

Summary
Frontotemporal dementia (FTD) therapy development is hamstrung by a lack of susceptibility, diagnostic, and prognostic biomarkers. Blood neurofilament light (NfL) shows promise as a biomarker, but studies have largely focused only on core FTD syndromes, often grouping patients with different diagnoses. To expedite the clinical translation of NfL, we avail ARTFL LEFFTDS Longitudinal Frontotemporal Lobar Degeneration (ALLFTD) study resources and conduct a comprehensive investigation of plasma NfL across FTD syndromes and in presymptomatic FTD mutation carriers. We find plasma NfL is elevated in all studied syndromes, including mild cases; increases in presymptomatic mutation carriers prior to phenoconversion; and associates with indicators of disease severity. By facilitating the identification of individuals at risk of phenoconversion, and the early diagnosis of FTD, plasma NfL can aid in participant selection for prevention or early treatment trials. Moreover, its prognostic utility would improve patient care, clinical trial efficiency, and treatment outcome estimations.
Keywords: behavioral variant frontotemporal dementia, biomarker, corticobasal syndrome, neurofilament light, plasma, primary progressive aphasia, presymptomatic, progressive supranuclear palsy, Richardson’s syndrome
Graphical abstract
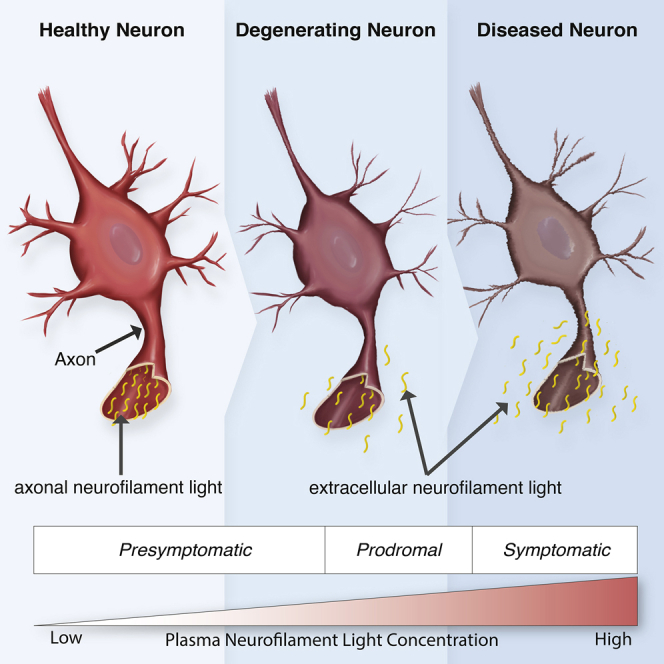
Highlights
-
•
Plasma NfL levels increase prior to frontotemporal dementia (FTD) symptom onset
-
•
Plasma NfL can facilitate an earlier FTD diagnosis
-
•
Plasma NfL levels associate with clinical indicators of FTD disease severity
-
•
Plasma NfL shows promise as a susceptibility and prognostic biomarker for FTD
Gendron et al. show that plasma neurofilament light (NfL) increases before symptom onset in individuals with a genetic risk of frontotemporal dementia (FTD) and associates with clinical indicators of disease severity. Plasma NfL thus holds promise as a susceptibility and prognostic biomarker that can improve patient care and FTD treatment development.
Introduction
Frontotemporal dementia (FTD) is a term for disorders marked by behavior, language, executive function, and/or motor impairments. Approximately 30% of FTD cases are genetic; mutations in chromosome 9 open reading frame 72 (C9orf72), progranulin (GRN), or microtubule-associated protein tau (MAPT) most often being the cause.1 While FTD syndromes can have overlapping symptoms,2 the dominant presenting feature in patients with behavioral variant FTD (bvFTD), the most common FTD syndrome, is a change in personality or behavior associated with executive dysfunction. Individuals with nonfluent and agrammatic variant primary progressive aphasia (nfvPPA) experience nonfluent speech output, agrammatism, and telegraphic speech, while those with semantic variant PPA (svPPA) develop word loss and object knowledge and comprehension deficits. The FTD spectrum also includes bvFTD plus amyotrophic lateral sclerosis (FTD-ALS) and the parkinsonian disorders corticobasal syndrome (CBS) and progressive supranuclear palsy-Richardson syndrome (PSP-RS).
Because of substantial clinical heterogeneity within and among FTD syndromes, establishing methods to predict prognosis is an important endeavor. Prognostic biomarkers improve drug development by ensuring balanced patient groups (e.g., equal proportions of slow and fast progressors) in clinical trial treatment arms and by providing a means to estimate therapeutic benefit. Studies that investigated cerebrospinal fluid (CSF) neurofilament light (NfL), a measure of neuroaxonal damage, as a biomarker of FTD severity or progression found that higher CSF NfL generally predicts worse brain atrophy and neuropsychological performance as well as shorter survival.3, 4, 5, 6, 7, 8, 9, 10, 11 Yet a blood biomarker is more practical and economical, especially when monitoring biomarkers longitudinally. Most of the largely cross-sectional studies examining the prognostic power of blood NfL for FTD focused on bvFTD, nfvPPA, and svPPA,5,12, 13, 14, 15, 16, 17, 18, 19, 20 with studies on CBS and PSP-RS being rare.21, 22, 23 In only some analyses did blood NfL correlate with indicators of disease severity. The discrepant findings among studies may result from small cohort sizes and clinical variability among FTD syndromes. Indeed, several studies grouped patients with different syndromes together to increase statistical power, but doing so may mask associations of interest or incorrectly ascribe findings to a particular diagnostic group. There is thus a need for more rigorous investigations on the prognostic utility of blood NfL for FTD syndromes if this biomarker is to become clinically useful.
Blood NfL may also improve patient care and treatment development by aiding in the earlier diagnosis of FTD and monitoring presymptomatic disease progression in individuals with FTD-causing gene mutations. Therapies are expected to be most effective when initiated early in the disease course and, until there is a biomarker to forecast phenoconversion, recruiting presymptomatic mutation carriers to prevention or early treatment trials will remain challenging. Fortunately, findings suggest that blood NfL increases as presymptomatic FTD mutation carriers approach prodromal and disease stages.20,24, 25, 26 However, because the number of phenoconverters in each study was small, additional investigations are crucial if we are to establish whether NfL represents a viable phenoconversion biomarker.
Toward expediting the translation of plasma NfL to the clinic and creating a major informational database for FTD investigators and the scientific community, we undertook a comprehensive cross-sectional and longitudinal study to evaluate the staging and prognostic utility of plasma NfL across FTD syndromes. To do so, we availed the resources of the ARTFL LEFFTDS Longitudinal Frontotemporal Lobar Degeneration (ALLFTD) (http://www.allftd.org) study, which allowed us to measure plasma NfL in the largest series of well-characterized presymptomatic FTD mutation carriers and patients with bvFTD, nfvPPA, svPPA, CBS, or PSP-RS.
Results
Patient characteristics
We measured baseline plasma NfL in clinically normal, mutation-negative individuals in kindreds with an FTD-causing mutation (controls; n = 144), presymptomatic individuals with a C9orf72 repeat expansion or a GRN or MAPT mutation (n = 85), and patients with sporadic or genetic bvFTD (n = 289), nfvPPA (n = 72), svPPA (n = 84), CBS (n = 89), or PSP-RS (n = 124). Patients with FTD-ALS (n = 25), ALS (n = 12), and mild cognitive or behavioral changes (mild cognitive impairment [MCI]) (n = 57) were included for comparison in some analyses. Demographic and clinical data are presented in Table 1. Table S1 indicates the number of presymptomatic phenoconverters, non-converters, and individuals in other phenotype groups with baseline and longitudinal NfL measures and for whom rates of change in NfL could be determined.
Table 1.
