

Abstract
Epigenetic regulation is an important factor in glucose metabolism, but underlying mechanisms remain largely unknown. Here we investigated epigenetic control of systemic metabolism by bromodomain-containing proteins (Brds), which are transcriptional regulators binding to acetylated histone, in both intestinal cells and mice treated with the bromodomain inhibitor JQ-1. In vivo treatment with JQ-1 resulted in hyperglycemia and severe glucose intolerance. Whole-body or tissue-specific insulin sensitivity was not altered by JQ-1; however, JQ-1 treatment reduced insulin secretion during both in vivo glucose tolerance testing and ex vivo incubation of isolated islets. JQ-1 also inhibited expression of fibroblast growth factor (FGF) 15 in the ileum and decreased FGF receptor 4–related signaling in the liver. These adverse metabolic effects of Brd4 inhibition were fully reversed by in vivo overexpression of FGF19, with normalization of hyperglycemia. At a cellular level, we demonstrate Brd4 binds to the promoter region of FGF19 in human intestinal cells; Brd inhibition by JQ-1 reduces FGF19 promoter binding and downregulates FGF19 expression. Thus, we identify Brd4 as a novel transcriptional regulator of intestinal FGF15/19 in ileum and FGF signaling in the liver and a contributor to the gut-liver axis and systemic glucose metabolism.
Introduction
Epigenetic regulation of chromatin structure and transcriptional control is an important determinant of glucose metabolism and diabetes risk (1). One mediator of epigenetic control is histone acetylation, which mediates chromatin accessibility to transcriptional factors and coactivators (2). Histone acetyltransferases (HAT) and histone deacetylases (HDAC) are responsible for adding and removing the acetyl groups to histones, respectively. Moreover, modulation of histone acetylation alters systemic metabolism. For example, mice with heterozygous deficiency of the HAT CREBBP remain insulin sensitive, despite lipodystrophy (3). Conversely, HDAC deletion in muscle increases lipid oxidation, energy expenditure, and insulin resistance (4,5). Thus, histone acetylation plays an important role in energy expenditure and glucose metabolism.
One target for experimental and therapeutic manipulation of histone acetylation is the bromodomain and extra-terminal domain (BET) family of proteins, including bromodomain-containing proteins (Brd) 2, Brd3, Brd4, and BrdT in mammals (6). These “readers” have two tandem bromodomains that recognize and bind acetylated lysine of histones or nonhistone targets. While Brds are recognized for their potential in cancer therapeutics, Brds also influence metabolism. For example, mice with genetic disruption of Brd2 have severe obesity but normal glucose metabolism, potentially via increased peroxisome-proliferator-activated receptor-γ (PPAR-γ) activity (7). Brd4 binds to enhancers regulating adipogenesis and myogenesis (8), coactivates nuclear factor-κB (9,10), and modulates oxidative phosphorylation (11). In adult mice, ablation of Brd4 reduces cellular diversity in the small intestine (12), suggesting Brd4 may influence metabolism via the intestinal endocrine system.
The intestine is increasingly recognized as an important regulator of systemic glucose and lipid metabolism in humans, as demonstrated by potent effects of bariatric/metabolic surgery to improve glucose metabolism and even reverse type 2 diabetes (T2D) (13–15). While increased secretion of incretin hormones and favorable changes in the microbiome and luminal bile acid species are likely contributors, the fibroblast growth factor (FGF) 15/19 pathway has also emerged as an important signaling mediator linking bile acid metabolism and the intestine to control of systemic and hepatic metabolism (13,16,17).
Mouse FGF15 and its human ortholog FGF19 share ∼50% amino acid identity and have similar physiological functions to regulate intestine-to-liver cross talk (18). In the liver, FGF15/19 binds to and activates the FGF receptor 4 (FGFR4)/β-klotho complex and downstream signaling, leading to repression of bile acid synthesis (19). FGF15/19 also exerts potent effects on systemic glucose metabolism, reducing blood glucose via inhibition of hepatic gluconeogenesis, increased glycogen synthesis, and increased insulin-independent glucose disposal (20). Additionally, FGF15/19 reduces hepatic lipogenesis (21) and decreases Agouti-related peptide/neuropeptide Y neuronal activity in the hypothalamus (20,22). FGF19 levels are reduced in humans with T2D (23), markedly increased after bariatric surgery (13), and further increased in individuals who develop postbariatric hypoglycemia (24). Thus, the FGF15/19 axis is responsive to intestinal tract manipulation during adult life. We previously demonstrated that the bile acid-Fgf15 axis is disrupted in mice with programmed risk for T2D due to undernutrition (UN) exposure during prenatal life (25). Together these data suggest that the FGF15/19 axis may be an effector system that integrates environment signals to regulate systemic and hepatic metabolism, potentially via epigenetic regulation in the intestine. We therefore hypothesized that bromodomain-dependent transcription could modulate the gut-liver axis via the Fgf15/19 axis, altering both hepatic and systemic metabolism.
Research Design and Methods
Animal Care and Studies
Male CD-1 mice obtained from Envigo (South Easton, MA) were housed (three to five per cage) at 24°C under a 12-h light/dark cycle, with free access to food and water. Intestine-specific Fgf15-knockout (KO) mice were generated on the C57BL/6J background by breeding Fgf15flox/flox to VilCreERT2 mice and treating with tamoxifen as described (26). CAGrtTA3/+; TtG-shRen and CAGrtTA3/+; TtG-shBRD4 mice were fed with 625 mg/kg doxycycline-containing food pellets (Harlan Teklad) for 2 weeks (12). All animal experiments were approved by the local institutional animal care committees and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Metabolic Parameters
Glucose tolerance was assessed after an intraperitoneal injection of glucose (1 or 2 g/kg body wt) after a 16-h fast. Blood glucose was measured via tail vein sampling at the indicated time points. Insulin tolerance was assessed after an intraperitoneal injection of insulin (0.5 units/kg body wt; Humulin-R, Lilly) after a 4-h fast. Plasma insulin, glucagon, and Fgf15 were measured using an ELISA kit (Crystal Chem, R&D, and Cloud-Clone Corp., respectively). Plasma total cholesterol was measured using a colorimetric assay (Cell Biolabs).
