

Abstract
The actomyosin cytoskeleton serves as a key regulator of the integrity and remodeling of epithelial barriers by controlling assembly and functions of intercellular junctions and cell‐matrix adhesions. Although biochemical mechanisms that regulate the activity of non‐muscle myosin II (NM‐II) in epithelial cells have been extensively investigated, little is known about assembly of the contractile myosin structures at the epithelial adhesion sites. UNC‐45A is a cytoskeletal chaperone that is essential for proper folding of NM‐II heavy chains and myofilament assembly. We found abundant expression of UNC‐45A in human intestinal epithelial cell (IEC) lines and in the epithelial layer of the normal human colon. Interestingly, protein level of UNC‐45A was decreased in colonic epithelium of patients with ulcerative colitis. CRISPR/Cas9‐mediated knock‐out of UNC‐45A in HT‐29cf8 and SK‐CO15 IEC disrupted epithelial barrier integrity, impaired assembly of epithelial adherence and tight junctions and attenuated cell migration. Consistently, decreased UNC‐45 expression increased permeability of the Drosophila gut in vivo. The mechanisms underlying barrier disruptive and anti‐migratory effects of UNC‐45A depletion involved disorganization of the actomyosin bundles at epithelial junctions and the migrating cell edge. Loss of UNC‐45A also decreased contractile forces at apical junctions and matrix adhesions. Expression of deletion mutants revealed roles for the myosin binding domain of UNC‐45A in controlling IEC junctions and motility. Our findings uncover a novel mechanism that regulates integrity and restitution of the intestinal epithelial barrier, which may be impaired during mucosal inflammation.
Keywords: adherens junctions, cell migration, epithelial barriers, myosins, tight junctions
Abbreviations
- AJ
adherens junctions
- CD
Crohn's disease
- ECM
extracellular matrix
- FA
focal adhesion
- FM‐AFM
frequency‐modulated acoustic atomic force microscopy
- FRET
Forster resonance energy transfer
- IBD
inflammatory bowel disease
- IEC
intestinal epithelial cell
- MTT
(3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide)
- NM‐II
non‐muscle myosin II
- PLA
proximity ligation assay
- TEER
transepithelial electrical resistance
- TJ
tight junctions
- TPR
tetratricopeptide repeat
- UC
ulcerative colitis
- UCS
UNC‐45/Cro1/She4p
1. INTRODUCTION
Formation of intercellular junctions represents a key molecular event that is required for epithelial cell differentiation, apico‐basal polarization and establishment of the paracellular barrier. 1 It is also essential for tissue morphogenesis by enabling the orchestrated movement, expansion and folding of epithelial sheets. 2 , 3 Although cell–cell engagement results in the assembly of several adhesive structures, the apically located tight junctions (TJ) and adherens junctions (AJ) are arguably the most important regulators of epithelial barriers and homeostasis. 4 , 5 , 6 , 7 TJ and AJ are elaborate multiprotein complexes that include transmembrane adhesive proteins connected to various intracellular scaffolds. Transmembrane AJ (e.g., E‐cadherin, nectins) and TJ (claudins, occludin, junctional adhesion molecule A, etc.) proteins at the opposing plasma membranes mediate cell–cell adhesion and epithelial barrier formation. 4 , 5 , 6 , 7 The intracellular scaffolds that include members of a ‘zonula occludens’ (ZO) protein family at TJ and different catenin proteins at AJ, orchestrate assembly, clustering and remodeling of transmembrane adhesive modules of epithelial junctions. 4 , 5 , 6 , 7
AJ and TJ functions in regulating epithelial barriers and tissue morphogenesis depend on their ability to sense and transduce mechanical forces. 8 , 9 Such ability is determined by coupling of junctional complexes to the cortical actomyosin cytoskeleton. In simple differentiated epithelia, the most noticeable cytoskeletal structure is a circumferential F‐actin belt that is directly associated with AJ and TJ. 10 , 11 This belt is enriched with a key actin motor protein, non‐muscle myosin II (NM‐II). It has been firmly established that NM‐II activity mediates both passive tension, required for maintenance of epithelial barrier integrity and contractile forces that mediate junctional remodeling (disassembly and reassembly). 8 , 9 , 12
Superresolution microscopy imaging revealed highly ordered periodic distribution of NM‐II clusters in the perijunctional F‐actin belt of epithelial tissues that resembles the sarcomeric organization of actomyosin units in muscle cells. 10 , 13 Although such high‐order organization is likely to be important for the spatiotemporal NM‐II interactions with actin filaments and force generation, little is known about the mechanisms regulating the ordered, myofibril‐like assembly of contractile NM‐II units in the perijunctional actomyosin belt. Incorporation of myosin II molecules into myofibrils is known to be orchestrated with the folding of the myosin motor domain and requires cytosolic molecular chaperones. 14 UNC‐45 belongs to the evolutionary conserved UCS (UNC‐45/Cro1/She4p) protein family, and is the most studied myosin chaperone regulating a variety of actomyosin driven processes in high eukaryotes. 14 , 15 Vertebrate organisms express two different paralogs, UNC‐45A and UNC‐45B, which have ~55% of the sequence identity. Whereas UNC‐45A is ubiquitously expressed in different tissues, UNC‐45B is mainly expressed in muscle cells. 16 UNC‐45 contains three different domains: the N‐terminal tetratricopeptide repeat (TPR) domain, the central region and the C‐terminal USC domain. 14 , 15 , 17 The USC domain directly binds to myosin II motor and regulates myosin folding. 18 , 19 , 20 , 21 The TPR domain interacts with heat shock protein 90, which serves as co‐chaperones assisting UNC‐45‐mediated myosin folding. 19 , 20 , 22 Studies in Caenorhabditis elegans revealed that intermolecular interactions between the TPR and USC domains are essential for formation of UNC‐45 oligomers. 20 , 21 There, oligomerization of this chaperone into linear molecular arrays promotes the assembly of folded myosin II molecules into highly ordered myofilaments. Notably, UNC‐45A has been recently shown to bind and destabilize microtubules. 23 , 24 , 25 Such microtubule‐destabilizing activity requires the TPR domain but appears to be independent of myosin binding by UNC‐45A. 23
The majority of functional studies of UNC‐45 were conducted in model organisms such as C. elegans and Drosophila and focused on muscle development and contractility. 14 , 15 , 17 Little is known about the functions of this chaperone in mammalian non‐muscle cells. Previously described phenotypes of UNC‐45A depletion in these systems include attenuation of growth cone development in neurons, 26 as well as modulation of proliferation and motility of different cancer cells. 27 , 28 , 29 , 30 However, UNC‐45A involvement in the regulation of epithelial barrier integrity and remodeling has remained largely unknown.
We are filling these crucial knowledge gaps by reporting that UNC‐45A acts as a positive regulator of barrier properties both in the human intestinal epithelial monolayers as well as in the Drosophila gut. Such barrier enhancing activity of this chaperone involves controlling the assembly of apical junctions and the perijunctional actomyosin cytoskeleton. We also observed that UNC‐45A regulates the repair of injured intestinal epithelium by modulating cell‐matrix adhesions and promoting wound healing. Together, our findings signify this myosin chaperone as a novel regulator of the establishment and remodeling of epithelial barriers.
2. MATERIALS AND METHODS
2.1. Antibodies and other reagents
The following monoclonal (mAb) and polyclonal (pAb) antibodies were used to detect apical junction, cytoskeletal and matrix adhesion proteins: anti‐E‐cadherin (BD Biosciences, San Jose, CA, Cat# 610181, RRID:AB_397580) and β‐catenin mAbs (BD Biosciences, Cat# 610154, RRID:AB_397555); anti‐cleaved PARP (Cell Signaling Technology, Beverly, MA, Cat# 9541, RRID:AB_331426), cleaved caspase3 (Cell Signaling Technology Cat# 9664, RRID:AB_2070042), and NM‐IIC pAbs (Cell Signaling Technology Cat# 3405, RRID:AB_2148340); anti‐ZO‐1 (Thermo Fisher Scientific, Waltham, MA, Cat# 40‐2200, RRID:AB_2533456) and phosphorylated paxillin pAbs (Thermo Fisher Scientific Cat# 44‐722G, RRID:AB_2533733); anti‐occludin (Proteintech, Rosemont, IL, Cat# 13409‐1‐AP, RRID:AB_2156308), GFP (Proteintech Cat# 50430‐2‐AP, RRID:AB_11042881), and anti‐UNC‐45A pAbs (Proteintech Cat# 19564‐1‐AP, RRID:AB_10644335); anti‐E‐cadherin pAb (R & D System, Minneapolis, MN, Cat# AF748, RRID:AB_355568); anti‐NM‐IIA pAb (Biolegend, San Diego, CA, Cat# 909801, RRID:AB_2565100); anti‐α‐tubulin mAb (Millipore‐Sigma, San Louis, MO, Cat# T6199, RRID:AB_477583); anti‐NM‐IIA mAb (Abnova, Walnut, CA, Cat# H00004627‐M03, RRID:AB_606627). Alexa Fluor‐488‐conjugated donkey anti‐rabbit (Cat# A‐21206, RRID:AB_2535792), Alexa Fluor‐555‐conjugated donkey anti‐mouse (Cat# A‐31570, RRID:AB_2536180) and Alexa Fluor‐488‐conjugated donkey anti‐goat (Cat# A‐11055, RRID:AB_2534102) secondary antibodies were obtained from Thermo Fisher Scientific. Horseradish peroxidase‐conjugated goat anti‐rabbit (Cat# 170‐6515, RRID:AB_11125142) and anti‐mouse (Cat# 170‐6516, RRID:AB_11125547) secondary antibodies were obtained from Bio‐Rad Laboratories (Hercules, CA). Antibodies used for immunoblotting of Drosophila lysates included β‐tubulin (Cell Signaling Technology Cat# 2146, RRID:AB_2210545) and Unc‐45 (gift from Dr. Sanford Bernstein, San Diego State University). 31 Others reagents were obtained from Millipore‐Sigma.
2.2. Drosophila Stocks
Standard laboratory strains w1118 and unc‐4503692 /+ were obtained from Bloomington Drosophila Stock Center. unc‐45EY03043‐33 /+ was a generous gift from the Bernstein laboratory at San Diego State University.
2.3. Cell culture and calcium switch
SK‐CO15 human colonic epithelial cells 32 were a gift from Dr. Enrique Rodriguez‐Boulan (Weill Medical College of Cornell University, New York, NY). HT‐29cf8, a well‐differentiated clone of HT‐29 cells 33 , 34 was provided by Dr. Judith M. Ball (College of Veterinary and Biomedical Sciences, Texas A&M University, College Station, TX). Caco‐2 (Cat# HTB‐37, RRID:CVCL_0025), T84 (Cat# CCL‐248, RRID:CVCL_0555), Colo320 (Cat# CCL‐220, RRID:CVCL_0219) and SW620 (Cat# CCL‐227, RRID:CVCL_0547) cell lines were obtained from the American Type Culture Collection (Manassas, VA). HT‐29cf8 and SK‐CO15 cell lines were authenticated by the small tandem repeat profiling by the LabCorp (Burlington, NC). All cultured cells were mycoplasma free according to the mycoplasma PCR detection assay (PromoCell, Heidelberg, Germany). HT‐29cf8, and SK‐CO15 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS), HEPES, non‐essential amino acids, and penicillin‐streptomycin antibiotic. The cells were seeded on collagen‐coated Transwell filters (Corning Incorporated, Corning, NY), or glass coverslips for permeability measurements and immunolabeling experiments and on 6‐well plastic plates for other functional and biochemical studies. The calcium switch assay was performed as previously described. 35 , 36 , 37 Briefly, well‐differentiated SK‐CO15 cell monolayers were subjected to overnight extracellular calcium depletion by incubating in the Eagle's minimum essential medium for suspension culture (Thermo Fisher Scientific) supplemented with 5 µM CaCl2, 10 mM HEPES, and 10% dialyzed FBS, pH 7.4. To induce junctional reassembly, the cells were returned to a normal cell culture medium with high (~1.8 mM) calcium for the indicated time.