Subject characteristics according to phenotype groups
| Variable | Median (minimum, maximum) or no. (%) of subjects |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Controls (n = 144) | Presymptomatic mutation carriers (n = 85) | bvFTD (n = 289) | nfvPPA (n = 72) | svPPA (n = 84) | CBS (n = 89) | PSP-RS (n = 124) | MCI (n = 57) | FTD-ALS (n = 25) | ALS (n = 12) | |
| Age at baseline (years) | 53 (40, 80) | 49 (40, 71) | 62 (32, 85) | 70 (49, 86) | 66 (50, 88) | 68 (40, 87) | 69 (48, 82) | 60 (27, 82) | 61 (45, 75) | 61 (48, 70) |
| Gender (male) | 49 (34.0%) | 41 (48.2%) | 170 (58.8%) | 33 (45.8%) | 43 (51.2%) | 46 (51.7%) | 63 (50.8%) | 29 (50.9%) | 14 (56.0%) | 8 (66.7%) |
| Age at symptom onset (years) | NA | NA | 57 (26, 80) | 64 (44, 81) | 60 (17, 81) | 64 (32, 82) | 64 (47, 79) | 53 (20, 78) | 60 (35, 69) | 59 (48, 69) |
| Symptom duration at baseline (years) | NA | NA | 5 (0, 32) | 4 (1, 12) | 6 (1, 34) | 4 (0, 32) | 5 (1, 20) | 3 (0, 54) | 3 (1, 15) | 2 (0, 10) |
| Mutation status | ||||||||||
| None | 144 (100.0%) | 0 (0.0%) | 188 (67.6%) | 62 (89.9%) | 78 (94.0%) | 75 (89.3%) | 115 (98.3%) | 28 (50.0%) | 17 (68.0%) | 4 (33.3%) |
| C9orf72 | 0 (0.0%) | 35 (41.2%) | 43 (15.5%) | 1 (1.4%) | 2 (2.4%) | 2 (2.4%) | 1 (0.9%) | 15 (26.8%) | 8 (32.0%) | 8 (66.7%) |
| GRN | 0 (0.0%) | 26 (30.6%) | 15 (5.4%) | 6 (8.7%) | 1 (1.2%) | 5 (6.0%) | 0 (0.0%) | 7 (12.5%) | 0 (0.0%) | 0 (0.0%) |
| MAPT | 0 (0.0%) | 22 (25.9%) | 28 (10.1%) | 0 (0.0%) | 0 (0.0%) | 1 (1.2%) | 1 (0.9%) | 6 (10.7%) | 0 (0.0%) | 0 (0.0%) |
| C9orf72 and GRN | 0 (0.0%) | 2 (2.4%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| Other | 0 (0.0%) | 0 (0.0%) | 4 (1.4%) | 0 (0.0%) | 2 (2.4%) | 1 (1.2%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| Unknown | 0 | 0 | 11 | 3 | 1 | 5 | 7 | 1 | 0 | 0 |
| Years of education | 16 (12, 22) | 16 (10, 22) | 16 (6, 26) | 16 (10, 24) | 16 (12, 21) | 16 (12, 26) | 16 (12, 24) | 16 (9, 20) | 16 (12, 20) | 16 (12, 18) |
| CDR + NACC-FTLD global score | ||||||||||
| 0 | 144 (100.0%) | 85 (100.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 2 (2.2%) | 1 (0.8%) | 1 (1.8%) | 1 (4.0%) | 3 (25.0%) |
| 0.5 | 0 (0.0%) | 0 (0.0%) | 19 (6.6%) | 27 (37.5%) | 11 (13.1%) | 24 (27.0%) | 22 (17.7%) | 56 (98.2%) | 2 (8.0%) | 5 (41.7%) |
| 1 | 0 (0.0%) | 0 (0.0%) | 96 (33.2%) | 31 (43.1%) | 48 (57.1%) | 38 (42.7%) | 49 (39.5%) | 0 (0.0%) | 9 (36.0%) | 1 (8.3%) |
| 2 | 0 (0.0%) | 0 (0.0%) | 148 (51.2%) | 12 (16.7%) | 24 (28.6%) | 22 (24.7%) | 42 (33.9%) | 0 (0.0%) | 10 (40.0%) | 3 (25.0%) |
| 3 | 0 (0.0%) | 0 (0.0%) | 26 (9.0%) | 2 (2.8%) | 1 (1.2%) | 3 (3.4%) | 10 (8.1%) | 0 (0.0%) | 3 (12.0%) | 0 (0.0%) |
| Number of individuals with longitudinal plasma samples | 79 | 58 | 39 | 10 | 2 | 14 | 10 | 18 | – | – |
| Number of plasma visits | ||||||||||
| 2 | 41 (51.9%) | 27 (46.6%) | 23 (59.0%) | 5 (50.0%) | 2 (100.0%) | 9 (64.3%) | 6 (60.0%) | 7 (38.9%) | – | – |
| 3 | 30 (38.0%) | 19 (32.8%) | 12 (30.8%) | 4 (40.0%) | 0 (0.0%) | 5 (35.7%) | 3 (30.0%) | 6 (33.3%) | – | – |
| 4 | 8 (10.1%) | 12 (20.7%) | 4 (10.3%) | 1 (10.0%) | 0 (0.0%) | 0 (0.0%) | 1 (10.0%) | 5 (27.8%) | – | – |
| Time from baseline to last plasma sampling (years) | 1.9 (0.5, 3.8) | 2.0 (0.7, 3.8) | 1.2 (0.5, 3.2) | 0.2 (1.1, 3.1) | 1.1 (1.0, 1.3) | 0.8 (0.4, 3.1) | 0.8 (0.5, 3.2) | 2.1 (1.0, 3.4) | – | – |
Other mutations include an intermediate C9orf72 expansion (n = 1 CBS patient), a likely pathogenic GRN variant (n = 1 bvFTD patient), and TARDBP mutations (n = 3 bvFTD patients and n = 2 svPPA patients). Data for age at symptom onset and for symptom duration at baseline were not available for one patient with nfvPPA, two patients with MCI, and two patients with FTD-ALS, CDR + NACC-FTLD, CDR Dementia Staging Instrument plus behavior and language domains from the National Alzheimer’s Disease Coordinating Center FTLD module. See also Table S1.
Associations of baseline plasma NfL concentrations with age, gender, and symptom duration (time from symptom onset to plasma collection) are shown in Figures 1 and S1A–S1C and in Table S2. After correction was made for multiple testing, increased NfL was associated with older age in controls, presymptomatic mutation carriers, and patients with bvFTD, CBS, or MCI (p ≤ 0.013) and with shorter symptom duration in patients with bvFTD (p = 0.002). NfL was higher in females than in males for controls and bvFTD patients (p ≤ 0.001). Because of these associations, we discuss below data from analyses adjusted for age and gender and, when relevant, also for symptom duration and education. Data from unadjusted analyses are provided in Tables S3–S6, S8, and S11–S13.
Figure 1.

Plasma NfL concentrations associate with age, gender, and symptom duration in some phenotype groups
Associations of baseline NfL concentrations with age (A), gender (B), and symptom duration (C) assessed using linear regression models adjusted for age, gender, and symptom duration. β coefficients (β), 95% confidence intervals (CIs), and p values are shown for significant associations, where p < 0.025 is considered significant for controls and presymptomatic mutation carriers and p <0.0167 is considered significant for symptomatic groups. In (B), black horizontal bars represent median NfL concentrations. Gray circles represent 11 bvFTD patients and five CBS patients with unknown mutation status, one bvFTD patient with a likely pathogenic GRN variant and three with a TARDBP mutation, and one CBS patient with an intermediate C9orf72 repeat expansion. NfL in plasma samples was measured in duplicate, and the mean concentration of replicates is shown on the base 10 logarithm scale. See Figures S1A–S1C to view NfL concentrations on the linear scale and also see Table S2. The number of individuals per group (n) is shown in figure panels and in Tables 1 and S1.
Plasma NfL is elevated in FTD and discriminates controls from patients with mild to severe impairment
We compared baseline plasma NfL concentrations between controls and presymptomatic carriers with all groups (Figures 2A and S1D; Tables S3 and S4). Presymptomatic individuals had higher NfL compared with controls (p < 0.001), which may be driven by their eventual phenoconversion. Indeed, compared to controls and to the 43 mutation carriers who remained asymptomatic for at least 1 year from baseline, the 14 presymptomatic carriers who later phenoconverted had higher NfL (p < 0.001; Figures 2B and S1E; Table S5). NfL was also higher in non-converters than in controls (p = 0.006). The 28 presymptomatic individuals for whom conversion status could not be determined because of limited follow-up data (Table S1) were excluded from these analyses. For phenoconverters, median time from baseline to conversion was 1.3 years (range: 1.0–2.8 years), whereas median time from baseline to last follow-up for non-converters was 2.2 years (range: 1.0–4.8 years).
Figure 2.

Baseline plasma NfL is elevated in presymptomatic mutation carriers and all symptomatic groups
(A) Comparison of baseline plasma NfL between healthy controls or presymptomatic mutation carriers and symptomatic groups.
(B) Comparison of baseline NfL between presymptomatic carriers who phenoconverted and controls or presymptomatic carriers who remained asymptomatic for at least 1 year.
(C) Comparison of baseline NfL between controls or presymptomatic carriers and patients in symptomatic groups with a CDR + NACC-FTLD global score of 0 or 0.5.
(D) Heatmap showing AUCs comparing controls to the indicated groups that include all individuals of a given group (all subjects) or only those with an CDR + NACC-FTLD global score of 0 or 0.5 (mildly impaired) from unadjusted or age- and gender-adjusted analyses.
(E) Comparison of baseline plasma NfL between non-mutation carriers and mutation carriers for clinically normal individuals and patients with bvFTD or MCI. Data in (E) do not include the two presymptomatic carriers with a mutation in both C9orf72 and GRN. p values are from analysis adjusted for age and gender.
∗∗∗p < 0.001 and ∗∗p < 0.01, comparison to controls; ###p < 0.001 and ##p = 0.003, comparison to presymptomatic carriers; ǂǂǂp < 0.001, comparison with presymptomatic non-converters. Horizontal bars represent median NfL concentrations. NfL in plasma samples was measured in duplicate, and the mean concentration of replicates is shown on the base 10 logarithm scale. See Figures S1D and S1E to view NfL concentrations on the linear scale. Relating to (A)–(D), see also Tables S3–S7. Relating to (E), see also Tables S8 and S9. The number of individuals per group (n) is shown in figure panels and in Tables 1 and S1.
Baseline NfL was also higher in each symptomatic group than in controls, and it was higher in all groups except MCI in comparison to presymptomatic carriers (p < 0.001; Figures 2A and S1D; Table S3). Results were similar after stratification by symptom duration (Table S4). Compared to NfL levels in controls or presymptomatic individuals, NfL was elevated even for patients with only questionable or minimal impairment as determined by a global score of 0 or 0.5 on the CDR Dementia Staging Instrument plus behavior and language domains from the National Alzheimer’s Disease Coordinating Center Frontotemporal Lobar Degeneration module (CDR + NACC-FTLD)27 (p ≤ 0.003; Figure 2C; Table S6).
