

Abstract
Radiographic axial spondyloarthritis (also known as ankylosing spondylitis [AS]) is a chronic immune-mediated arthritis characterized by inflammation of the axial skeleton, peripheral joints, and entheses. It is estimated that 1 in every 200 people are affected by AS, making it an important healthcare and socioeconomic issue. In this review, we aim to explore the current understanding of AS risk factors and provide a comprehensive update. Multiple search strings were used to identify articles of interest published in PubMed between January 1, 2013, and February 1, 2021. On the basis of the literature review and analysis, we present up-to-date information on the risk factors of developing AS and our viewpoints on disease onset and progression. Multiple genetic and nongenetic risk factors have been suggested in the onset of AS. HLA-B27 is known to have a strong association with the disease, but other genes have been implicated in disease development. Aside from genetics, other factors are thought to be involved; up to 70% of patients with AS have subclinical intestinal inflammation, suggesting that the origin of the disease may be in the gut. The exact mechanism by which AS onset begins is most likely complex and multifactorial.
Keywords: Ankylosing spondylitis, Genetics, HLA-B27, Pathogenesis, Radiographic axial spondyloarthritis, Risk factors
Introduction
Radiographic axial spondyloarthritis (axSpA, also known as ankylosing spondylitis [AS]) is a chronic, progressive, immune-mediated arthritis characterized by the absence of rheumatoid factor and presence of inflammation of the axial skeleton, peripheral joints, entheses, and extra-articular sites such as the eye and bowel. AS is perhaps one of the best recognized forms of spondyloarthritis (SpA), which comprises axSpA, peripheral SpA (including psoriatic arthritis, reactive arthritis, and arthropathy of inflammatory bowel disease), and undifferentiated SpA. Radiographic sacroiliitis is the key distinguishing feature of AS although patients usually report symptoms such as back pain for several years before radiographic changes are observed [1]. The presentation of axSpA without radiographic sacroiliitis denotes nonradiographic axSpA (nr-axSpA) [2]. In 2009–2010, the National Health and Nutrition Examination Survey estimated that the prevalence of axSpA among adults in the USA varies from 0.9 to 1.4% [3]. Approximately, 1 in 200 people are affected by AS and over one in 100 by axSpA (although there are marked variations in estimates, which are thought to be partially due to differences in study methodology and criteria used) and the number of AS cases in Europe and Asia is estimated to be 1.30–1.56 million and 4.63–4.98 million, respectively [4]. The onset of AS usually occurs before the age of 45 years [5], when adults are in their peak productive years, and patients experience limited physical function, significant loss of work productivity, and a decreased quality of life during this period after disease onset [6]. Thus AS is an important healthcare and socioeconomic issue.
Great recent progress has been made in elucidating the pathogenesis of AS, which has led to a better understanding of risk factors and disease causation, and to the development of targeted treatments. Genetic studies have made a significant contribution to our understanding of AS [7]. Gut involvement in patients with AS is also common, and as such, the role of the intestinal microbiome in AS onset is an active area of research [8]. The role of other environmental triggers (i.e., infection, mechanical stress) has also provided important clues [9], and changes in the immune system composition and function have also been noted in patients with AS vs healthy controls [10].
Overall, it remains unclear how interactions between genes, microbes, mechanical stress, gender, and other environmental and lifestyle factors predispose patients to the development of AS. The exact mechanisms are complex and multifactorial and the research agenda continues forward. Recognizing the risk factors, as well as understanding gene-gene and gene-environment interactions, may offer valuable insights into the etiology of AS and have important implications for diagnosis and treatment strategies. Numerous reviews have recently been published on the genetics of AS, and several systematic literature reviews have been published on AS susceptibility [11–15], but these reviews only considered one or two individual risk factors. Thus, the aim of this review is to provide a more in-depth update on current research and comprehensively outline risk factors associated with AS.
Methods
Search strategy
The design of the present analysis was based on a systematic literature review using theoretical recommendations suggested by The PRISMA Group [16]. Multiple search strings using Medical Subject Headings (MeSH) and MeSH major topics (majr) were used to identify articles of interest published in PubMed between January 1, 2013, and February 1, 2021. The primary search included the terms (“Spondylitis, Ankylosing”[majr] AND “Spondylitis, Ankylosing/etiology”[MeSH]) OR (“Spondylitis, Ankylosing”[majr] AND “Spondylitis, Ankylosing/ethnology”[MeSH]) OR (“Spondylitis, Ankylosing”[majr] AND “risk factors”[MeSH]) OR (“Spondylitis, Ankylosing”[majr] AND “sex distribution”[MeSH]), OR (“Spondylitis, Ankylosing”[majr] AND “Klebsiella” MeSH]). Two individuals independently reviewed the titles and available abstracts to retrieve potentially relevant studies, and an adjudicator made the final decision on any difference in reviewer opinion. The full texts and bibliographies of relevant English-language articles were evaluated for specific data relating to risk factors in patients with AS. Case studies and comments were not included. Additional articles were identified from the bibliographies of the articles from the primary search or through secondary searches using related terms and included based on clinical expert opinion. Only those manuscripts reporting studies deemed relevant to the objectives of this study, along with those recommended by clinical experts, were included.
Study inclusion and characteristics
According to the defined search strategy, the literature search identified 1066 articles, which are summarized in Fig. 1. Through examination of the title and abstract, 500 studies were identified for full-text analysis. Of these, 436 were excluded due to lack of relevance with the study objectives, leaving 64 articles from the initial searches for inclusion. These were supplemented with an additional 45 articles of interest from the authors, including several preceding the time period of the review felt important to this review and five added that were published after the search was conducted while the article was being prepared for publication as well as secondary analyses of the literature. Overall, a total of 128 articles were included in this review.
Fig. 1.
PRISMA flow diagram
Results
Risk factors for the development of AS
We identified multiple genetic, nongenetic, and stochastic risk factors in the development of AS; these are summarized in Table 1 and detailed herein.
Table 1.
Summary of AS risk factors
| Genetic |
| HLA-B27 [17–38] |
| MHC genes other than HLA-B27 [12, 18, 25, 28, 38–46] |
| ERAP1/2 [7, 25, 47–56] |
| SNPs within or adjacent to other genes (detailed in Table 3) [7, 11, 12, 40, 45, 47–70] |
| Gut microbiota and associated factors [71–74] |
| Altered gut microbiota, most notably a presence of [8, 75–79]: • Lachnospiraceae • Prevotellaceae • Ruminococcaceae • Rikenellaceae • Porphyromonadaceae • Bacteroidaceae |
| Lack of gut-induced protection by absence of breast feeding [80] |
| Infections |
| Klebsiella pneumoniae [81–87] (not established) |
| Respiratory tract infections and tonsillitis during childhood hospitalization [88] |
| Mechanical stress [89–93] |
| Gender at birth |
| Male [27, 94–102] |
| Social and lifestyle factors |
| Having older siblings [103] |
| Vitamin D deficiency [15, 104] |
| Smoking (including e-cigarettes) [105–108] |
| Other |
| Altered immune system and functionality [109–112]: • Lower number of Tregs [111] • Lower IL-10 production by CD4+ T cells but higher IL-6 production by CD8+ T cells [111] • Increased MIF levels in serum and synovial fluid [112] |
IL, interleukin; MIF, macrophage migration inhibitory factor; SNP, single-nucleotide polymorphism; Treg, regulatory T cell
Genetics
AS is widely regarded as an inherited disease, with over 90% of the risk of development attributed to HLA-B27 [113]. HLA-B27 positivity is present in 85 to 95% of White and Han Chinese patients and over 86% of Hispanic patients with AS [17, 18], although only ≤ 8% of the general population overall [19]. In fact, only approximately 5% of HLA-B27–positive individuals in the general population have SpA [20]. In the Middle East and North Africa, and in American Black patients, the prevalence of HLA-B27 among patients with AS ranges from approximately 50 to 84% [18, 21]. AS cases that do not involve HLA-B27 comprise > 10%, and twin concordance rates are not 100% [113]. Concordance rates of 63% and 27%, respectively, were reported for HLA-B27-positive monozygotic (17 of 27 patients) and dizygotic (4 of 15 patients) twins [113]. However, HLA-B27 is still considered an important factor that is highly associated with the development of AS, particularly for the magnitude and severity of bone marrow edema lesions in the sacroiliac joints in early disease [22]. The HLA-B27 family has a high degree of genetic polymorphism and consists of 328 alleles and 231 protein subtypes ranging from HLA-B*27:01 to HLA-B*27:232 (the subtype HLA-B*27:22 was found to be in error and was withdrawn); these subtypes differ from each other in only a few amino acids, which may alter the peptide-binding specificity of the molecule [23, 114]. The most common HLA-B27 subtype, the ancestral subtype HLA-B*27:05, is distributed ubiquitously worldwide, is found in all races and ethnicities, and is strongly associated with AS [18, 24, 25]. Specific populations have other HLA-B*27 subtypes that are positively associated with AS (Table 2) such as the “major” or most common subtypes HLA-B*27:02 (in people of European, Chinese, and Mediterranean or Northern African ancestry), HLA-B*27:04 (in Eastern Asia and China), HLA-B*27:07 (in Western Asia), and HLA-B*27:15 (derived from HLA-B*27:04 and found in China). Other less rare HLA-B27 subtypes that are not associated with AS include HLA-B*27:03 (in West Africa), HLA-B*27:06 (found in Southeast Asia and derived from HLA-B*27:04) [26], and HLA-B*27:09 (found primarily in Sardinia) [24], the latter two rarely, if ever, occurring in patients with AS.
Table 2.
MHC genes associated with the development of AS and associated risks
| MHC class | Populations | Associated/increased risks |
|---|---|---|
| HLA-B*27 | Overall [17, 18, 21, 22, 24, 25, 27–36, 113] | AS |
| HLA-B*27:02 | White [18, 25] | AS |
| Chinese [18] | ||
| Mediterranean [21] | ||
| HLA-B*27:03 | West African [24, 39] | AS |
| HLA-B*27:04 | Chinese [18, 24] | AS, AS with peripheral joint involvement in Chinese patients with AS [27] |
| Eastern Asian [24] | ||
| HLA-B*27:05 | White [18, 25 | AS |
| Chinese [18, 24] | ||
| Eastern Asian [18] | ||
| HLA-B*27:06 | Southeast Asian [24, 26] | Negatively associated with AS [26] |
| HLA-B*27:07 | Western Asian [24] | |
| HLA-B*27:08 | Southern Asian [24] | AS |
| HLA-B*27:09 | Sardinia [24] | Negatively associated with AS [24] |
| HLA-B*27:15 | Chinese [18] | AS |
| HLA-B27/HLA-B*40 (B60 when defined by alloantisera) | Dutch [36], US white, black, Han Chinese [18], European white [25] | AS |
| HLA-B*07, B*15, B*35 | White, black, Han Chinese [18], Colombians [38] | Negatively associated with AS but positively with peripheral SpA [36] |
| HLA-A*02:01 | European and American white [25, 40] | AS |
| HLA-C*12:02:02 | Taiwanese [43] | AS |
| HLA-DRB1*15:01/DQB1*06:02 | White [18, 25, 28, 45] | Negatively associated with AS, positively with uveitis |
| HLA-DPA1/DPB1 (DPB1*03:01) | US and European whites [18, 25, 40, 44, 45] | AS |
AS, ankylosing spondylitis; MHC, major histocompatibility complex
Other HLA-B27 subtypes are rare, representing amino acid substitutions derived from HLA-B*27:05 and its major subtypes (Fig. 2). Studies show that those positive for specific HLA-B27 subtypes have an increased risk of developing AS and specific AS manifestations, including peripheral joint involvement [25, 27] and uveitis [28] in some populations.