2.4. Primary human intestinal epithelial cells
Deidentified, full‐thickness, intestinal surgical resection specimens obtained from inflammatory bowel diseases (IBDs) patients with active Crohn's disease (CD), ulcerative colitis (UC) and non‐IBD controls (diverticulosis or non‐affected margins of resected colorectal cancer) were provided by the Biorepository Core of the Cleveland Digestive Diseases Research Core Center. Primary colonic epithelial cells were isolated based on a previously described protocol. 38 Briefly, resected samples were cut into approximately 1.5 cm long strips and washed with HEPES‐buffered Hanks' balanced salt solution without calcium and magnesium (HBSS−). Colonic epithelial cells were isolated by sequential incubation of the tissue strips in HBSS− containing 10 mM dithiothreitol for 30 min followed by incubation in HBSS− containing 1 mM EDTA (pH 7.4) under constant agitation for 3 h at room temperature. Detached epithelial cells were centrifuged for 10 min at 3000 rpm. Cells were washed once in ice‐cold HBSS−, resuspended into RIPA buffer and homogenized. The described studies conform to the US Federal policy for the protection of human subjects. The study was approved by the Institutional Review Board of Cleveland Clinic Foundation as a minimal risk investigation.
2.5. CRISPR‐Cas9 mediated knockout of UNC‐45A and generation of UNC‐45A overexpressing intestinal epithelial cell
A stable knockout of UNC‐45A in HT‐28cf8 and SK‐CO15 intestinal epithelial cell (IEC) was carried out using a CRISPR‐Cas9 technology. The single‐guide (sg) RNA sequences used for knocking out UNC‐45A in HT‐29cf8 cells were: sgRNA1, forward 5′‐CACCGTCTCTAGGGCTTGG CTCCGC‐3′ and reverse 5′‐ AAACGCGGAGCCAAGCCC TAGAGAC‐3′; and sgRNA2 forward 5′‐CACCGGGGGC GTCGCGTCCAGACCC‐3′ and reverse 5′‐AAACGGGTC TGGACGCGACGCCCCC‐3′. For UNC‐45A knockout in SK‐CO15 cells the forward 5′‐CACCGGCAGGCTTCTCA GAACCTGG‐3′ and reverse 5′‐ AAACCCAGGTTCTGAG AAGCCTGCC‐3′ sgRNAs were used. The single guide oligonucleotides were phosphorylated, annealed and cloned into the BsmBI site of a lentiCRISPR v2 vector (RRID:Addgene_52961) according to a published protocol. 39 , 40 Obtained constructs were verified by sequencing. Transfer plasmids possessing annealed sgRNAs were transformed into recombination‐deficient Stbl3 bacteria and amplified plasmids were isolated from the bacteria using Qiagen Midi prep plasmid isolation kit. Lentiviruses were produced by transfecting HEK‐293T cells with the transfer lentiCRISPR v2 plasmids and packaging plasmids pLTR‐G (RRID:Addgene_17532) and pCD/NL‐BH*DDD (RRID:Addgene_17531). Viral supernatants were collected 48 h and 72 h after transfection and used to infect IEC. After 24 h of the transduction, the lentivirus‐containing medium were replaced with fresh cell culture medium containing puromycin (10 µg/ml) and puromycin‐resistant cells were collected after 7‐day selection. After puromycin selection, single clone isolation was performed in HT‐28cf8 and SK‐CO15 cells by using a cell dilution protocol and visually screening 96‐wells plates for single cell identification. When colonies reach 70% confluence, cells were harvested and the UNC‐45A depletion was analyzed by immunoblotting.
SK‐CO15 cell lines were generated to stable express either full length UNC‐45A (UNC‐45A‐GFP), its N‐terminal truncated ΔTPR‐GFP (residues 147‐971), or C‐terminally truncated ΔUCS‐GFP (residues 1‐539). Cloning of the full length and deletion mutant UNC‐45A constructs into pEGFP‐N3 vector (Takara Bio, Inc, Kusatsu, Japan) has been previously described. 30 IEC were transfected using Lipofectamine 2000 (Thermo Fisher Scientific). 48 h after transfection, fresh medium containing G418 antibiotic (0.5 mg/ml) was added. After 7 days of antibiotic selection, GFP expressing cells were sorting using FACSAria™ fusion flow cytometer (BD Bioscience). Briefly, cultured cells were harvested and resuspended in PBS with 1% of FBS. After sorted cells were collected in DMEM with 10% FBS and the overexpression of full length and deletion mutant UNC‐45A was analyzed by immunoblotting.
2.6. Measurement of epithelial barrier permeability
Transepithelial electrical resistance (TEER) of HT‐29cf8, and SK‐CO15 IEC monolayers cultured on collagen‐coated Transwell filters (Corning Incorporated) was measured using an EVOM2 Volt Ohm Meter (World Precision Instruments, Sarasota, FL). The resistance of cell‐free collagen‐coated filters was subtracted from each experimental point. A transmonolayer dextran flux assay was performed as previously described. 41 , 42 IEC monolayers cultured on Transwell filters were apically exposed to 1 mg/ml of FITC‐labeled dextran (4000 Da) in HEPES‐buffered Hanks' balanced salt solution with calcium and magnesium (HBSS+). After 120 min of incubation, samples were collected from the lower chamber and FITC fluorescence intensity was measured using a SpectraMax M2 plate reader (Molecular Devices, San Jose, CA), at excitation and emission wavelengths 485 and 544 nm, respectively. The amount of FITC‐dextran translocated across the epithelial cell monolayer was calculated based on a calibration curve using Prism 9.10 software (GraphPad, La Jolla, CA).
2.7. Immunoblotting analysis and immunoprecipitation
IEC monolayers were scraped and homogenized using a Dounce homogenizer in RIPA buffer (20 mM Tris, 50 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1% sodium deoxycholate, 1% Triton X‐100 (TX‐100), and 0.1% SDS, pH 7.4) containing protease inhibitor cocktail and phosphatase inhibitor cocktails 2 and 3 (Millipore‐Sigma). 15 adult Drosophila males and females of different genotypes were harvested at 10 days post‐enclosure, crushed in high salt lysis buffer (300 mM NaCl, 50 mM Tris pH 7.5, 1 mM EDTA, 0.1%Triton, 10% glycerol, 1 mM dithiothreitol, and a complete mini EDTA‐free protease tablet), and incubated on ice for 15 min. The obtained total cell lysates were cleared by centrifugation (20 min at 14 000× g), diluted with 2x SDS sample loading buffer, and boiled. SDS‐polyacrylamide gel electrophoresis was conducted using a standard protocol with equal amounts of total protein (10 or 20 μg of protein from human cells or 30 µg of protein per genotype for Drosophila extracts) loaded per each lane. The separated proteins were transferred to nitrocellulose membranes and the membranes were blocked with 5% non‐fat milk. The blocked membranes were incubated overnight with primary antibodies, and then exposed to HRP‐conjugated secondary antibodies for 1 h. The labeled proteins were visualized using a standard enhanced chemiluminescence solution and X‐ray films. For immunoblotting of Drosophila proteins, bands were quantified from films, using a BioRad ChemiDocTM XRS+ Molecular Imager and Image LabTM software, with “Protein Gel” settings, using identically sized windows to calculate Absolute volumes, as directed by the manufacturer. All bands were first normalized to respective loading controls, prior to comparison between lanes.
Immunoprecipitation (IP) was performed as previously described. 43 Briefly, cells were homogenized in the IP buffer (50 mM 1,4‐piperrazinediethanesulfonic acid, 50 mM HEPES, 1 mM EDTA, 2 mM MgSO4, 1% TX‐100, and 0.5% Igepal, pH 7.0) containing protease inhibitor and phosphatase inhibitor cocktails 2 and 3. After centrifuged, supernatants were precleared with protein A‐sepharose beads (GenScript Biotech, Piscataway, NJ). Precleared supernatants were then incubated overnight at 4°C with 5 µg of anti‐NM‐IIA pAb or control rabbit immunoglobulin. Immunocomplexes were recovered by incubation with protein A‐sepharose beads for 2 h at 4°C. After three washes with IP buffer, beads were boiled with 2x SDS sample loading buffer and pelleted by centrifugation. 20 µl of supernatants were loaded into SDS‐polyacrylamide gel electrophoresis using a standard protocol as described above.
2.8. Immunofluorescence labeling, confocal microscopy, and image analysis
To visualize the structure of epithelial junctions and localization of UNC‐45A and NM‐II heavy chains, cultured IEC monolayers were fixed and permeabilized with 100% methanol for 20 min at −20°C. To visualize the actin cytoskeleton, cells were fixed with 4% PFA for 20 min and permeabilized with 0.5% Triton X‐100 for 5 min at room temperature. Fixed samples were blocked for 60 min in HBSS+ containing 1% bovine serum albumin, followed by a 60‐min incubation with primary antibodies. Then, samples were washed and incubated with Alexa‐Fluor‐488–conjugated donkey anti‐rabbit and Alexa‐Fluor‐555–conjugated donkey anti‐mouse secondary antibodies for 60 min, rinsed with blocking buffer, and mounted on slides with ProLong Antifade mounting reagent (Thermo Fisher Scientific). For the actin cytoskeleton labeling, fixed/permeabilized cells were incubated for 1 h with Alexa‐Fluor‐488‐conjugated phalloidin. Immunofluorescence labeled cell monolayers were imaged using a Leica SP8 confocal microscope (Wentzler, Germany). The Alexa Fluor 488 and 555 signals were acquired sequentially in frame‐interlace mode, to eliminate cross talk between channels.
For the quantification of the junctional lengths in the calcium switch experiments, four images were taken per slide and the total length of junctional labeling per image was measured by using ImageJ software (National Institute of Health, Bethesda, MD). To quantify thickness of the perijunctional myosin belt, four images were taken per slide and the width of 50 cell–cell contacts were measured by ImageJ and averaged to produce a single data point. Quantification of focal adhesions (FA) was performed by counting the number of dots on p‐paxillin images using ImageJ. Number of FA in three different images was averaged to produce a single data point.
2.9. Proximity ligation assay
Proximity ligation assay (PLA) was performed by using a DUOLINK kit (Millipore‐Sigma) according to the manufacturer's instructions. Briefly, IEC monolayers were fixed in ice‐cold methanol and incubated for 1 h with a DUOLINK blocking solution at 37°C. After two washes, anti‐UNC‐45A polyclonal and anti‐NM‐IIA monoclonal antibodies diluted in a DUOLINK antibody diluent were added and incubated for 1 h at 37°C. As a negative control, anti‐UNC‐45A antibody was added alone, without the anti‐NM‐IIA antibody. Next, the slides were washed and incubated for 1 h with secondary antibodies (DUOLINK anti‐rabbit PLA‐plus probe and DUOLINK anti‐mouse PLA‐minus probe). The labeling was completed by consequent incubation with a ligation solution for 30 min and an amplification solution for 100 min at 37°C. Labeled slides were mounted with the Prolong Antifade mounting medium with DAPI. Images were acquired using a Leica SP8 confocal microscope and processed using ImageJ. For quantification, four images per slide were taken and the total number of dots was measured by using ImageJ software and divided by the number of cells.
