

Abstract
Introduction
COVID-19 induces venous, arterial and microvascular thrombosis, involving several pathophysiological processes. In patients with severe COVID-19 without macrovascular thrombosis, escalating into high-dose prophylactic anticoagulation (HD-PA) or therapeutic anticoagulation (TA) could be beneficial in limiting the extension of microvascular thrombosis and forestalling the evolution of lung and multiorgan microcirculatory dysfunction. In the absence of data from randomised trials, clinical practice varies widely.
Methods and analysis
This is a French multicentre, parallel-group, open-label, randomised controlled superiority trial to compare the efficacy and safety of three anticoagulation strategies in patients with COVID-19. Patients with oxygen-treated COVID-19 showing no pulmonary artery thrombosis on computed tomography with pulmonary angiogram will be randomised to receive either low-dose PA, HD-PA or TA for 14 days. Patients attaining the extremes of weight and those with severe renal failure will not be included. We will recruit 353 patients. Patients will be randomised on a 1:1:1 basis, and stratified by centre, use of invasive mechanical ventilation, D-dimer levels and body mass index. The primary endpoint is a hierarchical criterion at day 28 including all-cause mortality, followed by the time to clinical improvement defined as the time from randomisation to an improvement of at least two points on the ordinal clinical scale. Secondary outcomes include thrombotic and major bleeding events at day 28, individual components of the primary endpoint, number of oxygen-free, ventilator-free and vasopressor-free days at day 28, D-dimer and sepsis-induced coagulopathy score at day 7, intensive care unit and hospital stay at day 28 and day 90, and all-cause death and quality of life at day 90.
Ethics and dissemination
The study has been approved by an ethical committee (Ethics Committee, Ile de France VII, Paris, France; reference 2020-A03531-38). Patients will be included after obtaining their signed informed consent. The results will be submitted for publication in peer-reviewed journals.
Trial registration number
Keywords: COVID-19, Anticoagulation, Respiratory infections, Thromboembolism
Strengths and limitations of this study.
This randomised controlled trial may contribute to establish solid recommendations with a high level of evidence on the best anticoagulation strategy to limit the extension of microvascular thrombosis and to forestall the evolution of lung and multiorgan microcirculatory dysfunction in patients with severe COVID-19 without initial macrovascular thrombosis.
Eligibility criteria differ from those retained by previous published studies on anticoagulation strategies in patients with COVID-19 given the systematic pre-randomisation screening for macrothrombosis and the exclusion of obese and renal failure patients to minimise baseline bleeding risk.
One limitation of the trial is that it is not blinded.
Introduction
Background and rationale
COVID-19, a respiratory viral infection caused by SARS-CoV-2, may predispose patients to thrombotic complication1 incurred by a combination of intense inflammation, platelet activation and endothelial dysfunction leading to respiratory distress and high mortality.2–4
The incidence of macrovascular thrombotic events varies from 10% to 30% in COVID-19 hospitalised patients depending on the type of thrombosis, arterial or venous, and the severity of the illness.2–4 Based on observational data of patients receiving routine low-dose prophylactic anticoagulation (LD-PA), several institutions released guidance statements recommending escalated anticoagulant doses to prevent macrovascular thrombotic events.5 6 In these recommendations, high-dose PA (HD-PA) and therapeutic anticoagulation (TA) can be employed either empirically or based on various criteria like body mass index or D-dimer concentration.5–7 However, other conflicting recommendations challenge this approach.6 8
Microvascular thrombotic events are another major concern in COVID-19 patients. A large review screened the autopsy findings of COVID-19-related deaths and reported the presence of microthrombi in small pulmonary vessels.9 COVID-19-induced endothelitis and coagulopathy across vascular beds of different organs precipitate widespread microvascular thrombosis.2 10 11 Thus, in critically ill COVID-19 patients without initial macrovascular thrombotic event, HD-PA or TA could be beneficial in limiting the extension of microvascular thrombosis and forestalling lung and multiorgan microcirculatory dysfunction.
To date, no randomised clinical trial has evaluated the best anticoagulation strategy in patients with severe COVID-19, in whom an initial macrovascular thrombotic event is systematically excluded. It seems important to rationalise and compare anticoagulation strategies in this population.
Hypothesis
Our hypotheses are formulated in patients who have severe COVID-19 pneumonia and are macrovascular-thrombosis free to assess: (1) First, that TA and HD-PA strategies mitigate microthrombosis, and each thwarts COVID-19 progression to respiratory failure and multiorgan dysfunction, thus decreases mortality and disease duration, as compared with LD-PA; (2) second, that TA outperforms HD-PA in this setting.
Objectives
Primary objective
The main objective is to compare the efficacy of the three strategies (LD- PA, HD-PA and TA) in reducing mortality and time to clinical improvement.
Secondary objectives
The secondary objectives are to compare the benefits and risks of the three strategies (LD-PA, HD-PA and TA) in terms of: (1) mortality, morbidity and organ dysfunction; (2) thrombotic events, bleeding events and net clinical benefit.
Ancillary study
An ancillary study will assess clinical and biological characteristics of severe COVID-19 pneumonia with or without pulmonary embolism to establish a scoring system for COVID-19-related pulmonary embolism diagnosis.