When making baseline NfL pairwise comparisons between symptomatic groups, no difference in NfL levels was detected among bvFTD, nfvPPA, svPPA, CBS, or PSP-RS (Figures 2A and S1D; Table S7). However, NfL was higher in ALS (p < 0.001) than in any other group except FTD-ALS, and it was lower in MCI (p < 0.001) than in any other group.
We examined whether baseline NfL could distinguish controls from symptomatic groups by estimating the area under receiver operating characteristic (AUC) curves. Age- and gender-adjusted AUC values were lowest for MCI (AUC = 0.68). AUCs for other symptomatic groups ranged from 0.82 to 0.97, indicating good to excellent discriminatory ability (Figure 2D; Table S3). When determining whether baseline NfL could distinguish presymptomatic carriers from symptomatic groups, we noted the lowest adjusted AUC value for MCI (AUC = 0.57), with other adjusted AUC estimates ranging from 0.69 to 0.93 (Table S3). Results were similar when we stratified by symptom duration (Table S4) and when we considered only patients with a global FTLD-CDR score of 0 or 0.5 (Figure 2D; Table S6). Finally, we assessed whether baseline NfL distinguishes presymptomatic phenoconverters from non-converters or controls. We found moderate to good discriminatory ability; NfL levels differentiated between controls and converters with an adjusted AUC of 0.85 and between non-converters and converters with an adjusted AUC of 0.78. A NfL cutoff of >10 pg/mL identified 57.1% of converters, 20.8% of controls, and 30.2% of non-converters.
Baseline plasma neurofilament by mutation status
We examined baseline NfL by mutation status for cohorts having at least 30% of individuals with a mutation in C9orf72, GRN, or MAPT (i.e., clinically normal individuals [controls or presymptomatic mutation carriers] and patients with MCI or bvFTD; Figure 2E; Table S8). The previously seen increase in NfL in presymptomatic individuals compared to controls appeared most prominent for C9orf72 (p = 0.004) and MAPT (p < 0.001) mutation carriers, with a weaker increase for GRN carriers (p = 0.066). However, NfL levels did not differ among mutation groups (Table S9). Compared to bvFTD patients with no mutation, only GRN carriers had higher NfL (p < 0.001; Table S8). NfL was also higher in GRN carriers with bvFTD than in C9orf72 and MAPT carriers (p < 0.001; Table S9). There was no difference in NfL between MCI patients with or without a mutation (Table S8).
Plasma NfL correlates with indicators of disease severity
To determine the prognostic potential of plasma NfL, we evaluated, for each FTD spectrum disorder separately and for all groups combined, associations of baseline NfL with baseline indicators of clinical severity (CDR + NACC-FTLD sum of boxes [sb]), global cognitive function (Montreal Cognitive Assessment [MoCA]), social changes (Social Norms Questionnaire [SNQ]), language deficits (Northwestern Anagram Test [NAT]; Multilingual Naming Test [MINT]; phonemic fluency; category fluency), and executive dysfunction (Digit Span Forward and Backward; Trail Making Test Part B [Trails B]). Test scores by group are provided in Table S10.
Higher NfL associated with worse performance on all tests for all FTD groups combined and for bvFTD alone (p ≤ 0.003; Tables 2 and S11). Significant (p < 0.005) or nominally significant (p < 0.05) associations with NfL were seen for nfvPPA (Digit Span Forward), svPPA (CDR + NACC-FTLDsb, MoCA, NAT, MINT, phonemic and category fluency, and Digit Span Backward), CBS (CDR + NACC-FTLDsb, phonemic fluency, and Digit Span Backward), PSP-RS (MoCA, SNQ, phonemic and category fluency, Digit Span Forward and Backward, and Trails B), and MCI (CDR + NACC-FTLDsb and MoCA). For these smaller phenotype groups, where power to detect associations is lower, estimated β coefficients were similar or stronger than those observed for the larger bvFTD group.
Table 2.
Associations of baseline NfL with indicators of disease severity
| Association between NfL and: |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phenotype group | CDR + NACC- FTLDsb |
MoCA | SNQ | NAT | MINT | Phonemic fluency | Category fluency | Digit span forward | Digit span backward | Trails B |
| All FTD groups | n = 658 | n = 593 | n = 543 | n = 483 | n = 563 | n = 579 | n = 579 | n = 582 | n = 576 | n = 441 |
| β (95% CI) | 0.04 (0.03, 0.06)a | −2.22 (−2.79, −1.65)a | −0.92 (−1.19, −0.64)a | −0.88 (−1.15, −0.61)a | −2.13 (−2.96, −1.30)a | −4.10 (−4.87, −3.33)a | −4.00 (−4.94, −3.07)a | −0.50 (−0.73, −0.27)a | −0.81 (−1.04, −0.58)a | 15.87 (6.66, 25.09)a |
| p value | <0.001a | <0.001a | <0.001a | <0.001a | <0.001a | <0.001a | <0.001a | <0.001a | <0.001a | <0.001a |
| bvFTD | n = 289 | n = 267 | n = 236 | n = 215 | n = 251 | n = 255 | n = 252 | n = 263 | n = 257 | n = 179 |
| β (95% CI) | 0.06 (0.04, 0.09)a | −2.33 (−3.10, −1.56)a | −1.11 (−1.51, -−0.72)a | −1.05 (−1.43, −0.68)a | −2.31 (−3.27, −1.35)a | −5.00 (−6.03, −3.96)a | −5.37 (−6.64, −4.09)a | −0.50 (−0.82, −0.18)a | −0.96 (−1.30, −0.62)a | 18.73 (6.34, 31.13)a |
| p value | <0.001a | <0.001a | <0.001a | <0.001a | <0.001a | <0.001a | <0.001a | 0.002a | <0.001a | 0.003a |
| nfvPPA | n = 72 | n = 61 | n = 57 | n = 57 | n = 57 | n = 54 | n = 56 | n = 58 | n = 58 | n = 50 |
| β (95% CI) | 0.03 (−0.01, 0.07) | −2.33 (−4.74, 0.07) | −1.05 (−2.21, 0.10) | −0.71 (−1.88, 0.46) | −0.09 (−2.63, 2.45) | −2.30 (−4.88, 0.28) | −1.68 (−5.66, 2.30) | −1.16 (−1.84, −0.48)a | −0.74 (−1.52, 0.04) | 33.22 (−1.91, 68.34) |
| p value | 0.17 | 0.057 | 0.074 | 0.23 | 0.94 | 0.079 | 0.40 | 0.001a | 0.063 | 0.063 |
| svPPA | n = 84 | n = 80 | n = 69 | n = 70 | n = 67 | n = 79 | n = 75 | n = 71 | n = 73 | n = 74 |
| β (95% CI) | 0.06 (0.01, 0.10)b | −2.92 (−4.89, −0.95)a | −1.06 (−2.12, 0.00) | −1.46 (−2.34, −0.57)a | −4.97 (−7.83, −2.11)a | −2.83 (−5.66, −0.00)b | −4.87 (−7.68, −2.05)a | −0.87 (−1.80, 0.07) | −1.16 (−2.02, −0.29)b | 15.03 (−11.92, 41.98) |
| p value | 0.009b | 0.004a | 0.051 | 0.002a | <0.001a | 0.050b | 0.001a | 0.069 | 0.009b | 0.27 |
| CBS | n = 89 | n = 71 | n = 76 | n = 59 | n = 75 | n = 76 | n = 78 | n = 77 | n = 76 | n = 56 |
| β (95% CI) | 0.05 (0.01, 0.09)b | −1.33 (−2.88, 0.23) | 0.04 (−0.48, 0.56) | −0.45 (−1.13, 0.23) | −0.29 (−1.11, 0.53) | −2.46 (−4.57, −0.35)b | −0.33 (−2.88, 2.22) | −0.03 (−0.76, 0.70) | −0.63 (−1.18, −0.07)b | 5.83 (−20.79, 32.45) |
| p value | 0.024b | 0.094 | 0.88 | 0.19 | 0.49 | 0.023b | 0.80 | 0.94 | 0.027b | 0.66 |
| PSP-RS | n = 124 | n = 114b | n = 105b | n = 82 | n = 113 | n = 115 | n = 118 | n = 113 | n = 112 | n = 82 |
| β (95% CI) | 0.03 (−0.00, 0.06) | −1.58 (−2.93, −0.24)b | −0.71 (−1.32, −0.10)b | −0.17 (−0.90, 0.56) | −1.01 (−2.30, 0.28) | −2.89 (−4.79, −1.00)a | −2.95 (−4.89, −1.00)a | −0.73 (−1.30, −0.16)b | −0.71 (−1.20, −0.22)b | 35.81 (12.61, 59.02)a |
| p value | 0.051 | 0.022b | 0.024b | 0.64 | 0.12 | 0.003a | 0.003a | 0.012b | 0.005b | 0.003a |
| MCI | n = 57 | n = 57 | n = 56 | n = 49 | n = 56 | n = 56 | n = 56 | n = 57 | n = 57 | n = 56 |
| β (95% CI) | 0.29 (0.01, 0.57)b | −0.93 (−1.83, −0.03)b | −0.37 (−0.97, 0.24) | 0.03 (−0.45, 0.50) | −0.93 (−2.21, 0.36) | −0.55 (−2.52, 1.42) | −2.06 (−4.53, 0.40) | 0.08 (−0.50, 0.66) | −0.06 (−0.68, 0.57) | 5.16 (−10.37, 20.70) |
| p value | 0.040b | 0.044b | 0.23 | 0.91 | 0.15 | 0.58 | 0.10 | 0.78 | 0.85 | 0.51 |
β values, 95% CIs, and p values result from linear regression models that were adjusted for age, gender, symptom duration, and years of education. β values are interpreted as the change in the mean value of the given disease indicator for each doubling of NfL level. The FTD group includes patients with bvFTD, nfvPPA, svPPA, CBS, or PSP-RS. See also Tables S10–S12.
