

Summary
Localized prostate cancer exhibits multiple genomic alterations and heterogeneity at the proteomic level. Single-cell technologies capture important cell-to-cell variability responsible for heterogeneity in biomarker expression that may be overlooked when molecular alterations are based on bulk tissue samples. This study aims to identify prognostic biomarkers and describe the heterogeneity of prostate cancer and the associated microenvironment by simultaneously quantifying 36 proteins using single-cell mass cytometry analysis of over 1.6 million cells from 58 men with localized prostate cancer. We perform this task, using a high-dimensional clustering pipeline named Franken to describe subpopulations of immune, stromal, and prostate cells, including changes occurring in tumor tissues and high-grade disease that provide insights into the coordinated progression of prostate cancer. Our results further indicate that men with localized disease already harbor rare subpopulations that typically occur in castration-resistant and metastatic disease.
Keywords: single-cell proteomics, prostate cancer, cellular heterogeneity
Graphical abstract
Highlights
-
•
Single-cell proteomics of localized prostate cancer defines disease heterogeneity
-
•
Malignant and benign prostate tissues differ in rare cell-type proportional shifts
-
•
T cells and proliferating macrophages are associated with high-grade PCa
-
•
Rare CD15+ epithelial cells are amplified in high-grade PCa
De Vargas Roditi et al. describe prostate cancer heterogeneity at the proteomic level by simultaneously quantifying 36 proteins using single-cell mass cytometry (n = 58). High-dimensional clustering indicates that men with hormone-naive localized prostate cancer already harbor rare cell subpopulations that typically occur in castration-resistant and metastatic disease.
Introduction
The treatment of localized prostate cancer is based on clinicopathological information including Gleason score, prostate-specific antigen (PSA) levels, stage, and patient age.3 While the majority of patients with localized disease can be cured, some men recur with metastatic disease4 because of microscopic spread. The observed heterogeneity of outcomes might be explained by heterogeneity within tumors,5 which is missed by the current grading system, and new prognostic biomarkers are of utmost importance.
Several potential biomarkers including gene fusions, mutations, epigenetic heterogeneity, and proteins have been studied.6 Technological advances in proteomics now allow both exploration of the proteome for biomarkers and assessment of the heterogeneity of biomarker expression. However, analysis of a whole tissue core misses important cell-to-cell variability. In this study, we performed mass cytometry analysis of dissociated single cells from prostatectomies of 58 patients with tumors at varying grades and UICC (Union Internationale Contre le Cancer) stages using a set of 36 metal-tagged antibodies that recognize surface markers, enzymes, transcription factors, and markers of functional readouts selected to facilitate characterization of the phenotypic diversity of prostate tumors and their microenvironment. The power to comprehensively analyze heterogeneity of tumors by simultaneously measuring dozens of markers in hundreds of thousands to millions of cells makes mass cytometry the ideal tool to characterize single-cell subpopulations present in prostate or other tumors including those rare populations that cannot be detected with lower parametricity or lower throughput methods.
Although mass cytometry has single-cell resolution capabilities, there are statistical challenges involved in analyzing such high-dimensional data. State-of-the-art clustering methods either underperform in precision and recall or require long runtimes and prohibitive computational resources.2 To address this issue we developed an unsupervised, single-cell clustering approach, Franken, combining speed and performance. Use of Franken to quantify the phenotypic diversity of single cells in prostate tumor samples identified progression-related single-cell phenotypes. We detected immune landscape features unique to patients with high-grade prostate cancer, reflected by higher frequencies of macrophage and T cell phenotypes than observed in patients with intermediate grade disease. Further, we observed tumor-specific prostate epithelial phenotypes, including AR-negative and/or PSA-negative phenotypes typically associated with resistance to adrogen deprivation therapy (ADT) and castration resistant prostate cancer (CRPC),7, 8, 9 and rare CD15+ phenotypes.
Results
Clustering of high-dimensional mass cytometry data defines molecular profiles of prostate subpopulations
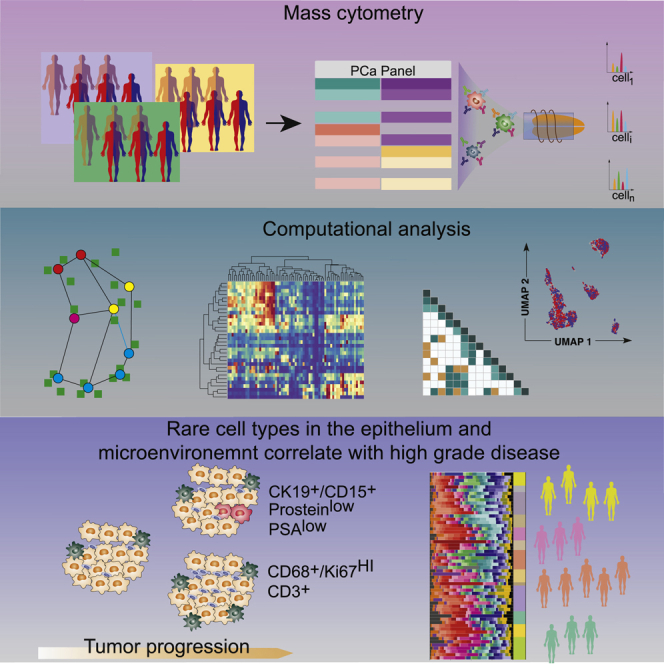
Using mass cytometry, we profiled tumor samples from 58 prostate cancer patients with localized, hormone-naive tumors, including 24 patients with the International Society of Urological Pathology (ISUP) grade II (Gleason score 3 + 4), 22 grade III cases (Gleason score 4 + 3), and 12 patients with grade V prostate carcinomas (Gleason scores 4 + 5, 5 + 4, or 5 + 5) (Figure 1A). No tissue was collected from low-grade tumors, and grades II and III are henceforth referred to as the intermediate-grade group, and grade V tumors are referred to as the high-grade group. Fresh tissue samples were collected from 58 prostate cancer patients after surgical resection of the index tumor lesions (the most extensive with the highest Gleason score). For 17 patients (29%), paired samples were taken from a macroscopically visible tumor mass and adjacent benign prostatic tissue (ABPT); remaining samples are referred to as random prostatic tissue (RPT). The presence of prostate cancer was confirmed histologically after examining the opposite side of the specimen, first in the frozen section and then after paraffin embedding; ABPT specimens were taken from the contralateral transitional zone of the prostate and never from the peripheral one, where tumor is more likely to be located. Single-cell suspensions from all prostate tissue samples and from 10 cell lines, including prostate cancer, stromal, and immune cells (key resources table), were barcoded, pooled, and stained with a 36-antibody prostate cancer-centric panel, before mass cytometry acquisition. The antibody panel was designed to quantify markers that identify prostate epithelial cells, cells of the stroma and immune microenvironment, and markers of proliferation and survival (Figure 1B). Data for a total of 1,670,117 live cells were generated.
Figure 1.

Schematic of method for characterization of primary prostate cancer tissue using mass cytometry
(A) The patient cohort consisted of 58 primary prostate cancer cases. For 16 patients, tumor and adjacent benign prostatic tissue (ABPT) samples were available. The remaining samples were from randomly selected regions from a prostatectomy without tumor assessment. Samples were analyzed by mass cytometry, and data were analyzed using Franken.
(B) Markers used to categorize prostate epithelial cells as luminal, basal, or transitional, and markers used to identify tumor cells, cells from the microenvironment, and functional features, such as proliferation, apoptosis, or hypoxia.
The high dimensionality of this dataset represented a challenge for data visualization and clustering. To address this task, we developed an efficient computational pipeline called Franken (Figure S1A). The pipeline begins by building a large self-organizing map10, 11, 12 (SOM), which is used to fit all of the data. The SOM nodes in the original high-dimensional space are used to build a mutual nearest neighbor graph using the Tanimoto similarity13 (also known as extended Jaccard similarity) followed by Walktrap graph clustering.1 Although all results presented in this paper were obtained in an unsupervised manner, the pipeline offers the option to define the chosen number of clusters (STAR Methods).
We demonstrated the F1 performance and scalability of Franken when compared to state-of-the-art methods on CyTOF data obtained from two independent celullar datasets: (1) a real-world healthy bone marrow dataset1 and (2) data from 10 cell lines stained with our prostate-centric antibody panel (around 200,000 cells each; Figures S1B–S1E and STAR Methods). We also showed that our pipeline is robust to its parameters' choice and scalable up to dozens of millions of cells in a synthetic dataset (Figures S1F and S1G and STAR Methods). Franken could be run on up to 40 million cells (which was computationally infeasible for other methods) in the time it would take Phenograph14 to analyze one million cells. Franken also ran over 20 times faster than the state-of-the-art single-cell RNA sequencing clustering technique, Seurat.
Franken’s scalability and ability to resolve rare metaclusters made it suitable to explore our new prostate cancer patient dataset containing 1,670,117 cells. Analysis of the prostate cancer dataset using Franken identified a total of 55 clusters (Figure 2A). It must be noted that discrete labels do not easily apply to datasets with continuous expression such as single-cell data, where a clear cutoff between cell states does not necessarily exist. Nonetheless, to obtain clusters that were qualitatively different in terms of marker combination (which markers were expressed, instead of how much), Franken clusters were further merged into 33 metaclusters using hierarchical clustering of Pearson correlation dissimilarities with average linkage. Based on marker expression profiles, we defined 14 epithelial, 16 immune, one stromal, and one endothelial phenotype (Figure S2A). We also identified one cluster that was mostly negative for all 36 markers in the panel (denoted as NE01). This may represent a cell type not characterized by the markers in our panel or simply outliers and was excluded from further analysis. All metaclusters were annotated using a two-letter and two-digit identifier ranked by decreasing metacluster size (TC01 > TC02 > TC03 > …) for each cell category. The total number of cells in a cluster ranged from a few hundred (437 cells in EP01) to hundreds of thousands (391,554 in TC01; Figure S2B).
Figure 2.

Prostate cancer samples have similar overlapping phenotypic profiles
(A) Heatmap of scaled mean signal of marker expression in 55 Franken clusters; numbers are colored according to metaclusters resulting from hierarchical clustering merging (using Pearson correlation dissimilarities) of Franken clusters. Bar plot below the heatmap corresponds to the number of cells found in each cluster.