Methods and analysis
Trial design
This is a French multicentre, parallel-group, open-label, randomised controlled superiority trial to compare the efficacy and safety of three anticoagulation strategies (LD- PA, HD-PA and TA) in patients with COVID-19 pneumonia. The trial protocol follows the Standard Protocol Items: Recommendations for Interventional Trial (SPIRIT) reporting guidelines.
Study setting
The study will be conducted in 31 units (23 intensive care units (ICUs) and 8 conventional hospital wards) in 23 hospitals in France (list of the study sites in online supplemental appendix A).
bmjopen-2021-059383supp001.pdf (20.3KB, pdf)
Eligibility criteria
Inclusion criteria
Adult patients (age ≥18 years) admitted to hospital will be eligible as soon as they meet all of the following criteria:
Severe COVID-19 pneumonia, defined by: (1) new pulmonary parenchymal infiltrate and (2) positive Reverse Transcription (RT)-PCR (either upper or lower respiratory tract) for SARS-CoV-2 and (3) WHO ordinal scale ≥5.12;
Provide written informed consent as per the French law (patient, next of kin or differed consent if an emergency case).
Non-inclusion criteria
Patients presenting any of the following criteria will not be included:
Pregnant or breastfeeding women.
Post partum (6 weeks).
Attaining the extremes of body weight (<40 kg or >100 kg).
Hospital admission of more than 72 hours (if the WHO ordinal scale is 5 at the time of inclusion) or ICU admission of more than 72 hours (if the WHO ordinal scale is 6 or more at time of inclusion).
Clinical need for TA.
Bleeding related to haemostasis disorders, acute clinically significant bleeding, presence of active gastrointestinal ulcer or any organic lesion with high risk for bleeding.
Platelet count <50 x 10∧9/L.
Within 15 days of recent surgery, within 24 hours of spinal or epidural anaesthesia;
A history of intracranial haemorrhage, large acute ischaemic stroke, known intracranial malformation or neoplasm, acute infectious endocarditis.
Severe renal failure (creatinine clearance <30 mL/min).
Iodine allergy.
Hypersensitivity to heparin or its derivatives including low-molecular-weight heparin (LMWH).
A history of type II heparin-induced thrombocytopaenia (HIT).
Chronic oxygen supplementation.
Moribund patient or death expected from an underlying disease during the current admission.
Patient deprived of liberty and persons subject to institutional psychiatric care.
Patients under guardianship or curatorship.
Participation in another interventional research on anticoagulation.
Intervention
All patients hospitalised with a positive RT-PCR (either upper or lower respiratory tract) for COVID-19 (SARS-CoV-2) in the participating centres will be systematically screened every day looking for inclusion and non-inclusion criteria. The number of patients who do not meet the inclusion criteria will be reported prospectively in a paper register by each of the participating centres. A patient identification number as well as the reason for non-inclusion will be noted (local register of non-inclusion in each of the concerned centres).
Inclusion (D0) is performed as soon as possible, within 72 hours of hospital admission (if the WHO ordinal scale is 5 at time of inclusion,12) or within 72 hours of ICU admission (if the WHO ordinal scale is 6 or more at time of inclusion12).
Chest CT with pulmonary angiogram (CTPA) should be performed within 72 hours before (or up to 24 hours after) inclusion; If CTPA is performed within 7 days of inclusion and the likelihood of pulmonary artery thrombosis is deemed unchanged by the clinician, the result of that CTPA might be considered at time of inclusion (figure 1).
Figure 1.
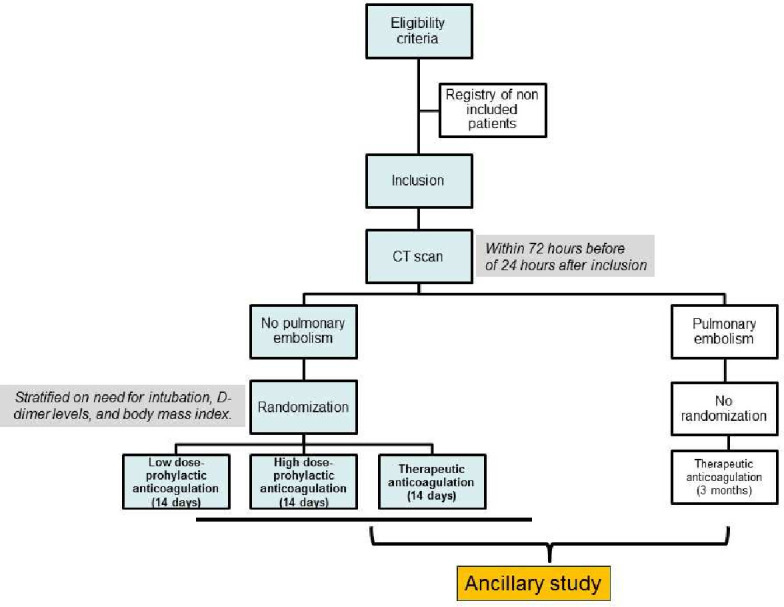
Experimental schema.
If the CTPA reveals pulmonary artery thrombosis, the patient will receive TA following current guidelines13 and will not be randomised.
