

Abstract
Tumor redox stimulus-responsive nanoparticulate drug delivery systems (nano-DDSs) have attracted considerable attention due to their thermodynamically stable microstructures and well-controlled drug release properties. However, drug-loading nanoparticle conformation and redox-triggered drug release mechanisms at the molecular level remain unclear. Herein, doxorubicin-conjugated polymers were constructed using disulfide bonds as linkages (PEG–SS–DOX), which loaded photosensitizer chlorin e6 (Ce6). We integrated multiple scale dynamic simulations (density functional theory (DFT) calculation, atomistic molecular dynamics (MD) simulation and dissipative particle dynamics (DPD) simulations) to elucidate the assembly/drug release dynamic processing. First, it was revealed that the emergence of the calculated bond flexible angle of disulfide bonds facilitated the assembly behavior and improved the stability of conformation. Sorted by the binding model, hydrogen bonding accounted for the major interactions between polymers and photosensitive drugs. DPD simulations were further delved into to acquire knowledge regarding the drug-free self-aggregation and Ce6-loaded assembly mechanism. The results show that nano-assembly conformation not only depended on the concentration of polymers, but also were associated with the polymer–drug ratio. Different from dicarbon bond-bridging polymers, disulfide bonds would contribute to the breakage of the polymer and the rapid release of DOX and Ce6. Our findings provide deep insights into the influence of redox-responsive chemical linkages and offer theoretical guidance to the rational design of specific stimulus-responsive nano-DDSs for cancer therapy.
Schematic of disulfide/dicarbide-bridged DOX polymer-encapsulated photosensitive drugs Ce6: a case of computational simulations on the redox-responsive chemo-photodynamic drug delivery system.
Introduction
Malignant tumor remains a fatal disease threatening human health. Chemotherapy is the most common treatment against cancer.1 However, low clinical efficacy along with serious adverse effects has led to unsatisfactory therapeutic effects.2 Therefore, nanoparticulate drug delivery systems (nano-DDSs) have been developed to resolve the above-mentioned problems. The success of several anticancer nano-formulations (e.g. Abraxane and Doxil) in the treatment of diverse cancers lends significant therapeutic promise as it translates from bench to bedside.3,4 However, despite distinctly reduced toxicity, the chemotherapeutic nano-DDSs alone cannot fully satisfy the clinical requirements, and the combinational treatment is still essential to enhance the therapeutic effectiveness.
Apart from chemotherapy, photodynamic therapy (PDT) as a clinically approved therapeutic regimen, holds an appealing prospect against cancer.5,6 Photosensitizer (PS) exposure upon laser irradiation would generate abundant toxic reactive oxygen species (ROS) to kill the tumor without systemic side-effects.7 Combining chemotherapy and PDT provides tremendous advantages: (i) multiple anti-cancer mechanisms against multi-drug resistance; (ii) reduce unwanted side-effects via reducing the dosage; and (iii) remarkable synergistic clinical outcomes. Despite these beneficial aspects, it was a huge challenge to guarantee synchronous chemotherapeutic drug and PS delivery for achieving high-efficiency chemo-photodynamic treatment. The conventional nano-DDSs have been utilized to co-encapsulate two types of drugs for combination therapy. On account of different physicochemical properties between chemical drugs and PSs, the conventional formulations suffered from low drug loading, premature leakage in vivo and material-associated toxicity.8,9 Therefore, it is critical to rationally design nano-DDSs for high-efficiency co-loading, co-delivery and on-demand release of drugs and PSs.