β, regression coefficient; CI, confidence interval; CDR + NACC-FTLDsb, CDR Dementia Staging Instrument plus behavior and language domains from the National Alzheimer’s Disease Coordinating Center FTLD module sum of boxes; MINT, Multilingual Naming Test; MoCA, Montreal Cognitive Assessment; NAT, Northwestern Anagram Test; SNQ, Social Norms Questionnaire; Trails B, Trail Making Test Part B.
p < 0.005 is considered as statistically significant after correcting for multiple testing.
Nominally significant p < 0.05.
We also evaluated whether baseline NfL associates with rates of change in indicators of disease severity for individuals with longitudinal clinical data spanning at least 1 year from baseline. This allowed the study of bvFTD patients alone and patients with bvFTD, nfvPPA, svPPA, CBS, and PSP-RS combined (Table S12). In bvFTD patients, increased baseline NfL associated with a faster longitudinal decline in performance for the CDR + NACC-FTLDsb, MoCA, and category fluency test (p ≤ 0.001). Nominally significant associations were noted for the MINT (p = 0.030) and Trails B (p = 0.018). Findings were similar for the combined group of FTD syndromes.
Plasma NfL increases throughout presymptomatic and symptomatic disease phases
In Figure 3A, we show longitudinal NfL profiles for controls, presymptomatic carriers, and patients with MCI, bvFTD, PPA (nfvPPA and svPPA), or parkinsonian disorders (CBS and PSP-RS). For individuals with an NfL measurement at least 1 year from baseline, we compared temporal NfL trajectories across groups by determining the rate of NfL concentration change per year (Figure 3B; Table S13). The rate of NfL change was greater for presymptomatic carriers who later phenoconverted, MCI patients, bvFTD patients, and the combined group of patients with nfvPPA or svPPA than for controls (p < 0.001). No difference in the rate of NfL change was seen between presymptomatic phenoconverters and patient groups, but phenoconverters had a nominally significant higher rate of NfL change than non-converters (p = 0.008; Figure 3B; Table S14). We thus assessed whether rate of NfL change can distinguish converters from non-converters or controls. We found that the rate of NfL change distinguished controls from converters with an adjusted AUC of 0.76 (95% confidence interval [CI]: 0.58–0.93) and distinguished non-converters from converters with an adjusted AUC of 0.74 (95% CI: 0.57–0.92). A cutoff value of >2.5 pg/mL of NfL per year identified 50.0% of converters, 4.4% of controls, and 5.7% of non-converters. Combining information from rate of NfL change and baseline NfL to differentiate between converters and non-converters was not more effective than using the individual measures themselves, as evidenced by an unchanged AUC.
Figure 3.

Plasma NfL increases throughout presymptomatic and symptomatic disease phases
(A) Longitudinal NfL concentrations are depicted for the indicated phenotype groups. NfL in plasma samples was measured in duplicate, and the mean concentration of replicates is shown on the base 10 logarithm scale.
(B) For individuals with one or more serial NfL measurements at least 1 year from baseline, we show comparisons of rate of change in NfL concentration per year for controls, all presymptomatic mutation carriers (all PreSx), presymptomatic carriers who did not convert (non-conv), those that did phenoconvert (phenoconv), and patients with MCI, bvFTD, PPA, or parkinsonian disorders. p values from analysis comparing rates of NfL change between the indicated group and either controls, non-converters, or phenoconverters after adjusting for age and gender are shown.
The number of individuals per group (n) is shown in figure panels and in Tables 1 and S1. See also Tables S13 and S14.
We next evaluated longitudinal NfL concentrations and rates of NfL change by mutation status in presymptomatic individuals and bvFTD patients (Figures 4A and 4B; Table S13). Nominally significant increases in the rate of NfL change were noted for all presymptomatic carriers combined (p = 0.029) and GRN mutation carriers alone (p = 0.012) when compared to controls. No difference in the rate of NfL change was seen among presymptomatic C9orf72, GRN, or MAPT mutation carriers (Table S15). For bvFTD, all mutation carriers combined, and MAPT carriers alone, had faster rates of NfL increases than did controls (p < 0.001; Table S13). Comparisons of rates of NfL change in bvFTD patients with no mutation or with a GRN mutation to controls were not examined since these groups had only four individuals with a second NfL measure more than 1 year from baseline. Finally, we show in Figure 5 the temporal profiles of NfL for individuals whose clinical diagnosis changed during their disease course.
Figure 4.

Longitudinal profiles and rates of NfL change in presymptomatic individuals and patients with bvFTD according to mutation status
(A) Longitudinal NfL concentrations are depicted for clinically normal individuals and bvFTD patients with no mutation or a mutation in C9orf72, GRN, or MAPT. NfL in plasma samples was measured in duplicate, and the mean concentration of replicates is shown on the base 10 logarithm scale.
(B) For individuals with one or more serial NfL measurements at least 1 year from baseline, we show comparisons of rate of change in NfL concentration per year between controls and presymptomatic mutation carriers or bvFTD patients according to mutation status. Patients with bvFTD without a mutation (n = 4) or with a GRN mutation (n = 4) were not included in the analysis. p values from analysis comparing rates of NfL change between the indicated group and controls after adjusting for age and gender are presented.
Longitudinal data for the presymptomatic carrier with a mutation in both C9orf72 and GRN are not included in (A) or (B). The number of individuals per group (n) is shown in figure panels and in Tables 1 and S1. See also Tables S13 and S15.
Figure 5.

Temporal trajectories of plasma NfL in presymptomatic individuals and individuals with MCI who phenoconverted according to mutation status
Shown, according to mutation status, are longitudinal NfL concentrations for 23 mutation carriers who phenoconverted (i.e., 14 from presymptomatic to symptomatic and nine from MCI to bvFTD). For five individuals, plasma NfL was not available from the follow-up visit at which their diagnosis changed; these individuals are marked by a white circle partially shaded in black for clinically normal individuals later diagnosed with MCI or by a gray square partially shaded in black for a patient with MCI subsequently diagnosed with bvFTD. NfL in plasma samples was measured in duplicate, and mean concentrations are shown on the base 10 logarithm scale. CN, clinically normal; PD, Parkinson’s disease.
Discussion
Through this cross-sectional and longitudinal study of the largest cohort of patients with sporadic or genetic bvFTD, nfvPPA, svPPA, CBS, or PSP-RS and clinically normal individuals with or without FTD-causing mutations, we show that plasma NfL increases prior to phenoconversion in presymptomatic mutation carriers; is significantly elevated in all studied FTD spectrum disorders, including mild cases; and associates with multiple indicators of disease severity.
Our observation that plasma NfL is higher in bvFTD, nfvPPA, svPPA, CBS, and PSP-RS compared with controls is consistent with prior reports.13,14,21, 22, 23 We saw no difference in baseline NfL among FTD syndromes. While no previous study compared blood NfL among all syndromes, those that did perform group comparisons observed comparable NfL concentrations between nfvPPA and svPPA15,18 or among nfvPPA, svPPA, and bvFTD.13 Thus, while NfL does have utility in confirming the presence of neurodegeneration, it is insufficient to discriminate between FTD syndromes.
That plasma NfL was elevated in FTD patients presenting with only mild symptoms and can distinguish these patients from healthy controls suggests that NfL may inform the diagnosis of questionable cases and allow a more rapid diagnosis. One study estimates that almost half of FTD patients are diagnosed more than 1 year after first symptoms.28 Diagnostic workups for patients routinely include imaging and neuropsychological testing, but the additional implementation of plasma NfL has the potential to reduce diagnostic delay and thereby improve the care of patients and allow their earlier participation in clinical trials.
Plasma NfL may also prove useful in detecting disease progression in presymptomatic mutation carriers. We show that, compared to measures in controls or presymptomatic mutation carriers who did not phenoconvert, baseline plasma NfL and rates of NfL change were higher in presymptomatic carriers before phenoconversion. These data validate and extend prior cross-sectional24 and longitudinal20,25,26 studies. For example, Rojas et al.24 found that median baseline plasma NfL concentrations were higher in presymptomatic carriers who showed phenoconversion in a cohort comprising a subset of individuals in the present study, and van der Ende et al.20 similarly reported higher baseline serum NfL in presymptomatic converters than in non-converters. Moreover, in a study by Saracino et al.,25 four presymptomatic C9orf72 expansion carriers with elevated rates of change in plasma NfL moved to the prodromal and symptomatic disease stage at follow-up. Yet four other presymptomatic individuals with high baseline NfL or rate of NfL change did not phenoconvert during the examined time frame. While data from these studies and ours indicate that plasma NfL could facilitate the identification of presymptomatic mutation carriers approaching phenoconversion, they also suggest that additional traits, such as mutation status, brain atrophy, and neuropsychological test scores, will likely be needed to better approximate when phenoconversion will occur and to select suitable individuals at risk of phenoconversion to participate in clinical trials designed to prevent or delay symptom onset and progression.
As with phenoconverters, patients with MCI, bvFTD, or PPA, but not patients with parkinsonian disorders, had higher rates of NfL change than did controls. Though longitudinal data were relatively limited, our data suggest that NfL generally increases over time in patients with MCI, bvFTD, and PPA. Were routine plasma NfL measures to be incorporated into the standard of care for FTD patients, baseline NfL or its rate of change could be compared pre- and post-treatment for individuals enrolled in clinical trials and conceivably serve as a pharmacodynamic biomarker of therapeutic response.