If the CTPA does not show pulmonary artery thrombosis, the patient will be randomised to receive either LD-PA, HD-PA or TA for 14 days (or until hospital discharge or weaning of supplemental oxygen for 48 consecutive hours, whichever comes first). If the patient has no pulmonary artery thrombosis but presents clinical signs of deep venous thrombosis at inclusion, complete duplex ultrasound (CDUS) of the lower extremities will be performed.14 If the CDUS demonstrates deep venous thrombosis, the patient will receive TA according to current guidelines and will not be randomised; if the CDUS is negative, the patient will be randomised.
LD-PA, HD-PA and TA will be initiated immediately in all patients after randomisation using LMWH, tinzaparin at a dose of 3500 IU/24 hours, 7000 IU/24 hours or 175 IU/kg/24 hours, respectively. If tinzaparin is not available, enoxaparin can be used at a dose of 4000 IU/24 hours, 4000 IU/12 hours and 100 IU/kg/12 hours, respectively.
In case renal failure (creatinine clearance <30 mL/min) happens after randomisation or if a patient needs invasive, high bleeding risk procedure, better replace LMWH by a continuous intravenous infusion of unfractioned heparin as follows: (1) LD-PA: 100 IU/kg/24 hours; (2) HD-PA: 200 IU/kg/24 hours; (3) TA: 500 IU/kg/24 hours, adapted to the anti-Xa activity (target between 0.3 and 0.6 IU/mL) as per current guidelines.
After day 14, or hospital discharge, or in case TA is clinically indicated, or serious anticoagulation-related adverse event occurs, the trial anticoagulation strategy will be discontinued. Pursuing further anticoagulation treatment will be left at the discretion of the attending physicians.
In all groups, current recommendations for the management of COVID-19 pneumonia will be followed, including the use of dexamethasone.15
Criteria and procedures of premature withdrawal of a participant from the study
In compliance with the conventional management of severe COVID-19 pneumonia, anticoagulation will be discontinued if one of the following happens:
Major bleeding event (MBE) according to the International Society on Thrombosis and Haemostasis (ISTH) definition.
Large acute ischaemic stroke.
Skin necrosis at the injection site.
Type II HIT.
Allergic reaction.
Hospital discharge prior to day 14.
The TA strategy will be temporarily interrupted if any of the following conditions arises before terminating the treatment period (14 days from randomisation); the study drug will be resumed at least 6 hours after the resolution of the anomaly:
Clinical indication for TA.
Indication for lumbar puncture, spinal or epidural anaesthesia.
Indication for surgery.
Follow-up visits
The trial clinical examination is part of the daily practice. Parameters collected in the study are those usually collected during the management of patients with severe COVID-19 pneumonia. The trial follow-up visits are at day 7, day 28 and day 90.
If the patient is still hospitalised at day 28 and day 90, data will be collected from the patient’s medical records with the possible assistance of a clinical research technician (CRT). If the patient is discharged:
The CRT will collect the medical records from the clinical departments where the patient stayed; these will be analysed by the investigator who included the patient.
-
The CRT will collect data on the patient’s vital status and occurrence of serious adverse events during the follow-up period:
(If necessary) telephone the patient (three different attempts, days and times over 15 days).
(If necessary) telephone the physician in charge of the patient during the follow-up period.
(If necessary) telephone the patient’s treating or referring physician(s).
(If necessary) contact the town hall of the patient’s birthplace.
Endpoints
Primary endpoint
The primary endpoint is a hierarchical criterion assessed at day 28 and includes all-cause mortality followed by the time to clinical improvement. It is calculated in such a manner that death constitutes a worse outcome than delay of clinical improvement.
The time (days) to clinical improvement is defined as the time from randomisation to an improvement of at least two points (from the status at randomisation), using a seven-category ordinal scale derived from the WHO recommended instrument,12 as proposed by Coa et al16 (table 1). Since all included patients will at least require oxygen supplementation, live discharge from hospital will represent in itself a 2-point decrease in the 7-point scale, that is, clinical improvement.
Table 1.
Seven-category ordinal scale derived from the who recommended instrument (proposed by Coa et al16)
| Status of patient | Description | Points |
| Not hospitalised | Resumption of normal activities | 1 |
| Unable to resume normal activities | 2 | |
| Hospitalised | Not requiring supplemental oxygen | 3 |
| Requiring supplemental oxygen | 4 | |
| Intensive care unit | Requiring nasal high-flow oxygen therapy, non-invasive mechanical ventilation or both | 5 |
| Requiring invasive mechanical ventilation, Extracorporeal membrane oxygenation (ECMO) or both | 6 | |
| Death | Death | 7 |
Secondary endpoints
Secondary endpoints will include the following:
-
Efficacy on morbi-mortality and organ function
Individual components of the hierarchical primary endpoint, including time to clinical improvement and all-cause death at day 28.
All-cause death at day 90.
Score on WHO ordinal scale and 7-point ordinal scale at day 28.
D-dimers and Sepsis-Induced Coagulopathy Score (see detailed definition in table 2) at day 7.
Percentage of patients needing invasive mechanical ventilation at day 28.
Number of days alive and supplemental oxygen-free at day 28.
Number of days alive and mechanical ventilator-free at day 28.
Number of days alive and vasopressor-free at day 28.
Length of ICU stay at day 28 and day 90.
Length of hospital stay at day 28 and day 90.
Quality of life assessed using a quality-of-life questionnaire (EuroQol 5-Dimension 5-Level, EQ-5D-5L)17 at day 90.
