 ubiquitination and subsequent degradation. As expected, our results showed that the ubiquitination levels of SESN2 were increased, and also the interaction of SESN2 and MDM2 was enhanced in DOX-treated cardiomyocytes. All these changes were reversed by SIRT1 activation (Fig. 7C–E and Supplementary Fig. S6A). However, the inhibitory effect of RES on DOX-enhanced interaction of SESN2 and MDM2 was blocked by Sirt1 knockout or knockdown (Supplementary Figs. S6B and C). Furthermore, knockdown of Mdm2 eliminated DOX-downregulated SESN2 expression in primary cardiomyocytes, suggesting that MDM2 acts as a key E3 ligase to ubiquitinate SESN2 (Fig. 7F and Supplementary Fig. S6D).
Another noteworthy finding of our study is that the cardioprotective effect of SIRT1 activation was independent of mTOR in response to DOX-induced toxicity. Unexpectedly, the phosphorylation levels of AMPKα and mTOR were both downregulated in DOX-treated mice hearts and cardiomyocytes (Fig. 5A and B), despite AMPK is generally considered as an upstream negative regulator of mTOR [48]. Consistent with our results, some studies demonstrated that mTOR inhibition was a primary contributor to DOX-induced cardiotoxicity [35,49,50]. Conversely, others found that the phosphorylation levels of cardiac mTOR were increased by DOX stimulation [51,52]. These controversial reported findings might be related to the specific species and dosages of DOX. More importantly, the mTOR pathway is very complex with various upstream signaling and downstream effector molecules. Therefore, the effect of alteration in mTOR expression on DOX-induced cardiotoxicity needs to be further investigated. Lastly, we did not evaluate female Sirt1flox/flox and Sirt1-CKO mice because different and extended reproductive strategies are required to produce cohorts of both male and female mice and respective Sirt1flox/flox litters. Although DOX-related cardiotoxicity is currently thought to be more prevalent in males [53,54], it is critical to determine the impact of sex hormones on SIRT1/SESN2-regulated cardioprotective effect in the follow-up study.
In summary, our current study revealed that the activation of SIRT1 could improve DOX-induced cardiotoxicity through preventing oxidative damage and apoptosis. This is partly due to increased SESN2 expression by reducing MDM2-mediated SESN2 ubiquitination and subsequently increased AMPKα phosphorylation levels, as illustrated in Fig. 7G. Therefore, it is important to develop pharmacological interventions targeting SIRT1/SESN2 signaling pathway for the prevention and treatment of DOX-induced cardiotoxicity.
Author's contribution
J.W(a)., J.W(b). and M.X. performed the experiments and analyzed the data. J.G. designed the study and supervised the project. J.G., J.W(a)., Y.T., J.Z., J.W(b)., M.X., G.L., J.L., Q.L. and Y.G. participated in the discussion and edited the manuscript. All authors contributed to the article and approved the submitted version.
Declaration of competing interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by Qilu Young Scholar's Program of Shandong University (21330089963007) and the Natural Science Foundation of Shandong Province (ZR2021MH330). We thank Translational Medicine Core Facility of Shandong University for consultation and instrument availability that supported this work.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102310.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Vejpongsa P., Yeh E.T. Prevention of anthracycline-induced cardiotoxicity: challenges and opportunities. J. Am. Coll. Cardiol. 2014;64:938–945. doi: 10.1016/j.jacc.2014.06.1167. [DOI] [PubMed] [Google Scholar]