In Vivo Treatment With the Brd4 Inhibitor JQ-1
The Brd4 inhibitor JQ-1 was synthesized and purified in the laboratory of Dr. Jun Qi (Dana-Farber Cancer Institute). For in vivo experiments, a stock solution (50 mg/mL in DMSO) was diluted to a working concentration of 5 mg/mL in 10% hydroxypropyl β-cyclodextrin (Sigma-Aldrich). Mice were injected at a dose of 25 or 50 mg/kg given intraperitoneally. Vehicle controls were given an equal amount of DMSO in 10% hydroxypropyl β-cyclodextrin. For in vitro experiments, JQ-1 was dissolved in DMSO and added to cells at indicated concentrations, with an equal volume of DMSO as control.
Adenoviral Overexpression of FGF19
Mice received a single tail vein injection of 3 × 1011 vector genome adeno-associated virus (AAV)-FGF19 or a control virus encoding green fluorescent protein (GFP; AAV-GFP). FGF19 cDNA was subcloned into a pAAV-EF1α vector as previously shown (27). After 16 days of treatment, tissues were collected.
Quantitative Real-Time PCR
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA), and cDNA was synthesized using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. Quantitative real-time PCR was performed using SYBR Green (Bio-Rad, Hercules, CA). Expression was normalized by Rn18s (18S rRNA), Rpl13a, or 36B4. Primer sequences are provided in Supplementary Table 2.
Microarray Analysis
Total RNA was isolated from liver tissue from three representative mice per group. RNA quality was assessed using Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA), and samples were processed for microarray analysis (Affymetrix Mouse Gene 2.0 ST, Molecular Phenotyping Core, Joslin Diabetes Center).
Bioinformatics Analysis
For liver transcriptomics and metabolomics data sets, principal component analysis (PCA) revealed that the first principal component was an extraneous source of variation, so it was accounted for as a covariate in linear modeling (28). Metabolomics/lipidomics sample weights were unbiasedly estimated (29) and used in linear modeling. Linear modeling differential analysis was done with the R package limma (30). Nominal P values were corrected for multiple testing using the false discovery rate (FDR). Transcriptomic pathway and transcription factor prediction analysis was done using the ROAST (rotation gene set testing) method (31), with pathways defined by Reactome (32).
Immunohistochemistry
Pancreata were dissected and fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned. Sections were stained using hematoxylin and eosin or immunostained for lysozyme (1:1000; Abcam, ab108508) and insulin (1:100; Cell Signaling Technology, no. 4590). Islet area was calculated from analysis of >100 islets per mouse (Adobe Photoshop).
Cell Culture
The human intestinal cell line HT-29 (ATCC, Manassas, VA) was maintained in McCoy’s 5A medium (Thermo Fisher Scientific) supplemented with 10% FBS, 1% PenStrep (Thermo Fisher Scientific) in a humidified atmosphere with 5% CO2 at 37°C. For knockdown of BRD4, HT-29 cells were transfected with 20 nmol/L siRNA SMARTpools targeting BRD4 or scrambled control using DharmaFECT transfection reagent (Dharmacon) diluted in OptiMEM (15 μL DharmaFECT/1 mL OptiMEM).
Chromatin Immunoprecipitation-PCR
Chromatin immunoprecipitation (ChIP) was performed on HT-29 cells cultured in the presence or absence of JQ-1 (250 nmol/L) for 24 h. Chromatin pooled from ∼1 × 106 HT-29 cells was used for each immunoprecipitation. HT-29 cells were fixed directly on the dish with 1% formaldehyde for 10 min, followed by quenching with 0.125 mol/L glycine for 5 min. Chromatin was extracted, followed by shearing on a Tekmar Sonic Disruptor (Cincinnati, OH) (three cycles, 80% Amp and 6-s pulse, 5 min on/off). The sonicated chromatin was immunoprecipitated with 5 μg of antibody (anti-BRD4; Bethyl, no. A301-985A) bound to Dynabeads (Invitrogen), as previously described (33). Cross-linking was reversed in immunoprecipitates and input chromatin samples prior to purification of genomic DNA. Target and nontarget regions of genomic DNA were amplified by PCR or quantitative real-time RT-PCR using SYBR Green chemistry. Enrichment was calculated as a percentage of input DNA for each sample. ChIP-PCR primer sequences are shown in Supplementary Table 2. ChIP-PCR primers for MYC were previously described (34).
Statistical Analysis
Data are expressed as mean ± SEM. One-way ANOVA and repeated-measures ANOVA, followed by multiple comparison tests (Bonferroni/Dunn method), were used where applicable. The Student t test was used to analyze the differences between two groups. Differences were considered significant at P < 0.05.
Data and Resource Availability
Data are available upon reasonable request from the authors.
Results
Brd4 Inhibition by JQ-1 Decreases Body Weight and Induces Hyperglycemia
To determine the effect of Brd4 inhibition on body weight and glucose metabolism, we treated CD-1 mice with JQ-1, a Brd inhibitor with high selectivity for Brd4 (25 mg/kg intraperitoneally for 11 days) (Fig. 1A). This dose was previously shown to be without toxicity during long-term treatment (35). Weight was modestly reduced (Fig. 1B), with modest effects on food intake in one cohort (vehicle: 5.4 ± 0.2 g/day; JQ-1: 3.7 ± 1.1 g/day), but not in subsequent cohorts treated with the same dose of JQ-1. Blood glucose was significantly increased in JQ-1–treated mice versus vehicle after a 4-h fast (Fig. 1C). Likewise, JQ-1–treated mice were severely glucose intolerant during intraperitoneal glucose tolerance testing (Fig. 1D and E), with nearly twofold increase in glucose area under the curve (AUC) (P < 0.01).