Efficacy on thrombotic events: percentage of patients with at least one thrombotic event at day 28, including ischaemic stroke, non-cerebrovascular arterial thrombotic event, deep venous thrombosis, pulmonary embolism or central venous catheter-related deep venous thrombosis.
-
Tolerance to anticoagulation
Percentage of patients with at least one MBE at day 28, according to the ISTH definition.
Percentage of patients with at least one life-threatening bleeding event at day 28 according to the Randomized Evaluation of Long-term anticoagulant therapY (RE-LY) definition.
Percentage of patients with any bleeding event, whether major or minor, at day 28, with minor bleedings being all non-MBE.
Percentage of patients with HIT at day 28.
Table 2.
Sepsis-induced coagulopathy score25
| Variable | Points | |
| INR | ≤1.2 | 0 |
| >1.2 to 1.4 | 1 | |
| >1.4 | 2 | |
| Platelet count, ×109 /L | ≥150 | 0 |
| 100 to <150 | 1 | |
| <100 | 2 | |
| Total SOFA score* | 0 | 0 |
| 1 | 1 | |
| ≥2 | 2 | |
*Summation of the SOFA respiratory, cardiovascular, hepatic and renal score components.
INR, International Normal Ratio; SOFA, Sequential Organ Failure Dysfunction.
Classification of the severity of thrombotic and bleeding events will be carried out by an independent adjudication committee.
Sample size and its statistical justification
The required number of participants to be randomised is 300 patients (from 353 included). Estimates, derived from prior studies led in similar populations,16 showed that a sample of at least 300 patients (100 per group) suffices to achieve ≥80% power that is required to detect a statistically significant difference in the ranked composite primary endpoint. The analyses rely on two-sided alpha of 0.017 using Bonferroni correction for multiple testing considering three pairwise comparisons between the randomised arms. Sample size calculation assumed having day 28 mortality of 24%, 21% and 18%, and time to clinical improvement of 16±3 days (SD), 14±3 days and 12±3 days, with LD-PA, HD-PA and TA, respectively. We hypothesise that the rate of positive CTPA would be 15%.18 19 For such, we aim to include 353 patients in order to randomise 300.
Sample size calculation also considered the pairwise comparisons between the groups. For each performed comparison, 5000 samples were simulated using R software. For the first component of the hierarchical primary endpoint (mortality), survival curves were simulated based on a Weibull distribution using the R package simsurv. For the second component of the hierarchical primary endpoint (time to clinical improvement) assessed in alive patients, two different approaches, taking into account the distribution of this parameter, were used to test the robustness of results in relation with the retained hypotheses. First, a normal distribution was hypothesised with means±SD of 16±3, 14±3 and 12±3 days in LD-PA, HD-PA and TA, respectively. Second, incidence curves of clinical improvement were simulated based on Weibull distribution using the R package simsurv, with survival medians of 16, 14 and 12 days in LD-PA, HD-PA and TA groups, respectively. With both approaches, 5% of patients were systematically identified through simulation as alive patients at day 28 but without achieving clinical improvement, which is consistent with Cao et al.16 SD and mean number of days to clinical improvement, as well as shape and scale parameters of Weibull survival curves simulations were determined from the study of Cao et al.16 considering median (IQR) survival time and Kaplan-Meier curves. Within each sample/pairwise comparison, an individual score is calculated by comparing each patient in one group with all patients in the second group (23). These scores are then compared between groups using Mann-Whitney/Wilcoxon test in each of the 5000 samples, and the p value of each test is recorded. For each pairwise comparison, the percentage of tests with a p<0.017 is calculated, which gives an estimate of the achieved statistical power.
Recruitment
The expected duration of patients enrolment is 18 months starting from April 2021. The chronogram of the study is as follows: (1) December 2020: winning industrial grant award; (2) December 2020: promotion by Assistance Publique-Hôpitaux de Paris (AP-HP); (3) March 2021: approval by an independent ethics committee; (4) April 2021–October 2022: inclusion of patients; (5) 2022–2023: end of inclusions, monitoring by the participating centres and research work by the investigators; cleaning and closure of the database; blind review to screen for protocol violation, to define intention-to-treat (ITT) and per-protocol (PP) analysis populations; (6) 2022–2023: data analysis, writing the manuscript and submission for publication.
Allocation of intervention and data management
After signing the consent by the patient or their relative, all inclusion/exclusion criteria will be checked by the investigator before randomisation. Centralised blocked randomisation on the basis of a 1:1:1 ratio will be prepared by the Clinical Research Unit before the start of the trial. Randomisation will be carried out in balanced blocks and stratified by hospital centre and according to the following criteria at inclusion: need for intubation (yes or no), D-dimer levels (more or less than 3 µg/mL) and body mass index (more or less than 30 kg/m2). Patients will be randomised electronically on logging to the centralised electronic case report form (e-CRF) website ‘Cleanweb’ provided by Telemedicine technologies.