Smart nano-DDSs, which respond to intratumoral biological characteristics, have been developed for on-demand drug release at the tumor regime.10 Compared with normal tissues, overexpressed reactive oxygen species (ROS) and glutathione (GSH) resulted in the redox-heterogeneous intratumoral microenvironment.11 Disulfur bonds, as a well-known redox-responsive chemical linkage, had been generously introduced into the design of tumor-specific nano-DDSs for cancer therapy.12–14 Furthermore, a large number of studies suggested that the assembly capacity of nano-DDSs strengthened, followed by the insertion of this bridging group. The flexible disulfide bond linker reduced the rigidity of the molecular structure and balanced the interactions among intermolecules.12 Therefore, a small disulfide bond played a big role in the assembly stability of nano-DDSs and controlled drug release at tumor sites. Although great progress of disulfur bond-based nano-DDSs had been reported, the knowledge underlining the drug-free assembly formation, drug loading and release mechanisms at the molecular level are still scarce. It is of great importance for researchers to study the basis for the rational design of smart nano-DDSs via both experiments and theoretical calculations.
Herein, we attempted to construct the redox-responsive chemo-photodynamic nanoparticles (NPs), that is, a disulfur bond-bridging doxorubicin (DOX)-grafted PEG polymer (PEG–S–S–DOX), encapsulating the photosensitive drug chlorin e6 (Ce6). A systematic investigation containing density functional theory (DFT) calculations, atomistic molecular dynamics (MD) simulations and dissipative particle dynamics (DPD) simulations was utilized to reveal the dynamic behavior of assembly/release at the microscopic scale in this study. The dihedral angles and partial charges were compared to explain the differences between dicarbon and disulfur bonds. We investigated the intermolecular interactions between dicarbon-/disulfur bond-bridging polymers and Ce6. DPD simulations were performed to elucidate the self-aggregation of drug-free PEG–S–S–DOX NPs and the mesoscopic morphologies of Ce6-loading NPs at different polymer/drug ratios and concentrations. Furthermore, the redox-responsive drug release mechanism of Ce6-loading NPs was also evaluated. This research would be necessary for guiding the clinical application of smart nano-DDSs in rational formulation design through the computational simulation strategy.
Results and discussion
Disulfur/dicarbon bond characteristics
According to previous reports, the flexible bond angle of the disulfur bond could enhance the structural stability of nanoassemblies. While the dihedral angle of the disulfur bond is closed to 90°, the established structure would be the most stable.12 Thus, the dihedral angle of the disulfur/dicarbon bond was first calculated, as shown in Fig. 1: –C–CC–C– (179.9°) and –C–SS–C– (85.9°). Compared with the dicarbon bond, the disulfur bond approaches 90°, which would be able to balance the intermolecular interactions to form stable nanoassemblies. Furthermore, we investigated the possible broken bond mechanism though computing the bind energy/partial charge of the disulfur/dicarbon bond. From the thermodynamic view, the lower bind energy (ΔG < 0) represented a chemical bond less susceptible to breakage.15,16 As shown in Fig. 1, the calculated bind energy values of disulfur/dicarbon bonds were as follows: –C–CC–C– (−471.3 kcal mol−1) <–C–SS–C–(−262.5 kcal mol−1). The partial charge of both bonds could be calculated via DFT calculations. The disulfur bond (−0.040) had a lower charge potential than the dicarbon bond (0.044), thereby undergoing electrophilic substitution more easily. These results suggested that the simple insertion of disulfur bond would enhance the assembly stability and trigger the sensitive broken process.
Fig. 1. Partial charges and dihedral angles of the disulfur bond/dicarbon bond, the molecular docking between the disulfur bond/dicarbon bond-contained polymers and photosensitive drug Ce6.

Polymer–drug interactions via molecular docking
Based on the above-mentioned mechanisms, we constructed novel DOX-conjugated disulfur bond-bridging PEG (PEG–S–S–DOX) NPs to encapsulate the hydrophobic photosensitive drugs Ce6 for the chemo-photodynamic cancer therapy. First, the molecular docking was conducted to reveal Ce6–carrier interactions at the molecular level. The schematic representation of the binding model is shown in Fig. 1. These results demonstrated that disulfur bond-contained polymers (kcal mol−1) had stronger interactions with Ce6 than dicarbon bond-contained polymers (kcal mol−1). The flexible disulfur bond-based polymer was easy to bend and ultimately assembled the stable conformation via multiple hydrogen bonds and hydrophobic interactions, indicating that these redox-responsive nanoassemblies were promising for further investigation.