Fig. 2.
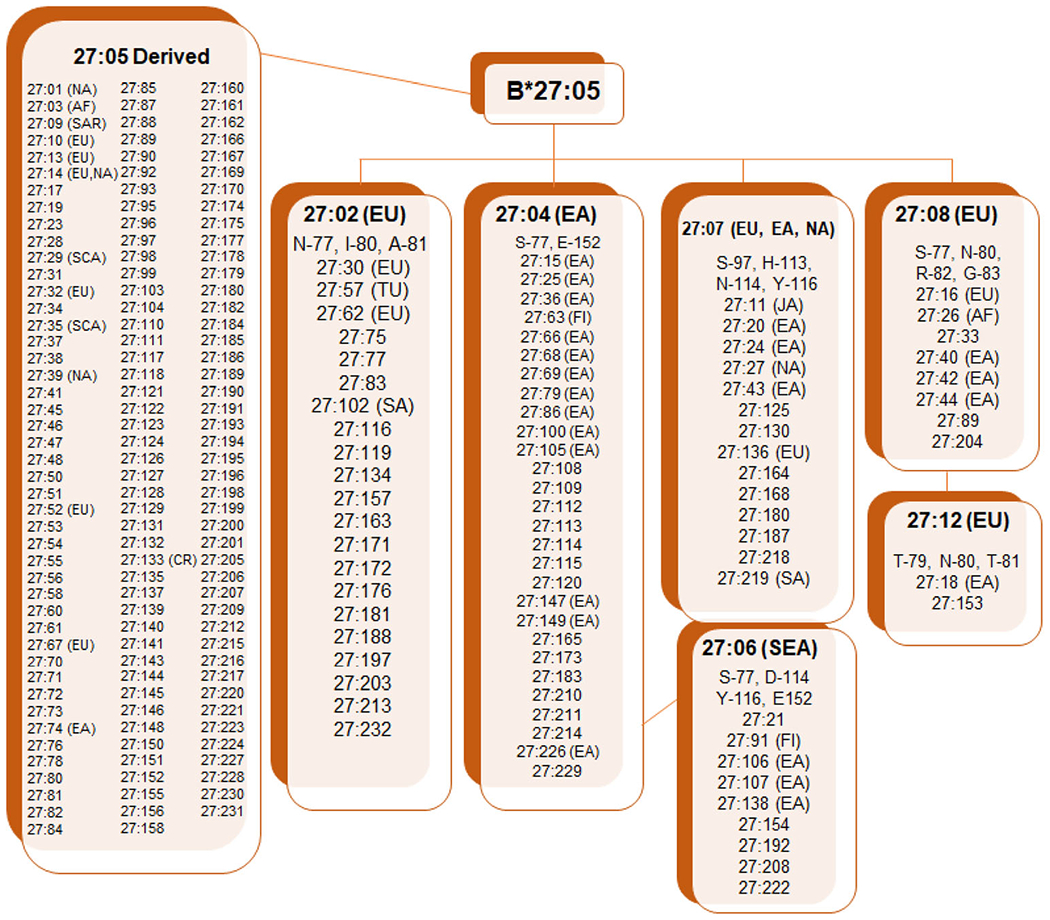
Derivation of main and rare HLA-B27 subtypes from founder HLA-B*27 allele B*27:05. AF, African; EA, Eastern Asian; EU, European; FI, Filipino; JA, Japanese; NA, North American; SA, South American; SCA, Scandinavian; SEA, Southeast Asian; TUR, Turkic. Subtypes where ethnicity origin was not designated derived from individuals where ethnicity was not recorded. Source: https://www.ebi.ac.uk/ipd/imgt/hla/allele.html; accessed September 27, 2020
The role that HLA-B27 plays in the pathogenesis of AS is a subject of much investigation. Six mechanisms have been proposed:
Presentation of an “arthritogenic” peptide [29]. HLA-B27, a major histocompatibility complex (MHC) class I molecule, presents endogenous peptides, such as those from viruses, bacteria, neoplastic, or “self” peptides that have been degraded intracellularly in lysosomes to the αβ T cell receptor on CD8+ T lymphocytes or to the killer immunoglobulin (KIR) receptor on natural killer (NK) cells. Despite a great deal of work that has gone into identifying a peptide specific for SpA, this has proven to be an elusive target. That said, CD8+ T lymphocytes have a role in AS pathogenesis, and recent data do suggest AS patients have a reduced cytotoxic CD8+ T cell profile in their peripheral blood, and an enrichment in the inflamed joint [30].
HLA-B27 heavy chains have the rather unique tendency to misfold in the endo/ ; plasmic reticulum (ER) compared to other HLA-B alleles [31–33]. HLA-B27 misfolding (i.e., incorrect folding and loading of peptides) has been postulated as one reason for genetic susceptibility [31]. One study demonstrated that the AS-associated HLA-B27 subtypes B*27:02, B*27:05, and B*27:07 differed from the non–AS-associated B*27:06 and B*07:02 alleles by a greater tendency to accumulate in intracellular ER-derived vesicles, at high expression levels examining cells from SpA patients or HLA–B27/human β2-microglobulin (hβ2m)–transgenic rats [32, 33]. This misfolding and the accumulation of misfolded HLA-B27 heavy chains in the ER results in ER-associated degradation of the heavy chains and leads to a proinflammatory unfolded protein response, which activates the innate immune response and upregulates proinflammatory cytokines such as interferon gamma and interleukin (IL)-23, as well as other cytokines, especially those in the T helper 17 (Th17) pathway [31–33].
HLA-B27 heavy chains have a striking tendency to self-adhere and form homodimers by virtue of having a cysteine residue at position 67 in the « 1 domain (and elsewhere). These homodimers have been detected at the cell surface and are recognized by KIR and leukocyte immunoglobulin-like receptors. How and if homodimerization affects predisposition to AS is unclear, especially in that HLA-B27 subtypes that are associated with AS (HLA-B*27:02, B*27:04, B*27:05, B*27:07) and those not disease-associated (B*27:06, B*27:09) share this property, with the exception of B*27:03, which does not efficiently-self adhere [34].
d. HLA-B27–positive individuals exhibit alteration of intracellular invasion and killing of arthritogenic bacteria. This is especially seen for reactive arthritis, where impaired intracellular killing of causative microorganisms has been described, leading to intracellular bacterial persistence and upregulated cytokine production [35].
HLA-B27 itself, either through the trimolecular complex of B27 heavy chain, α2 microglobulin, and peptide, or of free B27 heavy chains or homodimers (or peptides derived therefrom) are recognized as antigenic by the T cell receptor (or peptides bound therein) on CD4+ T lymphocytes, generating an autoimmune response [36].
HLA-B27–positive individuals have an altered microbiome, which influences disease susceptibility (discussed below).
MHC genes other than HLA-B27
Other HLA-B alleles have also been implicated with AS susceptibility, albeit to a much lesser extent than HLA-B27. The development of AS has been positively associated with HLA-B*40 and negatively associated with HLA-B*07, B*35, and B*57, as demonstrated in studies of Whites, Han Chinese, and Blacks [18, 25, 37, 38]. HLA-B*15 favors the development of peripheral vs axial spondyloarthritis [39]. In HLA-B27-negative West Africans, HLA-B*14:03 may render susceptibility to AS [38]; this subtype was not observed in African Americans [18]. HLA-A*02:01 was independently linked to AS susceptibility in a large multinational imputation analysis [40]. DNA sequencing implicated the MHC related gene MICA in a large cohort of White American patients with AS and confirmed in a Han Chinese cohort [41]; however this was not confirmed in a much larger imputation study [42]. Similarly, HLA-C alleles were associated with AS in a study of Taiwanese patients [43], although this was not seen in a larger study of American White patients after correcting for linkage with HLA-B27 [18]. More compelling were associations of AS with MHC class II alleles, including HLA-DRB1 and especially with alleles at HLA-DPA1 and DPB1 [18, 28, 44, 45]. Correlations with other MHC loci, such as TAP and TNF [12, 46], are likely explained by linkage to HLA-B27 haplotypes.
Non-MHC genes
Genome-wide association studies have identified and characterized the role of > 100 susceptibility genes or loci outside the MHC locus genes for AS, Crohn’s disease, ulcerative colitis, and psoriasis, which are summarized in Fig. 3. Especially important are the genes endoplasmic reticulum aminopeptidase 1 (ERAP1) and interleukin 23 receptor (IL23R); ERAP1 is more frequently identified in patients with AS who are HLA-B27 positive than in those who are HLA-B27 negative. AS susceptibility genes that have been identified by genomewide association and gene chip studies, including ERAP1, are summarized in Supplemental Table 1 [7, 11, 12, 40, 44, 47–69]. Of particular note is a recent study comparing 2752 patients with AS with acute anterior uveitis (AAU) and 3836 patients with AS without AAU; novel AAU-associated associations were discovered [45] (Supplemental Table 1). A major achievement of these studies relates to the identification of important biological pathways that are likely responsible for AS pathogenesis. This identification has led to the discovery of novel therapeutic targets. Understanding genetic differences in AS pathogenesis may allow better patient and treatment matching.
Fig. 3.