Non-identifying data will be entered into the e-CRF via a web browser by a trained investigator or research assistant at each centre. The participating centres have access to e-CRF forms via a web-based data collection system (unique identification and password by user). Patients’ follow-up and work schedule are detailed in the study Gantt chart (table 3). The e-CRF was devised by the principal investigator and the scientific supervisor of the study in collaboration with the data manager of the clinical research unit, Henri Mondor Hospital AP-HP. CRF and data dictionary (containing variables coding and definitions) are saved and archived in the clinical research unit—Henri Mondor secured servers. Paper CRF are available in the documentation provided at each site. eCRF (CleanWeb Telemedecine) uses the secured computer servers of AP-HP. The computer files used for this research are implemented in compliance with the French (amended ‘Informatique et Libertés’ law governing data protection) and European (General Data Protection Regulation, GDPR) regulations. The sponsor already obtained authorisation of CNIL (French Data Protection Agency) before implementing any data processing involving data required for this research (Ref.:MLD/MFI/AR215255 AUTORISATION). Database quality control is undertaken by Data manager of the clinical research unit—Henri Mondor Hospital, AP-HP (missing data, range checks on quantitative values, date chronology check; R-Project computer programming) and put at the disposal of the investigation team. Data management procedures are validated by the clinical research nit quality specialist and recorded in their secured servers and on paper.
Table 3.
Study Gantt chart (work schedule)
| Procedures and assessments (C=care; R=research) |
Day 0 (inclusion) |
Day 1 (randomisation) |
Day 7 | Day 2–14 | Day 15–28 (or hospital discharge) |
Day 90 ±10 days (end of study) |
| Inclusion and non-inclusion criteria | R | |||||
| Enrolment | ||||||
| Informed consent | R | |||||
| CTPA | C | |||||
| Intervention | ||||||
| Low-dose prophylactic anticoagulation strategy | C | C | ||||
| High-dose prophylactic anticoagulation strategy | C | C | ||||
| Therapeutic anticoagulation | C | C | ||||
| Assessments | ||||||
| Characteristics of the patient* | C | |||||
| Seven-category ordinal scale and its components† | C | C | C | |||
| D-dimers and platelet count | C | C | C | |||
| Sepsis coagulopathy score and its components‡ | C | C | ||||
| Adverse event | C | C | C | R | ||
| ICU stay and hospital stay | R | R | ||||
| Vital status | C | C | C | R |
*Characteristics of patients include age, gender, height, weight, severity score indicated by the Simplified Acute Physiological Score II and the Sepsis-related Organ Failure Assessment score, pre-existing conditions (chronic cardiovascular, respiratory, renal, liver or gastric diseases, arterial hypertension, diabetes mellitus, thrombotic or bleeding event, stroke, neoplasia, positive serology for HIV, solid-organ transplantation), treatments of COVID-19 at baseline, baseline organ support.
†Derived from the WHO scale.12
‡International normal ratio, platelet count, Sepsis-related Organ Failure Assessment score.25
CTPA, CT with pulmonary angiogram; ICU, intensive care unit.
Statistical methods
All analyses will be performed by the study statistician according to a predefined statistical analysis plan, using Stata V.16.1 (StataCorp) and R V.4.0.3 (R Foundation for Statistical Computing, Vienna, Austria). A two-tailed p<0.05 should indicate statistical significance.
In compliance with the SPIRIT statement, a flow diagram will describe the progress of the three groups of patients throughout the different phases of the trial (enrolment, allocation, received interventional agents, follow-up and data analysis). The analysis will be performed on an ITT basis. In case of premature interruption or withdrawal from the study, patients will not be substituted. Missing values will be described and, according to their nature and frequency, multiple imputation methods will be applied. A PP analysis will be conducted as the trial sensitivity analysis since it excludes patients wrongly randomised or who did not receive the allocated intervention.
Comparative analysis will systematically be done with (main analysis) and without adjustment for randomisation stratification factors. There is no intention to perform interim analysis. The primary endpoint analysis will be done on the ITT population whereas supportive analyses on the PP population. The latter aim is to investigate PP-excluded patients and their impact on ITT analysis, and eventually to check whether similar results can be obtained for a robust interpretation. All secondary endpoints analyses will be conducted on both ITT and PP populations to assess the robustness of the results.
Descriptive analysis
Descriptive statistical analyses will be conducted on the whole study population, in particular the randomised groups to describe their general and baseline characteristics, demographics, history, as well as numbers of premature study withdrawals. Quantitative variables will be presented as mean (±SD) or median (25–75th percentiles) according to the normality of their distribution as assessed by Shapiro-Wilk tests and graphical methods. Qualitative variables will be presented as numbers (%).
Analysis of the primary endpoint
The prespecified primary endpoint will be a ranked composite score that incorporates death and time to clinical improvement, calculated in such a manner that death constitutes a worse outcome than delayed clinical improvement. Each patient will be compared with every other patient in the study and assigned a score (equality: 0, win:+1, loss: −1) for each pairwise comparison based on who fared better. If a patient survived and the other did not, the first will be attributed +1 and the latter −1 for that pairwise comparison. If both patients in the pairwise comparison survived, the scoring will depend on who needed more time (days) to clinically improve: fewer days mean a score of +1, and more days mean a score of −1. If both patients survived and had the same number of days to clinical improvement, or if both patients died, both will score 0 for that pairwise comparison. For each patient, scores of all pairwise comparisons will be summed to obtain a cumulative score. These cumulative scores will be ranked and compared between the three groups via non-parametric Mann-Whitney test.