(B) UMAP map of 23,200 (400 per patient) cells colored by cellular metacluster as indicated in (A).
(C) UMAP map of 23,200 (400 per patient) cells colored by patient.
(D and E) Boxplots of frequencies of the main cell types across all 58 samples from tumor, ABPT, and RPT. Significant changes were seen between tumor and ABPT in the proportion of granulocytes (two-sided Wilcoxon signed rank paired test, p = 0.008). N = 17.
(E) Boxplots of frequencies of the main cell types in samples from all 58 patients in our cohort stratified by intermediate- and high-grade tumors. Changes in luminal and T cell compartments are significant according to a two-sided Mann-Whitney-Wilcoxon test (p = 0.028 and 0.014, respectively). Intermediate N = 46 and high grade N = 12.
To project the high-dimensional data into a two-dimensional representation, we used the UMAP (Uniform Manifold Approximation and Projection) method for dimensionality reduction visualization.15 UMAP showed that our analysis recapitulated the main cell-type compartments within the prostate (Figure 2B); also detected were rarer cell states such as apoptotic cells (TR02, AE01, AI01, MA03, and TC03). Franken was capable of resolving very rare populations present at frequencies as low as 1/5,000 (PR-high metacluster EP01; Figure S2C). To ensure good separability of each class (metacluster) and quality of the final metaclustering, we trained a logistic regression classifier with lasso regularization (using 5-fold cross-validation to identify the regularization parameter λ; Figures S2D and S2E). Most metaclusters could be predicted with higher than 99% accuracy, and the lowest at 93%. A few metaclusters (TR03, GR03, MA01) showed unexpected marker co-expressions (CK19+/CD15+, CD15+/CD3+, CD68+/CD3+ respectively), and to ensure these did not result from doublets, DNA was plotted against event length (Figures S2F and S2H).
All detected clusters contained cells from ABPT and tumor regions (Figure S2I). This suggested that tumor cells were present within the ABPT tissue and/or that ABPT tissue was present inside the tumor mass. This was expected due to the way prostate tumors infiltrate the prostate as well as intrinsic limitations of the macroscopic-based sample collection procedure (which could not ensure the adjacent regions were 100% tumor-free). Alternatively, this could suggest our custom panel missed markers to allow such distinction.
Luminal cells were the most abundant cell type in the prostate and corresponded on average (across all 58 patients) to 32% of a patient’s sample. This is in line with findings from Chen et al.16 who concluded from a scRNA-seq experiment that luminal is the dominant epithelial type for most prostate tumors. They also observed that most clusters consisted of cells from multiple patients, as was the case in our dataset. T cells were the second most abundant population (24% on average). When combined, cells from the immune compartment and other cells of the tumor microenvironment made up over half the cells (54% on average) found within samples of this cohort (Figure S2J). Franken clustering identified a range of prostate epithelial phenotypes including a single basal cell phenotype characterized by CK5 and CK14 expression (BA01), four transitional epithelial phenotypes expressing a combination of CK7 and CK19 (TR01-04), and seven epithelial luminal phenotypes (LU01-07) defined by the expressed CK8, CK18. Cellular metaclusters that contained a combination of CK7 or CK19 and CK8 or CK18 were annotated as luminal epithelial cells. Only CK7- and CK19-positive metaclusters with very low to no CK8 and CK18 expression were denoted as transitional epithelial cells. Luminal epithelial cells also expressed a combination (or varying expression intensities) of AR, PSA, prostein, synaptophysin, AMACR, EZH2, PTEN, and Nkx3.1. These were weakly or not expressed in transitional or basal cell metaclusters.
In the microenvironment, five different T cell phenotypes were detected expressing CD3 and CD45, and five macrophage phenotypes were characterized by CD68 and CD45 expression. Also detected were three granulocytic (expressing CD24 and/or CD15), one stromal (characterized by SMA and S100A4), one B cell (CD20-expressing), and one endothelial (CD31-expressing) metaclusters. Unlike in previous studies,17 we did not observe patient-specific batch effects, which could have led to each patient clustering separately from one another. We found that each metacluster contained a mixture of cells from most patients as illustrated in the UMAP visualization (Figure 2C).
There was considerable overlap in the single-cell phenotypes present within paired tumor and ABPT regions (UMAP; Figure 2D). Samples were stratified according to tumor, ABPT, and RPT across cell types, and we found significantly lower frequency of granulocytes in tumor regions than in ABPT (Figure 2D). Visualization of cells from intermediate (ISUP grades II and III) and high-grade tumors (ISUP grade V) also revealed significant overlap (UMAP; Figure 2E) and indicated lower frequencies of luminal cells and higher frequencies of T cells in samples from patients with high- versus intermediate-grade disease (Figure 2D).
Immune landscape differs between tumor and benign adjacent tissue and across prostate cancer ISUP grade
The UMAP visualization of 23,200 cells (400 per patient) revealed the expression patterns of markers associated with the microenvironment (Figures 3A and S2K). We validated both observations by quantifying T cells (CD3+) and granulocytes (CD15+) in a tissue microarray (Figure S2J and STAR Methods) that included formalin-fixed paraffin-embedded tissues from all patients in the cohort. Confirming the mass cytometry data, we observed higher densities of T cells in high-grade (Figures 3B and 3C) than in intermediate prostate tumors and lower densities of granulocytes in tumor regions than in ABPT regions (Figures 3D and 3E).
Figure 3.

Stratification of samples reveals prostate tissue changes associated with tumor and advanced disease
(A) UMAP of 23,200 cells (400 per patient) colored by expression of indicated marker.
(B) Representative tissue sample stained for CD3 from a tissue microarray generated from prostate samples from the same cohort analyzed by mass cytometry.
(C) Densities of T cells as determined by CD3 staining (p = 0.042).
(D) Representative tissue sample stained for CD15 from a tissue microarray generated from prostate samples from the same cohort analyzed by mass cytometry.
(E) Densities of granulocytes as determined by CD15 staining (p = 0.058). Scale bar, 50 μm.
(F) Proportion of T cell metaclusters in ABPT and tumor samples across patients with paired samples (p = 0.066, 0.169, 0.023, 0.002, and 0.332 for TC01–05, respectively). N = 17.
(G) Summary table of clusters that were significant when comparing ABPT and tumor samples (N = 17 for both groups). Metaclusters enriched in ABPT are colored in blue, while those enriched in tumor samples are colored in red. Comparison between intermediate- and high-grade patient samples (for combined tumor/ABPT; intermediate N = 46 and high grade N = 12). Metaclusters enriched in patients with high-grade disease are colored in dark red. Only significant relationships are colored, and remaining comparisons are shown in gray.
(H) Proportion of macrophage metaclusters in ABPT and tumor samples across patients with paired samples (p = 0.095, 0.169, 0.515, 0.0004, and 0.014 for MA01–05, respectively). N = 17. In panels (F) and (H), dots are colored by disease severity (intermediate versus high grade). In all boxplots, boxes illustrate the IQR (25th to 75th percentile), the median is shown as the middle band, and the whiskers extend to 1.5 times the IQR from the top (or bottom) of the box to the furthest datum within that distance. Statistical testing between dependent paired tumor and ABPT samples was done using a Wilcoxon signed rank paired-sample statistical tests (two-sided). Independent intermediate- and high-grade samples were tested using a two-sided Wilcoxon rank-sum test. Number of patients in each group is indicated by N.
We identified multiple subpopulations of some immune cell types. By focusing on the comparison of each individual metacluster, we found that two T cell clusters were significantly enriched in tumor samples (TC03 and TC04, apoptotic T cells and proliferating T cells, respectively) when compared to the adjacent ABPT regions (Figure 3F). Notably, proliferating T cells were also enriched in high-grade tumors and in high-grade patient samples when tumor and adjacent tissue were mixed (Figures 3G and S3A), suggesting that this T cell phenotype is enriched throughout the prostate of patients with high-grade disease and not only in the core of the tumor.
The overall frequencies of macrophages were not significantly different between tumor and tumor-adjacent (ABPT) samples (Figure 2D). However, two macrophage metaclusters were enriched in tumor samples (MA04 and MA05, proliferating macrophages and CD45low macrophages, respectively). These same metaclusters were further enriched in high-grade patient samples (Figure S3B). The overall macrophage proportion was lower in tumor samples than in tumor-adjacent samples, highlighting the importance of analyzing such a complex dataset at single-cell resolution to reveal that rare macrophages phenotypes can change in the opposite trend to the overall macrophage population. The majority of macrophages are localized in the prostate stroma, but their density is greater in tumorigenic regions.18,19 This is a confounding factor when comparing macrophage frequencies across tumor grades, since lower grade tumors have a greater proportion of stroma than high-grade tumors, resulting in a higher frequency of stroma-infiltrating macrophages (Figure 3H).
In summary, our clustering analysis identified changes in the cellular phenotypes present in the prostate tumor microenvironment compared to adjacent ABPT regions. Distinct macrophage phenotypes were associated with prostate tumors and with the stroma rich ABPT regions. Overall, the cell-type compositions of the tumor microenvironments differed with tumor grade, except for granulocytes, which were decreased in tumor regions regardless of grade (Figure S3D). Although the tumor microenvironment of the intermediate sub-cohort was characterized mostly by a relative decrease of immune phenotypes compared with ABPT regions, we found the opposite in the high-grade sub-cohort where multiple immune phenotypes were enriched, notably highly proliferative macrophage and T cell phenotypes.