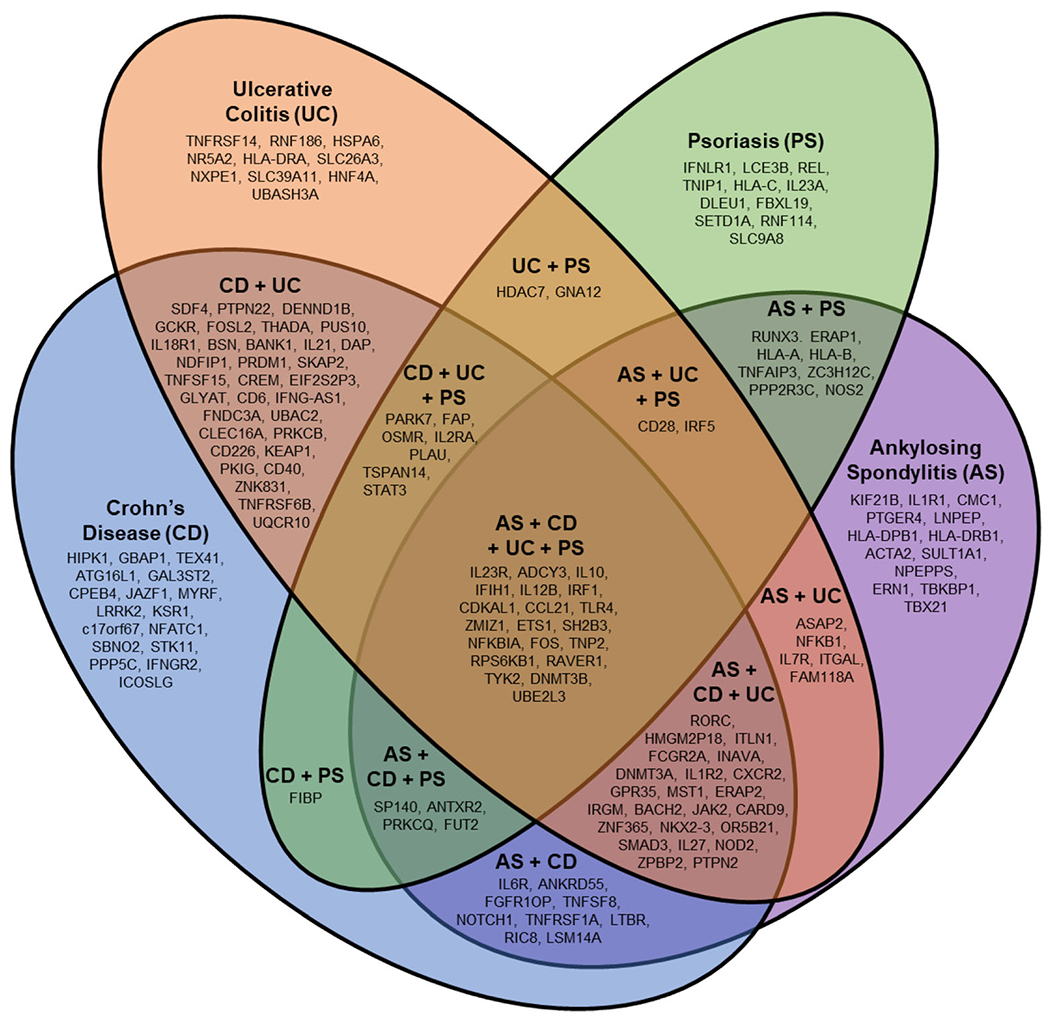
Overlap between AS, Crohn’s disease, ulcerative colitis, and psoriasis susceptibility genes. ACTA2, alpha actin 2, smooth muscle aortic; ADCY3, adenylate cyclase 3; ANKRD55, ankyrin repeat domain-containing protein 55; ANTXR2, anthrax toxin receptor 2; ASAP2, ArfGAP with SH3 domain, ankyrin repeat and PH domain 2; ATG16L1, autophagy16-like 1; BACH2, BTB and CNC homology 2; BANK1, B cell scaffold protein with ankyrin repeats-1; BSN, bassoon mouse, homolog of (zinc finger 231); C17orf67, chromosome 17 open reading frame 67; CARD9, caspase recruitment domain family member 9; CCL21, C-C motif chemokine ligand 21; CD6, CD6 molecule; CD28, CD28 molecule; CD40, CD40 molecule; CD226, CD226 molecule; CDKAL1, CDK5 regulatory subunit associated protein 1 like 1; CLEC16A, C-type lectin domain containing 16A; CMC1, C-X9-C motif containing 1; CPEB4, cytoplasmic polyadenylation element binding protein 4; CREM, cAMP responsive element modulator; CXCR2, C-X-C motif chemokine receptor 2; DAP, death-associated protein; DENND1B, DENN domain containing 1B; DLEU1, deleted in lymphocyte leukemia 1; DNMT3A, DNA methyltransferase 3 alpha; DNMT3B, DNA methyltransferase 3 beta; EIF2S2P3, eukaryotic translation initiation factor 2 subunit 2 beta pseudogene 3; ERAP1, endoplasmic reticulum aminopeptidase 1; ERAP2, endoplasmic reticulum aminopeptidase 2; ERN1, endoplasmic reticulum to nucleus signaling 1; ETS1, ETS proto-oncogene 1, transcription factor; FAM118A, family with sequence similarity 118 member A; FAP, fibroblast activation protein alpha; FBXL19, F-box and leucine rich repeat protein 19; FCGR2A, Fc fragment of IgG receptor IIa; FGFR1OP, FGFR1 oncogene partner; FIBP, FGF1 intracellular binding protein; FNDC3A, fibronectin type III domain containing 3A; FOS-V, Finkel-Biskis-Jinkins murine osteosarcoma viral oncogene homolog; FOSL2, FOS-related antigen 2; FUT2, fucosyltransferase 2; GAL3ST2, galactose-3-O-sulfotransferase 2; GBAP1, glucosylceramidase beta pseudogene 1; GCKR, glucokinase regulator; GLYAT, glycine-N-acyltransferase; GNA12, G protein subunit alpha 12; GPR35, G protein-coupled receptor 35; HDAC7, histone deacetylase 7; HIPK1, homeodomain interacting protein kinase 1; HLA-A, human eukocyte antigen A; HLA-B, human leukocyte antigen B; HLA-C, human leukocyte antigen C; HLA-DP, human leukocyte antigen DP; HLA-DRA, human leukocyte antigen DR alpha; HLA-DRB1, human leukocyte antigen DR beta 1; HMGN2P18, high mobility group nucleosomal binding domain 2 pseudogene 18; HNF4A, hepatocyte nuclear factor 4-alpha; HSPA6, heat shock 70kd protein 6; ICOSLG, inducible T cell costimulator ligand; IFIH1, interferon-induced helicase C domain-containing protein 1; IFNG-AS1, interferon gamma; IFNGR2, interferon gamma receptor 2; IFNLR1, interferon lambda receptor 1; IL1R1, interleukin 1 receptor type 1; IL1R2, interleukin 1 receptor type 2; IL2RA, interleukin 2 receptor alpha; IL6R, interleukin 6 receptor; IL7R, Interleukin 7 receptor; IL10, interleukin 10; IL12B, interleukin 12B (IL12 p40 subunit); IL18R1, interleukin 18 receptor 1 (alpha chain); IL21, interleukin 21; IL23A, interleukin 23 alpha (IL12 p19 subunit); IL23R, interleukin 23 receptor; IL27, interleukin 27; INAVA, innate immunity activator; IRF1, interferon regulatory factor 1; IRF5, interferon regulatory factor 5; IRGM, immunity-related GTPase family, M; ITGAL, integrin alpha-L; ITLN1, intelectin 1; JAK2, Janus kinase 2; JAZF1, JAZF zinc finger 1; KEAP1, kelch like ECH associated protein 1; KIF21B, kinesin family member 21B; KSR1, kinase suppressor of ras 1; LCE3B, late cornified envelope 3B; LNPEP, leucyl-cystinyl aminopeptidase; LRRK2, leucine rich repeat kinase 2; LSM14A, LSM14A mRNA processing body assembly factor; LTBR, lymphotoxin B receptor; MST1, macrophage stimulating 1; MYRF, myelin regulatory factor; NDFIP1, neural precursor cell expressed, developmentally downregulated 4 family-interacting protein 1; NFATC1, nuclear factor of activated t cells, cytoplasmic, calcineurin-dependent 1; NFIP1; Nedd4 Family Interacting Protein 1;NFKB1, nuclear factor kappa-b, subunit 1; NFKBIA, nuclear factor of kappa light chain gene enhancer in b cells inhibitor, alpha; NKX2-3, NK2 homeobox 3; NOD2, nucleotide-binding oligomerization domain protein 2; NOS2, nitric oxide synthase 2A; NOTCH1, NOTCH, Drosophila, homolog of, 1; NPEPPS, aminopeptidase, puromycin-sensitive; NR5A2, nuclear receptor subfamily 5 group A member 2; NXPE1, neurexophilin and PC-esterase domain family member 1; OR5B21, olfactory receptor family 5 subfamily B member 21; OSMR, oncostatin M receptor; PARK7, Parkinsonism associated deglycase; PKIG, cAMP-dependent protein kinase inhibitor gamma; PLAU, plasminogen activator, urokinase; PPP2R3C, protein phosphatase 2 regulatory subunit B double prime gamma; PPP5C, protein phosphatase 5 catalytic subunit; PRDM1, PR domain-containing protein 1; PRKCB, protein kinase C beta; PRKCQ, protein kinase C theta; PTGER4, prostaglandin E receptor 4, EP4 subtype; PTPN2, protein tyrosine phosphatase, non-receptor type 2; PTPN22, protein tyrosine phosphatase, non-receptor type 22; PUS10, pseudouridylate synthase 10; RAVER1, RAVER1, mouse, homolog of; RIC8B, RIC8, C. elegans, homolog of, B; RNF114, ring finger protein 114; RNF186, ring finger protein 186; RORC, RAR-related orphan receptor C; RPS6KB1, ribosomal protein S6 kinase, 70-Kd, 1; RUNX3, runt-related transcription factor 3; SBNO2, strawberry notch, Drosophila, homolog of, 2; SDF4, stromal cell derived factor 4; SETD1A, SET domain-containing protein 1A; SH2B3, SH2B adaptor protein 3; SKAP2, SRC kinase-associated phosphoprotein 2; SLC9A8, solute carrier family 9 (zinc transporter), member 8; SLC26A3, solute carrier family 26 (zinc transporter), member 26; SLC39A11, solute carrier family 39 (zinc transporter), member 11; SMAD3, mothers against decapentaplegic, Drosophila, homolog of, 3; SP140, nuclear body protein SP140; STAT3, signal transducer and activator of transcription 3; STK11, serine/threonine protein kinase 11; SULT1A2, sulfotransferase family 1A, cytosolic-phenol preferring member 2; TBKBP1, tank binding kinase binding protein 1; TBX21, T-box 21; TEX41, testis expressed 41; THADA, thyroid adenoma associated gene; TLR4, Toll receptor 4; TNFAIP3, tumor necrosis factor alpha-induced protein 3; TNFRSF1A, tumor necrosis factor receptor superfamily, member 1A; TNFRSF6B, tumor necrosis factor receptor superfamily, member 6B; TNFRSF14, tumor necrosis factor receptor superfamily, member 14; TNFSF8, tumor necrosis factor ligand superfamily, member 8; TNFSF15, tumor necrosis factor ligand superfamily, member 15; TNIP1, TNFAIP3-interacting protein 1; TNP2, transition protein 2; TSPAN14, tetraspanin 14; TYK2, tyrosine kinase 2; UBAC2, UBA domain-containing protein 2; UBASH3, ubiquitin-associated and SH3 domain-containing protein A; UBE2L3, ubiquitin conjugating enzyme 2EL 3; UQCR10, ubiquinol-cytochrome c reductase complex, 7.2 kd subunit; ZPBP2, zona pellucida-binding protein 2; ZC3H12C, zinc finger CCCH domain-containing protein 12C; ZMIZ1, Zinc finger MIZ-domain containing 1; ZNF365, zinc finger protein 365; ZNF831, melanoma, cutaneous malignant-susceptibility to 1. Not shown are genetic markers from noncoding regions: AC008697.1, Homo sapiens chromosome 5 clone CIT978SKB_70D3; AC020743.4, Homo sapiens chromosome 7 clone RP11-813 K3; AL031590.1, Homo sapiens chromosome 22, clone 232D4; AP001057.1, uncharacterized LOC107983952; CTD-2260A1, DNA marker; NPM1P17, nucleophosmin 1 pseudogene 17; RP1-66C13.4; RP11-24F11.2; RP11-129 J12.1; RP11-300 J18.1; RP11-1C1.5; RP11-84D1.2; and RP11-672A2.7
Gut microbiota and associated factors
On the basis of the current understanding of AS pathogenesis, it is most likely that the microbiome plays a role, especially in those who are already genetically susceptible. This is supported by current human research as well as studies carried out in animal models. The introduction of commensal bacteria such as Bacteroides vulgatus into the transgenic animals led to the development of arthritis [71]. Additionally, the transfer of the HLA-B27–transgenic rats from their sterile environment to a conventional rat colony led to the development of SpA symptoms [71]. In a cross-sectional study assessing the relationship between disease activity and infections among Mexican patients with different forms of SpA, more infections were found to occur among those with HLA-B27 positivity, particularly enteric infections [72], thus supporting a role for genetics and microbial infection in AS development. HLA-B27 may render susceptibility to AS by altering the gut microbiome and displaying a separate and divergent array of peptides in the gut, thereby introducing a microenvironment that leads to microbial imbalance, inflammation, and subsequent overproduction of IL-23 and other proinflammatory mediators [73], an effect that is even seen in HLA-B27 positive “healthy controls”[74]. Additionally, gut permeability is increased among patients with AS and their first-degree relatives as well as in experimental animal models, which perhaps allows for a greater systemic exposure to potentially pathogenic gut microbes [115]. In this regard, Paneth cells, a subset of specialized secretory host-defense epithelial cells located in the small intestines, have been shown to secrete IL-23 and activate key IL-23 responsive cells such as group 3 innate lymphoid cells (ILC3), γδ T cells, and mucosal-associated invariant T (MAIT) cells, which recirculate from the gut to sites of inflammation important in SpA pathogenesis, such as the entheses [116].