To estimate the prognostic power of NfL for each FTD syndrome, we examined associations of baseline plasma NfL with indicators of global cognitive function, social change, language deficits, and executive dysfunction. In the largest group of bvFTD patients, higher NfL associated with worse performance at baseline on all assessments and with a faster longitudinal decline in performance on the CDR + NACC-FTLDsb, the MoCA, and the category fluency test. For the smaller phenotype groups of nfvPPA, svPPA, CBS, and PSP-RS, associations were observed for some, but not all, assessment scores. Of note, however, the estimated β coefficients (a measure of effect strength) for these groups were similar or stronger than those for bvFTD. Moreover, because each syndrome is characterized, at least initially, by a predominant clinical trait, it is expected that associations of NfL with only some disease indicators would be observed for a given group. Comparing our findings with prior observations is complicated by differences in clinical assessments, cohort sizes, and the grouping of patients with different FTD syndromes, which may mask associations otherwise seen for a given syndrome. For example, in a cohort of patients with an FTD syndrome having underwent multiple psychometric assessments, serum NfL only correlated with executive dysfunction measures, but these did not survive correction for multiple testing.12 In contrast, another study found serum NfL associated with worse CDRsb, but not with Mini-Mental State Exam (MMSE) scores in mutation carriers with an FTD syndrome.5 These findings differ from our observed associations of NfL with CDR + NACC-FTLDsb and MoCA scores (an alternative cognition test to the MMSE) and measures of executive dysfunction in some phenotype groups. Data from the few studies that examined a particular FTD syndrome are more in line with our findings. For instance, NfL was reported to associate with the extent of semantic impairment in svPPA patients,19 with MMSE and CDR + NACC-FTLDsb scores in bvFTD patients,14 and with MMSE scores and cognitive disability in PSP-RS patients.22
Strengths of our study include evaluating the largest series of plasma NfL from well-characterized patients with bvFTD, nfvPPA, svPPA, PSP-RS, and CBS along with pre-presymptomatic mutation carriers and controls; performing all NfL measurements at one site to allow direct group comparisons; including cross-sectional and longitudinal assessments of plasma NfL; and examining the prognostic potential of NfL separately for each syndrome. Of equal if not more importance, our NfL data, along with extensive clinical data beyond what are shown in the present study, are available to the scientific community through ALLFTD.
Our data support the utility of plasma NfL for improving patient care and overcoming major barriers in clinical trial design, which we hope will expedite the discovery of effective FTD treatments. By facilitating the estimation of phenoconversion in individuals at genetic risk of FTD, and their earlier diagnosis, plasma NfL would aid in the selection of participants for prevention or early treatment trials. As a marker of disease severity and progression, NfL would also enable a more balanced stratification of patients with slowly or rapidly progressing disease in clinical trial treatment arms. This would improve clinical trial efficiency and the evaluation of treatment outcomes. The latter is a key challenge when conducting FTD clinical trials because of the heterogeneity among FTD patients.29 Overall, we show that plasma NfL represents a promising susceptibility and prognostic biomarker for FTD. In addition to these important observations, our major informational database comprising cross-sectional and longitudinal NfL, demographic, genetic, clinical, and neuropsychological data is sure to ignite new lines of investigation on FTD spectrum disorders.
Limitations of the study
Participants enrolled in this study may not represent the general FTD patient population, and diagnoses were made based on clinical assessments and were not neuropathologically confirmed. Also, the relatively small sample sizes of some groups may have resulted in a lack of power to detect some associations, raising the possibility of a type II error (i.e., a false-negative finding).
ALLFTD consortium members
Brian S. Appleby, Sami Barmada, Yvette Bordelon, Hugo Botha, Danielle Brushaber, David Clark, Giovanni Coppola, Ryan Darby, Katrina Devick, Dennis Dickson, Kelley Faber, Anne Fagan, Julie A. Fields, Ralitza Gavrilova, Daniel Geschwind, Jill Goldman, Jonathon Graff-Radford, Ian Grant, David T. Jones, Kejal Kantarci, Diana Kerwin, David S. Knopman, John Kornak, Walter Kremers, Maria Lapid, Argentina Lario Lago, Peter Ljubenkov, Diane Lucente, Ian R. Mackenzie, Scott McGinnis, Carly Mester, Bruce L. Miller, Peter Pressman, Rosa Rademakers, Vijay K. Ramanan, E. Marisa Ramos, Katherine P. Rankin, Meghana Rao, Katya Rascovsky, Rodolfo Savica, William Seeley, Adam M. Staffaroni, Jeremy Syrjanen, Jack Taylor, Lawren VandeVrede, Sandra Weintraub, and Bonnie Wong
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human plasma | ALLFTD (ARTFL-LEFFTDS Longitudinal Frontotemporal Lobar Degeneration: a multisite research consortium). https://www.allftd.org/data | |
| Critical commercial assays | ||
| NF-light™ | Quanterix | Cat#103186 |
| Other | ||
| Simoa HD-1 Analyzer® instrument | Quanterix | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Tania Gendron (Gendron.tania@mayo.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Protocol approvals and patient consents
Study participants were recruited through two North American multicenter observational studies: Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS; ClinicalTrials.gov NCT02372773) and Advancing Research and Treatment in Frontotemporal Lobar Degeneration (ARTFL; ClinicalTrials.gov NCT02365922). Participants or their caregivers provided written informed consent. For each participating center, study procedures were approved by their local Institutional Review Board committee.
Human subject characteristics
Human subject characteristics are provided in Table 1, and include data on gender, age at symptom onset, age at baseline plasma collection, age from symptom onset to baseline plasma collection and mutation status for each diagnostic group. Clinically defined phenotypes of the 981 participants are comprised of 144 clinically normal, mutation-free individuals who were in kindreds with known FTD-related gene mutations, 85 clinically normal individuals with an FTD-causing mutation (referred to as presymptomatic mutation carriers), 289 patients with bvFTD, 72 with nfvPPA, 84 with svPPA, 89 with CBS, 124 with PSP-RS, 25 with FTD/ALS, 12 with ALS and 57 with MCI [either mild cognitive impairment (MCI-cog, n = 38) or mild behavioral changes (MCI-beh, n = 19)]. Individuals in these phenotype groups were classified as previously reported based on widely accepted published criteria for each disorder.27 Mildly symptomatic participants who experienced a cognitive change compared with their previous level of functioning, had mild impairment in one or more domains of cognition on neuropsychological assessment but were still independent in functional abilities, and did not meet the criteria for dementia30 were classified as MCI-cog. MCI-beh was applied to participants exhibiting behavior/comportment/personality changes but not having dementia nor meeting criteria for probable bvFTD.
Clinical procedures
Study participants underwent annual standardized evaluations that included neurologic assessment, caregiver or companion interview and neuropsychological testing. Clinical scales included CDR + NACC-FTLD global and sum of boxes scores, which provide global measures of clinical severity,27 and the MoCA, a 30-point cognitive screening tool assessing visuospatial, semantic, phonemic and fluent language, working memory, recall, attention, and orientation.31 The following neuropsychological tests were also included:
Social Norms Questionnaire (SNQ)
This test evaluates the participant’s ability to identify appropriate and inappropriate behaviors in several hypothetical scenarios.32
Northwestern Anagram Test (NAT)
Participants must organize words to create 10 grammatically correct sentences that describe a picture stimulus. This test, believed to detect grammatic impairments, allows sentence production to be assessed independently of speech production, word-finding difficulties, or working memory capacity.33
The Multilingual Naming Test (MINT)
This is a 32-item object picture naming task. The total score includes items named correctly with semantic, but not phonemic, cues.34
Verbal fluency phonemic test
In two separate 60 s trials, participants must generate as many words as possible that begin with the letter “F” or “L”. The outcome is the total correct words summed across both trials.35
Verbal semantic/category test
In two separate 60 s trials, participants must generate as many words as possible that belong to the categories “animals” or “vegetables”. The outcome is the total correct words summed across both trials.
Digit span forward and backward
Participants are read a sequence of numbers that become increasingly longer, and must repeat the same sequence back to the examiner in order (forward span) or in reverse order (backward span).
Trail Making Test Part B
This test of executive function consists of 24 circles on a piece of paper; half of the circles have the numbers 1–12 in them, and half have the letters A-L. The participant has to draw a line from one circle to the next in ascending order alternating between circles with numbers and circles with letters (i.e., 1-A-2-B-3-C-4-D-5-E …).36
A breakdown of test scores by group is provided in Table S10.
Method details
Plasma neurofilament concentration determination
Participant blood samples were collected in ethylene diamine tetra-acetic acid tubes, centrifuged at 1,500 g at 4°C for 15 min, and the resulting plasma was aliquoted and stored at −80°C at NCRAD. Aliquots were shipped to the Mayo Clinic in Jacksonville, FL. All samples were assayed on the same instrument and by the same person in a blinded fashion. NfL concentrations in plasma were measured with the NF-Light digital immunoassay (Quanterix, Cat#103186, Lot 501992) using the HD-X Analyzer per the manufacturer’s protocol. In brief, samples were thawed on ice, mixed thoroughly by low-speed vortexing and centrifuged at 10,000 g for 5 min before transferring samples to 96-well plates. Samples were diluted 1:4 by the instrument and tested in duplicate. In addition to participant plasma samples, each run included 8 calibrators and 2 quality control samples provided with the kits, as well as a pooled reference sample provided by NCRAD. When the concentration of NfL in a sample exceeded the upper limit of the calibration curve, it was retested after first diluting it 1:4 or 1:8 at the bench, followed by an onboard 1:4 dilution. Concentrations were interpolated from the standard curve using a 4-parameter logistic curve fit (1/y2 weighted).