- 2.Liang X., Wang S., Wang L., Ceylan A.F., Ren J., Zhang Y. Mitophagy inhibitor liensinine suppresses doxorubicin-induced cardiotoxicity through inhibition of Drp1-mediated maladaptive mitochondrial fission. Pharmacol. Res. 2020:104846. doi: 10.1016/j.phrs.2020.104846. [DOI] [PubMed] [Google Scholar]
- 3.Cappetta D., Esposito G., Piegari E., Russo R., Ciuffreda L.P., Rivellino A., Berrino L., Rossi F., De Angelis A., Urbanek K. SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int. J. Cardiol. 2016;205:99–110. doi: 10.1016/j.ijcard.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z., Wang M., Liu J., Ye J., Jiang H., Xu Y., Ye D., Wan J. Inhibition of TRPA1 attenuates doxorubicin-induced acute cardiotoxicity by suppressing oxidative stress, the inflammatory response, and endoplasmic reticulum stress. Oxid. Med. Cell. Longev. 2018:5179468. doi: 10.1155/2018/5179468. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang C., Feng Y., Qu S., Wei X., Zhu H., Luo Q., Liu M., Chen G., Xiao X. Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in mice through SIRT1-mediated deacetylation of p53. Cardiovasc. Res. 2011;90:538–545. doi: 10.1093/cvr/cvr022. [DOI] [PubMed] [Google Scholar]
- 6.Gu J., Fan Y.Q., Zhang H.L., Pan J.A., Yu J.Y., Zhang J.F., Wang C.Q. Resveratrol suppresses doxorubicin-induced cardiotoxicity by disrupting E2F1 mediated autophagy inhibition and apoptosis promotion. Biochem. Pharmacol. 2018;150:202–213. doi: 10.1016/j.bcp.2018.02.025. [DOI] [PubMed] [Google Scholar]
- 7.Wang S., Wang Y., Zhang Z., Liu Q., Gu J. Cardioprotective effects of fibroblast growth factor 21 against doxorubicin-induced toxicity via the SIRT1/LKB1/AMPK pathway. Cell Death Dis. 2017;8 doi: 10.1038/cddis.2017.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sundaresan N.R., Pillai V.B., Gupta M.P. Emerging roles of SIRT1 deacetylase in regulating cardiomyocyte survival and hypertrophy. J. Mol. Cell. Cardiol. 2011;51:614–618. doi: 10.1016/j.yjmcc.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winnik S., Auwerx J., Sinclair D.A., Matter C.M. Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur. Heart J. 2015;36:3404–3412. doi: 10.1093/eurheartj/ehv290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alcendor R.R., Gao S., Zhai P., Zablocki D., Holle E., Yu X., Tian B., Wagner T., Vatner S.F., Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 11.Lu T.M., Tsai J.Y., Chen Y.C., Huang C.Y., Hsu H.L., Weng C.F., Shih C.C., Hsu C.P. Downregulation of Sirt1 as aging change in advanced heart failure. J. Biomed. Sci. 2014;21:57. doi: 10.1186/1423-0127-21-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu D., Ma Z., Xu L., Zhang X., Qiao S., Yuan J. PGC1alpha activation by pterostilbene ameliorates acute doxorubicin cardiotoxicity by reducing oxidative stress via enhancing AMPK and SIRT1 cascades. Aging (Albany NY) 2019;11:10061–10073. doi: 10.18632/aging.102418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L., Quan N., Sun W., Chen X., Cates C., Rousselle T., Zhou X., Zhao X., Li J. Cardiomyocyte-specific deletion of Sirt1 gene sensitizes myocardium to ischaemia and reperfusion injury. Cardiovasc. Res. 2018;114:805–821. doi: 10.1093/cvr/cvy033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S., Zhu Z., Xue M., Yi X., Liang J., Niu C., Chen G., Shen Y., Zhang H., Zheng J., et al. Fibroblast growth factor 21 protects the heart from angiotensin II-induced cardiac hypertrophy and dysfunction via SIRT1. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2019;1865:1241–1252. doi: 10.1016/j.bbadis.2019.01.019. [DOI] [PubMed] [Google Scholar]