Analysis of secondary endpoints
Comparisons between randomised groups at given timepoints will be conducted using χ2 or Fisher exact tests, according to expected numbers in crossings, for categorical variables, and using t-test or non-parametric Mann-Whitney test (pairwise comparisons), and analysis of variance or Kruskal-Wallis tests (comparisons of >2 groups) for quantitative variables, as appropriate. Pairwise comparisons within groups (across timepoints) will be conducted using tests for paired data, that is, McNemar test for qualitative data, and t-tests for paired data or Wilcoxon signed ranks for continuous data, as appropriate.
Individual components of the composite primary endpoint will be assessed as secondary endpoints, and those include all-cause mortality at day 28 and number of days to clinical improvement. For such, calculation of time-to-event endpoints based on follow-up censored data will be employed, taking into account the competing risks of hospital discharge (for mortality evaluation) and death (for time to clinical improvement). Kaplan-Meier survival curves and cumulative incidence curves will be plotted for each treatment group, and Fine-Gray regression model will be used to calculate sub-HRs along with their 95% CIs and corresponding p values.
Analyses of independent determinants of quantitative secondary endpoints will be performed using multivariable linear regression model adjusting for baseline characteristics. As for global longitudinal analysis, we will use generalised linear regression mixed model to test interactions between timepoints, groups and prespecified predictors while entering patient level as a random effect to take into consideration the hierarchical structure of repeated data.
Tolerance analysis will examine the intervention-related adverse events, according to their period of appearance and the concerned randomised group, to compare rates and time of occurrence.
Data monitoring
The trial steering committee (principal investigator, senior investigator and methodologist) will supervise the progression and monitoring of the study. Research assistants will regularly monitor all centres on site to check protocol adherence and accuracy of the recorded data. An investigator at each centre will be responsible for daily patient screening, patient enrolment, adherence to protocol and completion of the eCRF. Since the three treatment strategies are currently used in routine practice, no data safety monitoring board was required by the ethical committee.
Patient and public involvement
Patients and/or the public were not involved in the development of this study.
Ethics and dissemination
Ethical approval
The study has been approved by an independent ethics committee (Ethics Committee, Ile de France VII, Paris, France) under the registration number 2020-A03531-38.
Consent to participate
Patients will be included after signing a written informed consent (online supplemental appendix B). If the patient is not able to understand the information given in the consent, they can be included if a next of kin consents or helps obtain the consent. Eligible patients unable to receive information and for whom a substitute decision-maker is not present, can still be included through a process of deferred consent. After recovery, the patient’s agreement to stay in the trial will be sought.
bmjopen-2021-059383supp002.pdf (97.9KB, pdf)
Confidentiality
Data will be handled according to the French law on data protection and the European GDPR. All original records will be archived at the trial sites for 15 years.
Funding and sponsorship
This study was funded by a grant from LEO Pharma. The sponsor is AP-HP (Délégation à la Recherche Clinique et à l’Innovation).
Access to data
Investigators will make the documents and individual data required for monitoring, quality control and audit of the study available to dedicated persons, in fulfilment with the law.
Dissemination policy
Findings will be published in peer-reviewed journals and presented at national and international meetings. Communications, reports and publication of the results of the study will be placed under the responsibility of the principal investigator–coordinator of the study and the steering committee. Reporting will adhere to the SPIRIT statement, and rules of publication will follow the international recommendations as for The Uniform Requirements for Manuscripts (ICMJE, April 2010) (SPIRIT checklist, online supplemental appendix C).
bmjopen-2021-059383supp003.pdf (51.9KB, pdf)
Discussion
Currently, there are no randomised controlled trials investigating the best anticoagulation strategy to manage microvascular thrombosis and to hinder the evolution of lung and multiorgan microcirculatory dysfunction in patients with COVID-19 without initial macrovascular thrombosis.
Recent trials have studied various anticoagulation strategies using heparin in COVID-19 patients.8 In the Iranian INSPIRATION trial,20 Sadeghipour et al compared the efficacy of standard LD-PA (40 mg enoxaparin once a day) with weight-based, higher dose-PA (1 mg/kg enoxaparin) in severe COVID-19 patients admitted to ICU. Higher dose-PA did not result in a significant difference in the primary outcome (a composite of adjudicated venous or arterial thrombosis, indication for extracorporeal membrane oxygenation, or mortality within 30 days), as compared with the standard-dose PA. Additionally, the risk of bleeding was similar between the two groups. An international, multiplatform, randomised clinical trial combined data from patients who had already been enrolled in a conventional randomised trial (ACTIV-4a) and in two response-adaptive randomisation trials (REMAP-CAP and ATTACC). They found that the potential benefits and risks of TA versus standard PA (at a lower or higher dose based on local practice) depended on the initial severity of patients.21 22 In critically ill patients, TA did not improve the primary outcome of organ support–free days at day 21 and was associated with more major bleedings (3.8% vs 2.3%) as compared with PA.22 In non-critically ill patients, TA appeared to increase the probability of survival to hospital discharge with reduced use of cardiovascular or respiratory organ support. However, major bleeding occurred in 1.9% of the patients receiving TA and in 0.9% of those receiving PA.21
Our ANTICOVID study differs from these studies in several methodological and clinical aspects. The inclusion criteria differ as CTPA is systematically (ANTICOVID) vs non-systematically (INSPIRATION, REMAPCAP, ACTIV-4, ATTACC) performed to exclude macrothrombosis, which is de facto an indication for curative anticoagulation. By excluding macrothrombosis before randomisation, ANTICOVID will provide an answer to the specific question of microthrombosis. On the other hand, and in contrast to other trials, ANTICOVID explicitly excludes patients with renal failure (creatinine clearance <30 mL/min), which has been entangled as an independent risk factor for bleeding in critically ill patients requiring TA.23 Additionally, ANTICOVID excludes patients attaining the extremes of body weights, for whom LMWH dosage has not been assessed. In particular, obese patients, since they have a lower proportion of lean body mass in relation to their big total body weight. As a result, determining LMWH dosage based on total body weight could cause supra-therapeutic anticoagulation.24 ANTICOVID will allow evaluation of anticoagulation dose escalation in a population with a minimal baseline bleeding risk. Eventually, our study is the only one to investigate in separate arms, lower and higher prophylactic doses, and compare them with curative anticoagulation. For all of the above, ANTICOVID trial is needed in order to explore the lowest effective dose (given the bleeding risk of anticoagulation) and to answer the key question of dose escalation anticoagulation in COVID-19 patients without initial macrovascular thrombosis.