Malignant and benign prostate tissues diverge in rare phenotypes
Matched tumor and ABPT samples exhibited overall similar single-cell phenotypic profiles, and phenotypic profiles were similar across patients. These similarities were likely due to the presence of benign tissue in both tumor and ABPT samples, whereas patient-specific phenotypes are related to heterogeneous, de-regulated malignant cells.20 We found that every sample from every patient, including tumor and ABPT samples, contained basal cells (BA01) as well as CK7+/CK19+ live and apoptotic transitional epithelial cells (TR01 and TR02, respectively). All patient samples also contained a variety of luminal epithelial cells (LU01–LU07) containing varying combinations of luminal markers CK8/18, AR, PSA, prostein, Nkx3.1, and in some cases the co-expression of CK19 and CK7 (LU02 and LU04, respectively). However, cell types co-expressing both CK7 and CK19 expressed little to no CK8/18 (TR01-04). Stem cell marker CD24 and neuroendocrine marker synaptophysin showed highest expression in luminal epithelial cells. We carried out statistical comparisons for epithelial metaclusters and summarized the significant relative enrichment results across all metaclusters (Figures 4A, S3, and Table S1) for comparisons between patient grade groups (top row; NI = 46, NH = 12), tumor and ABPT (middle row; NI,H = 17), and high- versus intermediate-grade tumor regions only (bottom row; NI = 10, NH = 7). We identified the enrichment of apoptotic epithelial cells in tumor versus ABPT regions, which was irrespective of tumor grade (Figures 4A, S3E, and S3F). We also found that luminal metaclusters were typically enriched in ABPT regions and/or in intermediate-stage patient samples. In particular, prostein-high and AR-low metaclusters (LU03 and LU05; p values = 0.0001 and 0.026 respectively) were depleted in tumors versus ABPT samples (Figures 4A, S3G, and S3H). The depletion of prostein-high phenotypes was even more pronounced in high-grade compared to intermediate tumors (Figure 4A; bottom row). It is possible that during tumor progression, regulation of differentiation programs is lost, and PSAs are no longer expressed, supporting the hypothesis that aggressive tumor cells are de-differentiated. A rare SMA-positive luminal cell type (LU06; p value = 0.012) was characteristic of patients with intermediate disease, irrespective of tumor or ABPT region, although this was strongly influenced by the contribution of one specific patient to this cell type. The only luminal metacluster enriched in high-grade patients was a rare PSA-low, CD15+, CD24+ (an adhesion protein previously identified as a cancer stem cell marker21), and AR-high cell type (LU07; p value = 0.028; Figures 4A and S3H). Two additional CD15+ cell populations were identified, TR03 and TR04, among transitional epithelial metaclusters. Both were increased in tumor and high-grade patient samples, though significant enrichment could only be detected in TR03 (p value = 0.003), which co-expressed CK19 and CK7 (Figures 4A and S3J), while TR04 may be a more common precursor with lower cytokeratin expression. TR03 and TR04 also expressed a low amount of basal markers CK5/14.
Figure 4.

Characterization of epithelial tumor clusters and patient groups
(A) Bar indicating which metaclusters were significantly enriched between three pairs of conditions: (top bar) high- and intermediate-grade samples, irrespective of tumor status; (middle bar) tumor versus ABPT; (bottom bar) intermediate- and high-grade tumor regions only. All significant nominal p values are indicated given their level of significance for the figure, and those still significant after Bonferroni correction are indicated by a dark black outline. p values for all tested hypotheses are given in Table S1.
(B) Pairwise Tanimoto similarity of intermediate- (I) and high-grade (H) tumor (T) and benign tumor-adjacent (TA) samples for metaclusters in the microenvironment and epithelium. Color intensity and the size of the circle are proportional to the Tanimoto similarity.
(C) Correlation of metaclusters across 17 tumor patient samples. Correlations in the paired adjacent benign tissue that were lost in tumor are indicated by an L in the correlation plot, while correlations that were gained are indicated by a G. Only Spearman correlations with a significance level <0.05 are shown to exclude spurious correlations. Metacluster labels are colored to reflect cell types as in Figure 2A.
(D) UMAP projections of 23,200 cells (400 cells per patient) colored by expression of indicated epithelial and prostate-specific markers. Maximum signal (=1) is shown in gray.
(E) CD15 and p63 co-stained showing CD15 expression in epithelial cells from patients with acinar (left) and with ductal (middle) carcinoma as well as absence of CD15 in normal glands (right) showing basal cell layer expressing p63. Scale, 25 μm.
(F) Number of patients with cells belonging to a specific metacluster. Colors and labels matched to panels (A) and (C).
(G) Grouped patient samples represented by proportion of metaclusters. Colors in bar plot reflect those on panels (A) and (C).
When interpreting changes in tumor and ABPT (Figure 4A, middle bar) in combination with tumor-only changes across grades (Figure 4A, bottom bar), we found that in some metaclusters (LU03, MA04, TC04) the effect between tumor and ABPT was stronger than the effect between patient grades. The depletion of LU03 and enrichment of MA04 and TC04 in tumors was observed across high- and intermediate-grade patients, but an effect could still be observed between the different grade groups. In other cases (AE01, GR01, GR02, and TC03), no effect could be detected between grades, only between tumor and ABPT samples, suggesting that enrichment (AE01 and TC03; p values = 0.003 and 0.022) or depletion (GR01 and GR02; p values = 0.044 and 0.0004) of these cellular phenotypes happens in tumors of patients irrespective of grade. Lastly, there were metaclusters that changed less significantly between tumor and ABPT (LU05, TR03, and MA05; p values = 0.025, 0.096, and 0.014) and more across grades (p values = 0.004, 0.026, and 0.0007). This suggests that although there may have been a difference between tumor and ABPT regions, a difference also existed between the tumor regions of intermediate- and high-grade tumors. These cellular phenotypes suggest a possible progressive change in the prostate, where some metaclusters are lowest in ABPT, higher in intermediate-grade tumors, and even higher in high-grade tumors (or the reverse with highest expression in ABPT and progressive loss in intermediate-, then high-grade tumors). We were also interested in integrating information across all cellular phenotypes in the epithelium and in the microenvironment (Figure 4B). We took the mean metacluster proportion across all 17 patients for which we had tumor and ABPT (tumor adjacent) samples. We calculated the Tanimoto similarity between intermediate-grade tumor-adjacent regions (ITAs), intermediate-grade tumors (ITs), high-grade tumor-adjacent regions (HTAs), and high-grade tumors (HTs). We found that in the epithelium, tumor-adjacent regions in intermediate and high-grade are most similar to one another, while ITs and HTs bear more similarity to each other than to their benign adjacent regions. However, in the microenvironment, HTAs are more similar to ITs than to their paired HTs, suggesting that HTs may have progressed from ITs, but with further changes in epithelial cellular phenotypes, while the tumor-adjacent microenvironment remains similar to that of an IT (Figure 4B).
Next, we analyzed correlations between metaclusters in tumor samples across the 17 patients for which we had paired tumor and ABPT samples (Figure 4C). We restricted the analysis to Spearman correlations with a significance level <0.05 for both correlations across tumor and ABPT samples. We compared correlations in the tumor to those in the paired adjacent benign tissue and found that while some correlations were lost in tumor (indicated by an L in the correlation plot; Figure 4C), others were gained (indicated by a G; Figure 4C). For example, luminal epithelial cell types LU03 and LU05 were uncorrelated in benign tissue but correlated in tumors. Indeed, we had found that both of these prostein-high metaclusters were depleted in tumors, and we found their decrease was correlated with one another. We found that while transitional epithelial metaclusters TR01 and TR02 (CK7+/CK19+ cells and CK7+/CK19+ apoptotic cells, respectively) were correlated in both tumor and adjacent benign prostate tissue, the strong correlation between TR03 and TR04 (CD15+/CK7+/CK19+ and CD15+/CK-low cells respectively) was only present in tumor regions. New correlations also appeared between cell types of the microenvironment. For example, GR02 and GR03 (CD15-low and CD3+ granulocytes, respectively) were newly correlated in tumor regions. We also observed that most T cell types became correlated in tumors, although they were uncorrelated in adjacent benign tissue. Anti-correlations were also gained. Notably, MA04, MA05 (proliferating and CD45-low macrophages), and TC04 (proliferating T cells) became anti-correlated with LU03 in tumor. This result suggests that the increase of these macrophage and T cell metaclusters may be related to the depletion of this, likely benign, prostein-high luminal cell type.
While many new correlations and anti-correlations were gained in tumor regions, we also found that previously (anti-)correlated cell types in benign tissue became uncorrelated in tumor samples. Most macrophage metaclusters were correlated in benign tumor-adjacent samples but no longer in tumor. Basal cells were correlated with TR03, but this was not the case in tumor samples anymore, likely due to the de-regulated, malignant expansion of these transitional epithelial cells. Apoptotic epithelial cells (AE01) were correlated with transitional metaclusters TR01, TR02, and TR04 in tumor-adjacent regions, but these correlations were lost in tumors. The main transitional epithelial metacluster TR01 used to be anti-correlated with various, likely benign, luminal cell types (LU01, LU03, LU05). It is believed that these transitional cells may originate from basal cells and are precursors of luminal cells, and therefore a balance between the population of CK7+/CK19+ cells and CK8/18 exists in a healthy scenario. However, our results support that this balance is disrupted in tumor regions.
The expression of CD15 in various malignant prostate epithelial cell populations (LU07, TR03, and TR04) was surprising, although high-dimensional mapping using UMAP had already revealed CD15-high expressing cells in regions of epithelial marker expression such as E-cadherin and EpCAM, luminal markers CK8 and CK18, and transitional epithelial markers CK7 and CK19 (Figures 3A and 4D). Immunohistochemistry confirmed the presence of CD15 in both ductal and acinar carcinoma of the prostate from patients who were shown to have this rare population by mass cytometry (one representative acinar and one ductal patient sample are shown in Figure 4E). This adhesion molecule is typically used as a granulocyte marker and plays important roles in cell adhesion22 and migration.23 Although previously observed in other carcinomas24, 25, 26 and demonstrated to be a marker of propagating tumor cells,27 cells expressing CD15 had not been previously detected in prostate tumors. To assess the clinical relevance of this CD15+/CK19+ subpopulation (metacluster TR03), we analyzed two other TMA cohorts with 374 patients (example core shown in Figure S4B) with localized disease (336) and metastatic disease (38) and found that the proportion of patients with CD15-positive cells increased with disease severity (low, intermediate, and high ISUP grades) and was highest among metastatic cases (Figure S4C). Survival analysis did not yield any differences for this cohort, but the number of CD15+/CK19+ cases was very low. Only 5% positive cases were detected (19 patients out of 374, for which survival data were only available for 11), while via CyTOF most patients contained this cellular phenotype and 12% (7 out of 58) showed an enrichment. However, even in enriched cases, TR03 represented on average 0.6%–1.1% of cells in a patient, and overall, across all patients, this cellular phenotype represented on average 0.3% of cells, making it difficult to identify a lot of positive cases in a TMA spot with diameter 0.06 mm; here the number of cells is substantially lower than can be detected via high-throughput mass cytometry. It is very likely many CD15-positive patients were missed in the TMA analysis, which impaired survival analysis.