Figure 1.

JQ-1 induces hyperglycemia in vivo. A: Experimental protocol. GTT, glucose tolerance test; ITT, insulin tolerance test; ip, intraperitoneal. B: Body weight changes during JQ-1 treatment (n = 5–6). C: Fasting blood glucose levels (4-h fasting, day 10; n = 5–6). Veh, vehicle. Blood glucose levels (D) and AUC (E) during ipGTT (day 6). Plasma insulin (F) and glucagon (G) levels during ipGTT. H: Hematoxylin and eosin (H&E) and insulin staining of pancreatic sections. Scale bar, 200 and 100 μm. I: Islet size (n = 5–6; >100 islets/mouse). ipITT (day10) (J) and glycerol tolerance test (day 4) (K). Data are expressed as means ± SEM. *P < 0.05, **P < 0.01 vs. vehicle-treated mice (Veh).
We next analyzed potential contributors to hyperglycemia in JQ-1–treated mice. While fasting insulin did not differ, insulin levels were decreased in JQ-1–treated mice after glucose injection (50% reduction at 15 min, P < 0.05) (Fig. 1F). Glucagon levels were similarly reduced by glucose in both vehicle- and JQ-1–treated mice (Fig. 1G). JQ-1 had no effect on pancreatic islet size (Fig. 1H, quantified in Fig. 1I), but insulin content was decreased in JQ-1–treated mice (Supplementary Fig. 1A), potentially due to the sustained in vivo hyperglycemia. To determine the direct effect of JQ-1 on pancreatic islets, we measured glucose-stimulated insulin secretion (GSIS) in isolated islets treated ex vivo with JQ-1. Insulin release was significantly decreased under both low (2.9 mmol/L) and high (20.2 mmol/L) glucose conditions, but insulin content was unchanged (Supplementary Fig. 1B and C).
There was no difference in systemic insulin sensitivity, assessed by both insulin tolerance test (Fig. 1J) and whole-body Rd during hyperinsulinemic-euglycemic clamp testing (Supplementary Fig. 2A–J). In addition, glucose uptake into skeletal muscle, white adipose tissue, and brown adipose tissue, measured at the end of the clamp, did not differ between control and JQ-1–treated mice (Supplementary Fig. 2O–Q). Consistent with these results, expression of key metabolic genes in skeletal muscle and adipose tissue were unchanged in JQ-1–treated mice (Supplementary Fig. 3). Likewise, there was no change in plasma adiponectin, a Brd4 target in 3T3-L1 adipocytes (36), suggesting that adipose tissue is not a major target of JQ-1 in vivo (Supplementary Fig. 3L).
We next examined effects of JQ-1 on hepatic glucose metabolism. There was a 1.6-fold greater increase in peak glucose in JQ-1–treated mice after injection of the gluconeogenic substrate glycerol (1.4-fold increase in AUC, P < 0.05) (Fig. 1K and L). Fasting hyperglycemia without change in fasting insulin, unchanged systemic insulin sensitivity, and increased glycemic response to glycerol suggested increased gluconeogenesis in vivo in JQ-1–treated mice. This was not related to impaired hepatic insulin action in vivo, as hepatic insulin action measured during the clamp did not differ (Supplementary Fig. 2K–N), and liver Akt phosphorylation did not differ in JQ-1–treated mice (Supplementary Fig. 4A and B). Similarly, basal and insulin-stimulated Akt phosphorylation were unaffected by incubation of primary hepatocytes with JQ-1 (Supplementary Fig. 4C and D); basal and insulin-stimulated insulin receptor, Akt, Foxo1, and S6 phosphorylation were also unchanged in AML12-cultured hepatocytes treated with JQ-1 for 16 h (Supplementary Fig. 4E–H). Together, these data suggested that mechanisms independent of insulin signaling mediated the in vivo hyperglycemia induced by JQ-1.
JQ-1 Alters Hepatic Gene Expression and Sterol Metabolism
To identify potential molecular mediators of fasting hyperglycemia in vivo, we analyzed liver gene expression and found 1,471 genes were upregulated and 1,036 genes were downregulated by JQ-1 (FDR < 0.25). Of these, 45% were previously shown to be loci occupied by Brd4 in ChIP-seq in HepG2 hepatoma cells, suggesting they were direct targets of Brd4 inhibition. Pathway analysis using gene set enrichment analysis (GSEA) and Reactome pathways (Supplementary Table 1) revealed upregulation of multiple genes related to glucose production (Supplementary Fig. 5A), including Pepck (Supplementary Fig. 5C and D). Effects on lipid metabolism in JQ-1–treated mice were striking (Supplementary Fig. 5B), with upregulation of genes linked to triglyceride biosynthesis, lipogenesis/fatty acid synthesis (e.g., Fasn), β-oxidation (e.g., Ppara), and cholesterol biosynthesis (e.g., Srebp2, Hmgcr) confirmed by PCR (Supplementary Fig. 5E–K).
Given these prominent patterns of lipid-related gene expression in JQ-1–treated mice, we analyzed hepatic and plasma lipids by mass spectrometry (37). Of 54 triglyceride species measured in the liver, 42 were increased in JQ-1–treated mice (13 with P < 0.05, FDR < 0.25), while 12 were decreased (5 with P < 0.05, FDR < 0.25) (Supplementary Fig. 6A). Similar patterns across the spectrum of triglyceride chain length were observed in plasma (Supplementary Fig. 6B). By contrast, cholesterol levels were reduced by 28% in the liver and 52% in plasma (P < 0.05) in JQ-1–treated mice (Supplementary Fig. 6C and D).