The mean %CV of duplicate NfL measurements was 4.39%. Across 15 runs, the mean concentration of the first quality control sample was 4.01 pg/ml and the inter-assay %CV was 8.16%, the mean concentration of the second quality control sample was 150.18 pg/ml and the inter-assay %CV was 5.17%, and the mean concentration of the reference sample was 15.25 pg/ml and the inter-assay %CV was 7.56%.
Among the samples we tested, NfL concentrations for 155 samples had previously been determined at Quanterix using their NF-Light digital immunoassay assayed on an HD-1 Analyzer.24 The mean inter-site %CV of NfL concentrations was 14.37%, with NfL concentrations from each group showing a near perfect correlation (Pearson’s r = 0.99, p < 0.0001).
Quantification and statistical analysis
In all statistical association analyses, the baseline NfL concentration was examined on the base 2 logarithm scale due to its skewed distribution. All covariates that were adjusted for in multivariable regression models were pre-defined. Unstandardized β coefficients and 95% CIs were estimated. The number of subjects (n) in each group for a given statistical analysis is reported in the appropriate corresponding tables and figures. All statistical tests were two-sided and performed using SAS (version 9.4; SAS Institute, Inc., Cary, North Carolina).
Associations of plasma NfL with age, gender and symptom duration
Separately for each phenotype group, associations of baseline NfL with age, gender and symptom duration (the latter for analysis of patients only) were assessed using linear regression models that were adjusted for the pre-defined covariates of age, gender and symptom duration (Figures 1A–1C and S1A–S1C, Table S2). After applying a Bonferroni correction for multiple testing, p < 0.025 (controls and presymptomatic mutation carriers) or p < 0.0167 (symptomatic groups) were considered as statistically significant.
Comparisons of baseline NfL concentrations among phenotype groups
Comparisons of baseline NfL concentrations between controls and nine separate groups as well as between presymptomatic mutation carriers and eight separate groups, and when stratifying by symptom duration, were made using linear regression models (Figures 2A and S1D, Tables S3 and S4). Unadjusted models were first examined, followed by multivariable models that were adjusted for age and gender. β coefficients and 95% CIs were estimated and are interpreted as the difference in mean NfL concentration (on the base 2 logarithm scale) between the two groups of interest. p < 0.0056 (comparisons between controls and other groups) or p < 0.0063 (comparisons between presymptomatic mutation carriers and other groups) were considered as significant after utilizing a Bonferroni correction for multiple comparisons.
Comparisons of baseline NfL concentrations between controls and presymptomatic mutation carriers who did or did not phenoconvert after baseline, and between phenoconverters and non-converters, were made using linear regression models that were adjusted for age and gender (Figures 2B and S1E, Table S5).
Comparisons of baseline NfL between controls or presymptomatic mutation carriers and five separate groups of patients with a CDR + NACC-FTLD global score of 0 (CBS, n = 2; PSP-RS, n = 1) or 0.5 (n = 106) were made using linear regression models. Unadjusted models were first examined, followed by multivariable models that were adjusted for age and gender (Table S6). β coefficients and 95% CIs were estimated and are interpreted as the difference in mean NfL concentration (on the base 2 logarithm scale) between the two groups of interest. p < 0.01 was considered as significant after applying a Bonferroni correction for multiple comparisons.
Comparisons of baseline NfL among eight symptomatic groups (Figures 2A and S1D, Table S7) were made using linear regression models that were adjusted for age, gender and symptom duration. p < 0.0018 was considered as significant after utilizing a Bonferroni correction for multiple comparisons.
Comparisons of baseline NfL concentrations according to mutation status
Comparisons of baseline NfL among individuals with no mutation and individuals with a mutation in either C9orf72, GRN or MAPT were analyzed for three phenotype groups (clinically normal, bvFTD or MCI) using linear regression models that were adjusted for age, gender and symptom duration (the latter for analysis of patients only) (Figure 2E, Tables S8 and S9). p < 0.0083 (six pair-wise comparisons for clinically normal individuals), or p < 0.0071 (seven pair-wise comparisons bvFTD patients) were considered as significant after utilizing a Bonferroni correction for multiple comparisons.
Determination of rates of change in NfL concentrations and in indicators of disease severity
For subjects with longitudinal NfL measurements and for whom the first and last measurements were at least 1 year apart, we estimated the rate of change in NfL per year, separately for each individual, by extracting the β coefficient from a linear regression model where NfL was the dependent variable and time since initial NfL measure was the independent variable (a logarithm transformation of NfL was not utilized in this part of the analysis). All available NfL measures for a given subject were utilized in these linear regression models. This same strategy was utilized to calculate rate of change in disease indicators per year.
Determination of the discriminatory power of baseline NfL
To assess the ability of baseline NfL to discriminate between controls or presymptomatic mutation carriers and other phenotype groups (Figure 2D, Tables S3, S4, and S6), we estimated area under the ROC curve (AUC) values along with 95% CIs. An AUC equal to 1.0 indicates perfect predictive ability for a given model, whereas an AUC equal to 0.5 represents predictive ability equal to chance. Unadjusted AUCs were initially estimated, however as age and gender differed between comparison groups and were also associated with baseline NfL concentrations, these AUC estimates are influenced by age and gender and are therefore biased. Therefore, we calculated age/gender-adjusted AUC estimates for subjects involved in a given comparison by first extracting the residuals from a linear regression model where baseline NfL is the dependent variable and both age and gender and independent variables, thereby essentially normalizing by age and gender. We then compared these model residuals between the two groups of interest to estimate AUC for a given pairwise comparison in our adjusted analyses.
We used the same strategy as above to test the ability of baseline NfL level and rate of change in NfL level per year to discriminate presymptomatic mutation carriers who phenoconverted after baseline from those who did not convert and controls. To assess whether utilizing both rate of change in NfL over time and baseline NfL to differentiate converters from non-converters resulted in improved predictive ability when compared to examining the individual measures themselves, we estimated the increase in AUC that was observed when comparing the following two logistic regression models, where the outcome was converter/non-converter status, and only subjects with a measure for rate of change in NfL over time were included: (1) covariates of age, gender and baseline NfL level, and (2) covariates of age, gender, baseline NfL level and rate of change in NfL level over time.
Associations of baseline plasma NfL with indicators of disease severity
Associations of baseline NfL with baseline disease indicators (Tables 2 and S11) and with rate of change over time (per year) in disease indicators (Table S12) were examined using linear regression models. Unadjusted and multivariable models were evaluated, where multivariable models were adjusted for age, gender, symptom duration and years of education. β coefficients and 95% CIs were estimated and are interpreted as the change in the mean outcome measure for each doubling in NfL concentration. p < 0.005 was considered as significant after utilizing a Bonferroni adjustment for multiple testing separately for each phenotype group. Disease severity characteristics according to phenotype group are shown in Table S10.
Comparisons of rate of change in NfL concentrations between controls or presymptomatic mutation carriers and other phenotype groups and by mutation status
We compared rate of change in NfL concentrations per year vs. controls for different phenotype groups using linear regression models that were adjusted for age and gender, where p < 0.0039 was considered as significant after applying a Bonferroni correction for multiple testing (Table S13). β coefficients and 95% confidence CIs were estimated and are intreated as the difference in the mean rate of change in NfL level per year between the given two groups. Additionally, rate of change in NfL level per year was compared among presymptomatic non-converters and phenoconverters as well as phenotype groups using linear regression models that were adjusted for age, gender and, for comparisons among patient groups, symptom duration. p < 0.0033 were considered as statistically significant after applying a Bonferroni correction for multiple comparisons (Table S14). Finally, rate of change in NfL level per year was compared according to mutation status among presymptomatic mutation carriers using linear regression models that were adjusted for age and gender. p < 0.0167 was considered as statistically significant after utilizing a Bonferroni correction for multiple testing (Table S15).
Additional resources
Participants were enrolled through the Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects study (ClinicalTrials.gov NCT02372773) and the Advancing Research and Treatment in Frontotemporal Lobar Degeneration study (ClinicalTrials.gov NCT02365922).
Acknowledgments
Data collection and dissemination were supported by the ALLFTD Consortium (U19AG063911; funded by the National Institute on Aging [NIA] and the National Institute of Neurological Diseases and Stroke [NINDS]) and the former ARTFL and LEFFTDS Consortia (ARTFL: U54NS092089, funded by the NINDS and National Center for Advancing Translational Sciences; LEFFTDS: U01AG045390, funded by the NIA and NINDS). The manuscript was reviewed by the ALLFTD Executive Committee. Samples from the National Centralized Repository for Alzheimer’s Disease and Related Dementias (NCRAD), which receives government support under a cooperative agreement grant (U24AG021886) awarded by the NIA, were used in this study. We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible. This work was also supported by the Association for Frontotemporal Degeneration (L.P.) and the NINDS (R35NS097273 [L.P.], P01NS084974 [T.F.G. and L.P.], and P01NS099114 [T.F.G. and L.P.]).