The assembly/release mechanisms of NPs though DPD simulation
The DPD simulation was used to understand the assembly/dissociation mechanisms at the macroscopic time and space scale.15,17 Herein, the schematic representation of the coarse grained model is shown in Fig. 2.
Fig. 2. Schematic of the coarse grain scheme for the redox-responsive chemo-photodynamic NPs: the bead model of PEG–S–S–DOX for which seven beads were defined to constitute one PEG molecule “P”, two beads represent the disulfide bond “S”, one DOX molecule was divided into seven beads “D”. One single photosensitive drug Ce6 molecule was designated to show eight beads “C”.

As illustrated in Fig. 3, the equilibrium conformation of self-assembled PEG–S–S–DOX NPs was studied at different (1%, 5%, 10%, 20% and 40%) concentrations. It was observed that 1% concentration PEG–S–S–DOX could spontaneously construct “core–shell” NPs, that is, hydrophilic PEG encapsulated the hydrophobic DOX into the core of nano-assembly. Following the increase in the concentration of polymers (20%), precipitation could occur. Thus, the NPs at 10% concentration were selected to investigate the drug loading capacity of this polymer.
Fig. 3. The equilibrium morphologies and local sectional view of drug-free DOX–S–S–PEG NPs under various concentrations (1%, 5%, 10%, 20% and 40%).

Moreover, the photosensitive drug Ce6-loaded core–shell nanoassemblies can be observed from Fig. 4. At a 5 : 1 or 1 : 1 carrier/drug ratio, the hydrophobic Ce6 along with DOX from the polymer were entrapped into the core of NPs; the hydrophilic PEG chains were outside the aqueous phase. However, an evident separated phase in the water box was found under the 1 : 5 carrier/drug ratio, demonstrating that high drug-loaded NPs were unstable. The 1 : 1 NPs were chosen as the model, which was used to further evaluate the drug release mechanism.
Fig. 4. The equilibrium morphologies and local sectional view of photosensitive drug Ce6-loading NPs at various polymer/drug ratios (5 : 1, 1 : 1 and 1 : 5).

The Ce6 and DOX release process from redox-responsive drug-loaded nanoassemblies under a high redox environment were simulated via DPD simulations. At 0 steps, Ce6 was loaded into the core of PEG–S–S–DOX NPs. Under the simulated tumor environment (high redox condition), disulfide bonds from polymers broke. Drug-loaded NPs start at swelling instantly. Then, the solvation effect could destruct the hydrogen bonds. Also, Ce6 along with DOX were released out of this system exposed to water. Fig. 5B also shows the dynamic process of non-responsive NPs. In the beginning, the Ce6-loading NPs were randomly mixed at 0 steps. These non-responsive nano-aggregations would be unchanged constantly.
Fig. 5. The release process of Ce6-contained redox-responsive (A) and non-responsive (B) NPs at different steps.

Conclusion
In our study, we investigated the assembly/release mechanisms of redox-responsive chemo-photodynamic NPs for cancer therapy via multiple-scale computational simulations. The results revealed that the insertion of a smart bisulfide bond with the flexible dihedral angle would turn the photosensitive drug Ce6 and the DOX–S–S–PEG polymer into nano-assemblies with high-assembly stability and on-demand drug release. Moreover, Ce6-contained NPs via DPD simulations showed that 1 : 1 or 1 : 5 drug/polymer ratios could form shell–core NPs, with the hydrophobic Ce6 loaded in the hydrophilic DOX–S–S–PEG polymer shell. Hydrogen bond and hydrophobic interactions between drugs and polymers were found to be critical determinants in facilitating the assembly formation. Moreover, the bisulfide bond-bridging NPs could effectively release the drugs (DOX and Ce6) under high redox microenvironment, whereas dicarbon bond-bridging NPs would constantly prevent drug release. This research demonstrated the superiority of smart bisulfide bond NPs in drug delivery at the molecular level, providing the theoretical perspectives for the design of novel stimulus-responsive chemo-photodynamic nanomedicines against multiple malignant cancers.