- 15.Morrison A., Chen L., Wang J., Zhang M., Yang H., Ma Y., Budanov A., Lee J.H., Karin M., Li J. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. Faseb. J. 2015;29:408–417. doi: 10.1096/fj.14-258814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang P., Lan R., Guo Z., Cai S., Wang J., Wang Q., Li Z., Li Z., Wang Q., Li J., et al. Histone demethylase JMJD3 mediated doxorubicin-induced cardiomyopathy by suppressing SESN2 expression. Front. Cell Dev. Biol. 2020;8:548605. doi: 10.3389/fcell.2020.548605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang P., Wang L., Lu J., Hu Y., Wang Q., Li Z., Cai S., Liang L., Guo K., Xie J., et al. SESN2 protects against doxorubicin-induced cardiomyopathy via rescuing mitophagy and improving mitochondrial function. J. Mol. Cell. Cardiol. 2019;133:125–137. doi: 10.1016/j.yjmcc.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Li R., Huang Y., Semple I., Kim M., Zhang Z., Lee J.H. Cardioprotective roles of sestrin 1 and sestrin 2 against doxorubicin cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2019;317:H39–H48. doi: 10.1152/ajpheart.00008.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatlidede E., Sehirli O., Velioglu-Ogunc A., Cetinel S., Yegen B.C., Yarat A., Suleymanoglu S., Sener G. Resveratrol treatment protects against doxorubicin-induced cardiotoxicity by alleviating oxidative damage. Free Radic. Res. 2009;43:195–205. doi: 10.1080/10715760802673008. [DOI] [PubMed] [Google Scholar]
- 20.Gu J., Song Z.P., Gui D.M., Hu W., Chen Y.G., Zhang D.D. Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in lymphoma nude mice by heme oxygenase-1 induction. Cardiovasc. Toxicol. 2012;12:341–349. doi: 10.1007/s12012-012-9178-7. [DOI] [PubMed] [Google Scholar]
- 21.Costantino S., Akhmedov A., Melina G., Mohammed S.A., Othman A., Ambrosini S., Wijnen W.J., Sada L., Ciavarella G.M., Liberale L., et al. Obesity-induced activation of JunD promotes myocardial lipid accumulation and metabolic cardiomyopathy. Eur. Heart J. 2019;40:997–1008. doi: 10.1093/eurheartj/ehy903. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J., Cheng Y., Gu J., Wang S., Zhou S., Wang Y., Tan Y., Feng W., Fu Y., Mellen N., et al. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clin. Sci. (Lond.) 2016;130:625–641. doi: 10.1042/CS20150623. [DOI] [PubMed] [Google Scholar]
- 23.Gu J., Wang S., Guo H., Tan Y., Liang Y., Feng A., Liu Q., Damodaran C., Zhang Z., Keller B.B., et al. Inhibition of p53 prevents diabetic cardiomyopathy by preventing early-stage apoptosis and cell senescence, reduced glycolysis, and impaired angiogenesis. Cell Death Dis. 2018;9:82. doi: 10.1038/s41419-017-0093-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang C., Huang Z., Gu J., Yan X., Lu X., Zhou S., Wang S., Shao M., Zhang F., Cheng P., et al. Fibroblast growth factor 21 protects the heart from apoptosis in a diabetic mouse model via extracellular signal-regulated kinase 1/2-dependent signalling pathway. Diabetologia. 2015;58:1937–1948. doi: 10.1007/s00125-015-3630-8. [DOI] [PubMed] [Google Scholar]
- 25.Gu J., Cheng Y., Wu H., Kong L., Wang S., Xu Z., Zhang Z., Tan Y., Keller B.B., Zhou H., et al. Metallothionein is downstream of Nrf2 and partially mediates sulforaphane prevention of diabetic cardiomyopathy. Diabetes. 2017;66:529–542. doi: 10.2337/db15-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai Y., Cui W., Xin Y., Miao X., Barati M.T., Zhang C., Chen Q., Tan Y., Cui T., Zheng Y., et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. J. Mol. Cell. Cardiol. 2013;57:82–95. doi: 10.1016/j.yjmcc.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y., Feng W., Xue W., Tan Y., Hein D.W., Li X.K., Cai L. Inactivation of GSK-3beta by metallothionein prevents diabetes-related changes in cardiac energy metabolism, inflammation, nitrosative damage, and remodeling. Diabetes. 2009;58:1391–1402. doi: 10.2337/db08-1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z., Wang S., Zhou S., Yan X., Wang Y., Chen J., Mellen N., Kong M., Gu J., Tan Y., et al. Sulforaphane prevents the development of cardiomyopathy in type 2 diabetic mice probably by reversing oxidative stress-induced inhibition of LKB1/AMPK pathway. J. Mol. Cell. Cardiol. 2014;77:42–52. doi: 10.1016/j.yjmcc.2014.09.022. [DOI] [PubMed] [Google Scholar]