Effects on bile acid pathway regulatory and synthetic genes were striking, including a twofold increase in Shp and 1.8-fold increase in Cyp7a1, the rate-limiting enzyme in classical bile acid synthesis, in JQ-1–treated liver (Supplementary Fig. 5L–P). Consistent with these patterns, there was a significant increase in plasma downstream primary bile acids, with a fivefold increase in glycochenodeoxycholic acid (P < 0.05, FDR = 0.02) and a threefold increase in glycocholic acid (P < 0.05, FDR = 0.14); cholate and deoxycholate were unchanged.
JQ-1 Alters Hepatic FGF Receptor Signaling
The complex pattern of glucose, lipid, sterol, and bile acid gene expression and metabolism in JQ-1–treated mice raised the possibility that FGF receptor-dependent signaling might be reduced. Consistent with this hypothesis, 10 of 14 top-ranking pathways downregulated in the liver were related to FGFR signaling (FDR < 0.25) (Fig. 2A and B), with reduced expression of both Fgfr4 and 1 (Fig. 2C). Indeed, expression of Fgfr4 as determined by PCR was decreased by 27% (P < 0.01), with a similar trend for its coreceptor β-klotho (17% lower, P = 0.07). In addition, FGFR signaling was significantly reduced, as demonstrated by a 46% reduction in phosphorylation of FGF receptor substrate 2 (Frs2) in liver protein extracts of JQ-1–treated mice (Fig. 2D and E). In primary hepatocytes, treatment with FGF19 reduced glucose production, as predicted, but acute incubation with JQ-1 did not substantially change this response (Supplementary Fig. 4I), suggesting that reduced in vivo FGFR signaling was primarily mediated by indirect systemic effects, potentially hormonal in origin.
Figure 2.

JQ-1 reduces hepatic Fgfr signaling. A–C: Microarray analysis in the liver was performed in JQ-1 vs. mice vehicle (Veh)-treated mice (n = 3 per group). Minus log10 (P value) (A) and upregulated or downregulated gene numbers (B) in pathways related to FGFR signaling. C: Heat map representing log2 fold change (JQ-1 vs. vehicle) in gene expression in pathways related to FGFR ligand binding and activation. D and E: Western blot analysis of total and phosphorylated (p) FRS2 in liver lysates. β-actin is shown as a loading control. **P < 0.01 vs. vehicle (Veh).
Brd4 Inhibition by JQ-1 Reduces Ileal Expression of Fgf15
Downregulation of FGFR-mediated signaling in vivo suggests that JQ-1 modulates the FGF15/19 regulatory axis. Fgf15/19 is secreted into the bloodstream by intestinal enterocytes in response to bile acid-mediated activation of FXR transcriptional activity. Systemic effects of Fgf15 include inhibition of bile acid synthesis, cholesterol metabolism, gluconeogenesis, and lipogenesis in the liver (38,39), and additional improvements in glucose metabolism mediated via the hypothalamus (40). We thus hypothesized that JQ-1 altered systemic metabolism via reduced intestinal secretion of Fgf15. To test this possibility, we first confirmed robust expression of Brd4 in the ileum (Fig. 3A). While there was no change in expression of the bile acid transporter Asbt (Slc10a2), the bile acid receptor Tgr5, or Fxr in the ileum of JQ-1–treated mice (not shown), we observed profound reductions in expression of multiple FXR target genes. Expression of Fgf15 and Shp was reduced by 90% and 95% (P < 0.01 for all), with a similar trend for Slc51a and Slc51b (Fig. 3C–F). In agreement, plasma Fgf15 was decreased by 29% in JQ-1–treated mice (Fig. 3G).
Figure 3.

JQ-1 decreases ileal Fgf15 expression without the change in overall villus structure. A: Western blot analysis of Brd4 in ileum and liver lysates. B: Schematic of bile acid signaling in ileal enterocytes. Expression levels of genes related to bile acid signaling in the ileum (Fgf15) (C), Nr0b2 (SHP) (D), Slc51a (OSTα) (E), and Slc51b (OSTβ) (F) (n = 5–6). Expression levels were normalized by Rpl13 expression. G: Plasma Fgf15 level in JQ-1 vs. vehicle (Veh)-treated mice (n = 5–6). *P < 0.05, **P < 0.01 vs. vehicle (Veh)-treated mice. Data are expressed as means ± SEM. H: Hematoxylin and eosin staining of intestinal sections. Scale bar, 100 μm.
Previous studies have shown that RNA interference–mediated silencing of Brd4 reduces intestinal cellular diversity (12), potentially via reduced stem cell differentiation (41). To test whether alterations in specific intestinal cell subtypes could potentially contribute to JQ-1–mediated reductions in Fgf15, we analyzed intestinal histology and cell type markers. While there was no change in overall villus structure in JQ-1–treated mice (Fig. 3H), there were reduced numbers of Paneth cells and reduced expression of the Paneth cell marker Reg4, as previously reported (12) (Supplementary Fig. 7A). However, expression of the microbicidal functional marker α-defensin cryptdin-4 (Crp4) did not differ (Supplementary Fig. 7G and K) (42). Moreover, there was no change in expression of markers for enterocytes (keratin 20 [Krt20]), enteroendocrine cells (hepatocyte nuclear factor 1α [Hnf1a]), neuroendocrine cells (chromogranin A [ChgA]), goblet cells (mucin 2 [Muc2]), stem cells (leucine-rich orphan G-protein-coupled receptor 5 [Lgr5]), or intestinal neuroendocrine peptides (Gcg or Pyy). Thus, JQ-1–mediated reduction in Fgf15 expression was not related to alterations in cell type distribution (Supplementary Fig. 7B–K).