Author contributions
B.F.B., A.L.B., T.F.G., L.P., and H.R. contributed to the conception and/or design of the study. A.C.B., B.C.D., K.D.-R., D.R.G., N.G., N.R.G.-R., M.G., E.D.H., G.-Y.R.H., D.J.I., D.I.K., G.C.L., I.L., J.C.M., M.F.M., C.U.O., B.P., A.R., E.D.R., J.C.R., M.C.T., and Z.K.W. contributed through patient evaluations, collecting patient samples, and/or clinical data. L.K.F. and H.W.H. contributed to the study design and clinical data. T.F. contributed through protocol development and management and biospecimen collection. T.F.G. measured NfL concentrations, and A.R.B. assisted with sample processing. O.P. conducted patient evaluations and provided guidance on the selection of disease indicators. Statistical analyses were performed by M.G.H. and L.J.W. Literature review was undertaken by T.F.G. and A.M.V. The manuscript was written by T.F.G. and M.G.H. and was reviewed by all authors.
Declaration of interests
A.C.B. is site PI for the Alector INFRONT-3 trial. A.L.B. receives research support from NIH (R01AG038791, R01AG073482, and U24AG057437), Rainwater Charitable Foundation, Association for Frontotemporal Degeneration, Bluefield Project to Cure Frontotemporal Dementia, Alzheimer’s Drug Discovery Foundation, and the Alzheimer’s Association. He has served as a consultant for Alector, AGTC, Arkuda, Arvinas, AZTherapies, GSK, Oligomerix, Ono, Roche, Samumed, Stealth, Third Rock, Transposon, TrueBinding, and Wave and received research support from Biogen, Eisai, and Regeneron. B.F.B. has served as an investigator for clinical trials sponsored by Biogen, Alector, and EIP Pharma. He receives royalties from a published book entitled Behavioral Neurology of Dementia (Cambridge Medicine, 2009, 2017), serves on the Tau Consortium Scientific Advisory Board, and receives research support from the NIH. B.C.D. consults for Acadia, Arkuda, Axovant, Lilly, Biogen, Merck, Novartis, and Wave LifeSciences; has Elsevier editorial duties with payment (Neuroimage: Clinical and Cortex); and receives royalties from Oxford University Press and Cambridge University Press. K.D.-R. has research funding from Biogen and Lawson Health Research Institute and receives consultant fees from Biogen and educational fees from MedBridge. D.R.G. consults for Biogen, Fujirebio, and Amprion and is on the DSMB for Cognition Therapeutics. M.G. is participating in treatment trials sponsored by Alector, Prevail, and Passage Bio and is a consultant to Takeda, Passage Bio, and Biogen. N.G. has or is participating in clinical trials of anti-dementia drugs sponsored by Bristol Myers Squibb, Lilly/Avid Radiopharmaceuticals, Janssen, Novartis, Pfizer, and Wyeth. N.R.G.-R. has taken part in multicenter studies funded by Biogen, AbbVie, and Lilly. G.-Y.R.H. has received research support from Anavax, Biogen, and Roche. I.L. received support from Roche, Abbvie, Biogen, EIP-Pharma, and Biohaven Pharmaceuticals; was member of a Lundbeck Advisory Board; and receives salary from the University of California, San Diego and as Chief Editor of Frontiers in Neurology. J.C.M. participates on a speaker forum for Biogen and receives research support from Biogen, Eisai, Eli Lilly, Green Valley, and Novartis. C.U.O. is a consultant with Alector and Acadia and receives research funding from Alector. L.P. is a consultant for Expansion Therapeutics. E.D.R. receives funding from NIH, Alzheimer’s Drug Discovery Foundation, Bluefield Project, and Alector; consults for Biogen, AVROBIO, and AGTC; and owns intellectual property related to tau. J.C.R. is a site PI for Eli Lilly and Eisai clinical trials and receives research support from NIH K23AG059888. M.C.T. participates in clinical trials with Biogen, Avanex, UCB, and Janssen. Z.K.W. is supported by the NIH/NIA and NIH/NINDS (1U19AG063911, FAIN: U19AG063911), Mayo Clinic Center for Regenerative Medicine, Mayo Clinic in Florida Focused Research Team Program, the gifts from The Sol Goldman Charitable Trust, the Donald G. and Jodi P. Heeringa Family, the Haworth Family Professorship in Neurodegenerative Diseases fund, and The Albertson Parkinson’s Research Foundation. He serves as PI or co-PI on Biohaven Pharmaceuticals, Inc. (BHV4157-206 and BHV3241-301); Neuraly, Inc. (NLY01-PD-1); and Vigil Neuroscience, Inc. (VGL101–01.001) grants. He serves as co-PI of the Mayo Clinic APDA Center for Advanced Research and as an external advisory board member for Vigil Neuroscience, Inc. All other authors report no competing interests.
Published: April 19, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100607.
Contributor Information
Tania F. Gendron, Email: Gendron.Tania@mayo.edu.
Leonard Petrucelli, Email: Petrucelli.Leonard@mayo.edu.
ALLFTD consortium:
Brian S. Appleby, Sami Barmada, Yvette Bordelon, Hugo Botha, Danielle Brushaber, David Clark, Giovanni Coppola, Ryan Darby, Katrina Devick, Dennis Dickson, Kelley Faber, Anne Fagan, Julie A. Fields, Ralitza Gavrilova, Daniel Geschwind, Jill Goldman, Jonathon Graff-Radford, Ian Grant, David T. Jones, Kejal Kantarci, Diana Kerwin, David S. Knopman, John Kornak, Walter Kremers, Maria Lapid, Argentina Lario Lago, Peter Ljubenkov, Diane Lucente, Ian R. Mackenzie, Scott McGinnis, Carly Mester, Bruce L. Miller, Peter Pressman, Rosa Rademakers, Vijay K. Ramanan, E. Marisa Ramos, Katherine P. Rankin, Meghana Rao, Katya Rascovsky, Rodolfo Savica, William Seeley, Adam M. Staffaroni, Jeremy Syrjanen, Jack Taylor, Lawren VandeVrede, Sandra Weintraub, and Bonnie Wong
Supplemental information
Data and code availability
-
•
De-identified human/patient clinical, demographic and plasma NfL data are available from ALLFTD upon request. Investigators are required to complete the Request Clinical Data form on the request portal (https://www.allftd.org/data) and to review the data sharing and publication policy. Data that could identify a participant are not provided. Data requests are reviewed quarterly and generally fulfilled approximately four weeks after they are approved depending on the complexity of the request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact and ALLFTD.
References
- 1.Greaves C.V., Rohrer J.D. An update on genetic frontotemporal dementia. J. Neurol. 2019;266:2075–2086. doi: 10.1007/s00415-019-09363-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosen H.J., Boeve B.F., Boxer A.L. Tracking disease progression in familial and sporadic frontotemporal lobar degeneration: recent findings from ARTFL and LEFFTDS. Alzheimers Dement. 2020;16:71–78. doi: 10.1002/alz.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sjogren M., Rosengren L., Minthon L., Davidsson P., Blennow K., Wallin A. Cytoskeleton proteins in CSF distinguish frontotemporal dementia from AD. Neurology. 2000;54:1960–1964. doi: 10.1212/wnl.54.10.1960. [DOI] [PubMed] [Google Scholar]
- 4.Scherling C.S., Hall T., Berisha F., Klepac K., Karydas A., Coppola G., Kramer J.H., Rabinovici G., Ahlijanian M., Miller B.L., et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann. Neurol. 2014;75:116–126. doi: 10.1002/ana.24052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meeter L.H., Dopper E.G., Jiskoot L.C., Sanchez-Valle R., Graff C., Benussi L., Ghidoni R., Pijnenburg Y.A., Borroni B., Galimberti D., et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann. Clin. Transl. Neurol. 2016;3:623–636. doi: 10.1002/acn3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alcolea D., Vilaplana E., Suarez-Calvet M., Illan-Gala I., Blesa R., Clarimon J., Llado A., Sanchez-Valle R., Molinuevo J.L., Garcia-Ribas G., et al. CSF sAPPbeta, YKL-40, and neurofilament light in frontotemporal lobar degeneration. Neurology. 2017;89:178–188. doi: 10.1212/WNL.0000000000004088. [DOI] [PubMed] [Google Scholar]
- 7.Meeter L.H.H., Vijverberg E.G., Del Campo M., Rozemuller A.J.M., Donker Kaat L., de Jong F.J., van der Flier W.M., Teunissen C.E., van Swieten J.C., Pijnenburg Y.A.L. Clinical value of neurofilament and phospho-tau/tau ratio in the frontotemporal dementia spectrum. Neurology. 2018;90:e1231–e1239. doi: 10.1212/WNL.0000000000005261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsson B., Portelius E., Cullen N.C., Sandelius A., Zetterberg H., Andreasson U., Hoglund K., Irwin D.J., Grossman M., Weintraub D., et al. Association of cerebrospinal fluid neurofilament light protein levels with cognition in patients with dementia, motor neuron disease, and movement disorders. JAMA Neurol. 2019;76:318–325. doi: 10.1001/jamaneurol.2018.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delaby C., Alcolea D., Carmona-Iragui M., Illan-Gala I., Morenas-Rodriguez E., Barroeta I., Altuna M., Estelles T., Santos-Santos M., Turon-Sans J., et al. Differential levels of Neurofilament Light protein in cerebrospinal fluid in patients with a wide range of neurodegenerative disorders. Sci. Rep. 2020;10:9161. doi: 10.1038/s41598-020-66090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abu-Rumeileh S., Mometto N., Bartoletti-Stella A., Polischi B., Oppi F., Poda R., Stanzani-Maserati M., Cortelli P., Liguori R., Capellari S., et al. Cerebrospinal fluid biomarkers in patients with frontotemporal dementia spectrum: a single-center study. J. Alzheimers Dis. 2018;66:551–563. doi: 10.3233/JAD-180409. [DOI] [PubMed] [Google Scholar]