Materials
The copyrights of the softwares (Schrödinger and Materials Studio) belong to Shenyang Pharmaceutical University. They have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Simulation methods
Density functional theory (DFT) calculation and molecular docking
We performed the DFT method to calculate molecule A and molecule B (Fig. 6); all DFT calculations were performed using Jaguar module in Schrödinger.20 DFT calculations were performed in the 6-311(d,p)+ base set and in M06-2x method.18,19 After optimization, we calculated the bond energy and torsion of the dicarbon bond in molecule A and the disulfur bond in molecule B as well as partial charges of molecules A and B. We also predicted the binding mode and binding energy between disulfur/dicarbon bond-bridging polymers and DOX in the molecular docking method. Docking was performed using Glide module in Schrödinger; disulfur/dicarbon bond-contained polymers were suggested as receptors; and DOX was docked onto the polymer; and binding energy was calculated.
Fig. 6. (A) Disulfur bond-contained chain; (B) dicarbon bond-contained chain.

DPD simulation
DPD is a mesoscopic simulation method for studying soft matter systems over greater length and time scales.15,17 It was introduced by Hoogerbrugge and Koelman in 1992, and was successfully used to study the self-assembly and phase separation processes of polymer systems.21 In general, a DPD bead represents a group of atoms or a volume of fluid that is large on the atomistic scale but still macroscopically small. For simplicity, the beads in simulation are equal in mass and volume, and the bead mass m, the radius of interaction cutoff rc, and the thermal energy are set as the units of the simulations, i.e., m = rc = kBT ≡ 1, so the time unit 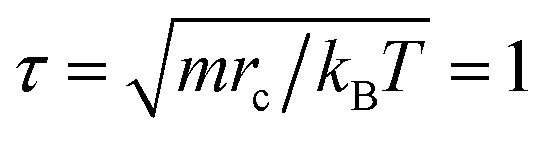 . The force acting on each bead is at least composed of three parts: conservative force FC, dissipative force FD, and random force FR.
. The force acting on each bead is at least composed of three parts: conservative force FC, dissipative force FD, and random force FR.
| Fij = FCij + FDij + FRij | 1 |
They are given by the following:
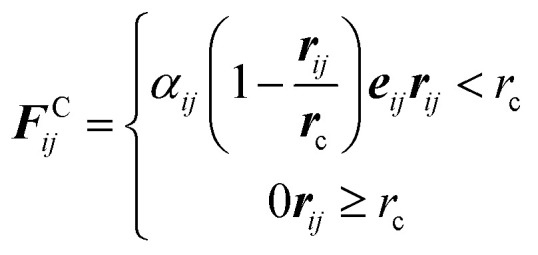 |
2 |
| FDij = −γωD(rij)(vij·eij)eij | 3 |
| FRij = σωR(rij)ξijΔt−1/2eij | 4 |
Here rij = ri − rj, rij = |rij|, eij = rij/rij, vij = vi − vj, ξij is a random number with zero mean and unit variance. According to Espaeñol and Warren, ωD and ωR are two weight functions for the dissipative and random forces, with
| ωD(rij)=[ωR(rij)]2, σ2 = 2γkBT | 5 |
We choose a simple form of ωD and ωR following Groot and Warren:22
 |
6 |
The friction parameter is γ = 4.5. The time evolution of interacting beads obeys Newton's equations of motion.23 The canonical ensemble simulations are performed in a three-dimensional box of size Lx × Ly × Lz = 30 × 30 × 30. The integration time step Δt = 0.02. The coarse-grained scheme for this system is shown in Fig. 2.