JQ-1 Inhibits Brd4 Binding to the Fgf19 Promoter in Human Intestinal Cells
Marked downregulation of ileal expression of Fgf15 in JQ-1–treated mice suggested that JQ-1 might directly contribute to reduced Fgf15 signaling and systemic metabolic effects. To test this hypothesis at a cellular level, we used the human intestinal cell line HT-29, which constitutively expresses FGF19 at a high level (43); cells were treated with 500 nmol/L JQ-1 for 24 h. While there was no change in BRD4 expression (Fig. 4A), expression of the BRD4 target MYC was reduced by 83% with JQ-1 (P < 0.01), as predicted (Fig. 4B). Expression of FGF19 was reduced by 96% (P < 0.01), with similar dramatic downregulation of SHP (89% reduction, P < 0.01) (Fig. 4C and D). There was no change in FXR or SLC51B expression (not shown), consistent with prior evidence indicating BRD4 does not directly bind FXR or SLC51B (GSE73319 [44] and ENCSR514EOE [45]). Moreover, secretion of FGF19 into culture medium was completely abolished by JQ-1, with >98% decrease in conditioned medium (P < 0.01) (Fig. 4E).
Figure 4.

BRD4 binds promoter regions of FGF19 and SHP to modulate expression in human intestinal cells. A–E: HT-29 cells were treated with 250 or 500 nmol/L JQ-1 or DMSO (Veh) for 12–24 h. Gene expression levels of BRD4 (A), MYC (B), FGF19 (C), and NR0B2 (SHP) (D) (n = 4). The levels were normalized by those of RN18S. E: FGF19 level in conditioned media (n = 4). F–H: HT-29 cells were treated with siRNA for BRD4 and JQ-1. Gene expression levels of BRD4 (F), and FGF19 (G) (n = 3). The levels were normalized by those of 36B4. H: Primer design for ChIP assays for FGF19 and NR0B2 (SHP) promoter regions. I and J: Cells were incubated with JQ-1 or DMSO (Veh) for 24 h and then harvested for the ChIP assay. The precipitated DNA was analyzed by PCR (I) and real-time PCR (J) (n = 4). Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01 vs. vehicle (Veh)-treated mice. N.D., not detected.
To confirm the contribution of BRD4 to the regulation of FGF19 expression, HT-29 cells were transfected with BRD4 siRNA. In BRD4 siRNA–treated cells, expression of FGF19 was reduced by 40–46% (P < 0.01) (Fig. 4F). As expected, JQ-1 had no effect on BRD4 mRNA level in control or siRNA-treated cells (Fig. 4F). FGF19 mRNA levels were significantly decreased by BRD4 siRNA treatment (Fig. 4G). However, the impact of JQ-1 to decrease FGF19 expression was greater, and this effect did not differ between control and siRNA-treated cells (Fig. 4G).
To determine whether JQ-1 mediated reduction in expression of FGF19 and SHP was mediated by modulation of BRD4 binding to promoter regions of these genes, we performed ChIP-PCR analysis using an anti-BRD4 antibody (Fig. 4H–J) (34). As expected, BRD4 bound to its target MYC (control); BRD4 also bound robustly to promoter sequences of both FGF19 and SHP (Fig. 4I and J). This was markedly inhibited by JQ-1, with a >90% reduction in binding (Fig. 4I and J). Thus, JQ-1 inhibits BRD4 binding to the FGF19 promoter in HT-29 cells.
Overexpression of FGF19 Reverses Glucose Intolerance Induced by JQ-1
To determine the requirement for systemic FGF15/19 signaling in mediating the metabolic effects of JQ-1, we took two distinct approaches, asking whether Fgf19 would reverse the impact of JQ-1 on glucose metabolism and whether JQ-1 systemic effects had additional Fgf15-independent mechanisms.
First, we treated mice with AAV-FGF19 or GFP control vectors, and then treated mice with JQ-1 (25 mg/kg, intraperitoneally) for 10 days (Fig. 5A). Plasma FGF19 was not detectable in AAV-GFP controls, but was readily detected in plasma of AAV-FGF19 mice at 3 weeks after AAV injection (Fig. 5B). Consistent with prior studies (20,46), AAV-FGF19–treated mice had lower body weight and blood glucose at baseline (Fig. 5C), but weight did not change during JQ-1 treatment in either the GFP or FGF19 groups (Fig. 5C). As with prior cohorts, JQ-1 increased blood glucose in AAV-GFP control mice as early as 4 days after the onset of treatment; by contrast, no change in glucose was observed in AAV-FGF19 mice. Glucose tolerance testing at day 10 confirmed the significant impairment in JQ-1 treated mice (35% increase in glucose AUC, P < 0.01) (Fig. 5D). Strikingly, JQ-1–mediated glucose intolerance was fully reversed in mice overexpressing FGF19 (Fig. 5D and E). The FGF19-mediated reversal of glucose intolerance was not associated with alterations in expression of the gluconeogenic genes Pck (Pepck), G6pc, or Nr0b2 (Shp) (Supplementary Fig. 8) and was independent of insulin, as both basal and glucose-stimulated insulin levels were reduced in FGF19-overexpressing mice, in accord with the lower plasma glucose levels (Fig. 5F).
Figure 5.

Overexpression of FGF19 reverses hyperglycemia induced by JQ-1. A: Experimental protocol. GTT, glucose tolerance test; ip, intraperitoneal. B: Plasma FGF19 levels in AAV-GFP– and AAV-FGF19–treated mice. C: Body weight in AAV-GFP– and AAV-FGF19–treated mice during JQ-1 treatment. Blood glucose levels (D) and AUC (E) during ipGTT (day10). F: Plasma insulin levels during ipGTT. Data are expressed as means ± SEM. *P < 0.05, **P < 0.01 vs. AAV-GFP–treated mice (n = 9–10). N.D., not detected.
Second, we treated mice with intestine (INT)-specific ablation of Fgf15 (Fgf15INT-KO) with JQ-1. Consistent with prior cohorts (Supplementary Fig. 9), intestinal Fgf15 expression was reduced in wild-type (WT) mice, with no further reduction in Fgf15INT-KO mice. Surprisingly, JQ-1 effects to induce glucose intolerance were similar in WT and Fgf15INT-KO mice (Supplementary Fig. 9A and B). Likewise, intestinal expression of Shp was equally inhibited by JQ-1 in both WT and Fgf15INT-KO mice (Supplementary Fig. 9D), suggesting additional Fgf15-independent effects of bromodomain inhibition on intestinal gene expression and systemic glucose metabolism, potentially involving regulation of Shp or other Fxr targets.