The gut microbiome
Several families of gut bacteria have been associated with AS development in humans, including Lachnospiraceae, Prevotellaceae, Rikenellaceae, Porphyromonadaceae, Ruminococcaceae, and Bacteroidaceae [8], and these AS-associated microbial families have been linked to fecal calprotectin levels, a marker of intestinal inflammation, but not to other clinical parameters [75]. A metagenomics study analyzed gut microbial DNA from 211 Chinese individuals and found that patients with AS had an increased load of Prevotella melaninogenica, Prevotella copri, and Prevotella sp. C561, and decreases in Bacteroides sp. [76]. It is noteworthy that the Bifidobacterium genus, which is commonly used in probiotics, accumulates in patients with AS [76]. Another study, again in Chinese AS patients, confirmed previous reports of gut dysbiosis in AS, and TNFi therapy was correlated with a restoration the perturbed microbiome that was observed in untreated AS cases compated to that of healthy controls [77].
Asymptomatic intestinal inflammation, usually involving the terminal ileum, is also known to occur in a large proportion of patients with AS (57 to 70%) and is especially apparent in those with peripheral arthritis, again suggesting a link between the gut and AS [117]. However, no evidence of an exact connection of AS with subclinical gut inflammation has been found to date [118]. The overproduction of IL-23 by Paneth cells lining the epithelium of the small intestine has been implicated in regulating gut mucosal immunity. Among patients with AS and Crohn’s disease, a marked upregulation of IL-23p19 transcripts was observed in the terminal ileum, suggesting an association between polymorphisms in the IL-23 receptor and gut inflammation [119].
Gut-induced protection through breastfeeding
Only one study to date has examined breastfeeding history in patients with AS [80]. This retrospective case-control study suggested that breastfeeding has a protective effect on the development of AS [80]. Patients with AS were breastfed less often than healthy controls. Patients with AS and HLA-B27 positivity were breastfed less often than their siblings who did not have AS as well as unrelated, healthy controls, which suggested that breastfeeding may decrease the familial prevalence of AS, perhaps through factors induced in the gut for those who were breastfed.
Infections
Klebsiella pneumoniae
Klebsiella was implicated in AS pathogenesis when an increased fecal carriage was found in patients with “active” disease [81]. Subsequently, another study reported that patients with HLA-B27 positivity had lower in vitro lymphocyte responsiveness to Klebsiella antigens [82]. However, these early findings were not confirmed by others [120, 121].
Nevertheless, molecular mimicry between Klebsiella (and other enterobacteria) capsular antigens and HLA-B27 was proposed with the discovery of cross-reactivity between antigens in several Gram-negative microorganisms and lymphocytes of patients who were HLA-B27 positive [83]. Patients with SpA were also reported to have an increased frequency of antibodies to a homologous region shared by HLA-B27 and Klebsiella nitrogenase compared with HLA-B27–positive controls [84]. This suggested that AS represents an autoimmune response directed against HLA-B27 that was initially induced against nitrogenase proteins of K. pneumoniae. However, this finding could not be independently confirmed [122]. Other reports postulated that active AS was characterized instead by elevated IgA antibodies to various enterobacteria in both AS and AAU regardless of HLA-B27 status [123, 124], raising doubts about the molecular mimicry theory. Alternatively, modification by a Klebsiella K43 plasmid-derived soluble cell wall factor of specific MHC-associated gene products was implicated in the pathogenesis of the HLA-B27–linked arthropathies [85], which likewise could not be confirmed by others [125, 126]. Studies of patients with AS or their affected or unaffected family members with familial AS have not demonstrated a specific K. pneumoniae antibody response for AS [127, 128].
By the middle of the 2000s, the lack of any consistent compelling story implicating Klebsiella in AS susceptibility or severity caused the interest in further researching this topic to wane; however, there has been a recent systematic review [14], a report demonstrating Klebsiella protein antibody responsiveness persisting in patients with AS [86], and another addressing the popularity of “low starch” diets, as well as “anti-Klebsiella” dietary supplements [87], albeit with little evidence of their effect on disease activity. Of note, previous results implicating Klebsiella in the gut have not been confirmed in recent studies of the gut microbiome [8, 78, 127].
Infections during childhood hospitalization
In a Swedish national case-control study, childhood hospitalization (with infections) was associated with later development of AS [88]. The study included 2453 patients with AS and 10,257 control subjects, of whom 17.4% and 16.3%, respectively, had been hospitalized with an infection before 17 years of age [88]. Appendicitis was associated with a decreased risk of AS, whereas rates of respiratory tract infections and tonsillitis, respectively, were increased in patients with AS vs controls. There were no associations between AS and any other type of infection.
Mechanical stress
McGonagle and colleagues first proposed an enthesitis-based model for the pathogenesis of SpA, whereby interactions between biomechanical factors and the innate immune response may lead to disease [89]. In a mouse model in which a 69-base pair deletion comprising the tumor necrosis factor (TNF) AU-rich elements (ARE) yielded TNFΔARE-mutant mice with chronic inflammatory arthritis and Crohn’s disease, mechanical stress was found to be involved in the development of enthesitis in the Achilles’ tendon [90], where hind limb unloading could efficiently prohibit the development of enthesitis in those sites. In more recent work [91] examining human spinal entheseal tissue, Vδ1 and Vδ2 subsets of T lymphocytes were shown to be tissue resident cells with inducible IL-17A production and that the Vδ1 subset does so independently of IL-23R expression.
It is well established that patients with AS who are engaged in physically demanding jobs are more likely to experience permanent or temporary work disability [92], especially in occupations requiring dynamic flexibility (i.e., the ability to repeatedly bend, stretch, twist, or reach), because patients with AS tend to have more functional limitations than those whose past jobs required little or no dynamic flexibility [92]. This finding was underscored by a recent study showing that radio-graphic progression of AS was higher in blue-collar vs white-collar workers [93]. This would suggest that a young patient with AS, in evaluating his/her future employment, consider avoiding those with physically demanding tasks.
Gender at birth
Despite the equivalent prevalence of HLA-B27 between men and women [19], AS reportedly affects more men than women (approximately 2:1 ratio, although this varies greatly between studies) [94, 95], and this gender disparity is not seen in nraxSpA, where there is relatively equal proportion of men and women, or even a female preponderance [3]. Male gender has been implicated as a risk factor for progression from nr-axSpA to radiographic axSpA, but this has not been examined in longitudinal studies [7, 96, 97].
There also exists a gender-at-birth difference in the severity of AS, the type of clinical manifestations, and response to treatment [93, 97]. A Swedish study reported the higher prevalence of anterior uveitis among men with AS vs women and of peripheral arthritis and psoriasis among women with AS vs men [94]. Furthermore, men with a family history of AS are at a higher risk of developing AS than both women with a family history and men without a family history of AS [27]. The differences observed in men are, in part, due to genetic factors predisposing them to AS development, and to immunological and lifestyle differences (e.g., smoking, diet).
The onset of AS is reportedly earlier among men than among women, which leads to a more rapid diagnosis [98]; however, women experience more delay in receiving an AS diagnosis [99]. Furthermore, compared with women, men have lower disease activity as measured by Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) and Ankylosing Spondylitis Disease Activity Score, and a better quality of life (Ankylosing Spondylitis Quality of Life Questionnaire) but have worse spinal mobility (Bath Ankylosing Spondylitis Metrology Index) and a more severe radiographic involvement (Bath Ankylosing Spondylitis Radiology Index) [100]. With regard to clinical AS manifestations, women usually have more peripheral arthritis and an increased prevalence of arthritis, dactylitis, and enthesitis compared to men [100]. Women also report lower response rates to anti–TNF treatment than men [100]. However, data appear conflicting. In a small prospective cohort study (N = 216) using data from the Outcome in AS International Study, no gender-at-birth–attributable differences in physical function or disease activity over time were found; however, radiographic damage was more severe in men [101]. This supports previous reports documenting that men with longstanding AS had more severe radiographic changes compared with women [100]. Despite this, men report a better quality of life than women over time [101]. Additionally, distinct sexual dimorphism in the activation status of the immune system in patients with AS, particularly in the Th17 axis, has been observed, possibly explaining clinical gender-related differences among men and women with AS [102]. This dimorphism may suggest gender-specific AS treatment.
Other genetic differences (IL-22 copy number variants, rs11428092 and rs10208769 in USP34, and IRGM) have also been observed between the genders (Supplemental Table 1).
Social and lifestyle factors
Older siblings
Using the Swedish National Patient Register to identify patients with AS, Lindström and colleagues examined the impact of perinatal characteristics and/or presence of older siblings on the risk of developing AS [103]. Having older siblings was strongly associated with greater risk of developing AS and birth weight below 3000 g, but not low birth weight (i.e., < 2500 g), was weakly associated with the risk of AS development. As this was a single study, the findings are not conclusive and would need to be repeated and confirmed in other cohorts.
Vitamin D levels
Although there has been little focus on the potential immunomodulatory role of vitamin D in AS, current data suggest that it may have significant effects on both innate and acquired immunity and that its deficiency may be associated with both susceptibility and disease severity in AS and other autoimmune diseases [15]. Higher levels of serum vitamin D have been associated with a decreased risk of AS and decreased disease activity [15, 104].
