

Summary
Although tobacco use is an independent adverse prognostic feature in HPV(+) oropharyngeal squamous cell carcinoma (OPSCC), the biologic features associated with tobacco use have not been systematically investigated. We characterized genomic and immunologic features associated with tobacco use through whole exome sequencing, mRNA hybridization, and immunohistochemical staining in 47 HPV(+) OPSCC tumors. Low expression of transcripts in a T cell-inflamed gene expression profile (TGEP) was associated with tobacco use at diagnosis and lower overall and disease-free survival. Tobacco use was associated with an increased proportion of T > C substitutions and a lower proportion of expected mutational signatures, but not with increases in mutational burden or recurrent oncogenic mutations. Our findings suggest that rather than increased mutational burden, tobacco’s primary and clinically relevant association in HPV(+) OPSCC is immunosuppression of the tumor immune microenvironment. Quantitative assays of T cell infiltration merit further study as prognostic markers in HPV(+) OPSCC.
Subject areas: Oncology, Immunology, Genomics
Graphical abstract
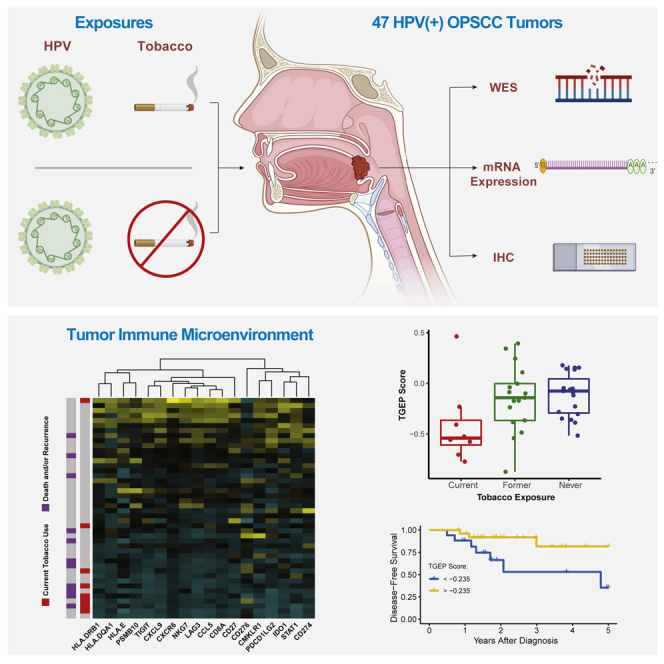
Highlights
-
•
Biologic features of tobacco use in HPV(+) OPSCC not previously described in a systematic fashion
-
•
Tobacco use was not associated with increased mutational burden or oncogenic mutations
-
•
Tobacco use was associated with low T-cell infiltration of primary TIME
-
•
In HPV(+) OPSCC, the major biologic effect of tobacco exposure is an immunosuppressed TIME
Oncology; Immunology; Genomics
Introduction
Oropharyngeal squamous cell carcinoma (OPSCC) is among the most common cancers of the upper aerodigestive tract. An estimated 60–70% of new OPSCC diagnoses in the United States are attributable to oncogenic human papillomavirus (HPV) exposure (Chaturvedi et al., 2011). The incidence of HPV(+) OPSCC is rising, and OPSCC has surpassed cervical cancer as the most common HPV-related malignancy in the United States (Ang et al., 2010; Chaturvedi et al., 2011; Centers for Disease Control and Prevention, 2019).
HPV(+) OPSCC is both molecularly and clinically distinct from the HPV(−) disease, for which tobacco and/or alcohol use are the key causative exposures. Although HPV(−) OPSCC often portends a grim prognosis, HPV(+) OPSCC outcomes are comparably favorable, with approximately 75–80% of HPV(+) OPSCC patients surviving five years after diagnosis (Ang et al., 2010; Sinha et al., 2018). For most patients who respond to treatment, the morbidity associated with surgery, radiation therapy, and/or platinum-based chemotherapy is substantial and often lifelong. It is not clear that HPV(+) OPSCC requires the aggressive treatment approaches used in the HPV(−) disease to produce the favorable oncologic outcomes observed in the HPV(+) population. For this reason, treatment deintensification for this patient group is a major focus of recent and ongoing clinical trials in HPV(+) OPSCC (Wahle and Zevallos, 2020). The unique challenges in the future of HPV(+) OPSCC treatment are reducing iatrogenic morbidity through treatment deintensification in low-risk individuals while also improving outcomes in the high-risk patients with an aggressive disease not cured with the current standard of care therapies. Importantly, both tasks rely on accurately predicting a high- or low-risk disease.
Tobacco use is well established as an independent adverse prognostic factor in HPV(+) OPSCC (Hafkamp et al., 2008; Ang et al., 2010; Gillison et al., 2012; Lin et al., 2013; Fakhry et al., 2014; Elhalawani et al., 2020). Multiple reports have demonstrated that tobacco use is associated with an increased risk of disease-specific adverse outcomes, suggesting that tobacco users’ attenuated prognosis is not entirely attributable to competing causes of mortality (Hafkamp et al., 2008; Maxwell et al., 2010; Gillison et al., 2012; Elhalawani et al., 2020). Tobacco users with HPV(+) OPSCC represent an intermediate prognostic group, between tobacco-naïve HPV(+) patients with an excellent prognosis and HPV(−) OPSCC patients whose prognosis is poor (Ang et al., 2010; Gillison et al., 2012; Elhalawani et al., 2020). Despite this, the molecular features associated with tobacco use in HPV(+) OPSCC are poorly defined. In this study, we hypothesized that tobacco influences two biologic processes that could contribute to more aggressive disease and worse outcomes in HPV(+) OPSCC. First, tobacco may be associated with increased somatic mutational burden and/or a distinct array of oncogenic mutations, contributing to a more aggressive disease. Despite tobacco’s canonical role as a mutagen in head and neck squamous cell carcinoma (HNSCC), recent work does not suggest that tobacco exposure is associated with increased mutational burden when HPV is the causative exposure (Gillison et al., 2018). Second, tobacco exposure may be associated with changes in the tumor immune microenvironment (TIME) that produce relative immunosuppression and a more aggressive disease course. This hypothesis is supported by a recent report demonstrating lower CD8+ immunohistochemical (IHC) staining in tobacco-exposed HPV(+) tumors (Kemnade et al., 2020). No prior investigation has systematically evaluated the molecular features associated with tobacco, comparing both its mutational and tumor immune effects. Using a multi-omics approach, we tested both of these hypotheses in a cohort of HPV(+) OPSCC patients. We evaluated somatic mutational burden with whole exome sequencing (WES) and investigated the TIME by measuring the expression of immune-related mRNA transcripts and IHC staining.
Results
Demographic and clinical characteristics of our cohort are displayed in Table 1. The majority of patients had a history of tobacco exposure, with 19 (40.4%) being former users and 9 (19.1%) being current users. Consistent with established demographic trends in HPV(+) OPSCC, the median age at diagnosis was 58 years old (range 45–79) and the majority of patients were White males. No significant differences in demographic and American Joint Committee on Cancer (AJCC) 8th edition staging parameters were present based on tobacco exposure groups. Our cohort included patients treated with primary surgery (76.6%) and primary chemoradiation therapy (CRT) (23.4%). The median time to loss of follow-up or death was 1.9 years (range 0.23–7.8 years). Eight patients (17.0%) in the cohort had disease recurrence and six (12.8%) died during the study period. Overall, survival (OS) and disease-free survival (DFS) were both significantly associated with a clinical disease stage (Figures S1A and S1B; both Log rank p < 0.001). There were no OS or DFS differences related to the tobacco use status (Figures S1C and S1D; Log rank p = 0.220 and 0.650, respectively), which is an expected finding given our cohort’s size.
Table 1.