The interaction parameter αij in eqn (2) is a constant, which describes the maximum repulsion between interacting beads. It takes the value of α = 25 for the beads of the same type. This value reflects the incompressibility of typical fluid at room temperature. In fact, Δαij = αij − 25 can be mapped to the Flory–Huggins χ parameter through Δαij = 3.27χij for the system with reduced number density ρ = 3.24 Also, the parameter χ can be “translated” from the solubility parameters by using eqn (7):
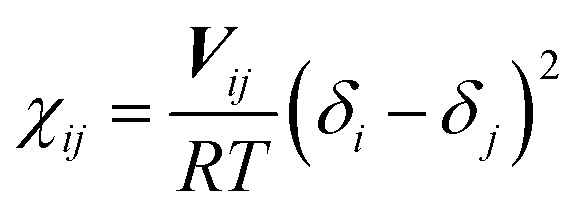 |
7 |
Here, Vij is the average molar volume of the bead, R is the gas constant, T is the absolute temperature, and δi and δj are the solubility parameters for the chemical entities i and j, respectively, which can be calculated with molecular dynamic (MD) simulations. All MD simulations are performed using the Materials Studio Program (Accelrys Inc.). First, an amorphous cell was used to construct molecular aggregation models. Then, 50 000 steps energy minimization are applied followed with 10 ns equilibrium MD with COMPASS in 298 K and NPT ensemble.25 Finally, cohesive energy density and solubility parameters are calculated by using Forcite module. In this simulation, there are three types of the polymer chain, i.e., one type for PEG (for short as “P”), one type for DOX (for short as “D”), and one type for disulfide (for short as “S”). There are three other particles in this system, i.e., one type for photosensitive drug Ce6 (for short as “C”), one type for the carbon–carbon bond (for short as “T”), and one type for solvent water (for short as “W”). Because DOX and Ce6 can carry electric charge in the acid system, so we use D+ and C+ to represent DOX and Ce6 in the acid system. The interaction parameters in this simulation between different kinds of beads are shown in Table 1.
The interaction parameters between different kinds of beads.
| α | P | S | D | D + | C | C + | T | W |
|---|---|---|---|---|---|---|---|---|
| P | 25 | 25 | 28 | 25 | 29 | 25 | 25 | 28 |
| S | 25 | 27 | 25 | 27 | 27 | — | 30 | |
| D | 25 | — | 25 | — | 26 | 37 | ||
| D + | 25 | — | 26 | — | 28 | |||
| C | 25 | — | 27 | 39 | ||||
| C + | 25 | — | 25 | |||||
| T | 25 | 31 | ||||||
| W | 25 |
Conflicts of interest
The authors declare no competing financial interests.
Supplementary Material
Acknowledgments
This work was supported by the Young Talents Project of Huzhou Central Hospital (No. 2020YC09).
References
- Smith R. A. Andrews K. S. Brooks D. Fedewa S. A. Manassaram-Baptiste D. Saslow D. Brawley O. W. Wender R. C. Ca-Cancer J. Clin. 2017;67(2):100–121. doi: 10.3322/caac.21392. [DOI] [PubMed] [Google Scholar]
- Bocci G. Kerbel R. S. Nat. Rev. Clin. Oncol. 2016;13(11):659–673. doi: 10.1038/nrclinonc.2016.64. [DOI] [PubMed] [Google Scholar]
- Barenholz Y. C. J. Controlled Release. 2012;160(2):117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- Green M. R. Manikhas G. M. Orlov S. Afanasyev B. Ann. Oncol. 2006;17(8):1263–1268. doi: 10.1093/annonc/mdl104. [DOI] [PubMed] [Google Scholar]