Ileal Expression of Fgf15 Is Decreased in Mice Exposed to Intrauterine UN
Exposures experienced during key developmental periods can increase risk for chronic disease such as T2D, potentially via nutrient-sensitive epigenetic mechanisms. We have previously demonstrated that exposure to maternal UN during late gestation results in glucose intolerance and T2D with aging (47). UN-exposed offspring have dysregulation of bile acid metabolism during early life and are resistant to the weight-lowering and insulin-sensitizing effects of bile acid supplementation during adult life (25,48). We thus asked whether UN-exposed offspring mice had altered expression of the bile acid signaling and Brd4 targets Fgf15 and Shp in the ileum. As previously shown (47), offspring of dams subjected to food restriction from embryonic day 12.5 to birth (Supplementary Fig. 10A) had higher body weight and blood glucose compared with control mice (Supplementary Fig. 10B and C). In the ileum of UN mice, expression of Slc51a, Slc51b, Fxr, Asbt (Slc10a2), or Tgr5 did not differ; however, expression of Fgf15 and Shp was reduced by 48% and 67% in UN-exposed offspring (P < 0.05 for both) (Supplementary Fig. 10E–K).
Discussion
Our study provides new evidence supporting epigenetic regulation of intestinal function and metabolism and identification of new gene targets of Brd4 regulation. Prior studies have demonstrated that genetic or pharmacologic inhibition of Brd4-dependent signaling (12,41) can modulate intestinal cell populations. While we also find that JQ-1 decreased the number of Reg4+ Paneth cells, there was no change in the Paneth functional marker Crp4 (42) (Supplementary Fig. 7), ileal expression of cytokines (data not shown), or markers of additional intestinal cell populations. Thus, effects of JQ-1 in our experimental conditions do not appear to require alterations in Paneth cell-linked host defense systems or cell type distribution. Rather, our data point to new targets of Brd4 action in the intestine—including regulation of enterocyte FGF15/19 expression—with secondary effects on hepatic and systemic glucose and lipid metabolism (Fig. 6).
Figure 6.
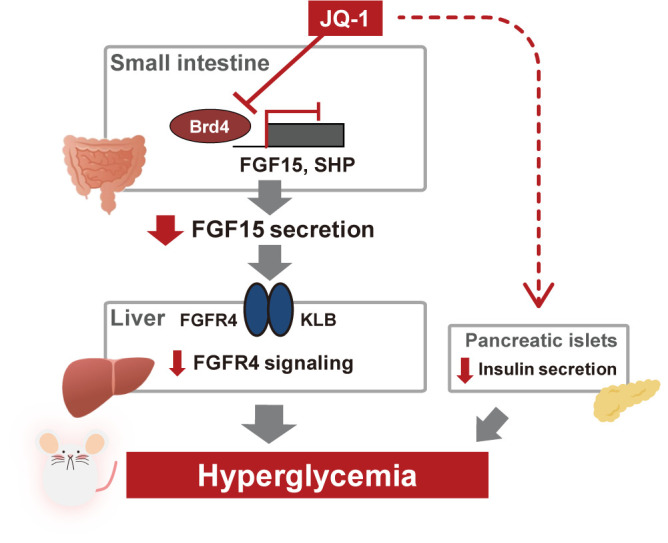
Hypothetical model illustrating how inhibition of Brd4 induced hyperglycemia in vivo.
FGF15/19 expression and secretion in the intestine can be regulated by bile acid binding to the luminal bile acid receptor Tgr5 or the nuclear receptor Fxr. We observed no change in expression of Tgr5, the apical bile acid transporter Asbt, Fxr, or Rxr; as expected with reduced Fgf15/19 signaling, plasma levels of some bile acid species were significantly increased in JQ-1–treated mice. We cannot exclude the possibility that luminal signaling via bile acids or other metabolites could contribute to reduced intestinal Fgf15 expression and secretion in response to JQ-1 in vivo. However, our data in cultured cells indicate that JQ-1 exerts a direct, cell-autonomous effect on transcription and secretion of FGF15/19 via inhibition of BRD4 binding.
Our data suggest that inhibition of FGF15/19 is a dominant mediator of in vivo metabolic effects of JQ-1. Several key findings are consistent with reduction in hepatic signaling by Fgf15 in JQ-1 treated mice: 1) downregulation of Fgfr4 and β-klotho (Klb) pathways and reduced signaling, as indicated by reduced Frs2 phosphorylation (Fig. 2D), 2) increased hepatic glucose production, as indicated by increased glucose after glycerol injection (Fig. 1K and L) and increased gluconeogenic gene expression (Supplementary Fig. 5), and 3) increased expression of lipogenic genes (e.g., Fasn), with increased plasma and liver triglycerides. Finally, JQ-1–induced hyperglycemia was fully reversed with adenoviral-mediated restoration of systemic FGF19 levels.
We acknowledge that JQ-1 may have additional FGF15/19-independent effects, as supported by our findings that JQ-1 was able to induce glucose intolerance even in mice with intestine-specific knockout of Fgf15. In the intestine, JQ-1 reduced expression of several FXR target genes, including Shp. Brd4 was recently demonstrated to be a cofactor for FXR in the liver (49). However, effects of short-term JQ-1 treatment in primary hepatocytes in our experiments were small in magnitude, suggesting that cell-autonomous effects were not dominant. Neither systemic insulin sensitivity nor hepatic insulin signaling was affected by short-term treatment with JQ-1, indicating insulin resistance was not a major contributor to hyperglycemia.