2.10. FRET biosensors
A previously developed and validated Forster resonance energy transfer (FRET)‐based tension biosensor 44 was used to measure mechanical tension applied to E‐cadherin complexes. This sensor was expressed in HT‐29cf8 cells using adenovirus. The adenovirus was constructed by subcloning the existing E‐cadherin tension sensor into the pShuttle‐CMV vector (RRID:Addgene_16403), followed by recombination into the pAdEasy1 vector (RRID:Addgene_16400). Adenovirus was prepared using standard protocols. 45
HT‐29cf8 cells expressing the E‐cadherin tension sensor were grown on Transwell inserts (Corning Incorporated). Inserts were cut out, flipped over, and living cells were imaged through glass bottom dishes using a plan‐apochromat 40x water immersion NA 1.1 objective lens on an inverted Zeiss LSM 710 laser scanning microscope (Carl Zeiss AG, Oberkochen, Germany). All the images were acquired at 458 nm excitation wavelength from an Argon laser source. Using the online‐unmixing mode, both mTFP1 (donor) and mEYFP (acceptor) channels were collected via spectral unmixing as previously described. 46 Intensity images were further processed and analyzed using a custom Python code, which involved background subtraction and removal of saturated pixels, as previously described. 46 The FRET index images were obtained by taking the ratio of the acceptor fluorophore channel (mEYFP) to the donor fluorophore channel (mTFP1), which was then multiplied with the binary image masks that outlined the cell‐cell junctions to inspect FRET pixels of interest. When comparing multiple groups, all pixels of interest were aggregated following which, an upper and lower bound for the intensity was chosen to exclude dim pixels.
2.11. Acoustic frequency‐modulated atomic force microscopy
Epithelial viscoelastic properties measurements in control and UNC‐45A‐depleted HT‐29cf8 epithelial cell monolayers by noncontact acoustic frequency modulation atomic force microscopy (FM‐AFM). 47 , 48 HT‐29cf8 cells were seeded on glass‐bottom petri dishes and cultured for 7–10 days to achieve monolayer confluency. FM‐AFM experiments were performed using a Bruker Bioscope Catalyst Atomic Force Microscope system (Bruker, Billerica, MA) mounted on an inverted Axiovert 200M optical microscope (Carl Zeiss AG) equipped with a Confocal Laser Scanning Microscope 510 Meta (Carl Zeiss AG), and using a 40x objective lens (0.95 NA, Plan‐Apochromat; Carl Zeiss AG). During experiments, cell monolayers were maintained at 37°C using a heating sample stage (Bruker). A modified AFM microcantilever with an attached 25 µm polystyrene bead (Novascan Technologies, Ames, IA) was used for all FM‐AFM measurements. Using the thermal tune fluctuations method built in the AFM system, the calibrated spring constant were between 0.6 and 1.4 N/m. Next, explants were placed on the AFM sample stage and tapping mode was selected. Then, the cantilever tune curve mode was engaged and the cantilever driving frequency was chosen to be the largest frequency peak near the cantilever natural resonance frequency (predetermined by the thermal tune method). After gently engaging the cell monolayer apical surface, the cantilever tune was launched, and the cantilever was positioned at 8 µm away from the epithelium. Then, a frequency sweep was recorded, and the cantilever phase lag corrected to be π/2. Next, the acoustically vibrating bead is moved from the 8 to 1 µm gap distance by 500 nm intervals. A frequency sweep is recorded for each interval. Supracellular apical epithelial tension and viscosity calculations were performed using a custom‐made MATLAB script (MathWorks, Natick, MA). The apical epithelial mechanics model based on lubrication theory for linearized Stokes flow used herein can be accessed for detailed description. 48
2.12. Scratch wound assay
IECs were plated into Culture‐Insert 3 Well in µ‐Dish (Ibidi USA, Fitchburg, WI) and allowed to grow to confluence. A three‐well silicone gasket was removed and the bottom of the dish was marked to define the position of the wound. The monolayers were supplied with fresh cell culture medium and images of a cell‐free area at the marked region were acquired at 0 and 24 h after the gasket removal using a Keyence BZ‐X710 microscope (Keyence, Osaka, Japan). The wound area of three different wounds was measures and the percentage of wound closure was calculated using Image J.
2.13. Boyden chamber migration assay
Boyden chamber migration assay was performed using Transwell 6.5‐mm membrane inserts with 8.0‐µm pores (Corning Incorporated). The membrane inserts were coated with 15 µg/cm2 of collagen I. Cells were detached from the plate using a TrypLE Express solution (Thermo Fisher Scientific), resuspended in serum‐free medium, and added to the Transwell upper chamber at the density of 5000 HT‐29cf8 cells and 6000 SK‐CO15 cells per chamber. A complete cell culture medium containing 10% FBS as a chemoattractant was added to the lower chamber and cells were allowed to migrate for 16 h at 37°C. Membrane inserts were fixed with methanol and non‐migrated cells were removed from the top of the filter using a cotton swab. The cells remained at the bottom of the filter were labeled with DAPI nuclear stain and images using the Keyence BZ‐X710 microscope. Three images per membrane insert were taken and number of DAPI‐positive cells in each image was counted using the Image J and averaged to yield a single data point.
2.14. Extracellular matrix adhesion assay
Cells were detached from the plate, counted with a T20 automated cell counter (Bio‐Rad), and resuspended in the complete medium. Ten thousand (10 000) HT‐29cf8 cells and 7500 SK‐CO15 cells were seeded per well in a 24‐well plate coated with rat collagen I and were allowed to adhere for 30 min at 37°C. After incubation, unattached cells were aspirated and the wells were gently washed with HBSS+ buffer. The attached cells were fixed with methanol and stained using a DIFF stain kit (Electron Microscopy Sciences, Hatfield, PA). Images of adherent cells were captured using the Keyence BZ‐X710 microscope. Three images were taken for each well and number of adhered cells in each image was counted using the Image J and averaged to yield a single data point.
2.15. Cell proliferation assay
Cell proliferation was examined by the MTT assay involving the conversion of the water‐soluble MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) into insoluble formazan. Cells were cultured on 98‐well plates, and at different times post‐plating, their medium was replaced with 100 µl of fresh culture medium containing 50 µg/ml of MTT. MTT added to 100 µl of medium alone was included as a negative control. Cells were incubated at 37°C for 4 h and the generated formazan was dissolved by adding 50 µl of DMSO with 10 min incubation at 37°C. The absorbance of formazan solution was measured using the SpectraMax M2 plate reader at 540 nm.
2.16. Collagen contraction assay
Collagen contraction assay was performed as previously described. 49 Briefly, IEC were detached from the plate, counted with a T20 automated cell counter, and resuspended in DMEM medium to have 6000 HT‐29cf8 cells or 5000 SK‐CO15 cells per 200 µl of cell suspension. The cell suspension (200 µl) was mixed with 400 µl of collagen I solution (3 mg/ml) and 7 µl of 1 M NaOH solution, and 500 µl of this mixture was added per well of a 24‐well plate. After cells/collagen mixture solidified during 30 min incubation at 37°C, 500 µl of cell culture medium was added and the gels were detached from the plastic surface by using a pipet tip. The floating collagen gels were cultured at 37°C in 5% CO2 for 4 days. Images of the gels were acquired using Epson V500 photo scanner and gel area was measured using ImageJ.
2.17. Drosophila UNC‐45 mutants and Smurf assay
Flies were aged on standard medium for 20 days prior to performing the Smurf assay. 25 flies per vial were used during the assay. 2.5% (wt/vol) Brilliant Blue FCF dye solution (Millipore‐Sigma) was prepared in 5% sucrose solution. Small pieces of kimwipes were saturated with dye in empty vials and 20‐day‐old flies were placed into the vials for 24 h. 200 µl of dye was used per vial. A Smurf fly was counted when total body dye coloration was observed after 24 h. A Leica S6 E Greenough stereomicroscope with 6.3:1 zoom was used to sort and identify Smurf flies. The average percentage of Smurf flies per vial was counted for a total of 5 vials per genotype. Statistical significance was calculated using the Mann–Whitney test.
2.18. Statistical analysis
All data are expressed as means ± standard error (SE) from at least three biological replicates, except for FM‐AFM experiments. FM‐AFM data are presented as mean ± standard deviation (SD) from three independent biological replicates. Statistical analysis was performed by using two‐tailed unpaired student t‐test to compare the results obtained with two experimental groups. When more than two samples were compared, a one‐way ANOVA with Bonferroni's post‐hoc test was used (to compare knockout with two different UNC‐45A sgRNAs; or overexpression of full‐length UNC‐45A and different deletion mutants; with the control). p Values < .05 were considered statistically significant. All statistical analysis was performed using GraphPad Prism 9.10 program.
3. RESULTS
3.1. Depletion of UNC‐45A impairs formation of the epithelial barrier and assembly of apical junctions
Immunoblotting analysis of a panel of IEC lines demonstrated abundant expression of UNC‐45A in well‐differentiated HT‐29cf8, SK‐CO15, T84 and Caco‐2 cells (Figure S1A). In confluent HT‐29cf8 and SK‐CO15 cell monolayers, this cytoskeletal chaperone accumulated at the epithelial cell apex in a close vicinity to apical junctions (Figure S1B,C, arrows). To gain insights into functional roles of UNC‐45A, we used CRISPR/Cas9‐mediated gene editing to downregulate expression of this chaperone in HT‐29cf8 and SK‐CO15 human colonic epithelial cells. In HT‐29cf8 cells, the two most efficient sgRNAs resulted in 94%–99% decrease in UNC‐45A protein expression after clonal selection (Figure 1A). In SK‐CO15 cells, we selected a UNC‐45A‐deficient clone with ~97% downregulation of the targeted protein (Figure 1B). In both IEC lines, loss of UNC‐45A did not alter expression of two major epithelial myosin II motors, NM‐IIA and NM‐IIC (Figure 1A).
FIGURE 1.
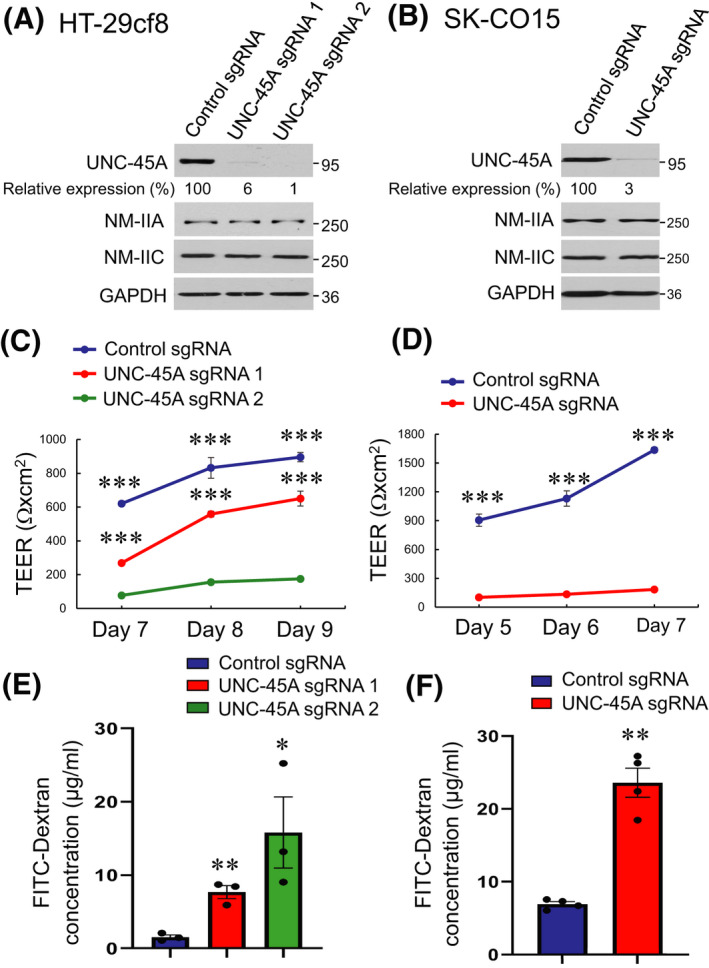
Loss of UNC‐45A expression increases paracellular permeability of intestinal epithelial cell monolayers. (A, B) Immunoblotting analysis of UNC‐45A and NM‐II isoform expression in HT‐29cf8 (A) and SK‐CO15 (B) human intestinal epithelial cells with CRISPR/Cas9 mediated knockout of UNC‐45A. (C, D) Transepithelial electrical resistance (TEER) of control and UNC‐45A‐depleted HT‐29cf8 (C) and SK‐CO15 (D) cell monolayers at different times post‐plating. (E, F) Transmonolayer flux of FITC‐dextran in control and UNC‐45A‐depleted IEC monolayers on day 9 (HT‐29cf8, E) and day 7 (SK‐CO15, F) post‐plating. Data are presented as means ± SE (n = 3 for HT‐29cf8 and n = 4 for SK‐CO15 cells); *p < .05, **p < .005, ***p < .0005 compared with the control sgRNA groups. (A–F) Data are representative of at least three independent experiments
Effects of UNC‐45A knockout on the development of the epithelial barrier were examined by measuring TEER and transmonolayer flux of FITC‐labeled dextran. UNC‐45A‐depleted HT‐29cf8 and SK‐CO15 cells developed significantly lower TEER (Figure 1C,D) and had higher dextran flux (Figure 1E,F) compared with the control cell lines, which indicates compromised barrier integrity. Such barrier dysfunction was accompanied by focal disruption of apical junctions that was observed by immunofluorescence labeling of specific markers of both AJ (E‐cadherin) and TJ (ZO‐1 and occludin) (Figure 2). While in control cell monolayers AJ and TJ proteins displayed a continuous ‘chicken wire’ labeling pattern (Figure 2A,B, arrows), this pattern was disrupted due to mislocalization of junctional proteins in UNC‐45A‐depleted IEC (Figure 2A,B, arrowheads). Our initial studies with pooled UNC‐45A‐depleted HT‐29cf8 and SK‐CO15 cells, demonstrated disruption of the epithelial barrier and AJ/TJ disassembly, similar to the effects observed in the selected clones of these cells (data not shown). Hence, the observed effects do not represent clone‐specific peculiarities of UNC‐45A depleted cells. Loss of UNC‐45A also resulted in altered cell morphology, manifested by transformation from the orthogonal to the elongated cell shape in HT‐29cf8 monolayers and increased cell size in SK‐CO15 monolayers (Figure 2A,B).