Smoking cigarettes and e-cigarettes
Current smoking (but not a history of smoking) appears to be a risk factor for AS and is linked to disease activity level in those with AS. This is not surprising, given the known proinflammatory and prooxidative effects of smoking, especially in contributing to disease onset in a genetically predisposed individual and to the evolution of nr-axSpA into AS [105]. In the population-based Nord-Trøndelag health study, incident AS was associated with current smoking and hypertension [105]. Present smoking was significantly associated with incident self-reported AS in logistic regression adjusted for potential confounders. Furthermore, a meta-analysis evaluating the associations between smoking and disease outcome among Chinese patients with AS showed that current, former, or ever smokers had significantly higher BASDAI and worse functional capacity than non-smokers [106], and among French AxSpA patients smoking was independently associated with more MRI-detected SI joint inflammation at each visit over a 5-year period of follow-up [107]. Although no data are available for patients with AS who smoke e-cigarettes, data from a murine model of arthritis found that nicotine exacerbated inflammatory arthritis [108], thus suggesting that nicotine-containing products, including e-cigarettes, may have deleterious effects on patients with AS.
Other risk factors
Immunological aspects have been an area of research for their clear involvement in disease pathogenesis. A wide variety of immune cell types have been suggested to be involved in AS development [109, 110]. In a flow cytometry analysis of peripheral blood from patients with AS, HLA-B27+ individuals without AS and healthy controls, the percentage of regulatory T cells in patients with AS was lower than that in healthy individuals [111]. Furthermore, patients with AS had lower expression of negative checkpoint regulators such as PD-1 and Tim-3 on T lymphocytes, and lower IL-10 production by CD4+ T cells, with higher IL-6 production by CD8+ T cells compared with healthy individuals; these data suggest negative regulation of the immune response is impaired in patients with AS [111]. In another study, compared with healthy volunteers and patients with rheumatoid arthritis and osteoarthritis, patients with AS had significantly higher levels of baseline serum macrophage migration inhibitory factor (MIF), which independently predicted AS progression [112]. High MIF levels were detected predominantly in the synovial fluid of patients with AS, and the primary producers of MIF (i.e., macrophages and Paneth cells) were enriched in the intestines [112]. MIF production led to the generation of TNF-α in monocytes and activated β-catenin in osteoblasts and induced osteoblastic mineralization [112].
Limitations
The specific keyword searches on which this systematic review was based would omit papers that were not directly relevant. However, with further searches and inclusion of manuscripts deemed of interest, we believe that this review provides comprehensive insight into the current understanding of risk factors in AS. The search methodology covered a limited period (January 1, 2013, and February 1, 2021). However, this was supplemented with papers of key interest from the last several decades based on clinical expert opinion. PubMed was the only database used and only articles in the English language were included.
Summary
Genetic predisposition fails to fully explain the cause of AS, and this has led to a strong effort to identify additional predisposing factors. The inappropriate expression of HLA-B27 and ERAP1 peptides to adaptive immune cells may compromise recognition of self-antigens, rendering susceptibility to AS development [55]. In the B27 rat model of AS, rats housed under germ-free conditions did not develop AS-like symptoms, thus demonstrating that microbes, at least in this model, are important for disease occurrence [71]. The gut microbiota may therefore become a potential target for AS treatment in the future. Mechanical stress may also play an important role in triggering enthesitis and driving radiographic progression in AS [90, 91].
The numerous investigations and studies carried out within the last decade have markedly improved our understanding of the pathogenesis and risk factors of AS and have facilitated the development of new treatment strategies. HLA-B27 remains the most important genetic risk factor in AS, followed by IL23R and ERAP1 [7, 70]. In addition to (or in combination with) genetic aspects, various triggering factors have been implicated in the development of AS. Despite concerns regarding some aspects of microbial involvement in AS, overall, available data strongly suggest involvement of the gut microbiome in the pathogenesis of AS [75–77, 79]. The characterization of the species composition of the microbiota associated with AS and the exact mechanism for the functional role of intestinal microbiome in disease onset and progression should be the focus of more research in the future; the role of mechanical stress in triggering/worsening AS is also an active area of investigation.
Despite the progress being made in understanding the functions and potential pathogenic roles of some of these risk factors, much work remains to better comprehend their complex interactions. Further studies with longitudinal designs are required to better understand whether optimizing vitamin D levels in AS is a potential disease-modifying intervention [15]. Smoking (including smoking of e-cigarettes) should be discouraged in those at a higher risk of developing AS (i.e., those with a family history of AS or who are HLA-B27 positive). The role of mechanical entheseal stress on disease or flare triggering should be considered, especially in planning future employment. The exact trigger of AS onset and achieving even more efficient control through eliminating known risk factors (wherever possible), along with improved disease-modifying treatments, will help reduce the burden and severity of AS.
Supplementary Material
Key Points.
It remains unclear how interactions between genes, microbes, mechanical stress, gender, and other environmental and lifestyle factors predispose patients to the development of ankylosing spondylitis (AS).
The exact mechanisms of AS are complex and multifactorial which will require much future research
Recognizing the risk factors, as well as understanding gene-environment interactions, may offer valuable insights into the etiology of AS and have important implications for diagnosis and treatment strategies
Acknowledgements
The authors thank Kheng Bekdache, PhD, of Health Interactions Inc, Hamilton, NJ, USA, and Shelley Maria Lindley, PhD, of SciMentum Ltd, London, UK, for providing medical writing support/editorial support.
Funding
Editorial support for this review article was sponsored by Novartis Pharmaceuticals Corporation. This review was funded by Novartis Pharmaceuticals Corporation, East Hanover, New Jersey, in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s10067-021-05679-7) contains supplementary material, which is available to authorized users.
Data availability Not applicable.
Ethics approval Not applicable.
Consent to participate Not applicable.
Consent for publication Not applicable.
Disclosures None.
References
- 1.Ostergaard M, Lambert RG (2012) Imaging in ankylosing spondylitis. Ther Adv Musculoskelet Dis 4:301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rudwaleit M, van der Heijde D, Landewe R, Listing J, Akkoc N, Brandt J et al. (2009) The development of Assessment of SpondyloArthritis International Society classification criteria for axial spondyloarthritis (part II): validation and final selection. Ann Rheum Dis 68:777–783 [DOI] [PubMed] [Google Scholar]
- 3.Reveille JD, Witter JP, Weisman MH (2012) Prevalence of axial spondylarthritis in the United States: estimates from a cross-sectional survey. Arthritis Care Res (Hoboken) 64:905–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dean LE, Jones GT, MacDonald AG, Downham C, Sturrock RD, Macfarlane GJ (2014) Global prevalence of ankylosing spondylitis. Rheumatology (Oxford) 53:650–657 [DOI] [PubMed] [Google Scholar]
- 5.Braun A, Saracbasi E, Grifka J, Schnitker J, Braun J (2011) Identifying patients with axial spondyloarthritis in primary care: how useful are items indicative of inflammatory back pain? Ann Rheum Dis 70:1782–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dagfinrud H, Kjeken I, Mowinckel P, Hagen KB, Kvien TK (2005) Impact of functional impairment in ankylosing spondylitis: impairment, activity limitation, and participation restrictions. J Rheumatol 32:516–523 [PubMed] [Google Scholar]
- 7.Ellinghaus D, Jostins L, Spain SL, Cortes A, Bethune J, Han B et al. (2016) Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet 48:510–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costello ME, Ciccia F, Willner D, Warrington N, Robinson PC, Gardiner B et al. (2015) Brief report: intestinal dysbiosis in ankylosing spondylitis. Arthritis Rheumatol 67:686–691 [DOI] [PubMed] [Google Scholar]
- 9.Zhu W, He X, Cheng K, Zhang L, Chen D, Wang X, Qiu G, Cao X. Weng X (2019) Ankylosing spondylitis: etiology, pathogenesis, and treatments. Bone Res 7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simone D, Al Mossawi MH, Bowness P (2018) Progress in our understanding of the pathogenesis of ankylosing spondylitis. Rheumatology (Oxford) 57(suppl 6):vi4–vi9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han R, Xia Q, Xu S, Fan D, Pan F (2018) Interleukin-23 receptor polymorphism (rs10889677 A/C) in ankylosing spondylitis: meta-analysis in Caucasian and Asian populations. Clin Chim Acta 477: 53–59 [DOI] [PubMed] [Google Scholar]
- 12.Qian Y, Wang G, Xue F, Chen L, Wang Y, Tang L, Yang H (2017) Genetic association between TAP1 and TAP2 polymorphisms and ankylosing spondylitis: a systematic review and meta-analysis. Inflamm Res 66:653–661 [DOI] [PubMed] [Google Scholar]
- 13.Yang M Zou Y, Bai Y, Li M (2015) The programmed cell death 1 gene polymorphisms (PD 1.3 G/A, PD 1.5 C/T and PD 1.9 C/T) and susceptibility to ankylosing spondylitis: a meta-analysis. J Orthop Sci 20:55–63 [DOI] [PubMed] [Google Scholar]