Demographic and clinical characteristics of the study cohort
| Current |
Former |
Never |
p value | Total |
|
|---|---|---|---|---|---|
| (N = 9) | (N = 19) | (N = 19) | (N = 47) | ||
| Age at diagnosis | |||||
| Mean (SD) | 57.6 (5.22) | 61.3 (10.6) | 56.9 (9.07) | 0.312 | 58.8 (9.26) |
| Decade of diagnosis | |||||
| 50–59 | 6 (66.7%) | 7 (36.8%) | 7 (36.8%) | 0.212 | 20 (42.6%) |
| 60–69 | 3 (33.3%) | 5 (26.3%) | 4 (21.1%) | 12 (25.5%) | |
| 40–49 | 0 (0%) | 2 (10.5%) | 6 (31.6%) | 8 (17.0%) | |
| 70–79 | 0 (0%) | 5 (26.3%) | 2 (10.5%) | 7 (14.9%) | |
| Gender | |||||
| Female | 1 (11.1%) | 3 (15.8%) | 1 (5.3%) | 0.828 | 5 (10.6%) |
| Male | 8 (88.9%) | 16 (84.2%) | 18 (94.7%) | 42 (89.4%) | |
| Race/Ethnicity | |||||
| Asian | 0 (0%) | 1 (5.3%) | 0 (0%) | 1 | 1 (2.1%) |
| Caucasian | 9 (100%) | 18 (94.7%) | 19 (100%) | 46 (97.9%) | |
| Primary tumor subsite | |||||
| Base of Tongue | 3 (33.3%) | 9 (47.4%) | 8 (42.1%) | 0.838 | 20 (42.6%) |
| Tonsil | 6 (66.7%) | 9 (47.4%) | 11 (57.9%) | 26 (55.3%) | |
| Overlapping Sites | 0 (0%) | 1 (5.3%) | 0 (0%) | 1 (2.1%) | |
| Clinical T stage | |||||
| T0 | 1 (11.1%) | 2 (10.5%) | 0 (0%) | 0.462 | 3 (6.4%) |
| T1 | 2 (22.2%) | 4 (21.1%) | 3 (15.8%) | 9 (19.1%) | |
| T2 | 2 (22.2%) | 7 (36.8%) | 12 (63.2%) | 21 (44.7%) | |
| T3 | 3 (33.3%) | 3 (15.8%) | 3 (15.8%) | 9 (19.1%) | |
| T4 | 1 (11.1%) | 3 (15.8%) | 1 (5.3%) | 5 (10.6%) | |
| Clinical N stage | |||||
| N0 | 1 (11.1%) | 2 (10.5%) | 0 (0%) | 0.166 | 3 (6.4%) |
| N1 | 4 (44.4%) | 5 (26.3%) | 10 (52.6%) | 19 (40.4%) | |
| N2 | 3 (33.3%) | 12 (63.2%) | 7 (36.8%) | 22 (46.8%) | |
| N3 | 1 (11.1%) | 0 (0%) | 2 (10.5%) | 3 (6.4%) | |
| Clinical M stage | |||||
| M0 | 9 (100%) | 18 (94.7%) | 19 (100%) | 1 | 46 (97.9%) |
| M1 | 0 (0%) | 1 (5.3%) | 0 (0%) | 1 (2.1%) | |
| Overall clinical stage | |||||
| I | 3 (33.3%) | 4 (21.1%) | 8 (42.1%) | 0.658 | 15 (31.9%) |
| II | 4 (44.4%) | 12 (63.2%) | 8 (42.1%) | 24 (51.1%) | |
| III | 2 (22.2%) | 2 (10.5%) | 3 (15.8%) | 7 (14.9%) | |
| IV | 0 (0%) | 1 (5.3%) | 0 (0%) | 1 (2.1%) | |
All patients were p16(+) and had untreated primary OPSCC.
WES of tumor-normal pairs was performed for all 47 patients in our cohort, with a median depth of sequencing of 71x for tumor and 95x for normal samples. We identified 12 genes that met the criteria for statistically significant mutations in our cohort (MuSiC False Discovery Rate (FDR) < 0.1). These included multiple genes identified in HPV(+) OPSCC in previous publications (Lawrence et al., 2015; Gillison et al., 2018), including PIK3CA, ZNF750, FGFR3, PTEN, TRAF3, AND FBXW7 (Figure 1; Tables S1A and S1B). Moreover, this approach identified mutations in genes that have not previously been noted as statistically significant in HPV(+) OPSCC, including B2M, IFI27, AK5, METTL24, IQCG, and SMARCAL1. Additional recurrent mutations within functional networks included AKT1, CUX1, FGF2, FGF8, HLA-B, KMT2D, NRAS, PIK3R1, PLEC, SYNE1, and SYNE2 (MUFFINN probabilistic score >0.5, Table S1C).
Figure 1.

Somatic mutations in HPV(+) OPSCC
Center panel: Waterfall plot of somatic mutations for each patient. Each row represents an individual recurrently mutated gene, and each column represents a patient. Mutated genes are ranked from most frequently mutated (top) to the least (bottom). Colored boxes indicate mutated genes, with colors corresponding to mutational consequences. Black circles or triangles represent the number of changes (amplifications or deletions, respectively, with log2 ratio greater than +/− 0.5) at each locus. Top panel: mutational frequency is displayed for each patient, representing the total number of nonsynonymous single nucleotide variations after filtration. Left panel: mutated genes are annotated by the mutational significance classifiers used in the study. Right panel: the frequency of mutations in each gene as a proportion of the total cohort size. Bottom panel: Clinical annotation for each patient.
Many observed number of alterations (CNAs) were consistent with previous reports and included amplifications on 1q, 3q, 5p, 8q, 12p, 19q, and 20q and deletions on 3p, 9q, 11q, 14q, and 16q (Figure S2) (Lawrence et al., 2015; Gillison et al., 2018). Many focal amplifications and deletions not described for HPV(+) OPSCC in previous publications met the criteria for statistical significance (GISTIC q < 0.01), of which the most notable were an amplification of 7p22.1, containing RAC1, and a deletion of 13.q14.2, containing RB1 (Table S1D and S1E).
Tobacco use is associated with differences in single base substitutions, but not mutational burden or recurrent oncogenic mutations
Because of tobacco’s causative role in HNSCC at multiple sites, we hypothesized that WES would reveal additional somatic mutational burden in patients with tobacco use history. Despite its wide variability in the tumor mutational burden (TMB) throughout the cohort, ranging from 0.04 to 30.1 mutations/Mb, there were no significant differences in TMB related to the history of tobacco use regardless of how we categorized tobacco use, a finding consistent with prior work (Gillison et al., 2018). We observed no difference in TMB comparing tobacco-exposed versus never-exposed users (Wilcoxon p = 0.614), current versus former plus never tobacco users (Wilcoxon p = 0.642) or by comparing current, former, and never tobacco use groups separately (Figure S3A, Kruskal–Wallis p = 0.655). Moreover, TMB did not significantly differ in patients who had adverse clinical outcomes including death and/or recurrence (Figure S3B, Wilcoxon p = 0.738).
We sought to identify differences in mutational profiles based on tobacco exposure. In a subset of patients with sufficiently high mutational burden (total nonsynonymous SNVs >45, N = 39 patients, 83.0% of cohort), we examined the rate of single nucleotide substitutions and their trinucleotide contexts. Consistent with previous reports of HPV(+) OPSCC, tumors were dominated by C > T transitions and C > G transversions (Figure 2A) (Gillison et al., 2018) Tobacco-exposed tumors had a 2.3-fold greater median proportion of T > C transitions in any trinucleotide context compared to tumors of never tobacco users (Figure 2F, Wilcoxon p = 0.015, FDR = 0.088). Tobacco exposure was not associated with significant differences in the remaining substitution classes (Figures 2B–2E and 2G).
Figure 2.

Tobacco exposure is associated with differences in trinucleotide substitution classes
(A) Consistent with previous reports, mutations in our cohort predominantly consisted of C > T transitions and C > G transversions.
(B–G) Each class of nucleotide substitution is displayed as a proportion of total mutations.
(F) Tobacco exposed tumors demonstrated a 2.3-fold greater median proportion of T > C transitions compared with tumors of never tobacco users (∗ Wilcoxon p < 0.05 and FDR <0.1, ns = not significant).
Single base substitution signatures were determined for each sample and compared based on tobacco use status (Alexandrov et al., 2013). Consistent with prior reports, three mutational signatures were dominant (Figure 3A). Signature #1 was present in 92.3% of samples and reflects spontaneous 5-methylcytosine deamination associated with aging. Signatures #2 and #13, which reflect mutations attributable to apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) activity, were present in 74.3% of patients. The median proportion of mutations accounted for by signatures #1, #2, and #13 combined was 66.8% (range 9.2–91.7%). Together, these dominant signatures accounted for a significantly higher proportion of mutations in tumors of never tobacco users when compared with tobacco-exposed tumors (Figure 3B, Wilcoxon p = 0.026).
Figure 3.

Aging and APOBEC-related mutational signatures are dominant in HPV(+) OPSCC but account for a lower proportion of mutations in tobacco users
(A) Heatmap of mutational signatures demonstrates the dominance of signatures #1 (Aging-related), #2 and 13 (both APOBEC-related) in HPV(+) OPSCC. Despite high reported rates of tobacco use in the cohort, mutations attributable to tobacco-related signatures #4 and #29 were absent.
(B) The proportion of signature #1, #2, and #13 mutations is significantly greater in never tobacco users, whereas (C) the proportion of uncategorized mutations is significantly greater in those reporting current or former tobacco use (∗ Wilcoxon p < 0.05).