- 29.Yoon J.J., Son C.O., Kim H.Y., Han B.H., Lee Y.J., Lee H.S., Kang D.G. Betulinic acid protects DOX-triggered cardiomyocyte hypertrophy response through the GATA-4/calcineurin/NFAT pathway. Molecules. 2020;26 doi: 10.3390/molecules26010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smeets P.J., Teunissen B.E., Planavila A., de Vogel-van den Bosch H., Willemsen P.H., van der Vusse G.J., van Bilsen M. Inflammatory pathways are activated during cardiomyocyte hypertrophy and attenuated by peroxisome proliferator-activated receptors PPARalpha and PPARdelta. J. Biol. Chem. 2008;283:29109–29118. doi: 10.1074/jbc.M802143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka R., Umemura M., Narikawa M., Hikichi M., Osaw K., Fujita T., Yokoyama U., Ishigami T., Tamura K., Ishikawa Y. Reactive fibrosis precedes doxorubicin-induced heart failure through sterile inflammation. ESC Heart Fail. 2020;7:588–603. doi: 10.1002/ehf2.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong Z., Umemura A., Sanchez-Lopez E., Liang S., Shalapour S., Wong J., He F., Boassa D., Perkins G., Ali S.R., et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fialkow L., Wang Y., Downey G.P. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic. Biol. Med. 2007;42:153–164. doi: 10.1016/j.freeradbiomed.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 34.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X., Hu C., Kong C.Y., Song P., Wu H.M., Xu S.C., Yuan Y.P., Deng W., Ma Z.G., Tang Q.Z. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020;27:540–555. doi: 10.1038/s41418-019-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li S., Wang W., Niu T., Wang H., Li B., Shao L., Lai Y., Li H., Janicki J.S., Wang X.L., et al. Nrf2 deficiency exaggerates doxorubicin-induced cardiotoxicity and cardiac dysfunction. Oxid. Med. Cell. Longev. 2014:748524. doi: 10.1155/2014/748524. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wen S.Y., Tsai C.Y., Pai P.Y., Chen Y.W., Yang Y.C., Aneja R., Huang C.Y., Kuo W.W. Diallyl trisulfide suppresses doxorubicin-induced cardiomyocyte apoptosis by inhibiting MAPK/NF-kappaB signaling through attenuation of ROS generation. Environ. Toxicol. 2018;33:93–103. doi: 10.1002/tox.22500. [DOI] [PubMed] [Google Scholar]
- 38.Ghosh J., Das J., Manna P., Sil P.C. The protective role of arjunolic acid against doxorubicin induced intracellular ROS dependent JNK-p38 and p53-mediated cardiac apoptosis. Biomaterials. 2011;32:4857–4866. doi: 10.1016/j.biomaterials.2011.03.048. [DOI] [PubMed] [Google Scholar]
- 39.Quan N., Sun W., Wang L., Chen X., Bogan J.S., Zhou X., Cates C., Liu Q., Zheng Y., Li J. Sestrin2 prevents age-related intolerance to ischemia and reperfusion injury by modulating substrate metabolism. Faseb. J. 2017;31:4153–4167. doi: 10.1096/fj.201700063R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang D., Han B., Baiyun R., Lv Z., Wang X., Li S., Lv Y., Xue J., Liu Y., Zhang Z. Sulforaphane attenuates hexavalent chromium-induced cardiotoxicity via the activation of the Sesn2/AMPK/Nrf2 signaling pathway. Metallomics. 2020;12:2009–2020. doi: 10.1039/d0mt00124d. [DOI] [PubMed] [Google Scholar]