- 14.Zhang L, Zhang YJ, Chen J, Huang XL, Fang GS, Yang LJ, Duan Y, Wang J (2018) The association of HLA-B27 and Klebsiella pneumoniae in ankylosing spondylitis: a systematic review. Microb Pathog 117:49–54 [DOI] [PubMed] [Google Scholar]
- 15.Zhao S, Duffield SJ, Moots RJ, Goodson NJ (2014) Systematic review of association between vitamin D levels and susceptibility and disease activity of ankylosing spondylitis. Rheumatology (Oxford) 53:1595–1603 [DOI] [PubMed] [Google Scholar]
- 16.Moher D, Liberati A, Tetzlaff J, Altman DG, Group P (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA Statement. Open Med 3:e123–e130 [PMC free article] [PubMed] [Google Scholar]
- 17.Jamalyaria F, Ward MM, Assassi S, Learch TJ, Lee M, Gensler LS, Brown MA, Diekman L, Tahanan A, Rahbar MH, Weisman MH, Reveille JD (2017) Ethnicity and disease severity in ankylosing spondylitis a cross-sectional analysis of three ethnic groups. Clin Rheumatol 36:2359–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reveille JD, Zhou X, Lee M, Weisman MH, Yi L, Gensler LS, Zou H, Ward MM, Ishimori ML, Learch TJ, He D, Rahbar MH, Wang J, Brown MA (2019) HLA class I and II alleles in susceptibility to ankylosing spondylitis. Ann Rheum Dis 78:66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reveille JD, Hirsch R, Dillon CF, Carroll MD, Weisman MH (2009) The prevalence of HLA-B27 in the US: data from the US National Health and Nutrition Examination Survey. Arthritis Rheum 64:1407–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akkoc N, Khan MA (2005) Overestimation of the prevalence of ankylosing spondylitis in the Berlin study: comment on the article by Braun et al. Arthritis Rheum 52:4048–4049 author reply 4049–4050 [DOI] [PubMed] [Google Scholar]
- 21.Ziade NR (2017) HLA B27 antigen in Middle Eastern and Arab countries: systematic review of the strength of association with axial spondyloarthritis and methodological gaps. BMC Musculoskelet Disord 18:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennett AN, McGonagle D, O’Connor P, Hensor EM, Sivera F, Coates LC et al. (2008) Severity of baseline magnetic resonance imaging-evident sacroiliitis and HLA-B27 status in early inflammatory back pain predict radiographically evident ankylosing spondylitis at eight years. Arthritis Rheum 58:3413–3418 [DOI] [PubMed] [Google Scholar]
- 23.Galocha B, López de Castro JA (2010) Mutational analysis reveals a complex interplay of peptide binding and multiple biological features of HLA-B27. J Biol Chem 285:39180–39190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dashti N, Mahmoudi M, Aslani S, Jamshidi A (2018) HLA-B*27 subtypes and their implications in the pathogenesis of ankylosing spondylitis. Gene 670:15–21 [DOI] [PubMed] [Google Scholar]
- 25.Cortes A, Pulit SL, Leo PJ, Pointon JJ, Robinson PC, Weisman MH, Ward M, Gensler LS, Zhou X, Garchon HJ, Chiocchia G, Nossent J, Lie BA, Førre Ø, Tuomilehto J, Laiho K, Bradbury LA, Elewaut D, Burgos-Vargas R, Stebbings S, Appleton L, Farrah C, Lau J, Haroon N, Mulero J, Blanco FJ, Gonzalez-Gay MA, Lopez-Larrea C, Bowness P, Gaffney K, Gaston H, Gladman DD, Rahman P, Maksymowych WP, Crusius JBA, van der Horst-Bminsma IE, Valle-Oñate R, Romero-Sánchez C, Hansen IM, Pimentel-Santos FM, Inman RD, Martin J, Breban M, Wordsworth BP, Reveille JD, Evans DM, de Bakker PIW, Brown MA (2015) Major histocompatibility complex associations of ankylosing spondylitis are complex and involve further epistasis with ERAP1. Nat Comrnun 6:7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Gaalen FA (2012) Does HLA-B*2706 protect against ankylosing spondylitis? A meta-analysis. Int J Rheum Dis 15:8–12 [DOI] [PubMed] [Google Scholar]
- 27.Lin H, Gong YZ (2017) Association of HLA-B27 with ankylosing spondylitis and clinical features of the HLA-B27-associated ankylosing spondylitis: a meta-analysis. Rheumatol Int 37:1267–1280 [DOI] [PubMed] [Google Scholar]
- 28.Robinson PC, Claushuis TA, Cortes A, Martin TM, Evans DM, Leo P et al. (2015) Genetic dissection of acute anterior uveitis reveals similarities and differences in associations observed with ankylosing spondylitis. Arthritis Rheumatol 67:140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faham M, Carlton V, Moorhead M, Zheng J, Klinger M, Pepin F, Asbury T, Vignali M, Emerson RO, Robins HS, Ireland J, Baechler-Gillespie E, Inman RD (2017) Discovery of T cell receptor β motifs specific to HLA-B27-positive ankylosing spondylitis by deep repertoire sequence analysis. Arthritis Rheumatol 69: 774–784 [DOI] [PubMed] [Google Scholar]
- 30.Gracey E, Yao Y, Qaiyum Z, Lim M, Tang M, Inman RD (2020) Altered cytotoxicity profile of CD8+ T cells in ankylosing spondylitis. Arthritis Rheumatol 72:428–434 [DOI] [PubMed] [Google Scholar]
- 31.Navid F, Holt V, Colbert RA (2021) The enigmatic role of HLA-B*27 in spondyloarthritis pathogenesis. Semin Immunopathol. 10.1007/s00281-021-00838-z [DOI] [PubMed] [Google Scholar]
- 32.Jeanty C, Sourisce A, Noteuil A, Jah N, Wielgosik A, Fert I, Breban M, André C (2014) HLA-B27 subtype oligomerization and intracellular accumulation patterns correlate with predisposition to spondyloarthritis. Arthritis Rheumatol 66:2113–2123 [DOI] [PubMed] [Google Scholar]
- 33.Jah N, Jobart-Malfait A, Emroza K, Noteuil A, Chiocchia G, Breban M, André C (2020) HLA–B27 Subtypes Predisposing to Ankylosing Spondylitis Accumulate in an Endoplasmic Reticulum–Derived Compartment Apart From the Peptide-Loading Complex. Arthritis Rheumatol 72:1534–1546 [DOI] [PubMed] [Google Scholar]
- 34.Lim Kam Sian TCC, Indumathy S, Halim H, Greule A, Cryle MJ, Bowness P, Rossjohn J, Gras S, Purcell AW, Schittenhelm RB (2019) Allelic association with ankylosing spondylitis fails to correlate with human leukocyte antigen B27 homodimer fomration. J Biol Chem 294:20185–20195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sahlberg AS, Ruuska M, Colbert RA, Granfors K, Penttinen MA (2012) Altered PKR signalling and C/EBPβ expression is associated with HLA-B27 expression in monocytic cells. Scand J Immunol 75:184–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boyle LH, Goodall JC, Opat SS, Gaston JS (2001) The recognition of HLA-B27 by human CD4(+) T lymphocytes. J Immunol 167:2619–2624 [DOI] [PubMed] [Google Scholar]
- 37.van Gaalen FA, Verduijn W, Roelen DL, Böhringer S, Huizinga TW, van der Heijde DM et al. (2013) Epistasis between two HLA antigens defines a subset of individuals at a very high risk for ankylosing spondylitis. Ann Rheum Dis 72:974—978 [DOI] [PubMed] [Google Scholar]
- 38.Londono J, Santos AM, Peña P, Calvo E, Espinosa LR, Reveille JD et al. (2015) Analysis of HLA-B15 and HLA-B27 in spondyloarthritis with peripheral and axial clinical patterns. BMJ Open 5:e009092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.López-Larrea C, Mijiyawa M, González S, Fernández-Morera JL, Blanco-Gelaz MA, Martínez-Borra J, López-Vázquez A (2002) Association of ankylosing spondylitis with HLA-B*1403 in a West African population. Arthritis Rheum 46:2968–2971 [DOI] [PubMed] [Google Scholar]
- 40.Cortes A, Hadler J, Pointon JP, Robinson PC, Karaderi T, Leo P et al. (2013) Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet 45:730–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou X, Wang J, Zou H, Ward MM, Weisman MH, Espitia MG, Xiao X, Petersdorf E, Mignot E, Martin J, Gensler LS, Scheet P, Reveille JD (2014) MICA, a gene contributing strong susceptibility to ankylosing spondylitis. Ann Rheum Dis 73:1552–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cortes A, Gladman D, Raychaudhuri S, Cui J, Wheeler L, Brown MA et al. (2018) Imputation-based analysis of MICA alleles in the susceptibility to ankylosing spondylitis. Ann Rheum Dis 77: 1691–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang CM, Wang SH, Jan Wu YJ, Lin JC, Wu J, Chen JY (2017) Human leukocyte antigen C*12:02:02 and killer immunoglobulin-like receptor 2DL5 are distinctly associated with ankylosing spondylitis in the Taiwanese. Int J Mol Sci 18:1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Díaz-Peña R, Castro-Santos P, Aransay AM, Brüges-Armas J, Pimentel-Santos FM, López-Larrea C (2013) Genetic study confirms association of HLA-DPA1(*)01:03 subtype with ankylosing spondylitis in HLA-B27-positive populations. Hum Immunol 74:764–767 [DOI] [PubMed] [Google Scholar]
- 45.Huang XF, Li Z, De Guzman E, Robinson P, Gensler L, Ward MM et al. (2020) Genomewide association study of acute anterior uveitis indentifies new susceptibility loci. Invest Ophthalmol Vis Sci 61:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma B, Yang B, Guo H, Wang Y, Zhang D, Zhang Y, Xiao Z (2013) The association between tumor necrosis factor alpha promoter polymorphisms and ankylosing spondylitis: a meta-analysis. Hum Immunol 74:1357–1362 [DOI] [PubMed] [Google Scholar]
- 47.Reveille JD, Sims AM, Danoy P, Evans DM, Leo P, Pointon JJ et al. (2010) Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat Genet 42:123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu J, Pu W, Li Y, Ma Y, Zhu Q, Wan W, Yang C, Wang X, Chen X, Zhou X, Reveille JD, Jin L, Zou H, Wang J (2019. *) Genetic association of non-MHC region with ankylosing spondylitis in a Chinese population. Ann Rheum Dis 78:852–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin Z, Bei JX, Shen M, Li Q, Liao Z, Zhang Y, Lv Q, Wei Q, Low HQ, Guo YM, Cao S, Yang M, Hu Z, Xu M, Wang X, Wei Y, Li L, Li C, Li T, Huang J, Pan Y, Jin O, Wu Y, Wu J, Guo Z, He P, Hu S, Wu H, Song H, Zhan F, Liu S, Gao G, Liu Z, Li Y, Xiao C, Li J, Ye Z, He W Xiu D, Shen L, Huang A, Wu H, Tao Y, Pan X, Yu B, Tai ES, Zeng YX, Ren EC, Shen Y, Liu J, Gu J (2011) A genome-wide association study in Han Chinese identifies new susceptibility loci for ankylosing spondylitis. Nat Genet 44:73–77 [DOI] [PubMed] [Google Scholar]
- 50.Tang Y, Yang P, Wang F, Xu H, Zong SY (2018) Association of polymorphisms in ERAP1 and risk of ankylosing spondylitis in a Chinese population. Gene 646:8–11 [DOI] [PubMed] [Google Scholar]
- 51.Cinar M, Akar H, Yilmaz S, Simsek I, Karkucak M, Sagkan RI, Pekel A, Erdem H, Avci IY, Acikel C, Musabak U, Tunca Y, Pay S (2013) A polymorphism in ERAP1 is associated with susceptibility to ankylosing spondylitis in a Turkish population. Rheumatol Int 33:2851–2858 [DOI] [PubMed] [Google Scholar]