Overall, 0.1%, 0.3%, and 0.1% cells were found across the whole dataset from LU07, TR03, and TR04, respectively. Additionally, these rare CD15-expressing metaclusters were among the 14 observed in a subset of the patient cohort (Figure 4F). The remaining majority of metaclusters (29) were represented across all patients. In conclusion, our methodology identified a prostate tumor subpopulation, which may also characterize a distinct patient subtype.
Although elevated PSA levels are typically associated with localized prostate cancer, luminal and transitional phenotypes found enriched in tumor or high-grade samples had very little or no PSA expression (AE01, TR03, and LU07). Prostate cancer cells that express low levels or no PSA may be a self-renewing, tumor-propagating cell population that resists ADT.8 Similarly, not all phenotypes increased in tumor regions were high in AR. While AR overexpression is associated with advanced disease and was found in one of the metaclusters enriched in HTs (LU07), an AR-low phenotype was also enriched in high-grade samples (TR03). Loss of AR has been associated with resistance to ADT,9 and our results support that malignant phenotypes are not necessarily high in AR. We also found that AR-low metaclusters could be distinguished between benign and malignant by the presence of prostein and PSA. AR-low phenotypes that were PSA- and prostein-high were benign (LU03 and LU05), while malignant TR03, which was low in AR, was also low in PSA and prostein.
Rare cellular phenotypes define patient subgroups
After having described the different cellular phenotypes present in prostate tumors, we wondered whether certain metaclusters (or combinations) could characterize patient groups. We clustered patients according to metacluster proportions using hierarchical clustering with Pearson correlation dissimilarity. We found many small groups across our 58 patient cohort, and some could be characterized by the enrichment or depletion of a handful of phenotypes (Figure 4G). After statistical testing, we were able to define which cell types significantly defined a patient group (Figures 4G and S4D). Notably, patient group 1 consisted of patients with enrichment of transitional epithelial cells expressing CD15 (TR03 and TR04). The former, as had already been observed in our study, was enriched in HTs. Patient group 2 showed significant enrichment of prostein-high phenotype LU05. Group 3 consisted of patients with the highest proportion of apoptotic epithelial cells (AE01), previously associated with malignant tissue. One patient was characteristically different from all others due to the highest enrichment of SMA, metacluster LU06, and clustered separately from the rest constituting group 5. A high proportion of LU07 (CD15+, AR-high luminal cells), which we had already shown as enriched in HTs (Figure 4A), was characteristic of patient group 8. LU04, a CK7+/CK8/18 luminal cell type, was enriched in patients in group 9, although it is not clear the role of this metacluster as it was not enriched/depleted in tumors nor did it differ according to tumor grade. Group 10 was marked by the enrichment of proliferating macrophages (MA04) as well as CD45low macrophages (MA05), which were increased in tumor regions compared to adjacent benign tissue and in the case of MA05 were even further enriched in the tumor regions of patients with high-grade disease. These two populations may be related to each other as they strongly correlated both in a tumor and benign tissue samples (Figure 4C). Patient groups 11, 12, and 13 consisted of patients with a rather mixed cell type composition, or very few patients and very weak enrichment or depletion of markers; it is not clear whether these consist of cohesive patient groups.
The long-term effect that these phenotypes may have on survival remains to be determined. We showed that few single-cell phenotypes can further stratify patients beyond their ISUP grade and may represent treatment targets for personalized treatment.
Discussion
We performed single-cell analysis of 1,670,117 cells from 58 prostate cancer patients by mass cytometry using a newly developed computational pipeline that provides a combination of high-dimensional clustering performance and speed. Of note is the fact that most prostate cancer patients differed mainly in rare cell types. Based on our results, we hypothesize that cellular phenotypes associated with resistance to hormone ablation therapy are also found in treatment-naive prostate cancer patients. This is consistent with the big bang theory as has been shown by Sottoriva et al. in colorectal cancer. In addition, we found alterations in the microenvironment, namely T cells and proliferating macrophages, which were associated with aggressive disease.28 Of note, we identified a rare CD15+ epithelial phenotype that was increased in a subset of patients with high-grade prostate cancer.
In this study, we have described 33 prostate cellular phenotypes including 14 epithelial and 18 cell types from the microenvironment. Tumors and surrounding ABPT had considerable similarity: 9 of the 33 metaclusters (27%) were present at significantly different frequencies between the two regions. Although this was much higher than would be expected at random for a confidence level of 5% (1.65 out of 33), almost two-thirds of cell types and states detected (24 out of 33) were shared at similar proportions in tumor and ABPT tissue. Most epithelial differences between tumor and ABPT and tumor grades involved luminal cell types with the exception of one important transitional epithelial phenotype (the CD15-high metacluster TR03), suggesting that prostate tumorigenesis is strongly affected by an interplay of luminal phenotypes. A scRNA-seq study earlier this year by Chen et al.16 noted that a transitional epithelial cells signature is associated with better survival, when this signature was applied to TCGA data. This is in line with our findings if considering the most abundant transitional epithelial cell subpopulations (TR01 and TR02), but their conclusion, due to the comparatively lower throughput (36,424 cells across 12 patients), overlooks rare subpopulations.
Prostein-high phenotypes were depleted in tumor regions and even more so in HTs; this suggests that during tumorigenesis there is selection for poorly differentiated prostate cell types. Tumor-enriched phenotypes all contained EpCAM. High levels of EpCAM expression at both mRNA and protein levels were previously reported in prostate cancer tissues and cell lines.29,30 Our analysis showed that both AR-high/PSA-low (LU07) and AR-low/PSA-low (TR03) cells were present in localized, hormone-naive prostate tumors even though such phenotypes had previously been associated only with castration-resistant disease after ADT or metastatic disease.7, 8, 9 Cells in the AR-high/PSA-low cluster also overexpressed Nkx3.1. Loss of prostein15 and PSA expression,31 Nkx3.1 overexpression,32 and AR overexpression or amplification33, 34, 35 have been shown to be common in castration-resistant disease states. Furthermore, men with localized high-grade prostate cancer but low PSA show inferior cancer survival.36 It remains to be determined whether these rare cells with the properties of tumors resistant to ADT are capable of dissemination and are responsible for disease progression after prostatectomy. Surprisingly, two phenotypes enriched in high-grade patients expressed CD15. After analyzing an additional 374 patients’ TMA samples, we also found that CD15+/CK19+ prostate epithelial cells were further enriched in metastatic disease. CD15 plays an important role in cell adhesion and migration,22,23 and CD15-expressing cells have been identified in other tumor types as having stem-like potential but not yet in prostate cancer24, 25, 26 and might represent a biomarker for aggressive phenotypes with a higher metastatic potential.
The tumor microenvironments were similar in both tumor and the neighboring ABPT regions for patients of different tumor grades with the exception of granulocytes, which were present at lower levels in tumors regardless of grade. Other rarer immune cell types changed both between tumor and ABPT regions as well as across tumor grades. In particular, we observed that one proliferating T cell (TC04) and two macrophage (proliferating MA04 and CD45-low MA05) phenotypes were enriched in tumor regions and were further enriched in HTs.
The microenvironments of tumors from the intermediate sub-cohort had lower frequencies of immune phenotypes compared to the ABPT regions, but we found the opposite in the high-grade sub-cohort. In HTs, there was an enrichment of multiple immune phenotypes compared to the ABPT regions. It is currently unclear whether ITs progress to high-grade disease. If such a progression happens, our analysis suggests that the hyperplasia and expansion of the epithelial compartment might precede alterations in the tumor microenvironment, or it may be that these differences are reflective of disease stage. We proceeded to analyze the overall changes in the microenvironment and epithelium by integrating information across all metaclusters in these two compartments and estimating the Tanimoto similarity between HTs and ITs as well as high-grade and intermediate-grade tumor-adjacent ABPT regions (HTA and ITA, respectively). We found further evidence of a possible progression from intermediate to HTs suggested by the similarity of the microenvironments of ITs and high-grade ABPT regions. Some of the rare cell types in the microenvironment, which we found to be enriched in tumors from both grade groups and further enriched in high-grade patients, may represent putative targets that can be used to prevent the progression of the disease.
Tumors do not grow in isolation; cancerous cells require support from the microenvironment. Accessory cells have been successfully targeted with therapy.37, 38, 39 We found that most immune metaclusters were present at similar frequencies or were decreased in the tumor compared to the adjacent ABPT regions with the notable exceptions of rare T cell and macrophages cellular phenotypes. Both monocyte infiltration and macrophage proliferation are necessary for macrophage maintenance during tumor growth,40 and in breast cancer, proliferating macrophages are associated with high tumor grade, hormone receptor negativity, and poor clinical outcome.41 However, macrophage counting based on immunohistochemical analysis had not led to any consensus on the prognostic significance of tumor-associated macrophages in prostate cancer.18,42 In our prostate cancer cohort, certain macrophage phenotypes (MA04 and MA05) were enriched in prostate tumors and even more so in high-grade patients. Taken together with findings that tumor-associated macrophages (TAMs) are capable of proliferation,41 our data suggest that in addition to therapy that inhibits differentiation of TAMs from circulating monocytes, blocking the proliferation of macrophages may represent an additional therapeutic avenue to pursue in order to slow the development of high-grade disease in prostate cancer patients. Our clustering analysis identified that not all macrophage phenotypes changed frequency in tumor compared to ABPT regions. This suggests the presence of separate cancer- versus stroma-infiltrating macrophage phenotypes that may have opposing influences on tumorigenesis,18,20 highlighting the importance of investigating macrophage infiltration in prostate cancer.