Our study has several limitations. Anticoagulation assignment was open-label given the overburdened, resource-limited healthcare system during the pandemic. Time to clinical improvement, the second component of the hierarchical primary endpoint, may be too subjective, thus liable to performance bias. Detection bias could occur if potential events (especially incidental thromboses) were less likely to be investigated in patients receiving TA than in those receiving LD-PA or HD-PA. The opposite could be true for bleeding events. Reporting bias is unlikely for the primary outcome given that (1) all cause death is objective and (2) ICU hospitalisation and type of ventilatory support determining time to clinical improvement are unambiguously supported by medical records. Nonetheless, an independent clinical events committee will blindly adjudicate all relevant outcomes. Both ICU and non-ICU patients are eligible, so our future results should not be compared directly to those of other trials limited only to critically ill patients or to non-ICU patients. We will not include obese patients and patients with renal failure, which limits the generalisability of the results to all COVID-19 inpatients. Finally, we will not take into account symptoms duration in the analysis neither quantify microvascular thrombosis on CTPA.
In summary, ANTICOVID trial is an open label randomised controlled trial testing the efficacy of three routinely used anticoagulation strategies (LD-PA, HD-PA and TA) in limiting the extension of microvascular thrombosis in severe COVID-19 patients without initial macrovascular thrombosis. The trial targets a well-selected population (notably at lower risk of bleeding), with a suitable primary objective and experimental design, to provide a robust response (lowest effective dose with respect to the bleeding risk of anticoagulation). Therefore, this trial may help establish international recommendations with a high level of evidence for the efficacy and safety of anticoagulation dose escalation needed to improve outcomes in severe COVID-19 patients.
Supplementary Material
Acknowledgments
The authors thank Professor Nadia Aissaoui, MD.PhD (Department of Intensive Care, Hôpital Cochin, Assistance Publique-Hôpitaux de Paris, Paris, France), and Professor Matthieu Schmidt, MD.PhD (Department of Intensive Care, Hôpital Pitié-Salpêtrière, Assistance Publique-Hôpitaux de Paris, Paris, France) for being part of the independent endpoint adjudication committee. We thank Yasmina Moussaoui (Unité de Recherche Clinique Henri Mondor, Hôpitaux Universitaires Henri Mondor-Albert Chenevier, AP-HP, Créteil, France) for her valuable work.
Footnotes
Twitter: @AlexRobert84, @Denis_Doyen
Contributors: VL and AMD in collaboration with all authors designed the study and wrote the manuscript together. EA provided substantial contributions to the conception and design of the study, and wrote the statistical analysis plan and estimated the sample size. VL, DC, NH, BM, HA-O; FB, SC, AR, EV, MF, DD, MM, SP, EN-S, BS, NZ, S-AP, MD, WJ, EM, J-FT, MT, KR, SG, SB, GV, MF, EA and AMD contributed for drafting the work, revising it critically for important intellectual content and approved the final version of the manuscript. VL, DC, NH, BM, HA-O; FB, SC, AR, EV, MF, DD, MM, SP, EN-S, BS, NZ, S-AP, MD, WJ, EM, J-FT, MT, KR, SG, SB, GV, MF, EA and AMD gave their agreement to be accountable for all aspects of the work, and ensure the accuracy and integrity of any part of it.
Funding: This study was funded by a grant from LEO Pharma. The sponsor is Assistance Publique—Hôpitaux de Paris, AP-HP (Délégation à la Recherche Clinique et à l’Innovation, DRCI).
Competing interests: AMD reports lectures for Leo Pharma. EA reports personal fees from GBT, personal fees from Hemanext, both unrelated to the present study. GV received research grant from Bio-Mérieux, SOS Oxygène, Janssen, all unrelated to the present study; and advisory board fees from BioMérieux that are unrelated to the present study. VL receives advisory board fees from Amomed, unrelated to the present study.
Patient and public involvement: Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Ethics statements
Patient consent for publication
Not applicable.