- 41.Yan J., Yan J.Y., Wang Y.X., Ling Y.N., Song X.D., Wang S.Y., Liu H.Q., Liu Q.C., Zhang Y., Yang P.Z., et al. Spermidine-enhanced autophagic flux improves cardiac dysfunction following myocardial infarction by targeting the AMPK/mTOR signalling pathway. Br. J. Pharmacol. 2019;176:3126–3142. doi: 10.1111/bph.14706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Canto C., Gerhart-Hines Z., Feige J.N., Lagouge M., Noriega L., Milne J.C., Elliott P.J., Puigserver P., Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lear T.B., Lockwood K.C., Ouyang Y., Evankovich J.W., Larsen M.B., Lin B., Liu Y., Chen B.B. The RING-type E3 ligase RNF186 ubiquitinates Sestrin-2 and thereby controls nutrient sensing. J. Biol. Chem. 2019;294:16527–16534. doi: 10.1074/jbc.AC119.010671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar A., Shaha C. RBX1-mediated ubiquitination of SESN2 promotes cell death upon prolonged mitochondrial damage in SH-SY5Y neuroblastoma cells. Mol. Cell. Biochem. 2018;446:1–9. doi: 10.1007/s11010-017-3267-7. [DOI] [PubMed] [Google Scholar]
- 45.Nihira N.T., Ogura K., Shimizu K., North B.J., Zhang J., Gao D., Inuzuka H., Wei W. Acetylation-dependent regulation of MDM2 E3 ligase activity dictates its oncogenic function. Sci. Signal. 2017;10 doi: 10.1126/scisignal.aai8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cappetta D., De Angelis A., Sapio L., Prezioso L., Illiano M., Quaini F., Rossi F., Berrino L., Naviglio S., Urbanek K. Oxidative stress and cellular response to doxorubicin: a common factor in the complex milieu of anthracycline cardiotoxicity. Oxid. Med. Cell. Longev. 2017:1521020. doi: 10.1155/2017/1521020. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang A.J., Zhang J., Xiao M., Wang S., Wang B.J., Guo Y., Tang Y., Gu J. Molecular mechanisms of doxorubicin-induced cardiotoxicity: novel roles of sirtuin 1-mediated signaling pathways. Cell. Mol. Life Sci. 2021;78:3105–3125. doi: 10.1007/s00018-020-03729-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sciarretta S., Forte M., Frati G., Sadoshima J. New insights into the role of mTOR signaling in the cardiovascular system. Circ. Res. 2018;122:489–505. doi: 10.1161/CIRCRESAHA.117.311147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hang P., Zhao J., Sun L., Li M., Han Y., Du Z., Li Y. Brain-derived neurotrophic factor attenuates doxorubicin-induced cardiac dysfunction through activating Akt signalling in rats. J. Cell Mol. Med. 2017;21:685–696. doi: 10.1111/jcmm.13012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sahu R., Dua T.K., Das S., De Feo V., Dewanjee S. Wheat phenolics suppress doxorubicin-induced cardiotoxicity via inhibition of oxidative stress, MAP kinase activation, NF-kappaB pathway, PI3K/Akt/mTOR impairment, and cardiac apoptosis. Food Chem. Toxicol. 2019;125:503–519. doi: 10.1016/j.fct.2019.01.034. [DOI] [PubMed] [Google Scholar]
- 51.Wang X., Li C., Wang Q., Li W., Guo D., Zhang X., Shao M., Chen X., Ma L., Zhang Q., et al. Tanshinone IIA restores dynamic balance of autophagosome/autolysosome in doxorubicin-induced cardiotoxicity via targeting beclin1/LAMP1. Cancers (Basel) 2019;11 doi: 10.3390/cancers11070910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee Y., Kwon I., Jang Y., Cosio-Lima L., Barrington P. Endurance exercise attenuates doxorubicin-induced cardiotoxicity. Med. Sci. Sports Exerc. 2020;52:25–36. doi: 10.1249/MSS.0000000000002094. [DOI] [PubMed] [Google Scholar]
- 53.Cadeddu Dessalvi C., Pepe A., Penna C., Gimelli A., Madonna R., Mele D., Monte I., Novo G., Nugara C., Zito C., et al. Sex differences in anthracycline-induced cardiotoxicity: the benefits of estrogens. Heart Fail. Rev. 2019;24:915–925. doi: 10.1007/s10741-019-09820-2. [DOI] [PubMed] [Google Scholar]
- 54.Meiners B., Shenoy C., Zordoky B.N. Clinical and preclinical evidence of sex-related differences in anthracycline-induced cardiotoxicity. Biol. Sex Differ. 2018;9:38. doi: 10.1186/s13293-018-0198-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.