Tobacco-related mutational signatures were largely absent within our cohort. Signature #4, which is associated with tobacco and common in HNSCC (Alexandrov et al., 2013), was not responsible for any proportion of mutations in our study population. Signature #29, associated with chewing tobacco use, was present in one patient accounting for 8.2% of mutations in this individual. Signature #16 mutations have been previously correlated with tobacco exposure in HPV(−) disease (Gillison et al., 2018) but, as expected, these were not significantly associated with tobacco use (Wilcoxon p = 0.418) in this study’s HPV(+) OPSCC cohort. We also considered the proportion of mutations that could not be categorized into one of the 30 mutational signatures. Uncategorized mutations were present in 94.9% of samples and accounted for a minority of mutations in most patients (median 6.0% of mutations, range 0–20.6%). Uncategorized mutations accounted for a significantly greater proportion of mutations in tobacco-exposed tumors (Figure 3C, Wilcoxon p = 0.017). This difference approached a statistical significance with current, former, and never tobacco users compared separately (Kruskal–Wallis p = 0.052).
In an exploratory analysis, we compared the frequency of recurrent somatic mutations in the whole cohort by tobacco use groups. There were no significant differences in the occurrence of any single mutated gene based on tobacco use groups (Table S1F). We hypothesized that mutations typically observed in HPV(−) HNSCC may be induced by tobacco use. To test this, we examined the rate of canonical HPV(−) HNSCC mutations that were significantly mutated in TCGA(Table S1G) (Lawrence et al., 2015). These mutations individually occurred at a low rate in our cohort with no single gene having more than an 8.5% mutation rate. Collectively, canonical HPV(−) mutations occurred at a rate of 32.1% of those with a history of tobacco use compared with 15.8% of those with no tobacco history (Fisher’s Exact p = 0.31).
Targeted gene expression profiling reveals that T cell-inflamed TIME is inversely associated with current tobacco use
Given the lack of significant differences in tumor-associated variants by the tobacco use status, we then hypothesized that tobacco use would be associated with discernible differences in the TIME. We utilized a 760-gene Nanostring immuno-oncology panel to measure expression of immune-cell related transcripts. Of the 47 tumors available for analysis, three samples were excluded because they did not pass quality control review owing to low binding density and/or low efficiency hybridization.
We performed unsupervised hierarchical clustering of the 760 transcripts for all patients. This approach divided patients into two clusters which differed by disease-free survival (Figure 4). A cluster of 10 low-risk patients was identified, none of which had death or recurrence, with all adverse events concentrated in the remaining 34 patients (Log rank p = 0.069, HR = 3.703, 95% CI 0.905–15.15). We observed that the 10-patient low-risk cluster was enriched for a cluster of 202 genes containing T cell-related transcripts (Figure 4, column cluster 4), including T cell-related cell surface markers, members of the T cell receptor complex, granzymes, and IFN-λ and JAK-STAT pathway members (Table S2D). Over representation analysis (ORA) revealed that relative to all transcripts in the panel, this cluster was significantly enriched for multiple biological processes related to adaptive immunity. Importantly, adaptive-immune related processes were identified in all functional databases we queried (Table S2D). To validate these findings, IHC staining of primary tumors was performed using anti-CD3, CD4, and CD8 antibodies. The 10-patient low-risk cluster had a significantly greater degree of T cell infiltration (Figures S4A–S4C, all Wilcoxon FDR <0.01), confirming that the enrichment for adaptive immune-related transcripts in this cluster correlates with protein expression of T cell surface markers. ORA provided relatively less support for biological processes identified in clusters #1–3, with only clusters #2 and #3 having significant biological processes identified, and only in one of the databases queried. Full gene lists and ORA results are available in Tables S2A–S2C.
Figure 4.

Unsupervised clustering of 760 transcripts reveals a low-risk patient group with a high expression of T cell-related transcripts
Heatmap of gene expression displays a high expression of transcripts in column cluster 4 in a subset of patients without death and/or recurrence. ORA of transcripts in this cluster revealed that cluster 4 was significantly enriched for multiple biological processes related to adaptive immunity. Patients in the high-risk patient cluster who had a lower expression of cluster 4 transcripts displayed an increased risk of death and/or recurrence (HR = 3.703, 95% CI 0.905–15.15, Log rank p = 0.069).
To further characterize and expand on the functional significance of T cell related differences suggested by unsupervised clustering, we utilized a previously published and validated 18-gene T cell-inflamed gene expression profile (TGEP) score (Ayers et al., 2017; Cristescu et al., 2018). A high score reflects existing cytotoxic activity in the TIME and has been validated in multiple tumor types, including large cohorts of HNSCC patients, as a predictor of response to PD-1 inhibitors (Ayers et al., 2017). Figure 5C displays the spectrum of transcript expression contributing to TGEP scores in our cohort. TGEP scores showed significant positive correlations with the IHC expression of CD3+, CD4+ and PD-L1 (Figures S5A–S5C, all Spearman correlation FDR <0.1); the strongest and most significant correlation was with CD8+ cells (Figure 5B, Spearman Rho = 0.728, FDR <0.001), suggesting that TGEP scores serve as a reliable indicator of a CD8+ T cell-inflamed TIME.
Figure 5.

Tobacco use at the time of diagnosis is associated with decreased T cell infiltration of the primary TIME
(A) Heatmap displaying the TGEP transcript expression for each patient. Patients are ranked from high (top) to low (bottom) based on their TGEP scores.
(B) TGEP scores were significantly associated with CD8+ IHC staining (Spearman Rho = 0.728, p < 0.001).
(C) TGEP scores differ significantly by tobacco use, with current tobacco users having the lowest scores (∗ Kruskal–Wallis p < 0.05).
(D) Tobacco history reveals differences in TGEP scores (high vs. low) and mutational frequency (high vs. low) by tobacco use groups (Fisher’s exact p < 0.05).
(E) In never tobacco users, there was a significant positive correlation between TGEP scores and mutational frequency. This correlation was not present in current or former tobacco users (∗∗ Spearman p < 0.01).
TGEP scores significantly differed based on the current, former, or never tobacco use groups (Figure 5C, Kruskal–Wallis p = 0.029), with current tobacco users having lower scores than never or former users (Dunn’s test, p = 0.028 and 0.085, respectively). Tobacco’s effect on the TIME was also evident when TGEP scores were analyzed in combination with tumor mutational frequency as has been previously described (Cristescu et al., 2018). We used previously published thresholds for TGEP (score greater than −0.318) and mutational frequency (greater than 86 somatic, non-synonymous variants per sample) that have been previously shown to predict response to PD-1 inhibition in HNSCC alone or in combination with TGEP scores (Cristescu et al., 2018). These thresholds divided patients into four groups based on high and low TGEP score and mutational frequency values. Compared with former or never tobacco users, current tobacco users disproportionately distributed into groups where one or both of TGEP scores or mutational frequency were low (Figure 5D, Fisher’s exact p = 0.018). Although current tobacco users had significantly lower TGEP scores, Figures 5A, 5C, and 5D demonstrate that low TGEP scores were not exclusive to current tobacco users. Never tobacco users demonstrated a significant positive correlation between the TGEP score and mutational frequency (Figure 5E, Spearman Rho = 0.612, p = 0.006). This positive correlation was not present in former users (Figure 5E, Spearman Rho = 0.010, p = 0.974) and or current tobacco users (Figure 5E, Spearman Rho = −0.524, p = 0.197). Taken together, our results suggest that varying degrees of T cell-related mRNA and protein expression are present within HPV(+) OPSCC primary tumors, and that the low expression of these markers is associated with tobacco use at diagnosis.
T cell-inflamed TIME is associated with survival and treatment response
Similar to our unsupervised clustering results, associations between T cell inflammation and treatment outcomes were observed using TGEP scores. A single patient with distant metastases at diagnosis was removed from survival analyses. We performed cut point analysis, which revealed that at a TGEP score threshold of −0.235, high TGEP scores were significantly associated with improved OS and DFS (Figures 6A and 6B; Log rank p = 0.004 and 0.025, respectively). 60.5% of the cohort had TGEP scores above this threshold. Among the 39.5% of the cohort with scores below this threshold, 6 (35.2%) were current tobacco users, 5 (29.5%) were former users, and 6 (35.2%) were never users. The expressions of CD3, CD4, CD8, and PD-L1 were all significantly greater in samples with TGEP scores above this threshold (Figures 6C–6F, all Wilcoxon FDR <0.1). In a multivariate analysis adjusting for the patient age and disease stage at diagnosis, DFS was independently associated with the TGEP score (Table S2, Cox proportional hazards, HR 4.52, 95% CI 1.04–19.7, p = 0.04), whereas neither stage nor age were significant in this model. Multivariate analysis was not performed using OS because all mortalities occurred in patients with TGEP < −0.235.
Figure 6.

TGEP scores are significantly associated with clinical outcomes
(A) OS was significantly reduced in patients with a TGEP < −0.235 (Log rank p < 0.01).