- 11.Ljubenkov P.A., Staffaroni A.M., Rojas J.C., Allen I.E., Wang P., Heuer H., Karydas A., Kornak J., Cobigo Y., Seeley W.W., et al. Cerebrospinal fluid biomarkers predict frontotemporal dementia trajectory. Ann. Clin. Transl Neurol. 2018;5:1250–1263. doi: 10.1002/acn3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohrer J.D., Woollacott I.O., Dick K.M., Brotherhood E., Gordon E., Fellows A., Toombs J., Druyeh R., Cardoso M.J., Ourselin S., et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016;87:1329–1336. doi: 10.1212/WNL.0000000000003154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilke C., Preische O., Deuschle C., Roeben B., Apel A., Barro C., Maia L., Maetzler W., Kuhle J., Synofzik M. Neurofilament light chain in FTD is elevated not only in cerebrospinal fluid, but also in serum. J. Neurol. Neurosurg. Psychiatry. 2016;87:1270–1272. doi: 10.1136/jnnp-2015-312972. [DOI] [PubMed] [Google Scholar]
- 14.Steinacker P., Anderl-Straub S., Diehl-Schmid J., Semler E., Uttner I., von Arnim C.A.F., Barthel H., Danek A., Fassbender K., Fliessbach K., et al. Serum neurofilament light chain in behavioral variant frontotemporal dementia. Neurology. 2018;91:e1390–e1401. doi: 10.1212/WNL.0000000000006318. [DOI] [PubMed] [Google Scholar]
- 15.Matias-Guiu J.A., Gomez-Pinedo U., Forero L., Pytel V., Cano F., Moreno-Ramos T., Cabrera-Martin M.N., Matias-Guiu J., Gonzalez-Rosa J.J. Plasma neurofilament light chain in primary progressive aphasia and related disorders: clinical significance and metabolic correlates. J. Alzheimers Dis. 2019;72:773–782. doi: 10.3233/JAD-190838. [DOI] [PubMed] [Google Scholar]
- 16.Katisko K., Cajanus A., Jaaskelainen O., Kontkanen A., Hartikainen P., Korhonen V.E., Helisalmi S., Haapasalo A., Koivumaa-Honkanen H., Herukka S.K., et al. Serum neurofilament light chain is a discriminative biomarker between frontotemporal lobar degeneration and primary psychiatric disorders. J. Neurol. 2020;267:162–167. doi: 10.1007/s00415-019-09567-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cajanus A., Katisko K., Kontkanen A., Jaaskelainen O., Hartikainen P., Haapasalo A., Herukka S.K., Vanninen R., Solje E., Hall A., et al. Serum neurofilament light chain in FTLD: association with C9orf72, clinical phenotype, and prognosis. Ann. Clin. Transl Neurol. 2020;7:903–910. doi: 10.1002/acn3.51041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinacker P., Semler E., Anderl-Straub S., Diehl-Schmid J., Schroeter M.L., Uttner I., Foerstl H., Landwehrmeyer B., von Arnim C.A., Kassubek J., et al. Neurofilament as a blood marker for diagnosis and monitoring of primary progressive aphasias. Neurology. 2017;88:961–969. doi: 10.1212/WNL.0000000000003688. [DOI] [PubMed] [Google Scholar]
- 19.Heller C., Chan E., Foiani M.S., Todd E., Russell L.L., Greaves C.V., Heslegrave A.J., Warren J.D., Zetterberg H., Bocchetta M., et al. Plasma glial fibrillary acidic protein and neurofilament light chain are measures of disease severity in semantic variant primary progressive aphasia. J. Neurol. Neurosurg. Psychiatry. 2020:2020. doi: 10.1136/jnnp-2020-325085. [DOI] [PubMed] [Google Scholar]
- 20.van der Ende E.L., Meeter L.H., Poos J.M., Panman J.L., Jiskoot L.C., Dopper E.G.P., Papma J.M., de Jong F.J., Verberk I.M.W., Teunissen C., et al. Serum neurofilament light chain in genetic frontotemporal dementia: a longitudinal, multicentre cohort study. Lancet Neurol. 2019;18:1103–1111. doi: 10.1016/S1474-4422(19)30354-0. [DOI] [PubMed] [Google Scholar]
- 21.Rojas J.C., Karydas A., Bang J., Tsai R.M., Blennow K., Liman V., Kramer J.H., Rosen H., Miller B.L., Zetterberg H., et al. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann. Clin. Transl Neurol. 2016;3:216–225. doi: 10.1002/acn3.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donker Kaat L., Meeter L.H., Chiu W.Z., Melhem S., Boon A.J.W., Blennow K., Zetterberg H., van Swieten J.C. Serum neurofilament light chain in progressive supranuclear palsy. Parkinsonism Relat. Disord. 2018;56:98–101. doi: 10.1016/j.parkreldis.2018.06.018. [DOI] [PubMed] [Google Scholar]
- 23.Hansson O., Janelidze S., Hall S., Magdalinou N., Lees A.J., Andreasson U., Norgren N., Linder J., Forsgren L., Constantinescu R., et al. Blood-based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology. 2017;88:930–937. doi: 10.1212/WNL.0000000000003680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rojas J.C., Wang P., Staffaroni A.M., Heller C., Cobigo Y., Wolf A., Goh S.M., Ljubenkov P.A., Heuer H.W., Fong J.C., et al. Plasma neurofilament light for prediction of disease progression in familial frontotemporal lobar degeneration. Neurology. 2021;96:e2296–e2312. doi: 10.1212/WNL.0000000000011848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saracino D., Dorgham K., Camuzat A., Rinaldi D., Rametti-Lacroux A., Houot M., Clot F., Martin-Hardy P., Jornea L., Azuar C., et al. Plasma NfL levels and longitudinal change rates in C9orf72 and GRN-associated diseases: from tailored references to clinical applications. J. Neurol. Neurosurg. Psychiatry. 2021;92:1278–1288. doi: 10.1136/jnnp-2021-326914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilke C., Reich S., van Swieten J.C., Borroni B., Sanchez-Valle R., Moreno F., Laforce R., Graff C., Galimberti D., Rowe J.B., et al. Stratifying the presymptomatic phase of genetic frontotemporal dementia by serum NfL and pNfH: a longitudinal multicentre study. Ann Neurol. 2021;91:33–47. doi: 10.1002/ana.26265. [DOI] [PubMed] [Google Scholar]
- 27.Miyagawa T., Brushaber D., Syrjanen J., Kremers W., Fields J., Forsberg L.K., Heuer H.W., Knopman D., Kornak J., Boxer A., et al. Utility of the global CDR((R)) plus NACC FTLD rating and development of scoring rules: data from the ARTFL/LEFFTDS Consortium. Alzheimers Dement. 2020;16:106–117. doi: 10.1002/alz.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Besser L.M., Galvin J.E. Diagnostic experience reported by caregivers of patients with frontotemporal degeneration. Neurol. Clin. Pract. 2020;10:298–306. doi: 10.1212/CPJ.0000000000000738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boxer A.L., Gold M., Feldman H., Boeve B.F., Dickinson S.L., Fillit H., Ho C., Paul R., Pearlman R., Sutherland M., et al. New directions in clinical trials for frontotemporal lobar degeneration: methods and outcome measures. Alzheimers Dement. 2020;16:131–143. doi: 10.1016/j.jalz.2019.06.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albert M.S., DeKosky S.T., Dickson D., Dubois B., Feldman H.H., Fox N.C., Gamst A., Holtzman D.M., Jagust W.J., Petersen R.C., et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coleman K.K., Coleman B.L., MacKinley J.D., Pasternak S.H., Finger E.C. Detection and differentiation of frontotemporal dementia and related disorders from alzheimer disease using the montreal cognitive assessment. Alzheimer Dis. Assoc. Disord. 2016;30:258–263. doi: 10.1097/WAD.0000000000000119. [DOI] [PubMed] [Google Scholar]
- 32.Rankin K. NINDS; 2008. Social Norms Questionaire. Domain Specific Tasks of Executive Function. [Google Scholar]
- 33.Weintraub S., Mesulam M.M., Wieneke C., Rademaker A., Rogalski E.J., Thompson C.K. The northwestern anagram test: measuring sentence production in primary progressive aphasia. Am. J. Alzheimers Dis. Other Demen. 2009;24:408–416. doi: 10.1177/1533317509343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gollan T.H., Weissberger G.H., Runnqvist E., Montoya R.I., Cera C.M. Self-ratings of spoken language dominance: a multi-lingual naming test (MINT) and preliminary Norms for young and aging Spanish-English bilinguals. Biling (Camb Engl) 2012;15:594–615. doi: 10.1017/S1366728911000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grossman M. The non-fluent/agrammatic variant of primary progressive aphasia. Lancet Neurol. 2012;11:545–555. doi: 10.1016/S1474-4422(12)70099-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Llinas-Regla J., Vilalta-Franch J., Lopez-Pousa S., Calvo-Perxas L., Torrents Rodas D., Garre-Olmo J. The Trail making test. Assessment. 2017;24:183–196. doi: 10.1177/1073191115602552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
De-identified human/patient clinical, demographic and plasma NfL data are available from ALLFTD upon request. Investigators are required to complete the Request Clinical Data form on the request portal (https://www.allftd.org/data) and to review the data sharing and publication policy. Data that could identify a participant are not provided. Data requests are reviewed quarterly and generally fulfilled approximately four weeks after they are approved depending on the complexity of the request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact and ALLFTD.