References
- 1.Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020;135:2033–40. 10.1182/blood.2020006000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y, Xiao M, Zhang S, et al. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med 2020;382:e38. 10.1056/NEJMc2007575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Danzi GB, Loffi M, Galeazzi G, et al. Acute pulmonary embolism and COVID-19 pneumonia: a random association? Eur Heart J 2020;41:41. 10.1093/eurheartj/ehaa254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Samkari H, Karp Leaf RS, Dzik WH, et al. COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020;136:489–500. 10.1182/blood.2020006520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Susen S, Tacquard CA, Godon A. Propositions Du GFHT/GIHP pour Le traitement anticoagulant pour La Prévention du Risque thrombotique CheZ un patient hospitalisé avec COVID-19 2020.
- 6.Bikdeli B, Madhavan MV, Jimenez D, et al. COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-up: JACC state-of-the-art review. J Am Coll Cardiol 2020;75:2950–73. 10.1016/j.jacc.2020.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Susen S, Tacquard CA, Godon A, et al. Prevention of thrombotic risk in hospitalized patients with COVID-19 and hemostasis monitoring. Crit Care 2020;24:364. 10.1186/s13054-020-03000-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Recommandations d’experts portant sur la prise en charge en réanimation des patients infectés SARS-CoV2. SRLF-SFAR -GFRUP-SPILF-SPLF.. Mise en oeuvre avec la mission COREB nationale 2020. [Google Scholar]
- 9.Maiese A, Manetti AC, La Russa R. Autopsy findings in COVID-19-related deaths: a literature review. Forensic Sci Med Pathol. [Epub ahead of print: 7 October 2020]. 10.1007/s12024-020-00310-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. The Lancet 2020;395:1417–8. 10.1016/S0140-6736(20)30937-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular Endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med 2020;383:120–8. 10.1056/NEJMoa2015432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection . A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect Dis 2020;20:e192–7. 10.1016/S1473-3099(20)30483-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konstantinides SV, Meyer G, Becattini C. Esc guidelines for the diagnosis and management of acute pulmonary embolism developed in collaboration with the European respiratory Society (ERS): the task force for the diagnosis and management of acute pulmonary embolism of the European Society of cardiology (ESC). Eur Respir J 2019;2019:54. [DOI] [PubMed] [Google Scholar]
- 14.Laurence N, Cronan John J, Lilly Michael P. Ultrasound for lower extremity deep venous thrombosis. Circulation 2018;137:1505–15. [DOI] [PubMed] [Google Scholar]
- 15.Keyt H. WHO recommends corticosteroids for patients with severe or critical COVID-19. Ann Intern Med 2021;174:JC2. 10.7326/ACPJ202101190-002 [DOI] [PubMed] [Google Scholar]
- 16.Cao B, Wang Y, Wen D, et al. A trial of Lopinavir-Ritonavir in adults hospitalized with severe Covid-19. N Engl J Med 2020;382:1787–99. 10.1056/NEJMoa2001282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brooks R. EuroQol: the current state of play. Health Policy 1996;37:53–72. 10.1016/0168-8510(96)00822-6 [DOI] [PubMed] [Google Scholar]
- 18.COVID-ICU Group on behalf of the REVA Network and the COVID-ICU Investigators . Clinical characteristics and day-90 outcomes of 4244 critically ill adults with COVID-19: a prospective cohort study. Intensive Care Med. [Epub ahead of print: 29 October 2020]. 10.1007/s00134-020-06294-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suh YJ, Hong H, Ohana M. Pulmonary embolism and deep vein thrombosis in COVID-19: a systematic review and meta-analysis. Radiology 2020:203557. 10.1148/radiol.2020203557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.INSPIRATION Investigators . Effect of Intermediate-Dose vs standard-dose prophylactic anticoagulation on thrombotic events, extracorporeal membrane oxygenation treatment, or mortality among patients with COVID-19 admitted to the intensive care unit. JAMA. [Epub ahead of print: 18 March 2021]. 10.1001/jama.2021.4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ATTACC Investigators, ACTIV-4a Investigators, REMAP-CAP Investigators, et al. Therapeutic anticoagulation with heparin in Noncritically ill patients with Covid-19. N Engl J Med 2021;385:790–802. 10.1056/NEJMoa2105911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.REMAP-CAP Investigators, ACTIV-4a Investigators, ATTACC Investigators, et al. Therapeutic anticoagulation with heparin in critically ill patients with Covid-19. N Engl J Med 2021;385:777–89. 10.1056/NEJMoa2103417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown RB, Klar J, Teres D, et al. Prospective study of clinical bleeding in intensive care unit patients. Crit Care Med 1988;16:1171–6. 10.1097/00003246-198812000-00001 [DOI] [PubMed] [Google Scholar]
- 24.Clark NP. Low-Molecular-Weight heparin use in the obese, elderly, and in renal insufficiency. Thromb Res 2008;123 Suppl 1:S58–61. 10.1016/j.thromres.2008.08.005 [DOI] [PubMed] [Google Scholar]
- 25.Iba T, Nisio MD, Levy JH, et al. New criteria for sepsis-induced coagulopathy (sic) following the revised sepsis definition: a retrospective analysis of a nationwide survey. BMJ Open 2017;7:e017046. 10.1136/bmjopen-2017-017046 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2021-059383supp001.pdf (20.3KB, pdf)
bmjopen-2021-059383supp002.pdf (97.9KB, pdf)
bmjopen-2021-059383supp003.pdf (51.9KB, pdf)