(B–F) (B) DFS was significantly reduced in patients with a TGEP < −0.235.(Log rank p < 0.05). IHC staining was significantly lower for patients with TGEP scores < −0.235 compared with those with scores > −0.235 for the following markers: (C) CD3, (D) CD4, (E) CD8, and (F) PD-L1 (Wilcoxon ∗p < 0.05, ∗∗∗p < 0.001, all FDR <0.1).
To ensure tissue specimen sources did not represent a source of bias in these results, we compared samples by primary treatment modality. We did not identify differences in TGEP scores (Figure S5D, Welch’s T test, p = 0.19, mean difference 0.13, 95% CI –0.07 to 0.33), nor did we identify categorical differences using the −0.235 TGEP score threshold (Fisher’s exact p = 0.31). We sought to determine if our observed trends related to tobacco and clinical outcomes were present in an independent cohort. We examined expression of the 18 transcripts in the TGEP score in p16-positive OPSCC patients from The Cancer Genome Atlas (TCGA) using normalized RNA sequencing data. In this cross-platform analysis, unsupervised clustering of the TGEP transcript expression revealed that current tobacco users and patients with death and/or recurrence were similarly concentrated among patients with a low expression of TGEP transcripts in both our study cohort and a TCGA cohort with the same inclusion criteria (Figures S6A and S6B, respectively).
Discussion
This study represents the most comprehensive investigation to date of the biology associated with tobacco use in HPV(+) OPSCC, a disease for which tobacco use represents a strong, independent, adverse prognostic factor (Ang et al., 2010). Using an integrative genomic approach, we have identified multiple biologic differences present in primary HPV(+) OPSCC tumors related to tobacco exposure. Most importantly, we demonstrate that current tobacco use is associated with decreased T cell infiltration of HPV(+) OPSCC tumors. Our WES results suggest this effect is present despite a lack of significant differences in TMB or recurrent oncogenic mutations associated with tobacco use. Further, low T cell infiltration was associated with decreased OS and DFS, regardless of tobacco use. Taken together, our results suggest that the primary and clinically relevant feature associated with tobacco use in HPV(+) OPSCC is decreased T cell infiltration of the primary TIME. These findings may have important implications for treatment stratification in HPV(+) OPSCC, particularly in the era of treatment deintensification.
The immunosuppressive molecular effects of tobacco exposure have been previously demonstrated using a variety of experimental methods. In case-control studies and experimental work using human PBMCs, tobacco exposure has been associated with cytokine alterations with particular relevance to both antiviral and antitumor immune responses, including decreases in IFN-y, IL-2, TNF, IL-15, and IL-16(van Dijk et al., 1998; Mian et al., 2009b; Mian et al., 2009a; Maet al., 2010; Shiels et al., 2014; Liu et al., 2019). Experimental evidence using animal models exposed to tobacco smoke also support this hypothesis, with multiple studies showing that tobacco exposure results in systemic T cell anergy and impaired immune responses to a variety of challenges, including viruses (Geng et al., 1995, 1996; Kalra et al., 2000; Feng et al., 2011; Schierl et al., 2014). Other groups have demonstrated impairment of NK and dendritic cells in response to tobacco exposure (Vassallo et al., 2005; Lu et al., 2007; Hao et al., 2013; Alkhattabi et al., 2018). These studies provide biological rationale for the association between tobacco and poor immune infiltrate identified in our study.
The effect of tobacco exposure on the TIME has been investigated in multiple human malignancies, with varying results related to cancer histologic classification and tumor site. Multiple groups have described a similar inverse relationship between tobacco exposure and immune infiltration in HPV(−) HSNCC (Foy et al., 2017; Desrichard et al., 2018; Iglesia et al., 2020). In particular, Iglesia et al. demonstrated that current tobacco use was associated with decreased T cell infiltration and IFN signaling in HPV(−) HNSCC tumors. Conversely, the effect of tobacco has been associated with an inflamed TIME in lung SCC (Desrichard et al., 2018). This difference in lung SCC may be related to tobacco-induced pulmonary inflammation that has been demonstrated outside of the context of cancer (Hodge et al., 2007). There is a paucity of data on the effect of tobacco exposure on HPV(+) OPSCC, with a single recent report demonstrating decreased CD8+ IHC staining in tobacco-exposed primary HPV(+) OPSCC tumors (Kemnade et al., 2020). Our findings are consistent with this previous study and elucidate the immunosuppressive effect of tobacco exposure on HPV(+) OPSCC phenomenon in greater biologic detail using multiple mRNA hybridization probes, validating in IHC, and controlling for mutational burden using our WES data. Our results, along with another recent study in HPV(−) HNSCC, suggest that the immunosuppressive effect of tobacco use at the time of diagnosis may be a feature shared by HNSCC tumors regardless of HPV status (Iglesia et al., 2020). Beyond SCC, tobacco has been associated with immunosuppression in primary colorectal carcinoma, another highly immunogenic tumor (Hamada et al., 2018; Fujiyoshi et al., 2020).
Further, tobacco exposure influences HPV-specific immune responses. Multiple groups have demonstrated that tobacco use is associated with impaired immune responses and clearance of oncogenic HPV infections in human female genital infections (Giuliano et al., 2002; Koshiol et al., 2006; Simen-Kapeu et al., 2008). In the oropharynx specifically, the current tobacco use is strongly associated with an increased incidence of oral HPV16 infections (Fakhry et al., 2014b). These findings are important given the putative oncologic benefits of HPV in the context of HNSCC. HPV(+) OPSCC tumors display greater T cell infiltration compared with HPV(−) HNSCC, a finding that may contribute to superior oncologic outcomes in HPV(+) disease (Jung et al., 2013; Mandal et al., 2016; Masterson et al., 2016). The results of the present study are important, given clinical evidence demonstrating that tobacco users with HPV(+) OPSCC have an attenuated oncologic prognosis compared with non-tobacco users (Hafkamp et al., 2008; Ang et al., 2010; Lin et al., 2013; Fakhry et al., 2014a; Chen et al., 2020). Based on our results, we speculate that this attenuated prognosis is related to tobacco-induced immunosuppression, which diminishes the immunologic benefit of having an HPV(+) tumor, forcing HPV(+) OPSCC tobacco users into an intermediate prognostic group in between tobacco-naïve HPV(+) OPSCC and HPV(−) OPSCC. Prospective studies are needed to precisely define the systemic and local immunologic features associated with tobacco use in HPV(+) OPSCC and how these features intersect with oncologic outcomes.
The TGEP score used in this study provided a useful method to quantitatively describe the spectrum of T cell-inflammation in our cohort. This score has been validated in large patient cohorts as a predictor of response to PD-1 inhibitors, is agnostic to tumor-type, and was developed on the same NanoString RNA hybridization platform used in this study (Ayers et al., 2017; Cristescu et al., 2018). Its utility in stratifying patients by response to PD-1 inhibitors likely lies in its ability to identify which patients generated an initial antitumor immune response (Ayers et al., 2017). The transcripts present in the TGEP characterize different components of this response: neoantigen presentation, chemoattraction, and infiltration of cytotoxic T cells and professional antigen-presenting cells, IFN-λ signaling, and the resulting upregulation of immune checkpoints (Ayers et al., 2017; Spranger and Gajewski, 2018). We demonstrated robust correlations between TGEP scores and T cell infiltration as measured by IHC staining. Like IHC staining, TGEP scores may be derived from formalin fixed paraffin embedded samples. However, when compared with IHC, TGEP scores are quantitative, operator-independent, and reliant on hybridization of numerous probes rather than the binding of a single antibody.
An important finding from our investigation is that overlap between low TGEP scores and tobacco use was incomplete, with a subset of former or never tobacco users also having low TGEP scores. These results suggest that current tobacco use is not required to achieve this high-risk phenotype. In our cohort, low TGEP scores were associated with lower overall and disease-free survival, including when adjusting for the age and disease stage. We speculate that gene expression profiles like TGEP, which provide quantitative measures of TIME inflammation, may provide useful objective data for risk stratification in clinical trials aimed at HPV(+) OPSCC treatment deintensification and/or candidacy for immunotherapeutics. The results of this study merit further investigation in a larger cohort where the prognostic contributions of tobacco and TGEP can be more accurately quantified and compared.
Our WES data also provide important insights related to tobacco exposure in HPV(+) OPSCC. The results of this study were consistent with the largest genomics study to date of HPV(+) OPSCC patients, which demonstrated that although mutational burden increased with heavy tobacco exposure in HPV(−) tumors, there was no difference in mutational burden in HPV(+) OPSCC on the basis of tobacco exposure (Gillison et al., 2018). Similarly, we were unable to identify a higher rate of any particular mutation in tobacco users, including canonical HPV(−) mutations (Gillison et al., 2018). We acknowledge that we were not powered to detect small effect sizes and that a much larger sequencing cohort of HPV(+) OPSCC patients will be needed to definitively determine if mutational profiles differ on the basis of tobacco use.