- Ni K. Luo T. Lan G. Culbert A. Song Y. Wu T. Jiang X. Lin W. Angew. Chem., Int. Ed. 2020;59(3):1108–1112. doi: 10.1002/anie.201911429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian P. Warloe T. Berg K. Moan J. Kongshaug M. Giercksky K. Nesland J. M. Cancer. 2010;65(2):235–251. doi: 10.1002/(sici)1097-0142(19970615)79:12<2282::aid-cncr2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Liu Z. Cao T. Xue Y. Li M. Zhang W. Angew. Chem., Int. Ed. 2020;59(9):3711–3717. doi: 10.1002/anie.201914434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee D. Fong L. Zhang Y. Adv. Drug Delivery Rev. 2008;60(15):1627–1637. doi: 10.1016/j.addr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Zhang W. Shen J. Su H. Mu G. Sun J. H. Tan C. P. Liang X. J. Ji L. N. Mao Z. W. ACS Appl. Mater. Interfaces. 2016:13332–13340. doi: 10.1021/acsami.6b03881. [DOI] [PubMed] [Google Scholar]
- Dong L. Fang Y. Xiong F. Ning G. Theranostics. 2016;6(9):1306–1323. doi: 10.7150/thno.14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wa Ng Q. Sun M. Li D. Li C. Sun J. Theranostics. 2020;10(12):5550–5564. doi: 10.7150/thno.42633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. Liu D. Zheng Q. Zhao Q. Zhang H. Ma Y. Fallon J. Fu Q. Haynes M. Lin G. Zhang R. Wang D. Yang X. Zhao L. He Z. Liu F. Nano Lett. 2014;14(10):5577–5583. doi: 10.1021/nl502044x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W. Luo C. Yang J. Sun B. Zhao D. Liu Y. Wang Y. Yang W. Kan Q. Sun J. He Z. J. Controlled Release. 2018;285:187–199. doi: 10.1016/j.jconrel.2018.07.010. [DOI] [PubMed] [Google Scholar]
- Sun B. Cong L. Han Y. Zhang X. Sun J. Nano Lett. 2018;18(6):3643–3650. doi: 10.1021/acs.nanolett.8b00737. [DOI] [PubMed] [Google Scholar]
- Sun M. Zhang X. Gao Z. Liu T. Luo C. Zhao Y. Liu Y. He Z. Wang J. Sun J. Nanoscale. 2019;11(9):3864–3876. doi: 10.1039/C8NR07014H. [DOI] [PubMed] [Google Scholar]
- Sun M. Wu C. Fu Q. Di D. Kuang X. Wang C. He Z. Wang J. Sun J. Int. J. Pharm. 2016:238–246. doi: 10.1016/j.ijpharm.2016.01.062. [DOI] [PubMed] [Google Scholar]
- Sun M. Li B. Li Y. Liu Y. Liu Q. Jiang H. He Z. Zhao Y. Sun J. Int. J. Pharm. 2016:185–195. doi: 10.1016/j.ijpharm.2016.11.049. [DOI] [PubMed] [Google Scholar]
- Chahkandi B. Chahkandi M. J. Mol. Model. 2020;26(6):151. doi: 10.1007/s00894-020-04382-9. [DOI] [PubMed] [Google Scholar]
- Filho A. H. d. S. Candeias F. S. da Silva S. C. Vicentini F. C. Assumpção M. H. M. T. Brown A. de Souza G. L. C. J. Mol. Model. 2020;26(11):309. doi: 10.1007/s00894-020-04567-2. [DOI] [PubMed] [Google Scholar]
- Lin H. Hu B. He X. Mao J. Wang Y. Wang J. Zhang T. Zheng J. Peng Y. Zhang F. Biochem. Pharmacol. 2020;171:113733. doi: 10.1016/j.bcp.2019.113733. [DOI] [PubMed] [Google Scholar]
- Hoogerbrugge P. J. Koelman J. Epl. 2007;19(3):155. doi: 10.1209/0295-5075/19/3/001. [DOI] [Google Scholar]
- Search H.; Contact A.; Iopscience M.; Address I. P.1995
- Groot R. D. Warren P. B. J. Chem. Phys. 1998;107(11):4423–4435. doi: 10.1063/1.474784. [DOI] [Google Scholar]
- Groot R. D. Madden T. J. J. Chem. Phys. 1998;108(20):8713–8724. doi: 10.1063/1.476300. [DOI] [Google Scholar]
- Sun H. Jin Z. Yang C. Akkermans R. L. C. Robertson S. H. Spenley N. A. Miller S. Todd S. M. J. Mol. Model. 2016;22(2):47. doi: 10.1007/s00894-016-2909-0. [DOI] [PubMed] [Google Scholar]