FIGURE 2.
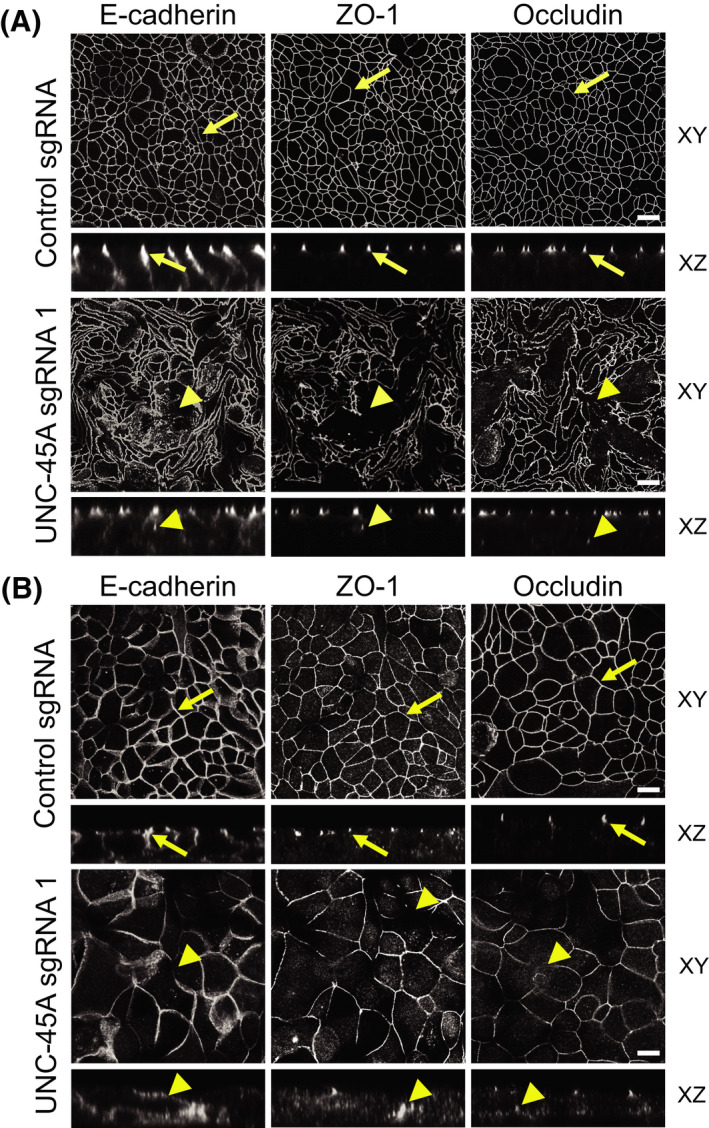
Downregulation of UNC‐45A expression impairs assembly of epithelial adherens and tight junctions. Confocal microscopy images of control and UNC‐45A‐depleted HT‐29cf8 (A) and SK‐CO15 (B) cell monolayers immunolabeled for different AJ (E‐cadherin) and TJ (ZO‐1, occludin) proteins. Arrows indicate intact AJ and TJ in control cell monolayers. Arrowheads point to junctional disassembly in UNC‐45A‐depleted cells. Scale bars, 20 µm. Images shown are representative of at least three independent experiments with multiple images taken per slide
3.2. Loss of UNC‐45A disrupts assembly of the perijunctional actomyosin cytoskeleton
Because assembly and barrier properties of epithelial junctions depend on their association with the actomyosin cytoskeleton, we rationalized that loss of UNC‐45A could compromise AJ/TJ integrity by impairing assembly of the perijunctional actomyosin belt. To test this hypothesis, we visualized junction‐associated actin filaments and NM‐II motors using fluorescence labeling and confocal microscopy. Consistent with previously published data, 35 , 36 control IEC monolayers displayed a prominent perijunctional F‐actin belt, enriched in both NM‐IIA and NM‐IIC (Figure 3A,B, arrows). In contrast, this F‐actin belt was either markedly distorted or poorly assembled in UNC‐45A knockout HT‐29cf8 and SK‐CO15 cells, respectively (Figure 3A,B, arrowheads). Furthermore, loss of UNC‐45A significantly diminished recruitment of both NM IIA and NM IIC to the circumferential F‐actin belt (Figure 3A,B, arrowheads).
FIGURE 3.
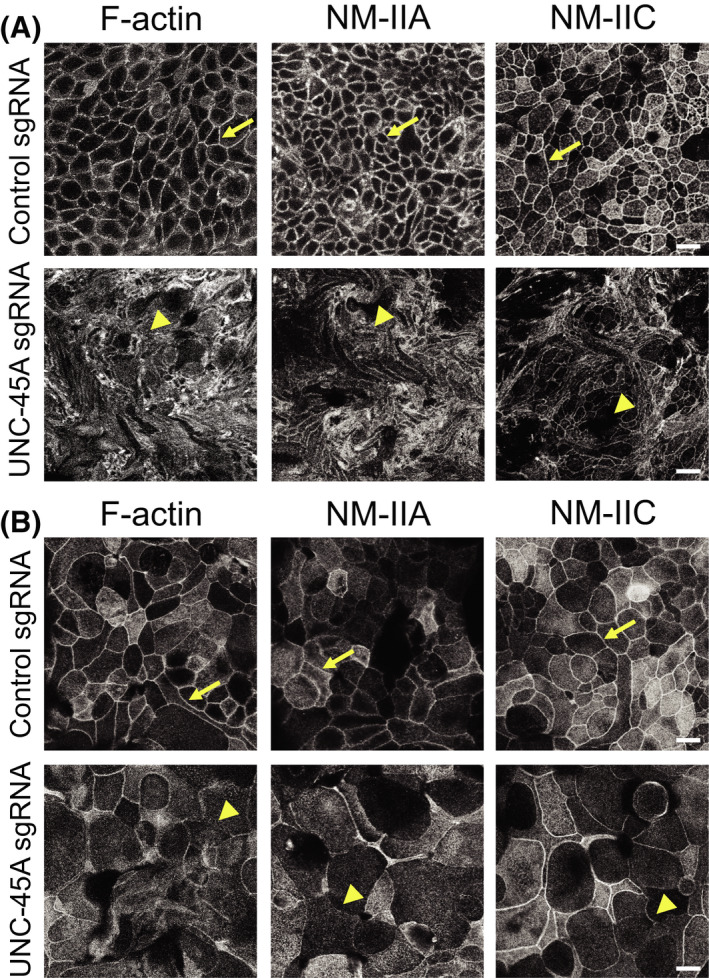
Loss of UNC‐45A expression impairs organization of the perijunctional actomyosin belt in intestinal epithelial cell monolayers. Confocal microscopy images of control and UNC‐45A‐depleted HT‐29cf8 (A) and SK‐CO15 (B) cell monolayers fluorescently labeled for F‐actin and major epithelial myosin II motors, NM‐IIA and NM‐IIC. Arrows indicate perijunctional actomyosin structures in control IEC. Arrowheads show disruption of perijunctional actomyosin bundles in UNC‐45A‐depleted IEC. Scale bars, 20 µm. Representative of at least three independent experiments with multiple images taken per slide
Importantly, our data indicate physical interactions between UNC‐45A and NM‐IIA, which is the predominant NM‐II paralog in IEC. 34 , 50 NM‐IIA was co‐immunoprecipitated with UNC‐45A from SK‐CO15 cell lysates (Figure S2A). Furthermore, a PLA detected extensive interactions between UNC‐45A and NM‐IIA in intact HT‐29cf8 and SK‐CO15 cell monolayers, with such interactions being greatly diminished after UNC‐45A depletion (Figure S2B,C).
3.3. Loss of UNC‐45A attenuates reassembly of apical junctions and the perijunctional cytoskeleton during calcium switch
The impaired structure and permeability of the IEC barrier may not be specific consequences of UNC‐45A depletion and could also reflect the delayed establishment of epithelial monolayers due to decreased cell proliferation. Indeed, loss of UNC‐45A significantly delayed growth of SK‐CO15, but not HT‐29cf8 cells without causing cell apoptosis (Figure S3A–D). To establish the specific effects of UNC‐45A depletion on AJ/TJ assembly, we used a classical calcium switch model in confluent IEC monolayers that enables selective examination of junctional dynamics. 35 , 36 , 37 The described experiments were performed in SK‐CO15 cells, because HT‐29cf8 cells did not readily restore TJ integrity in this model. Re‐addition of extracellular calcium caused rapid (within 5 h) AJ and TJ reassembly in control SK‐CO15 cell monolayers (Figure 4A arrows, B). In contrast, restoration of apical junctions was significantly delayed in UNC‐45A‐deficient cells (Figure 4A, arrowheads, B). Calcium driven AJ/TJ reassembly in control IEC was accompanied by reformation of the sharp, NM‐IIA and NM‐IIC‐containing circumferential actomyosin belt (Figure 4C arrows, D). The assembly of this belt was inhibited in UNC‐45A deficient IEC that displayed a loose, non‐condensed arrays of parallel myosin bundles at the intercellular contacts (Figure 4C arrowheads, D).
FIGURE 4.