In summary, new biomarkers are needed to identify which men qualify for active surveillance or need aggressive treatment. Understanding the cellular complexity of prostate tumors and their microenvironments is key to the development of new diagnostic and treatment strategies. Here, we provide a description of prostate tissue heterogeneity at the single-cell level and describe differences between tumors and the neighboring benign regions as well as across tumor grades. We identify two CD15-high phenotypes enriched in high-grade patients as well as changes to the microenvironment in rare macrophage and T cell phenotypes associated with tumor regions and high-grade disease. We also identify in men with localized disease, epithelial subpopulations associated with advanced castration-resistant disease. The alterations to the epithelium and microenvironment should be further explored to guide development of new diagnostic and treatment paradigms for prostate cancer and to understand which cellular phenotypes in primary prostate cancer need to be detected and may change treatment decisions.
Limitations of the study
Our study provides the largest scale analysis of prostate cancer at the single-cell proteomics level. While it provides evidence that rare cellular types associated with advanced, hormone-refractory disease pre-exist in hormone-naive patients, it has its limitations; Our patient cohort did not allow for a survival analysis as patients with localized disease have an expected survival of 5–10 years. Additionally, we found that tumor and non-tumor regions differed in rare cell types. This made it difficult to employ bulk RNA sequencing in survival analysis as well. Furthermore, we discovered a rare proliferating macrophage and T cell subpopulations as well as a rare CD15+ cell type that is enriched in tumor and advanced disease, but further work is needed to investigate the role played by these cell types in the context of prostate tumorigenesis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-human-AMACR2 (13H4) | Thermo Fischer | Cat# MA5-14576, RRID:AB_10985819 |
| anti-human-Androgen Receptor AR (D6F11) | Cell Signaling Technologies | Cat# 5153; RRID:AB_10692774 |
| anti-human-Carbonic Anhydrase IX (polyclonal_CA9_AF2188) | R&D Systems | Cat# AF2188; RRID:AB_416562 |
| anti-human-CD15 (HI98) | Biolegend | Cat# 301902; RRID:AB_314194 |
| anti-human-CD20 (H1(FB1)) | Becton Dickinson | Cat# 555677; RRID:AB_396030 |
| anti-human-CD24 (ML5) | Becton Dickinson | Cat# 555426; RRID:AB_395820 |
| anti-human-CD3 (UCHT1) | E-Biosciences | Cat# 300402; RRID:AB_314056 |
| anti-human-CD31 (HC1/6) | Millipore | Cat# CBL468-K; RRID:AB_1586934 |
| anti-human-CD326 (EpCAM) (9C4) | Biolegend | Cat# 324202; RRID:AB_756076 |
| anti-human-CD44 (IM7) | Biolegend | Cat# 550538; RRID:AB_393732 |
| anti-human-CD45 (HI30) | Biolegend | Cat# 304002; RRID:AB_314390 |
| anti-human-CD68 (KP1) | E-Biosciences | Cat# 333802; RRID:AB_1089058 |
| anti-human-Cleaved Caspase3 (C92-605) | Becton Dickinson | Cat# 559565; RRID:AB_397274 |
| anti-human-cleaved PARP (F21-852) | Becton Dickinson | Cat# 552596; RRID:AB_394437 |
| anti-human-Cytokeratin 19 (Troma-III) | Dev Studies Hybridoma Bank | Cat# MABT913 RRID: AB_2133570 |
| anti-human-Cytokeratin 5 (EP1601Y) | Abcam | Cat# ab52635; RRID:AB_869890 |
| anti-human-Cytokeratin 7 (RCK105) | Becton Dickinson | Cat# 550507; RRID:AB_2134456 |
| anti-human-Cytokeratin 8/18 (C51) | Cell Signaling Technologies | Cat# 4546; RRID:AB_2134843 |
| anti-human-E-Cadherin (24E10) | Cell Signaling Technologies | Cat# 3195; RRID:AB_10694492 |
| anti-human-ERG (EPR3864(2)) | Abcam | Cat# ab174739, RRID:AB_2905642 |
| anti-human-EZH2 (SP129) | Spring Bioscience | Cat# 5246; RRID:AB_10694683 |
| anti-human-fap (polyclonal_FAP) | R&D Systems | Cat# AF3715; RRID:AB_2102369 |
| anti-human-FSP1 / S100A4 (NJ-4F3-D1) | Biolegend | Cat# 370002 RRID: AB_2566630 |
| anti-human-Glucocorticoid Receptor (D6H2L) | Cell Signaling Technologies | Cat# 12041, RRID:AB_2631286 |
| anti-human-H3K27me3 (C36B11) | Cell Signaling Technologies | Cat# 9733; RRID:AB_2616029 |
| anti-human-Histone H3 (HTA28) | Biolegend | Cat# 641002, RRID:AB_1227659 |
| anti-human-Keratin 14 (KRT14) (polyclonal_PA5-16722) | Thermo Fischer | Cat# PA5-99310, RRID:AB_2818243 |
| anti-human-Keratin Epithelial (AE3) | EMD Millipore | Cat# MAB1611, RRID:AB_2134409 |
| anti-human-Ki-67 (B56) | Becton Dickinson | Cat# 550609, RRID:AB_393778 |
| anti-human-NKX3.1 (EPR14970) | Abcam | Cat# ab186413, RRID:AB_2905641 |
| anti-human-p53 (EPR17343) | Abcam | Cat# ab179477; RRID:AB_2737134 |
| anti-human-pan Cytokeratin (AE1) | Millipore | Cat# MAB1612; RRID:AB_2132794 |
| anti-human-Progesterone Receptor (YR85) | Abcam | Cat# ab32085; RRID:AB_777452 |
| anti-human-Progesterone Receptor A/B (PR-2C5) | Thermo Fisher | Cat# 18-0172, RRID:AB_86695 |
| anti-human-Prostein (E-5) | Santa Cruz | Cat# sc-390873, RRID:AB_2905640 |
| anti-human-PSA (D6B1) | Cell Signaling Technologies | Cat# 5365, RRID:AB_2797609 |
| anti-human-PSMA (YPSMA-1) | Abcam | Cat# ab19071, RRID:AB_444751 |
| anti-human-PTEN (138G6) | Cell Signaling Technologies | Cat# 9559; RRID:AB_390810 |
| anti-human-SMA (1A4) | Abcam | Cat# ab8207; RRID:AB_306356 |
| anti-human-Synaptophysin (YE269) | Abcam | Cat# ab187259, RRID:AB_2905639 |
| anti-human-Vimentin (EPR3776) | Abcam | Cat# ab92547; RRID:AB_10562134 |
| Biological samples | ||
| Prostate Cancer tumor tissue samples | University Hospital Zurich | N/A |
| PBMC | Blutspende Zürich | N/A |
| Fibroblasts, foreskin | gift from the laboratory of Dr. Robert A. Weinberg at the Massachusetts Institute of Technology | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Antibody Stabilizer PBS | Candor Bioscience | Cat# 131 050 |
| Bis(2,2′-bipyridine)-4′-methyl-4-carboxybipyridine-ruthenium-N-succidimyl ester-bis(hexafluorophosphate) (96Ru, 98-102Ru, 104Ru) | Sigma Aldrich | Cat# 96631 |
| Bismuth trichloride (209Bi) | Sigma Aldrich | Cat# 450723 |
| maleimidomono-amido-DOTA (mDOTA) | Macrocyclics | Cat# B272 |
| Cell-ID Intercalator-Ir | Fluidigm | Cat# 201192B |
| DMSO | Sigma Aldrich | Cat# D2438 |
| EDTA | StemCell Technologies, Inc. | Cat# EDS-100G |
| EQ Four Element Calibration Beads | Fluidigm | Cat# 201078 |
| FcR Blocking Reagent, human | Miltenyi Biotec | Cat# 130-059-901 |
| Indium (113In, 115In) | Fluidigm | N/A |
| Isothiocyanobenzyl-EDTA | Dojindo Laboratories | M030-10 |
| Lanthanide (III) metal isotopes as chloride salts | Fluidigm | N/A |
| Paraformaldehyde, 16 % w/v | Electron Microscopy Sciences | Cat# 15710 |
| Saponin | Sigma Aldrich | Cat# S7900 |
| Yttrium (89Y) | Sigma Aldrich | N/A |
| RPMI 1640 Medium | Thermo Fisher | Cat# 21875-034 |
| Tumor Dissociation Kit, human | Miltenyi Biotec | Cat #130-095-929 |
| heat-inactivated FBS | Thermo Fisher | Cat #10500064 |
| Cis-Diamminplatinum (II) Dichloride | TCI | Cat# D3371 |
| Maxpar X8 Multimetal Labeling Kit | Fluidigm | Cat# 201300 |
| Deposited data | ||
| Mass cytometry data | Mendeley |
https://doi.org/10.17632/5k5xfj 626k.1 |
| Experimental model: Cell lines | ||
| VCaP | ATCC | Cat# CRL-2876 |
| PC-3 | ATCC | Cat# CRL-1435 |
| LNCaP | ATCC | Cat# CRL-1740 |
| Du145 | ATCC | Cat# HTB-81 |
| 22Rv1 | ATCC | Cat# CRL-2505 |
| T47D | ATCC | Cat# HTB-133 |
| MDA-MB-231 | ATCC | Cat# HTB-26 |
| HMLE | gift from the laboratory of Dr. Robert A. Weinberg at the Massachusetts Institute of Technology | N/A |
| Software and algorithms | ||
| Franken algorithms | Zenodo/Github | https://zenodo.org/badge/latestdoi/340344455 |
| Cytobank | Kotecha et al., 2010 | https://www.cytobank.org/ |
| CATALYST | Chevrier et al., 2018 | http://bioconductor.org/packages/release/bioc/html/CATALYST.html |
| R Version 3.4.1 | R Core Team (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. | https://www.r-project.org/ |
| MATLAB | MATLAB, 2018 (R2018b), Natick, Massachusetts: The MathWorks Inc. | https://www.mathworks.com/products/matlab.html |
| UMAP 0.2.7 | McInnes et al.,2018 | https://cran.r-project.org/web/packages/umap/index.html |
| Seurat 3 | Butler et al., 2019 | https://satijalab.org/seurat/ |
| FlowSOM v1.22.0 | Van Gassen et al., 2015 | https://bioconductor.org/packages/FlowSOM/ |
| Phenograph/CYT3 | Levine et al., 2015 | https://github.com/dpeerlab/cyt3 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peter J. Wild (peter.wild@kgu.de).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Patient samples
The Ethics Committee of the Canton of Zurich approved all procedures involving human prostate material (KEK-ZH-No. 2008-0040). All patients were part of the Zurich Prostate Cancer Outcomes Cohort (ProCOC) study,43,44 and each patient signed an informed consent form. Prostatectomy samples were taken from 58 prostate cancer patients from the ProCOC cohort between 2015 and 2017. Tumors were of a range of ISUP grades. No clinical or histological status was used in the selection of the cohort. Staging and grading was performed using World Health Organization and ISUP criteria45). Twenty-four patients had ISUP grade II (Gleason score 3 + 4), 22 had ISUP grade III (Gleason score 4 + 3), and 12 had ISUP grade V prostate carcinoma (Gleason scores 4 + 5, 5 + 4, and 5 + 5).