We did identify mutational differences related to tobacco use in our cohort in other analyses. We found that tobacco exposed tumors had higher rates of T > C substitutions, lower rates of APOBEC and aging-related mutational signatures, and a greater share of mutations that could not be classified into a COSMIC signature. Importantly, these effects were observed in both current and former tobacco users, whereas tobacco’s associations with the TIME were observed predominately in current tobacco users. The finding that our cohort totally lacked mutational signature 4, which is associated with tobacco use, is not unexpected. Compared with tobacco-naïve tumors, signature 4 is significantly increased in tobacco exposed lung SCC, lung adenocarcinoma, and larynx SCC, but remains largely absent in oral cavity, pharynx, and esophageal SCC as well as bladder cancer (Alexandrov et al., 2016). Among cancers without signature 4, significant increases T > C substitutions have been observed in tobacco-exposed oral cavity and bladder tumors (Alexandrov et al., 2016). Based on our findings and previous genomic studies, we conclude that while tobacco exposure in HPV(+) OPSCC is associated with discernible differences in somatic mutations, there remains no clear evidence that this affects genomic features such as mutational burden or recurrent oncogenic mutations. Although they have an intermediate prognosis, available evidence does not support the hypothesis that tobacco-exposed HPV(+) tumors represent a mutational intermediary between tobacco naive HPV(+) OPSCC and HPV(−) OPSCC.
We identified multiple genes that have yet to be described as significantly mutated in HPV(+) OPSCC. B2M variants were previously noted for HNSCC and HPV(+) OPSCC specifically in TCGA, although criteria for statistical significance were not met (Lawrence et al., 2015). B2M was identified using both of our computational tools for assessing mutational significance. B2M is a component of the MHC-I complex and serves an indispensable role in antigen presentation (D’Urso et al., 1991). Another identified novel mutated gene with potential biologic relevance was SMARCAL1, which codes for an annealing helicase that limits genomic damage at stalled replication forks (Bansbach et al., 2009). Like p53, SMARCAL1 is also phosphorylated by ATM serine/threonine kinase, whose gene is deleted in recurrent 11q22 number of losses in HPV(+) OPSCC (Lawrence et al., 2015). Unlike B2M, SMARCAL1 was only called a significant variant by one computational tool. Further work is needed to clarify the role of SMARCAL1 relative to other known sources of genomic instability in HPV(+) OPSCC. The biological relevance of the other identified mutations AK5, IFI27, IQGC, and METTL24 in HPV(+) OPSCC is not clear and will also require further investigation.
In summary, we have shown that current tobacco use is associated with decreased immune infiltration in HPV(+) OPSCC and not with significant mutational differences by tobacco exposure status. Although we identified tobacco-associated differences in single base substitutions and mutational signatures, tobacco was not associated with increased TMB or differences in recurrent oncogenic mutations. TGEP scores are associated with OS and DFS in HPV(+) OPSCC and may represent a quantitative clinical assay for use in future risk stratification efforts. We conclude that in HPV(+) OPSCC, the primary and clinically relevant association with tobacco use is decreased T cell infiltration of the TIME. This works suggests that immunosuppression may account for the increased oncologic risk observed in tobacco users with HPV(+) OPSCC.
Limitations of the study
The present study has several limitations not previously mentioned. Our study’s most significant limitation was that we relied on a retrospective review of the medical record to determine tobacco exposure. Although we confidently classified all patients into current, former, or never tobacco status in all patients, determining pack years was not possible in many cases. This limitation may be avoided in future studies through administration of a tobacco survey at the time of enrollment. Even with these measures in place, biases associated with patient-reported tobacco use are never entirely avoided, a fact that underscores the importance of identifying objective biomarkers of high-risk disease that do not rely on patient-reported behavioral data. We acknowledge that although our patients’ tumors were well characterized using multiple modalities, our cohort’s size represents a limitation. To address this, we demonstrated that p16(+) OPSCC current smokers in TCGA had a similarly low expression of TGEP transcripts. We acknowledge that larger studies are needed to precisely quantify the risk associated with low tumor T cell infiltration in this disease. We also acknowledge that although the mRNA hybridization panel provides a platform that is more readily converted into a clinical assay, it is targeted toward certain biological processes, including immunity, making its value for discovery limited when compared with unbiased approaches such as RNA sequencing. Although the TGEP score we used has had extensive prior validation and correlated well with IHC staining in our cohort, other experimental approaches will be needed to better define the mechanisms associated with smoking-related immunosuppression in HPV(+) OPSCC.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Jose Zevallos (jpzevallos@wustl.edu).
Materials availability
This study did not generate unique reagents.
Experimental model and subject details
Patient characteristics and sample acquisition
All patients were treated at the Washington University Siteman Cancer Center. After informed consent, primary tumor samples were collected prospectively from patients with newly diagnosed, biopsy proven, p16-positive OPSCC as part of the Washington University tumor banking protocol. The tumor acquisition protocol and correlative studies were approved by the Washington University Human Research Protection Office. A total of 47 patients were included in the study. Patient characteristics are summarized in Table 1.
Method details
Clinical data acquisition
Demographic and clinical data were collected from medical records. Tobacco use information was available for all patients such that they could be assigned to one of three tobacco use categories: current tobacco users were those using tobacco at the time of their cancer diagnosis, former users were those who previously used tobacco but quit using tobacco prior to diagnosis, and never users were those who denied ever using tobacco. Dates of diagnosis, death, or recurrence (local, regional, or distant) were recorded for each patient and used to calculate overall and disease-free survival (OS and DFS, respectively).
Whole exome sequencing
Sample preparation
Genomic DNA was isolated from 47 paired tumor/normal samples using Qiagen DNeasy Blood and Tissue Kits. Genomic DNA was fragmented to 100-300bp and libraries were constructed using the KAPA HTP Library Kits (KAPA Biosystems). A single low input sample used the Swift Accel-NGS 2S DNA Library Kit (Swift BioSciences) for library construction. Samples were then sequenced to a target depth of 120x per sample on the Illumina NovaSeq platform (S4, 2 x 151 bp reads).
Sequence alignment, somatic variant calling, filtering, and significance
Exome sequencing data was processed with the February 2019 version of the common workflow language somatic_exome pipeline developed at the McDonnell Genome Institute (https://github.com/genome/analysis-workflows). Pipeline execution was performed using Cromwell. Tracking of sample metadata and analysis results was performed using the McDonnell Genome Institute’s Genome Modeling System (Griffith et al., 2015). Briefly, reads were aligned with bwa-mem (0.7.15) to version GRCh38 of the human genome reference supplemented with HLA decoy sequences (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/GRCh38_reference_genome/). Duplicate reads were marked with picard (2.18.1) and a base quality score recalibration applied with GATK (3.6) (Auwera et al., 2013). Variant calling was performed using the union of calls from Strelka (2.9.9) (Saunders et al., 2012), MuTect (3.6) (Cibulskis et al., 2013), VarScan (2.4.2) (Koboldt et al., 2013) and Pindel (0.2.5b8) (Ye et al., 2009) and variants were annotated with the Variant Effect Predictor (Ensembl 93) (McLaren et al., 2016). Read counting of variants was performed via bam-readcount (0.7).
In order to obtain a final variant list, pipeline variants were further refined as follows. The variant list was filtered in R such that a candidate variant must have had > 4 variant supporting reads in the tumor sample, tumor variant allele frequency > 0.05, normal read depth > 20, and normal variant allele frequency <= 0.05. Non-coding and synonymous variants were removed from the candidate variant list. Using features described in deepSVR (Ainscough et al., 2018), variants were then classified as “Somatic” or “Failed” using an Extreme Gradient Boosting classifier via the XGB (booster = "gbtree", eta = 0.1, min_child_weight=1, max_depth = 8, gamma=0, subsample=1, scale_pos_weight=1, eval_metric = "auc") R library with a binary logistic classification objective. The classifier was trained on a subset of manually reviewed variants from the cohort. Five-fold cross validation of the model yielded an AUC of 0.85. Variants with a binary logistic probability between 0.35 and 0.65 were further refined via manual review (Barnell et al., 2019). Finally, variants within TTN or Mucin genes or variants exhibiting a gnomAD (Karczewski et al., 2020) minor allele frequency > 0.10 were removed.
Mutational significance was determined using MuSiC (Dees et al., 2012) with default settings on the filtered variant list. Potential cancer genes were prioritized via MUFFINN (Cho et al., 2016) using the HumanNet V1 network and the ndmax method. Genes with a probabilistic score > 0.5 were selected as candidate cancer genes of interest.
DNA mutational signatures
The R library deconstructSigs (Rosenthal et al., 2016) was used to identify patterns in the somatic variants and their surrounding nucleotide context that support specific DNA mutational signatures in each sample. The somatic variants of each sample were compared to version 2 of the COSMIC mutational signature reference, which contains 30 unique signatures (Alexandrov et al., 2013). Briefly, a final variant list was constructed as described above except the gnomAD allele frequency filter was not applied and synonymous variants were included in the final variant list for this analysis. Samples with less than 45 total nonsynonymous SNVs (N=8) were omitted from the analysis. The “exome2genome” normalization method within deconstructSigs was used to obtain COSMIC signature weights for each sample.