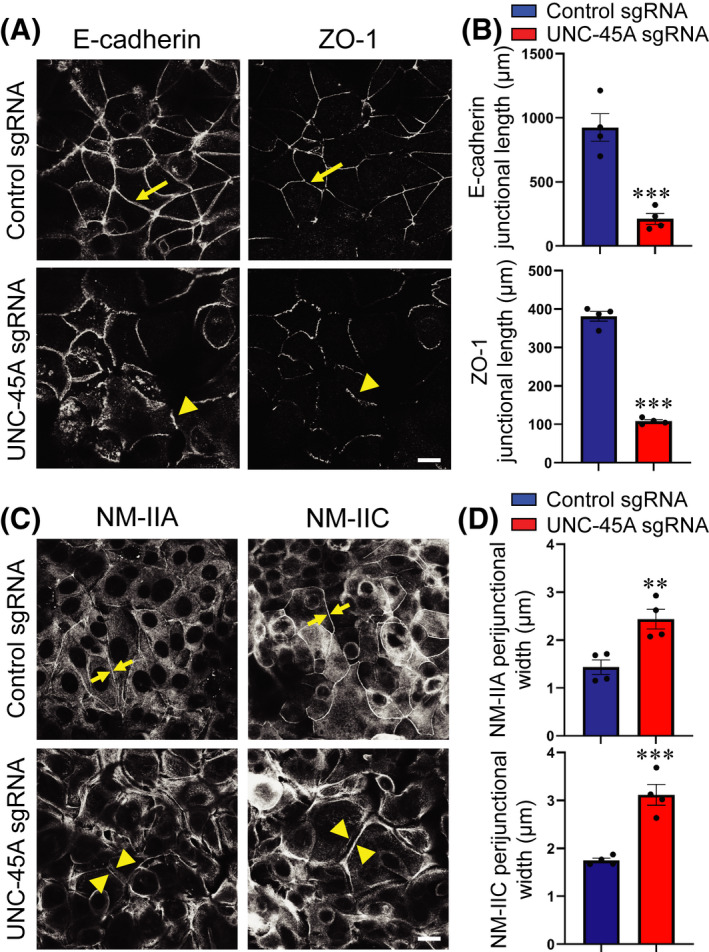
Depletion of UNC‐45A attenuates AJ/TJ reassembly and reformation of the perijunctional myosin belt during the extracellular calcium switch. (A, B) Immunolabeling of E‐cadherin and ZO‐1 in control and UNC‐45A knockout SK‐CO15 cells after 5 h of extracellular calcium repletion. Representative confocal microscopy images (A) and quantification of junctional length (B) is shown. Arrows indicate AJ/TJ reassembly in control IEC. Arrowheads show poorly assembled apical junctions in UNC‐45A knockout IEC. Each dot represents an averaged junctional length measured in four different images. (C, D) Immunolabeling of NM‐IIA and NM‐IIC in control and UNC‐45A knockout SK‐CO15 cells after 5 h of extracellular calcium repletion. Representative confocal microscopy images (C) and quantification of the width of the perijunctional NM‐II belt (D) is shown. Arrows indicate restoration of the actomyosin belt in control IEC. Arrowheads show attenuated reassembly of perijunctional NM‐II bundles in UNC‐45A knockout IEC. Each dot represents an averaged width of 200 cell–cell contacts in four different images. Means ± SE (n = 4); **p < .005, ***p < .0005 compared with the control sgRNA groups. Scale bar 20 µm. (A–D) Data are representative of three independent experiments
3.4. Loss of UNC‐45A inhibits intestinal epithelial cell migration
Apical junctions are critical not only for the establishment of epithelial barriers, but also for the repair of injured cell monolayers, by controlling collective migration of epithelial cells. 2 , 3 Given the observed role of UNC‐45A in regulating junctional integrity, we sought to examine if this chaperone also influences on IEC motility. Loss of UNC‐45A significantly delayed collective migration of HT‐29cf8 and SK‐CO15 cell monolayers in a wound healing assay (Figure 5A–D). Furthermore, depletion of this chaperone caused a marked inhibition of transfilter migration of individual IEC in the Boyden Chamber (Figure 5E–H). The latter result uncouples the effects of UNC‐45A inhibition on epithelial junctional integrity and cell migration, and suggests independent functions of this chaperone in stationary and motile IEC. To gain insights into the mechanism of UNC‐45A regulation of IEC migration, we examined whether it controls cell adhesion to the extracellular matrix (ECM), which represents a key step of cell motility. 39 , 51 UNC‐45A depletion significantly increased adhesion of HT‐29cf8 and SK‐CO15 to collagen I (Figure 5I–L). Such hyperadhesiveness of UNC‐45A‐deficient IEC was accompanied by the increased number of FA that mediate cell–ECM interactions (Figure S4A,B) and decreased abundance of actomyosin cables at the migrating cell edge (Figure S4C, arrowhead).
FIGURE 5.
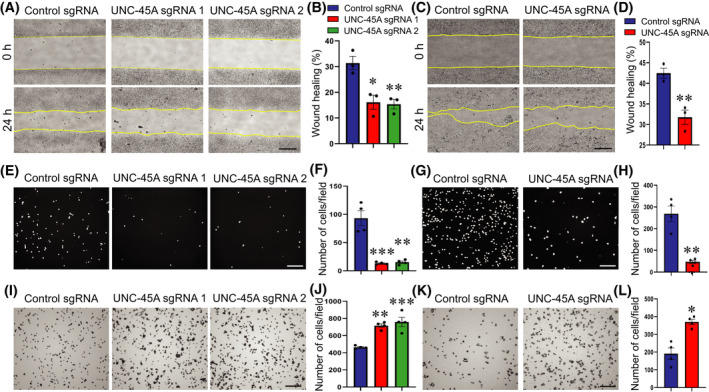
Loss of UNC‐45A expression inhibits epithelial cell migration and increases cell extracellular matrix adhesion. (A–D) Wound healing assay in control and UNC‐45A‐depleted HT‐29cf8 (A, B) and SK‐CO15 (C, D) cell monolayers. Representative wound images (A, C) and quantification of wound closure (B, D) are shown. (E–H) Boyden Chamber migration assay of control and UNC‐45A‐knockout HT‐29cf8 (E, F) and SK‐CO15 (G, H) cells. Representative images of migrated cells stained with DAPI (E, G) and quantification of transfilter cell migration (F, H) are shown. (I–L) Collagen I matrix adhesion assay of control and UNC‐45A‐knockout HT‐29cf8 (I, J) and SK‐CO15 (K, L) cells. Representative images of attached cells (I, K) and quantification of cells adhered after 30 min incubation (J, L) are shown. Mean ± SE (n = 3); *p < .05, **p < .005; ***p < .0005, compared with the control sgRNA groups. Scale bars, 200 µm. (A–D) Data are representative of three independent experiments, (E–K) data are representative of two experiments
3.5. Loss of UNC‐45A inhibits intestinal epithelial cell supracellular tension and contractility
Our data indicate that UNC‐45A controls both IEC apical junctions and cell–matrix interactions by regulating proper assembly of actomyosin structures associated with different adhesion complexes. Because NM‐II strengthens epithelial junctions and focal adhesions by generating mechanical forces, we hypothesized that loss of UNC‐45A would diminish mechanical forces in IEC. We tested this hypothesis by examining cytoskeletal forces in control and UNC‐45A‐deficient IEC by using three experimental approaches. FM‐AFM was used to examine force generation at the apical IEC surface, including apical junctions. 48 Mechanical forces at lateral cell‐cell contacts were measured by using an E‐cadherin based mechanosensitive FRET sensor. 44 , 52 Forces generated at ECM adhesions were examined using a collagen contraction assay.
Loss of UNC‐45A significantly decreased the tension and viscosity of the apical surface of HT‐29cf8 cell monolayers as measured by FM‐AFM (Figure 6A,B), thereby suggesting decreased contractile forces. By contrast, downregulation of this cytoskeletal chaperone did not show effects on forces applied to E‐cadherin based lateral cell–cell contacts (Figure 6C,D). Finally, loss of UNC‐45A significantly inhibited collagen gel contraction by embedded HT‐29cf8 (Figure 6E,F) and SK‐CO15 (data not shown), which indicates diminished actomyosin contractile forces at cell‐ECM contacts. Together, these data suggest that UNC‐45A is essential for generation of NM‐II‐driven contractile forces at specific plasma membrane compartments of IEC.
FIGURE 6.
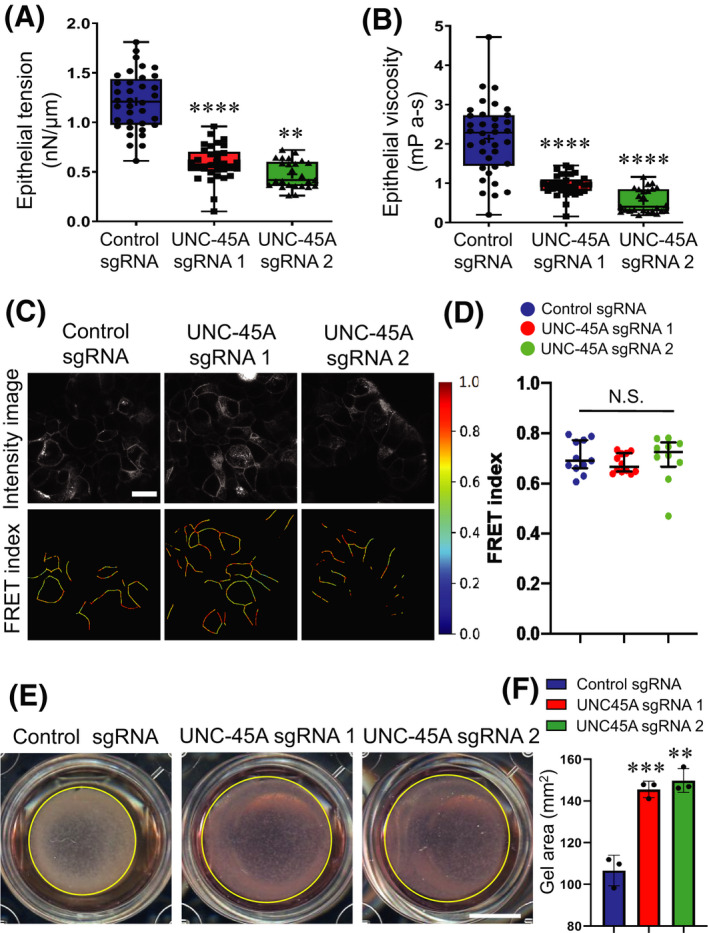
UNC‐45A regulates mechanical forces in intestinal epithelial cells. (A, B) Viscoelastic properties, epithelial tension (A) and viscosity (B) measurements in control and UNC‐45A‐depleted HT‐29cf8 cell monolayers by noncontact frequency modulation atomic force microscopy (FM‐AFM). Means ± SD; number of cells for control sgRNA n = 35, UNC‐45A sgRNA1 n = 31, and UNC‐45A sgRNA2 n = 26; **p < .005; ****p < .0001, compared with the control sgRNA group. (C, D) FRET analysis of control and UNC‐45A knockout HT‐29cf8 cells expressing an E‐cadherin FRET‐based biosensor. Representative images (C) and the scatter dot plot (D) shows FRET index measurements across monolayers. The horizontal line (black) in the scatter dot plot denotes the median of the FRET index estimates and the whiskers denote the largest observation within 1.5 × interquartile range (IQR) (representative data shown is from one experiment out of a total of four independent experiments; N.S, not significant, p > .05). Scale Bar, 20 µm. (E, F) Contraction of collaged I gel with embedded control or UNC‐45A‐depleted HT‐29cf8 cells. Representative images of contracted gels (E) and quantification of the gel area after 4 days of contraction (F) are shown. Scale bar, 50 mm. Means ± SE (n = 4); **p < .005; ***p < .0005, compared with the control sgRNA group. (A–F) Data are representative of three independent experiments
3.6. Expression of the myosin‐binding domain of UNC‐45A selectively perturbs barrier properties and migration of intestinal epithelial cell monolayers
While conventional myosins are the most well‐known folding clients for UNC‐45A, this chaperone has additional myosin‐independent activities, such as interacting with tubulin and modulating microtubule dynamics. 23 , 24 , 25 Since altered dynamics of microtubules could affect epithelial barrier integrity and cell migration, we sought to examine whether this mechanism contributes to abnormal junctional assembly and wound healing caused by UNC‐45A knockout. First, we visualized microtubules in differentiated SK‐CO15 and HT‐29cf8 cell monolayers using immunofluorescence labeling and confocal microscopy but did not observe significant alterations in the apical microtubule meshwork following UNC‐45A depletion (Figure S5A, arrows). Next, we acutely disrupted microtubules in confluent control and UNC‐45A depleted SK‐CO15 monolayers by treating with 30 μM nocodazole for 4 h. Such microtubule depolymerization did not eliminate the disruptive effects of UNC‐45A knockout on the IEC barrier (Figure S5B,C), suggesting that such effects may be microtubule independent. Finally, we created SK‐CO15 cells stably expressing either full length UNC‐45A (UNC‐45A‐GFP), its N‐terminal truncated ΔTPR‐GFP (residues 147‐971), or C‐terminally truncated ΔUCS‐GFP (residues 1‐539) (Figure 7A). Expression of an empty GFP‐containing vector serves as a control. Due to technical difficulties in generating stable IEC lines, we were unable to perform rescue experiments by introducing these mutants into UNC‐45A knockout IEC. Instead, we sought to examine possible dominant negative effects of the mutants after their expression in wild type SK‐CO15 cells that have endogenous UNC‐45A. Epithelial permeability measurements revealed that overexpression of the full‐length UNC‐45A did not affect the integrity of IEC barrier (Figure 7B,C). Interestingly, expression of the ΔTPR mutant that binds to NM‐II, but not microtubules, markedly diminished TEER and increased FITC‐dextran flux (Figure 7B,C), suggesting a potential dominant negative effects. By contrast, the ΔUCS mutant that interacts with microtubules, but lacks the myosin‐folding activity modestly attenuated TEER development without affecting the FITC‐dextran flux (Figure 7B,C). Similar results were obtained using the calcium switch assay where expression of the ΔTPR, but not the ΔUCS mutant (or full length UNC‐45A) selectively attenuated AJ and TJ reassembly (Figure 7D,E). In agreement with its effects on IEC barrier and junctions, expression of the ΔTPR UNC‐45A mutant significantly inhibited collective migration SK‐CO15 cells in the wound healing experiments (Figure S6A,B). Overexpression of full‐length UNC‐45A did not affect wound healing, whereas expression of its ΔUCS mutant had a slight inhibitory effect on cell migration that did not reach significant difference (Figure S6A,B). Together, our data suggest that UNC‐45A‐dependent regulation of IEC barrier, junctional assembly and cell migration is determined by the NM‐II binding ability of this chaperone and does not involve targeting microtubules.