While there were no differences in pancreatic islet size or insulin staining, glucose-stimulated insulin levels were lower in JQ-1–treated mice, and we identified cell-autonomous effects of JQ-1 to reduce insulin secretion in isolated islets. While BRD4 can modulate senescence-associated genes in islets (48), our data suggest that effects of JQ-1 on insulin secretion are not likely to be the dominant contributor to hyperglycemia in our model, as FGF19 overexpression reversed JQ-1–mediated glucose intolerance while also further reducing insulin secretion.
We acknowledge additional mechanisms independent of systemic levels of Fgf15/19 must contribute to the impact of JQ-1 on systemic metabolism, given persistent glucose intolerance in Fgf15INT-KO mice treated with JQ-1. We hypothesize these may be linked to the marked alterations in hepatic signaling through the Fgfr and hepatic lipid metabolism observed in JQ-1–treated mice, but additional experiments will be required to test this possibility.
In summary, inhibition of the bromodomain protein Brd4 by JQ-1 reduces BRD4 binding at the promoter regions of FGF15/19 and SHP, leading to downregulation of FGF15/19 and SHP expression in the ileum. Reduced Fgf15- and Fgfr-dependent signaling in the liver, together with reductions in insulin secretion, contribute to hyperglycemia mediated by JQ-1. Moreover, hyperglycemia and glucose intolerance are fully reversed with in vivo viral expression of FGF19. Our data also suggest additional Fgf15/19-independent contributors to the impact of JQ-1. Given the ubiquitous nature of bromodomain regulatory complexes, it is not surprising that inhibition of Brd4-mediated transcription would have additional systemic, hepatic, or extrahepatic metabolic effects (8,48) contributing to hyperglycemia induced by JQ-1. Future studies will be required to dissect additional mechanisms—beyond intestinal regulation of Fgf15/19—underlying metabolic effects of the inhibition of bromodomain regulatory complexes.
Our results also raise the possibility that epigenetic regulation in the ileum may contribute to glucose intolerance in T2D or that caused by environmental factors such as intrauterine UN (25,47). Further detailed studies of epigenetic changes induced by environmental factors will be required to determine whether modulation of ileal epigenetic transcriptional regulation could be a potential target for reducing diabetes risk and improving systemic metabolism.
Article Information
Acknowledgments. The authors thank NGM for providing adenoviral FGF19 and GFP, and thank Susan Bonner-Weir, Joslin Diabetes Center and Harvard Medical School, Boston, MA, for helpful discussion.
Funding. Grant support was received from National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant DK106193 (to M.E.P.), R01DK101043 (to R.G.), and P30 DK036836 (Joslin Diabetes Research Center), National Cancer Institute grant R01CA142106, and Eunice Kennedy Shriver National Institute of Child Health and Human Development grant R01HD093540 (to J.Q.). C.K. was supported by Sunstar Foundation and Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (KAKENHI) Grant Number JP19K17971, and S.O. was supported by the Prince Mahidol Award Foundation.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. C.K., V.E., V.M.S., L.Z., S.O., Y.Y., J.C.-W., C.M., E.I., J.D., S.S., J.K., L.G., X.S., R.E.G., C.A-M., P.C., S.D.T., D.S., D.A.-C., L.W., J.Q., and M.-E.P. performed experiments. C.K., L.Z., and X.S. analyzed the data. C.K. and M.-E.P. designed the study. C.K. and M.-E.P. wrote the manuscript. V.M.S., L.Z., S.O., Y.Y., J.C.-W., C.M., E.I., J.D., S.S., J.K., L.G., X.S., R.E.G., L.W., and J.Q. reviewed and edited the manuscript. M.E.P. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
C.K. is currently affiliated with YCI Laboratory for Metabolic Epigenetics, RIKEN Center for Integrative Medical Sciences, Kanagawa, Japan.
This article contains supplementary material online at https://doi.org/10.2337/figshare.19064309.
References
- 1. Rosen ED, Kaestner KH, Natarajan R, et al. Epigenetics and epigenomics: implications for diabetes and obesity. Diabetes 2018;67:1923–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol 2015;16:258–264 [DOI] [PubMed] [Google Scholar]
- 3. Yamauchi T, Oike Y, Kamon J, et al. Increased insulin sensitivity despite lipodystrophy in Crebbp heterozygous mice. Nat Genet 2002;30:221–226 [DOI] [PubMed] [Google Scholar]
- 4. Gaur V, Connor T, Sanigorski A, et al. Disruption of the class IIa HDAC corepressor complex increases energy expenditure and lipid oxidation. Cell Rep 2016;16:2802–2810 [DOI] [PubMed] [Google Scholar]
- 5. Hong S, Zhou W, Fang B, et al. Dissociation of muscle insulin sensitivity from exercise endurance in mice by HDAC3 depletion. Nat Med 2017;23:223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marmorstein R, Zhou MM. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb Perspect Biol 2014;6:a018762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang F, Liu H, Blanton WP, Belkina A, Lebrasseur NK, Denis GV. Brd2 disruption in mice causes severe obesity without Type 2 diabetes. Biochem J 2009;425:71–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee JE, Park YK, Park S, et al. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat Commun 2017;8:2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol Cell Biol 2009;29:1375–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mauro C, Leow SC, Anso E, et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol 2011;13:1272–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barrow JJ, Balsa E, Verdeguer F, et al. Bromodomain inhibitors correct bioenergetic deficiency caused by mitochondrial disease complex I mutations. Mol Cell 2016;64:163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bolden JE, Tasdemir N, Dow LE, et al. Inducible in vivo silencing of Brd4 identifies potential toxicities of sustained BET protein inhibition. Cell Rep 2014;8:1919–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bozadjieva N, Heppner KM, Seeley RJ. Targeting FXR and FGF19 to treat metabolic diseases-lessons learned from bariatric surgery. Diabetes 2018;67:1720–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Holst JJ, Gribble F, Horowitz M, Rayner CK. Roles of the gut in glucose homeostasis. Diabetes Care 2016;39:884–892 [DOI] [PubMed] [Google Scholar]