Cell lines
Human epithelial cell lines were purchased from the American Type Culture Collection (ATCC) and were propagated and maintained according to the supplier’s instructions. Cell lines included PC-3, VCaP, Du145, LNCaP, 22Rv1, and T47D. HMLE were a gift from the laboratory of Dr. Robert A. Weinberg at the Massachusetts Institute of Technology and were cultured with the MEGM bullet kit (Lonza). Fibroblasts were a gift from the laboratory of Prof. Silvio Hemmi at the University of Zurich and were cultured in DMEM medium (Sigma Aldrich) supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, and 10% fetal bovine serum (FBS).
Peripheral blood mononuclear cells (PBMCs) from healthy donors were obtained from the Zurich Blood transfusion Service and were isolated by histopaque (Sigma Aldrich) density gradient centrifugation.
Method details
Sample collection and microarray construction
Immediately after surgery, native radical prostatectomy specimens were transferred to the frozen section lab on ice (4°C) and were processed within 15 min in the Department of Pathology and Molecular Pathology, University Hospital Zurich. The first slice after dissection of the apex was quartered and snap frozen in four separate blocks for biobanking within the ProCOC study. Fresh tumor and ABPT tissue were taken from the second slice after dissection of the apex without destruction of surgical margins and the pseudocapsule. After formalin fixation overnight, the rest of the specimen was embedded in paraffin. Hematoxylin and eosin-stained sections of the four frozen blocks were sliced for immediate evaluation regarding tumor load and margins in synopsis with the standard formalin-fixed paraffin-embedded histology to control for the representativeness of tissue sampling for mass cytometry.
Following evaluation of tissue sections by uropathologists (NJR, JHR, PJW) a tissue microarray (TMA) containing two ABPT and two tumor regions from all patients in the selected cohort was generated as previously described.46 For TMA construction, representative tumor areas of the second and third slice of radical prostatectomy specimens were chosen, as close as possible to the area of tissue sampling for mass cytometry. Figure S5 shows H&E images of the selected regions.
Fresh tissue preparation
After surgical resection and based on the aforementioned real-time frozen sections, the index tumor lesions (the most extensive with the highest Gleason score) were immediately harvested and transferred to precooled MACS tissue storage solution (Miltenyi Biotec) and shipped at 4°C. To better select the index lesion, only cases in which a tumor nodule was also macroscopically visible were selected, ultimately resulting in a cohort with higher ISUP grades. Only tumors that had a tumor cell content of at least 80% after histological evaluation of the fresh frozen sample were selected, and a representative piece of tissue (at least 4 × 4 × 1 mm) was excised with a normal blade. Tissue processing was completed within 24 h of collection.
For the dissociation of tissues to single cells, the tissue was minced using surgical scalpels and further disintegrated using the Tumor Dissociation Kit, human (Miltenyi Biotech) and the gentleMACS Dissociator (Miltenyi Biotech) according to the manufacturer’s instructions. The resulting single-cell suspensions were filtered through sterile 70-μm and 40-μm cell strainers and stained for viability with 25 μM cisplatin (Enzo Life Sciences) in a 1-min pulse before quenching with 10% FBS (Fienberg et al., 2012). Cells were then fixed with 1.6% paraformaldehyde (Electron Microscopy Sciences) for 10 min at room temperature and stored at −80°C.
Mass cytometry barcoding
To ensure homogenous staining, 0.3 × 106 to 0.8 × 106 cells from each tumor sample were barcoded as previously described using a 126-well barcoding scheme consisting of unique combinations of four out of nine barcoding reagents.47 Metals included palladium (105Pd, 106Pd, 108Pd, 110Pd, Fluidigm) chelated to 1-(4-Isothiocyanatobenzyl)ethylenediamine-N,N,N EN Etetraacetic acid (Isothiocyanobenzyl-EDTA, Dojino) and indium (113In and 115In, Fluidigm), yttrium (89Y, Sigma Aldrich), rhodium (103Rh, Fluidigm), and bismuth (209Bi, Sigma Aldrich) chelated to 1,4,7,10-tetraazacy-clododecane-1,4,7-tris-acetic acid 10-maleimide ethylacetamide (mDOTA, Dojino) following standard procedures.45 The chelated barcoding reagents were titrated for equivalent staining intensities; the final concentrations were adjusted to 20 nM (209Bi), 100 nM (105-110Pd, 115In, 89Y), 200 nM (113In), or 2 μM (103Rh). Cells were barcoded using the transient partial permeabilization protocol.48 Cells were washed with 0.03% saponin in PBS (Sigma Aldrich) and incubated for 30 min at room temperature with 200 μL of mass tag barcoding reagents. Cells were then washed twice with PBS plus saponin and twice with cell staining medium (CSM, PBS with 0.5% bovine serum albumin and 2mM EDTA).
Antibodies and antibody labeling
The supplier, clone, and metal tag for each antibody used in this study are listed in the key resources table. Antibody labeling with the indicated metal tag was performed using the MaxPAR antibody conjugation kit (Fluidigm). After metal conjugation, the concentration of each antibody was assessed using a Nanodrop (Thermo Scientific). The concentration was adjusted to 200 μg/mL and stored in Candor Antibody Stabilizer. All conjugated antibodies were titrated for optimal concentration for use with prostate tissues. Antibody usage in this study was managed using the AirLab cloud-based platform.49
Antibody staining and mass cytometry data collection
After barcoding, pooled cells were incubated with FcR blocking reagent (Miltenyi Biotech) for 10 min at 4°C. Samples were stained with 100 μL of the antibody panel per 106 cells for 60 min at 4°C. Cells were washed twice in CSM and resuspended in 1 mL of nucleic acid Ir-Intercalator (Fluidigm) overnight at 4°C. Cells were then washed once in CSM, once in PBS, and twice in water. Cells were then diluted to 0.5 × 106 cells/mL in H2O containing 10% of EQ™ Four Element Calibration Beads (Fluidigm). Samples were placed on ice until analysis. Data were acquired on an upgraded Helios CyTOF 2 mass cytometer using the Super Sampler (Victorian Airship) introduction system.
Immunohistochemistry
For immunohistochemical validation studies anti-CD3 (mouse monoclonal, clone LN10, Leica Microsystems) and anti-CD15 (mouse monoclonal, clone Carb-3, Agilent Dako) antibodies were used. Automated platforms were used for in situ protein expression analyses of CD15 (Ventana Benchmark CD15), and CD3 (Leica Bond-Max).
Quantification and statistical analysis
Mass cytometry data analysis
Individual .fcs files collected from each set of samples were concatenated using the .fcs concatenation tool from Cytobank, and data were normalized using the executable MATLAB version of the Normalizer tool.50 Individual samples were debarcoded using the CATALYST R/Bioconductor package51 (Figure S8 and Table S2). Debarcoded files were compensated for channel crosstalk using single-stained polystyrene beads as previously described.51
CyTOF data was analyzed by initially applying an arcsinh transformation with a cofactor of 5 (newdatai = arcsinh(datai/5)). The UMAP algorithm52 was applied to the high-dimensional data from 23,200 (400 per patient) cells taken at random from across the patient cohort using default parameters (perplexity, 30; theta, 0.5) to facilitate visualization in two dimensions. The pre-processed data were analyzed using the Franken algorithm as described in detail below. All analysis was done using R version 3.4.1
The Franken pipeline
The initial step of the Franken pipeline uses a SOM11,12 to over-cluster the preprocessed data into a large number of nodes. Prostate patient data was pooled from all patient samples (1,670,117 cells) and 400 SOM nodes were used (SOM grid dimensions were x = 20 and y = 20). Next, a mutual k-nearest neighbor graph (k = 6) was built between the SOM nodes using the Tanimoto similarity,13 which unlike the binary version, can be applied to continuous or discrete non-negative features and retains the sparsity property of the cosine while allowing discrimination of collinear vectors. Lastly, the resulting graph is clustered using a random-walk-based graph clustering technique called Walktrap, using the implementations available in package igraph53 in R. Walktrap is a graph partitioning technique that requires a choice of random walk steps. To increase our pipeline’s robustness, this procedure is applied for a range of random walk steps and the smallest step that maximizes the graph’s modularity is chosen.
According to the thorough review and comparison of community detection algorithms by Yang et al.,54 Walktrap is amongst the best performing algorithms for both large and small networks regardless of whether the mixing parameter is high or low. Although Yang et al. found that for large mixing parameters most algorithms failed to detect the community structure, Walktrap was able to do it. Another advantage of Walktrap is that it is possible (although not necessary) for a user to define the number of communities one wishes to find in the data. This allows the user to decide exactly how many clusters they wish to find, although the method is by default run in an unsupervised way. Although Walktrap is not the fastest method for large networks, the network size in Franken is never large due to the initial SOM-building step.
Benchmarking Franken against other methods for additional datasets
To test the performance of Franken, we compared it to two state-of-the-art clustering methods for mass cytometry data, Phenograph14 and FlowSOM.12 All three methods were used to cluster data obtained from two celullar datasets: (i) a real-world healthy bone marrow dataset1 and (ii) data from 10 cell lines stained with our prostate-centric antibody panel (Supplemental Figures S1B–S1F). The three methods were also applied to synthetic datasets generated from the simulation of high-dimensional gaussians (Figure 1G). The ground truth for the healthy bone marrow cellular phenotypes had been previously manually annotated in Bendall et al.1 while the ground truth for the cell lines dataset was known from their cell line origin. We calculated precision and recall (as shown by F1 scores according to Weber et al.2) for each phenotype in each dataset (Figures S1B, S1C and S1E). Franken was able to recover the most phenotypes in both datasets, resulting in the least phenotypes with zero F1 scores. After repeating the F1 estimates for multiple runs (with different random seeds) of each method, on average, Franken performed as well or better than the other methods. Franken requires the input of three parameters: the SOM grid dimensions (x and y which multiplied correspond to the number of nodes used to build the SOM) and k neighbors (the number of neighbors used to decide whether two nodes are connected by an edge in the mutual nearest neighbor graph); a SOM size of 400 (x = y = 20) and k = 6 were used in our simulations of all three datasets. Franken results were robust to the choice of its parameters (Figure S1F).