FIGURE 7.
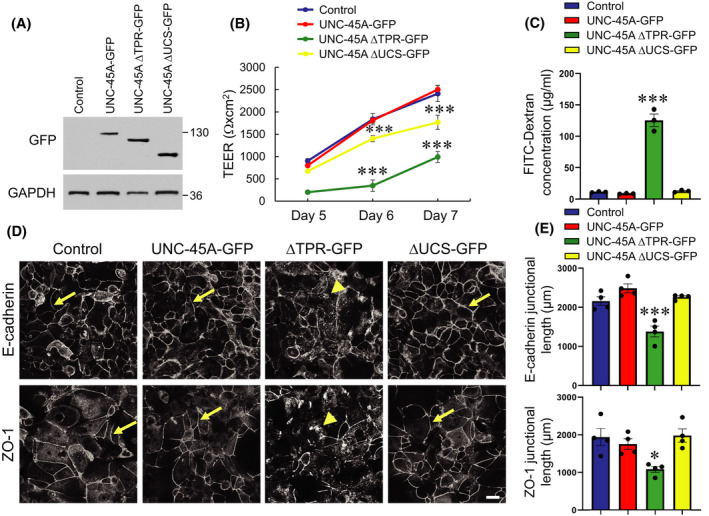
Expression of N‐terminal domain deletion mutant UNC‐45A disrupts epithelial barrier integrity and attenuates adherens junction and tight junction assembly. (A) Immunoblotting analysis of SK‐CO15 expressing either GFP‐tagged full‐length UNC‐45A or two different deletion mutants of UNC‐45A, ΔTPR‐GFP (N‐terminal truncated), or ΔUCS‐GFP (C‐terminally truncated). (B) Transepithelial electrical resistance (TEER) of control SK‐CO15 cell monolayers and cells expressing UNC‐45A constructs at different times post‐plating. (C) Transmonolayer flux of FITC‐dextran in control SK‐CO15 cell monolayers and cells expressing different UNC‐45A constructs on day 7 post‐plating. (D, E) Immunolabeling of E‐cadherin and ZO‐1 in control SK‐CO15 cell monolayers and cells expressing different UNC‐45A constructs after 5 h of extracellular calcium repletion. Representative confocal microscopy images (D) and quantification of junctional length (E) is shown. Arrows indicate AJ/TJ reassembly in control, full length UNC‐45A, and the ΔUCS mutant expressing IEC. Arrowheads show poorly reassembled apical junctions in the ΔTPR mutant expressing cells. Means ± SE (n = 3 for B,C; n = 4 for D); *p < .05, ***p < .0005. Scale bar, 20 µm. (A–E) Data are representative of three independent experiments
3.7. UNC‐45 regulates permeability of Drosophila gut
To validate the in vivo relevance of our finding in IEC monolayers we sought to determine the effects of lowering UNC‐45 expression on permeability of the Drosophila gut. Two mutant fly strains with monoallelic depletion of UNC‐45 were examined since homozygous deletion of this chaperone results in Drosophila lethality. 31 Immunoblotting analysis demonstrated approximately 30%–50% reduction of UNC‐45 expression in tissues of mutant flies (Figure 8A). Aged flies were fed with a sucrose solution containing Brilliant Blue dye, and then scored as to whether their intestines were leaky, based on the whether or not dye had leaked into extra‐intestinal tissues (“Smurf” phenotype) (Figure 8B). 53 , 54 Increased numbers of Smurf flies were observed in both 20 day‐old females and males of the unc‐4503692/+ mutant genotype characterized by 50% UNC‐45 depletion (Figure 8C). Another mutant genotype with less pronounced UNC‐45 downregulation, unc‐45EY03043‐33/+, showed a tendency toward increased gut permeability, however numbers of Smurf flies did not reach statistical significance (Figure 8C). These data suggest that UNC‐45 expression could serve as a “rheostat” controlling permeability of the Drosophila gut.
FIGURE 8.
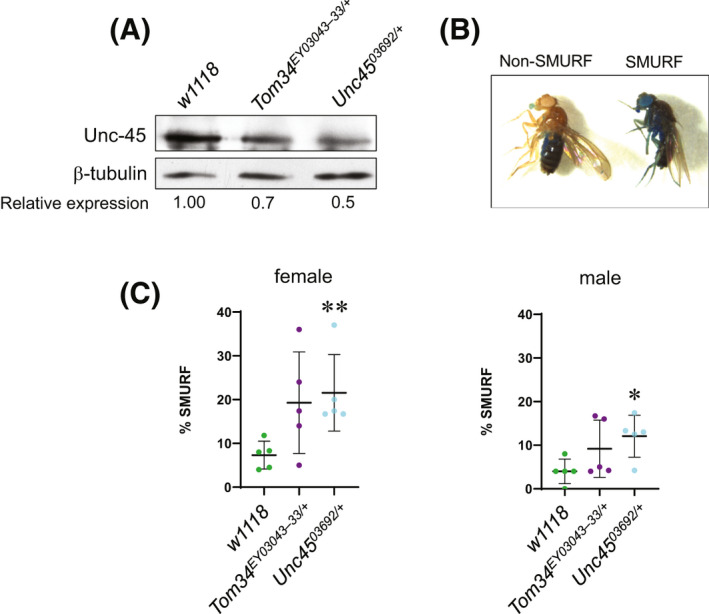
Reduced expression of Unc‐45 in Drosophila increases intestinal permeability. (A) Immunoblotting analysis of Unc‐45 protein in wild‐type (w1118) or heterozygous unc‐45 mutant (unc‐45EY03043‐33 or unc‐4503692) Drosophila melanogaster adult flies. β‐tubulin was used as a loading control. (B) Pictures of adult Drosophila females categorized as non‐Smurf (blue dye localized to intestine and proboscis) or Smurf (blue dye spread from intestine to the majority of tissues). (C) Quantitation of the percentage of flies exhibiting the Smurf phenotype (right picture in B) for each gender of each genotype analyzed in A. Each dot represents an individual experiment containing 25 flies. *p < .05, **p < .01
3.8. UNC‐45A expression is downregulated in the colonic epithelium of ulcerative colitis patients
Immunofluorescence labeling and immunoblotting detected abundant expression of UNC‐45A in normal human colonic mucosa and isolated primary human colonic epithelial cells (Figure S7A,B). In normal colonic mucosa UNC‐45A was enriched at the apical pole of IEC where it colocalized with epithelial junctions (Figure S7A, arrow). Because previous studies reported disrupted barrier properties and altered NM‐II activity in the intestinal mucosa during IBD, 55 , 56 we sought to compare UNC‐45A expression in patients with active CD, UC, and non‐IBD controls. Immunofluorescence labeling and immunoblotting analysis demonstrated marked decrease of UNC‐45A protein level in colonic epithelial cells of UC patients, both in full thickness tissue sections (Figure S7A, arrowhead) and isolated colonic epithelial cells (Figure S7B,C). By contrast, colonic epithelial expression and localization of UNC‐45A protein appeared normal in the intestinal mucosa of CD patients (Figure S7A–C).
4. DISCUSSION
NM‐II is a crucial cytoskeletal motor that regulates assembly and functions of epithelial apical junctions and basal ECM adhesions. 12 , 57 , 58 Although molecular mechanisms that govern NM‐II ATPase activity and its dynamic interactions with actin filaments have been extensively investigated, much less is known about the regulation of NM‐II organization into higher‐order contractile structures associated with epithelial adhesive complexes. In this study we describe a novel mechanism controlling NM‐II‐dependent assembly of apical junctions and ECM adhesions that is essential for the integrity and repair of the intestinal epithelial barrier. This mechanism involves a cytoplasmic myosin chaperone, UNC‐45A (Figure 9). Although an essential role for UNC‐45A in controlling myosin folding and function was originally recognized more than two decades ago, 59 its involvement in the regulation of intercellular adhesions and tissue barriers has not been previously investigated.
FIGURE 9.
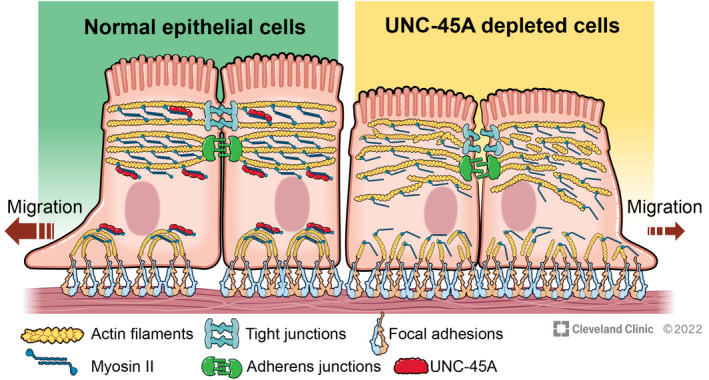
Schematic diagram of the proposed UNC‐45A roles in regulating intestinal epithelial junctions and cell migration. In control intestinal epithelial cells, UNC‐45A promotes myofilament assembly to create major contractile actomyosin structures, such as the perijunctional belt and focal adhesion associated stress‐fibers. The perijunctional belt is essential for tight junction/adherens junction integrity and barrier establishment. The actomyosin stress fibers mediate remodeling of focal adhesions and cell motility. Loss of UNC‐45A impairs assembly of the perijunctional actomyosin belt resulting in junctional disruption. It also diminishes formation of stress fibers that are essential for focal adhesion remodeling, thereby impeding epithelial cell migration. Illustration by David Schumick, BS, CMI. Reprinted with the permission, Cleveland Clinic Foundation © 2022. All Rights Reserved
Our data suggest that UNC‐45A regulates two major permeability pathways in IEC monolayers. One is a paracellular pore pathway selectively permeable to small ions, and the other is a leak pathway permeated by large molecules. 4 Disruption of the pore and the leak pathways in UNC‐45A deficient IEC cells was revealed by decreased TEER and increased FITC‐dextran flux, respectively (Figure 1C–F). The described defects in barrier permeability were associated with disruption of AJ and TJ integrity and mislocalization of junctional proteins (Figure 2). Importantly, loss of UNC‐45A markedly attenuated epithelial junctional reassembly during extracellular calcium switch (Figure 4A,B), which signifies specific regulatory roles of this chaperone at IEC apical junctions.