- 52.Wen YF, Wei JC, Hsu YW, Chiou HY, Wong HS, Wong RH et al. (2014) rs10865331 associated with susceptibility and disease severity of ankylosing spondylitis in a Taiwanese population. PLoS One 9:e104525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Evans DM, Spencer CC, Pointon JJ, Su Z, Harvey D, Kochan G et al. (2011) Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet 43:761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A et al. (2007) Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet 39:1329–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seregin SS, Rastall DP, Evnouchidou I, Aylsworth CF, Quiroga D, Kamal RP et al. (2013) Endoplasmic reticulum aminopeptidase-1 alleles associated with increased risk of ankylosing spondylitis reduce HLA-B27 mediated presentation of multiple antigens. Autoimmunity 46:497–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson PC, Costello ME, Leo P, Bradbury LA, Hollis K, Cortes A, Lee S, Joo KB, Shim SC, Weisman M, Ward M, Zhou X, Garchon HJ, Chiocchia G, Nossent J, Lie BA, Førre Ø, Tuomilehto J, Laiho K, Jiang L, Liu Y, Wu X, Elewaut D, Burgos-Vargas R, Gensler LS, Stebbings S, Haroon N, Mulero J, Fernandez-Sueiro JL, Gonzalez-Gay MA, Lopez-Larrea C, Bowness P, Gafney K, Gaston JSH, Gladman DD, Rahman P, Maksymowych WP, Xu H, van der Horst-Bruinsma IE, Chou CT, Valle-Oñate R, Romero-Sánchez MC, Hansen IM, Pimentel-Santos FM, Inman RD, Martin J, Breban M, Evans D, Reveille JD, Kim TH, Wordsworth BP, Brown MA (2015) ERAP2 is associated with ankylosing spondylitis in HLA-B27-positive and HLA-B27-negative patients. Ann Rheum Dis 74: 1627–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuan Y, Ma Y, Zhang X, Han R, Hu X, Yang J, Wang M, Guan SY, Pan G, Xu SQ, Jiang S, Pan F (2019) Genetic polymorphisms of G protein-coupled receptor 65 gene are associated with ankylosing spondylitis in a Chinese Han population: a case-control study. Hum Immunol 80:146–150 [DOI] [PubMed] [Google Scholar]
- 58.Ruan WF, Xie JT, Jin Q, Wang WD, Ping AS (2018) The diagnostic and prognostic role of interleukin 12B and interleukin 6R gene polymorphism in patients with ankylosing spondylitis. J Clin Rheumatol 24:18–24 [DOI] [PubMed] [Google Scholar]
- 59.Wang NG, Wang DC, Tan BY, Wang F, Yuan ZN (2015) TNF-α and IL10 polymorphisms interaction increases the risk of ankylosing spondylitis in Chinese Han population. Int J Clin Exp Pathol 8: 15204–15209 [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang X, Li X, Han R, Chen M, Yuan Y, Hu X, Wang M, Li R, Yang X, Xia Q, Ma Y, Yang J, Tong J, Xu S, Xu J, Shuai Z, Pan F (2017) Copy number variations of the IL-22 gene are associated with ankylosing spondylitis: a case-control study in Chinese Han population. Hum Immunol 78:547–552 [DOI] [PubMed] [Google Scholar]
- 61.Xia Q, Wang M, Yang X, Li X, Zhang X, Xu S, Shuai Z, Xu J, Fan D, Ding C, Pan F (2017) Autophagy-related IRGM genes confer susceptibility to ankylosing spondylitis in a Chinese female population: a case-control study. Genes Immun 18:42–17 [DOI] [PubMed] [Google Scholar]
- 62.Yang X, Li M, Wang L, Hu Z, Zhang Y, Yang Q (2015) Association of KIF21B genetic polymorphisms with ankylosing spondylitis in a Chinese Han population of Shandong Province. Clin Rheumatol 34:1729–1736 [DOI] [PubMed] [Google Scholar]
- 63.Zhai Z, Wang Z, Wang L, Chen S, Ren H, Wang D (2018) Relationship between inducible NOS single-nucleotide polymorphisms and hypertension in Han Chinese. Herz 43:461–465 [DOI] [PubMed] [Google Scholar]
- 64.Lee YH, Bae SC, Kim JH, Song GG (2015) Meta-analysis of genetic polymorphisms in programmed cell death 1. Associations with rheumatoid arthritis, ankylosing spondylitis, and type 1 diabetes susceptibility. Z Rheumatol 74:230–239 [DOI] [PubMed] [Google Scholar]
- 65.Liu X, Hu LH, Li YR, Chen FH, Ning Y, Yao QF (2011) Programmed cell death 1 gene polymorphisms is associated with ankylosing spondylitis in Chinese Han population. Rheumatol Int 31:209–213 [DOI] [PubMed] [Google Scholar]
- 66.Aita A, Basso D, Ramonda R, Moz S, Lorenzin M, Navaglia F, Zambon CF, Padoan A, Plebani M, Punzi L (2018) Genetics in TNF-TNFR pathway: A complex network causing spondyloarthritis and conditioning response to anti-TNFα therapy. PLoS One 13:e0194693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsui FW, Tsui HW, Akram A, Haroon N, Inman RD (2014) The genetic basis of ankylosing spondylitis: new insights into disease pathogenesis. Appl Clin Genet 7:105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang M, Xin L, Cai G, Zhang X, Yang X, Li X, Xia Q, Wang L, Xu S, Xu J, Shuai Z, Ding C, Pan F (2017) Pathogenic variants screening in seventeen candidate genes on 2p15 for association with ankylosing spondylitis in a Han Chinese population. PLoS One 12:e0177080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lau MC, Keith P, Costello ME, Bradbury LA, Hollis KA, Thomas R, Thomas GP, Brown MA, Kenna TJ (2017) Genetic association of ankylosing spondylitis with TBX21 influences T-bet and proinflammatory cytokine expression in humans and SKG mice as a model of spondyloarthritis. Ann Rheum Dis 76:261–269 [DOI] [PubMed] [Google Scholar]
- 70.Brown MA, Wordsworth BP (2017) Genetics in ankylosing spondylitis - Current state of the art and translation into clinical outcomes. Best Pract Res Clin Rheumatol 31:763–776 [DOI] [PubMed] [Google Scholar]
- 71.Rath HC, Herfarth HH, Ikeda JS, Grenther WB, Hamm TE, Balish E et al. (1996) Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J Clin Invest 98:945–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martínez A, Pacheco-Tena C, Vázquez-Mellado J, Burgos-Vargas R (2004) Relationship between disease activity and infection in patients with spondyloarthropathies. Ann Rheum Dis 63:1338–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rosenbaum JT, Davey MP (2011) Time for a gut check: evidence for the hypothesis that HLA-B27 predisposes to ankylosing spondylitis by altering the microbiome. Arthritis Rheum 63:3195–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Asquith M, Sternes PR, Costello ME, Karstens L, Diamond S, Martin TM, Li Z, Marshall MS, Spector TD, Cao KA, Rosenbaum JT, Brown MA (2019) HLA Alleles Associated With Risk of Ankylosing Spondylitis and Rheumatoid Arthritis Influence the Gut Microbiome. Arthritis Rheumatol 71:1642–1650 [DOI] [PubMed] [Google Scholar]
- 75.Klingberg E, Magnusson MK, Strid H, Deminger A, Ståhl A, Sundin J et al. (2019) A distinct gut microbiota composition in patients with ankylosing spondylitis is associated with increased levels of fecal calprotectin. Arthritis Res Ther 21:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wen C, Zheng Z, Shao T, Liu L, Xie Z, Le Chatelier E et al. (2017) Quantitative metagenomics reveals unique gut microbiome biomarkers in ankylosing spondylitis. Genome Biol. 18:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yin J, Stemes PR, Wang M, Song J, Morrison M, Li T, Zhou L, Wu X, He F, Zhu J, Brown MA, Xu H (2020) Shotgun metagenomics reveals an enrichment of potentially crossreactive bacterial epitopes in ankylosing spondylitis patients, as well as the effects of TNFi therapy upon microbiome composition. Ann Rheum Dis 79:132–140 [DOI] [PubMed] [Google Scholar]
- 78.Breban M, Beaufrère M, Glatigny S (2019) The microbiome in spondyloarthritis. Best Pract Res Clin Rheumatol 33:101495. [DOI] [PubMed] [Google Scholar]
- 79.Rosenbaum JT, Asquith MJ (2016) The microbiome: a revolution in treatment for rheumatic diseases? Curr Rheumatol Rep 18:62. [DOI] [PubMed] [Google Scholar]
- 80.Montoya J, Matta NB, Suchon P, Guzian MC, Lambert NC, Mattei JP, Guis S, Breban M, Roudier J, Balandraud N (2016) Patients with ankylosing spondylitis have been breast fed less often than healthy controls: a case-control retrospective study. Ann Rheum Dis 75:879–882 [DOI] [PubMed] [Google Scholar]
- 81.Ebringer R, Cooke D, Cawdell DR, Cowling P, Ebringer A (1977) Ankylosing spondylitis: klebsiella and HL-A B27. Rheumatol Rehabil 16:190–196 [DOI] [PubMed] [Google Scholar]
- 82.Seager K, Bashir HV, Geczy AF, Edmonds J, de Vere-Tyndall A (1979) Evidence for a specific B27-associated cell surface marker on lymphocytes of patients with ankylosing spondylitis. Nature 277:68–70 [DOI] [PubMed] [Google Scholar]
- 83.Ebringer A (1983) The cross-tolerance hypothesis, HLA-B27 and ankylosing spondylitis. Br J Rheumatol 22(4 Suppl 2):53–66 [DOI] [PubMed] [Google Scholar]
- 84.Schwimmbeck PL, Yu DT, Oldstone MB (1987) Autoantibodies to HLA B27 in the sera of HLA B27 patients with ankylosing spondylitis and Reiter’s syndrome. Molecular mimicry with Klebsiella pneumoniae as potential mechanism of autoimmune disease. J Exp Med 166:173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Geczy AF, Alexander K, Bashir HV, Edmonds JP (1980) Characterization of a factor(s) present in Klebsiella culture filtrates that specifically modifies an HLA-B27-associated cell-surface component. J Exp Med 152(2 Pt 2):331 s–340 s [PubMed] [Google Scholar]
- 86.Puccetti A, Dolcino M, Tinazzi E, Moretta F, D’Angelo S, Olivieri I et al. (2017) Antibodies directed against a peptide epitope of a Klebsiella pneumoniae-derived protein are present in ankylosing spondylitis. PLoS One 12:e0171073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rashid T, Wilson C, Ebringer A (2015) Raised incidence of ankylosing spondylitis among Inuit populations could be due to high HLA-B27 association and starch consumption. Rheumatol Int 35:945–951 [DOI] [PubMed] [Google Scholar]
- 88.Lindström U, Exarchou S, Lie E, Dehlin M, Forsblad-d’Elia H, Askling J et al. (2016) Childhood hospitalisation with infections and later development of ankylosing spondylitis: a national case-control study. Arthritis Res Ther 18:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McGonagle D, Stockwin L, Isaacs J, Emery P (2001) An enthesitis based model for the pathogenesis of spondyloarthropathy. additive effects of microbial adjuvant and biomechanical factors at disease sites. J Rheumatol 28:2155–2159 [PubMed] [Google Scholar]
- 90.Jacques P, Lambrecht S, Verheugen E, Pauwels E, Kollias G, Armaka M, Verhoye M, van der Linden A, Achten R, Lories RJ, Elewaut D (2014) Proof of concept: enthesitis and new bone formation in spondyloarthritis are driven by mechanical strain and stromal cells. Ann Rheum Dis 73:437–4454 [DOI] [PubMed] [Google Scholar]