Franken requires minimal computational resources, and runtimes were very fast when evaluated on the two benchmark datasets containing around 200,000 cells (Figure S1E). While FlowSOM was slightly faster than Franken, it performed very poorly in F1 scores compared to both Franken and Phenograph. FlowSOM was run using the default parameters chosen by the authors as optimal (SOM nodes 100). However, as FlowSOM and Franken share the SOM-building step we also tested FlowSOM using the same 400-node SOM grid size and the F1 scores were equivalent while the runtime was increased, no longer making FlowSOM superior in speed, therefore we chose to use the author’s default parameters for benchmarking which were more favorable to the FlowSOM method.
Although the F1 scores from Phenograph were comparable to Franken’s, their scalability varied greatly. After testing Franken on several synthetic datasets of sizes varying from 20 thousand to 40 million, we showed that one could analyze 40 million cells with Franken in the equivalent time taken to analyze 1 million cells using Phenograph (Figure S1G). Franken can also be applied to single-cell RNA sequencing data, therefore we also compared Franken’s scalability with the state-of-the-art method for single-cell RNA sequencing Seurat and could show that Franken was far superior in scalability (Figure S1G). Franken could cluster 40 million cells in the half of the time taken to cluster 3 million cells with Seurat. As Phenograph and Seurat require far larger computational resources they could not be run on the larger datasets beyond 1 and 3 million respectively.
Other computational methods
PhenoGraph runs included in Figure S1 were performed using the MATLAB (R2018b) implementation using the GUI CYT3 as the matlab implementation was the only one which allowed different random seeds to be used in each run. Default parameters were used: k nearest neighbors = 30. PhenoGraph runs in Figure S2 were performed using its implementation in R (Rphenograph) for its ease in including it in scripts instead of manually running the MATLAB GUI. FlowSOM and Seurat runs were performed using their implementation in R and default parameters. All software versions are provided in the key Resources Table.
TMA analysis
Immunohistochemistry applied to TMA was used to validate single-cell mass cytometry data. The open-source software QuPath55 was used to quantify cell types in TMA. CD3+ cells were quantified using an automated detection procedure, and CD15+ cells were manually selected by a pathologist (J.H.R.).
Acknowledgments
The research was supported by a European Research Council PrECISE project from the European Union Horizon 2020 research and innovation program under grant agreement No. 668858. We thank the Wild and Bodenmiller lab members as well as Dr. Andreas Moor for useful discussions and valuable feedback.
Author contributions
L.D.V.R., B.B., and P.W. conceived the study. P.W., B.B., A.J., and L.D.V.R designed the antibody panel and A.J. performed all antibody validation and data acquisition experiments. L.D.V.R. designed and developed Franken and performed data analysis. L.D.V.R and S.C tested algorithm in multiple datasets. T.H., C.P., and C.D.F. provided clinical samples. L.D.V.R. and P.W. developed TMA. P.W., P.B., J.H.R., A.T., and L.D.V.R. performed the TMA IHC image analysis. L.D.V.R., B.B., and P.W. performed biological analysis and interpretation with input from J.H.R and H.W.J.; L.D.V.R, B.B., and P.W. wrote the manuscript with input from F.C., H.W.J, S.C., and C.D.F.
Declaration of interests
P.J.W. has received consulting fees and honoraria (institutional/personal) for lectures by Bayer, Janssen-Cilag, Novartis, Roche, MSD, Astellas Pharma, Bristol-Myers Squibb, Thermo Fisher Scientific, Molecular Health, Sophia Genetics, Qiagen, and Astra Zeneca. L.D.V.R. is an employee at Monte Rosa Therapeutics. All other authors have declared no competing interests.
Published: April 19, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100604.
Contributor Information
Bernd Bodenmiller, Email: bernd.bodenmiller@imls.uzh.ch.
Peter J. Wild, Email: peter.wild@kgu.de.
Supplemental information
Data and code availability
-
•
The single-cell data supporting the findings of this study including raw.fcs files from primary samples and cell lines have been deposited at Mendeley and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
The published bone marrow CyTOF data used in the clustering methods benchmarking (Figure S1) is from Bendall et al.1 and publicly available at http://cytobank.org/nolanlab/reports. Pre-processin is detailed in Weber et al.2 Comparison of clustering methods for high-dimensional single-cell flow and mass cytometry data. Cytometry Part A, 89(12):1084-1096.
-
•
Code for above mentioned data pre-processing pipeline (from Weber et al.) can be found at: https://github.com/lmweber/cytometry-clustering-comparison
-
•
The Franken package and related all code is available at Github and is publicly available as of the date of publication with corresponding Zenodo DOI provided in key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
References
- 1.Bendall S.C., Simonds E.F., Qiu P., Amir el-AD., Krutzik P.O., Finck R., Bruggner R.V., Melamed R., Trejo A., Ornatsky O.I., et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weber L.M., Robinson M.D. Comparison of clustering methods for high-dimensional single-cell flow and mass cytometry data. Cytometry A. 2016;89:1084–1096. doi: 10.1002/cyto.a.23030. [DOI] [PubMed] [Google Scholar]
- 3.Dess R.T., Suresh K., Zelefsky M.J., Freedland S.J., Mahal B.A., Cooperberg M.R., Davis B.J., Horwitz E.M., Terris M.K., Amling C.L., et al. Development and validation of a clinical prognostic stage group system for nonmetastatic prostate cancer using disease-specific mortality results from the international staging collaboration for cancer of the prostate. JAMA Oncol. 2020;6:1912–1920. doi: 10.1001/jamaoncol.2020.4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Penney K.L., Stampfer M.J., Jahn J.L., Sinnott J.A., Flavin R., Rider J.R., Finn S., Giovannucci E., Sesso H.D., Loda M., et al. Gleason grade progression is uncommon. Cancer Res. 2013;73:5163–5168. doi: 10.1158/0008-5472.CAN-13-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo T., Li L., Zhong Q., Rupp N.J., Charmpi K., Wong C.E., Wagner U., Rueschoff J.H., Jochum W., Fankhauser C.D., et al. Multi-region proteome analysis quantifies spatial heterogeneity of prostate tissue biomarkers. Life Sci. Alliance. 2018;1:e201800042. doi: 10.26508/lsa.201800042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bluemn E.G., Coleman I.M., Lucas J.M., Coleman R.T., Hernandez-Lopez S., Tharakan R., Bianchi-Frias D., Dumpit R.F., Kaipainen A., Corella A.N., et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell. 2017;32:474–489.e6. doi: 10.1016/j.ccell.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qin J., Liu X., Laffin B., Chen X., Choy G., Jeter C.R., Calhoun-Davis T., Li H., Palapattu G.S., Pang S., et al. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012;10:556–569. doi: 10.1016/j.stem.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Z.G., Mathew P., Yang J., Starbuck M.W., Zurita A.J., Liu J., Sikes C., Multani A.S., Efstathiou E., Lopez A., et al. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. J. Clin. Invest. 2008;118:2697–2710. doi: 10.1172/JCI33093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohonen T., Somervuo P. How to make large self-organizing maps for nonvectorial data. Neural Netw. 2002;15:945–952. doi: 10.1016/s0893-6080(02)00069-2. [DOI] [PubMed] [Google Scholar]
- 11.Kohonen T. The self-organizing map. Proc. IEEE. 1990;78:1464–1480. [Google Scholar]
- 12.Van Gassen S., Callebaut B., Van Helden M.J., Lambrecht B.N., Demeester P., Dhaene T., Saeys Y. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A. 2015;87:636–645. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 13.Strehl A., Ghosh J. Value-based customer grouping from large retail data-sets. Data Mining Knowledge Discov. Theor. Tools, Technol. Ii. 2000;4057:33–42. [Google Scholar]
- 14.Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir el-AD., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R., et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen S., Zhu G., Yang Y., Wang F., Xiao Y.T., Zhang N., Bian X., Zhu Y., Yu Y., Liu F., et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat. Cell Biol. 2021;23:87–98. doi: 10.1038/s41556-020-00613-6. [DOI] [PubMed] [Google Scholar]
- 16.Jackson H.W., Fischer J.R., Zanotelli V.R.T., Ali H.R., Mechera R., Soysal S.D., Moch H., Muenst S., Varga Z., Weber W.P., Bodenmiller B. The single-cell pathology landscape of breast cancer. Nature. 2020;578:615–620. doi: 10.1038/s41586-019-1876-x. [DOI] [PubMed] [Google Scholar]
- 17.Shimura S., Yang G., Ebara S., Wheeler T.M., Frolov A., Thompson T.C. Reduced infiltration of tumor-associated macrophages in human prostate cancer: association with cancer progression. Cancer Res. 2000;60:5857–5861. [PubMed] [Google Scholar]
- 18.Cao J., Liu J., Xu R., Zhu X., Zhao X., Qian B.Z. Prognostic role of tumour-associated macrophages and macrophage scavenger receptor 1 in prostate cancer: a systematic review and meta-analysis. Oncotarget. 2017;8:83261–83269. doi: 10.18632/oncotarget.18743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagner J., Rapsomaniki M.A., Chevrier S., Anzeneder T., Langwieder C., Dykgers A., Rees M., Ramaswamy A., Muenst S., Soysal S.D., et al. A single-cell Atlas of the tumor and immune ecosystem of human breast cancer. Cell. 2019;177:1330-e18. doi: 10.1016/j.cell.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ricardo S., Vieira A.F., Gerhard R., Leitão D., Pinto R., Cameselle-Teijeiro J.F., Milanezi F., Schmitt F., Paredes J. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011;64:937–946. doi: 10.1136/jcp.2011.090456. [DOI] [PubMed] [Google Scholar]