Several lines of evidence suggest that loss of UNC‐45A disrupts the integrity and remodeling of epithelial junctions by impairing assembly and contractility of the perijunctional actomyosin cytoskeleton. Thus, UNC‐45A was found to physically interact with NM‐IIA in IEC (Figure S2), and its loss markedly disrupted organization of the circumferential actomyosin belt associated with steady‐state and reassembling junctions (Figures 3 and 4). This phenocopy barrier disruption and attenuated AJ/TJ reassembly that was previously described in human epithelial cells after either pharmacologic inhibition of NM‐II motors, or genetic depletion of NM‐IIA. 35 , 36 , 60 Because the sarcomeric organization of NM‐II filaments within the perijunctional belt resembles organization of muscles myofibrils, 10 , 13 the observed disorganization of the perijunctional actomyosin belt in UNC‐45A‐deficient IEC resembles impaired myofibril assembly caused by loss of this chaperone in cultured muscle cells and model organisms. 16 , 18 , 31
Consistent with disordered organization of the perijunctional actomyosin cytoskeleton, loss of UNC‐45A reduced myosin‐dependent forces at apical junctions. Such force reduction was revealed by decreased epithelial tension and viscosity of the apical surface of UNC‐45A‐depleted IEC (Figure 6A,B). Because the applied AFM technique measures mechanical properties of epithelial monolayers with depth up to 2.5 µm, our experiments most likely detected decreased mechanical forces applied to the entire apical junctional complex consisting of TJ and AJ in UNC‐45A‐depleted cell. This is in agreement to a previous FM‐AFM study demonstrating that changes of apical tension and viscosity in polarized epithelial monolayers largely depend on altered contractility of the perijunctional actomyosin belt. 48 Surprisingly, no changes in local mechanical forces have been observed by using FRET‐based E‐cadherin tension sensor (Figure 6C,D). It is important to consider that the E‐cadherin tension sensor measures average force per E‐cadherin molecule, contrasting to the applied atomic force microscopy technique, which measures the overall mechanical force state between cells. Furthermore, this tension sensor incorporates both into mature AJ and underlying E‐cadherin clusters localized along the lateral plasma membrane. Unaltered properties of the E‐cadherin tension sensor in UNC‐45A‐depleted cells could indicate that this cytoskeletal chaperone does not regulate mechanical forces applied to the lateral cell–cell contacts in columnar IEC monolayers.
Our experiments using deletion mutant expression also suggests that UNC‐45A regulates epithelial barrier integrity and junction remodeling in the NM‐II‐dependent fashion. Indeed, expression of the N‐terminal deletion mutant that retains the myosin‐binding domain (ΔTPR‐UNC‐45A) selectively disrupted IEC barrier and attenuated AJ and TJ reassembly (Figure 7). Consistently, a previous study demonstrated that expression of this mutant resulted in the assembly of abnormal stress fibers in osteosarcoma cells. 30 What are the mechanisms by which the myosin‐interacting ΔTPR UNC‐45A mutant disrupts the IEC barrier integrity and remodeling? While the C‐terminal UCS domain is sufficient to fold myosin in vitro, 19 the entire UNC‐45A structure containing both the UCS and TPR domains is required to build the sarcomeric organization of muscle myofibrils. Both domains are involved in intermolecular interactions that mediate UNC‐45 oligomerization into periodic tandem modules thereby providing a template for orchestrated self‐assembly of multiple NM‐II molecules. 20 Therefore, the exogenous ΔTPR mutant is likely to compete for NM‐II binding with the endogenous full‐length chaperone, preventing assembly of high‐order myosin filaments. By contrast, we did not find strong evidence that interactions with microtubules are essential for UNC‐45A‐dependent regulation of IEC junctions. Indeed, UNC‐45A depletion increased epithelial permeability even after microtubule depolymerization (Figure S5B,C). Furthermore, expression of the UNC‐45A mutant that interacts with microtubules, but not NM‐II, failed to induce robust dominant negative effects on permeability of IEC barrier and AJ/TJ reassembly (Figure 7). This is consistent with our previous study indicating that microtubules are dispensable for apical junction assembly in IEC 61 and further signifies NM‐II‐dependent regulation of epithelial barrier integrity by UNC‐45A.
UNC‐45A appears to play dual functional roles in IEC by both regulating barrier integrity, and regulating cell migration. Generally, the roles of UNC‐45A in mammalian cell migration remain controversial. Depletion of this chaperone was shown to inhibit motility of breast and ovarian cancer cells, 27 , 29 but promoted migration of osteosarcoma and melanoma cells. 30 , 62 This resembles the reported opposite effects of NM‐II inhibition on cell migration under different experimental conditions 50 , 63 and may reflect complex regulatory effects of UNC‐45A‐driven myofilament assembly in motile cells. We observed that loss of UNC‐45A inhibited both collective migration of IEC sheets and individual cell passage through membrane pores (Figure 5A–H). Strikingly, the attenuated collective migration of UNC‐45A‐depleted IEC was coupled with increased cell adhesion to ECM and elevated number of FA (Figures 5I–L and S4A,B). Furthermore, hyperadhesiveness of UNC‐45A‐depleted IEC was accompanied by disorganization of F‐actin bundles at the migrating cell edge (Figure S4C) and decreased contractile forces at the ECM adhesions (Figure 6E,F). Decreased myosin‐driven forces are likely to prevent efficient FA remodeling thereby enhancing cell–ECM adhesion and attenuating the orchestrated directional movement of IEC monolayers. Likewise, inhibited NM‐II contractility could impair cell deformability, which is required for efficient migration through confined spaces, such as membrane pores. This could explain attenuated transfilter migration of UNC‐45A‐deficient IEC. Similar to the effects on the IEC barrier, UNC‐45A regulation of IEC migration appears to be dependent on its interactions with myosin motors, but not microtubules. This conclusion is supported by the unique anti‐migratory effect of exogenously expressed myosin‐binding mutant of UNC‐45A (Figure S6). Together, our results uncover roles for UNC‐45A in controlling multiple functions of NM‐II in the intestinal epithelium.
Drosophila UNC‐45 is an essential protein that is expressed at all developmental stages. Like human UNC‐45A, Drosophila UNC‐45 also colocalizes with non‐muscle myosin II. 31 Furthermore, unc‐45 mutant embryos display defective myosin accumulation in body wall muscles at 22 h post‐fertilization. 31 Our results demonstrate that the intestines of 20‐day old unc‐45 mutant adults display increased permeability, compared with wild‐type controls (Figure 8). Although the functions of UNC‐45 in myosin regulation have not been investigated in the adult Drosophila gut, it is possible that a conserved role for UNC‐45 in promoting myosin assembly at TJ and AJ exists in the Drosophila gut, and this contributes to the integrity of the adult Drosophila intestinal epithelial barrier.
In addition to characterizing novel roles of UNC‐45A in regulating barrier integrity and repair in IEC monolayers, our study suggests possible dysfunctions of this molecular chaperone during mucosal inflammation. Specifically, we found marked and selective downregulation of UNC‐45A protein expression in the colonic epithelium of UC patients (Figure S7). This chaperone was found to be upregulated during tumorigenesis 27 , 29 ; however, we present the first evidence for its altered expression under inflammatory conditions. Although we did not investigate molecular mechanisms of the disease‐associated downregulation of UNC‐45A, its expression is known to be tightly controlled in muscle cells via the ubiquitin‐proteosomal degradation system. 29 , 64 Interestingly, a loss‐of‐function mutation in UNC‐45A in humans resulted in chronic diarrhea, among other clinical symptoms. 65 This suggests that UNC‐45A function is essential for normal intestinal epithelial homeostasis, and the observed decrease in its expression during mucosal inflammation in UC patients could impair cytoskeleton‐dependent processes in IEC, including establishment of paracellular barrier and healing of mucosal wounds.
In conclusion, our study identified a novel mechanism that control actomyosin‐dependent integrity and remodeling of the intestinal epithelial barrier. This mechanism involves a myosin chaperone, UNC‐45A. UNC‐45A binds to NM‐IIA in cultured IEC and controls NM‐II assembly and contractility at epithelial adhesions. Through its myosin folding activity, this molecular chaperone plays a dual role in epithelial monolayers by regulating formation of apical junctions and cell migration (Figure 9). Loss of UNC‐45A expression during intestinal inflammation may promote leakiness of the gut barrier and impede mucosal restitution.
DISCLOSURES
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
Andrei I. Ivanov conceived the study; Alexander X. Cartagena‐Rivera performed atomic force microscopy studies; Vani Narayanan and Daniel E. Conway examined tensile forces using E‐cadherin FRET biosensor; Jaakko Lehtimäki and Pekka Lappalainen generated and provided UNC‐45A plasmids; Florian Rieder assisted with acquisition and analysis of human colonic samples; Afshin Khan, Bert I. Crawford and Michelle S. Longworth performed Drosophila experiments. All other experiments were carried out by Susana Lechuga. Susana Lechuga and Andrei I. Ivanov wrote the manuscript with input from the other authors.
Supporting information
Fig S1‐S7
ACKNOWLEDGMENTS
We thank Dr. Nayden Naydenov for help with isolating primary human colonic epithelial cells. This work was supported by National Institute of Health grants RO1 DK126702 and RO1 DK108278 to A.I. Ivanov; R35 GM119617 to D.E. Conway; R21 AI153780 to M.S. Longworth. M.S. Longworth was also supported by funding from Cure for IBD and a Velosano Award. A.X. Cartagena‐Rivera was supported by the National Institutes of Health (NIH) Intramural Research Program of the National Institutes of Biomedical Imaging and Bioengineering (grant ZIAEB000094) and the NIH Distinguished Scholars Program. Confocal microscopy was performed at the Lerner Research Institute Digital Imaging Microscopy Core.
Lechuga S, Cartagena‐Rivera AX, Khan A, et al. A myosin chaperone, UNC‐45A, is a novel regulator of intestinal epithelial barrier integrity and repair. FASEB J. 2022;36:e22290. doi: 10.1096/fj.202200154R
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are included in the article and its supplementary information files.