- 91.Cuthbert RJ, Watad A, Evangelos M, Fragkakis EM, Dunsmuir R, Loughenbury P, Almas Khan A et al. (2019) Evidence that tissue resident human enthesis γδT-cells can produce IL-17A independently of IL-23R transcript expression. Ann Rheum Dis 78:1559–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ward MM, Reveille JD, Learch TJ, Davis JC Jr, Weisman MH (2008) Occupational physical activities and long-term functional and radiographic outcomes in patients with ankylosing spondylitis. Arthritis Rheum 59:822–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ramiro S, Landewé R, van Tubergen A, Boonen A, Stolwijk C, Dougados M, van den Bosch F, van der Heijde D (2015) Lifestyle factors may modify the effect of disease activity on radiographic progression in patients with ankylosing spondylitis: a longitudinal analysis. RMD Open 1:e000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Exarchou S, Lindstrom U, Askling J, Eriksson JK, Forsblad-d’Elia H, Neovius M et al. (2015) The prevalence of clinically diagnosed ankylosing spondylitis and its clinical manifestations: a nationwide register study. Arthritis Res Ther 17:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee W, Reveille JD, Davis JC Jr, Learch TJ, Ward MM, Weisman MH (2007) Are there gender differences in severity of ankylosing spondylitis? Results from the PSOAS cohort. Ann Rheum Dis 66:633–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang R, Gabriel SE, Ward MM (2016) Progression of nonradiographic axial spondyloarthritis to ankylosing spondylitis: a population-based cohort study. Arthritis Rheumatol 68:1415–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ortega Castro R, Font Ugalde P, Castro Villegas MC, Calvo Gutiérrez J, Muñoz Gomariz E, Zarco Montejo P, Almodóvar R, Mulero Mendoza J, Torre-Alonso JC, Gratacós Masmitjá J, Juanola Roura X, Ariza Ariza R, Fernández Dapica P, Linares Ferrando LF, Brito Brito ME, Cuende Quintana E, Vázquez Galeano C, Moreno Ramos MJ, Giménez Úbeda E, Rodríguez Lozano JC, Fernández Prada M, Queiro Silva R, Moreno Ruzafa E, Júdez Navarro E, Más AJ, Medrano le Quement C, Ornilla E, Montilla Morales C, Pujol Busquets M, Clavaguera Poch T, Fernández-Espartero MC, Carmona Ortell L, Collantes Estévez E (2013) Different clinical expression of patients with ankylosing spondylitis according to gender in relation to time since onset of disease. Data from REGISPONSER. Reumatol Clin 9:221–225 [DOI] [PubMed] [Google Scholar]
- 98.Park JS, Hong JY, Park YS, Han K, Suh SW (2018) Trends in the prevalence and incidence of ankylosing spondylitis in South Korea, 2010-2015 and estimated differences according to income status. Sci Rep 8:7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jovaní V, Blasco-Blasco M, Ruiz-Cantero MT, Pascual E (2017) Understanding how the diagnostic delay of spondyloarthritis differs between women and men: a systematic review and metaanalysis. J Rheumatol 44:174–183 [DOI] [PubMed] [Google Scholar]
- 100.Landi M, Maldonado-Ficco H, Perez-Alamino R, Citera G, Arturi P et al. (2016) Gender differences among patients with primary ankylosing spondylitis and spondylitis associated with psoriasis and inflammatory bowel disease in an iberoamerican spondyloarthritis cohort. Medicine (Baltimore) 95:e5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Webers C, Essers I, Ramiro S, Stolwijk C, Landewé R, van der Heijde D, van den Bosch F, Dougados M, van Tubergen A (2016) Gender-attributable differences in outcome of ankylosing spondylitis: long-term results from the outcome in Ankylosing Spondylitis International Study. Rheumatology (Oxford) 55:419–428 [DOI] [PubMed] [Google Scholar]
- 102.Gracey E, Yao Y, Green B, Qaiyum Z, Baglaenko Y, Lin A, Anton A, Ayearst R, Yip P, Inman RD (2016) Sexual dimorphism in the Th17 signature of ankylosing spondylitis. Arthritis Rheumatol 68:679–683 [DOI] [PubMed] [Google Scholar]
- 103.Lindström U, Forsblad-d’Elia H, Askling J, Kristensen LE, Lie E, Exarchou S et al. (2016) Perinatal characteristics, older siblings, and risk of ankylosing spondylitis: a case-control study based on national registers. Arthritis Res Ther 18:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cai G, Wang L, Fan D, Xin L, Liu L, Hu Y, Ding N, Xu S, Xia G, Jin X, Xu J, Zou Y, Pan F (2015) Vitamin D in ankylosing spondylitis: review and meta-analysis. Clin Chim Acta 438:316–322 [DOI] [PubMed] [Google Scholar]
- 105.Videm V, Cortes A, Thomas R, Brown MA (2014) Current smoking is associated with incident ankylosing spondylitis – the HUNT population-based Norwegian health study. J Rheumatol 41:2041–2048 [DOI] [PubMed] [Google Scholar]
- 106.Zhang H, Wan W, Liu J, Dai S, Zou Y, Qian Q, Ding Y, Xu X, Ji H, He H, Zhu Q, Yang C, Ye S, Jiang L, Tang J, Tong Q, He D, Zhao D, Li Y, Ma Y, Zhou J, Mei Z, Chen X, Yuan Z, Zhang J, Wang X, Yang Y, Jin L, Gao Y, Zhou X, Reveille JD, Zou H, Wang J (2018) Smoking quantity determines disease activity and function in Chinese patients with ankylosing spondylitis. Clin Rheumatol 37:1605–1616 [DOI] [PubMed] [Google Scholar]
- 107.Nikiphorou E, Ramiro S, Sepriano A, Ruyssen-Witrand A, Landewé RBM, van der Heijde D (2020) Do smoking and socioeconomic factors influence Imaging Outcomes in Axial Spondyloarthritis? Five-Year Data From the DESIR Cohort. Arthritis Rheumatol 72:1855–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee J, Luria A, Rhodes C, Raghu H, Lingampalli N, Sharpe O, Rada B, Sohn DH, Robinson WH, Sokolove J (2017) Nicotine drives neutrophil extracellular traps formation and accelerates collagen-induced arthritis. Rheumatology (Oxford) 56:644–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ciccia F, Bombardieri M, Rizzo A, Principato A, Giardina AR, Raiata F, Peralta S, Ferrante A, Drago S, Cottone M, Pitzalis C, Triolo G (2010) Over-expression of Paneth cell-derived anti-microbial peptides in the gut of patients with ankylosing spondylitis and subclinical intestinal inflammation. Rheumatology (Oxford) 49:2076–2083 [DOI] [PubMed] [Google Scholar]
- 110.Zambrano-Zaragoza JF, Agraz-Cibrian JM, González-Reyes C, Durán-Avelar MJ, Vibanco-Pérez N (2013) Ankylosing spondylitis: from cells to genes. Int J Inflam 2013:501653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Duan Z, Gui Y, Li C, Lin J, Gober HJ, Qin J, Li D, Wang L (2017) The immune dysfunction in ankylosing spondylitis patients. Biosci Trends 11:69–76 [DOI] [PubMed] [Google Scholar]
- 112.Ranganathan V, Ciccia F, Zeng F, Sari I, Guggino G, Muralitharan J, Gracey E, Haroon N (2017) Macrophage migration inhibitory factor induces inflammation and predicts spinal progression in ankylosing spondylitis. Arthritis Rheumatol 69:1796–1806 [DOI] [PubMed] [Google Scholar]
- 113.Brown MA, Kennedy LG, MacGregor AJ, Darke C, Duncan E, Shatford JL et al. (1997) Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheum 40:1823–1828 [DOI] [PubMed] [Google Scholar]
- 114.EMBL-EBI. Allele Search Tool. 2020. [cited September 2020]; Available from: https://www.ebi.ac.uk/cgi-bin/ipd/imgt/hla/allele.cgi
- 115.Martínez-González O, Cantero-Hinojosa J, Paule-Sastre P, Gómez-Magán JC, Salvatierra-Ríos D (1994) Intestinal permeability in patients with ankylosing spondylitis and their healthy relatives. Br J Rheumatol 33:644–647 [DOI] [PubMed] [Google Scholar]
- 116.Sharif K, Bridgewood C, Dubash S, McGonagle D (2020) Intestinal and enthesis innate immunity in early axial spondyloarthropathy. Rheumatology (Oxford) 59(Suppl4):iv67–iv78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mielants H, Veys EM, Cuvelier C, de Vos M (1988) Ileocolonoscopic findings in seronegative spondylarthropathies. Br J Rheumatol 27(Suppl 2):95–105 [DOI] [PubMed] [Google Scholar]
- 118.Klingberg E, Olerod G, Hammarsten O, Forsblad-d’Elia H (2016) The vitamin D status in ankylosing spondylitis in relation to intestinal inflammation, disease activity, and bone health: a cross-sectional study. Osteoporos Int 27:2027–2033 [DOI] [PubMed] [Google Scholar]
- 119.Ciccia F, Bombardieri M, Principato A, Giardina A, Tripodo C, Porcasi R, Peralta S, Franco V, Giardina E, Craxi A, Pitzalis C, Triolo G (2009) Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum 60:955–965 [DOI] [PubMed] [Google Scholar]
- 120.Warren RE, Brewerton DA (1980) Faecal carriage of klebsiella by patients with ankylosing spondylitis and rheumatoid arthritis. Ann Rheum Dis 39:37–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stebbings S, Munro K, Simon MA, Tannock G, Highton J, Harmsen H, Welling G, Seksik P, Dore J, Grame G, Tilsala-Timisjarvi A (2002) Comparison of the faecal microflora of patients with ankylosing spondylitis and controls using molecular methods of analysis. Rheumatology (Oxford) 41:1395–1401 [DOI] [PubMed] [Google Scholar]
- 122.de Vries DD, Dekker-Saeys AJ, Gyodi E, Bohm U, Ivanyi P (1992) Absence of autoantibodies to peptides shared by HLA-B27.5 and Klebsiella pneumoniae nitrogenase in serum samples from HLA-B27 positive patients with ankylosing spondylitis and Reiter’s syndrome. Ann Rheum Dis 51:783–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mäki-Ikola O, Lehtinen K, Nissilä M, Granfors K (1994) IgM, IgA and IgG class serum antibodies against Klebsiella pneumoniae and Escherichia coli lipopolysaccharides in patients with ankylosing spondylitis. Br J Rheumatol 33:1025–1029 [DOI] [PubMed] [Google Scholar]
- 124.Kijlstra A, Luyendijk L, van der Gaag R, van Kregten E, Linssen A, Willers JM (1986) IgG and IgA immune response against klebsiella in HLA-B27-associated anterior uveitis. Br J Ophthalmol 70:85–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Trapani JA, McKenzie IF (1985) Klebsiella ‘modifying factor’: binding studies with HLA-B27+ and B27− lymphocytes. Ann Rheum Dis 44:169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ngo KY, Rochu D, D’Ambrosio AM, Muller JY, Lucotte G (1984) Klebsiella plasmid K21 is not involved in the aetiology of ankylosing spondylitis. Exp Clin Immunogenet 1:140–144 [PubMed] [Google Scholar]
- 127.Stone MA, Payne U, Schentag C, Rahman P, Pacheco-Tena C, Inman RD (2004) Comparative immune responses to candidate arthritogenic bacteria do not confirm a dominant role for Klebsiella pneumonia in the pathogenesis of familial ankylosing spondylitis. Rheumatology (Oxford) 43:148–155 [DOI] [PubMed] [Google Scholar]
- 128.Sprenkels SH, Van Kregten E, Feltkamp TE (1996) IgA antibodies against Klebsiella and other Gram-negative bacteria in ankylosing spondylitis and acute anterior uveitis. Clin Rheumatol 15(Suppl 1):48–51 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.