Copy number variant calling
Tumor ploidy aberrations were identified via CNVkit (0.9.6) (Talevich et al., 2016), from the somatic_exome pipeline described above. A somatic amplification or deletion was defined from the segmented log2 tumor/normal ratio output from cnvkit as +/- 0.5. Copy number plots were made with the R library GenVisR (1.18.1)(Skidmore et al., 2016). Significant amplified or deleted regions were identified with GISTIC (2.0.23)(Mermel et al., 2011) using default parameters.
Nanostring mRNA hybridization
RNA was isolated from 47 tumor samples using QIAGEN RNeasy kits. Transcripts were hybridized to a panel of 770 probes (Nanostring IO360). Hybridized transcripts were purified and immobilized on a sample cartridge. Counts of hybridized transcripts were read using the nCounter platform. Raw counts were normalized with the nSolver software (4.0) package using the geometric mean of both positive control probes and housekeeping probes. A background threshold count was set to 20. Normalized data was used for hierarchical clustering. Over representation analysis (ORA) was performed using WebGestalt (Liao et al., 2019). TGEP scores were calculated separately by renormalizing using available housekeeping probes and weighting transcript values as previously described (Ayers et al., 2017; Cristescu et al., 2018). Validation of TGEP score findings was performed by identifying p16(+) OPSCC cases from The Tumor Genome Atlas with available normalized RNAseq data (Lawrence et al., 2015). For a cross-platform comparison, z-scores were calculated for each of the 18 genes and unsupervised clustering was performed, followed by annotation with clinical data.
IHC
Tumor microarrays were assembled from formalin-fixed paraffin-embedded primary tumors. A clinical pathologist reviewed slides and selected areas containing tumor for inclusion in cores. Slides of 5-micron thickness were made and stained with Ventana antibodies for CD3 (2GV6), CD4 (SP35), CD8(SP57), and PD-L1(SP263) with hematoxylin counterstaining. A high-resolution digital scanner (NanoZoomer; Hamamatsu Photonics, Welwyn Garden City, UK) was used to create digitized bright field images from the slides at 20x magnification. Cores were not included in analyses if >30% of the core was missing or damaged during sectioning. The images were evaluated using digital analysis software (Visiopharm; Hoersholm, Denmark), which has been used previously for identifying T cells in the HNSCC TIME (Amin et al., 2020). Red-Blue-Green color filters were set which reliably distinguished 3,3′-diaminobenzidine (DAB) stained and hematoxylin stained cells without detecting unstained negative space. The total area stained by DAB or hematoxylin was calculated per core. Positive staining for each core was quantified and reported as the total DAB[+] area divided by the total cellular area (DAB[+] plus hematoxylin[+]). Example images are shown for anti-CD8 stained tumors in Figure S7.
Quantification and statistical analysis
Statistical analyses were performed in R (3.6.1) or GraphPad Prism (8.1.2). Additional R packages are detailed in the key resources table. Details of individual statistical tests are presented in the results section of this report. Shapiro Wilk tests were used to determine normality of distributions. Normally distributed continuous variables were compared using two-sided T-tests with Welch’s correction. Continuous variables without normal distributions were compared with two-sided Wilcoxon or Kruskal Wallis tests with Dunn’s multiple comparison tests. Spearman’s rank correlation coefficients were computed to determine degrees of correlation. Contingency testing was performed using Fisher’s exact tests. Univariate survival analyses were performed using Log-rank tests. Multivariate survival analysis was performed using Cox proportional hazards. False Discovery Rates were computed when multiple comparisons were performed together. Cut point analysis was performed using the R package survminer (0.4.2). Hierarchical clustering was performed using Euclidian distance with Ward’s linkage method.
Acknowledgments
Research reported in this publication was supported by a Genome Technology Access Center at the McDonnell Genome Institute (GTAC@MGI) Collaboration Pilot grant. Funding for this grant came from GTAC@MGI, the Dean of the School of Medicine, the Department of Arts and Sciences, the Institute of Clinical and Translational Sciences at the Washington University School of Medicine, and Illumina Inc.
Research reported in this publication was supported by National Cancer Institute of the National Institutes of Health under award number R01CA211939-01A1. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Research reported in this publication was supported by the National Institute on Deafness and Other Communication Disorders within the National Institutes of Health, through the “Development of Clinician/Researchers in Academic ENT” training grant, award number T32DC000022. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Research reported in this publication was supported by the Washington University Institute of Clinical and Translational Sciences grant UL1TR002345 from the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official view of the NIH.
VCS is supported by Career Development Award Number IK2 CX001953 from the United States (U.S.) Department of Veterans Affairs Clinical Sciences R&D (CSRD) Service.”
Author contributions
B.M.W., P.Z., A.M., O.L.G., M.G., and J.P.Z. conceived of and designed the study. A.M., O.L.G., M.G., and J.P.Z. supervised the investigation. B.M.W., P.Z., and R.R. acquired the data. B.M.W., Z.L.S., S.R.A., O.L.G., and M.G. performed the analysis. B.M.W. and P.Z. wrote the manuscript, which was reviewed and edited by R.R., Z.L.S., A.M., D.N.H., V.C.S., W.L.T., D.A., M.G., O.L.G., and J.P.Z.
Declaration of interests
J.P.Z. is a founder and equity shareholder in Droplet Biosciences, an equity shareholder in Summit Biolabs, a consultant for Merck, and receives clinical trial funding from Merck. R.J.R. receives licensing royalties from Washington University for technology licensed to Droplet biosciences. D.A. serves on the advisory boards of Pfizer, Celgene/BMS, Eli Lilly, Merck, and Cue Biopharma. He has received research funding from Aduro, Eli Lilly, Merck, Pfizer, Celgene/BMS, Novartis, Astra Zeneca, Atara Bio, Blueprint Medicine, Celldex, Kura, Exelixis, Innate, Sensei, Matrix BioMed, Hookipa, Co-Factor, Cue Biopharma, Medimmune, and Shanghai Denovo.
Published: May 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104216.
Supplemental information
7
(A) MuSiC Significantly Mutated Genes; (B) MuSiC Significance Compared with Prior Publications; (C) MUFFINN Probabalistic Scores; (D) GISTIC Amplifications; (E) GISTIC Deletions; (F) Comparison of Mutational Frequency – Recurrent HPV(+) Mutations; (G) Comparison of Mutational Frequency – TCGA HPV(−) Mutations.
(A – D) Genes and ORA Results for Figure 4 column clusters 1–4, respectively; (E) Survival Analysis – Cox Proportional Hazards Model of Disease-Free Survival.