REFERENCES
- 1. Green KJ, Getsios S, Troyanovsky S, Godsel LM. Intercellular junction assembly, dynamics, and homeostasis. Cold Spring Harb Perspect Biol. 2010;2:a000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gupta S, Yap AS. How adherens junctions move cells during collective migration. Fac Rev. 2021;10:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zulueta‐Coarasa T, Fernandez‐Gonzalez R. Tension (re)builds: biophysical mechanisms of embryonic wound repair. Mech Dev. 2017;144:43‐52. [DOI] [PubMed] [Google Scholar]
- 4. Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1:a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Furuse M. Molecular basis of the core structure of tight junctions. Cold Spring Harb Perspect Biol. 2010;2(1):a002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophysica Acta. 2008;1778:660‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meng W, Takeichi M. Adherens junction: molecular architecture and regulation. Cold Spring Harb Perspect Biol. 2009;1:a002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sluysmans S, Vasileva E, Spadaro D, Shah J, Rouaud F, Citi S. The role of apical cell‐cell junctions and associated cytoskeleton in mechanotransduction. Biol Cell. 2017;109:139‐161. [DOI] [PubMed] [Google Scholar]
- 9. Varadarajan S, Stephenson RE, Miller AL. Multiscale dynamics of tight junction remodeling. J Cell Sci. 2019;132:jcs229286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ebrahim S, Fujita T, Millis BA, et al. NMII forms a contractile transcellular sarcomeric network to regulate apical cell junctions and tissue geometry. Curr Biol. 2013;23:731‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hirokawa N, Tilney LG. Interactions between actin filaments and between actin filaments and membranes in quick‐frozen and deeply etched hair cells of the chick ear. J Cell Biol. 1982;95:249‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ivanov AI. Actin motors that drive formation and disassembly of epithelial apical junctions. Front Biosci. 2008;13:6662‐6681. [DOI] [PubMed] [Google Scholar]
- 13. Acharya BR, Wu SK, Lieu ZZ, et al. Mammalian diaphanous 1 mediates a pathway for E‐cadherin to stabilize epithelial barriers through junctional contractility. Cell Rep. 2017;18:2854‐2867. [DOI] [PubMed] [Google Scholar]
- 14. Hellerschmied D, Clausen T. Myosin chaperones. Curr Opin Struct Biol. 2014;25:9‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee CF, Melkani GC, Bernstein SI. The UNC‐45 myosin chaperone: from worms to flies to vertebrates. Int Rev Cell Mol Biol. 2014;313:103‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Price MG, Landsverk ML, Barral JM, Epstein HF. Two mammalian UNC‐45 isoforms are related to distinct cytoskeletal and muscle‐specific functions. J Cell Sci. 2002;115:4013‐4023. [DOI] [PubMed] [Google Scholar]
- 17. Ni W, Odunuga OO. UCS proteins: chaperones for myosin and co‐chaperones for Hsp90. Subcell Biochem. 2015;78:133‐152. [DOI] [PubMed] [Google Scholar]
- 18. Barral JM, Bauer CC, Ortiz I, Epstein HF. Unc‐45 mutations in Caenorhabditis elegans implicate a CRO1/She4p‐like domain in myosin assembly. J Cell Biol. 1998;143:1215‐1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barral JM, Hutagalung AH, Brinker A, Hartl FU, Epstein HF. Role of the myosin assembly protein UNC‐45 as a molecular chaperone for myosin. Science. 2002;295:669‐671. [DOI] [PubMed] [Google Scholar]
- 20. Gazda L, Pokrzywa W, Hellerschmied D, et al. The myosin chaperone UNC‐45 is organized in tandem modules to support myofilament formation in C. elegans . Cell. 2013;152:183‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hellerschmied D, Lehner A, Franicevic N, et al. Molecular features of the UNC‐45 chaperone critical for binding and folding muscle myosin. Nat Communicat. 2019;10:4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu L, Srikakulam R, Winkelmann DA. Unc45 activates Hsp90‐dependent folding of the myosin motor domain. J Biol Chem. 2008;283:13185‐13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Habicht J, Mooneyham A, Hoshino A, et al. UNC‐45A breaks the microtubule lattice independently of its effects on non‐muscle myosin II. J Cell Sci. 2021;134:jcs248815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Habicht J, Mooneyham A, Shetty M, et al. UNC‐45A is preferentially expressed in epithelial cells and binds to and co‐localizes with interphase MTs. Cancer Biol Ther. 2019;20:1304‐1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mooneyham A, Iizuka Y, Yang Q, et al. UNC‐45A is a novel microtubule‐associated protein and regulator of paclitaxel sensitivity in ovarian cancer cells. Mol Cancer Res. 2019;17:370‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iizuka Y, Mooneyham A, Sieben A, et al. UNC‐45A is required for neurite extension via controlling NMII activation. Mol Biol Cell. 2017;28:1337‐1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bazzaro M, Santillan A, Lin Z, et al. Myosin II co‐chaperone general cell UNC‐45 overexpression is associated with ovarian cancer, rapid proliferation, and motility. Am J Pathol. 2007;171:1640‐1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eisa NH, Jilani Y, Kainth K, et al. The co‐chaperone UNC45A is essential for the expression of mitotic kinase NEK7 and tumorigenesis. J Biol Chem. 2019;294:5246‐5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guo W, Chen D, Fan Z, Epstein HF. Differential turnover of myosin chaperone UNC‐45A isoforms increases in metastatic human breast cancer. J Mol Biol. 2011;412:365‐378. [DOI] [PubMed] [Google Scholar]
- 30. Lehtimaki JI, Fenix AM, Kotila TM, et al. UNC‐45a promotes myosin folding and stress fiber assembly. J Cell Biol. 2017;216:4053‐4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee CF, Melkani GC, Yu Q, et al. Drosophila UNC‐45 accumulates in embryonic blastoderm and in muscles, and is essential for muscle myosin stability. J Cell Sci. 2011;124:699‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Le Bivic A, Real FX, Rodriguez‐Boulan E. Vectorial targeting of apical and basolateral plasma membrane proteins in a human adenocarcinoma epithelial cell line. Proc Natl Acad Sci U S A. 1989;86:9313‐9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mitchell DM, Ball JM. Characterization of a spontaneously polarizing HT‐29 cell line, HT‐29/cl.f8. In Vitro Cell Dev Biol Anim. 2004;40:297‐302. [DOI] [PubMed] [Google Scholar]
- 34. Naydenov NG, Feygin A, Wang D, et al. Nonmuscle myosin IIA regulates intestinal epithelial barrier in vivo and plays a protective role during experimental colitis. Sci Rep. 2016;6:24161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ivanov AI, Bachar M, Babbin BA, Adelstein RS, Nusrat A, Parkos CA. A unique role for nonmuscle myosin heavy chain IIA in regulation of epithelial apical junctions. PLoS One. 2007;2:e658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ivanov AI, Hunt D, Utech M, Nusrat A, Parkos CA. Differential roles for actin polymerization and a myosin II motor in assembly of the epithelial apical junctional complex. Mol Biol Cell. 2005;16:2636‐2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Naydenov NG, Ivanov AI. Adducins regulate remodeling of apical junctions in human epithelial cells. Mol Biol Cell. 2010;21:3506‐3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roediger WE, Truelove SC. Method of preparing isolated colonic epithelial cells (colonocytes) for metabolic studies. Gut. 1979;20:484‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lechuga S, Amin PH, Wolen AR, Ivanov AI. Adducins inhibit lung cancer cell migration through mechanisms involving regulation of cell‐matrix adhesion and cadherin‐11 expression. Biochim Biophys Acta Mol Cell Res. 2019;1866:395‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang D, Naydenov NG, Dozmorov MG, Koblinski JE, Ivanov AI. Anillin regulates breast cancer cell migration, growth, and metastasis by non‐canonical mechanisms involving control of cell stemness and differentiation. Breast Cancer Res. 2020;22:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baranwal S, Naydenov NG, Harris G, et al. Nonredundant roles of cytoplasmic beta‐ and gamma‐actin isoforms in regulation of epithelial apical junctions. Mol Biol Cell. 2012;23:3542‐3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lechuga S, Baranwal S, Ivanov AI. Actin‐interacting protein 1 controls assembly and permeability of intestinal epithelial apical junctions. Am J Physiol Gastrointest Liver Physiol. 2015;308:G745‐G756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lechuga S, Baranwal S, Li C, et al. Loss of gamma‐cytoplasmic actin triggers myofibroblast transition of human epithelial cells. Mol Biol Cell. 2014;25:3133‐3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Borghi N, Sorokina M, Shcherbakova OG, et al. E‐cadherin is under constitutive actomyosin‐generated tension that is increased at cell‐cell contacts upon externally applied stretch. Proc Natl Acad Sci U S A. 2012;109:12568‐12573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509‐2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arsenovic PT, Ramachandran I, Bathula K, et al. Nesprin‐2G, a component of the nuclear LINC complex, is subject to myosin‐dependent tension. Biophys J. 2016;110:34‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cartagena‐Rivera AX, Le Gal S, Richards K, Verpy E, Chadwick RS. Cochlear outer hair cell horizontal top connectors mediate mature stereocilia bundle mechanics. Sci Adv. 2019;5:eaat9934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cartagena‐Rivera AX, Van Itallie CM, Anderson JM, Chadwick RS. Apical surface supracellular mechanical properties in polarized epithelium using noninvasive acoustic force spectroscopy. Nat Communicat. 2017;8:1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ngo P, Ramalingam P, Phillips JA, Furuta GT. Collagen gel contraction assay. Methods Mol Biol. 2006;341:103‐109. [DOI] [PubMed] [Google Scholar]
- 50. Babbin BA, Koch S, Bachar M, et al. Non‐muscle myosin IIA differentially regulates intestinal epithelial cell restitution and matrix invasion. Am J Pathol. 2009;174:436‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gupton SL, Waterman‐Storer CM. Spatiotemporal feedback between actomyosin and focal‐adhesion systems optimizes rapid cell migration. Cell. 2006;125:1361‐1374. [DOI] [PubMed] [Google Scholar]
- 52. Narayanan V, Schappell LE, Mayer CR, et al. Osmotic gradients in epithelial acini increase mechanical tension across E‐cadherin, drive morphogenesis, and maintain homeostasis. Curr Biol. 2020;30:624‐633.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rera M, Bahadorani S, Cho J, et al. Modulation of longevity and tissue homeostasis by the Drosophila PGC‐1 homolog. Cell Metab. 2011;14:623‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rera M, Clark RI, Walker DW. Intestinal barrier dysfunction links metabolic and inflammatory markers of aging to death in Drosophila. Proc Natl Acad Sci U S A. 2012;109:21528‐21533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Martini E, Krug SM, Siegmund B, Neurath MF, Becker C. Mend your fences: the epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol. 2017;4:33‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schoultz I, Keita AV. Cellular and molecular therapeutic targets in inflammatory bowel disease‐focusing on intestinal barrier function. Cells. 2019;8:1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Garrido‐Casado M, Asensio‐Juarez G, Vicente‐Manzanares M. Nonmuscle myosin II regulation directs its multiple roles in cell migration and division. Annu Rev Cell Dev Biol. 2021;37:285‐310. [DOI] [PubMed] [Google Scholar]
- 58. Zaidel‐Bar R, Zhenhuan G, Luxenburg C. The contractome—a systems view of actomyosin contractility in non‐muscle cells. J Cell Sci. 2015;128:2209‐2217. [DOI] [PubMed] [Google Scholar]
- 59. Venolia L, Waterston RH. The unc‐45 gene of Caenorhabditis elegans is an essential muscle‐affecting gene with maternal expression. Genetics. 1990;126:345‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Smutny M, Cox HL, Leerberg JM, et al. Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat Cell Biol. 2010;12:696‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ivanov AI, McCall IC, Babbin B, Samarin SN, Nusrat A, Parkos CA. Microtubules regulate disassembly of epithelial apical junctions. BMC Cell Biol. 2006;7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Krivosheeva IA, Filatova AY, Moshkovskii SA, Baranova AV, Skoblov MY. Analysis of candidate genes expected to be essential for melanoma surviving. Cancer Cell Int. 2020;20:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Naydenov NG, Lechuga S, Huang EH, Ivanov AI. Myosin motors: novel regulators and therapeutic targets in colorectal cancer. Cancers (Basel). 2021;13:741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hoppe T, Cassata G, Barral JM, et al. Regulation of the myosin‐directed chaperone UNC‐45 by a novel E3/E4‐multiubiquitylation complex in C. elegans . Cell. 2004;118:337‐349. [DOI] [PubMed] [Google Scholar]
- 65. Esteve C, Francescatto L, Tan PL, et al. Loss‐of‐function mutations in UNC45A cause a syndrome associating cholestasis, diarrhea, impaired hearing, and bone fragility. Am J Hum Genet. 2018;102:364‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S7
Data Availability Statement
The data that support the findings of this study are included in the article and its supplementary information files.