- 21.Forsyth K.D., Simpson A.C., Levinsky R.J. CD15 antibodies increase neutrophil adhesion to endothelium by an LFA-1-dependent mechanism. Eur. J. Immunol. 1989;19:1331–1334. doi: 10.1002/eji.1830190727. [DOI] [PubMed] [Google Scholar]
- 22.Jassam S.A., Maherally Z., Smith J.R., Ashkan K., Roncaroli F., Fillmore H.L., Pilkington G.J. TNF-α enhancement of CD62E mediates adhesion of non-small cell lung cancer cells to brain endothelium via CD15 in lung-brain metastasis. Neuro Oncol. 2016;18:679–690. doi: 10.1093/neuonc/nov248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kadota A., Masutani M., Takei M., Horie T. Evaluation of expression of CD15 and sCD15 in non-small cell lung cancer. Int. J. Oncol. 1999;15:1081–1089. doi: 10.3892/ijo.15.6.1081. [DOI] [PubMed] [Google Scholar]
- 24.Jang T.J., Park J.B., Lee J.I. The expression of CD10 and CD15 is progressively increased during colorectal cancer development. Korean J. Pathol. 2013;47:340–347. doi: 10.4132/KoreanJPathol.2013.47.4.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brooks S.A., Leathem A.J. Expression of the CD15 antigen (Lewis x) in breast cancer. Histochem. J. 1995;27:689–693. [PubMed] [Google Scholar]
- 26.Read T.A., Fogarty M.P., Markant S.L., McLendon R.E., Wei Z., Ellison D.W., Febbo P.G., Wechsler-Reya R.J. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell. 2009;15:135–147. doi: 10.1016/j.ccr.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Massoner P., Thomm T., Mack B., Untergasser G., Martowicz A., Bobowski K., Klocker H., Gires O., Puhr M. EpCAM is overexpressed in local and metastatic prostate cancer, suppressed by chemotherapy and modulated by MET-associated miRNA-200c/205. Br. J. Cancer. 2014;111:955–964. doi: 10.1038/bjc.2014.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sottoriva A., Kang H., Ma Z., Graham T.A., Salomon M.P., Zhao J., et al. A Big Bang model of human colorectal tumor growth. Nat Genet. 2015;47:209–216. doi: 10.1038/ng.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Went P.T., Lugli A., Meier S., Bundi M., Mirlacher M., Sauter G., Dirnhofer S. Frequent EpCam protein expression in human carcinomas. Hum. Pathol. 2004;35:122–128. doi: 10.1016/j.humpath.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 30.Xu J., Kalos M., Stolk J.A., Zasloff E.J., Zhang X., Houghton R.L., Filho A.M., Nolasco M., Badaró R., Reed S.G. Identification and characterization of prostein, a novel prostate-specific protein. Cancer Res. 2001;61:1563–1568. [PubMed] [Google Scholar]
- 31.Wang W., Epstein J.I. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am. J. Surg. Pathol. 2008;32:65–71. doi: 10.1097/PAS.0b013e318058a96b. [DOI] [PubMed] [Google Scholar]
- 32.Xu L.L., Srikantan V., Sesterhenn I.A., Augustus M., Dean R., Moul J.W., Carter K.C., Srivastava S. Expression profile of an androgen regulated prostate specific homeobox gene NKX3.1 in primary prostate cancer. J. Urol. 2000;163:972–979. [PubMed] [Google Scholar]
- 33.Bubendorf L., Kononen J., Koivisto P., Schraml P., Moch H., Gasser T.C., Willi N., Mihatsch M.J., Sauter G., Kallioniemi O.P. Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res. 1999;59:803–806. [PubMed] [Google Scholar]
- 34.Koivisto P., Kononen J., Palmberg C., Tammela T., Hyytinen E., Isola J., Trapman J., Cleutjens K., Noordzij A., Visakorpi T., Kallioniemi O.P. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57:314–319. [PubMed] [Google Scholar]
- 35.Watson P.A., Arora V.K., Sawyers C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer. 2015;15:701–711. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fankhauser C.D., Penney K.L., Gonzalez-Feliciano A.G., Clarke N.W., Hermanns T., Stopsack K.H., Fiorentino M., Loda M., Mahal B., Gerke T.A., et al. Inferior cancer survival for men with localized high-grade prostate cancer but low prostate-specific antigen. Eur. Urol. 2020;78:637–639. doi: 10.1016/j.eururo.2020.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mills C.D., Lenz L.L., Harris R.A. A breakthrough: macrophage-directed cancer immunotherapy. Cancer Res. 2016;76:513–516. doi: 10.1158/0008-5472.CAN-15-1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Restifo N.P., Dudley M.E., Rosenberg S.A. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat. Rev. Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mariathasan S., Turley S.J., Nickles D., Castiglioni A., Yuen K., Wang Y., Kadel E.E., Koeppen H., Astarita J.L., Cubas R., et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Franklin R.A., Liao W., Sarkar A., Kim M.V., Bivona M.R., Liu K., Pamer E.G., Li M.O. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921–925. doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campbell M.J., Tonlaar N.Y., Garwood E.R., Huo D., Moore D.H., Khramtsov A.I., Au A., Baehner F., Chen Y., Malaka D.O., et al. Proliferating macrophages associated with high grade, hormone receptor negative breast cancer and poor clinical outcome. Breast Cancer Res. Treat. 2011;128:703–711. doi: 10.1007/s10549-010-1154-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nonomura N., Takayama H., Nakayama M., Nakai Y., Kawashima A., Mukai M., Nagahara A., Aozasa K., Tsujimura A. Infiltration of tumour-associated macrophages in prostate biopsy specimens is predictive of disease progression after hormonal therapy for prostate cancer. BJU Int. 2011;107:1918–1922. doi: 10.1111/j.1464-410X.2010.09804.x. [DOI] [PubMed] [Google Scholar]
- 43.Umbehr M., Kessler T.M., Sulser T., Kristiansen G., Probst N., Steurer J., Bachmann L.M. ProCOC: the prostate cancer outcomes cohort study. BMC Urol. 2008;8:9. doi: 10.1186/1471-2490-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wettstein M.S., Saba K., Umbehr M.H., Murtola T.J., Fankhauser C.D., Adank J.P., Hofmann M., Sulser T., Hermanns T., Moch H., et al. Prognostic role of preoperative serum lipid levels in patients undergoing radical prostatectomy for clinically localized prostate cancer. Prostate. 2017;77:549–556. doi: 10.1002/pros.23296. [DOI] [PubMed] [Google Scholar]
- 45.Epstein J.I., Egevad L., Amin M.B., Delahunt B., Srigley J.R., Humphrey P.A. The 2014 international society of urological pathology (ISUP) consensus conference on gleason grading of prostatic carcinoma: definition of grading patterns and proposal for a new grading system. Am. J. Surg. Pathol. 2016;40:244–252. doi: 10.1097/PAS.0000000000000530. [DOI] [PubMed] [Google Scholar]
- 46.Mortezavi A., Hermanns T., Seifert H.H., Baumgartner M.K., Provenzano M., Sulser T., Burger M., Montani M., Ikenberg K., Hofstädter F., et al. KPNA2 expression is an independent adverse predictor of biochemical recurrence after radical prostatectomy. Clin. Cancer Res. 2011;17:1111–1121. doi: 10.1158/1078-0432.CCR-10-0081. [DOI] [PubMed] [Google Scholar]
- 47.Zunder E.R., Lujan E., Goltsev Y., Wernig M., Nolan G.P. A continuous molecular roadmap to iPSC reprogramming through progression analysis of single-cell mass cytometry. Cell Stem Cell. 2015;16:323–337. doi: 10.1016/j.stem.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Behbehani G.K., Thom C., Zunder E.R., Finck R., Gaudilliere B., Fragiadakis G.K., Fantl W.J., Nolan G.P. Transient partial permeabilization with saponin enables cellular barcoding prior to surface marker staining. Cytometry A. 2014;85:1011–1019. doi: 10.1002/cyto.a.22573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Catena R., Özcan A., Jacobs A., Chevrier S., Bodenmiller B. AirLab: a cloud-based platform to manage and share antibody-based single-cell research. Genome Biol. 2016;17:142. doi: 10.1186/s13059-016-1006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finck R., Simonds E.F., Jager A., Krishnaswamy S., Sachs K., Fantl W., Pe'er D., Nolan G.P., Bendall S.C. Normalization of mass cytometry data with bead standards. Cytometry A. 2013;83:483–494. doi: 10.1002/cyto.a.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chevrier S., Crowell H.L., Zanotelli V.R.T., Engler S., Robinson M.D., Bodenmiller B. Compensation of signal spillover in suspension and imaging mass cytometry. Cell Syst. 2018;6:612–e5. doi: 10.1016/j.cels.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maaten L.J.P.V.D., Hinton G.E. Visualizing high-dimensional data using t-SNE. J. Machine Learn. Res. 2008;9:2579–2605. [Google Scholar]
- 53.Csardi Gabor, Nepusz Tamas. The igraph software package for complex network research. InterJournal Complex Syst. 2006:1695. [Google Scholar]
- 54.Yang Z., Algesheimer R., Tessone C.J. A comparative analysis of community detection algorithms on artificial networks. Sci. Rep. 2016;6:30750. doi: 10.1038/srep30750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bankhead P., Loughrey M.B., Fernández J.A., Dombrowski Y., McArt D.G., Dunne P.D., McQuaid S., Gray R.T., Murray L.J., Coleman H.G., et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 2017;7:16878. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The single-cell data supporting the findings of this study including raw.fcs files from primary samples and cell lines have been deposited at Mendeley and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
The published bone marrow CyTOF data used in the clustering methods benchmarking (Figure S1) is from Bendall et al.1 and publicly available at http://cytobank.org/nolanlab/reports. Pre-processin is detailed in Weber et al.2 Comparison of clustering methods for high-dimensional single-cell flow and mass cytometry data. Cytometry Part A, 89(12):1084-1096.
-
•
Code for above mentioned data pre-processing pipeline (from Weber et al.) can be found at: https://github.com/lmweber/cytometry-clustering-comparison
-
•
The Franken package and related all code is available at Github and is publicly available as of the date of publication with corresponding Zenodo DOI provided in key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.