Data and code availability
Whole exome sequencing data have been deposited at the National Institute of Health (NIH) database of Genotypes and Phenotypes (dbGaP) and are publicly available as of the date of publication. NanoString data have been deposited at the NIH Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
No original code was written in preparation of this publication. Existing R packages used in this work are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ainscough B.J., Barnell E.K., Ronning P., Campbell K.M., Wagner A.H., Fehniger T.A., Dunn G.P., Uppaluri R., Govindan R., Rohan T.E., et al. A deep learning approach to automate refinement of somatic variant calling from cancer sequencing data. Nat. Genet. 2018;50:1735–1743. doi: 10.1038/s41588-018-0257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.L., et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L.B., Ju Y.S., Haase K., Van Loo P., Martincorena I., Nik-Zainal S., Totoki Y., Fujimoto A., Nakagawa H., Shibata T., et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618–622. doi: 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhattabi N., Todd I., Negm O., Tighe P.J., Fairclough L.C. Tobacco smoke and nicotine suppress expression of activating signaling molecules in human dendritic cells. Toxicol. Lett. 2018;299:40–46. doi: 10.1016/j.toxlet.2018.09.002. [DOI] [PubMed] [Google Scholar]
- Amin D., Richa T., Mollaee M., Zhan T., Tassone P., Johnson J., Luginbuhl A., Cognetti D., Martinez-Outschoorn U., Stapp R., et al. Metformin effects on FOXP3+ and CD8+ T cell infiltrates of head and neck squamous cell carcinoma. Laryngoscope. 2020;130:E490–E498. doi: 10.1002/lary.28336. [DOI] [PubMed] [Google Scholar]
- Ang K.K., Harris J., Wheeler R., Weber R., Rosenthal D.I., Nguyen-Tân P.F., Westra W.H., Chung C.H., Jordan R.C., Lu C., et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010;363:24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J., et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers M., Lunceford J., Nebozhyn M., Murphy E., Loboda A., Kaufman D.R., Albright A., Cheng J.D., Kang S.P., Shankaran V., et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansbach C.E., Bétous R., Lovejoy C.A., Glick G.G., Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009;23:2405–2414. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnell E.K., Ronning P., Campbell K.M., Krysiak K., Ainscough B.J., Sheta L.M., Pema S.P., Schmidt A.D., Richters M., Cotto K.C., et al. Standard operating procedure for somatic variant refinement of sequencing data with paired tumor and normal samples. Genet. Med. 2019;21:972–981. doi: 10.1038/s41436-018-0278-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention Cancers Associated with Human Papillomavirus, United States—2012–2016, USCS Data Brief, no 10 [Preprint] 2019. https://www.cdc.gov/cancer/uscs/about/data-briefs/no10-hpv-assoc-cancers-UnitedStates-2012-2016.htm
- Chaturvedi A.K., Engels E.A., Pfeiffer R.M., Hernandez B.Y., Xiao W., Kim E., Jiang B., Goodman M.T., Sibug-Saber M., Cozen W., et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011;29:4294–4301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.Y., Massa S., Mazul A.L., Kallogjeri D., Yaeger L., Jackson R.S., Zevallos J., Pipkorn P. The association of smoking and outcomes in HPV-positive oropharyngeal cancer: a systematic review. Am. J. Otolaryngol. 2020;41:102592. doi: 10.1016/j.amjoto.2020.102592. [DOI] [PubMed] [Google Scholar]
- Cho A., Shim J.E., Kim E., Supek F., Lehner B., Lee I. MUFFINN: cancer gene discovery via network analysis of somatic mutation data. Genome Biol. 2016;17:129. doi: 10.1186/s13059-016-0989-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu R., Mogg R., Ayers M., Albright A., Murphy E., Yearley J., Sher X., Liu X.Q., Lu H., Nebozhyn M., et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362 doi: 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dees N.D., Zhang Q., Kandoth C., Wendl M.C., Schierding W., Koboldt D.C., Mooney T.B., Callaway M.B., Dooling D., Mardis E.R., et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res. 2012;22:1589–1598. doi: 10.1101/gr.134635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrichard A., Kuo F., Chowell D., Lee K.W., Riaz N., Wong R.J., Chan T.A., Morris L.G.T. Tobacco smoking-associated alterations in the immune microenvironment of squamous cell carcinomas. J. Natl. Cancer Inst. 2018;110:1386–1392. doi: 10.1093/jnci/djy060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk A.P., Meijssen M.A., Brouwer A.J., Hop W.C., van Bergeijk J.D., Feyerabend C., Wilson J.H., Zijlstra F.J. Transdermal nicotine inhibits interleukin 2 synthesis by mononuclear cells derived from healthy volunteers. Eur. J. Clin. Invest. 1998;28:664–671. doi: 10.1046/j.1365-2362.1998.00344.x. [DOI] [PubMed] [Google Scholar]
- D’Urso C.M., Wang Z.G., Cao Y., Tatake R., Zeff R.A., Ferrone S. Lack of HLA class I antigen expression by cultured melanoma cells FO-1 due to a defect in B2m gene expression. J. Clin. Invest. 1991;87:284–292. doi: 10.1172/JCI114984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhalawani H., Mohamed A.S.R., Elgohari B., Lin T.A., Sikora A.G., Lai S.Y., Abusaif A., Phan J., Morrison W.H., Gunn G.B., et al. Tobacco exposure as a major modifier of oncologic outcomes in human papillomavirus (HPV) associated oropharyngeal squamous cell carcinoma. BMC Cancer. 2020;20:912. doi: 10.1186/s12885-020-07427-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhry C., Zhang Q., Nguyen-Tan P.F., Rosenthal D., El-Naggar A., Garden A.S., Soulieres D., Trotti A., Avizonis V., Ridge J.A., et al. Human papillomavirus and overall survival after progression of oropharyngeal squamous cell carcinoma. J. Clin. Oncol. 2014;32:3365–3373. doi: 10.1200/JCO.2014.55.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhry C., Gillison M.L., D'Souza G. Tobacco use and oral HPV-16 infection. JAMA. 2014;312:1465–1467. doi: 10.1001/jama.2014.13183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., Kong Y., Barnes P.F., Huang F.F., Klucar P., Wang X., Samten B., Sengupta M., Machona B., Donis R., et al. Exposure to cigarette smoke inhibits the pulmonary T-cell response to influenza virus and Mycobacterium tuberculosis. Infect Immun. 2011;79:229–237. doi: 10.1128/IAI.00709-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy J.P., Bertolus C., Michallet M.C., Deneuve S., Incitti R., Bendriss-Vermare N., Albaret M.A., Ortiz-Cuaran S., Thomas E., Colombe A., et al. The immune microenvironment of HPV-negative oral squamous cell carcinoma from never-smokers and never-drinkers patients suggests higher clinical benefit of Ido1 and PD1/PD-L1 blockade. Ann. Oncol. 2017;28:1934–1941. doi: 10.1093/annonc/mdx210. [DOI] [PubMed] [Google Scholar]
- Fujiyoshi K., Chen Y., Haruki K., Ugai T., Kishikawa J., Hamada T., Liu L., Arima K., Borowsky J., Väyrynen J.P., et al. Smoking status at diagnosis and colorectal cancer prognosis according to tumor lymphocytic reaction. JNCI Cancer Spectr. 2020;4:pkaa040. doi: 10.1093/jncics/pkaa040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y., Savage S.M., Johnson L.J., Seagrave J., Sopori M.L. Effects of nicotine on the immune response. I. Chronic exposure to nicotine impairs antigen receptor-mediated signal transduction in lymphocytes. Toxicol. Appl. Pharmacol. 1995;135:268–278. doi: 10.1006/taap.1995.1233. [DOI] [PubMed] [Google Scholar]
- Geng Y., Savage S.M., Razani-Boroujerdi S., Sopori M.L. Effects of nicotine on the immune response. II. Chronic nicotine treatment induces T cell anergy. J. Immunol. 1996;156:2384–2390. [PubMed] [Google Scholar]
- Gillison M.L., Zhang Q., Jordan R., Xiao W., Westra W.H., Trotti A., Spencer S., Harris J., Chung C.H., Ang K.K. Tobacco smoking and increased risk of death and progression for patients with p16-positive and p16-negative oropharyngeal cancer. J. Clin. Oncol. 2012;30:2102–2111. doi: 10.1200/JCO.2011.38.4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillison M.L., Akagi K., Xiao W., Jiang B., Pickard R.K., Li J., Symer D.E. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res. 2018;29:1–17. doi: 10.1101/gr.241141.118. [Preprint] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliano A.R., Sedjo R.L., Roe D.J., Harri R., Baldwi S., Papenfuss M.R., Abrahamsen M., Inserra P. Clearance of oncogenic human papillomavirus (HPV) infection: effect of smoking (United States) Cancer Causes Control. 2002;13:839–846. doi: 10.1023/a:1020668232219. [DOI] [PubMed] [Google Scholar]
- Griffith M., Griffith O.L., Smith S.M., Ramu A., Callaway M.B., Brummett A.M., Kiwala M.J., Coffman A.C., Regier A.A., Oberkfell B.J., et al. Genome modeling System: a knowledge management platform for genomics. PLoS Comput. Biol. 2015;11:e1004274. doi: 10.1371/journal.pcbi.1004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafkamp H.C., Manni J.J., Haesevoets A., Voogd A.C., Schepers M., Bot F.J., Hopman A.H., Ramaekers F.C., Speel E.J. Marked differences in survival rate between smokers and nonsmokers with HPV 16-associated tonsillar carcinomas. Int. J. Cancer. 2008;122:2656–2664. doi: 10.1002/ijc.23458. [DOI] [PubMed] [Google Scholar]