- 15. Mingrone G, Panunzi S, De Gaetano A, et al. Metabolic surgery versus conventional medical therapy in patients with type 2 diabetes: 10-year follow-up of an open-label, single-centre, randomised controlled trial. Lancet 2021;397:293–304 [DOI] [PubMed] [Google Scholar]
- 16. Degirolamo C, Sabbà C, Moschetta A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat Rev Drug Discov 2016;15:51–69 [DOI] [PubMed] [Google Scholar]
- 17. Kir S, Kliewer SA, Mangelsdorf DJ. Roles of FGF19 in liver metabolism. Cold Spring Harb Symp Quant Biol 2011;76:139–144 [DOI] [PubMed] [Google Scholar]
- 18. Markan KR, Potthoff MJ. Metabolic fibroblast growth factors (FGFs): mediators of energy homeostasis. Semin Cell Dev Biol 2016;53:85–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005;2:217–225 [DOI] [PubMed] [Google Scholar]
- 20. Morton GJ, Matsen ME, Bracy DP, et al. FGF19 action in the brain induces insulin-independent glucose lowering. J Clin Invest 2013;123:4799–4808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Owen BM, Mangelsdorf DJ, Kliewer SA. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol Metab 2015;26:22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu S, Marcelin G, Blouet C, et al. A gut-brain axis regulating glucose metabolism mediated by bile acids and competitive fibroblast growth factor actions at the hypothalamus. Mol Metab 2018;8:37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barutcuoglu B, Basol G, Cakir Y, et al. Fibroblast growth factor-19 levels in type 2 diabetic patients with metabolic syndrome. Ann Clin Lab Sci 2011;41:390–396 [PubMed] [Google Scholar]
- 24. Mulla CM, Goldfine AB, Dreyfuss JM, et al. Plasma FGF-19 levels are increased in patients with post-bariatric hypoglycemia. Obes Surg 2019;29:2092–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma H, Sales VM, Wolf AR, et al. Attenuated effects of bile acids on glucose metabolism and insulin sensitivity in a male mouse model of prenatal undernutrition. Endocrinology 2017;158:2441–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bozadjieva-Kramer N, Shin JH, Shao Y, et al. Intestinal-derived FGF15 protects against deleterious effects of vertical sleeve gastrectomy in mice. Nat Commun 2021;12:4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou M, Learned RM, Rossi SJ, Tian H, DePaoli AM, Ling L. Therapeutic FGF19 promotes HDL biogenesis and transhepatic cholesterol efflux to prevent atherosclerosis. J Lipid Res 2019;60:550–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leek JT, Scharpf RB, Bravo HC, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet 2010;11:733–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ritchie ME, Diyagama D, Neilson J, et al. Empirical array quality weights in the analysis of microarray data. BMC Bioinformatics 2006;7:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu D, Lim E, Vaillant F, Asselin-Labat M-L, Visvader JE, Smyth GK. ROAST: rotation gene set tests for complex microarray experiments. Bioinformatics 2010;26:2176–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fabregat A, Jupe S, Matthews L, et al. The reactome pathway knowledgebase. Nucleic Acids Res 2018;46:D649–D655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anand P, Brown JD, Lin CY, et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 2013;154:569–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rathert P, Roth M, Neumann T, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015;525:543–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature 2010;468:1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sakurai N, Inamochi Y, Inoue T, et al. BRD4 regulates adiponectin gene induction by recruiting the P-TEFb complex to the transcribed region of the gene. Sci Rep 2017;7:11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roberts LD, Souza AL, Gerszten RE, Clish CB. Targeted metabolomics. Curr Protoc Mol Biol 2012;Chapter 30:Unit 30.2–30.2.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim YC, Byun S, Zhang Y, et al. Liver ChIP-seq analysis in FGF19-treated mice reveals SHP as a global transcriptional partner of SREBP-2. Genome Biol 2015;16:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kliewer SA, Mangelsdorf DJ. Bile acids as hormones: the FXR-FGF15/19 pathway. Dig Dis 2015;33:327–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marcelin G, Jo YH, Li X, et al. Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism. Mol Metab 2013;3:19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakagawa A, Adams CE, Huang Y, et al. Selective and reversible suppression of intestinal stem cell differentiation by pharmacological inhibition of BET bromodomains. Sci Rep 2016;6:20390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ouellette AJ. Paneth cell α-defensins in enteric innate immunity. Cell Mol Life Sci 2011;68:2215–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vergnes L, Lee JM, Chin RG, Auwerx J, Reue K. Diet1 functions in the FGF15/19 enterohepatic signaling axis to modulate bile acid and lipid levels. Cell Metab 2013;17:916–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McCleland ML, Mesh K, Lorenzana E, et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J Clin Invest 2016;126:639–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lan T, Morgan DA, Rahmouni K, et al. FGF19, FGF21, and an FGFR1/β-klotho-activating antibody act on the nervous system to regulate body weight and glycemia. Cell Metab 2017;26:709–718.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jimenez-Chillaron JC, Hernandez-Valencia M, Reamer C, et al. Beta-cell secretory dysfunction in the pathogenesis of low birth weight-associated diabetes: a murine model. Diabetes 2005;54:702–711 [DOI] [PubMed] [Google Scholar]
- 48. Thompson PJ, Shah A, Apostolopolou H, Bhushan A. BET proteins are required for transcriptional activation of the senescent islet cell secretome in type 1 diabetes. Int J Mol Sci 2019;20:4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jung H, Chen J, Hu X, et al. BRD4 inhibition and FXR activation, individually beneficial in cholestasis, are antagonistic in combination. JCI Insight 2020;6:e141640. [DOI] [PMC free article] [PubMed] [Google Scholar]