- Hamada T., Nowak J.A., Masugi Y., Drew D.A., Song M., Cao Y., Kosumi K., Mima K., Twombly T.S., Liu L., et al. Smoking and risk of colorectal cancer sub-classified by tumor-infiltrating T cells. J. Natl. Cancer Inst. 2018;111:42–51. doi: 10.1093/jnci/djy137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J., Shi F.D., Abdelwahab M., Shi S.X., Simard A., Whiteaker P., Lukas R., Zhou Q. Nicotinic receptor β2 determines NK cell-dependent metastasis in a murine model of metastatic lung cancer. PLoS One. 2013;8:e57495. doi: 10.1371/journal.pone.0057495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge G., Nairn J., Holmes M., Reynolds P.N., Hodge S. Increased intracellular T helper 1 proinflammatory cytokine production in peripheral blood, bronchoalveolar lavage and intraepithelial T cells of COPD subjects. Clin. Exp. Immunol. 2007;150:22–29. doi: 10.1111/j.1365-2249.2007.03451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesia J.V., Slebos R.J., Martin-Gomez L., Wang X., Teer J.K., Tan A.C., Chung C.H. Effects of tobacco smoking on the tumor immune microenvironment in head and neck squamous cell carcinoma. Clin. Cancer Res. 2020;26:1474–1485. doi: 10.1158/1078-0432.CCR-19-1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung A.C., Guihard S., Krugell S., Ledrappier S., Brochot A., Dalstein V., Job S., de Reynies A., Noël G., Wasylyk B., et al. CD8-alpha T-cell infiltration in human papillomavirus-related oropharyngeal carcinoma correlates with improved patient prognosis. Int. J. Cancer. 2013;132:E26–E36. doi: 10.1002/ijc.27776. [DOI] [PubMed] [Google Scholar]
- Kalra R., Singh S.P., Savage S.M., Finch G.L., Sopori M.L. Effects of cigarette smoke on immune response: chronic exposure to cigarette smoke impairs antigen-mediated signaling in T cells and depletes IP3-sensitive Ca(2+) stores. J. Pharmacol. Exp. Ther. 2000;293:166–171. [PubMed] [Google Scholar]
- Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemnade J.O., Elhalawani H., Castro P., Yu J., Lai S., Ittmann M., Mohamed A.S.R., Lai S.Y., Fuller C.D., Sikora A.G., Sandulache V.C. CD8 infiltration is associated with disease control and tobacco exposure in intermediate-risk oropharyngeal cancer. Sci. Rep. 2020;10:243. doi: 10.1038/s41598-019-57111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D.C., Larson D.E., Wilson R.K. Using VarScan 2 for germline variant calling and somatic mutation detection. Curr. Protoc. Bioinformatics. 2013;44:15.4.1–15.4.17. doi: 10.1002/0471250953.bi1504s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiol J., Schroeder J., Jamieson D.J., Marshall S.W., Duerr A., Heilig C.M., Shah K.V., Klein R.S., Cu-Uvin S., Schuman P., et al. Smoking and time to clearance of human papillomavirus infection in HIV-seropositive and HIV-seronegative women. Am. J. Epidemiol. 2006;164:176–183. doi: 10.1093/aje/kwj165. [DOI] [PubMed] [Google Scholar]
- Lawrence M.S., Sougnez C., Lichtenstein L., Cibulskis K., Lander E., Gabriel S.B., Getz G., Ally A., Balasundaram M., Birol I., Bowlby R. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Wang J., Jaehnig E.J., Shi Z., Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47:W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B.M., Wang H., D'Souza G., Zhang Z., Fakhry C., Joseph A.W., Drake V.E., Sanguineti G., Westra W.H., Pai S.I. Long-term prognosis and risk factors among patients with HPV-associated oropharyngeal squamous cell carcinoma. Cancer. 2013;119:3462–3471. doi: 10.1002/cncr.28250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., Arimilli S., Savage E., Prasad G.L. Cigarette smoke preparations, not moist snuff, impair expression of genes involved in immune signaling and cytolytic functions. Sci. Rep. 2019;9:13390. doi: 10.1038/s41598-019-48822-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L.M., Zavitz C.C., Chen B., Kianpour S., Wan Y., Stämpfli M.R. Cigarette smoke impairs NK cell-dependent tumor immune surveillance. J. Immunol. 2007;178:936–943. doi: 10.4049/jimmunol.178.2.936. [DOI] [PubMed] [Google Scholar]
- Modestou M.A., Manzel L.J., El-Mahdy S., Look D.C. Inhibition of IFN-gamma-dependent antiviral airway epithelial defense by cigarette smoke. Respir. Res. 2010;11:64. doi: 10.1186/1465-9921-11-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal R., Şenbabaoğlu Y., Desrichard A., Havel J.J., Dalin M.G., Riaz N., Lee K.W., Ganly I., Hakimi A.A., Chan T.A., Morris L.G. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight. 2016;1:e89829. doi: 10.1172/jci.insight.89829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson L., Lechner M., Loewenbein S., Mohammed H., Davies-Husband C., Fenton T., Sterling J. CD8+ T cell response to human papillomavirus 16 E7 is able to predict survival outcome in oropharyngeal cancer. Eur J Cancer. 2016;67:141–151. doi: 10.1016/j.ejca.2016.08.012. [DOI] [PubMed] [Google Scholar]
- Maxwell J.H., Kumar B., Feng F.Y., Worden F.P., Lee J.S., Eisbruch A., Wolf G.T., Prince M.E., Moyer J.S., Teknos T.N., et al. Tobacco use in human papillomavirus-positive advanced oropharynx cancer patients related to increased risk of distant metastases and tumor recurrence. Clin. Cancer Res. 2010;16:1226–1235. doi: 10.1158/1078-0432.CCR-09-2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A., Flicek P., Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian M.F., Stämpfli M.R., Mossman K.L., Ashkar A.A. Cigarette smoke attenuation of poly I:C-induced innate antiviral responses in human PBMC is mainly due to inhibition of IFN-beta production. Mol. Immunol. 2009;46:821–829. doi: 10.1016/j.molimm.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Mian M.F., Pek E.A., Mossman K.L., Stämpfli M.R., Ashkar A.A. Exposure to cigarette smoke suppresses IL-15 generation and its regulatory NK cell functions in poly I:C-augmented human PBMCs. Mol. Immunol. 2009;46:3108–3116. doi: 10.1016/j.molimm.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Rosenthal R., McGranahan N., Herrero J., Taylor B.S., Swanton C. deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31. doi: 10.1186/s13059-016-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders C.T., Wong W.S., Swamy S., Becq J., Murray L.J., Cheetham R.K. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- Schierl M., Patel D., Ding W., Kochhar A., Adhami K., Zhou X.K., Granstein R.D. Tobacco smoke-induced immunologic changes may contribute to oral carcinogenesis. J. Invest. Med. : official Publ. Am. Fed. Clin. Res. 2014;62:316–323. doi: 10.2310/JIM.0000000000000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiels M.S., Katki H.A., Freedman N.D., Purdue M.P., Wentzensen N., Trabert B., Kitahara C.M., Furr M., Li Y., Kemp T.J., et al. Cigarette smoking and variations in systemic immune and inflammation markers. J. Natl. Cancer Inst. 2014;106 doi: 10.1093/jnci/dju294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simen-Kapeu A., Kataja V., Yliskoski M., Syrjänen K., Dillner J., Koskela P., Paavonen J., Lehtinen M. Smoking impairs human papillomavirus (HPV) type 16 and 18 capsids antibody response following natural HPV infection. Scand. J. Infect Dis. 2008;40:745–751. doi: 10.1080/00365540801995360. [DOI] [PubMed] [Google Scholar]
- Sinha P., Karadaghy O.A., Doering M.M., Tuuli M.G., Jackson R.S., Haughey B.H. Survival for HPV-positive oropharyngeal squamous cell carcinoma with surgical versus non-surgical treatment approach: a systematic review and meta-analysis. Oral Oncol. 2018;86:121–131. doi: 10.1016/j.oraloncology.2018.09.018. [DOI] [PubMed] [Google Scholar]
- Skidmore Z.L., Wagner A.H., Lesurf R., Campbell K.M., Kunisaki J., Griffith O.L., Griffith M. GenVisR: genomic visualizations in R. Bioinformatics. 2016;32:3012–3014. doi: 10.1093/bioinformatics/btw325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S., Gajewski T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer. 2018;18:139–147. doi: 10.1038/nrc.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talevich E., Shain A.H., Botton T., Bastian B.C. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. Plos Comput. Biol. 2016;12:e1004873. doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassallo R., Tamada K., Lau J.S., Kroening P.R., Chen L. Cigarette smoke extract suppresses human dendritic cell function leading to preferential induction of Th-2 priming. J. Immunol. 2005;175:2684–2691. doi: 10.4049/jimmunol.175.4.2684. [DOI] [PubMed] [Google Scholar]
- Wahle B., Zevallos J. Transoral robotic surgery and de-escalation of cancer treatment, otolaryngologic clinics of North America. Otolaryngol. Clin. North Am. 2020;53:981–994. doi: 10.1016/j.otc.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K., Schulz M.H., Long Q., Apweiler R., Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
7
(A) MuSiC Significantly Mutated Genes; (B) MuSiC Significance Compared with Prior Publications; (C) MUFFINN Probabalistic Scores; (D) GISTIC Amplifications; (E) GISTIC Deletions; (F) Comparison of Mutational Frequency – Recurrent HPV(+) Mutations; (G) Comparison of Mutational Frequency – TCGA HPV(−) Mutations.
(A – D) Genes and ORA Results for Figure 4 column clusters 1–4, respectively; (E) Survival Analysis – Cox Proportional Hazards Model of Disease-Free Survival.
Data Availability Statement
Whole exome sequencing data have been deposited at the National Institute of Health (NIH) database of Genotypes and Phenotypes (dbGaP) and are publicly available as of the date of publication. NanoString data have been deposited at the NIH Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
No original code was written in preparation of this publication. Existing R packages used in this work are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.